
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA single outer-sphere amino-acid substitution turns on the NO reactivity of a hemerythrin-like protein†
Therese
Albert
 a,
Natasha
Pence
b,
Fangfang
Zhong
b,
Ekaterina V.
Pletneva
b and
Pierre
Moënne-Loccoz
*a
a,
Natasha
Pence
b,
Fangfang
Zhong
b,
Ekaterina V.
Pletneva
b and
Pierre
Moënne-Loccoz
*a
aDepartment of Chemical Physiology and Biochemistry, School of Medicine, Oregon Health & Science University, 3181 SW Sam Jackson Park Road, Portland, Oregon 97239, USA. E-mail: moennelo@ohsu.edu
bDepartment of Chemistry, Dartmouth College, Hanover, New Hampshire 03755, USA
First published on 8th January 2025
Abstract
Mycobacterial hemerythrin-like proteins (HLPs) are important for the survival of pathogens in macrophages. Their molecular mechanisms of function remain poorly defined but recent studies point to their possible role in nitric oxide (NO) scavenging. Unlike any nonheme diiron protein studied so far, the diferric HLP from Mycobacterium kansasii (Mka-HLP) reacts with NO in a multistep fashion to consume four NO molecules per diiron center. HLPs are largely conserved across mycobacteria and we argued that comparative studies of distant orthologs may illuminate the role of the protein scaffold in this reactivity and yield intermediates with properties more favorable for detailed spectroscopic characterization. Herein, we show that HLP from Azotobacter vinelandii (Avi-HLP) requires a single T47F point mutation in the outer sphere of its diferric center to adopt a bridging μ-oxo diferric structure as in Mka-HLP and makes it reactive toward NO. Radical combination of NO with the μ-oxo bridge yields nitrite and a mixed valent Fe(III)Fe(II) cluster that further react with NO to produce a stable magnetically coupled Fe(III){FeNO}7 cluster. We report characterization of this stable cluster by electronic absorption, EPR, FTIR and resonance Raman spectroscopies and suggest ways Phe 46 (Mka numbering) might control the Fe(III) reduction potential and the NO reactivity of HLPs.
1 Introduction
Mycobacterial hemerythrin-like proteins (HLPs) are known to be transcriptionally upregulated upon internalization in mammalian macrophages, but their functions remain uncertain.1–3 As their name indicates, HLPs are structurally related to hemerythrins (Hrs) with a helical-bundle fold that arranges His and Glu sidechains for the anchoring of a nonheme diiron center.4–6 Hrs use five His, two bridging Glu and a bridging solvent molecule to bind the diiron site and tightly control the coordination of exogenous ligands and confine the function of member of this family to O2 transport or O2 sensing.7 HLPs contain the same HxxxE sequence motifs as Hrs, but they lack the HxxxH motif, potentially offering open coordination sites on both iron centers for a wider range of chemical reactivity and functionality. A recent crystal structure of Mycobacterium kansasii HLP (Mka-HLP) has revealed an endogenous Tyr sidechain within the coordination sphere of its diiron cluster (Fig. 1).8 A subsequent spectroscopic characterization of diferric Mka-HLP confirmed the presence of a tyrosinate ligand and a bridging μ-oxo group.9 | ||
| Fig. 1 Overlap of the X-ray structure of Mka-HLP (PDB ID: 6Q09, green) with the structural model of Avi-HLP (brown) (left), inside view of the diiron center showing the stacking of Phe46 (green) and His71 (blue), cis to Tyr54 (red), and (top right), and schematic view of expected difference in residues in the vicinity of the diiron cluster (first coordination sphere in red, second coordination sphere in green, and outer sphere in blue) (bottom right). | ||
While carboxylate bridged diiron proteins normally adopt ferric high-spin electronic configurations that are unreactive toward nitric oxide (NO), it is not the case for Mka-HLP. The diferric protein binds NO with sub-micromolar binding affinity for NO (Fig. S1†).9 Titrating diferric Mka-HLP with NO showed the gradual accumulation of EPR features characteristic of high-spin and low-spin {FeNO}7 species as described in the Enemark and Feltham nomenclature that counts valence electrons from the metal d and NO π* orbitals.10 Starting from the diferric protein, the overall reaction requires the oxidation of two NO molecules to nitrite, allowing reduction of the iron(III) center to iron(II) to bind one NO at each metal center. These observations have led to a putative reaction mechanism that involves an initial radical attack of NO at the diferric μ-oxo group to form a mixed valent Fe(III)-nitrite-Fe(II) that reacts further with NO to consume a total of four NO molecules and leaves the diiron site magnetically uncoupled with one low-spin (LS) {FeNO}7 and one high-spin (HS) {FeNO}7 centers (Scheme 1).9 A different reaction initiation, involving a transient {FeNO}6 complex with a Fe(II)-NO+ resonance form prone to nucleophilic attack by a water to produced nitrite, was proposed by Davison, Caranto, and coworkers.11 Titrations of diferric Mka-HLP with NO only show spectroscopic signatures from the starting diferric and uncoupled {FeNO}7 pair.9 Pre-steady state studies of Mka-HLP reaction with NO using stopped-flow and rapid-freeze-quenched (RFQ) approaches were also used but failed to provide evidence for any of the intermediate species proposed in Scheme 1.
 | ||
| Scheme 1 Proposed mechanism for the NO reaction of diferric Mka-HLP.9 | ||
Because mycobacterial HLPs are highly conserved, we decided to look beyond the Mycobacteriaceae family. Herein, we report results with variants of HLP from Azotobacter vinelandii (Avi-HLP). Sequence alignments of Avi-HLP and Mka-HLP (Fig. S2†) show only 30% identity for the two proteins but predict an identical set of four His, two Glu and one Tyr coordinating residues, and conserved second-sphere residues, with the only difference of having one Asp-to-Glu and one His-to-Asp substitutions for hydrogen-bond partners to coordinating sidechains (Fig. S2† and 1).
