
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe main factor that determines the formation-efficiencies of photochemically derived one-electron-reduced species†
Naoki
Hosokawa
a,
Kyohei
Ozawa
a,
Kazuhide
Koike
b,
Yusuke
Tamaki
 c and
Osamu
Ishitani
*d
c and
Osamu
Ishitani
*d
aDepartment of Chemistry, School of Science, Institute of Science Tokyo (Tokyo Institute of Technology), 2-12-1-NE-2 O-okayama, Meguro-ku, Tokyo 152-8550, Japan
bNational Institute of Advanced Industrial Science and Technology, Onogawa 16-1, Tsukuba, Ibaraki 305-8569, Japan
cNational Institute of Advanced Industrial Science and Technology, 4-2-1 Nigatake, Miyaginoku, Sendai, Miyagi 983-8551, Japan
dDepartment of Chemistry, Graduate School of Advanced Science and Engineering, Hiroshima University, 1-3-1 Kagamiyama, Higashi-Hiroshima, Hiroshima 739 8526, Japan. E-mail: iosamu@hiroshima-u.ac.jp
First published on 28th January 2025
Abstract
While the quantum yields of photosensitiser-derived one-electron-reduced species (OERSs) significantly impact the overall efficiencies of various redox-photosensitised photocatalytic reactions, the primary factors that influence them remain unclear. In this study, we systematically compared the photochemical formation quantum yields for OERSs associated with Ru(II) and Os(II) tris-diimine, cis, trans-[ReI(diimine)(CO)2(PR3)2]+, and cyclometalated Ir(III) complexes in the presence of the same 1,3-dimethyl-2-phenyl-2,3-dihydro-1H-benzo[d]imidazole (BIH) reductant. The reduction potentials of the excited metal complexes, the heavy-atom effects of the central metal ions, and the oxidation potentials and charges of their OERSs were examined, which reveals that the driving force for photoinduced electron-transfer is the most important factor that determines the quantum yields associated with photochemical OERS formation. For complexes with higher oxidation power in their excited states, the formation quantum yield of OERSs divided by the quenching efficiency of the excited state by BIH is greater. This finding suggests that a higher photoinduced electron-transfer exergonicity promotes electron transfer over larger excited-complex/BIH distances, which in turn enables more-efficient separation of the resulting OERSs and one-electron-oxidised BIH species.
1 Introduction
Redox photosensitised reactions, also known as photoredox catalytic reactions, have been widely used in various research fields, including organic synthesis,1 to produce hydrogen2 and to reduce CO2.3 These photocatalytic reactions involve reductive quenching where a redox photosensitiser (PS) is excited (to form PS*) and subsequently generates a one-electron-reduced species (OERS, PS˙−) through the reductive quenching of PS* by an electron donor via photoinduced electron-transfer. This process results in the production of PS˙−, which is capable of reducing a substrate and/or catalyst. The quantum yield for the process in which PS is converted into PS˙− (ΦOERS) plays a crucial role in determining efficiency (i.e., the quantum yield of the overall reaction).4For instance, two different Ru(II)-complexes have been investigated as PSs in photocatalytic CO2-reduction systems using the same catalyst, namely fac-[Re(dmb)(CO)3Br] (dmb = 4,4′-dimethyl-2,2′-bipyridine), and the same reductant, namely 1,3-dimethyl-2-phenyl-2,3-dihydro-1H-benzo[d]imidazole (BIH).5 The system with the [Ru(dmb)3]2+ photosensitiser exhibited a quantum yield for the formation of CO (ΦCO) of 0.44, while ΦOERS was determined to be 0.66 under the same reaction conditions, with the exception that the catalyst was absent. In contrast, the [Ru(dmb)2(pic)]+-containing system (pic = deprotonated picolinic acid) exhibited ΦCO and ΦOERS values of 0.10 and 0.083, respectively. The observed difference in ΦCO was attributed to differences in the ΦOERS values of the photosensitisers.
We previously investigated the generation of OERSs in Ru(II) and Os(II) tris-diimine mononuclear complexes, both with and without electron-donating or electron-withdrawing groups on the diimine ligands.6 For example, [Ru(bpy)3]2+ (bpy = 2,2′-bipyridine) was determined to have an ΦOERS of 1.1, while it was 0.16 for [Os(bpy)3]2+ using BIH (0.1 M) as the reductant.7 Even though [Ru(bpy)3]2+ and [Os(bpy)3]2+ are almost identical in size and possess the same charges and ligands, they exhibited significantly different ΦOERS values. This discrepancy is possibly ascribable to differences in the quenching efficiencies (ηq) of PS* by BIH, which represents the fraction of PS* quenched by BIH. Quenching is determined by the rate constant kq[BIH] in Scheme 1, which competes with radiative (kr) and nonradiative (knr) decay processes, as expressed by eqn (1).
 | (1) |
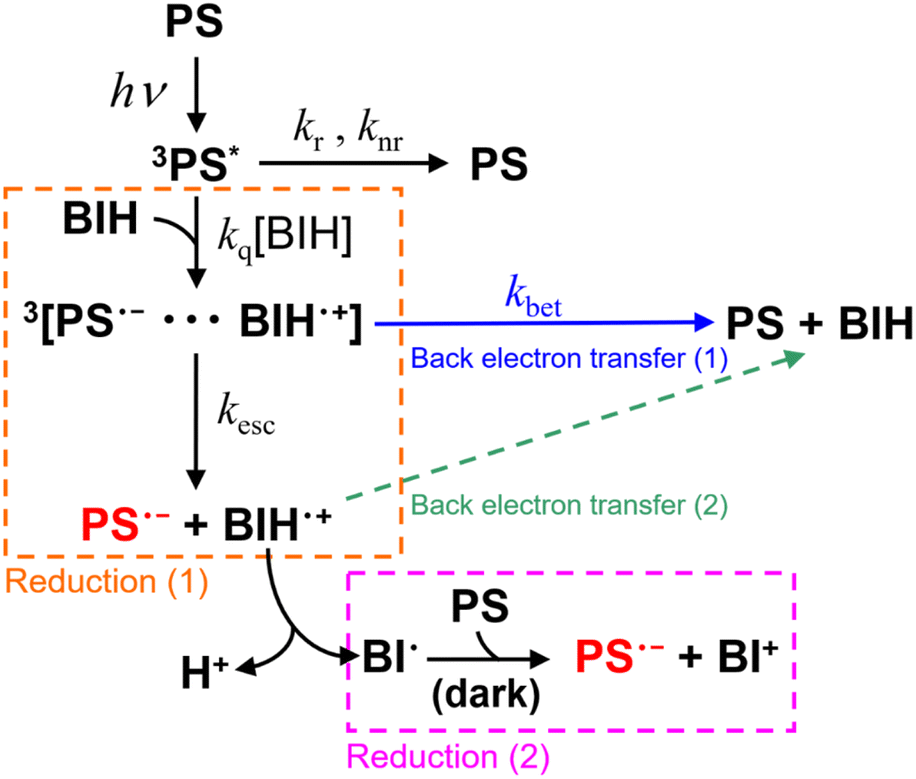 | ||
| Scheme 1 Photochemical reduction of a redox photosensitiser (PS) by BIH. | ||
We previously proposed two key factors that may influence ΦOERS based on the data acquired in experiments using the Ru(II) and Os(II) tris-diimine complexes with different photophysical and electrochemical properties; which are as follows:
1.1 The heavy-atom effect (spin–orbit coupling)
The triplet metal-to-ligand charge transfer (3MLCT) state is the lowest excited state in the Ru(II) or Os(II) tris-diimine complex.8,9 The geminate ion pair formed immediately following photoinduced electron-transfer comprises OERSs (PS˙−) and BIH˙+, and this ion pair ([PS˙−⋯BIH˙+] in Scheme 1) exists in a triplet state.10,11 Consequently, the back electron transfer within the geminate ion pair is a spin-forbidden transition because it requires a spin flip to transition from the triplet state to the singlet ground state.10–12 This back electron transfer process may be accelerated by the stronger heavy-atom effect (larger spin–orbit coupling) in an Os(II) complex, as the atomic number of the Os atom (spin–orbit coupling constant: 3531 cm−1) is much larger than that of the Ru atom (spin–orbit coupling constant: 1081 cm−1).13 As a result, the back electron transfer process may become more favourable, leading to a lower ΦOERS value.1.2 Driving force for photoinduced electron-transfer (−ΔGPET)
The driving force for the forward photoinduced electron transfer reaction (−ΔGPET) is determined by the reduction potential of PS*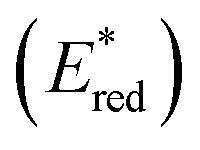 when the same electron donor (BIH) is used. For example, [Ru(bpy)3]2+ reportedly exhibits an
when the same electron donor (BIH) is used. For example, [Ru(bpy)3]2+ reportedly exhibits an 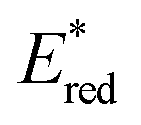 of +0.52 V (vs. Ag/AgNO3) in N,N′-dimethylacetamide (DMA), while that of [Os(bpy)3]2+ is +0.21 V; hence, the [Os(bpy)3]2+ system has a lower −ΔGPET value.6 Consequently, a shorter excited-[Os(bpy)3]2+/BIH distance is expected for photoinduced electron-transfer compared to the analogous distance in the [Ru(bpy)3]2+ system due to differences in the driving-force.14–16 This distance reflects the separation between the OERS and BIH˙+ within the geminate ion pair; a shorter OERS/BIH˙+ distance may lead to faster back electron transfer, as the electron-transfer rate is distance-dependent,17,18 resulting in a lower ΦOERS value.
of +0.52 V (vs. Ag/AgNO3) in N,N′-dimethylacetamide (DMA), while that of [Os(bpy)3]2+ is +0.21 V; hence, the [Os(bpy)3]2+ system has a lower −ΔGPET value.6 Consequently, a shorter excited-[Os(bpy)3]2+/BIH distance is expected for photoinduced electron-transfer compared to the analogous distance in the [Ru(bpy)3]2+ system due to differences in the driving-force.14–16 This distance reflects the separation between the OERS and BIH˙+ within the geminate ion pair; a shorter OERS/BIH˙+ distance may lead to faster back electron transfer, as the electron-transfer rate is distance-dependent,17,18 resulting in a lower ΦOERS value.
