
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA perspective on field-effect in energy and environmental catalysis
HuangJingWei
Li
ac,
Hongmei
Li
a,
Mengzhen
Du
bd,
Erjun
Zhou
 b,
Wan Ru
Leow
c and
Min
Liu
*a
b,
Wan Ru
Leow
c and
Min
Liu
*a
aHunan Joint International Research Center for Carbon Dioxide Resource Utilization, State Key Laboratory of Powder Metallurgy, School of Physics, Central South University, Changsha 410083, P. R. China. E-mail: minliu@csu.edu.cn
bCollege of Biological, Chemical Sciences and Engineering, Jiaxing University, Jiaxing, Zhejiang 314001, P. R. China
cInstitute of Sustainability for Chemicals, Energy and Environment (ISCE2), Agency for Science, Technology and Research (A*STAR), Singapore 627833, Singapore
dCollege of Chemical and Materials Engineering, Xuchang University, Xuchang, Henan 461000, P. R. China
First published on 18th December 2024
Abstract
The development of catalytic technologies for sustainable energy conversion is a critical step toward addressing fossil fuel depletion and associated environmental challenges. High-efficiency catalysts are fundamental to advancing these technologies. Recently, field-effect facilitated catalytic processes have emerged as a promising approach in energy and environmental applications, including water splitting, CO2 reduction, nitrogen reduction, organic electrosynthesis, and biomass recycling. Field-effect catalysis offers multiple advantages, such as enhancing localized reactant concentration, facilitating mass transfer, improving reactant adsorption, modifying electronic excitation and work functions, and enabling efficient charge transfer and separation. This review begins by defining and classifying field effects in catalysis, followed by an in-depth discussion on their roles and potential to guide further exploration of field-effect catalysis. To elucidate the theory-structure–activity relationship, we explore corresponding reaction mechanisms, modification strategies, and catalytic properties, highlighting their relevance to sustainable energy and environmental catalysis applications. Lastly, we offer perspectives on potential challenges that field-effect catalysis may face, aiming to provide a comprehensive understanding and future direction for this emerging area.
 HuangJingWei Li | Dr HuangJingWei Li received his MS degree from Xiangtan University in 2018 and then received his PhD degree from Central South University in 2022 under the supervision of Prof. Min Liu. In 2022–2024, he joined the Huazhong University of Science and Technology as a postdoc supervised by Prof. Tianyou Zhai and Prof. Youwen Liu. After that, he joined the A*STAR as a scientist under the guidance of Prof. Wan Ru Leow. His current research focuses on the photo/electrocatalytic conversion of organic molecules, especially the selective electrooxidation. |
 Min Liu | Prof. Min Liu received the BS (2004) and MS (2007) degrees from Hunan Normal University and PhD (2010) degree from the Institute of Electrical Engineering, Chinese Academy of Sciences. In 2010–2015, he joined the University of Tokyo with the guidance of Prof. Kazuhito Hashimoto and Prof.Kazunari Domen. In 2015–2017, he joined the University of Toronto as a postdoctoral fellow under the guidance of Prof. Edward Sargent. Starting from 2017, he is a professor in Central South University. His research interests focus on greenhouse gas reduction, include the resource utilization of perfluorocarbons and photo/electrocatalytic CO2 reduction. |
1. Introduction
Renewable energy conversion is widely recognized as the most promising route for achieving a sustainable society, which could address the issues of energy scarcity and environmental pollution resulting from excessive dependence on fossil fuels.1–4 In general, designing catalysts that are robust, efficient, and economical is essential to overcoming activation barriers and obtaining optimum performance.Recently, the field-effect has played a crucial role in catalytic reactions, which can significantly alter the processes of mass transfer, adsorption, and activation of reactants through the action of internal and external fields.5–9 Internal field effects are usually generated by the structural properties of the catalyst itself. For example, the tip geometry of catalysts can create a localized electric field on the surface, promoting an increase in the concentration of K+ ions on the surface, which facilitates the polarisation of carbon dioxide molecules and thus enhances the performance of the carbon dioxide reduction reaction (CO2RR).10 In addition to the catalyst, external fields such as light, magnetism, electricity, and strain are utilized. The external excitation effectively accelerated the charge and mass transfer, thus improving the reaction kinetics and thermodynamics. Such as the proton coupling in the OER process can be enhanced by applying an external electric field, which improves the reaction efficiency by changing the energy distribution and charge transfer.11 Furthermore, by loading Au particles on the Ni(OH)2 surface, Ye et al. exploited the localized plasma surface resonance (LPSR) effect of an external light field to promote the generation of more hot electrons,12 thereby enhancing the oxygen evolution reaction (OER) activity.
These research advances have demonstrated the increasing application of field-effect in electrocatalytic reactions covering a wide range of areas from water splitting,13–16 CO2RR,17–23 nitrogen reduction reaction (NRR),24–27 organic electrosynthesis,28–32 and biomass recycling.33–38 Such research has revealed that the performance of catalysts can be significantly enhanced by precise control of internal and external fields, providing new strategies for energy conversion and environmental applications. Therefore, writing a review to summarise the role and applications of field-effect in catalytic reactions will help to promote further developments in this area.
In this review, we will focus on summarising the definition, role, and corresponding advances in field-effect and perspectives for the future (Fig. 1). Firstly, the definition of field-effect will be discussed, including both physical and chemical perspectives, as well as the internal field and external field classification of field-effect. Subsequently, the role of field-effect will be explained, including influencing local concentration and mass transfer, adsorption and activation of reactants, charge excitation, and charge separation. Next, we present the progress of individual reactions such as water splitting, CO2RR, NRR, organic electrosynthesis, and biomass recycling, all of which are enhanced by field-effect. Finally, the current status of the field-effect is summarised and future directions are discussed. This review will further draw attention to the field-effect and its role, and provide valuable insights into the rational design of internal field catalysts and external field excitations that utilize the field-effect.
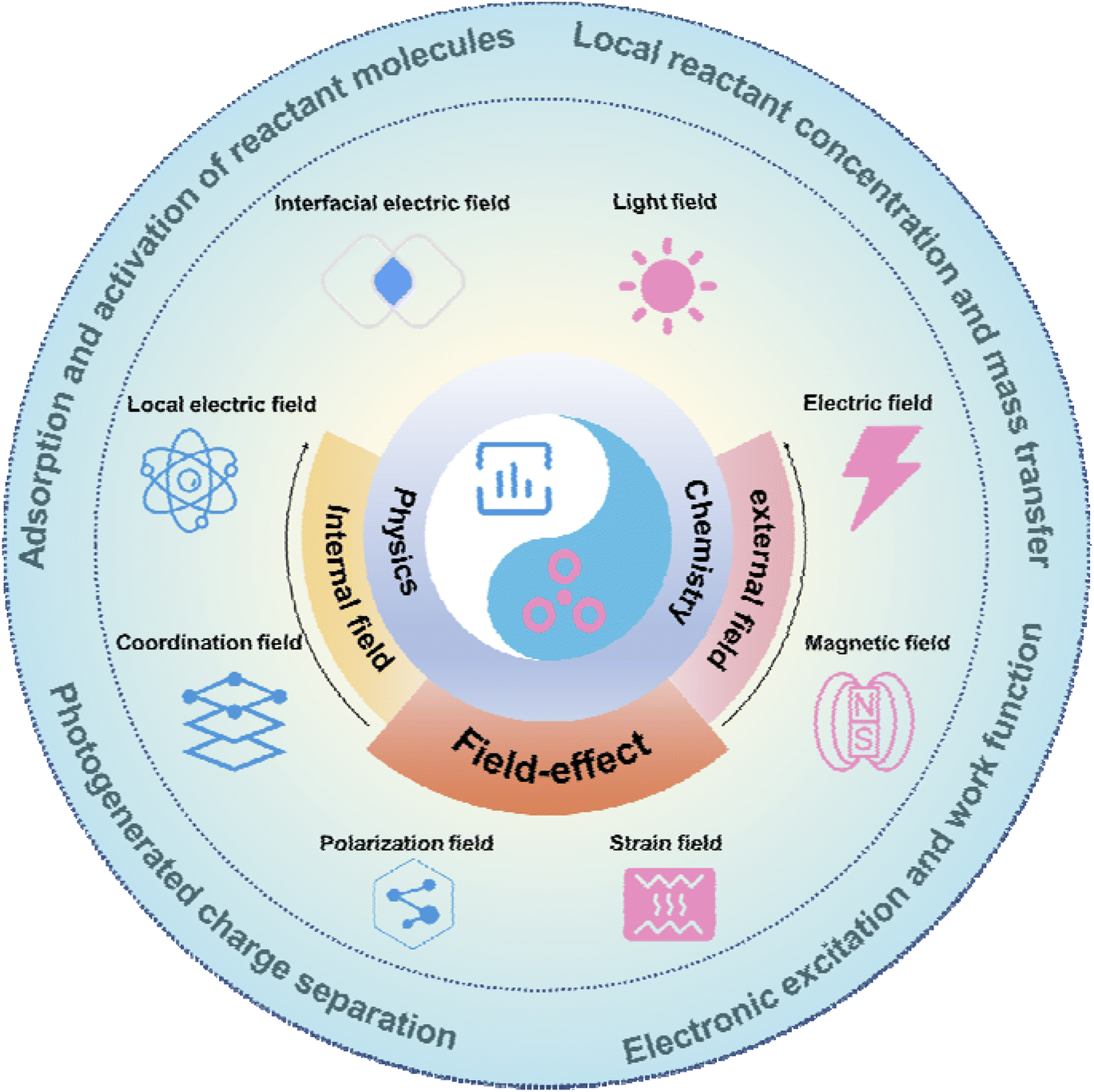 | ||
| Fig. 1 Schematic illustration of the contents of this review. | ||
2. Definition and classification of the field-effect
In physics, the term “field” refers to the spatial distribution of an object, and the field-effect arises from the influence of electric fields on semiconductors.39–41 The equilibrium of free electrons and holes in semiconductors is disrupted by the presence of an electric field. For example, in metal–oxide–semiconductor structures, different electric fields cause the attraction or repulsion of carriers near the semiconductor surface, leading to the accumulation or depletion of carriers. This vertical modulation of the electric field affecting semiconductor surface conductance is known as the field-effect.42–44 In chemistry, the field-effect involves intramolecular electrostatic interaction through space, where a substituent group generates an electric field that impacts a reaction center at another location.45,46 Overall, the field-effect provides a strategy for modulating material properties and reaction processes.At the microscopic level, the field-effect can lead to the directional arrangement of the internal charge of the substance, electronic excitation, and molecular polarization. Meanwhile, at the macroscopic level, the field-effect can affect the external reactant transport, adsorption, and activation.
2.1 Internal field
The field generated inside an object is uniformly referred to as an internal field. Such as the two-phase interfacial electric field can be derived from the formation of heterojunctions; the local electric field can be constructed through tip morphology optimization, elemental doping, and defect engineering; the coordination field can be modulated by incorporating electron-donating and electron-accepting groups/molecules; and the polarization field can be harnessed by leveraging the intrinsic polarization characteristics of the catalyst. In short, internal spatial interactions are utilized to alter the performance of the catalyst or catalytic reaction. Therefore, the aforementioned entities can be categorized as the interfacial electric field, local electric field, coordination field, and polarization field.For the measurement of the interface electric field, Zhu et al. has proposed that it can be measured from both theoretical and experimental perspectives: theoretically, the built-in electric field can be calculated through density functional theory (DFT); experimentally, the surface potential can be tested using Kelvin Probe Force Microscopy (KPFM) and zeta potential measurements (Fig. 2a–c).50 Furthermore, Li's group has developed a strategy to regulate the catalyst's surface/interface electric field by constructing heterojunctions. They have studied the impact of heterojunctions on the surface electric field and demonstrated the dependence of the surface electric field on the quantity and size of the internal heterojunction rectification interfaces. Increasing the size of metal nanoparticles within the range of 1–50 nanometers can significantly expand the area of influence of the built-in electric field, and further enlargement of the metal size will not further extend the range of influence.51,54 On the other hand, experimental evidence has shown that constructing more heterojunction structures within the catalyst can significantly enhance the surface electric field of the catalyst. Taking the nitrate reduction reaction as an example,51 the research team further explored the impact of the catalyst surface electric field during the catalytic process. By regulating the electric field strength and charge properties on the surface of the catalyst, the adsorption of substrate ions on the surface of the catalyst can be significantly enhanced (Fig. 2d). In addition, both experimental data and simulation results together indicate that the surface electric field of the catalyst plays an important role in reducing the reaction activation energy and promoting product desorption among other aspects (Fig. 2e). Constructing heterojunctions to regulate the surface electric field of the catalyst is very important and universal for improving the catalytic activity of the catalyst.
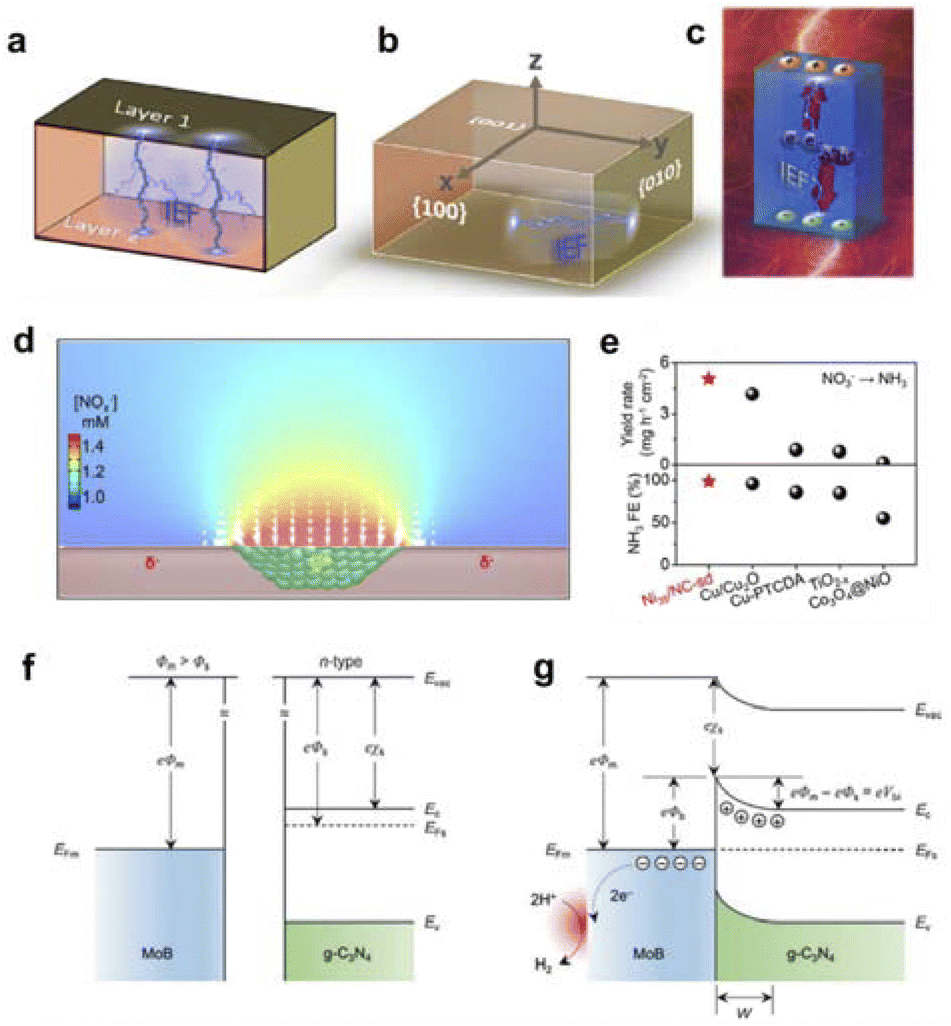 | ||
| Fig. 2 (a) IEF generation by orderly arranged unit cells. (b) IEF generation between different crystal faces within the unit cell. (c) IEF generation by photocatalysts induced by the external environment, where IEF refers to the internal electric field. Reproduced with permission from ref. 50 Copyright 2019 Wiley. (d) Finite element simulation of Schottky junction electric field enhancement of nitrate ion concentration. (e) Comparative plot of Schottky junction catalysts used for nitrate reduction performance. Reproduced with permission from ref. 51 Copyright 2021 Wiley. (f) Energy band structure of MoB and g-C3N4 before contacting. (g) Formation of the interfacial electric field after MoB/g-C3N4 contacting. Reproduced with permission from ref. 52 Copyright 2018 Wiley. | ||
Guo's group also constructed a MoB/g-C3N4 catalyst with the interfacial electric field based on the metal–semiconductor Schottky junction principle for hydrogen evolution reaction (HER).52 Through a series of spectroscopic tests, it was found that the introduction of the n-type semiconductor g-C3N4 resulted in an obvious charge redistribution at the MoB/g-C3N4 interface, increasing the local electron density on the MoB surface (Fig. 2f and g). The Schottky catalyst exhibited good HER activity with a Tafel slope of 46 mV dec−1 and an exchange current density of 17 μA cm−2, which was much higher than that of MoB. The first-principles calculations showed that the interfacial electric field could significantly promote the adsorption and activation of protons on the MoB surface, which was beneficial to the generation of surface hydrogen.
In photocatalysis, the built-in interface electric field can also be generated by constructing heterojunctions, which promotes the activity and selectivity of catalytic reactions. Yu's group utilized electrostatic self-assembly to form a Type II heterojunction of g-C3N4/ZnO, using the built-in electric field to suppress the recombination of photogenerated electron–hole pairs and promote the reduction of CO2 to methanol.56 The construction of heterojunctions can absorb the inherent characteristics of both substances, achieving complementary advantages. The built-in electric field formed at the interface between the two substances enhances light absorption and the separation of photogenerated carriers, thereby improving catalytic activity. This contributes to expanding the application of interface electric fields in energy conversion, improving catalytic efficiency, and reducing energy consumption.