Consistent with these alignments, our spectroscopic data show that the wild-type protein (WT-Avi-HLP) anchors an asymmetric diferric cluster with one ligating tyrosine sidechain, but in contrast to Mka-HLP, WT-Avi-HLP does not react with NO. This lack of reactivity coincides with the absence of a μ-oxo group bridging the two ferric ions. However, stabilization of a μ-oxo bridge and reactivity toward NO is gained upon a single Thr-to-Phe substitution of residue 47, corresponding to Phe46 in Mka-HLP. The X-ray structure of Mka-HLP shows Phe46 stacked against the coordinating His71 and within <4 Å from the bridging solvent molecule O444.8 The product of the NO reaction in T47F-Avi-HLP is spectroscopically characterized as a stable magnetically coupled Fe(III){FeNO}7 cluster. We compare this reactivity with the one seen in Mka-HLP and rationalize these results using reduction potentials of the diferric/mixed valent and mixed valent/diferrous pairs, and sidechain coordination malleability in diiron cluster proteins.
2 Experimental section
2.1. Primers, plasmid constructs, and site-directed mutagenesis
The sequence corresponding to the Avin_0300 gene was synthesized and cloned into the pET-20b(+) vector by GenScript with a His6-tag and TEV cleavage site added at the C-terminus. Point mutations were made using QuikChange kits (Agilent Technologies, Inc.) using primers synthesized by Invitrogen life technologies (USA) (Table S1†). Recombinant genes harboring the mutations were transformed into XL1-Blue Competent cells (Agilent Technologies, Inc). The DNA sequences of the plasmids were verified by the Vollum DNA Sequencing Core at OHSU. The plasmids were then transformed into E. coli BL21 (DE3) competent cells (Agilent Technologies. Inc.) for protein expression.2.2. Expression and purification of Avi-HLP
The WT and T47F variant were expressed in E. coli BL21(DE3) cells. Cells were grown overnight at 37 °C with continuous shaking at 250 rpm in LB medium with 100 μg mL−1 ampicillin and 100 mg L−1 ferrous ammonium sulfate before induced with 0.25 mM isopropyl β-D-1-thiogalactopyranoside. Growth was continued at 25 °C overnight before harvesting the cells at 4 °C. Cells pellets were resuspended in a 50 mM Tris, buffer at pH 7.5 containing 150 mM NaCl and 10 mM imidazole. DNase and phenylmethylsulfonyl fluoride (PMSF) protease inhibitor were added, and cells were lysed using an emulsifier (Avestin EmulsiFlex-C3, ATA Scientific Instruments). After removal of cell debris by centrifugation of the lysate, the supernatant was filtered using a 0.45 μm syringe filter and applied onto Ni-NTA column and washed with a 50 mM Tris buffer at pH 7.5 containing 150 mM NaCl and 20 mM imidazole. The Avi-HLP proteins were eluted with a buffer containing 250 mM imidazole, concentrated using 10 kDa molecular weight cutoff concentrators (Amicon), and dialyzed against a 50 mM Tris buffer at pH 7.5 containing 150 mM NaCl before storing the protein at −80 °C. The other Avi-HLP variants were produced using the same protocol, except the induction was performed at 37 °C and cells were grown for 5 h.2.3. Protein analysis
The purity of Avi-HLP WT and T47F proteins was confirmed by SDS-PAGE and total amino acid analyses (AAA Service Laboratory Inc., Damascus, Oregon). The protein concentration was determined using the total amino acid analysis data and led to a ε280 = 21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 900 M−1 cm−1 for oxidized Avi-HLP WT and T47F. The iron content was determined by ICP-OES with (PerkinElmer Optima 2000DV) and confirmed the expected stoichiometry of two irons per Avi-HLP protein. Size exclusion chromatography performed using a BioSil SEC pre-packed column (Bio-Rad) with a HPLC system (LaChrom Elite, Hitachi) showed Avi-HLP eluting as a dimer at neutral pH when using μM protein concentrations (Fig. S3†).
900 M−1 cm−1 for oxidized Avi-HLP WT and T47F. The iron content was determined by ICP-OES with (PerkinElmer Optima 2000DV) and confirmed the expected stoichiometry of two irons per Avi-HLP protein. Size exclusion chromatography performed using a BioSil SEC pre-packed column (Bio-Rad) with a HPLC system (LaChrom Elite, Hitachi) showed Avi-HLP eluting as a dimer at neutral pH when using μM protein concentrations (Fig. S3†).