The oxidation potential of the OERS, which influences the driving force for the back electron transfer reaction, is another potential factor. However, the data clearly show that this is not the primary factor that determines ΦOERS/ηq in systems using these Ru(II) and Os(II) tris-diimine complexes.6
Fig. 1 illustrates the relationship between −ΔGPET and ΦOERS/ηq for Ru(II) and Os(II) tris-diimine complexes, as determined in our previous study.6 Identifying whether the heavy-atom effect, −ΔGPET, or both play a dominant role in determining ΦOERS/ηq based on this relationship is difficult because all examined Os(II) complexes have more negative 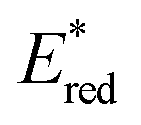 values (low −ΔGPET) and exhibit stronger heavy-atom effects than the corresponding Ru(II) complexes.
values (low −ΔGPET) and exhibit stronger heavy-atom effects than the corresponding Ru(II) complexes.
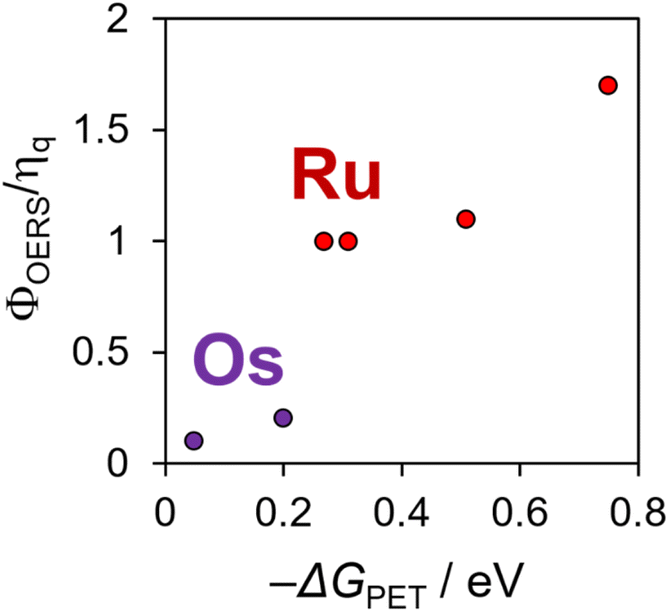 | ||
| Fig. 1 Φ OERS/ηq values of Ru(II) and Os(II) tris-diimine complexes using BIH as a reductant, as a function of the driving force for photoinduced electron-transfer: produced using the data reported in ref. 6. | ||
In this study, we examined the new Os(II) complex with a 4,4′-di(trifluoromethyl)-2,2′-bipyridine ligand (CF3bpy) [Os(CF3bpy)3]2+ (Chart 1), whose excited state has a more positive reduction potential 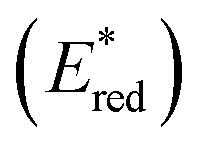 than not only the other Os(II) complexes but also most of the Ru(II) complexes. Additionally, we investigated a series of cis, trans-[Re(diimine)(CO)2(PR3)2]+ ([Re(PR3)2]+) and [Ir(C^N)2(diimine)]+ ([Ir(C^N)2(N^N)]+) type complexes as photosensitisers (PSs), as shown in Chart 1. The oxidation power of the excited Re(I) complexes, which have similar lowest triplet metal-to-ligand charge transfer (3MLCT) states as Ru(II) and Os(II) complexes,19 and excited Ir(III) complexes, whose lowest states are mixtures of 3MLCT and triplet ligand-to-ligand charge transfer (3LLCT) states,20 is generally stronger than that of the excited Os(II) tris-diimine complexes.21 Additionally, the atomic numbers of Re (75) and Ir (77) are close to that of Os (76). To systematically evaluate the effect of
than not only the other Os(II) complexes but also most of the Ru(II) complexes. Additionally, we investigated a series of cis, trans-[Re(diimine)(CO)2(PR3)2]+ ([Re(PR3)2]+) and [Ir(C^N)2(diimine)]+ ([Ir(C^N)2(N^N)]+) type complexes as photosensitisers (PSs), as shown in Chart 1. The oxidation power of the excited Re(I) complexes, which have similar lowest triplet metal-to-ligand charge transfer (3MLCT) states as Ru(II) and Os(II) complexes,19 and excited Ir(III) complexes, whose lowest states are mixtures of 3MLCT and triplet ligand-to-ligand charge transfer (3LLCT) states,20 is generally stronger than that of the excited Os(II) tris-diimine complexes.21 Additionally, the atomic numbers of Re (75) and Ir (77) are close to that of Os (76). To systematically evaluate the effect of 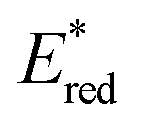 (−ΔGPET) on ΦOERS/ηq, we modified the electronic properties of the Re(I) and Ir(III) complexes by varying the phosphine ligands (PR3) in the Re(I) complexes and the bidentate ligands (C^N and N^N) in the Ir(III) complexes. Furthermore, we also investigated the impact of charge difference in determining ΦOERS/ηq because the Re(I) and Ir(III) complexes are singly (+1) charged, which differentiates them from the doubly (+2) charged Ru(II) and Os(II) diimine complexes.
(−ΔGPET) on ΦOERS/ηq, we modified the electronic properties of the Re(I) and Ir(III) complexes by varying the phosphine ligands (PR3) in the Re(I) complexes and the bidentate ligands (C^N and N^N) in the Ir(III) complexes. Furthermore, we also investigated the impact of charge difference in determining ΦOERS/ηq because the Re(I) and Ir(III) complexes are singly (+1) charged, which differentiates them from the doubly (+2) charged Ru(II) and Os(II) diimine complexes.
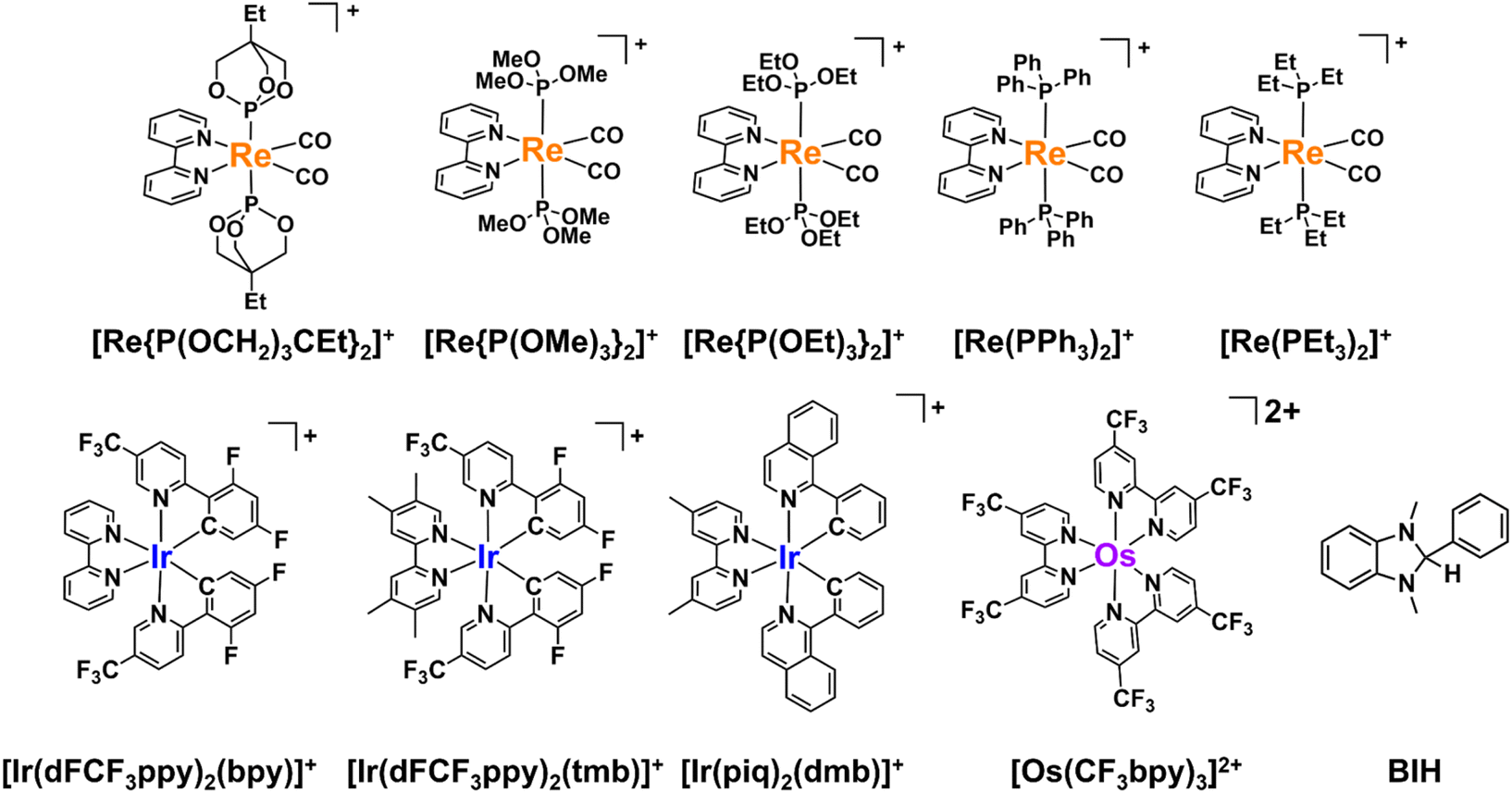 | ||
| Chart 1 Structures and abbreviations of the Re(I), Ir(III), and Ru(II) complexes, and the BIH used in this study. The counter anions of the complexes are PF6−. | ||
2 Results
2.1 Synthesis of the Re complexes and their photophysical and electrochemical properties
The [Re(PR3)2]+ complexes were synthesised using photo-ligand substitution reactions according to previous reports on the synthesis of similar Re(I) bisphosphine and bisphosphite complexes.22 The successful synthesis of [Re(PR3)2]+ was confirmed by 1H and 31P NMR spectroscopy, FTIR spectroscopy, ESI-MS spectroscopy, and elemental analysis, as described in the Experimental section.The FTIR spectra of all synthesised [Re(PR3)2]+ complexes exhibit two CO stretching vibrations (νCO) between 1800 and 2000 cm−1 (Table 1 and Fig. S1†). The [Re(PR3)2]+ complexes bearing phosphine ligands with larger Tolman's χ values23 displayed higher CO stretching vibrations (νCO), which indicates that PR3 ligands with stronger electron-withdrawing properties show lower π back donation from the central Re atom to the CO ligands because the χ value reflects the electron-withdrawing ability of the PR3 ligand. In other words, the central Re(I) ions in the [Re(PR3)2]+ complexes with higher χ values are clearly endowed with lower electron densities.