Our group, guided by the mechanism of field-induced reactant concentration, has developed an electro-deposition synthesis method for metal nanotips. Under the action of the tip discharge mechanism, an ultra-low overpotential can be applied to form a localized electric field at the tip of the nanotip (Fig. 3a and b). This causes cations in the electrolyte to enrich at the tip. The tip electric field-induced K+ aggregation effect can increase the local CO2 concentration near the catalytic active sites, making CO2 more easily adsorbed and activated, accelerating the kinetics of CO2 reduction, and ultimately enhancing the electrochemical reduction activity of CO2.10
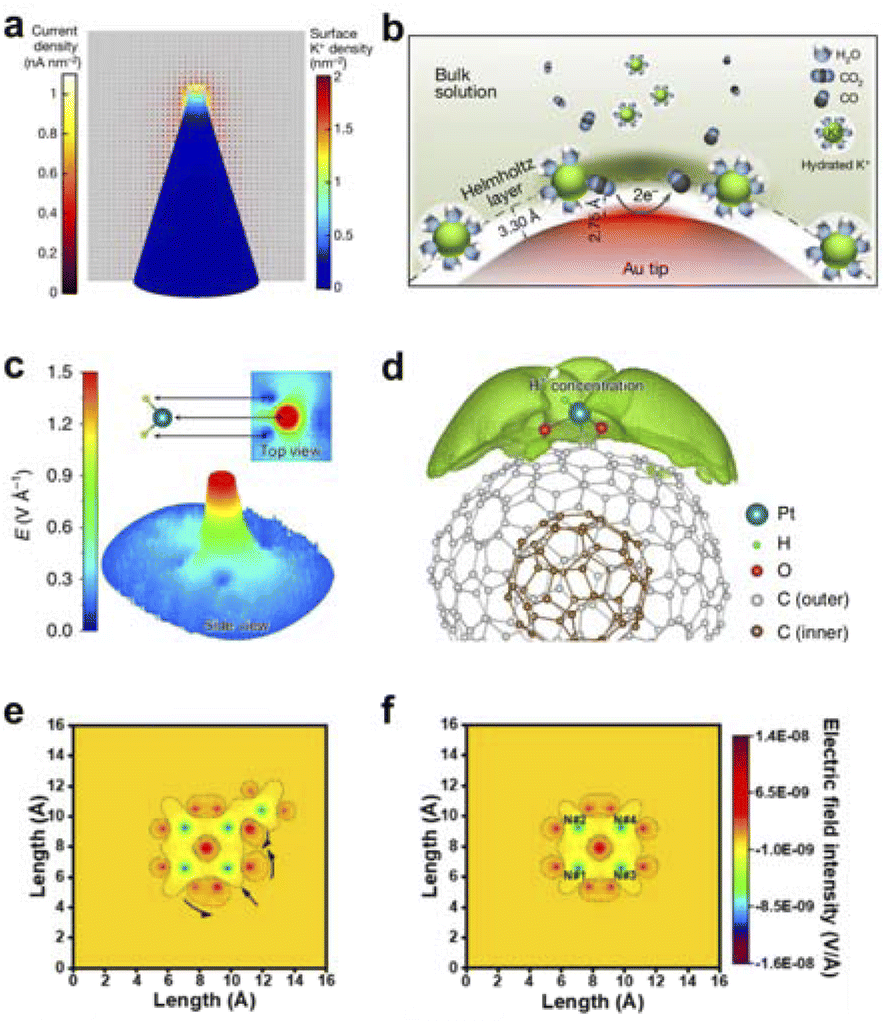 | ||
| Fig. 3 (a) Distribution of K+ density and current density on the surface of a gold nanotip with a tip radius of 5 nm. (b) Schematic representation of K+-assisted adsorption of CO2 molecules on the surface of the tip of a gold pinpoint electrode. Reproduced with permission from ref. 10 Copyright 2016 Springer Nature. (c) Simulation showing the local electric field of H2 at a tip-like Pt site with equilibrium potential. (d) A high concentration of protons is distributed around the Pt site induced by the localized pinpoint electric field of the Pt site. Reproduced with permission from ref. 60 Copyright 2019 Springer Nature. (e and f) localized electric field distributions of FeN4–PN due to different nitrogen doping. Reproduced with permission from ref. 61 Copyright 2021 American Chemical Society. | ||
Meanwhile, Song et al. utilized highly curved carbon carriers to anchor single atoms, simulating the active metal sites at corners and edges.60 The research demonstrated that single-atom Pt catalysts on onion-like nanocarbon carriers can achieve efficient electrocatalytic HER. Under the same loading conditions, the performance exceeded that of single-atom Pt catalysts supported by two-dimensional graphene sheets. DFT calculations further proved that the highly curved geometric structure concentrates electrons near Pt, generating an enhanced local electric field under the tip-induced conditions, which induces the enrichment of protons on the Pt surface and thus accelerates the electrocatalytic reaction process (Fig. 3c and d). Furthermore, our group designed iron single-atom catalysts with different types of nitrogen dopings.61 Theoretical calculations indicated that the introduction of peripheral impurity nitrogen would cause an inhomogeneous charge distribution in the active units. Among them, pyrrolic-N (PN), due to its strong electron-withdrawing ability, makes this inhomogeneous distribution more intense, thereby generating a local electric field (Fig. 3e and f). The subsequent theoretical analysis of several models demonstrated that FeN4–PN exhibits superior oxygen molecule activation performance compared to other models, indicating the effective enhancement of reactivity in this type of catalyst by the PN effect on the active site. The introduction of impurity nitrogen directly led to a local electric field with varying degrees of distortion.
Our group has improved the ORR activity of molecular catalysts by inducing charge localization through the coordination field. By using axial Fe–O coordination to induce the redistribution of electrons in a two-dimensional planar structure, coordination-induced charge localization breaks the charge symmetry of the active center, enhancing the oxygen reduction activity of the molecular catalyst.66 Meanwhile, Gu's team introduced F atoms into the Pd/N–C catalyst, creating a Pd surface coordination field environment richer in N, which improved catalytic activity and durability.62 Furthermore, the Sun group reported that on N-doped C materials generated by the pyrolysis of MOF, Pt single-atom sites were decorated by the atomic layer deposition method. With a lower coordination environment, the Pt single-atom structure tends to bind at the pyridine-type N sites, reducing the activation energy of the rate-determining step, and the Pt single-atom site exhibited high catalytic activity for oxygen reduction.65 Shao et al. used the connection between perovskite active sites and coordination environments to design electrocatalytic activity descriptors. For the first time, they used the principles of coordination chemistry to design perovskite HER activity descriptors and successfully developed a simple and reliable ionic electronegativity descriptor parameter. Using the ionic electronegativity design principle, several highly active perovskite HER catalysts were successfully screened.64 Fei and his colleague prepared a highly active and selective H2O2 electrosynthesis catalyst by adjusting the coordination number of Co–N–C catalysts and the surrounding oxygen functional groups.63 By synthesizing a low-coordinated Co–N2–C catalyst, the selectivity of electrocatalytic H2O2 reached 91.3% at a bias of 0.65. The high-coordinated Co–N4–C tends to undergo a 4-electron ORR to produce H2O. The selectivity of H2O2 can be adjusted by precisely controlling the coordination environment.
The polarization field is an efficient strategy for improving the bulk and surface charge separation of photocatalysts (Fig. 4a and b). During the photocatalytic process, photoexcited charge carriers are generated in bulk and migrate to the surface active sites to participate in redox reactions within hundreds of picoseconds. However, during this period, a large number of charges recombine rapidly in both the bulk and at the surface, especially the bulk charge recombination which only takes a few picoseconds. The polarization field can effectively promote bulk and surface charge separation.
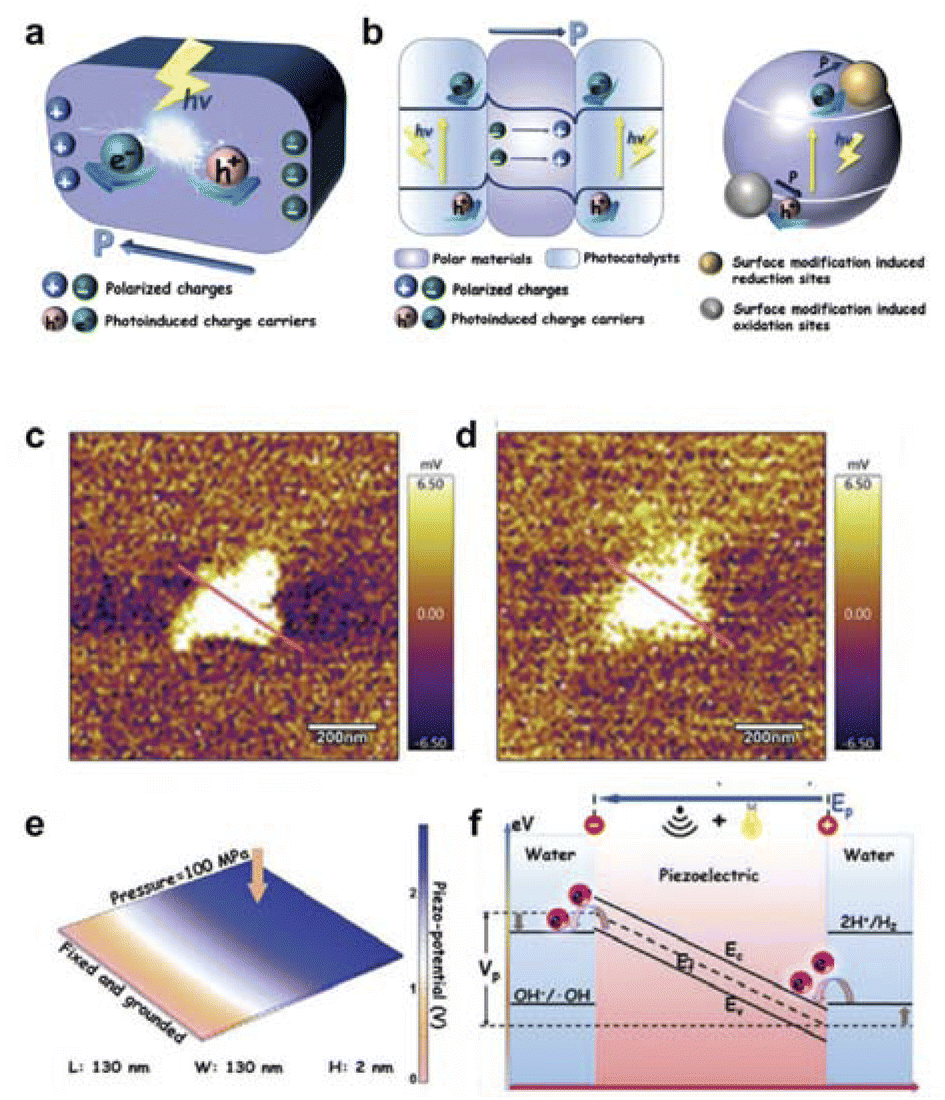 | ||
| Fig. 4 (a) Schematic diagram of the polarisation field promoting charge separation in the bulk phase. (b) The polarization field promoted surface charge separation. Reproduced with permission from ref. 67 Copyright 2019 Wiley. (c) KPFM surface potential map of UT-g-C3N4 in dark state. (d) KPFM surface potential map of UT-g-C3N4 under light conditions. (e) FEM simulation for the piezoelectric potential distribution of UT-g-C3N4. (f) The mechanism of the photo-improved piezocatalytic process. Reproduced with permission from ref. 70 Copyright 2021 Wiley. | ||
Hu et al. loaded a high work-function RuO2 co-catalyst on BiFeO3 nanoparticles, constructing a polarization field at the RuO2/BiFeO3 interface. This strategic modification facilitated efficient charge separation, resulting in a remarkable enhancement of the interfacial photovoltage increased by 3-fold times (from 3.5 mV to 9.5 mV), thereby significantly improving the performance of photocatalytic water oxidation reaction.69 In addition, Huang's group utilized two-dimensional piezoelectric materials to fabricate atomic-scale thickness g-C3N4 nanosheets. The non-piezoelectric bulk g-C3N4 exhibited strong piezoelectricity after becoming two-dimensional nanosheets due to the breakdown of symmetry (Fig. 4c and d).70 The catalyst was endowed with robust piezoelectric properties, a spacious energy capture region, and plentiful surface active sites. The strong piezoelectricity of g-C3N4 nanosheets is derived from the polarization induced by the uniform arrangement of polar heptazine ring units and the piezoelectric effect (Fig. 4e and f). Additionally, exceptional hydrogen production performance is facilitated by abundant reactive sites created through piezo-induced defects, along with enhanced charge migration under light irradiation. In brief, when spontaneous polarization occurs or due to external influence, the dipole moment causes positive and negative charges of polarization to appear on both sides of polar materials. This results in the formation of an electric field of polarization which facilitates the movement of photogenerated electrons and holes in opposite directions within photocatalysts, leading to effective separation of charges.
2.2 External field
External fields, such as light, electricity, magnetism, and strain, introduce additional non-contact factors that can significantly influence catalyst performance. These fields inject external energy into the catalytic system, intervening in the adsorption and desorption processes, as well as in electron excitation and charge separation within the catalytic reactions.71–74 In the following, we will introduce the characteristics of external fields such as the light, electric, magnetic, and strain fields, respectively.Utilizing solar energy, localized surface plasmon resonance (LSPR) has become an extremely attractive method for enhancing the electrocatalytic activity and selectivity of catalysts. LSPR excitation can induce hot electron and hole transfer, electromagnetic field enhancement, lattice heating, resonant energy transfer, and scattering, thereby promoting various photo/electrocatalytic reactions.79,80 The catalytic conversion efficiency strongly depends on the structure and composition of the plasmonic metal. Although extensive research has been conducted on LSPR-mediated electrocatalysis, there are currently three mainstream mechanisms.81–83
As shown in Fig. 5a, through electron–electron scattering and electron-phonon scattering, the relaxation of hot electrons causes a high lattice temperature in the plasmonic material, and the local electrolyte temperature gradually increases with this photothermal effect. This mechanism is used to improve reaction kinetics or current density. Fig. 5b shows the involvement of a highly enhanced electromagnetic field, which can activate nearby species or create vacancies in the conduction band of an adjacent semiconductor. The hot electrons are transferred from plasmonic nanostructures to neighboring species (Fig. 5c). This is crucial for both direct plasmonic metal-driven and indirect plasmonic-enhanced electrocatalytic processes.
 | ||
| Fig. 5 Main physical mechanisms of plasma metal nanostructures (a) photothermal phenomena. (b) Optical field enhancement of near-area electromagnetic fields. (c) Photo-induced electron–hole pair generation and hot electron injection. Reproduced with permission from ref. 79 Copyright 2021 Royal Society of Chemistry. (d) Optical image of an electrochemical micro–nano device. (e) Current density versus contact resistance is measured with the electric field. Reproduced with permission from ref. 84 Copyright 2016 Springer Nature. (f) Schematic diagram of the electric-field introduction into Pt SAs–MoS2, and the ΔGH in the 2H-MoS2 and Pt SAs–MoS2 models. (g) Charge analysis of Pt SAs–MoS2 under oriented external electric fields regulation. Reproduced with permission from ref. 85 Copyright 2022 Springer Nature. | ||
Jain et al. found that the enhancement of electrochemical processes by LSPR stems from the hot electrons generated by the d → sp transition in Au nanoparticles after light excitation, rather than the photothermal effect.86 LSPR predominantly amplifies faradaic processes while exerting minimal influence on non-faradaic processes. Moreover, Xia's group discovered that by leveraging the LSPR effect of Au nanorods, the hot electrons will inject into the conduction band of MoS2.87 This process increases the charge density of MoS2, aligning the energy levels of the catalyst more closely with those required for the HER. This indicates that hot electrons generated by the surface plasmon resonance of Au can be transferred to nearby semiconductor materials, making their surfaces electron-rich and accelerating the HER.
In summary, external light can enhance the energy state of charge carriers through the surface plasmon resonance effect, increase the local temperature of the catalyst through the photothermal effect, and increase the conductivity of the catalyst by photo-induced hot electron excitation. By leveraging these advantages of plasmonic nanostructures, plasmon-driven chemical reactions can be facilitated.
Electrocatalytic micro–nano devices offer multiple advantages for the study of electrochemical processes, including in situ revelation of structure–property relationships,92–94 external electric field modulation,44,95–97 identification of active sites,84,98 and regulation of single structural factors (such as defects, interfaces, phase transitions, etc.).99,100 These advantages enable the direct in situ probing of electrochemical processes.
Chhowalla et al. utilized micro–nano devices to compare the edge sites and in-plane sites of the metallic phase 1T-MoS2 and the semiconductor phase 2H-MoS2 nanosheets (Fig. 5d). They found that 1T-MoS2 exhibits higher HER activity due to its metallic phase. By regulating the in-plane sites of 2H-MoS2 with a gate voltage electric field, the HER activity of 1T-MoS2 can be achieved (Fig. 5e). The gate voltage electric field can effectively mitigate the contact resistance of the in-plane sites of 2H-MoS2, thereby enhancing its conductivity and aligning it with the HER activity exhibited by the metallic phase 1T-MoS2.84 In addition, Lou's group has also used micro–nano devices for HER activity analysis of 3R-NbS2 nanosheets and found that their in-plane active sites possess hydrogen adsorption and desorption properties similar to metallic Pt.98 Furthermore, Ding's team demonstrated the quantitative manipulation of localized catalytic sites of single-atom catalysts (SACs) immobilized on two-dimensional atomic crystals by a precisely controlled external electric field (Fig. 5f).85 Model SACs were systematically tested in microcells whose electrocatalytic performance could be dynamically tailored by the external electric field. The vertical electric field effectively modulates the charge distribution at the SA sites (Fig. 5g), polarises the frontier orbitals of the metal atoms and intermediates, and significantly alters the activation energies and reaction pathways in the catalytic process.
In conclusion, the gate electric field can regulate the conductivity of electrocatalysts, the adsorption strength of reaction intermediates on active sites, and the distribution of ions at the catalyst/electrolyte heterointerface through electrostatic coupling effects.
The magnetothermal effect, induced by the external application of a high-frequency alternating magnetic field on magnetic nanoparticles, exhibits localized heat generation properties that can elevate the surface temperature of the catalyst. The interaction between magnetic fields and local current density results in magneto-resistance (MR) and magnetohydrodynamic (MHD) effects, which induce macro- and micro-convections that facilitate bubble detachment from the electrode surface, mitigate ohmic polarization, reduce activation polarization, and alleviate concentration polarization. The Kelvin force effect enhances the mass transfer of paramagnetic oxygen, increasing the reaction rate near the electrode. Additionally, this will induce convection and reduce the diffusion layer thickness when the field gradient is perpendicular to the concentration gradient, increasing by limiting current. The Maxwell strain effect, resulting from the interaction between the magnetic field and dipole moment, induces magnetostrictive deformation in the shape of paramagnetic droplets. The orientation and strength of the magnetic field determine both the direction and extent of stretching, thereby influencing interfacial tension, contact angle, wettability, and adhesion force on solid surfaces. Consequently, the magnetic field exerts a distinct influence on both the shape of ionic clouds near electrodes and electrochemical electrode/electrolyte interfaces. The spin polarization effect allows the magnetic field to flip the spins of adsorbed intermediates on the catalyst surface, optimizing the reaction pathway and enhancing efficiency. This effect also alters the spin state of oxygen.