2.4. Preparation of samples for spectroscopic characterization
Except for the oxidized protein, all other sample preparations were performed in an anaerobic glovebox containing <1 ppm of O2 (Omnilab System, Vacuum Atmosphere Co). The reactivities of the diferric and mixed-valent or fully reduced proteins were examined using either purified NO gases or NO donors. Diethylamine (DEA) NONOate (ε250 = 6500 M−1 cm−1) and PROLI-NONOate (ε250 = 8400 M−1 cm−1) were purchased from Cayman Chemical. Stock solutions were prepared in 10 mM NaOH and used or discarded within a few hours. NO gas (98.5%, Aldrich) was further purified by incubation in a seal vessel with 1 M NaOH solution to remove N2O3 and NO2 impurities. Isotopically labeled NO gases (15NO and 15N18O, Cambridge Isotope Laboratory) were exclusively handled inside the glovebox and used without further purification. Precise concentrations of NO solutions were determined by titration against deoxymyoglobin and monitored by electronic absorption spectroscopy.2.5. Spectroscopic measurements
All UV-vis absorption measurement were performed on Cary 50 Varian spectrophotometers using quartz cuvettes, or measuring spectra directly from Raman capillaries, FTIR films, or EPR tubes. Typical protein concentrations for resonance Raman (RR) and FTIR experiments ranged from 1 to 3 mM protein. RR, FTIR and EPR spectra were collected as previously described.92.6. Quantification of nitrite production
Nitrite concentrations were determined using the Griess assay (modified Griess reagent, Sigma-Aldrich). The entire procedure was performed inside an anerobic glovebox containing <1 ppm of O2 (Omnilab System, Vacuum Atmosphere Co), as described previously.93 Results
3.1. The magnetically coupled diferric cluster of WT-Avi-HLP includes a tyrosinate ligand but it has no μ-oxo bridge and it does not react with NO
As-isolated Avi-HLP exhibits UV-vis absorption features at 497 (ε = 2700 M−1 cm−1) and 353 nm (ε = 8800 M−1 cm−1) (Fig. 2A) and is EPR silent as expected for an oxidized diiron protein with a magnetically coupled diferric iron cluster (data not shown). The RR spectra of diferric WT-Avi-HLP obtained with a 647 nm laser excitation show prominent tyrosinate modes consistent with the coordination of Tyr55, with ring C–C stretches at 1497 and 1595 cm−1, a tyrosinate C–O stretching doublet at 1274 and 1285 cm−1, a ring C–H bending vibration at 1168 cm−1, and a 526 and 545 cm−1 doublet with Fe-OTyr stretching character (Fig. 3A). These iron(III)-tyrosinate RR frequencies are nearly identical to those observed previously with Mka-HLP.9 However, in contrast with what was seen with Mka-HLP, switching the laser excitation to 514 nm does not results in the enhancement of new RR bands that could be assigned to symmetric and asymmetric diferric μ-oxo vibration (i.e., νs(Fe–O–Fe) between 400 and 550 cm−1 and νas(Fe–O–Fe) between 700 and 850 cm−1);12 such bands are also absent of the RR spectra of diferric Avi-HLP obtained with a 407 nm excitation (Fig. 3A).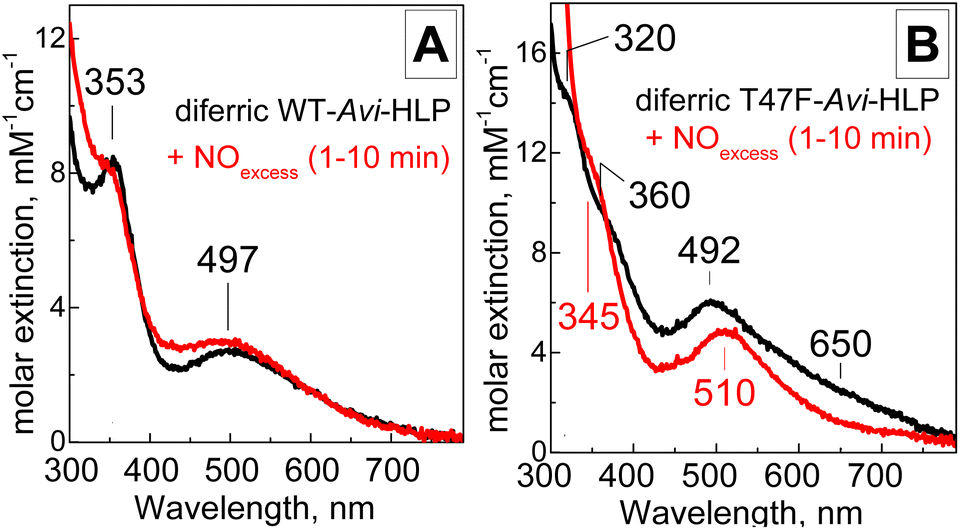 | ||
| Fig. 2 Room-temperature UV-vis spectra of diferric WT-Avi-HLP (A) and T47F-Avi-HLP (B) before (black) and after exposure to 2 mM NO (red). | ||
 | ||
| Fig. 3 Room-temperature RR spectra of diferric WT-Avi-HLP (A) and T47F-Avi-HLP (B). All RR spectra are normalized on the non-resonant Phe band at 1004 cm−1. | ||
Anaerobic exposure of diferric WT-Avi-HLP to NO, even at saturating NO concentrations, has minimal impact on its electronic absorption features (Fig. 2A) suggesting a lack of reactivity toward NO.