Cyclic voltammograms (CVs) of [Re(PR3)2]+ acquired in N,N′-dimethylacetamide (DMA) are shown in Fig. 2. Each complex exhibited a reversible redox wave attributable to the one-electron reduction of the bpy ligand in the reduction region (Fig. 2b),24 with potentials of up to −1.78 V (vs. Fc+/Fc). The half-wave potentials of [Re(PR3)2]+ (E1/2(PS/PS˙−)) listed in Table 2 are similar across the various complexes, and differ by no more than 70 mV. In contrast, the oxidation potentials (Ep(PS˙+/PS)) shown in Fig. 2a vary significantly among the complexes. Irreversible oxidation waves were observed in the CV oxidation region for [Re(PPh3)2]+ and [Re(PEt3)2]+, which bear PR3 ligands with relatively weak electron-withdrawing properties. However, oxidation waves were not observed for the other [Re(PR3)2]+ complexes because their oxidation potentials are more positive than the accessible potential window.
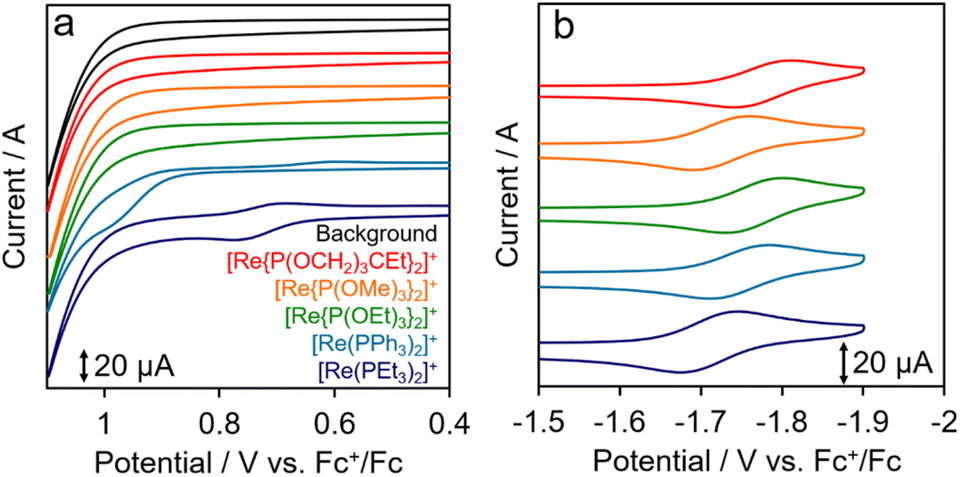 | ||
| Fig. 2 Cyclic voltammograms of [Re(PR3)2]+ (0.5 mM) acquired at 200 mV s−1 in Ar-saturated DMA containing Et4NBF4 (0.1 M) using a glassy-carbon working electrode (diameter: 3 mm) and a Pt-wire counter electrode: (a) oxidation and (b) reduction sides. | ||
| Entry | Complex | E 1/2 (PS/PS˙−)/V vs. Fc+/Fc | E 00 /eV | E 1/2 (PS*/PS˙−)/V vs. Fc+/Fc |
|---|---|---|---|---|
| a Determined by Franck–Condon analysis using emission spectra measured at 77 K (see details in the Experimental section). b Ref. 6. c Ref. 32. | ||||
| 1 | [Re{P(OCH2)3CEt}2]+ | −1.78 | 2.59 | +0.81 |
| 2 | [Re{P(OMe)3}2]+ | −1.73 | 2.43 | +0.71 |
| 3 | [Re{P(OEt)3}2]+ | −1.77 | 2.49 | +0.72 |
| 4 | [Re(PPh3)2]+ | −1.75 | 2.39 | +0.64 |
| 5 | [Re(PEt3)2]+ | −1.71 | 2.17 | +0.46 |
| 6 | [Ir(dFCF3ppy)2(bpy)]+ | −1.62 | 2.65 | +1.03 |
| 7 | [Ir(dFCF3ppy)2(tmb)]+ | −1.82 | 2.64 | +0.82 |
| 8 | [Ir(piq)2(dmb)]+ | −1.84 | 2.10c | +0.26 |
| 9 | [Os(CF3bpy)3]2+ | −1.17 | 1.75 | +0.58 |
| 10 | [Ru(4,4′-(COOMe)-bpy)3]2+ | −1.29 | 1.96 | +0.67 |
| 11 | [Ru(bpy)3]2+ | −1.73 | 2.16 | +0.43 |
| 12 | [Ru(dmb)3]2+ | −1.83 | 2.06 | +0.23 |
| 13 | [Ru(4,4′-(OMe)-bpy)3]2+ | −1.89 | 2.08 | +0.19 |
| 14 | [Os(bpy)3]2+ | −1.65 | 1.77 | +0.12 |
| 15 | [Os(dmb)3]2+ | −1.76 | 1.73 | −0.03 |
These results suggest that the electron-withdrawing properties of the PR3 ligand strongly affect the energy levels of the d orbitals of the central Re(I) atom (HOMO), while exerting only a minor influence on the energy levels of the π* orbital of the bpy ligand (LUMO).
The UV-vis absorption and emission spectra of [Re(PR3)2]+ acquired in DMA are shown in Fig. 3. The π–π* transitions of the bpy ligand correspond to absorption at λmax < 330 nm, with 1MLCT transitions appearing at λmax = 360–430 nm.19 All [Re(PR3)2]+ complexes were observed to emit at room temperature, which can be ascribed to the radiative decay of the 3MLCT excited state.19 With the exception of [Re(PPh3)2]+, emission maxima were observed at shorter wavelengths for [Re(PR3)2]+ complexes bearing PR3 ligands with higher Tolman's χ values (Fig. S2†). We previously reported that π–π interactions between the bpy ligand and the phenyl groups of the phosphine ligands in [Re(PPh3)2]+ resulted in emission at a shorter wavelength than that predicted by Tolman's χ value.25,26 Notably, a PR3 ligand with a higher χ value leads to lower d-orbital energies of the central Re(I) ion (HOMO) without significantly affecting the energy of the π* orbital of the bpy ligand (LUMO), as mentioned earlier.
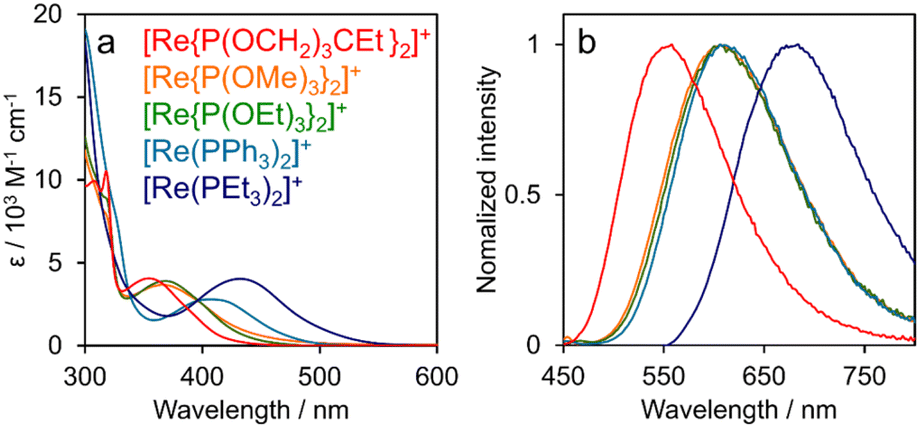 | ||
| Fig. 3 (a) UV-vis absorption and (b) emission spectra of [Re(PR3)2]+ in DMA acquired at room temperature. | ||
Emission lifetimes (τem) were determined at room temperature using the time-correlated single photon counting method (Fig. S3†), with values summarised in Table 3 along with emission quantum yields (Φem) measured at room temperature and the radiative (kr) and nonradiative (knr) decay rate constants calculated using τem and Φem.