Xu et al. has investigated the oxygen evolution reaction (OER) behavior of nickel-based catalysts under an in situ magnetic field.104 They utilized an electrochemical system they designed themselves, combined with a magnetometer that can finely adjust the magnetic field strength (0–1.4 T), to conduct experiments (Fig. 6a and b). The results indicated that the electrocatalytic activity of all materials increased with the enhancement of the magnetic field strength. Additionally, under the influence of the magnetic field, the oxidation peak current increased, suggesting that the electrical conductivity of the electrode was higher and the electron transfer efficiency was improved with the increase of the magnetic field strength. This also further indicates that the magnetic field can accelerate the multi-electron transfer process of the OER (Fig. 6c and d). Meanwhile, Gan et al. has explored the OER behavior of Ni3Fe nanoparticles on wood through in situ growth. They fabricated Ni3Fe-based carbonized wood electrodes, which exhibited excellent catalytic activity and magnetic response characteristics. The Ni3Fe nanoparticles exhibited high electron spin polarization under the influence of an external magnetic field, thereby acquiring a substantial charge density. This resulted in the aggregation of holes and a reduction in the energy barrier for electrochemical reactions, ultimately leading to excellent electrocatalytic performance.106
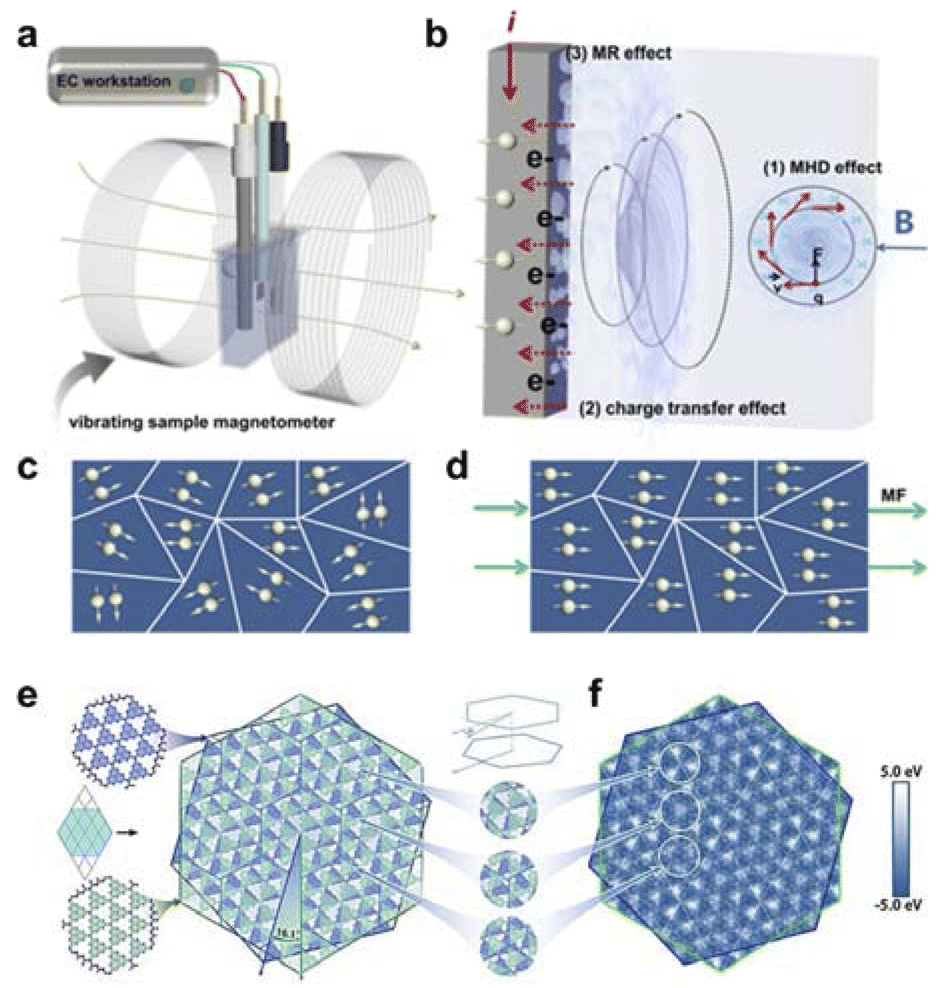 | ||
| Fig. 6 (a) Schematic diagram of the in situ tunable magnetic field coupled three-electrode OER test system. (b) Mechanism of magnetic field enhanced OER. (c and d) Effect of the presence or absence of magnetic field on the directional alignment of the dipole. Reproduced with permission from ref. 104 Copyright 2022 Royal Society of Chemistry. (e) Twisted g-C3N4 bilayer structure with an interlayer rotation angle of 16.1°. (f) Interlayer localized potentials showing some typical strain Mohr stripes. Reproduced with permission from ref. 105 Copyright 2021 Wiley. | ||
In summary, a magnetic field can enhance the overall performance of electrocatalytic reactions by accelerating the transport of materials in the electrolyte through the Lorentz force, increasing the local temperature of the electrocatalyst through the magneto-thermal effect, and regulating the reaction path of electrocatalysis by controlling the spin alignment.
Lu et al. studied the double-layer twisted g-C3N4 material and found that the interlayer twisting changes the symmetry of the material, leading to changes in the transition dipole moments between energy levels, thereby improving the material's response to visible light.105 The conduction band electrons generated by photoexcitation undergo interlayer transfer on an ultrafast timescale (∼120 fs), spatially separating from the photogenerated holes, which reduces the recombination chances of electrons and holes. At the same time, the strain-induced Moire potential produced by the interlayer twisting of the double-layer g-C3N4 material significantly regulates the adsorption energy of reaction intermediates at local sites on the material surface (Fig. 6e and f), thus affecting the surface overpotential and reducing the energy barriers for hydrogen and oxygen evolution reactions. Additionally, Guo's group discovered that encapsulating PdCu nanocrystals with an Ir shell consisting of four atomic layers on their surface can eliminate the impact of ligand effects found in traditional core–shell nanomaterials, resulting in PdCu/Ir core–shell nanocrystals with strain effects.109 Experimental results show that modifying the surface electronic structure of PdCu/Ir core–shell nanocrystals by applying compressive strain. Facilitate electron transfer from Ir to Pd, resulting in a reduction of adsorption energy for reaction intermediates. Theoretical calculations have confirmed that the tensile strain on the Ir shell reduces the adsorption strength for oxygen-containing intermediates and causes the d-band center to move downward, which is beneficial for promoting the formation of oxygen molecules. The electrochemical tests have confirmed that strain effects effectively enhance the electrocatalytic performance of core–shell structured nanocatalysts for OER and HER. Taken together, strain can regulate the d-band of active sites, including its energy level and density of states, thereby regulating its intrinsic activity. For early transition metals with a d-band below half-filling, tensile strain should have the opposite effect on the d-band center. Specifically, the band narrowing caused by tensile strain will move the d-band center downward, reducing the interaction with adsorbates.
In conclusion, it is feasible to establish internal fields originating from the catalyst itself, thereby exerting influence on catalytic reactions employing interface construction, morphological alterations, doping, regulation of coordination environment, polarization induction, and other solutions. External fields, as an auxiliary technique, can significantly improve the performance of catalysts through external light, electricity, magnetism, and strain factors. The mentioned strategies exhibit strong compatibility with conventional nano-engineering techniques, making them extremely valuable for developing and executing advanced catalytic systems with exceptional performance in the future. The role of field-effect in catalytic systems will be introduced below.
3. The role of field-effect
Currently, the field-effect has a wide range of applications in catalytic energy and environmental catalysis conversion, such as catalytic water splitting, CO2RR, NRR, organic electrosynthesis, and biomass recycling. A substantial amount of work has shown that the field-effect can influence the material properties of catalysts and the catalytic reaction process.111–114 This section mainly introduces the role of the field-effect in these catalytic reactions.3.1 Impact on local reactant concentration and mass transfer
The concentration and mass transfer of the reactants will seriously affect the smooth progress of the next reaction, while the field-effect can effectively gather the concentration and mass transfer of the reactants to promote the catalytic reaction process.As for water electrolysis, the slow kinetics of the OER leads to high energy consumption in electrochemical water splitting. Various strategies have been attempted to accelerate the OER rate, but it is challenging to control the transport of reactants, especially under high current densities where mass transfer factors dominate the reaction. The Yang group used NixFe1−x alloy nanocone arrays to facilitate the transport of reactants (Fig. 7a). They attempted to control the local electric field distribution by adjusting the morphology of the electrode, thereby optimizing the mass transfer process of reactants on the active sites. Finite element analysis indicated that high curvature tips can enhance the local electric field, thereby inducing a higher concentration of hydroxide ions (OH−) at the active sites and increasing the intrinsic OER activity. Experimental results showed that the nickel-iron nanocone array electrode with optimized alloy composition had a small overpotential of 190 mV at 10 mA cm−2 and 255 mV at 500 mA cm−2. By comparing samples with different tip curvature radii, the positive effect of the tip-enhanced local electric field on promoting mass transfer was also confirmed.115 At the same time, the group reported a selective enrichment effect of hydrogen ions on the surface of the Pt–Ni nanowire array with a conical structure, which greatly improved mass transfer efficiency and achieved excellent HER activity (Fig. 7b). The prepared the Pt–Ni nanowire array with a high-density nanocone structure using a unique magnetic field-assisted liquid-phase chemical deposition method. Finite element calculations showed that the high-density nanocones on the nanowire surface have a local electric field concentration effect. Under alkaline conditions, even with a high concentration of positively charged K+ and H+ competing, this distorted electric field on the surface can still significantly enrich H+. The calculation results showed that the enrichment effect of H+ is 8 times that of K+ (Fig. 7c and d). Experimental results showed that the HER activity of the thorny Pt–Ni nanowire array electrode (overpotentials of 23 mV and 71 mV at 10 mA cm−2 and 200 mA cm−2, respectively) was much higher than that of the smooth Pt–Ni nanowire array, confirming the role of the tip-enhanced local electric field in promoting mass transfer.116 Moreover, Jiang et al. synthesized the Ni/Ni0.2Mo0.8N catalyst, which exhibits a local electric field similar to that of a tip around the topmost Ni nanoparticles.118 This effect leads to an increased concentration of K+ ions within the inner Helmholtz plane, thereby improving the HER kinetics and surface reactant concentration. The Ni/Ni0.2Mo0.8N catalyst demonstrates excellent electrocatalytic performance for the HER reaction.
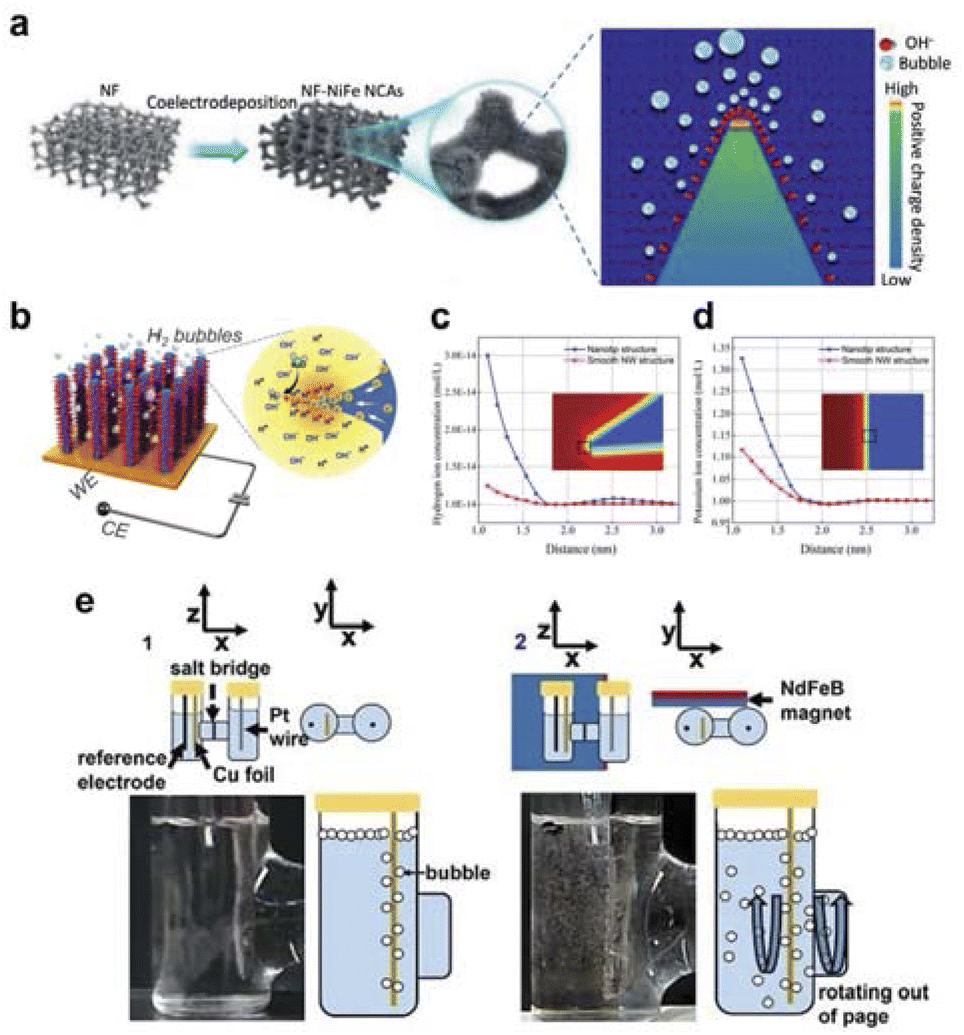 | ||
| Fig. 7 (a) Schematic of NixFe1−x alloy nanocone synthesis and the formation of local electric field. Reproduced with permission from ref. 115 Copyright 2021 Wiley. (b) Schematic of wattle-like Pt–Ni to enhance mass transfer. (c and d) Simulations of hydrogen and potassium ion concentrations at the tip and the plane. Reproduced with permission from ref. 116 Copyright 2021 Royal Society of Chemistry. (e) Actual diagram of the action of magnetic fields on electrocatalytic CO2RR to improve the mass transfer. Reproduced with permission from ref. 117 Copyright 2021 Elsevier. | ||
In the electrocatalytic CO2RR area, selectivity is not only dependent on the intrinsic reactivity of the catalyst but also on the transport of reactants to the catalyst. However, current methods for enhancing mass transport in CO2RR mostly rely on mechanical stirring or the use of gas diffusion electrodes, neither of which can eliminate concentration polarization. The Whitesides group proposed a new strategy for optimizing CO2RR, significantly improving mass transport and selectivity by utilizing fluid convection generated by a magnetic field.117 Compared to similar systems without a field or stirring, the current density is 1.3 times higher, and the selectivity relative to the hydrogen evolution reaction is 2.5 times higher, achieving high current density (>100 mA cm−2) and nearly 100% high faradaic efficiency (FE). A qualitative comparison was made between mass transport in two types of electrochemical H-cells (Fig. 7e). The presence of a magnetic field increases turbulence and mixing in the solution near the electrode, which facilitates convective mixing within the electrolyte and close to the electrode surface.
3.2 Influence the adsorption and activation of reactant molecules
The adsorption and activation of reactant molecules directly affect the reaction rate and activity. Field-effect influence the adsorption and activation of reactants.Xue's group developed a typical Mott–Schottky electrocatalyst through an electrospinning-carbonization strategy, which involved fixing Ni/CeO2 hetero-nanoparticles onto N-doped carbon nanofibers. The prepared Ni/CeO2 heterojunction effectively induces spontaneous electron redistribution, leading to the formation of a built-in electric field at the Mott–Schottky hetero-interface. This facilitates rapid charge transfer and reduces the energy barrier for water dissociation, thereby optimizing the chemical adsorption energy of reaction intermediates and ultimately accelerating reaction kinetics.119 Moreover, Yan et al. used two-dimensional NiPS3 nanosheets as precursors and carried out a thermal treatment, enabling the in situ growth of NiPS3/Ni2P epitaxial heterostructures.49 The built-in electric field at the NiPS3/Ni2P epitaxial interface enhances charge transfer, optimizing reactant adsorption and reducing the energy barrier for efficient electrocatalytic water splitting.
Additionally, Lou's group found that Cu ions coordinate with the hydroxyl groups of the HHTP ligand to form a honeycomb π-conjugated Cu-MOF layer, in which unsaturated coordination centers Cu1–O2 and saturated coordination centers Cu1–O4 can be observed.120 The Cu1–O2 center exhibits pronounced local charge polarization along the Cu–O–C bond, resulting in an asymmetric electron depletion region surrounding the Cu atom, while electrons are enriched in the neighboring O and C atoms (Fig. 8a). Local charge polarization is beneficial for strong coupling between the Cu1–O2 center and H2O molecules, thus facilitating the dissociation of H–O bonds and promoting HER. These unsaturated Cu1–O2 centers can serve as active centers for HER, effectively dissociating adsorbed H2O molecules into key *H intermediates, thereby giving the unsaturated Cu-MOF higher HER activity (Fig. 8b and c). Meanwhile, Cheng et al. designed a series of structurally clear and homogeneous metal active site d–π conjugated linear coordination polymers, serving as model catalysts.121 The coordination polymers examined in this study demonstrate the activation of dxz/dyz orbitals at the metal center through strong d–π conjugation. These orbitals not only serve as active sites for intermediate adsorption but also exhibit significant hybridization with ligand π orbitals, thereby amplifying the influence of ligand π electrons on the adsorption strength of oxygen-containing species at the metal center (Fig. 8d). It was found that the Co-tetra-aminobenzoquinone (Co-TABQ) coordination polymer has the most suitable adsorption strength for oxygen-containing intermediates, thus being closest to the top of the volcano of activity, showing the best electrocatalytic activity.
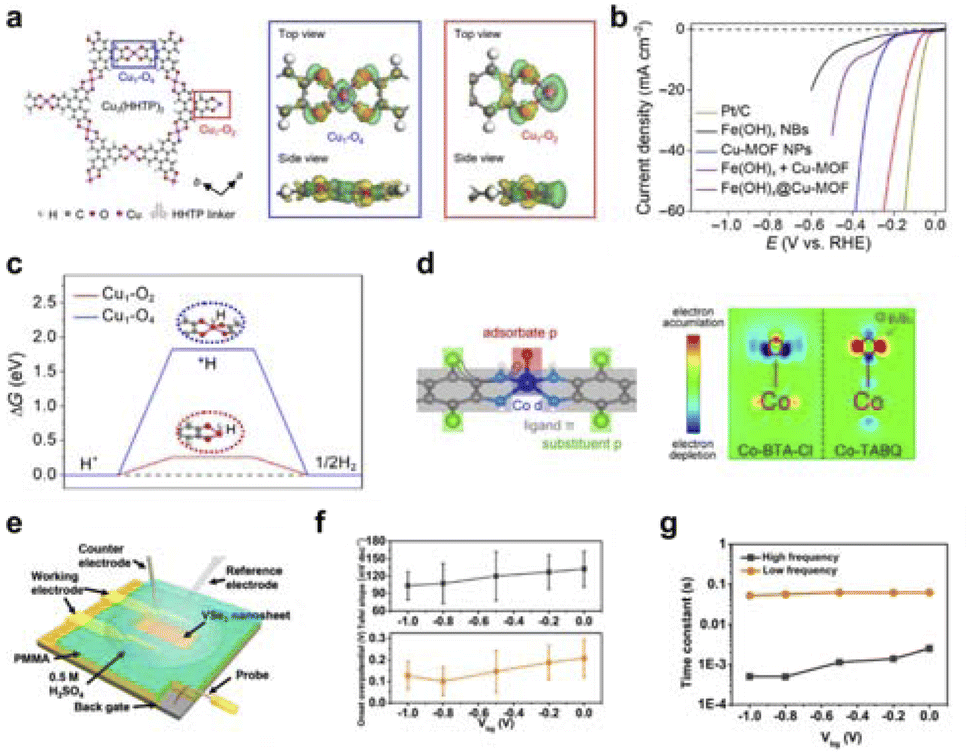 | ||
| Fig. 8 (a) Cu1–O2 coordination field construction. (b) Cu1–O2 coordination field electrocatalytic HER performance test. (c) Calculated free energy change of adsorbed *H on Cu sites of Cu1–O4 and Cu1–O2 centers. Reproduced with permission from ref. 120 Copyright 2021 Science. (d) Formation of coordination field by d–π conjugated linear coordination polymer to enhance adsorption. Reproduced with permission from ref. 121 Copyright 2021 Wiley. (e) Schematic diagram of VSe2 nanosheet micro-nanode device. (f) Back-gate voltage modulation of VSe2 nanosheet adsorption properties. (g) Variation of adsorption time constant for VSe2 nanosheets with back-gate voltage. Reproduced with permission from ref. 92 Copyright 2017 American Chemical Society. | ||
Furthermore, Hou et al. constructed two-dimensional carbon-based materials with coordinated unsaturated single-atom Cu–N2 structures (Cu–N2/GN) and coordinated saturated single-atom Cu–N4 structures (Cu–N4/GN) through a high-temperature coupling pyrolysis strategy.122 Electrocatalytic CO2 reduction performance studies showed that the coordinated unsaturated Cu–N2/GN catalyst exhibited better electrocatalytic reduction activity for CO2 than the coordinated saturated Cu–N4/GN. The adsorption of CO2 molecules on the coordinated unsaturated Cu–N2 structure significantly enhances compared to the coordinated saturated Cu–N4 structure. This is due to the shorter Cu–N bond length in the coordinated unsaturated Cu–N2 sites, which allows for quicker electron transfer from these sites to the adsorbed CO2 molecules. As a result, reaction intermediates are formed faster, leading to improved overall CO2 reduction to CO performance. As for the external field, Mai et al. used the vertical electric field generated by a field-effect transistor to regulate the adsorption and desorption characteristics of VSe2 nanosheets.92 They found that the transfer resistance decreased from 1.03 to 0.15 MΩ, and the adsorption process time was reduced from 2.5 × 10−3 to 5 × 10−4 S (Fig. 8e–g). Additionally, Frisbie's group also used the vertical electric field generated by a field-effect transistor to regulate the adsorption and desorption characteristics of MoS2 nanosheets.96 They discovered that the gate voltage affects the hydrogen adsorption and desorption energy of MoS2.