3.2. Substitution of the outer-sphere residue Thr47 to a Phe produces a Tyr-Fe(III)–O–Fe(III) cluster that reacts with NO
The electronic absorption spectrum of T47F-Avi-HLP is more complex than that of WT-Avi-HLP and more alike to what is observed with Mka-HLP.9 Specifically, absorption features seen as 497 and 353 nm for WT, are split in the mutant into doublets at 492 and 650 nm, and 320 and 360 nm, respectively (Fig. 2B).The RR spectra of diferric T47F-Avi-HLP obtained with a 647 nm excitation show nearly identical tyrosinate modes as seen with the WT protein, but switching the excitation to 514 nm results in the enhancement of bands at 430 and 859 cm−1 that are readily assigned to νs(Fe–O–Fe) and νas(Fe–O–Fe) modes, respectively (Fig. 3B).
Exposure of T47F-Avi-HLP to NO results in immediate changes in the electronic absorption spectra, with loss of the weak 650 nm tailing absorption feature in favor of a better defined visible band at 510 nm (Fig. 2B). Degassing NO-treated samples does not regenerate the spectrum of the diferric species, suggesting formation of a high-affinity NO adduct.
In contrast to previous findings with Mka-HLP,9 EPR spectra of NO-treated T47F-Avi-HLP did not show signals of either the HS or LS {FeNO}7 species. Instead, at all NO concentrations tested, the spectra are nearly featureless. Specifically, only a very weak and broad derivative signal around g = 1.5 is observed in the EPR spectra before being replaced by substoichiometric {FeNO}7 signals in the samples exposed to NO saturation for >1 h at room temperature before freezing (Fig. S4†). We tentatively assigned the g = 1.5 signal to an S = 1 state arising from antiferromagnetic coupling between the S = 5/2 Fe(III) and S = 3/5 {FeNO},7 by analogy with signals seen with nitrenes and other triplet species,13–15 and based on our vibrational analysis below that supports an Fe(III){FeNO}7 mixed valent cluster structure for the NO reaction product. Substoichiometric {FeNO}7 signals are likely to represent degraded diiron cluster states.
When DEA-NONOate which decays at neutral pH and room temperature to produce NO with a half-life of 15 minutes was used, FTIR spectra revealed the gradual build-up of a new absorption band at 1699 cm−1 supporting the formation of a {FeNO}7 species (Fig. 4A).16 The bandwidth of this signal at half-height is 17 cm−1, suggesting some inhomogeneous broadening. Low-temperature FTIR photolysis experiments with samples prepared with unlabeled and 15N-labeled NO gas, confirmed formation of the {FeNO}7 species. Dark minus illuminated difference spectra for product of the NO reaction show a broad positive feature centered at 1699 cm−1 which matches the ν(N–O) observed at room-temperature (Fig. 4B). This signal shifts to 1669 cm−1 with 15NO, confirming its assignment to a ν(N–O) from a {FeNO}7 species (Fig. 4B). A negative band at 1864 cm−1 that downshifts 33 cm−1 with 15NO is assigned to the photolyzed NO docked within the protein matrix. The intense differential signal centered at 1585 cm−1 and sharp positive features at 1651 and 1685 cm−1 are within the frequency range of symmetric carboxylate COO stretching frequencies and are accompanied by differential signals in the 1400 cm−1 region were symmetric COO stretches counterparts are expected (Fig. 4B). Similar differential signals seen previously in low-temperature photolysis FTIR spectra of Hrs(NO) adduct were assigned to carboxylate coordination rearrangement with confirmation by isotope sensitivity to global 13C-labeling.17 Annealing illuminated samples to 40 K confirms the reversibility of the photolytic process as identical FTIR difference spectra can be generated again at 30 K (Fig. S5†).
 | ||
| Fig. 4 Room-temperature FTIR spectra of the reaction mixture of diferric T47F-Avi-HLP with 10 mM DEA-NONOate (A) and 30 K photolysis FTIR difference spectra of the NO adducts prepared with unlabeled NO (black) and 15NO (red) (B). | ||
The product of the NO reaction was also analyzed by RR spectroscopy using a 457 nm laser excitation at room temperature. In the high-frequency region a weak signal at 1700 cm−1 downshifts to 1669 and 1636 cm−1 with 15NO and 15N18O (Fig. 5). This 1700 cm−1 frequency matches the ν(NO) mode detected at room temperature by FTIR, and its isotope sensitivity is within expectation for an N–O diatomic oscillator.
 | ||
| Fig. 5 Room-temperature RR spectra of the product of the reaction of diferric T47F-Avi-HLP with NO. The excitation wavelength used was 457 nm and the protein concentration 3 mM. | ||
Importantly, tyrosinate modes are also resonance-enhanced in the NO adduct and marginally upshifted relative to the spectra of the diferric protein, indicating that the Y55-Fe(III) ligation at the diiron cluster is retained after formation of the {FeNO}7 complex.