| Entry | Complex | χ | λ max /nm (ε/103 M−1 cm−1) | λ em /nm | τ em /ns | Φ em | E 00 /eV | k r/105 s−1 | k nr/105 s−1 |
|---|---|---|---|---|---|---|---|---|---|
| a Ref. 23. b Measured at room temperature. c Ref. 6. d Determined by Franck–Condon analysis using emission spectra measured at 77 K. e Ref. 32. | |||||||||
| 1 | [Re{P(OCH2)3CEt}2]+ | 30.7 | 355 (4.1) | 558 | 1300 | 0.35 | 2.59 | 2.7 | 4.9 |
| 2 | [Re{P(OMe)3}2]+ | 23.4 | 365 (3.9) | 606 | 340 | 0.052 | 2.43 | 1.5 | 28 |
| 3 | [Re{P(OEt)3}2]+ | 20.2 | 367 (3.9) | 606 | 300 | 0.051 | 2.49 | 1.7 | 31 |
| 4 | [Re(PPh3)2]+ | 12.8 | 408 (2.8) | 606 | 690 | 0.063 | 2.39 | 0.92 | 14 |
| 5 | [Re(PEt3)2]+ | 5.6 | 432 (4.1) | 684 | 47 | 0.004 | 2.17 | 0.85 | 210 |
| 6 | [Ir(dFCF3ppy)2(bpy)]+ | — | 380 (6.2) | 500 | 1700 | 0.77 | 2.65 | 4.5 | 1.3 |
| 7 | [Ir(dFCF3ppy)2(tmb)]+ | — | 378 (5.3) | 482 | 1800 | 0.81 | 2.64 | 4.6 | 1.1 |
| 8 | [Ir(piq)2(dmb)]+ | — | 444 (7.8) | 635 | 2600 | 0.34 | 2.10e | 1.2 | 2.4 |
| 9 | [Os(CF3bpy)3]2+ | — | 493 (14.1) | 794 | 36 | 0.009 | 1.75 | 2.5 | 280 |
| 10 | [Ru(4,4′-(COOMe)-bpy)3]2+ | — | 473 (23.8) | 653 | 1050 | 0.112 | 1.96 | 1.1 | 9 |
| 11 | [Ru(bpy)3]2+ | — | 456 (14.4) | 631 | 905 | 0.143 | 2.16 | 1.6 | 10 |
| 12 | [Ru(dmb)3]2+ | — | 462 (15.0) | 641 | 758 | 0.150 | 2.06 | 2.1 | 11 |
| 13 | [Ru(4,4′-(OMe)-bpy)3]2+ | — | 482 (12.8) | 680 | 190 | 0.032 | 2.08 | 1.7 | 50 |
| 14 | [Os(bpy)3]2+ | — | 482 (14.1) | 756 | 42 | 0.004 | 1.77 | 1.0 | 240 |
| 15 | [Os(dmb)3]2+ | — | 490 (14.5) | 778 | 24 | 0.005 | 1.73 | 2.1 | 410 |
The reduction potentials of the excited complexes (E1/2(PS*/PS˙−)) were calculated using eqn (2), where E00 is the excitation energy at the 0–0 transition.
| E1/2(PS*/PS˙−) = E1/2(PS/PS˙−) + E00 | (2) |
The emission spectrum of [Re(PR3)2]+ was acquired in DMA at 77 K in an attempt to determine E00; however no vibrational structure was observed in the spectrum, as reported previously for similar Re(I) complexes.27–29 Because we were unable to directly determine the vibrational quantum numbers for the high-frequency modes (νM) of [Re(PR3)2]+ using Franck–Condon analysis,30 we estimated E00 using the νM value reported for [Re(bpy)(CO)3Cl] (i.e., 1450 cm−1),31 which contains the same diimine ligand as [Re(PR3)2]+.
Table 2 summarises the ground-state reduction potential E1/2(PS/PS˙−), E00, and the reduction potential of the excited [Re(PR3)2]+ (E1/2(PS*/PS˙−)). The oxidation potential of BIH in DMA (i.e., E1/2(BIH˙+/BIH)) was determined using the rapid-scan cyclic voltammetry method and has been reported to be −0.11 V (vs. Fc+/Fc).6 We conclude that the reductive quenching of excited [Re(PR3)2]+ by BIH is thermodynamically favourable based on these results. The free energy changes for the photoinduced electron transfer from BIH to the excited [Re(PR3)2]+ (−ΔGPET) was calculated using eqn (3) and are summarised in Table 4:
| −ΔGPET = E1/2(PS*/PS˙−) − E1/2(BIH˙+/BIH) − wp + wr | (3) |
| Entry | Complex | K SV/103 M−1 | k q/109 M−1 s−1 | η q | −ΔGPETa/eV |
|---|---|---|---|---|---|
| a −ΔGPET = −E1/2 (BIH˙+/BIH) + E1/2 (PS*/PS˙−) − wp + wr: E1/2 (BIH˙+/BIH) = −0.11 V vs. Fc+/Fc; E1/2 (PS*/PS˙−) from Table 2; wp = 0.03 eV and wr = 0 eV.6 b Ref. 6. | |||||
| 1 | [Re{P(OCH2)3CEt}2]+ | 5.4 | 4.1 | 1.0 | 0.92 |
| 2 | [Re{P(OMe)3}2]+ | 1.9 | 5.4 | 1.0 | 0.82 |
| 3 | [Re{P(OEt)3}2]+ | 1.1 | 3.7 | 1.0 | 0.83 |
| 4 | [Re(PPh3)2]+ | 2.2 | 3.2 | 1.0 | 0.75 |
| 5 | [Re(PEt3)2]+ | 0.21 | 4.4 | 0.95 | 0.57 |
| 6 | [Ir(dFCF3ppy)2(bpy)]+ | 8.0 | 4.6 | 1.0 | 1.14 |
| 7 | [Ir(dFCF3ppy)2(tmb)]+ | 7.8 | 4.4 | 1.0 | 0.93 |
| 8 | [Ir(piq)2(dmb)]+ | 9.3 | 3.6 | 1.0 | 0.37 |
| 9 | [Os(CF3bpy)3]2+ | 3.8 | 10 | 0.97 | 0.66 |
| 10 | [Ru(4,4′-(COOMe)-bpy)3]2+ | 6.7 | 6.4 | 1.0 | 0.75 |
| 11 | [Ru(bpy)3]2+ | 2.4 | 2.6 | 1.0 | 0.51 |
| 12 | [Ru(dmb)3]2+ | 1.4 | 1.9 | 1.0 | 0.31 |
| 13 | [Ru(4,4′-(OMe)-bpy)3]2+ | 0.18 | 0.94 | 0.95 | 0.27 |
| 14 | [Os(bpy)3]2+ | 2.6 × 10−2 | 0.62 | 0.72 | 0.20 |
| 15 | [Os(dmb)3]2+ | 1.8 × 10−3 | 7.5 × 10−2 | 0.15 | 0.05 |
Fig. 4 displays a Stern–Volmer plot for [Re(PEt3)2]+ when BIH was used as the quencher, which led to a calculated Stern–Volmer constant (KSV) of 2.1 × 102 M−1 for this process. Good linear Stern–Volmer plots were obtained for the other [Re(PR3)2]+ complexes (Fig. S5†). The KSV values and quenching rate constants (kq) calculated using KSV and τem are summarised in Table 4. The fractions of excited [Re(PR3)2]+ quenched by 0.1 M BIH (i.e., ηq) were determined using kq and eqn (1) (Table 4).
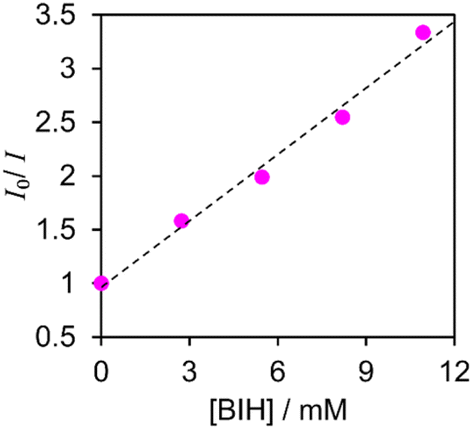 | ||
| Fig. 4 Stern–Volmer plot for [Re(PEt3)2]+ acquired in Ar-saturated DMA at room temperature in the presence of BIH. | ||
2.2 Synthesis of Ir complexes and their photophysical and electrochemical properties
The [Ir(C^N)2(N^N)]+ complexes were successfully synthesised according to previously reported methods32,33 and confirmed by NMR spectroscopy, ESI-MS spectroscopy, and elemental analysis, as described in the Experimental section.The UV-vis absorption and emission spectra of [Ir(C^N)2(N^N)]+ were acquired in DMA (Fig. S6†), which revealed that the C^N and N^N ligands exhibit bands for their π–π* transitions at λ < 370 nm.32 In addition, 1MLCT 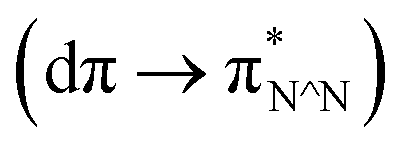 and 1LLCT
and 1LLCT 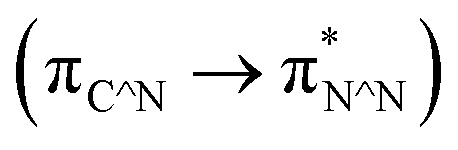 transitions were observed at λ = 370–530 nm, while 3MLCT and 3LLCT transitions appeared at λ = 450–480 nm (for [Ir(dFCF3ppy)2(bpy)]+ and [Ir(dFCF3ppy)2(tmb)]+) and λ = 520–600 nm (for [Ir(piq)2(dmb)]+).20,32 Each [Ir(C^N)2(N^N)]+ complex exhibited an emission spectrum at room temperature that originates from the radiative decay of a mixed 3LLCT and 3MLCT excited state (Fig. S6b†).20
transitions were observed at λ = 370–530 nm, while 3MLCT and 3LLCT transitions appeared at λ = 450–480 nm (for [Ir(dFCF3ppy)2(bpy)]+ and [Ir(dFCF3ppy)2(tmb)]+) and λ = 520–600 nm (for [Ir(piq)2(dmb)]+).20,32 Each [Ir(C^N)2(N^N)]+ complex exhibited an emission spectrum at room temperature that originates from the radiative decay of a mixed 3LLCT and 3MLCT excited state (Fig. S6b†).20
Vibrational structures were observed in the emission spectra acquired at low temperatures (Fig. S7†), which enabled the determination of E00 values using Franck–Condon analysis (Tables 2 and 3). Table 3 summarises the photophysical properties of [Ir(C^N)2(N^N)]+. Ground-state reduction potentials (E1/2(PS/PS˙−)) were determined by CV (Fig. S8†), while excited-state reduction potentials (E1/2(PS*/PS˙−)) were calculated using eqn (2) and are summarised in Table 2.
The E1/2(PS/PS˙−) values appear to be mainly influenced by the N^N ligand, as the first reduction in the ground state is attributable to the reduction of the N^N ligand.32 In contrast, E1/2(PS*/PS˙−) is significantly affected by the C^N ligand, which can be ascribed to the contribution of the π orbital of the C^N ligand to the lowest excited state. The photoinduced electron-transfer from BIH to the excited states of all [Ir(C^N)2(N^N)]+ complexes is also exergonic, similarly to the other metal complexes used in this research.
On the other hand, the excitation energy of the triplet excited state of BIH, with E00 = 3.04 eV (ESI†), is much higher than those of all PS* (E00 in Table 2). Therefore, energy transfer from PS* to BIH should be a very slow process and can be considered negligible for the following discussion.