3.3 Influence on electronic excitation and work function
The redox process is carried out mainly for the transfer and transport of electrons and migrate to the surface for the catalytic reaction. Field-effect can modulate the excitation of electrons and the work function to promote the nature of the catalytic reaction.Lau et al. used a Pt-based metal–support catalyst as the research system to systematically study the intrinsic correlation between work function and built-in electric field (Fig. 9a).123 By fine-tuning the disparity in work function between the metal and the substrate, a robust interface electric field was established at the interface. The specific potential difference at the interface of various materials can be obtained through KPFM testing (Fig. 9b). Further analysis showed that the interface electric field can control the excitation of electrons, thereby promoting the water electrolysis reaction. In summary, a Pt–CoOx Schottky interface with a strong built-in electric field can be constructed for efficient neutral electrolytic water hydrogen evolution.
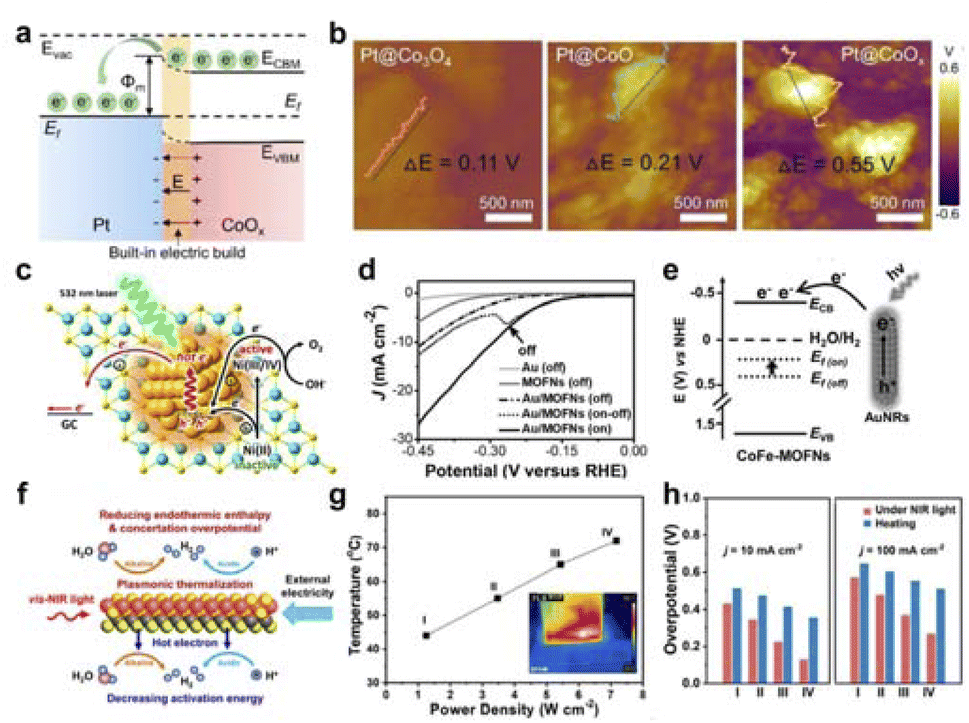 | ||
| Fig. 9 (a) Illustrations depicting the energy band arrangement of Schottky catalysts and the generation of electric fields at the interface. (b) KPFM tests of surface potential with different samples. Reproduced with permission from ref. 123 Copyright 2022 Wiley. (c) Schematic diagram of Au/Ni(OH)2 plasma excitation for OER. Reproduced with permission from ref. 12 Copyright 2016 American Chemical Society. (d) HER polarization curves of Au/CoFe-MOFN under light irradiation. (e) The energy level diagram illuminating hot-electron injection from AuNRs to CoFe-MOFNs. Reproduced with permission from ref. 124 Copyright 2019 Wiley. (f) Schematic diagram of Au/MXene LSPR-induced photothermal and hot-electron effect boosting catalytic activity. (g) Correlation between the electrode temperature and NIR light power. (h) Comparison of Au/MXene performance under NIR light and heating. Reproduced with permission from ref. 125 Copyright 2021 Wiley. | ||
Additionally, Ye's group used the surface plasmon resonance of Au to excite hot electrons and promote the transition from Ni(II) to Ni(IV) (Fig. 9c), thereby enhancing the OER performance of Ni(OH)2.12 Moreover, Jiang et al. used the LSPR effect to significantly accelerate the electrochemical process through hot electron injection.124 The LSPR effect of Au facilitated the injection of hot electrons from Au to CoFe-MOFNs under light illumination, elevating the Fermi level of CoFe-MOFNs and improving its energy level alignment with the H2O/H2 redox pair (Fig. 9d and e). As a result, the light illumination significantly reduced the activation energy for HER catalyzed by Au/CoFe-MOFN, effectively promoting the hydrogen evolution reaction. Meanwhile, Qiu's team studied the impact of LSPR effects on the electrocatalytic reaction process and the process intensification mechanism of MXene with different chemical compositions and crystal structures in an external electric field, using electrochemical hydrogen evolution as a probe reaction (Fig. 9f–h).125 They utilized femtosecond laser technology to discover that the LSPR effect of MXene in the visible to near-infrared spectral range resulted in significant photothermal conversion and sub-picosecond hot electron effects. The photothermal effect reduced overpotential for hydrogen evolution by supplying thermal energy and reducing concentration polarization, while the hot electron injection effect facilitated a decrease in activation energy for hydrogen evolution. Under different pH environments and light irradiation conditions, this coupled reinforcement mechanism could enhance the electrocatalytic hydrogen evolution activity of MXene by more than 5 times.
In the area of CO2RR, the LSPR of plasmonic nanostructures can capture low-energy photons to generate high-energy hot electrons and lower the activation barrier for CO2. The Zhang group synthesized plasmonic heterostructures by rationally assembling W18O49 nanowires onto Au nanoparticle-embedded TiO2 nanofibers.71 The Au/TiO2/W18O49 sandwich-like substructure in the plasmonic heterostructure becomes an active “hot spot” due to the interesting plasmonic coupling between the metal Au and the adjacent non-metallic W18O49 component. In the photocatalytic CO2 reduction process at a reaction temperature of 43 ± 2 °C, these active “hot spots” can not only strongly capture incident light to produce high-energy hot electrons, but also adsorb intermediate products of CO and protons through the bi-heteroactive centers (Au–O–Ti and W–O–Ti) in their heterointerface regions. The hot electrons, CO, and protons are simultaneously confined in the plasmonic active “hot spots”, accelerating the protonation of CO, thereby endowing the Au/TiO2/W18O49 plasmonic heterojunction with high photocatalytic activity (∼35.55 μmol g−1 h−1) and high selectivity (∼93.3%) for the preparation of methane. Furthermore, the Mai group using micro–nano devices as a model, controlled the work function of MoS2 by applying a vertical electric field formed by gate voltage, leading to faster electron transfer and accelerated catalytic reactions.95 At the same time, the Dong group adjusted the work function of WSe2 by gate voltage and found the electrical conductivity would be enhanced under high gate voltage conditions. This will have an impact on the Gibbs free energy for H.97
In the area of environmental science, Xie's research team adorned electrodes with metal nanoneedles.126 Due to the lightning rod effect, the electric field near the tip of the nanowire is greatly enhanced, exciting electron generation. Therefore, only a relatively low voltage needs to be applied to obtain a strong electric field at the nanowire tip for sterilization. The results showed that bacteria located at the tips of nanoneedles at both the positive and negative poles were rapidly inactivated during low-voltage treatment. The effectiveness of sterilization is directly related to the intensity of the localized enhanced electric field. When >90% of the nanoneedle bacteria were inactivated, no reactive oxygen species were generated. Rapid pore closure of the membrane under low voltage confirmed that the cells at the nanoneedle tip underwent electroporation of the cell membrane. Thus, electroporation caused by the locally enhanced electric field is the main mechanism for bacterial inactivation.
3.4 Affecting photogenerated charge separation
In photocatalytic reactions, apart from light absorption, a crucial step involves the separation of photogenerated electron–hole pairs. Numerous studies have demonstrated that the field-effect also influences the separation of these pairs, subsequently impacting catalytic performance.Zhu et al. successfully constructed a heterodimeric porphyrin structure by coupling meso-tetrakis(4-carboxyphenyl)porphyrin zinc (ZnTCPP) with meso-tetrakis (4-hydroxyphenyl) porphyrin (THPP), as shown in Fig. 10a.127 A novel ZnTCPP/THPP heterodimeric porphyrin structure was designed for photocatalytic H2 evolution through π–π stacking interactions, which could simultaneously achieve a strong interfacial electric field and appropriate interfacial matching between the two porphyrins. Under full-spectrum light, ZnTCPP/THPP achieved a high photocatalytic hydrogen evolution rate of 41.4 mmol h−1 g−1, which is about 5.1 times and 17.0 times higher than that of pure ZnTCPP and THPP, respectively. The significant enhancement in activity is mainly due to the huge interfacial electric field formed between the two porphyrins, which greatly promotes efficient charge separation and transfer. Additionally, a built-in interfacial electric field can be created by establishing a Z-scheme heterojunction structure (Fig. 10b).128 By precisely controlling the interfacial interactions, an effective Z-scheme interfacial electron transfer channel was established, which has a huge internal electric field, thus achieving efficient and stable photocatalytic overall water splitting performance. Moreover, the research group constructed a full-spectrum responsive donor–acceptor (D–A) supramolecular photocatalyst TPPS/C60–NH2.129 By adjusting the molecular dipole to form a huge built-in electric field, the charge separation efficiency was significantly increased, and a long-lived charge-separated state was formed, achieving efficient photocatalytic hydrogen production activity (Fig. 10c).
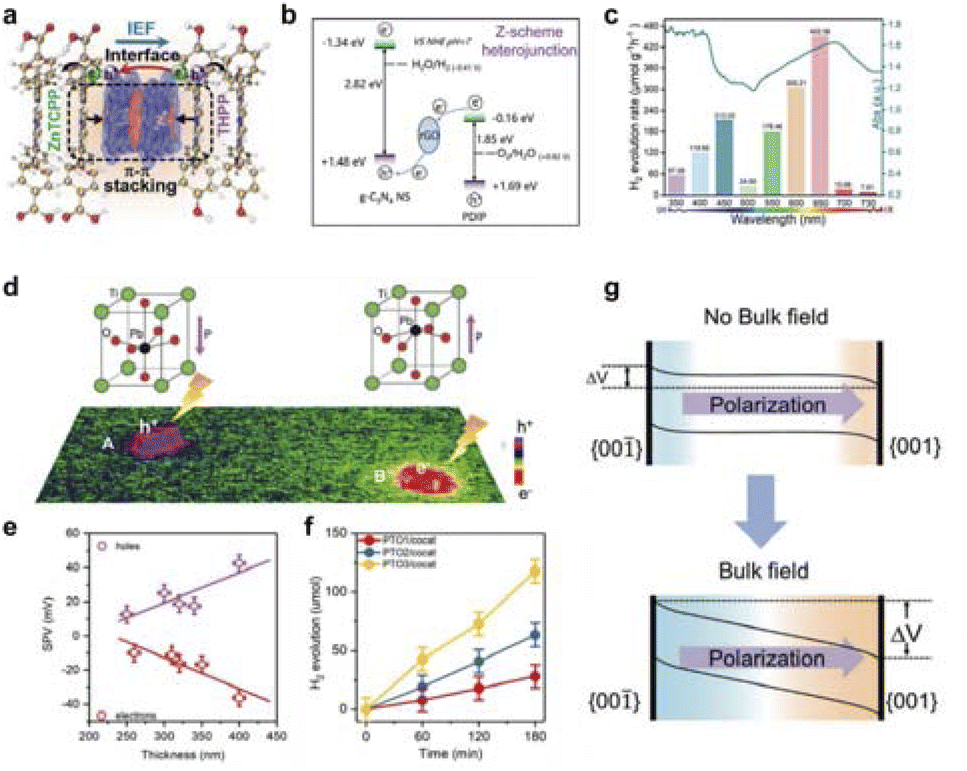 | ||
| Fig. 10 (a) Intramolecular built-in electric field to enhance photogenerated charge separation. Reproduced with permission from ref. 127 Copyright 2022 Wiley. (b) Construction of Z-scheme charge transfer mechanism. Reproduced with permission from ref. 128 Copyright 2021 Wiley. (c) Overlayer of wavelength-dependent hydrogen evolution and UV-vis absorption. Reproduced with permission from ref. 129 Copyright 2021 Wiley. (d) Image of ferroelectric field visualization. (e) Statistical SPV data of PTO3. (f) Photocatalytic hydrogen evolution activity of three PTO samples with cocatalysts. (g) Schematic representation of ferroelectric field affecting the energy band structure. Reproduced with permission from ref. 130 Copyright 2020 Wiley. | ||
Enhancing the separation efficiency of photogenerated electrons and holes is crucial for improving solar energy conversion efficiency in the photocatalytic process. Constructing an intrinsic electric field represents an effective approach to enhance charge separation. Fan et al. utilized the surface photovoltage imaging technique to investigate the mechanism of ferroelectric field in PbTiO3 single-domain ferroelectric photocatalytic particles, focusing on the separation of photogenerated charges (Fig. 10d).130 The results reveal that the depolarizing bulk electric field, stemming from ferroelectric spontaneous polarization, is the primary force for separating photogenerated charges and dictates their separation efficiency, rather than surface polarization as traditionally believed (Fig. 10e–g). The charge separation of ferroelectric materials can be enhanced by increasing the intensity of the depolarization field and minimizing the shielding field.
In summary, field-effect can influence the concentration and mass transfer of local reactants, adsorption and activation of reactant molecules, excitation of electrons and work function, and separation of photogenerated electron–hole pairs, thereby enhancing the activity of catalyst materials or catalytic reaction processes in the energy and environment catalysis.
4. Application of field-effect in energy and environment catalysis
While introducing the field effect and its role, the application of the field effect has also been widely explored to address key issues in energy and environmental catalysis. Despite extensive research over the years, there is ongoing work to enhance reaction rates, energy efficiency, and selectivity in these areas. The field-effect has garnered significant interest in photo- and electrocatalytic reactions, such as water splitting, CO2RR, NRR, organic electrosynthesis, and biomass recycling. This interest stems from the capacity of field-effect to modulate localized concentrations and mass transfer, activate reactant adsorption, excite electronics, and separate charges. This section offers a comprehensive review of the major advancements in field effects as they pertain to energy and environmental catalysis.4.1 Water splitting
The global consensus to address the pressing need for sustainable energy has led to a widespread pursuit of environmentally friendly energy sources. Hydrogen with high energy density and carbon-free properties make it a promising candidate for future energy sources. Water electrolysis, which utilizes abundant water as a reactant, offers an efficient and green method for hydrogen production. HER and OER are the two main reactions that enable water electrolysis technology. Research on establishing field effects for the above electrocatalytic reactions is progressing rapidly. Building field effects between noble metals and transition metal compounds provides a good opportunity to improve the performance of water electrolysis and to save the amount of noble metals. For example, the ferromagnet (FM) catalyst can enhance the reaction kinetics of spin-polarized oxygen evolution under the action of an external magnetic field. The single-area FM catalyst with a polarized magnetic area structure has better spin-polarized OER performance than the multi-area FM catalyst. At the same time, Huang group produced a net ferromagnetic moment by doping manganese in the antiferromagnetic RuO2, which can make more magnetic moments of Ru atoms parallel to each other when an external magnetic field is applied, constructing a new spin-polarized environment for OER to enhance its activity (Fig. 11a and b).131 Furthermore, Lu et al. used Ni1/MoS2 to form a distorted tetragonal crystal structure that facilitated ferromagnetic coupling with nearby S atoms and adjacent Ni1 sites, resulting in global room-temperature ferromagnetism (Fig. 11c and d).132 This coupling facilitates spin-selective charge transfer, resulting in the triplet state O2 in OER. In addition, a slight magnetic field of about 0.5 T increases the OER magnetic current by an order of magnitude. Magnetic fields can enhance the efficiency of electrocatalytic reactions by changing the flow pattern in the electrolyte and the mass transfer rate on the electrode surface.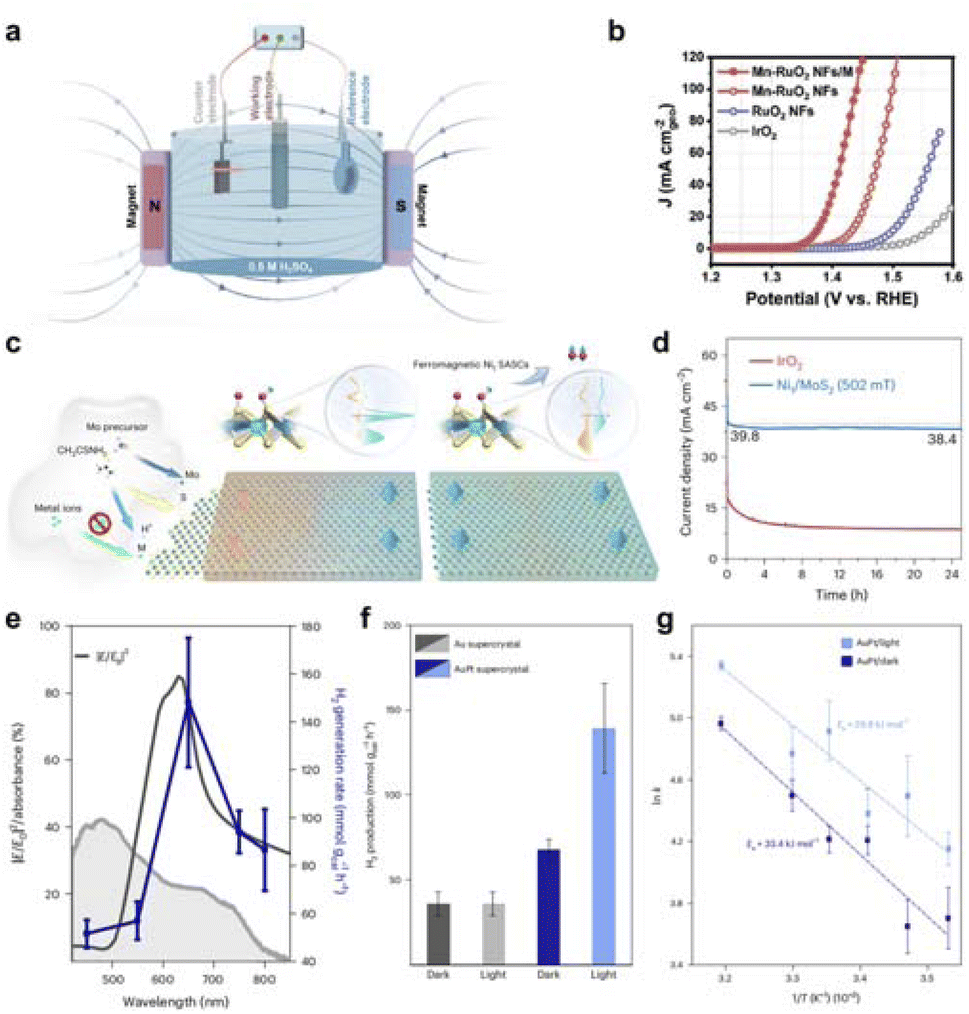 | ||
| Fig. 11 (a) Schematic illustration of a three-electrode system with applying a magnetic field. (b) LSV curves of Mn–RuO2 NFs/M, Mn–RuO2 NFs, RuO2 NFs, and IrO2 in 0.5 M H2SO4. Reproduced with permission from ref. 131 Copyright 2023 Wiley. (c) Illustration of the hydrothermal synthesis of M1/MoS2 SASCs under acidic conditions and the giant magnetic field enhancement of ferromagnetic SASCs for water splitting. (d) Current density versus time curves of Ni1/MoS2 under the magnetic field to compare with that of the IrO2 in water splitting cells. Reproduced with permission from ref. 132 Copyright 2023 Springer Nature. (e) Wavelength-dependent H2 production plotted simultaneously with the electric (E) field strength in the hotspots within the visible range and the weighted absorbance. (f) H2 generation rate normalized by the total mass of catalyst in both conditions, dark and light, for Au and AuPt supercrystals. (g) Revealing the impact of white-light illumination, the activation energy (Ea) for AuPt supercrystal decreased from 33.4 kJ mol−1 to 29.8 kJ mol−1 in the Arrhenius plot analysis. Reproduced with permission from ref. 133 Copyright 2023 Springer Nature. | ||
The charge transfer efficiency of the electrocatalyst depends on the intrinsic field/charge distribution around the atomic sites on the surfaces. Many advanced electrocatalysts have atomic defect structures on their surfaces, which can change the electric field/charge distribution of the electrocatalyst and improve the catalytic performance. The introduction of atomic defects on the electrocatalyst surface leads to the generation of aperiodic electric fields, which can significantly enhance the electric field effect at the reaction interface and help regulate the catalytic reaction kinetics. Xu et al. used a single layer of molybdenum disulfide (Mo2S) rich in atomic defects as a model catalyst, combined with advanced electron microscopy and differential phase contrast (DPC) technology, to observe the distribution of polarized electric fields around the inverse defects.134 At the same time, the distribution of polarized electric field around the inverse defect was observed using DPC technology. The polarization of the electric field of the antisite defect atoms directly leads to the appearance of asymmetric charge distribution, enhances the adsorption of H*, and optimizes the catalytic activity. Moreover, Mai et al. provided a new approach to fine-tune proton-electron coupling by integrating external electric fields into electrocatalysis using a strategy to regulate proton-electron coupling reactions.135 By cleverly optimizing the proton cycling within the α-MnO2 lattice, the modulation of the energy distribution of hydroxyl adsorption was achieved, leading to a significant enhancement of the oxygen precipitation performance.