The low-frequency region of the RR spectra for the NO adducts shows an isotope-sensitive band at 434 cm−1 (Fig. 5), which is assigned to a vibrational mode with Fe–NO stretching and Fe–N–O bending contributions based on prior studies of {FeNO}7 species.16,18 Comparison of these RR spectra to those of the diferric protein (gray trace in Fig. 5) reveals a complete loss of the νs(Fe–O–Fe) and νas(Fe–O–Fe) modes upon formation of the {FeNO}7 complex, confirming full conversion of the diferric cluster into what must be a mixed valent Fe(III){FeNO}7 cluster.
3.3. The T47F substitution in Avi-HLP stabilizes the mixed valent state relative to the oxidized and fully reduced states
After exposure to 10 mM dithionite for 2 h, the electronic absorption spectrum of WT-Avi-HLP is featureless in the visible region, suggesting full reduction of the diferric Fe(III)Fe(III) to the diferrous Fe(II)Fe(II) state (Fig. S6†). In contrast, anaerobic incubation of diferric WT-Avi-HLP with 10 mM ascorbate minimally impacts the electronic absorption spectra (Fig. S6†). The EPR analysis of these WT-Avi-HLP samples are nearly featureless with only trace population of mixed valent signals with characteristic resonances below g ∼ 2 (data not shown).Unlike the WT protein, T47F-Avi-HLP shows high accumulation of a Fe(III)Fe(II) mixed valent state under either of these conditions. The EPR signal is rhombic with g1 = 1.97, g2 = 1.94, and g3 = 1.69 (Fig. 6), very similar to the EPR signal seen with semi-methemerythrin,19 and with no evidence of heterogeneity, in contrast to partly-reduced Mka-HLP for which at least two sets of mixed-valent species were observed.9 Full reduction of T47F-Avi-HLP can be achieved after exposure for 2 h to 10 mM dithionite and 0.2 mM methyl viologen (Fig. S7†).
 | ||
| Fig. 6 EPR spectra of diferric T47F-Avi-HLP before (bottom trace) and after 2 h incubation with 10 mM dithionite (top trace), 2 h incubation with 10 mM ascorbate (second trace from the top) and 10 mM dithionite with 0.2 mM methyl viologen (third trace from the top). Experimental conditions: temperature, 5 K: microwave power, 0.2 mW; microwave frequency, 9.45 GHz; modulation frequency, 100 kHz; modulation amplitude, 10 G. | ||
Exposing the mixed-valent state of T47F-Avi-HLP to NO results in the appearance of the 510 nm absorption band as seen with the ferric protein with NO although it is a less stable complex as seen with the diferric protein (Fig. S8†). Titrating the mixed-valent state with NO shows the loss of its EPR signal at g < 2 concomitant with the appearance of the very weak and broad g ∼ 1.5 signal assigned to the Fe(III){FeNO}7 adduct (Fig. S9†). As with the diferric state, further exposure to excess NO leads to the appearance of substoichiometric {FeNO}7 signals at g ∼ 4 and g ∼ 2 (Fig. S10†). The RR and low-temperature FTIR photolysis spectra of the mixed-valent NO adduct are also similar to those obtained for the NO-treated diferric protein (Fig. S10 and S11†).
Filtrates of diferric and mixed valent proteins exposed to 1 atm NO for a few minutes before releasing the excess NO were analyzed for nitrite content using the Griess assay. While diferric T47F-Avi-HLP produced 1.1 equivalent of detectable nitrite, the mixed-valent protein produced less than 0.15 equivalent nitrite, within the range measured with negative controls.
 | ||
| Fig. 7 Room-temperature RR spectra of diferric WT-Avi-HLP (bottom trace) and T47 variants obtained with a 514 nm excitation. All RR spectra are normalized on the non-resonant Phe band at 1004 cm−1. | ||
The electronic absorption spectra of the T47V, T47L, T47W and T47Y diferric variants suggest that they are reactive toward NO, but, in contrast to T47F-Avi-HLP, they do not accumulate the 510 nm species corresponding to the mixed-valent Fe(III){FeNO}7 species (Fig. S14 and S15†).
4 Discussion
Sequence alignments of Mka-HLP and Avi-HLP predict conservation of the four His, two Glu, and one Tyr in the first coordination sphere of the diiron clusters. Structural modeling suggests that the hydrogen-bond partners to the sidechains of iron ligands which comprise the second coordination sphere are also conserved (Fig. S2†). The most noticeable difference in the vicinity of the active site is the substitution of residue Phe46 in Mka-HLP to Thr47 in Avi-HLP. In the X-ray structure of Mka-HLP, the phenyl ring of Phe46 points toward the diferric μ-oxo bridge and stacks against the imidazole ring of the Fe2 ligand His71.8 Models of Avi-HLP position the hydroxy group of Thr47 away from the core of the helical bundle, toward the protein surface. Our spectroscopic analysis shows that Avi-HLP anchors a magnetically coupled diferric cluster with one tyrosinate ligand in both WT and Thr47 variants. However, our data reveal that a bulky hydrophobic residue is required at residue 47 to stabilize a bridging μ-oxo group as seen in Mka-HLP. In WT Avi-HLP and the T47S and T47A variants, the diferric cluster is magnetically coupled but the bridge is protonated, either to the μ-hydroxo or μ-aqua forms.To our knowledge, our study represents the first example of a point mutation controlling the protonation state of a diferric bridging solvent molecule in a metalloprotein. How this control is executed, however, is not readily apparent. An increase in local hydrophobicity may favor a μ-oxo versus μ-hydroxo form. Steric interactions with His71 may also alter the coordination geometry around the Fe2(III) and its Lewis acidity to change the pKa of the bridge solvent molecule. Steric clashes could also affect Fe–O–Fe bond distance and bond angles. To gain further insights on this issue, attempts were made to generate F46 variants of Mka-HLP, but so far they have not produced stable holoproteins.