Linear Stern–Volmer plots for the [Ir(C^N)2(N^N)]+ complexes were acquired using BIH as the quencher (Fig. S9†), and the corresponding KSV, kq, ηq, and −ΔGPET values are summarised in Table 4.
2.3 Photophysical and electrochemical properties of [Os(CF3bpy)3]2+
The UV-vis absorption spectrum of [Os(CF3bpy)3]2+ is shown in Fig. S10a,† which reveals bands corresponding to π–π* transitions at λ ≤ 360 nm, 1MLCT transitions at λ = 360–560 nm, and 3MLCT transitions (S–T absorptions) at λ = 560–750 nm, which can be ascribed to the heavy-atom effect of the central Os ion. The emission spectrum of [Os(CF3bpy)3]2+ at room temperature is shown in Fig. S10b,† with the CV trace presented in Fig. S11.† The photophysical and electrochemical properties of [Os(CF3bpy)3]2+ are summarised in Table 2, in which wr = 0 eV and wp = 0.03 eV were used to calculate −ΔGPET (eqn (3)).6The excitation energy (E00 = 1.75 eV) was determined by Franck–Condon analysis of the emission spectrum at 77 K (Fig. S12†). The ground-state reduction (E1/2(PS/PS˙−)) and excited-state reduction (E1/2(PS*/PS˙−)) potentials of [Os(CF3bpy)3]2+ are also summarised in Table 2. The emission of [Os(CF3bpy)3]2+ was quenched reductively by BIH (Fig. S13†); corresponding data are listed in Table 4.
2.4 UV-vis absorption spectra of one-electron-reduced species (OERSs) derived from the complexes
The UV-vis and IR absorption spectra of OERSs of the various [Re(PR3)2]+ complexes were obtained using flow electrolysis. Fig. 5 shows changes in the absorption spectrum of [Re(PEt3)2]+ (as a typical example) along with the current observed during electrolysis at various applied potentials (Eapp). The UV-vis absorption spectrum changed continuously as Eapp was varied from −1.69 to −1.99 V (vs. Fc+/Fc) (Fig. 5a). Difference spectra, obtained by subtracting the spectrum acquired in the absence of an applied potential from those obtained at various potentials, are shown in Fig. 5b. | ||
| Fig. 5 (a) UV-vis absorption spectra, (b) difference spectra calculated using the spectra in panel (a), and (c) FTIR spectra acquired during the flow electrolysis of a DMA solution containing [Re(PEt3)2]+ (0.84 mM) and Et4NBF4 (0.1 mM). (d) ΔCurrents (blue) and Δabsorbances of the visible absorption at λ = 525 nm (green) and the IR absorption at νCO = 1986 cm−1 (purple) at various applied potentials. | ||
FTIR spectra of [Re(PEt3)2]+, acquired at potentials more negative than −1.49 V, exhibited weaker ground-state carbonyl stretching vibrations accompanied by new absorptions attributed to the carbonyl stretching vibrations of the OERS of [Re(PEt3)2]+ at lower frequencies (Fig. 5c).19 These changes were observed to be complete at Eapp = −1.99 V. Fig. 5d displays plots of observed current and absorbance at both 525 nm (visible region) and 1896 cm−1 (IR region) as a function of Eapp, all of which show similar trends, with plateaus observed from about −1.99 V. Additionally, approximately one electron per molecule of [Re(PEt3)2]+ (n ≈ 1) is transferred in the flowing solution at −1.99 V (see ESI†). These results clearly indicate that the spectra observed at Eapp = −1.99 V correspond to the OERS of [Re(PEt3)2]+, from which the molar extinction coefficient of the OERS (εOERS) of [Re(PEt3)2]+ was determined to be 4300 M−1 cm−1 at 525 nm. εOERS values for the other [Re(PR3)2]+ and [Ir(C^N)2(N^N)]+ complexes were obtained using the same method (Fig. S14–S17†). In contrast, the εOERS value of [Os(CF3bpy)3]2+ was determined using the optically transparent thin-layer electrochemical (OTTLE) method because it could not be determined using flow electrolysis (Fig. S18–S20†).
2.5 Photochemical formation of OERSs
Fig. 6a shows time-dependent visible absorption spectra of a DMA solution containing [Re(PEt3)2]+ (0.2 mM) and BIH (0.1 M) under Ar during irradiation at λmaxex = 436 nm using a Xe lamp with a bandpass filter. The absorption between 400 and 800 nm was observed to increase with time, as evidenced by the difference spectra shown in Fig. 6b, which are identical to those obtained using the flow electrolysis method (Fig. 5b). These results clearly demonstrate that the photochemical reduction of [Re(PEt3)2]+ proceeds selectively to give the OERS of [Re(PEt3)2]+.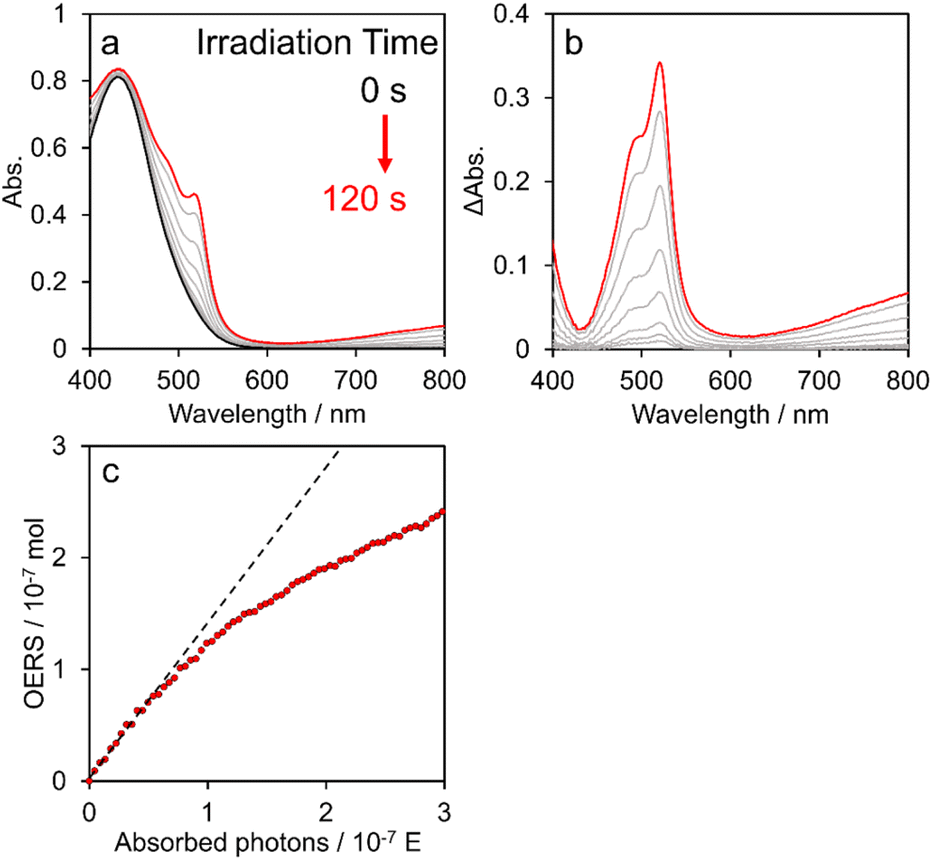 | ||
| Fig. 6 (a) Time-dependent visible absorption changes of an Ar-saturated DMA solution containing [Re(PEt3)2]+ (0.2 mM) and BIH (0.1 M) during irradiation with light at λex = 436 nm (5.2 × 10−9 E s−1) and (b) calculated difference absorption spectra before and after irradiation. (c) Relationship between the number of absorbed photons and the concentration of OERSs from [Re(PEt3)2]+ in solution. | ||
Fig. 6c shows the relationship between the calculated amount of OERSs produced from [Re(PEt3)2]+ during irradiation and the number of photons absorbed by the reaction solution; the formation rate exhibited linearity during the initial stage but then gradually decelerated, which can be attributed to the inner-filter effect of the produced OERS; specifically, the actual number of photons absorbed by ground state of [Re(PEt3)2]+ decreased owing to absorption by OERSs. Therefore, the quantum yield for OERS formation (ΦOERS) was determined from the slope of the relationship at the initial stage (Fig. 6c, black dashed line). This experiment was repeated three or four times to ensure accuracy. The obtained results led to a calculated quantum yield for the formation of the OERS from [Re(PEt3)2]+ of ΦOERS = 1.21 ± 0.02. The ΦOERS values for the other complexes were determined using the same method (Fig. S21–S23†) and are summarised in Table 5.