In addition, Cortés et al. proposed a two-dimensional bimetallic catalyst that combines platinum nanoparticles into a well-defined gold nanoparticle supercrystal, where Au nanoparticles with a size of 22 nm form the superlattice and the Au nanocrystals are separated by 3.5 nm Pt nanoparticles (Fig. 11e–g).133 The photocatalytic dehydrogenation of formic acid was performed by bimetallic supercrystals under visible light irradiation and solar irradiation, and the hydrogen production rate reached 139 mmol g−1 h−1. The authors observed that the enhancement of the catalytic activity of platinum nanoparticles was mainly related to the electric field intensity of the hot spot.
Moreover, Zhu's group studied a simple fast solution dispersion method to synthesize the supramolecular co-assembly catalyst of naphthalimide (NDINH)/perylene imide (PDINH).136 In the co-assembled structure, NDINH acts as an electron acceptor and creates a charge difference with maximum values at the contact interface between NDINH and PDINH. This behavior is similar to that of a donor–acceptor (D–A) catalyst, resulting in the formation of a strong local dipole. The presence of this dipole significantly enhances the built-in electric field driving force within the material, thereby facilitating carrier migration and separation. Our group increased the strength of the electric fields at a local and atomic level by introducing Mn atoms into the CoP tip.137 This will effectively increase the concentration of OH− on the catalyst surface, improve the rate at which mass is transferred, and optimize the energy barriers involved in the reaction process to facilitate efficient desorption of O2.
4.2 CO2 reduction
Catalytic CO2RR is a highly efficient technology for reducing atmospheric CO2 concentrations and achieving carbon recycling using clean electricity generated from renewable energy sources.In recent years, there has been significant interest in utilizing CO2RR technology for the production and storage of renewable energy to promote sustainable development.138–141 Despite great efforts have been devoted to the development of high performance CO2RR catalysts this area still has many changes such as high overpotentials, low Faraday efficiency, and poor single-product selectivity. The introduction of field-effect to generate photogenerated carriers, enhancement of localized mass transfer, and intermediate adsorption activation has been s feasible as a promising strategic reaction to facilitate catalytic CO2 reduction.
Atomic local doping induces local charge polarisation, which can affect the adsorption and activation of intermediates in CO2RR and promote performance. Our group found that the introduction of Sn atoms on the Ag substrate can construct a strong local electric field, which reduces the formation energy of neighboring atoms Ag–COOH and improves the reaction barrier of Ag–H.142 The Sn-induced local electric field state of Ag/Sn alloys was characterized using differential phase contrast scanning transmission electron microscopy (DPC-STEM) technique, which induced an enhancement of the electric field with a change in direction in the periphery of Sn (Fig. 12a and b). In situ IR and Raman characterization demonstrated that this strong local electric field can effectively promote the formation of CO on the surface of Ag/Sn alloys. As a result, the Ag/Sn presented a high CO yield (Fig. 12c). Meanwhile, Tan's team successfully created a robust and uneven local electric field in the Cu matrix by incorporating Ag and Sn heteroatoms.145 This local electric field facilitates CO2 molecule activation, stabilizes *COOH intermediates, and suppresses the HER, thus enhancing CO production during CO2RR. Additionally, our team discovered that O-doping can concentrate electrons in the p orbitals of a Ca single-atom catalyst (SAC) with an asymmetric Ca–N3O coordination.146 According to Mulliken charge analysis, O-doping increases the electron transfer from the Ca–N3O site (0.74e) to *COOH more than from Ca–N4 (0.68e), thereby enhancing CO2 activation. Moreover, Lum et al. found that a model system using single-atom catalysts (SACs) enables uniformly tunable electric field modulation.147 Ni SACs with optimized nanocurvature achieved high CO-partial current densities of ∼400 mA cm−2 in acidic media with >99% Faraday efficiency. Modulation of the interfacial electric field provides a powerful means of controlling the electrocatalyst activity. Importantly, the electric field can vary the binding energy of the adsorbate according to its polarisation rate and dipole moment, thus becoming independent of the scalar relationship that fundamentally limits performance.
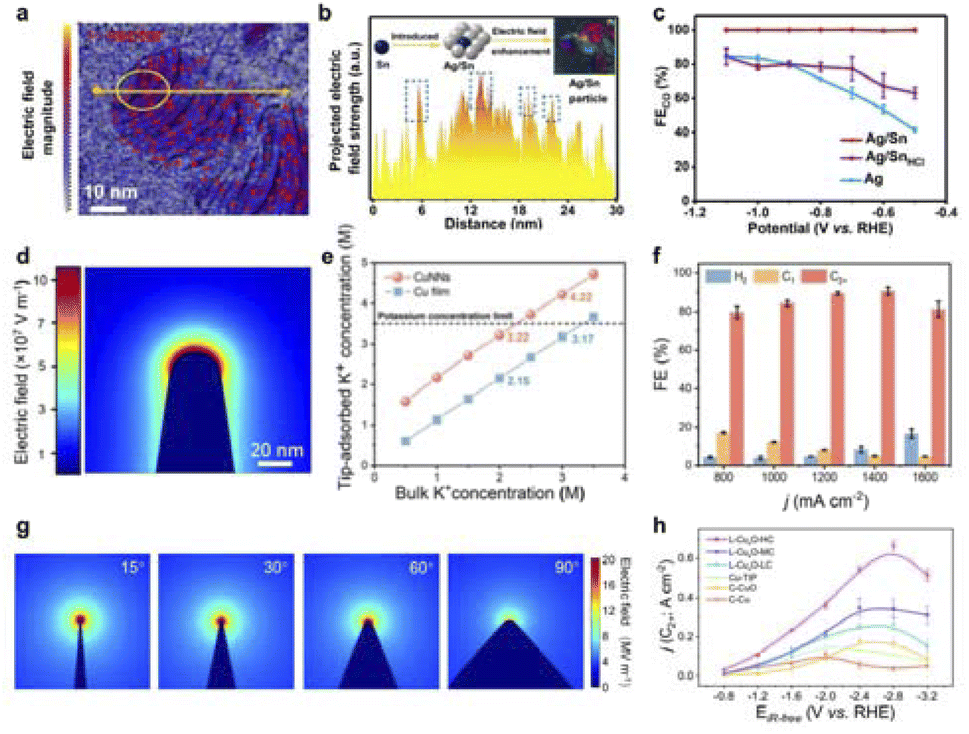 | ||
| Fig. 12 (a) Arrow-map representation of the strength and orientation of the electric field in the Ag/Sn catalyst. (b) Electric field intensity profile along the yellow line shown in (a). (c) FE of CO in H-cell. Reproduced with permission from ref. 142 Copyright 2022 Wiley. (d) Electric field distribution near the surface of CuNNs. (e) The adsorbed K+ concentration on the surface of CuNNs and a Cu film varies with different K+ concentrations in the bulk electrolyte. (f) The FE of CO2RR obtained at different current densities for CuNNs. Reproduced with permission from ref. 143 Copyright 2023 Wiley. (g) Electric field distribution near the surface of 15°, 30°, 60° and 90° on L–CuxO. (h) C2+ current density of each catalyst at different applied potentials. Reproduced with permission from ref. 144 Copyright 2023 Springer Nature. | ||
The sharp tip geometry induces a localized strong electric field that enhances electron transport and ion concentration to kinetically modulate the reaction microenvironment. Our team has found that Cu nanotip structures can generate extremely high local K+ concentrations, surpassing the 3.5 M threshold and reaching up to 4.22 M (Fig. 12d and e).143 This advancement significantly boosts C2+ product activity in the acidic electrocatalytic reduction of CO2 (Fig. 12f). In addition, Ye et al. proposed a laser-based method to engineer bipyramidal catalysts featuring adjustable tip curvature and enhanced nanoparticle interfaces (Fig. 12g and h).144 The Cu+/Cu2+ interfaces enriched the active sites for C2+ production by stabilizing *OC–COH intermediates, reducing the potential barrier and improving the thermodynamics of CO2RR.
Meanwhile, Xiong et al. reported efficient plasma-induced artificial photosynthesis without additional sacrificial agents using gold (Au) rods as plasma light-trapping units and copper–palladium (Cu–Pd) alloy shells as auxiliary catalysts.148 The Au-induced local electric field led to the emergence of new isolated states above the Fermi energy, as well as the divergence of the electron transfers within different molecular orbitals. The local electric field plays an irreplaceable role in efficient multiphoton absorption and selective energy transfer, resulting in excellent light-driven catalytic performance. Experiments revealed a CH4 yield of 0.55 mmol g−1 h−1 under 400 mW cm−2 full-spectrum light irradiation and an apparent quantum efficiency (AQE) of 0.38% under light irradiation at 800 nm wavelength.
4.3 NRR
The nitrogen reduction reactions (NRR) encompass the reduction of nitrogen fixation and nitrate reduction. Both reactions involve the reduction of elemental nitrogen and are integral components of NRR. The synthesis of green ammonia from H2O and N2, fueled by light and electricity, emerges as a viable alternative to the energy-intensive Haber–Bosch process. Nonetheless, the limited solubility of N2 in water and the formidable challenge of breaking the N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N triple bonds (940.95 kJ mol−1) pose significant barriers to the widespread implementation of nitrogen fixation reactions. The development of nitrogen fixation catalysts must overcome the challenges of low N2 solubility and difficult activation. Additionally, the release of NOx− from the combustion of fossil fuels and the discharge of agricultural and industrial wastewater contributes to the contamination of NOx− species in aquatic environments. The catalytic NOx− reduction reaction (NOx−RR), powered by renewable electricity, presents an eco-friendly approach for producing green ammonia and water quality remediation. The development of NOx−RR catalysts must overcome several critical challenges, such as the low nitrate concentration in actual wastewater and the sluggish reaction kinetics processes. The application of the field-effect strategy has enhanced the polarization of the NN triple bond and the local concentration of NO3− near the catalyst surface, thus promoting the activation of nitrogen molecules and NO3− mass transfer.26,149
N triple bonds (940.95 kJ mol−1) pose significant barriers to the widespread implementation of nitrogen fixation reactions. The development of nitrogen fixation catalysts must overcome the challenges of low N2 solubility and difficult activation. Additionally, the release of NOx− from the combustion of fossil fuels and the discharge of agricultural and industrial wastewater contributes to the contamination of NOx− species in aquatic environments. The catalytic NOx− reduction reaction (NOx−RR), powered by renewable electricity, presents an eco-friendly approach for producing green ammonia and water quality remediation. The development of NOx−RR catalysts must overcome several critical challenges, such as the low nitrate concentration in actual wastewater and the sluggish reaction kinetics processes. The application of the field-effect strategy has enhanced the polarization of the NN triple bond and the local concentration of NO3− near the catalyst surface, thus promoting the activation of nitrogen molecules and NO3− mass transfer.26,149
For example, Zhang et al. reported that interfacial polarization is an effective scheme to enhance NN cleavage to promote electrocatalytic ammonia synthesis.150 As a proof of concept, a protruded iron monatomic catalyst fixed on a MoS2 nanosheet generates an electric field to polarize N2 (Fig. 13a). The interfacial polarization facilitates the injection of electrons into the NN triple bond, thereby activating and breaking nitrogen molecules. Using 0.1 M KCl as the electrolyte, the faradaic efficiency of this electrocatalytic ammonia synthesis system at a working voltage of −0.2 V vs. RHE is 31.6% ± 2% (Fig. 13b). Additionally, Jiang et al. proposed that a single transition metal (TM) atom (i.e. BN/TM/G) sandwiched between hexagonal boron nitride (h-BN) and graphene flakes can serve as an effective SAC for electrochemical nitrogen fixation.152 These sandwich structures achieve stable, tunable interfacial polarization fields that enable TM atoms to supply electrons to neighboring B atoms as active sites. The results show that the partially occupied PZ orbitals of the B atom can form a B to N back bond with an antibond state of N2, thus weakening the N–N bond. The electric field on the surface of h-BN is neither strong nor weak, which further promotes the adsorption and activation of N2. Meanwhile, our group designed and prepared ordered silver nanotip array catalysts with different curvatures.25 The high curvature tip discharge effect generates a high-energy local electric field (Fig. 13c and d). This field shortens the distance between N2 and the catalyst, induces charge polarization on dinitrogen atoms and lowers the first protonation energy barrier of N2. As a result, this catalyst effectively promotes the adsorption and activation of inert N2 molecules. The detected N–N and N–H intermediates also prove that the NN bond breaks and hydrogenates under the drive of the local electric field. The reaction rate of nitrogen fixation was optimized to achieve a Faraday efficiency of 72.3 ± 4.0% (Fig. 13e).
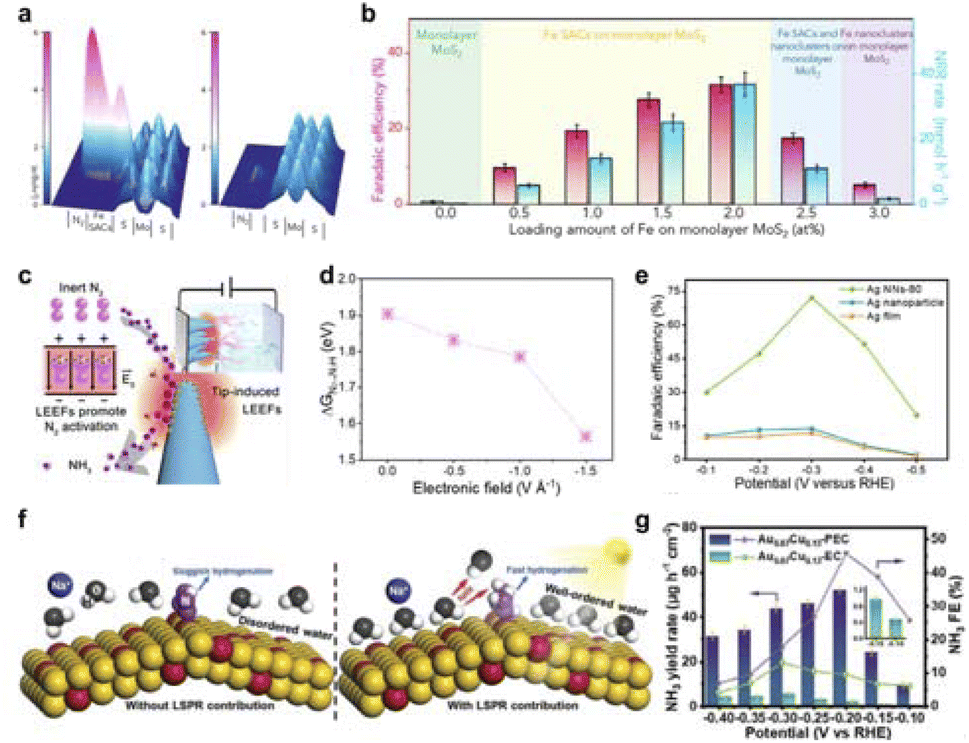 | ||
| Fig. 13 (a) 3D topographic potential distribution images of N2 adsorbed onto Fe SAC protrusion immobilized on MoS2 and pure MoS2. (b) Performance comparison of different samples at −0.2 V versus RHE in flow cell. Reproduced with permission from ref. 150 Copyright 2020 Elsevier. (c) Ag nanoneedle arrays are utilized to create localized enhanced electric fields (LEEFs) to facilitate the fracture of NN, thereby assisting the process of nitrogen fixation. (d) Potential-determining step (PDS) energy barrier of Ag NNs catalysts under various electric fields. (e) Faradaic efficiencies for Ag NNs-80, Ag nanoparticle, and Ag film at each applied potential for 2 h. Reproduced with permission from ref. 25 Copyright 2022 American Chemical Society. (f) Schematic showing water molecular behavior in the NRR process without (left) and with (right) light illumination, indicating LSPR-induced ordering of interfacial water molecules. (g) NH3 yield rate and FE of Au0.87Cu0.13-PEC and Au0.87Cu0.13-EC at different potentials. Reproduced with permission from ref. 151 Copyright 2024 Wiley. | ||
The pollution of nitrate (NO3−) in surface and groundwater is increasingly serious due to the utilization of nitrogenous fertilizer, and its removal has been a hot spot in the field of environmental protection. In this scenario, the electrocatalytic reduction of nitrate (NO3−RR) to ammonia (NH3) presents a promising pathway for both NO3− removal and NH3 synthesis. Li reported the use of a surface electric field induced by the Schottky barrier to promote the enrichment and fixation of all NOx− ions (including NO3− and NO2−) on the electrode surface by constructing high-density electron-deficient nickel nanoparticles in nitrogen-rich carbon, thus ensuring the ultimate selectivity for NH3.51 Meanwhile, Lu et al. constructed an internal electric field and successfully accumulated a relatively high concentration of NO3− ions near the surface area of the electrocatalyst, thus promoting mass transfer at ultra-low concentration for effective nitrate removal.153 By stacking CuCl (111) and rutile TiO2 layers to construct a model electrocatalyst, electron transfer from TiO2 to CuCl is successfully formed, thereby inducing the formation of an internal electric field. Molecular dynamics simulations and finite element analysis show that the formation of this built-in electric field can effectively trigger NO3− ions to accumulate at the interface around the electrocatalyst. In addition, the electric field can also increase the energy of the key reaction intermediate *NO, thus reducing the energy barrier of the determination step.