In contrast to Mka-HLP, diferric WT-Avi-HLP is unreactive toward NO, but the stabilization of a μ-oxo bridge in the T47F variant coincides with a gain in NO reactivity. These results are consistent with an initiation of the NO reaction through radical attack by NO to form nitrite and a mixed-valent Fe(II)Fe(III) cluster (Schemes 1 and 2). Such radical combination between NO and a μ-oxo group was originally proposed by Blumberg and Siegbahn in a theoretical study of the substrate inhibition reaction of cytochrome-c dependent denitrifying NO reductases.20 Build-up of radical character at the μ-oxo group bridging the heme-nonheme diferric cluster in the oxidized enzyme promotes radical attack by NO, leading to an inhibitory Fe(III)-nitrito-Fe(II) complex. A similar reactivity toward NO has also been observed in μ-oxo bridged heme-Fe(III)-O-Cu(II) model complexes.21 Recent experimental work with two denitrifying NO reductases brought further support to this inhibition mechanism.22
 | ||
| Scheme 2 Proposed mechanism for the NO reaction of diferric T47F-Avi-HLP. | ||
The initial NO attack of the diferric HLP proteins converts the μ-oxo group to a nitrito group, presumably reducing the Fe1(III) site since its reduction potential is expected to be higher than that of the tyrosinate-ligated Fe2(III) center.23,24 Upon reduction, the Fe1(II) is expected to adopt a HS configuration with an open coordination site and to present a high affinity site for NO binding, yielding the Fe2(III){Fe1NO}7 species in T47F-Avi-HLP we report here (Scheme 2).
Analysis of filtrates of this reaction with the Griess assay show that the nitrite group is released to the aqueous solution rather than remaining bound to the diiron cluster. Since the two iron centers remain magnetically coupled after the NO reaction, it is likely that the nitrito group is displaced by a solvent molecule to form a bridging μ-hydroxo group (Scheme 2). Such nitrite-to-hydroxide ligand substitution does not change the overall charge of the diiron cluster and can be expected to be favored at low nitrite concentrations.25,26
The T47F-Avi-HLP(NO) adduct reported here shows a ν(N–O) at 1699 cm−1 which is at the lower end of the frequency range typically seen for nonheme {FeNO}7 species. Synthetic model with strongly σ-donating ancillary ligand, including phenolate ligands, show similarly low ν(N–O) frequencies.27–30 Thus, this low ν(N–O) frequency could suggest NO binding at the Tyr-ligated Fe2 site. However, low ν(N–O) frequencies have also been reported for mononitrosyl adducts of Hrs and flavodiiron proteins, and assigned to semi-bridging NO-binding geometries.17 Perturbations of νas(COO) and νs(COO) frequencies from bridging carboxylates at the diiron clusters upon NO photodissociation are also comparable in the Fe2(III){Fe1NO}7 complex proposed for T47F-Avi-HLP(NO) and in the previously characterized Fe(II){FeNO}7 species in Hr(NO) and flavodiiron(NO).17
For diferric Mka-HLP, the Fe2(III){Fe1NO}7 species is not observed. Instead, two additional NO molecules are consumed to reduce the Fe2(III) center and release of a second nitrite ion, forming a second {FeNO}7 species. This latter {Fe2NO}7 species is readily observed along the {Fe1NO}7 species in the EPR spectra, implying a sufficiently large Fe–Fe distance to prevent strong dipole coupling between the two {FeNO}7 species.9 In contrast to Mka-HLP, the Fe2(III){Fe1NO}7 state observed in T47F-Avi-HLP reacts further with NO, only after prolonged exposure to saturating NO concentrations. This stabilization of the Fe2(III){Fe1NO}7 state is likely to reflect a lower reduction potential of the Fe2(III) center and a lower propensity to increase the Fe–Fe distance in T47F-Avi-HLP relative to that in Mka-HLP. Chemical reduction of the diferric proteins supports this hypothesis: reduction of the Fe2(III) center in T47F-Avi-HLP requires a stronger reductant than in Mka-HLP and it is reasonable to expect similar trends for the reduction of the Fe2(III){Fe1NO}7 species. The RR spectra of the tyrosinate-Fe2(III) chromophore in T47F-Avi-HLP and Mka-HLP do not support differences in OTyr-Fe2(III) bond strength or bond distance, which leaves the His71-Fe2(III) interaction as the likely modulator of the Fe2 reduction potential and of the malleability of the diiron cluster.