| Entry | Complex | −ΔGBETa/eV | Φ OERS | Φ OERS/2ηq |
|---|---|---|---|---|
| a −ΔGBET = −E1/2 (PS/PS˙−) + E1/2 (BIH˙+/BIH) − wp + wr; E1/2 (BIH˙+/BIH) = −0.11 V vs. Fc/Fc+; wp = 0 eV and wr = 0.03 eV.6 b Ref. 6. | ||||
| 1 | [Re{P(OCH2)3CEt}2]+ | 1.67 | 1.24 ± 0.03 | 0.6 |
| 2 | [Re{P(OMe)3}2]+ | 1.62 | 1.48 ± 0.08 | 0.75 |
| 3 | [Re{P(OEt)3}2]+ | 1.66 | 1.35 ± 0.08 | 0.7 |
| 4 | [Re(PPh3)2]+ | 1.64 | 1.30 ± 0.01 | 0.65 |
| 5 | [Re(PEt3)2]+ | 1.6 | 1.21 ± 0.02 | 0.65 |
| 6 | [Ir(dFCF3ppy)2(bpy)]+ | 1.55 | 1.72 ± 0.14 | 0.85 |
| 7 | [Ir(dFCF3ppy)2(tmb)]+ | 1.75 | 1.59 ± 0.06 | 0.8 |
| 8 | [Ir(piq)2(dmb)]+ | 1.76 | 1.21 ± 0.01 | 0.6 |
| 9 | [Os(CF3bpy)3]2+ | 1.09 | 1.00 ± 0.02 | 0.5 |
| 10 | [Ru(4,4′-(COOMe)-bpy)3]2+ | 1.21 | 1.7 | 0.85 |
| 11 | [Ru(bpy)3]2+ | 1.65 | 1.1 | 0.55 |
| 12 | [Ru(dmb)3]2+ | 1.75 | 1.0 | 0.5 |
| 13 | [Ru(4,4′-(OMe)-bpy)3]2+ | 1.81 | 1.0 | 0.5 |
| 14 | [Os(bpy)3]2+ | 1.57 | 0.16 | 0.11 |
| 15 | [Os(dmb)3]2+ | 1.68 | ∼0.01 | 0.05 |
3 Discussion
Fig. 7 shows a plot of log(kq) as a function of −ΔGPET that includes [Re(PR3)2]+, [Ir(C^N)2(N^N)]+, and [Os(CF3bpy)3]2+, as well as Ru(II) and Os(II) tris-diimine complexes reported previously.6 The inverted region described in the Marcus theory was not clearly observed even when −ΔGPETs were very large, e.g., [Ir(dFCF3ppy)2(tmb)]+ (−ΔGPET = 0.93 eV, kq = 4.4 × 109 M−1 s−1) and [Ir(dFCF3ppy)2(bpy)]+ (−ΔGPET = 1.14 eV, kq = 4.6 × 109 M−1 s−1). Such a “non-observed” inverted region (Rehm–Weller type plots34) was reported in many photoinduced electron-transfer systems between two independent molecules, and one of the main reasons of this is that larger −ΔGPET induces electron-transfer between PS* and the quencher even when they are separated by a larger distance.14–16 Therefore, it should be reasonable to assume that, in the photoinduced electron-transfer systems shown in Fig. 7, the system with a larger −ΔGPET value should proceed with a larger distance between PS* and BIH.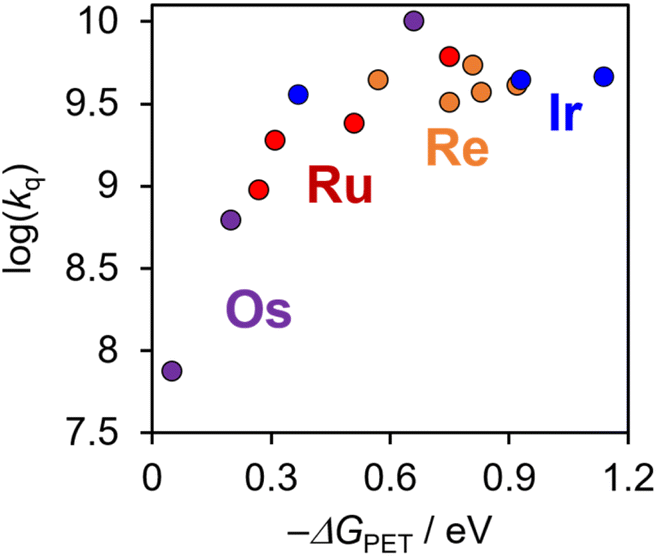 | ||
| Fig. 7 Relationship between −ΔGpet and kq for [Re(PR3)2]+ (orange), [Ir(C^N)2(N^N)]+ (blue), and reported Ru(II) (red) and Os(II) (purple) tris-(X2bpy) (X2bpy = 4,4′-X2-bpy) complexes and [Os(CF3bpy)3]2+. | ||
Notably, Fig. 7 also validates the use of the estimated E00 values for [Re(PR3)2]+, which were determined using the νM value reported for [Re(bpy)(CO)3Cl] (1450 cm−1), as discussed earlier.31
Many of the ΦOERS values listed in Table 5 exceed unity, which is reasonable because BIH can donate two electrons for each photon absorbed by the complex owing to the formation of BI˙, another effective electron donor that stems from the deprotonation of BIH˙+ following photoinduced electron-transfer between the excited complex and BIH (Scheme 2).35 The oxidation potential of BI˙ (i.e., Ep(BI+/BI˙)) is −2.14 V (vs. Fc+/Fc),6 which is more negative than the reduction potentials of the complexes (Table 2). BIH was reportedly oxidised and quantitatively converted into BI+, the two-electron oxidation product of BIH, in a previous study on photocatalytic systems involving a [Ru(diimine)3]2+-type photosensitiser.35 A similar reaction likely occurs during the photochemical formation of the OERSs of the complexes examined in this study (Reduction (1) and Reduction (2) in Scheme 1).
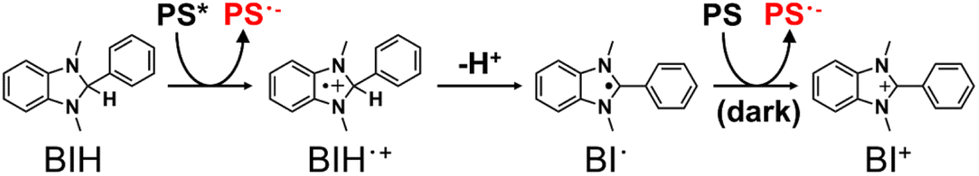 | ||
| Scheme 2 Two-electron transfer process from BIH to a photosensitiser (PS) following the absorption of one photon. | ||
Φ OERS divided by ηq (i.e., ΦOERS/ηq) is a suitable metric for evaluating the extent of back electron transfer from PS˙− to BIH˙+, as it is unaffected by radiative or non-radiative deactivation processes involving the excited state, as discussed in the Introduction. In addition, we previously reported ΦOERS/ηq values for [Os(X2bpy)3]2+ (X = H and Me) and [Ru(X2bpy)3]2+ (X = COOMe, H, Me, OMe) under the same experimental conditions,6 where we were able to separately observe two OERS-formation pathways in each case; namely, very fast photochemical reduction by BIH (Reduction (1) in Scheme 1) and a much slower electron-transfer process from BI˙ (Reduction (2)), using time-resolved absorption spectroscopy (TR-AB), owing to the large rate difference between these two processes.
Notably, nearly 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (53:47–51:49) reduction (1)/(2) ratios were observed, which indicates that back electron transfer between the OERS and BIH˙+ following separation from the geminate ion pair (Back electron transfer (2) in Scheme 1) rarely occurs despite irradiation with very strong laser light for a very short time (pulse width < 350 ps) (i.e., much higher concentrations of OERSs and BIH˙+ were present compared to the cases in which light of much-lower flux was irradiated in this study). We obtained a similar result in an analogous experiment in which [Re(PEt3)2]+ was used instead of the Ru(II) and Os(II) complexes, namely the ratio of 52:48 (Fig. S24†). Taken together, these results clearly reveal that back electron transfer from OERSs to BIH˙+ following separation from the geminate ion pair does not contribute to ΦOERS. In other words, the ratio between the rates of back electron transfer from the OERS to BIH˙+ (kbet) and separation from the geminate ion pair (kesc) controls ΦOERS/ηq. Consequently, the relationship expressed in eqn (4) can be used.
:1 (53:47–51:49) reduction (1)/(2) ratios were observed, which indicates that back electron transfer between the OERS and BIH˙+ following separation from the geminate ion pair (Back electron transfer (2) in Scheme 1) rarely occurs despite irradiation with very strong laser light for a very short time (pulse width < 350 ps) (i.e., much higher concentrations of OERSs and BIH˙+ were present compared to the cases in which light of much-lower flux was irradiated in this study). We obtained a similar result in an analogous experiment in which [Re(PEt3)2]+ was used instead of the Ru(II) and Os(II) complexes, namely the ratio of 52:48 (Fig. S24†). Taken together, these results clearly reveal that back electron transfer from OERSs to BIH˙+ following separation from the geminate ion pair does not contribute to ΦOERS. In other words, the ratio between the rates of back electron transfer from the OERS to BIH˙+ (kbet) and separation from the geminate ion pair (kesc) controls ΦOERS/ηq. Consequently, the relationship expressed in eqn (4) can be used.
 | (4) |
The back electron transfer from the OERS to BIH˙+ should be an almost diffusion controlled reaction owing to the high −ΔGBET values. However, the back transfer (2) is a negligible process in the photochemical formation of free OERS as described above. The main reason for this should be the much lower concentration of the OERS compared to BIH (0.1 M, BIH should work as a main proton acceptor) during irradiation. We reported the results and investigation in the cases of [Ru(bpy)3]2+ and [Os(bpy)3]2+.6 In the case of [Ru(bpy)3]2+ as an example, even in the TR-AB experiments, the concentration of OERSs was less than 25 × 10−6 M. The deprotonation process almost finished within 1 μs (kdp[BIH] = 6.1 × 106 s−1 where kdp is a rate constant of deprotonation). Therefore, the collision between the OERS and BIH˙+ cannot compete with the deprotonation process of BIH˙+. Accordingly, we use the ΦOERS/2ηq values listed in Table 5 in the following discussion.
[Re(PR3)2]+ and [Ir(C^N)2(N^N)]+ exhibited notably high ΦOERS/2ηq values ranging between 0.6 and 0.85 and are significantly higher than those of [Os(X2bpy)3]2+ (0.05 and 0.1). These ΦOERS/2ηq values are markedly different despite Re, Ir, and Os having similar atomic numbers (75, 77, and 76, respectively). Interestingly, [Ru(X2bpy)3]2+ exhibited similar ΦOERS/2ηq values (0.5–0.85) to those of [Re(PR3)2]+ and [Ir(C^N)2(N^N)]+ despite its significantly small atomic number (Ru, 44). Additionally, [Os(CF3bpy)3]2+ exhibited a relatively high ΦOERS/2ηq value (0.5). These results strongly suggest that the heavy-atom effect of the central metal ion does not play a major role in determining ΦOERS/2ηq. In other words, differences in spin–orbit coupling among Ru, Os, Ir, and Re do not significantly influence the rates of back electron transfer from the OERSs of these metal complexes to BIH˙+ in the geminate ion pairs formed immediately after photoinduced electron transfer between the excited metal complex and BIH. This observation is possibly ascribable to back electron transfer from the π* orbital (bpy ligand) of the complex to BIH˙+, which does not directly involve the orbitals of the central metal ion.10
The driving forces for the back-electron-transfer reactions from the OERSs of the various complexes to BIH˙+ (−ΔGBET) were determined using eqn (5) and are summarised in Table 5:
| −ΔGBET = −E1/2 (PS/PS˙−) + E1/2 (BIH˙+/BIH) − wp + wr | (5) |
[Re(PR3)2]+ and [Ir(C^N)2(N^N)]+ exhibit Coulomb terms wp between PS and BIH and wr between PS˙− and BIH˙+, which are zero owing to charge–shift reactions. On the other hand, [Os(CF3bpy)3]2+ exhibits the following Coulomb terms; wp = 0 eV and wr = 0.03 eV.6
We compared the ΦOERS/2ηq values for complexes with similar −ΔGBET values, which revealed that [Ru(bpy)3]2+ (−ΔGBET = 1.65 eV) exhibits a high ΦOERS/2ηq value of 0.55, while [Os(bpy)3]2+ (−ΔGBET = 1.57 eV) shows a significantly lower value of 0.1.6 Although [Os(dmb)3]2+ and [Re{P(OEt)3}2]+ have almost identical −ΔGBET values (1.68 eV), their ΦOERS/2ηq values differ markedly: 0.05 for [Os(dmb)3]2+ and 0.7 for [Re{P(OEt)3}2]+. These comparisons reveal that −ΔGBET affects ΦOERS/2ηq minimally in these complexes.