Very recently, Jain et al. demonstrated a plasmon-enhanced NO3−RR system based on Au NPs.154 Local surface plasmon resonance exists in gold nanoparticles, so they have the dual advantages of electrochemical nitrate reduction activity and visible light trapping ability. Plasma excitation of electrocatalysts can induce ammonia synthesis, and the activity of electrocatalysts is 15 times higher than that of conventional electrocatalysis. In addition to the hot electron promoting NRR catalytic reaction, another mechanism exists in local surface plasmon resonance.151 Using AuCu as the model catalyst (Fig. 13f), a strong local electric field will be generated near the catalyst surface under light. Electrostatic interactions at the interface cause water molecules to bind with cations from the solution. This binding weakens the intermolecular hydrogen bonds. Consequently, an ordered structure of water molecules forms near the catalyst surface. This entropy reduction process can enhance the energy conversion and increase the charge transfer efficiency and reaction rate. As a result, the catalyst exhibits an NH3 production rate of 52.09 gH−1 cm−2 and an FE of 45.82% at −0.2 V vs. RHE (Fig. 13g).
4.4 Organic electrosynthesis
Organic electrosynthesis is a method of synthesizing high-value organic compounds using electrochemical reactions.30,155 This method realizes the reduction and oxidation of substances through the gain and loss of electrons, avoids the use of oxidants and reducing agents in traditional chemical reactions, and has the characteristics of green, environmental protection and mild.156 The role of field-effect in organic electromechanical synthesis is mainly reflected in the intramolecular electrostatic interaction through space, that is, a substituent generates an electric field in space, which affects the reaction center in another place. The field-effect is related to the structure of the molecule, which can affect the acid–base and reaction rate of the compound.29,157 The field-effect can promote the accumulation of reactants on the electrode surface, thus accelerating the mass transfer process and improving the reaction rate and selectivity.Recently, starting with the analysis of the reaction process and possible side reactions, Zhang's team designed and synthesized NiCo2O4 nanotip with high curvature as the anode catalyst realized the bromine radical mediated epichlorohydrin (ECH) one-pot membrane free synthesis.158 The high curvature nanotip structure can accelerate the electrosynthesis of ECH by enhancing the electric field to enrich the reactants Br− and OH− ions (Fig. 14a and b). The NiCo2O4 nanotip has a low energy barrier for *OH formation and a high energy barrier for *OH oxidation, coupled with an isolated Br adsorption site. These characteristics enable it to efficiently produce OH− and Br− radical intermediates while suppressing the competitive oxygen and bromine evolution reactions (Fig. 14c). The NiCo2O4 nanotips have been shown to achieve high FE in ECH diaphragm-free electrosynthesis, which the FE reaches 67% at 1.3 V vs. Ag/AgCl (Fig. 14d). This performance significantly surpasses the profit target set by technical economic analysis, which requires an efficiency greater than 21%. At the same time, The NiCo2O4 nano-cone catalyst with high curvature, effectively suppresses side reactions of oxygen and chlorine precipitation, enhancing the Faraday efficiency of α,α-dichlorone production.160 The concentration of substrate Cl− and OH− enriched by the tip local electric field increased the reaction rate. Furthermore, Zhang et al. used a combination of nano-tip enhanced electric field and surfactant modified interface microenvironment to enable the electrocatalytic deuteration of aryl acetonitrile in D2O with 80% FE at −100 mA cm−2.159 The large electric field at the tip facilitates the transfer of aryl acetonitrile and the production of surfactants, which inhibit D2 evolution (Fig. 14e). This leads to an increase in aryl acetonitrile concentration with low activation energy.
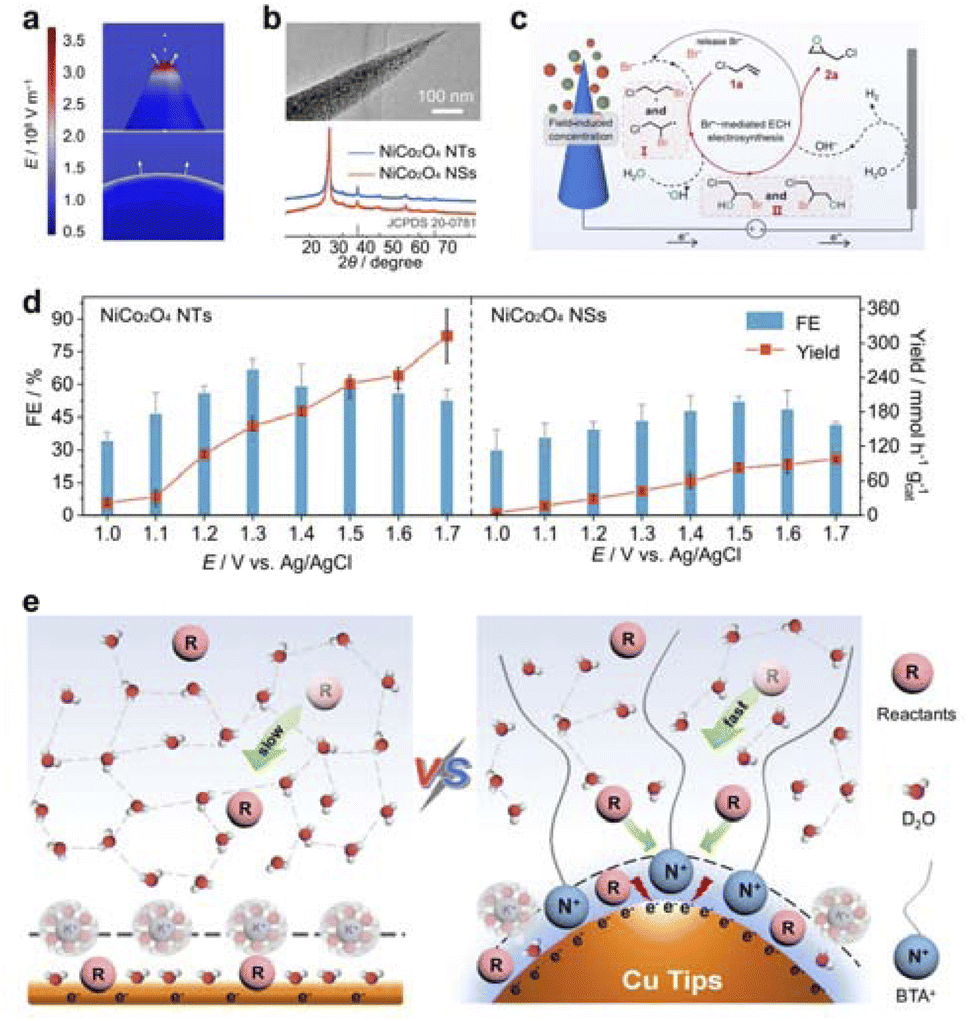 | ||
| Fig. 14 (a) Electric field on the surface of NiCo2O4 samples with different tip radii of 2 nm (top) and 40 nm (bottom). (b) TEM image and XRD patterns of NiCo2O4 NTs. (c) Proposed reaction mechanism. (d) Potential-dependent FEs and reaction rates toward ECH. Reproduced with permission from ref. 158 Copyright 2023 American Chemical Society. (e) The control strategy for interfacial microenvironment involves modifying the deuterophilic/deuterophobic property and local electric field to overcome the trade-off between high FE and current densities. Reproduced with permission from ref. 159 Copyright 2024 Springer Nature. | ||
Su et al. reported an electric field-enhanced ammoxylation system using a supported iron cluster catalyst (Fe/NC) that was able to efficiently synthesize nitrile from the corresponding aldehydes at ambient pressure at room temperature.161 The combination of an external electric field with the Fe/NC catalyst enhances ammonia activation and the dehydrogenation of imine intermediates. This synergy prevents the reverse reaction that leads to undesired aldehydes. In addition, Xiong et al. successfully constructed a light-driven catalytic CO2 hydrogenation platform by combining a centimeter-level Au sandwich plasmonic metamaterial absorber with a single copper atom-based alloy (Ag8Cu1) using template-assisted colloidal lithography.162 Due to the periodic metamaterial structure, the designed materials achieve ultra-wideband (370–1040 nm) light absorption with a strong local electric field. The intense local electric field fulfills several key functions in photocatalytic CO2 hydrogenation. This electric field facilitates the transfer of hot electrons, enhances CO2 activation, and lowers the potential barrier during the hydrogenation process. Under the synergistic action of photothermal catalysis and local electric field, the photocatalytic CO2 hydrogenation yields for 24 h reached 1106 and 301 mmol m−2, respectively. At the same time, they designed a novel Ni-based selective metamaterial absorber and utilized it as a photothermal catalyst for CO2 hydrogenation.163 The structure has selective absorption properties that enable efficient capture of sunlight and minimize heat loss through radiation, significantly amplifying the local photothermal temperature. The plasmon resonance enhances the local electric field and promotes the adsorption and activation of the reactants. Under the irradiation condition of 0.8 W cm−2, the CO2 conversion rate of the catalyst reached an unprecedented 516.9 mmol gcat h−1.
4.5 Biomass recycling
The biomass recycling refers to the conversion of biomass materials, such as agricultural and forestry waste as well as organic solid waste, into high-value chemicals through a series of sustainable processes. This process not only helps to reduce environmental pollution but also to achieve sustainable use of resources. Otherwise, biomass waste, including harmful drugs, bacteria, and microplastics, can pollute the environment. Recently, many studies have found that the field-effect plays an important role in the accumulation of harmful ions in sewage, the inactivation of harmful bacteria, and the activation of important intermediates.164–169For biomass degradation, especially the recycling of plastics, the field-effect can enhance the reaction activity. For instance, Zhu et al. demonstrated that a self-driven organic nanocrystal is capable of long-range electrophoresis for degradation by photoelectric field effect on the crystal surface.170 Perylene 3,4,9,10-tetracarboxylic acid dianhydride (PTCDA) nanocrystals can form an outer electric field (OEF) with a strength of 13.6 kV m−1 in the range of 25 μm on the nanocrystalline surface (Fig. 15a and b). OEF-driven PTCDA nanocrystals more effectively migrate to the surface of microplastics, facilitating solid–solid phase catalytic reactions that decompose the microplastics into CO2. This process achieves a notable decomposition rate of 196.8 mg h−1 for the microplastics (Fig. 15c). Moreover, Zhan et al. developed a general strategy to achieve >85% selective production of diesel olefins (C15–C28) from polyolefin waste plastics through a single reactive oxygen species (ROS).172 Modulating the generation of a single ROS with a tetrad (4-carboxyphenyl) porphyrin supramolecule (TCPP) containing different central metals shows that Zn–TCPP produces ˙O2− due to the strong internal electric field (IEF). Due to the positivity of hydrogen atoms and the high dissociation energy of C–H bonds, ˙O2− can promote the direct dehydrogenation of polyethylene (PE). Furthermore, Duan et al. designed an Au/Ni(OH)2 catalyst for the oxidation of glycerol.171 Initially, the lone pair electrons from the hydroxyl groups of glycerol form a σ bond with the 6p orbital of Au, and the o-hydroxyl group engages in a hydrogen bond with the surface hydroxyl groups of Ni(OH)2. This interaction effectively enriches the adsorption of glycerol molecules at the Au–Ni(OH)2 interface. As a result, the LSV activity of glycerol would be higher (Fig. 15d). Subsequently, under the influence of the surface electric field, the hydroxide ions (OH−) from the electrolyte are activated on the Au surface, generating adsorbed reactive oxygen species (OH*) (Fig. 15e and f). Concurrently, glycerol is oxidized to glyceraldehyde (GLD) and dihydroxyacetone (DHA). Finally, DHA/GLD is converted to lactic acid by a spontaneous base-catalyzed process under alkaline conditions.
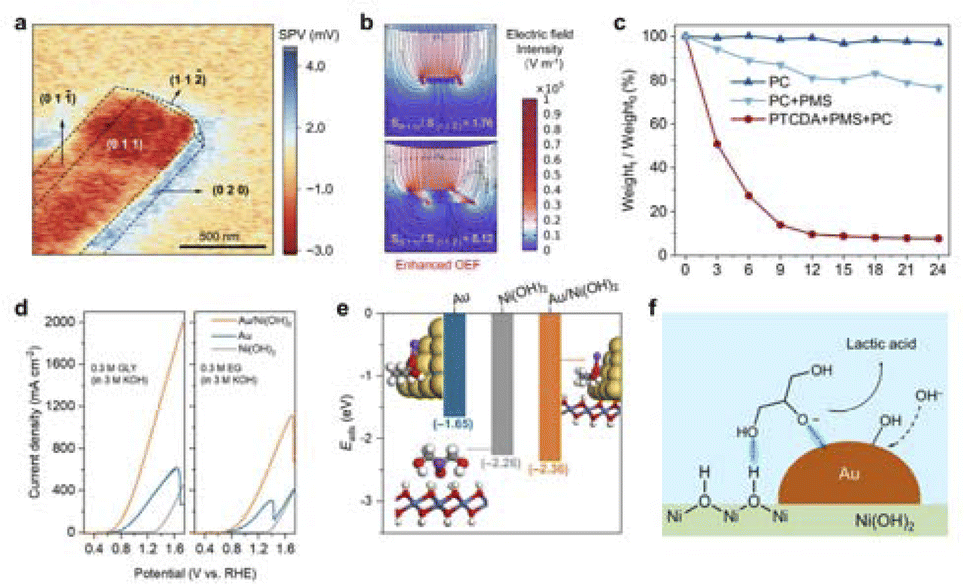 | ||
| Fig. 15 (a) Surface photovoltage microscope by kelvin probe force microscope on PTCDA nanocrystals. (b) Analysis of the electric field generated by light on the surface of PTCDA nanocrystals using finite element method (FEM). (c) The weight fluctuation pattern of microplastics made from polycarbonate (PC). Reproduced with permission from ref. 170 Copyright 2024 Springer Nature. (d) LSV curves depicting the electrooxidation of glycerol (GLY-left) and ethylene glycol (EG-right) using various catalysts. (e) Adsorption energies of GLY in the form of alkoxide on Au, Ni(OH)2, and Au/Ni(OH)2, respectively. (f) Schematic depiction of the adsorption arrangement of glycerol alkoxide on the interface between Au and Ni(OH)2. Reproduced with permission from ref. 171 Copyright 2023 American Chemical Society. | ||
As for the environmental treatment of wastewater, the field-effect has many applications. For example, Kim et al. designed double-sided gold nanorods/silica composite structures.173 LSPR harnessed by the catalyst converts photons from the visible and near-infrared sunlight regions into heat and hot electrons, which are instrumental in the advanced oxidation process for the degradation of organic micropollutants such as benzoic acid with persulfate. Moreover, Huang et al. used carbon nanotubes (CNTs) as a general method of electric field-induced bipolar electrodes to control electron transfer for efficient water purification.174 The electric field plays a crucial role in the electron transfer process. The energy barrier for electron transfer on CNTs can be lowered by the electric field. This effect induces the formation of bipolar electrodes by electrochemical polarisation of the CNTs. As a result, the electric field enhanced electron transfer and electrostatic interactions with reactants. Xie et al. developed a locally enhanced electric field treatment technology and applied it to water disinfection, enabling efficient inactivation of bacteria with low energy consumption.169 Locally enhanced electric field treatment (LEEFT) enables ultra-fast bacterial inactivation at the nanosecond level. In this process, the charge accumulation effect at the tip of the nanoneedle electrode causes nearby bacteria to be inactivated by ultrafine electroporation. LEEFT breaks the speed limit of traditional electric field treatment to inactivate bacteria, significantly reducing the required electric field strength and treatment time, and reducing the total energy consumption.
5. Summary and perspective
This review provides a groundbreaking and interdisciplinary overview of recent advances in field effects in energy and environmental catalysis, systematically covering the definition, classification, role, catalytic reaction applications, and outlook of field-effect. Field-effect enhanced catalysis represents a promising frontier in boosting the efficiency of catalytic reactions. Within catalytic systems, the field-effect facilitates mass transfer, modulates reaction pathways, enhances electronic excitation, and optimizes charge separation. This approach leverages a variety of internal fields—such as interfacial, local electric, coordinate, and polarization fields—as well as external fields like light, electric, magnetic, and strain fields to augment the performance of energy and environmental conversion reactions. Although much progress has been made, the field is still in its infancy and much remains to be explored by researchers.5.1 Deeper understanding of field-effect related mechanisms
Theoretically, the field-effect is capable of inducing multiple influences. For example, the gate electric field can simultaneously modulate the conductivity of the catalyst and the concentration distribution of charged substances in the catalyst-electrolyte. The light field can simultaneously increase the local temperature rise and enhance the internal electromagnetic field of the catalyst. The uniform magnetic field can simultaneously promote mass transfer and accelerate gas evolution from the electrodes. Furthermore, the alternating magnetic field can simultaneously modulate the temperature of the magnetic catalyst and the spin state of the reaction intermediate. In addition, the strain field can simultaneously change the temperature and the d-band center of metal-based catalysts. Currently, understanding and predicting the influences of a field-effect primarily employ finite element and first principles analysis, such as COMSOL and DFT, which are highly effective tools. However, understanding the multiple influences of field-effect is still far away. There is an urgent need for a deeper understanding of the mechanisms by which field-effect enhances catalytic performance. Advanced multiscale modeling techniques using artificial intelligence that integrate simulations from atomic to macroscopic scales maybe clarify the mechanism of field-effect.5.2 Principles of field-effect generation
The principle of the field effect in catalysis mainly involves the modulation of the electronic properties of the catalyst through an external electric field or a change in internal charge distribution within the catalyst. This modulation subsequently influences the activity and selectivity of the catalytic reaction. Rational design of catalysts with internal field effects, In general, non-centrosymmetric highly polar catalyst structures can produce strong internal field effects. Strategies such as elemental atom doping, defect engineering, heterojunction design, surface/interface interactions, and electronic structure modulation can generate internal field effects, thereby enhancing the catalytic process. In addition, the microstructural morphology design of catalysts is also crucial to realizing internal field effects. External field design utilizes non-directional external stimuli such as light, electricity, magnetism, and strain. Constructing external field effects is a favorable way to enhance the entire reaction process, including mass transfer, adsorption, and activation. However, internal and external fields are sometimes generated concurrently and interact with each other. The true principles of field effect generation still need further exploration.5.3 Application of the field-effect with in situ characterization
A deeper understanding of the mechanisms behind field-enhanced catalysis can significantly inform the design and optimization of reaction systems. Achieving this requires advancements in electrochemical in situ characterization methods and integrated analytical tools. For instance, in situ techniques like spectroscopy, microscopy, and electrochemical measurements enable observing structural changes in catalysts influenced by field effects, providing valuable insights into catalytic performance. Notably, Wei et al. recently applied in situ attenuated total reflectance-surface-enhanced infrared absorption spectroscopy (ATR-SEIRAS) to study plasma-enhanced ethanol oxidation at a molecular level, highlighting its utility in exploring catalytic processes in real-time.175 Presently, most research emphasizes in situ analysis of electrocatalytic reactions under light fields; however, there remains a need for techniques that can similarly elucidate reactions under various alternative fields. Integrating advanced materials characterization techniques with theoretical simulations is also essential to uncover catalytic reactions under field effects and identify precise reaction sites, ultimately guiding the rational design of catalysts to maximize their effectiveness. A systematic approach combining characterization, theoretical calculations, and experimental techniques will provide a comprehensive analysis of field-effect properties, serving as a scientific foundation for future research in this area.In conclusion, overcoming these challenges will allow for the rational design of internal field catalysts and more effective utilization of external fields to accelerate catalytic reactions. We hope this perspective will inspire more innovative research in field-effect catalysis and draw further attention to its potential in energy and environmental applications.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Author contributions
H. J. W. Li was responsible for conducting the literature review and drafting the initial manuscript. M. Liu was instrumental in conceiving the overarching concept, contributing to the overall planning, and participating in the writing and editing processes. H. Li, M. Du, E. Zhou, and W. R. Leow engaged in discussions and refined the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 22376222, 22002189, 52202125, and 52372253), Science and Technology Innovation Program of Hunan Province (Grant No. 2023RC1012), Central South University Research Programme of Advanced Interdisciplinary Studies (Grant No. 2023QYJC012). We are grateful for technical support from the High Performance Computing Center of Central South University.Notes and references
- M. Luo and S. Guo, Nat. Rev. Mater., 2017, 2, 17059 CrossRef.