5 Conclusions
In conclusion, the T47F-Avi-HLP variant reproduces the early steps of the reaction of WT Mka-HLP with NO, correlating the presence of a μ-oxo bridge at the diferric cluster with NO reactivity. Subsequent reaction steps observed in Mka-HLP corresponding to the reduction of the Fe2 site by NO to form a {Fe2NO}7 species do not occur in T47F-Avi-HLP. Steric interactions between Tyr54, His71, and Phe46 around the Fe2 site of Mka-HLP may contribute to this extended reactivity to NO. Analyses of other substitutions around the diiron site of Avi-HLP and of other HLP orthologs are underway to decipher structural properties controlling the uncoupling of the diiron centers and the formation of the HS and LS {FeNO}7 species first reported in mycobacterial HLPs.Data availability
All the experimental data are available in the ESI.†Author contributions
TH: conceptualization, formal analysis, data curation, writing – original draft preparation, writing – review & editing; NP: methodology, formal analysis, data curation; FZ: methodology, data curation; EVP: conceptualization, funding acquisition, writing – review & editing; PML: conceptualization, funding acquisition, writing – original draft preparation, writing – review & editing, supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by National Institute of Health through the grant GM147588 (P.M.L.) and Dartmouth College (Senior Faculty Grant to E. V.P). The authors thank Prof. Joshua Telser (Roosevelt University) for helpful discussions.Notes and references
- K. H. Rohde, R. B. Abramovitch and D. G. Russell, Mycobacterium tuberculosis invasion of macrophages: linking bacterial gene expression to environmental cues, Cell Host Microbe, 2007, 2(5), 352–364, DOI:10.1016/j.chom.2007.09.006.
- S. Homolka, S. Niemann, D. G. Russell and K. H. Rohde, Functional genetic diversity among Mycobacterium tuberculosis complex clinical isolates: delineation of conserved core and lineage-specific transcriptomes during intracellular survival, PLoS Pathog., 2010, 6(7), e1000988 CrossRef PubMed.
- Z. Ma, K. T. Strickland, M. D. Cherne, E. Sehanobish, K. H. Rohde, W. T. Self and V. L. Davidson, The Rv2633c protein of Mycobacterium tuberculosis is a non-heme di-iron catalase with a possible role in defenses against oxidative stress, J. Biol. Chem., 2018, 293(5), 1590–1595 CrossRef CAS PubMed.
- C. E. French, J. M. Bell and F. B. Ward, Diversity and distribution of hemerythrin-like proteins in prokaryotes, FEMS Microbiol. Lett., 2008, 279(2), 131–145 CrossRef CAS PubMed.
- C. Alvarez-Carreno, V. Alva, A. Becerra and A. Lazcano, Structure, function and evolution of the hemerythrin-like domain superfamily, Protein Sci., 2018, 27(4), 848–860 CrossRef CAS PubMed.
- Z. Ma, M. L. Caldas Nogueira, D. P. Marchi-Salvador and V. L. Davidson, Correlation of conservation of sequence and structures of mycobacterial hemerythrin-like proteins with evolutionary relationship and host pathogenicity, ACS Omega, 2020, 5(36), 23385–23392 CrossRef CAS PubMed.
- R. E. Stenkamp, Dioxygen and hemerythrin, Chem. Rev., 1994, 94, 715–726 CrossRef CAS.
- Z. Ma, J. Abendroth, G. W. Buchko, K. H. Rohde and V. L. Davidson, Crystal structure of a hemerythrin-like protein from Mycobacterium kansasii and homology model of the orthologous Rv2633c protein of M. tuberculosis, Biochem. J., 2020, 477(2), 567–581 CrossRef CAS PubMed.
- T. Albert and P. Moënne-Loccoz, Spectroscopic characterization of a diferric mycobacterial hemerythrin-like protein with unprecedented reactivity toward nitric oxide, J. Am. Chem. Soc., 2022, 144(38), 17611–17621 CrossRef CAS PubMed.
- J. H. Enemark and R. D. Feltham, Principles of structure, bonding, and reactivity for metal nitrosyl complexes, Coord. Chem. Rev., 1974, 13, 339–406 CrossRef CAS.
- Z. Ma, A. A. Holland, I. Szlamkowicz, V. Anagnostopoulos, M. L. Caldas Nogueira, J. D. Caranto and V. L. Davidson, The hemerythrin-like diiron protein from Mycobacterium kansasii is a nitric oxide peroxidase, J. Biol. Chem., 2022, 298(3), 101696 CrossRef CAS PubMed.
- J. Sanders-Loehr, W. D. Wheeler, A. K. Shiemke, B. A. Averill and T. M. Loehr, Electronic and Raman spectroscopic properties of oxo-bridged dinuclear iron centers in proteins and model compounds, J. Am. Chem. Soc., 1989, 111(21), 8084–8093 CrossRef CAS.
- G. Smolinsky, E. Wasserman and W. A. Yager, The E.P.R. of ground state triplet nitrenes, J. Am. Chem. Soc., 1962, 84(16), 3220–3221 CrossRef CAS.
- A. V. Akimov, Y. S. Ganushevich, D. V. Korchagin, V. A. Miluykov and E. Y. Misochko, The EPR spectrum of triplet mesitylphosphinidene: reassignment and new assignment, Angew Chem. Int. Ed. Engl., 2017, 56(27), 7944–7947 CrossRef CAS PubMed.
- D. W. N. Wilson, M. P. Franco, W. K. Myers, J. E. McGrady and J. M. Goicoechea, Base induced isomerisation of a phosphaethynolato-borane: mechanistic insights into boryl migration and decarbonylation to afford a triplet phosphinidene, Chem. Sci., 2020, 11(3), 862–869 RSC.