In contrast, a strong correlation was observed between the driving force for the photoinduced electron transfer reaction (−ΔGPET) and ΦOERS/2ηq for all complexes examined in this study (Fig. 8b); a higher driving force for photoinduced electron transfer consistently corresponds to a higher ΦOERS/2ηq value.
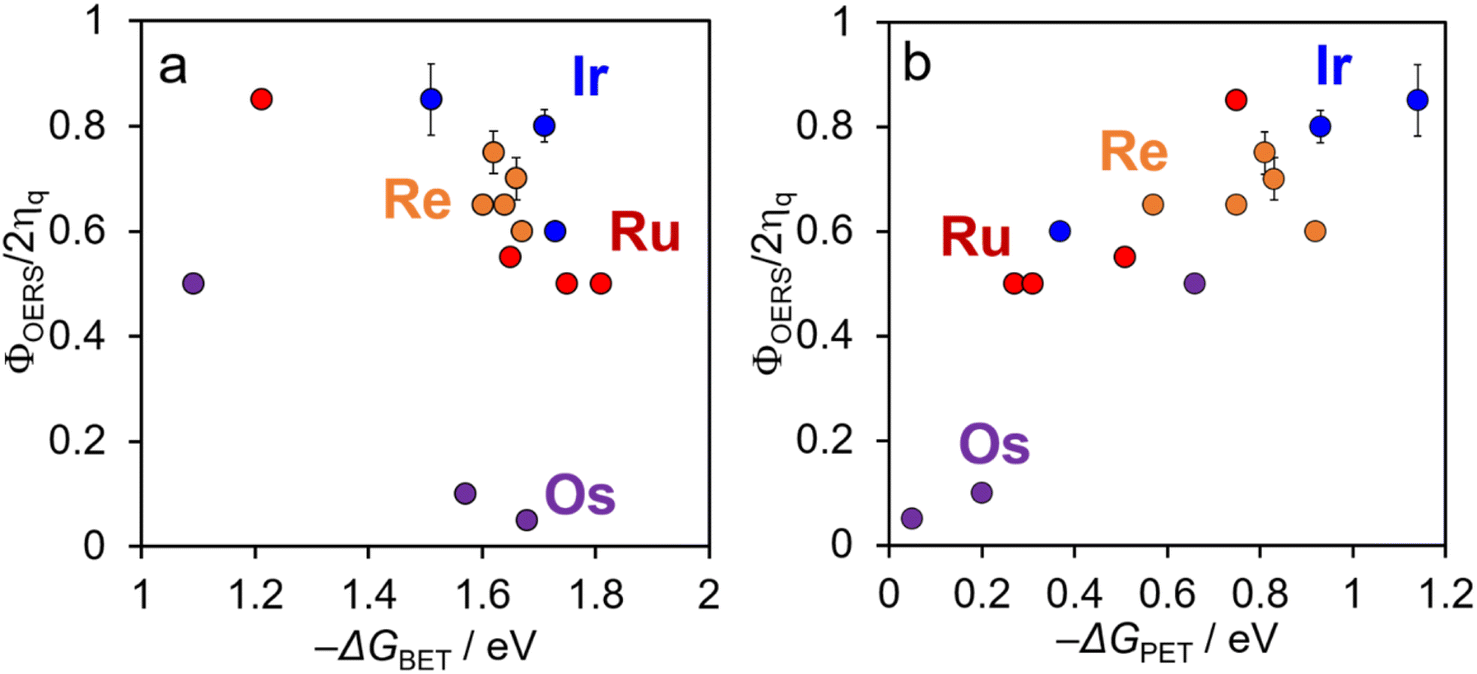 | ||
| Fig. 8 Plots of ΦOERS/2ηq for [Re(PR3)2]+ (orange), [Ir(C^N)2(N^N)]+ (blue), and Ru(II) (red), and Os(II) (purple) tris(diimine) complexes as a function of the driving force for (a) back-electron-transfer between OERSs and BIH˙+ (−ΔGBET) and (b) photoinduced electron-transfer (−ΔGPET). | ||
Φ OERS/2ηq is largely influenced by the efficiency of the back electron transfer process within the geminate ion pair formed immediately following photoinduced electron transfer. In other words, ΦOERS/2ηq is expected to depend on the rate of back electron transfer accompanied by a spin flip in the geminate ion pair. However, as shown in Fig. 8a, ΦOERS/2ηq is not strongly correlated with −ΔGBET, which provides a measure of the driving force for back electron transfer from the OERS of the metal complex to BIH˙+. In contrast, a larger −ΔGPET, which is a measure of the driving force for the photoinduced electron transfer process, does correlate with a higher ΦOERS/2ηq value. Taken together, these results suggest that the driving force for the photoinduced electron transfer from BIH to the excited state of the metal complex plays a crucial role in determining the back electron transfer rate between the OERSs of the metal complex and BIH˙+ (kbet) within the geminate ion pair.
According to Marcus theory,36 the back electron transfer rate, kbet, is expressed according to eqn (6) and (7):
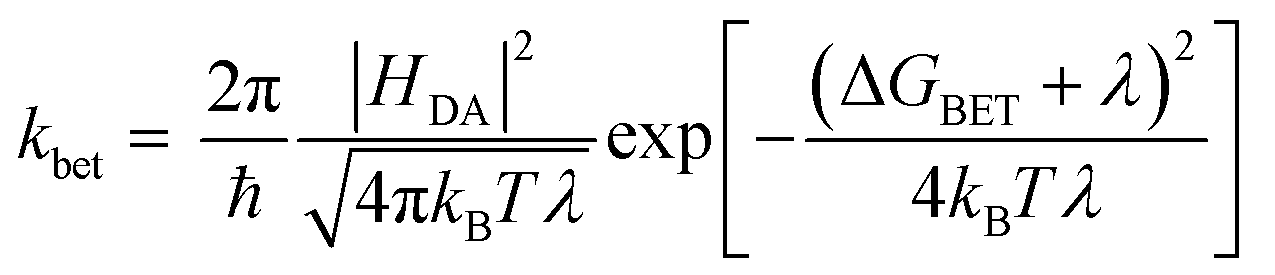 | (6) |
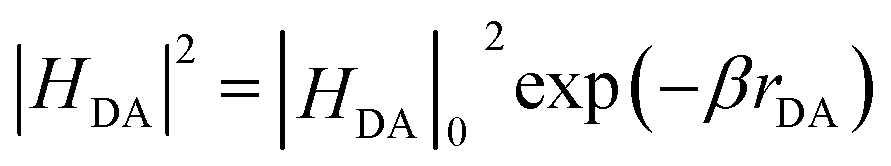 | (7) |
It was reported that Ru complexes with bulkier ligands exhibit slower photoinduced electron-transfer rates to methyl viologen compared to those with smaller ligands but with similar photooxidative powers. This observation can be ascribed to poorer donor/acceptor orbital overlap that suppresses electron transfer owing to the bulky substituent.37 As we discussed above for the systems reported in this paper, the “non-observed” inverted region in the photoinduced electron transfer reactions also supports that the stronger driving force (−ΔGPET) enables photoinduced electron-transfer to occur over a longer distance between the excited metal complex and BIH. This is because a larger −ΔGPET can compensate for weaker orbital coupling between the excited metal complex and BIH. Therefore, we reasonably expect that, in the geminate ion pair formed immediately after the photoinduced electron-transfer, a larger distance between the OERSs of the metal complex and BIH+ leads to poorer orbital overlap and a slower rate of back-electron-transfer from the OERS to BIH˙+ in the systems with similar −ΔGBET values.
The cage escape rate (kesc) was determined using the Eigen equation (eqn (8) and (9)):38
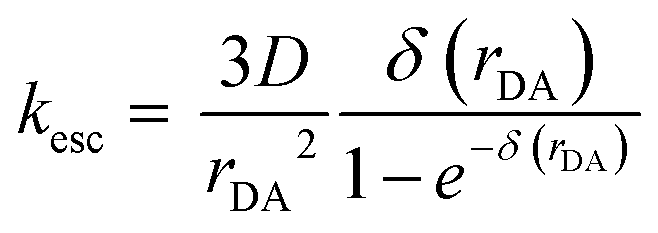 | (8) |
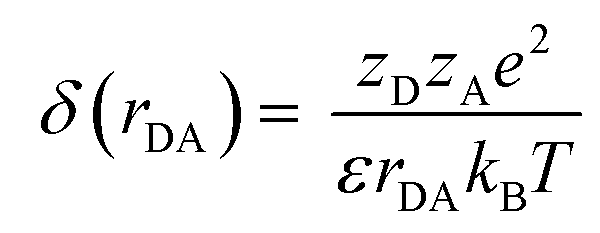 | (9) |
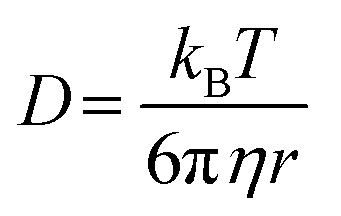 | (10) |
Based on these experimental results and analyses, we conclude that a larger −ΔGPET value primarily leads to a higher ΦOERS/2ηq value, indicative of more-efficient formation of free OERSs from the reductively quenched metal complex in the excited state. This conclusion is attributed to the larger distance between the excited metal complex and BIH during the photoinduced electron transfer process associated with the larger −ΔGPET value, which suppresses back electron transfer from the OERS to BIH˙+ within the geminate ion pair.