- Y. Zhou, F. Che, M. Liu, C. Zou, Z. Liang, P. De Luna, H. Yuan, J. Li, Z. Wang, H. Xie, H. Li, P. Chen, E. Bladt, R. Quintero-Bermudez, T.-K. Sham, S. Bals, J. Hofkens, D. Sinton, G. Chen and E. H. Sargent, Nat. Chem., 2018, 10, 974–980 CrossRef PubMed.
- C. Tian, R. Dorakhan, J. Wicks, Z. Chen, K.-S. Choi, N. Singh, J. A. Schaidle, A. Holewinski, A. Vojvodic, D. G. Vlachos, L. J. Broadbelt and E. H. Sargent, Nat. Catal., 2024, 7, 350–360 CrossRef.
- B. Belsa, L. Xia, V. Golovanova, B. Polesso, A. Pinilla-Sánchez, L. San Martín, J. Ye, C.-T. Dinh and F. P. García de Arquer, Nat. Rev. Mater., 2024, 9, 535–549 CrossRef.
- F. Li, C. Zhou and A. Klinkova, Phys. Chem. Chem. Phys., 2022, 24, 25695–25719 RSC.
- Z. Liu, W. R. Leow and X. Chen, Small Methods, 2018, 3, 1800295 CrossRef.
- J. Gargiulo, R. Berte, Y. Li, S. A. Maier and E. Cortes, Acc. Chem. Res., 2019, 52, 2525–2535 CrossRef.
- L. Chen, J. T. Ren and Z. Y. Yuan, Adv. Energy Mater., 2023, 13, 2203720 CrossRef.
- L. Luo, L. Xu, Q. Wang, Q. Shi, H. Zhou, Z. Li, M. Shao and X. Duan, Adv. Energy Mater., 2023, 13, 2301276 CrossRef.
- M. Liu, Y. Pang, B. Zhang, P. De Luna, O. Voznyy, J. Xu, X. Zheng, C. T. Dinh, F. Fan, C. Cao, F. P. G. de Arquer, T. S. Safaei, A. Mepham, A. Klinkova, E. Kumacheva, T. Filleter, D. Sinton, S. O. Kelley and E. H. Sargent, Nature, 2016, 537, 382–386 CrossRef PubMed.
- P. Wang, M. Yan, J. Meng, G. Jiang, L. Qu, X. Pan, J. Z. Liu and L. Mai, Nat. Commun., 2017, 8, 645 CrossRef PubMed.
- G. Liu, P. Li, G. Zhao, X. Wang, J. Kong, H. Liu, H. Zhang, K. Chang, X. Meng, T. Kako and J. Ye, J. Am. Chem. Soc., 2016, 138, 9128–9136 CrossRef PubMed.
- G. S. Karlberg, J. Rossmeisl and J. K. Nørskov, Phys. Chem. Chem. Phys., 2007, 9, 5158–5161 RSC.
- P. Christopher, H. Xin and S. Linic, Nat. Chem., 2011, 3, 467–472 CrossRef.
- Y. Shi, J. Wang, C. Wang, T.-T. Zhai, W.-J. Bao, J.-J. Xu, X.-H. Xia and H.-Y. Chen, J. Am. Chem. Soc., 2015, 137, 7365–7370 CrossRef.
- J. Wang, M. Yan, K. Zhao, X. Liao, P. Wang, X. Pan, W. Yang and L. Mai, Adv. Mater., 2016, 29, 1604464 CrossRef.
- L. D. Chen, M. Urushihara, K. Chan and J. K. Nørskov, ACS Catal., 2016, 6, 7133–7139 CrossRef.
- K. M. Choi, D. Kim, B. Rungtaweevoranit, C. A. Trickett, J. T. D. Barmanbek, A. S. Alshammari, P. Yang and O. M. Yaghi, J. Am. Chem. Soc., 2016, 139, 356–362 CrossRef.
- H. J. W. Li, H. Zhou, K. Chen, K. Liu, S. Li, K. Jiang, W. Zhang, Y. Xie, Z. Cao, H. Li, H. Liu, X. Xu, H. Pan, J. Hu, D. Tang, X. Qiu, J. Fu and M. Liu, Sol. RRL, 2019, 4, 1900416 CrossRef.
- P. An, L. Wei, H. Li, B. Yang, K. Liu, J. Fu, H. Li, H. Liu, J. Hu, Y.-R. Lu, H. Pan, T.-S. Chan, N. Zhang and M. Liu, J. Mater. Chem. A, 2020, 8, 15936–15941 RSC.
- W. Ni, Y. Xue, X. Zang, C. Li, H. Wang, Z. Yang and Y.-M. Yan, ACS Nano, 2020, 14, 2014–2023 CrossRef.
- H. Li, H. Zhou, Y. Zhou, J. Hu, M. Miyauchi, J. Fu and M. Liu, Chin. J. Catal., 2022, 43, 519–525 CrossRef.
- B. Yang, K. Liu, H. Li, C. Liu, J. Fu, H. Li, J. E. Huang, P. Ou, T. Alkayyali, C. Cai, Y. Duan, H. Liu, P. An, N. Zhang, W. Li, X. Qiu, C. Jia, J. Hu, L. Chai, Z. Lin, Y. Gao, M. Miyauchi, E. Cortés, S. A. Maier and M. Liu, J. Am. Chem. Soc., 2022, 144, 3039–3049 CrossRef.
- Y.-X. Lin, S.-N. Zhang, Z.-H. Xue, J.-J. Zhang, H. Su, T.-J. Zhao, G.-Y. Zhai, X.-H. Li, M. Antonietti and J.-S. Chen, Nat. Commun., 2019, 10, 4380 CrossRef PubMed.
- W. Liao, K. Liu, J. Wang, A. Stefancu, Q. Chen, K. Wu, Y. Zhou, H. Li, L. Mei, M. Li, J. Fu, M. Miyauchi, E. Cortés and M. Liu, ACS Nano, 2022, 17, 411–420 CrossRef PubMed.
- H. Wang, C. Zhang, B. Liu, W. Li, C. Jiang, Z. Ke, D. He and X. Xiao, Adv. Mater., 2024, 36, 2401036 Search PubMed.
- M. Zhang, Z. Zhang, S. Zhang, Z. Zhuang, K. Song, K. Paramaiah, M. Yi, H. Huang and D. Wang, ACS Catal., 2024, 14, 10437–10446 CrossRef.
- W. Donphai, N. Piriyawate, T. Witoon, P. Jantaratana, V. Varabuntoonvit and M. Chareonpanich, J. CO2 Util., 2016, 16, 204–211 CrossRef.
- S. Kiatphuengporn, P. Jantaratana, J. Limtrakul and M. Chareonpanich, Chem. Eng. J., 2016, 306, 866–875 CrossRef.
- A. Wiebe, T. Gieshoff, S. Möhle, E. Rodrigo, M. Zirbes and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 5594–5619 CrossRef PubMed.
- R. Michiels, Y. Engelmann and A. Bogaerts, J. Phys. Chem. C, 2020, 124, 25859–25872 CrossRef.
- J. Ryu and Y. Surendranath, J. Am. Chem. Soc., 2020, 142, 13384–13390 CrossRef PubMed.
- H. Li, F. Qin, Z. Yang, X. Cui, J. Wang and L. Zhang, J. Am. Chem. Soc., 2017, 139, 3513–3521 CrossRef PubMed.
- M. Černík, J. Nosek, J. Filip, J. Hrabal, D. W. Elliott and R. Zbořil, Water Res., 2019, 154, 361–369 CrossRef PubMed.
- P. Liu, J. Zhou, T. Wang, C. Yu, Y. Hong and X. Xie, Environ. Sci.: Nano, 2020, 7, 2021–2031 RSC.
- J. Zhou, T. Wang, W. Chen, B. Lin and X. Xie, Environ. Sci.: Nano, 2020, 7, 397–403 RSC.
- J. Zhou, T. Wang and X. Xie, Environ. Sci. Technol., 2020, 54, 14017–14025 CrossRef CAS PubMed.
- J. Zhou, C. Yu, T. Wang and X. Xie, J. Mater. Chem. A, 2020, 8, 12262–12277 RSC.
- J. Yao, W. Huang, W. Fang, M. Kuang, N. Jia, H. Ren, D. Liu, C. Lv, C. Liu, J. Xu and Q. Yan, Small Methods, 2020, 4, 2000494 CrossRef.
- C. H. Kim, Y. Wang and C. D. Frisbie, Anal. Chem., 2019, 91, 1627–1635 CrossRef PubMed.
- C. H. Kim and C. D. Frisbie, J. Am. Chem. Soc., 2016, 138, 7220–7223 CrossRef PubMed.
- M. Shur and M. Hack, J. Appl. Phys., 1984, 55, 3831–3842 CrossRef.
- L. Britnell, R. V. Gorbachev, R. Jalil, B. D. Belle, F. Schedin, A. Mishchenko, T. Georgiou, M. I. Katsnelson, L. Eaves, S. V. Morozov, N. M. R. Peres, J. Leist, A. K. Geim, K. S. Novoselov and L. A. Ponomarenko, Science, 2012, 335, 947–950 CrossRef.
- Y. Wang, C. H. Kim, Y. Yoo, J. E. Johns and C. D. Frisbie, Nano Lett., 2017, 17, 7586–7592 CrossRef PubMed.
- U. E. Steiner and T. Ulrich, Chem. Rev., 1989, 89, 51–147 CrossRef.
- S. Shaik and T. Stuyver, Effects of electric fields on structure and reactivity: new horizons in chemistry, Royal Society of Chemistry, 2021 Search PubMed.
- J. Low, J. Yu, M. Jaroniec, S. Wageh and A. A. Al-Ghamdi, Adv. Mater., 2017, 29, 201601694 CrossRef PubMed.
- J. Zhu, S. Pang, T. Dittrich, Y. Gao, W. Nie, J. Cui, R. Chen, H. An, F. Fan and C. Li, Nano Lett., 2017, 17, 6735–6741 CrossRef PubMed.
- Q. Liang, L. Zhong, C. Du, Y. Luo, J. Zhao, Y. Zheng, J. Xu, J. Ma, C. Liu, S. Li and Q. Yan, ACS Nano, 2019, 13, 7975–7984 CrossRef PubMed.
- Y. Guo, W. Shi and Y. Zhu, EcoMat, 2019, 1, e12007 CrossRef.
- P. Gao, Z. H. Xue, S. N. Zhang, D. Xu, G. Y. Zhai, Q. Y. Li, J. S. Chen and X. H. Li, Angew. Chem., Int. Ed., 2021, 60, 20711–20716 CrossRef CAS PubMed.
- Z. Zhuang, Y. Li, Z. Li, F. Lv, Z. Lang, K. Zhao, L. Zhou, L. Moskaleva, S. Guo and L. Mai, Angew. Chem., Int. Ed., 2018, 57, 496–500 CrossRef CAS.
- J. Hu, A. Al-Salihy, J. Wang, X. Li, Y. Fu, Z. Li, X. Han, B. Song and P. Xu, Adv. Sci., 2021, 8, e2103314 CrossRef.
- S. N. Zhang, P. Gao, L. H. Sun, J. S. Chen and X. H. Li, Chem.–Eur. J., 2022, 28, e202103918 CrossRef CAS.
- W. Zhang, L. Yang, Z. Li, G. Nie, X. Cao, Z. Fang, X. Wang, S. Ramakrishna, S. Long and L. Jiao, Angew. Chem., Int. Ed., 2024, 63, e202400888 CrossRef CAS.
- N. Nie, L. Zhang, J. Fu, B. Cheng and J. Yu, Appl. Surf. Sci., 2018, 441, 12–22 CrossRef CAS.
- F. Y. Gao, S. J. Hu, X. L. Zhang, Y. R. Zheng, H. J. Wang, Z. Z. Niu, P. P. Yang, R. C. Bao, T. Ma, Z. Dang, Y. Guan, X. S. Zheng, X. Zheng, J. F. Zhu, M. R. Gao and S. H. Yu, Angew. Chem., Int. Ed., 2020, 59, 8706–8712 CrossRef CAS PubMed.
- M. Jalali, R. S. Moakhar, T. Abdelfattah, E. Filine, S. S. Mahshid and S. Mahshid, ACS Appl. Mater. Interfaces, 2020, 12, 7411–7422 CrossRef CAS.
- J. Fu, K. Liu, K. Jiang, H. Li, P. An, W. Li, N. Zhang, H. Li, X. Xu, H. Zhou, D. Tang, X. Wang, X. Qiu and M. Liu, Adv. Sci., 2019, 6, 201900796 Search PubMed.
- D. Liu, X. Li, S. Chen, H. Yan, C. Wang, C. Wu, Y. A. Haleem, S. Duan, J. Lu, B. Ge, P. M. Ajayan, Y. Luo, J. Jiang and L. Song, Nat. Energy, 2019, 4, 512–518 CrossRef CAS.
- Y. Lin, K. Liu, K. Chen, Y. Xu, H. Li, J. Hu, Y.-R. Lu, T.-S. Chan, X. Qiu, J. Fu and M. Liu, ACS Catal., 2021, 11, 6304–6315 CrossRef CAS.
- J. Chang, G. Wang, M. Wang, Q. Wang, B. Li, H. Zhou, Y. Zhu, W. Zhang, M. Omer, N. Orlovskaya, Q. Ma, M. Gu, Z. Feng, G. Wang and Y. Yang, Nat. Energy, 2021, 6, 1144–1153 CrossRef CAS.
- H. Gong, Z. Wei, Z. Gong, J. Liu, G. Ye, M. Yan, J. Dong, C. Allen, J. Liu, K. Huang, R. Liu, G. He, S. Zhao and H. Fei, Adv. Funct. Mater., 2021, 32, 202106886 Search PubMed.
- D. Guan, J. Zhou, Y. C. Huang, C. L. Dong, J. Q. Wang, W. Zhou and Z. Shao, Nat. Commun., 2019, 10, 3755 CrossRef.
- Z. Song, Y. N. Zhu, H. Liu, M. N. Banis, L. Zhang, J. Li, K. Doyle-Davis, R. Li, T. K. Sham, L. Yang, A. Young, G. A. Botton, L. M. Liu and X. Sun, Small, 2020, 16, e2003096 CrossRef.
- K. Chen, K. Liu, P. An, H. Li, Y. Lin, J. Hu, C. Jia, J. Fu, H. Li, H. Liu, Z. Lin, W. Li, J. Li, Y.-R. Lu, T.-S. Chan, N. Zhang and M. Liu, Nat. Commun., 2020, 11, 4173 CrossRef CAS.
- F. Chen, H. Huang, L. Guo, Y. Zhang and T. Ma, Angew. Chem., Int. Ed., 2019, 58, 10061–10073 CrossRef CAS.
- L. Pan, S. Sun, Y. Chen, P. Wang, J. Wang, X. Zhang, J. J. Zou and Z. L. Wang, Adv. Energy Mater., 2020, 10, 2000214 CrossRef CAS.
- J. H. Shah, B. Huang, A. M. Idris, Y. Liu, A. S. Malik, W. Hu, Z. Zhang, H. Han and C. Li, Small, 2020, 16, e2003361 CrossRef.
- C. Hu, F. Chen, Y. Wang, N. Tian, T. Ma, Y. Zhang and H. Huang, Adv. Mater., 2021, 33, e2101751 CrossRef PubMed.
- X. Jiang, J. Huang, Z. Bi, W. Ni, G. Gurzadyan, Y. Zhu and Z. Zhang, Adv. Mater., 2022, 34, 2270106 CrossRef.
- Y. Zhang, C. Liang, J. Wu, H. Liu, B. Zhang, Z. Jiang, S. Li and P. Xu, ACS Appl. Energy Mater., 2020, 3, 10303–10316 CrossRef.
- S. Ezendam, M. Herran, L. Nan, C. Gruber, Y. Kang, F. Grobmeyer, R. Lin, J. Gargiulo, A. Sousa-Castillo and E. Cortes, ACS Energy Lett., 2022, 7, 778–815 CrossRef PubMed.
- C. Hu, S. Tu, N. Tian, T. Ma, Y. Zhang and H. Huang, Angew. Chem., Int. Ed., 2021, 60, 16309–16328 CrossRef PubMed.
- H.-J. Wang, J.-S. Lin, H. Zhang, Y.-J. Zhang and J.-F. Li, Acc. Mater. Res., 2022, 3, 187–198 CrossRef.
- E. Cortés, L. V. Besteiro, A. Alabastri, A. Baldi, G. Tagliabue, A. Demetriadou and P. Narang, ACS Nano, 2020, 14, 16202–16219 CrossRef PubMed.
- C. Wang, X.-G. Nie, Y. Shi, Y. Zhou, J.-J. Xu, X.-H. Xia and H.-Y. Chen, ACS Nano, 2017, 11, 5897–5905 CrossRef PubMed.
- Z. Zhang, C. Zhang, H. Zheng and H. Xu, Acc. Chem. Res., 2019, 52, 2506–2515 CrossRef PubMed.
- J. Zhao, S. Xue, R. Ji, B. Li and J. Li, Chem. Soc. Rev., 2021, 50, 12070–12097 RSC.
- H. J. W. Li, X. Zi, J. Wu, X. Wang, L. Zhu, Q. Wang, J. Han, J. Fu, H. Li, K. Huang, Y. Chen and M. Liu, Nano Res., 2024 DOI:10.26599/NR.2025.94907146.
- D. C. Ratchford, ACS Nano, 2019, 13, 13610–13614 CrossRef.
- E. Cortés, Adv. Opt. Mater., 2017, 5, 1700191 CrossRef.
- E. Cortés, Science, 2018, 362, 28–29 CrossRef.
- D. Voiry, R. Fullon, J. Yang, E. S. C. de Carvalho Castro, R. Kappera, I. Bozkurt, D. Kaplan, M. J. Lagos, P. E. Batson, G. Gupta, A. D. Mohite, L. Dong, D. Er, V. B. Shenoy, T. Asefa and M. Chhowalla, Nat. Mater., 2016, 15, 1003–1009 CrossRef CAS PubMed.
- Y. Pan, X. Wang, W. Zhang, L. Tang, Z. Mu, C. Liu, B. Tian, M. Fei, Y. Sun, H. Su, L. Gao, P. Wang, X. Duan, J. Ma and M. Ding, Nat. Commun., 2022, 13, 3063 CrossRef CAS.
- A. J. Wilson, V. Mohan and P. K. Jain, J. Phys. Chem. C, 2019, 123, 29360–29369 CrossRef CAS.
- Y. Shi, J. Wang, C. Wang, T. T. Zhai, W. J. Bao, J. J. Xu, X. H. Xia and H. Y. Chen, J. Am. Chem. Soc., 2015, 137, 7365–7370 CrossRef.
- P. Hu, M. Yan, X. Wang, C. Han, L. He, X. Wei, C. Niu, K. Zhao, X. Tian, Q. Wei, Z. Li and L. Mai, Nano Lett., 2016, 16, 1523–1529 CrossRef.
- M. Ding, Q. He, G. Wang, H. C. Cheng, Y. Huang and X. Duan, Nat. Commun., 2015, 6, 7867 CrossRef.
- Y. He, Q. He, L. Wang, C. Zhu, P. Golani, A. D. Handoko, X. Yu, C. Gao, M. Ding, X. Wang, F. Liu, Q. Zeng, P. Yu, S. Guo, B. I. Yakobson, L. Wang, Z. W. Seh, Z. Zhang, M. Wu, Q. J. Wang, H. Zhang and Z. Liu, Nat. Mater., 2019, 18, 1098–1104 CrossRef.