- N. Lehnert, E. Kim, H. T. Dong, J. B. Harland, A. P. Hunt, E. C. Manickas, K. M. Oakley, J. Pham, G. C. Reed and V. S. Alfaro, The biologically relevant coordination chemistry of iron and nitric oxide: electronic structure and reactivity, Chem. Rev., 2021, 121(24), 14682–14905 CrossRef CAS PubMed.
- T. Hayashi, J. D. Caranto, H. Matsumura, D. M. Kurtz Jr. and P. Moënne-Loccoz, Vibrational analysis of mononitrosyl complexes in hemerythrin and flavodiiron proteins: relevance to detoxifying NO reductase, J. Am. Chem. Soc., 2012, 134(15), 6878–6884 CrossRef CAS PubMed.
- S. Lu, E. Libby, L. Saleh, G. Xing, J. M. Bollinger Jr. and P. Moënne-Loccoz, Characterization of NO adducts of the diiron center in protein R2 of Escherichia coli ribonucleotide reductase and site-directed variants. Implications for the O2-activation mechanism, J. Biol. Inorg. Chem., 2004, 9, 818–827 CrossRef CAS PubMed.
- J. M. Nocek, D. M. Kurtz Jr., R. A. Pickering and M. P. Doyle, Oxidation of deoxyhemerythrin to semi-methemerythrin by nitrite, J. Biol. Chem., 1984, 259(20), 12334–12338 CrossRef CAS PubMed.
- M. R. Blomberg and P. E. M. Siegbahn, Mechanism for N2O generation in bacterial nitric oxide reductase: a quantum chemical study, Biochemistry, 2012, 51(25), 5173–5186 CrossRef CAS PubMed.
- S. Hematian, I. Kenkel, T. E. Shubina, M. Durr, J. J. Liu, M. A. Siegler, I. Ivanovic-Burmazovic and K. D. Karlin, Nitrogen oxide atom-transfer redox chemistry; mechanism of NO(g) to nitrite conversion utilizing mu-oxo heme-Fe(III)-O-Cu(II)(L) constructs, J. Am. Chem. Soc., 2015, 137(20), 6602–6615 CrossRef CAS PubMed.
- H. Matsumura, A. S. Faponle, P. L. Hagedoorn, T. Tosha, S. P. de Visser and P. Moënne-Loccoz, Mechanism of substrate inhibition in cytochrome-c dependent NO reductases from denitrifying bacteria (cNORs), J. Inorg. Biochem., 2022, 231, 111781 CrossRef CAS PubMed.
- C. H. Taboy, K. G. Vaughan, T. A. Mietzner, P. Aisen and A. L. Crumbliss, Fe3+ coordination and redox properties of a bacterial transferrin, J. Biol. Chem., 2001, 276(4), 2719–2724 CrossRef CAS PubMed.
- D. P. Hildebrand, D. L. Burk, R. Maurus, J. C. Ferrer, G. D. Brayer and A. G. Mauk, The proximal ligand variant His93Tyr of horse heart myoglobin, Biochemistry, 1995, 34(6), 1997–2005 CrossRef CAS PubMed.
- M. H. Baik, M. Newcomb, R. A. Friesner and S. J. Lippard, Mechanistic studies on the hydroxylation of methane by methane monooxygenase, Chem. Rev., 2003, 103(6), 2385–2419 CrossRef CAS PubMed.
- E. S. Traore and A. Liu, Charge maintenance during catalysis in nonheme iron oxygenases, ACS Catal., 2022, 12(10), 6191–6208 CrossRef CAS PubMed.
- C. A. Brown, M. A. Pavlosky, T. E. Westre, Y. Zhang, B. Hedman, K. O. Hodgson and E. I. Solomon, Spectroscopic and theoretical description of the electronic structure of S=3/2 iron-nitrosyl complexes and their relation to O2 activation by nonheme iron enzyme active sites, J. Am. Chem. Soc., 1995, 117(2), 715–732 CrossRef CAS.
- G. Villar-Acevedo, E. Nam, S. Fitch, J. Benedict, J. Freudenthal, W. Kaminsky and J. A. Kovacs, Influence of thiolate ligands on reductive N-O bond activation. Probing the O2 binding site of a biomimetic superoxide reductase analogue and examining the proton-dependent reduction of nitrite, J. Am. Chem. Soc., 2011, 133(5), 1419–1427 CrossRef CAS PubMed.
- A. Dey, J. B. Gordon, T. Albert, S. Sabuncu, M. A. Siegler, S. N. MacMillan, K. M. Lancaster, P. Moënne-Loccoz and D. P. Goldberg, A nonheme mononuclear {FeNO}7 complex that produces N2O in the absence of an exogenous reductant, Angew. Chem., Int. Ed., 2021, 60(39), 21558–21564 CrossRef CAS PubMed.
- H. T. Dong, S. Camarena, D. Sil, M. O. Lengel, J. Zhao, M. Y. Hu, E. E. Alp, C. Krebs and N. Lehnert, What is the right level of activation of a high-spin {FeNO}7 complex to enable direct N-N coupling? Mechanistic insight into flavodiiron NO reductases, J. Am. Chem. Soc., 2022, 144(36), 16395–16409 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc07529c |
| This journal is © The Royal Society of Chemistry 2025 |