4 Conclusions
This study investigated the formation efficiencies of free OERSs separated from their BIH˙+ counterpart in the solvated cage by examining a variety of metal complexes featuring Re(I), Ir(III), Ru(II), and Os(II) as central metal ions that are frequently used as redox photosensitisers (photoredox catalysts) in photochemical electron-transfer reactions. BIH was selected as the representative reductant because it is commonly employed in photocatalytic systems. We found that the driving force for photoinduced electron transfer (−ΔGPET) is the most critical factor that determines the ΦOERS/2ηq value of a mononuclear metal complex by examining how it responds to various factors; specifically, a larger −ΔGPET value leads to a higher ΦOERS/2ηq value. In contrast, the heavy-atom effect of the central metal ion was found to impact ΦOERS/2ηq minimally. The driving force for back electron transfer (−ΔGBET) and the charge of the complex have a weaker influence on ΦOERS/2ηq compared to −ΔGPET.We conclude that the distance between the excited metal complex and the electron donor (BIH) crucially determines the free-OERS formation efficiency based on our results and a theoretical investigation using Marcus theory for electron transfer between two independent molecules. An excited metal complex with a stronger oxidising power is more distant from the BIH during forward photoinduced electron transfer, which in turn suppresses the spin-flip-accompanied back electron transfer between the OERS and BIH˙+ within the solvated cage.
This finding is key to designing efficient redox photocatalytic systems that use molecular redox photosensitisers (photoredox catalysts) in homogeneous solutions. Specifically, a system with a larger −ΔGPET value between the excited state of the photosensitiser and the electron donor more-favourably delivers a high quantum yield in a photocatalytic reaction.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
NH performed almost all of the experiments and contributed to manuscript writing. KO's previous data on Ru complexes and most Os complexes shown in this paper were useful for this study as well. KK and YT contributed to discussion. OI designed this research, secured financial support, led the project, and guided the manuscript preparation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thanks JSPS (KAKENHI Grant Numbers: s) and the Iwatani Naoji Foundation for their financial support.Notes and references
- C. K. Prier, D. A. Rankic and D. W. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- K. Sakai and H. Ozawa, Coord. Chem. Rev., 2007, 251, 2753–2766 CrossRef CAS.
- Y. Tamaki and O. Ishitani, ACS Catal., 2017, 7, 3394–3409 CrossRef CAS.
- C. Wang, H. Li, T. H. Burgin and O. S. Wenger, Nat. Chem., 2024, 16, 1151–1159 CrossRef CAS PubMed.
- Y. Tamaki, K. Tokuda, Y. Yamazaki, D. Saito, Y. Ueda and O. Ishitani, Front. Chem., 2019, 7, 327 CrossRef CAS PubMed.
- K. Ozawa, Y. Tamaki, K. Kamogawa, K. Koike and O. Ishitani, J. Chem. Phys., 2020, 153, 154302 CrossRef CAS PubMed.
- The reason why ΦOERS exceeded 1 in the case of the Ru complex is that the electron donor (BIH) donates one electron during the photoinduced electron transfer process and after that, the generated BIH˙+ undergoes deprotonation to form BI˙, which can donate an additional electron to the complex in a dark reaction (Scheme 1)..
- A. Cannizzo, F. van Mourik, W. Gawelda, G. Zgrablic, C. Bressler and M. Chergui, Angew. Chem., Int. Ed., 2006, 45, 3174–3176 CrossRef CAS PubMed.
- O. Bräm, F. Messina, E. Baranoff, A. Cannizzo, M. K. Nazeeruddin and M. Chergui, J. Phys. Chem. C, 2013, 117, 15958–15966 CrossRef.
- J. Olmsted and T. J. Meyer, J. Phys. Chem., 1987, 91, 1649–1655 CrossRef CAS.
- S. Sasaki, A. Katsuki, K. Akiyama and S. TeroKubota, J. Am. Chem. Soc., 1997, 119, 1323–1327 CrossRef CAS.
- V. Glembockyte and G. Cosa, J. Am. Chem. Soc., 2017, 139, 13227–13233 CrossRef CAS PubMed.
- (a) S. Fraga, J. Karwowski and K. M. S. Saxena, Handbook of atomic data, Elsevier Scientific Publication Company, Amsterdam, New York, 1976 Search PubMed; (b) S. Koseki, N. Matsunaga, T. Asada, M. W. Schmidt and M. S. Gordon, J. Phys. Chem. A, 2019, 123, 2325–2339 CrossRef CAS PubMed.
- M. Tachiya and S. Murata, J. Phys. Chem., 1992, 96, 8441–8444 CrossRef CAS.
- M. A. Smitha, E. Prasad and K. R. Gopidas, J. Am. Chem. Soc., 2001, 123, 1159–1165 CrossRef CAS PubMed.
- A. Rosspeintner, D. R. Kattnig, G. Angulo, S. Landgraf and G. Grampp, Chem.–Eur. J., 2008, 14, 6213–6221 CrossRef CAS PubMed.
- L. T. Calcaterra, G. L. Closs and J. R. Miller, J. Am. Chem. Soc., 1983, 105, 670–671 CrossRef CAS.
- J. R. Winkler and H. B. Gray, J. Am. Chem. Soc., 2014, 136, 2930–2939 CrossRef CAS PubMed.
- O. Ishitani, M. W. George, T. Ibusuki, F. P. A. Johnson, K. Koike, K. Nozaki, C. J. Pac, J. J. Turner and J. R. Westwell, Inorg. Chem., 1994, 33, 4712–4717 CrossRef CAS.
- S. H. Wu, J. W. Ling, S. H. Lai, M. J. Huang, C. H. Cheng and I. C. Chen, J. Phys. Chem. A, 2010, 114, 10339–10344 CrossRef CAS PubMed.
- T. S. Teets, Y. Wu and D. Kim, Synlett, 2021, 33, 1154–1179 CrossRef.
- K. Koike, J. Tanabe, S. Toyama, H. Tsubaki, K. Sakamoto, J. R. Westwell, F. P. Johnson, H. Hori, H. Saitoh and O. Ishitani, Inorg. Chem., 2000, 39, 2777–2783 CrossRef CAS PubMed.
- C. A. Tolman, Chem. Rev., 1977, 77, 313–348 CrossRef CAS.
- H. Tsubaki, A. Sekine, Y. Ohashi, K. Koike, H. Takeda and O. Ishitani, J. Am. Chem. Soc., 2005, 127, 15544–15555 CrossRef CAS PubMed.
- H. Tsubaki, S. Tohyama, K. Koike, H. Saitoh and O. Ishitani, Dalton Trans., 2005, 385–395 RSC.
- T. Morimoto, M. Ito, K. Koike, T. Kojima, T. Ozeki and O. Ishitani, Chem.–Eur. J., 2012, 18, 3292–3304 CrossRef CAS PubMed.
- L. A. Worl, R. Duesing, P. Y. Chen, L. Dellaciana and T. J. Meyer, J. Chem. Soc., Dalton Trans., 1991, 849–858 RSC.
- K. Koike, N. Okoshi, H. Hori, K. Takeuchi, O. Ishitani, H. Tsubaki, I. P. Clark, M. W. George, F. P. A. Johnson and J. J. Turner, J. Am. Chem. Soc., 2002, 124, 11448–11455 CrossRef CAS PubMed.
- K. A. Walters and K. S. Schanze, The Spectrum, 1998, vol. 11, pp. 2–8 Search PubMed.
- G. H. Allen, R. P. White, D. P. Rillema and T. J. Meyer, J. Am. Chem. Soc., 1984, 106, 2613–2620 CrossRef CAS.
- J. V. Caspar, T. D. Westmoreland, G. H. Allen, P. G. Bradley, T. J. Meyer and W. H. Woodruff, J. Am. Chem. Soc., 1984, 106, 3492–3500 CrossRef CAS.
- (a) S. J. Liu, Q. Zhao, Q. L. Fan and W. Huang, Eur. J. Inorg. Chem., 2008, 2177–2185 CrossRef CAS; (b) Y. Kuramochi and O. Ishitani, Inorg. Chem., 2016, 55, 5702–5709 CrossRef CAS PubMed.
- P. Delgado, R. J. Glass, G. Geraci, R. Duvadie, D. Majumdar, R. I. Robinson, I. Elmaarouf, M. Mikus and K. L. Tan, J. Org. Chem., 2021, 86, 17428–17436 CrossRef CAS PubMed.
- D. Rehm and A. Weller, Isr. J. Chem., 1970, 8, 259–271 CrossRef CAS.
- Y. Tamaki, K. Koike, T. Morimoto and O. Ishitani, J. Catal., 2013, 304, 22–28 CrossRef CAS.
- R. A. Marcus, J. Chem. Phys., 1956, 24, 966–978 CrossRef CAS.
- T. Hamada, S. Tanaka, H. Koga, Y. Sakai and S. Sasaki, Dalton Trans., 2003, 692–698 RSC.
- M. Eigen, Z. Phys. Chem., 1954, 1, 176–200 CrossRef CAS.
- Y. S. Joshi, K. S. Kanse, D. N. Rander and A. C. Kumbharkhane, Indian J. Pure Appl. Phys., 2016, 54, 621–628 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, photophysical and electrochemical data of [Re(PR3)2]+, [Ir(C^N)2(N^N)]+, and related references [Os(CF3bpy)3]2+, and transient absorption spectroscopy results of a DMA solution containing [Re(PEt3)2]+ and BIH. See DOI: https://doi.org/10.1039/d4sc08268k |
| This journal is © The Royal Society of Chemistry 2025 |