- W. Wang, J. Qi, Z. Wu, W. Zhai, Y. Pan, K. Bao, L. Zhai, J. Wu, C. Ke, L. Wang, M. Ding and Q. He, Nat. Protoc., 2023, 18, 2891–2926 CrossRef PubMed.
- M. Yan, X. Pan, P. Wang, F. Chen, L. He, G. Jiang, J. Wang, J. Z. Liu, X. Xu, X. Liao, J. Yang and L. Mai, Nano Lett., 2017, 17, 4109–4115 CrossRef.
- X. Xu, M. Yan, X. Tian, C. Yang, M. Shi, Q. Wei, L. Xu and L. Mai, Nano Lett., 2015, 15, 3879–3884 CrossRef.
- J. Wan, W. Bao, Y. Liu, J. Dai, F. Shen, L. Zhou, X. Cai, D. Urban, Y. Li, K. Jungjohann, M. S. Fuhrer and L. Hu, Adv. Energy Mater., 2015, 5, 1401742 CrossRef.
- J. Wang, M. Yan, K. Zhao, X. Liao, P. Wang, X. Pan, W. Yang and L. Mai, Adv. Mater., 2017, 29, 1604464 CrossRef PubMed.
- Y. Wang, S. Udyavara, M. Neurock and C. D. Frisbie, Nano Lett., 2019, 19, 6118–6123 CrossRef PubMed.
- Z. Wang, H. H. Wu, Q. Li, F. Besenbacher, Y. Li, X. C. Zeng and M. Dong, Adv. Sci., 2019, 7, 201901382 Search PubMed.
- J. Zhang, J. Wu, X. Zou, K. Hackenberg, W. Zhou, W. Chen, J. Yuan, K. Keyshar, G. Gupta, A. Mohite, P. M. Ajayan and J. Lou, Mater. Today, 2019, 25, 28–34 CrossRef.
- Y. Yu, G. H. Nam, Q. He, X. J. Wu, K. Zhang, Z. Yang, J. Chen, Q. Ma, M. Zhao, Z. Liu, F. R. Ran, X. Wang, H. Li, X. Huang, B. Li, Q. Xiong, Q. Zhang, Z. Liu, L. Gu, Y. Du, W. Huang and H. Zhang, Nat. Chem., 2018, 10, 638–643 CrossRef PubMed.
- Y. Zhou, J. L. Silva, J. M. Woods, J. V. Pondick, Q. Feng, Z. Liang, W. Liu, L. Lin, B. Deng, B. Brena, F. Xia, H. Peng, Z. Liu, H. Wang, C. M. Araujo and J. J. Cha, Adv. Mater., 2018, 30, e1706076 CrossRef.
- L. T. Tufa, K. J. Jeong, V. T. Tran and J. Lee, ACS Appl. Mater. Interfaces, 2020, 12, 6598–6606 CrossRef PubMed.
- Y. Li, Z. Wang, Y. Wang, A. Kovács, C. Foo, R. E. Dunin-Borkowski, Y. Lu, R. A. Taylor, C. Wu and S. C. E. Tsang, Energy Environ. Sci., 2022, 15, 265–277 RSC.
- Q. Wang, L. I. Oldham, A. Giner-Requena, Z. Wang, D. Benetti, S. Montilla-Verdu, R. Chen, D. Du, T. Lana-Villarreal, U. Aschauer, N. Guijarro, J. R. Durrant and J. Luo, J. Am. Chem. Soc., 2024, 146, 34681–34689 CrossRef PubMed.
- Y. Zhang, P. Guo, S. Li, J. Sun, W. Wang, B. Song, X. Yang, X. Wang, Z. Jiang, G. Wu and P. Xu, J. Mater. Chem. A, 2022, 10, 1760–1767 RSC.
- X. Zhang, T. Wu, C. Yu and R. Lu, Adv. Mater., 2021, 33, e2104695 CrossRef PubMed.
- Y. Wang, Y. Shang, Z. Cao, K. Zeng, Y. Xie, J. Li, Y. Yao and W. Gan, Chem. Eng. J., 2022, 439, 135722 CrossRef.
- D. Voiry, H. Yamaguchi, J. Li, R. Silva, D. C. Alves, T. Fujita, M. Chen, T. Asefa, V. B. Shenoy, G. Eda and M. Chhowalla, Nat. Mater., 2013, 12, 850–855 CrossRef PubMed.
- Y. Miao, Y. Zhao, S. Zhang, R. Shi and T. Zhang, Adv. Mater., 2022, 34, e2200868 CrossRef.
- M. Li, Z. Zhao, Z. Xia, M. Luo, Q. Zhang, Y. Qin, L. Tao, K. Yin, Y. Chao, L. Gu, W. Yang, Y. Yu, G. Lu and S. Guo, Angew. Chem., Int. Ed., 2021, 60, 8243–8250 CrossRef.
- P. Strasser, S. Koh, T. Anniyev, J. Greeley, K. More, C. Yu, Z. Liu, S. Kaya, D. Nordlund, H. Ogasawara, M. F. Toney and A. Nilsson, Nat. Chem., 2010, 2, 454–460 CrossRef PubMed.
- X. Pan, X. Hong, L. Xu, Y. Li, M. Yan and L. Mai, Nano Today, 2019, 28, 100764 CrossRef.
- Y. Liu, H. Jiang and Z. Hou, Chem. Eur J., 2021, 27, 17726–17735 CrossRef.
- X. Jiang, Y. Chen, X. Zhang, F. You, J. Yao, H. Yang and B. Y. Xia, ChemSusChem, 2022, 15, e202201551 CrossRef PubMed.
- C. Wei and Z. J. Xu, Chin. J. Catal., 2022, 43, 148–157 CrossRef.
- P. Liu, B. Chen, C. Liang, W. Yao, Y. Cui, S. Hu, P. Zou, H. Zhang, H. J. Fan and C. Yang, Adv. Mater., 2021, 33, 2007377 CrossRef.
- A. Nairan, C. Liang, S.-W. Chiang, Y. Wu, P. Zou, U. Khan, W. Liu, F. Kang, S. Guo, J. Wu and C. Yang, Energy Environ. Sci., 2021, 14, 1594–1601 RSC.
- M. S. Kodaimati, R. Gao, S. E. Root and G. M. Whitesides, Chem Catal., 2022, 2, 797–815 CrossRef.
- B. Zhang, L. Zhang, Q. Tan, J. Wang, J. Liu, H. Wan, L. Miao and J. Jiang, Energy Environ. Sci., 2020, 13, 3007–3013 RSC.
- T. Li, J. Yin, D. Sun, M. Zhang, H. Pang, L. Xu, Y. Zhang, J. Yang, Y. Tang and J. Xue, Small, 2022, 18, 2106592 CrossRef.
- W. Cheng, H. Zhang, D. Luan and X. W. David Lou, Sci. Adv., 2021, 7, eabg2580 CrossRef.
- Y. Ni, L. Lin, Y. Shang, L. Luo, L. Wang, Y. Lu, Y. Li, Z. Yan, K. Zhang, F. Cheng and J. Chen, Angew. Chem., Int. Ed., 2021, 60, 16937–16941 CrossRef PubMed.
- W. Zheng, J. Yang, H. Chen, Y. Hou, Q. Wang, M. Gu, F. He, Y. Xia, Z. Xia, Z. Li, B. Yang, L. Lei, C. Yuan, Q. He, M. Qiu and X. Feng, Adv. Funct. Mater., 2019, 30, 1907658 CrossRef.
- L. Zhai, X. She, L. Zhuang, Y. Li, R. Ding, X. Guo, Y. Zhang, Y. Zhu, K. Xu, H. J. Fan and S. P. Lau, Angew. Chem., Int. Ed., 2022, 61, e202116057 CrossRef PubMed.
- S. S. Wang, L. Jiao, Y. Qian, W. C. Hu, G. Y. Xu, C. Wang and H. L. Jiang, Angew. Chem., Int. Ed., 2019, 58, 10713–10717 CrossRef.
- X. Wu, J. Wang, Z. Wang, F. Sun, Y. Liu, K. Wu, X. Meng and J. Qiu, Angew. Chem., Int. Ed., 2021, 60, 9416–9420 CrossRef PubMed.
- T. Wang, D. K. Brown and X. Xie, Nano Lett., 2021, 22, 860–867 CrossRef PubMed.
- J. Jing, J. Yang, W. Li, Z. Wu and Y. Zhu, Adv. Mater., 2022, 34, e2106807 CrossRef PubMed.
- X. Chen, J. Wang, Y. Chai, Z. Zhang and Y. Zhu, Adv. Mater., 2021, 33, 2007479 CrossRef CAS PubMed.
- J. Yang, J. Jing and Y. Zhu, Adv. Mater., 2021, 33, e2101026 CrossRef.
- Y. Liu, S. Ye, H. Xie, J. Zhu, Q. Shi, N. Ta, R. Chen, Y. Gao, H. An, W. Nie, H. Jing, F. Fan and C. Li, Adv. Mater., 2020, 32, e1906513 CrossRef.
- L. Li, J. Zhou, X. Wang, J. Gracia, M. Valvidares, J. Ke, M. Fang, C. Shen, J. M. Chen, Y. C. Chang, C. W. Pao, S. Y. Hsu, J. F. Lee, A. Ruotolo, Y. Chin, Z. Hu, X. Huang and Q. Shao, Adv. Mater., 2023, 35, 2302966 CrossRef CAS PubMed.
- T. Sun, Z. Tang, W. Zang, Z. Li, J. Li, Z. Li, L. Cao, J. S. Dominic Rodriguez, C. O. M. Mariano, H. Xu, P. Lyu, X. Hai, H. Lin, X. Sheng, J. Shi, Y. Zheng, Y.-R. Lu, Q. He, J. Chen, K. S. Novoselov, C.-H. Chuang, S. Xi, X. Luo and J. Lu, Nat. Nanotechnol., 2023, 18, 763–771 CrossRef CAS.
- M. Herran, S. Juergensen, M. Kessens, D. Hoeing, A. Köppen, A. Sousa-Castillo, W. J. Parak, H. Lange, S. Reich, F. Schulz and E. Cortés, Nat. Catal., 2023, 6, 1205–1214 CrossRef CAS.
- J. Xu, X.-X. Xue, G. Shao, C. Jing, S. Dai, K. He, P. Jia, S. Wang, Y. Yuan, J. Luo and J. Lu, Nat. Commun., 2023, 14, 7849 CrossRef CAS PubMed.
- X. Pan, M. Yan, Q. Liu, X. Zhou, X. Liao, C. Sun, J. Zhu, C. McAleese, P. Couture, M. K. Sharpe, R. Smith, N. Peng, J. England, S. C. E. Tsang, Y. Zhao and L. Mai, Nat. Commun., 2024, 15, 3354 CrossRef CAS PubMed.
- X. Xu, L. Meng, J. Zhang, S. Yang, C. Sun, H. Li, J. Li and Y. Zhu, Angew. Chem., Int. Ed., 2023, 63, e202308597 CrossRef PubMed.
- Q. Wang, Y. Gong, X. Zi, L. Gan, E. Pensa, Y. Liu, Y. Xiao, H. Li, K. Liu, J. Fu, J. Liu, A. Stefancu, C. Cai, S. Chen, S. Zhang, Y. R. Lu, T. S. Chan, C. Ma, X. Cao, E. Cortés and M. Liu, Angew. Chem., Int. Ed., 2024, 63, e202405438 CrossRef CAS PubMed.
- X.-Q. Wang, Q. Chen, Y.-J. Zhou, H.-M. Li, J.-W. Fu and M. Liu, Adv. Sens. Energy Mater., 2022, 1, 100023 CrossRef.
- J. Yu, J. Yin, R. Li, Y. Ma and Z. Fan, Chem Catal., 2022, 2, 2229–2252 CrossRef CAS.
- X. Zhao, M. Liu, Y. Wang, Y. Xiong, P. Yang, J. Qin, X. Xiong and Y. Lei, ACS Nano, 2022, 16, 19959–19979 CrossRef CAS PubMed.
- J. Hao, S. Xie, Q. Huang, Z. Ding, H. Sheng, C. Zhang and J. Yao, CCS Chem., 2023, 5, 2046–2058 CrossRef CAS.
- C. Cai, B. Liu, K. Liu, P. Li, J. Fu, Y. Wang, W. Li, C. Tian, Y. Kang, A. Stefancu, H. Li, C. W. Kao, T. S. Chan, Z. Lin, L. Chai, E. Cortés and M. Liu, Angew. Chem., Int. Ed., 2022, 61, e202212640 CrossRef PubMed.
- X. Zi, Y. Zhou, L. Zhu, Q. Chen, Y. Tan, X. Wang, M. Sayed, E. Pensa, R. A. Geioushy, K. Liu, J. Fu, E. Cortés and M. Liu, Angew. Chem., Int. Ed., 2023, 62, e202309351 CrossRef PubMed.
- W. Guo, S. Zhang, J. Zhang, H. Wu, Y. Ma, Y. Song, L. Cheng, L. Chang, G. Li, Y. Liu, G. Wei, L. Gan, M. Zhu, S. Xi, X. Wang, B. I. Yakobson, B. Z. Tang and R. Ye, Nat. Commun., 2023, 14, 7383 CrossRef.
- F. Xie, Z. Wang, C.-W. Kao, J. Lan, Y.-R. Lu and Y. Tan, Angew. Chem., Int. Ed., 2024, e202407661 Search PubMed.
- Q. Wang, M. Dai, H. Li, Y. R. Lu, T. S. Chan, C. Ma, K. Liu, J. Fu, W. Liao, S. Chen, E. Pensa, Y. Wang, S. Zhang, Y. Sun, E. Cortés and M. Liu, Adv. Mater., 2023, 35, 2300695 CrossRef PubMed.
- B. Wang, M. Wang, Z. Fan, C. Ma, S. Xi, L. Y. Chang, M. Zhang, N. Ling, Z. Mi, S. Chen, W. R. Leow, J. Zhang, D. Wang and Y. Lum, Nat. Commun., 2024, 15, 1719 CrossRef PubMed.
- C. Hu, X. Chen, J. Low, Y.-W. Yang, H. Li, D. Wu, S. Chen, J. Jin, H. Li, H. Ju, C.-H. Wang, Z. Lu, R. Long, L. Song and Y. Xiong, Nat. Commun., 2023, 14, 221 CrossRef PubMed.
- J. Yuan, W. Feng, Y. Zhang, J. Xiao, X. Zhang, Y. Wu, W. Ni, H. Huang and W. Dai, Adv. Mater., 2023, 36, 2303845 CrossRef.
- J. Li, S. Chen, F. Quan, G. Zhan, F. Jia, Z. Ai and L. Zhang, Chem, 2020, 6, 885–901 Search PubMed.
- S. Zhang, D. Chen, P. Chen, R. Zhang, Y. Hou, Y. Guo, P. Li, X. Liang, T. Xing, J. Chen, Y. Zhao, Z. Huang, D. Lei and C. Zhi, Adv. Mater., 2024, 36, 2310776 CrossRef PubMed.
- S. Tang, Q. Dang, T. Liu, S. Zhang, Z. Zhou, X. Li, X. Wang, E. Sharman, Y. Luo and J. Jiang, J. Am. Chem. Soc., 2020, 142, 19308–19315 CrossRef PubMed.
- W. J. Sun, H. Q. Ji, L. X. Li, H. Y. Zhang, Z. K. Wang, J. H. He and J. M. Lu, Angew. Chem., Int. Ed., 2021, 60, 22933–22939 CrossRef CAS PubMed.
- E. Contreras, R. Nixon, C. Litts, W. Zhang, F. M. Alcorn and P. K. Jain, J. Am. Chem. Soc., 2022, 144, 10743–10751 CrossRef CAS PubMed.
- N. Shida and S. Inagi, Chem. Commun., 2020, 56, 14327–14336 RSC.
- S. Guo, Y. Wu, C. Wang, Y. Gao, M. Li, B. Zhang and C. Liu, Nat. Commun., 2022, 13, 5297 CrossRef CAS PubMed.
- S. Ning, H. Ou, Y. Li, C. Lv, S. Wang, D. Wang and J. Ye, Angew. Chem., Int. Ed., 2023, 62, e202302253 CrossRef CAS PubMed.
- Y. Gao, M. Yan, C. Cheng, H. Zhong, B.-H. Zhao, C. Liu, Y. Wu and B. Zhang, J. Am. Chem. Soc., 2023, 146, 714–722 CrossRef PubMed.
- M. He, R. Li, C. Cheng, C. Liu and B. Zhang, Nat. Commun., 2024, 15, 5231 CrossRef CAS.
- J. Yao, R. Yang, C. Liu, B.-H. Zhao, B. Zhang and Y. Wu, ACS Cent. Sci., 2023, 10, 155–162 CrossRef PubMed.
- C. Wang, J. Li, T. Shao, D. Zhang, Y. Mai, Y. Li, F. Besenbacher, H. Niemantsverdriet, F. Rosei, J. Zhong and R. Su, Angew. Chem., Int. Ed., 2023, 62, e202313313 CrossRef CAS PubMed.
- T. Shao, X. Wang, H. Dong, S. Liu, D. Duan, Y. Li, P. Song, H. Jiang, Z. Hou, C. Gao and Y. Xiong, Adv. Mater., 2022, 34, 2311957 Search PubMed.
- S. Liu, X. Wang, Y. Chen, Y. Li, Y. Wei, T. Shao, J. Ma, W. Jiang, J. Xu, Y. Dong, C. Wang, H. Liu, C. Gao and Y. Xiong, Adv. Mater., 2024, 36, 2311957 CrossRef CAS.
- S. Bhattacharjee, M. Rahaman, V. Andrei, M. Miller, S. Rodríguez-Jiménez, E. Lam, C. Pornrungroj and E. Reisner, Nat. Synth., 2023, 2, 182–192 CrossRef CAS.
- F. Mo, J. Zhou, C. Yu, F. Liu, M. Jumili, Y. Wu and X. Xie, Water Res.:X, 2023, 21, 100206 CAS.
- A. Wang, M. Du, J. Ni, D. Liu, Y. Pan, X. Liang, D. Liu, J. Ma, J. Wang and W. Wang, Nat. Commun., 2023, 14, 6733 CrossRef CAS.
- B. Wu, Z. Li, Y. Zu, B. Lai and A. Wang, Water Res., 2023, 246, 120678 CrossRef CAS.
- J. Zhou, Y.-C. Hung and X. Xie, J. Hazard. Mater., 2023, 445, 130561 CrossRef CAS.
- T. Wang and X. Xie, Nat. Water, 2023, 1, 104–112 CrossRef CAS.
- Y. Guo, B. Zhu, C. Y. Tang, Q. Zhou and Y. Zhu, Nat. Commun., 2024, 15, 428 CrossRef CAS.
- Y. Yan, H. Zhou, S.-M. Xu, J. Yang, P. Hao, X. Cai, Y. Ren, M. Xu, X. Kong, M. Shao, Z. Li and H. Duan, J. Am. Chem. Soc., 2023, 145, 6144–6155 CrossRef CAS PubMed.
- S. Yue, Z. Zhao, T. Zhang, F. Li, K. Liu and S. Zhan, Angew. Chem., Int. Ed., 2024, e202406795 CAS.
- H. Wei, S. K. Loeb, N. J. Halas and J.-H. Kim, Proc. Natl. Acad. Sci. U. S. A, 2020, 117, 15473–15481 CrossRef.
- Z. Ge, X. Wang, X. Lei, W. Chen, Q. Guo, C. Lei, Y. Hu, Y.-G. Zhou, C. Feng and B. Huang, Environ. Sci. Technol., 2024, 58, 11843–11854 CrossRef.
- Y. Wei, Z. Mao, X. Y. Ma, C. Zhan and W. B. Cai, J. Phys. Chem. Lett., 2022, 13, 11288–11294 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2025 |