
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThiosemicarbazones as versatile photoswitches with light-controllable supramolecular activity†
Bengi
Sentürk
a,
Burkhard
Butschke
 b and
Fabian
Eisenreich
*a
b and
Fabian
Eisenreich
*a
aDepartment of Chemical Engineering and Chemistry, Institute for Complex Molecular Systems, Eindhoven University of Technology, 5600 MB, Eindhoven, The Netherlands. E-mail: f.r.eisenreich@tue.nl
bInstitute for Inorganic and Analytical Chemistry, University of Freiburg, Albertstr. 21, 79104 Freiburg, Germany
First published on 17th January 2025
Abstract
Using photoswitchable molecules to manipulate supramolecular interactions under light illumination has driven advancements in numerous fields, allowing for the strategic alteration of molecular systems. However, integrating the moiety responsible for these interactions into the photochromic scaffold can be complex and may hamper the switching efficiency. We thus explored a simple class of organic molecules, namely thiosemicarbazones, featuring both a photoisomerizable C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bond and a thiourea moiety capable of hydrogen bonding. The scalable two-step synthesis allowed us to prepare 23 thiosemicarbazones to systematically elucidate their optical properties. Attaching various functional groups, extended π-systems, and heterocycles enabled fine-tuning of their absorption profiles. UV light illumination converts thiosemicarbazones from the stable E-isomer to the metastable Z-isomer, exhibiting both negative and positive T-type photochromism, a wide range of thermal half-lives, PSS values up to 92%, and high fatigue resistance. Substituting the CN moiety with a pyridinyl group stabilizes the Z-isomer via intramolecular hydrogen bonding, confirmed by single-crystal X-ray analysis, and transforms thiosemicarbazones into bistable P-type photoswitches. Additionally, thiosemicarbazones dimerize or form aggregates through hydrogen bonding—a process that can be turned off or on with light. Overall, thiosemicarbazones offer tunable photochromic and supramolecular properties, rendering them a promising photoswitch for creating stimuli-responsive systems.
N double bond and a thiourea moiety capable of hydrogen bonding. The scalable two-step synthesis allowed us to prepare 23 thiosemicarbazones to systematically elucidate their optical properties. Attaching various functional groups, extended π-systems, and heterocycles enabled fine-tuning of their absorption profiles. UV light illumination converts thiosemicarbazones from the stable E-isomer to the metastable Z-isomer, exhibiting both negative and positive T-type photochromism, a wide range of thermal half-lives, PSS values up to 92%, and high fatigue resistance. Substituting the CN moiety with a pyridinyl group stabilizes the Z-isomer via intramolecular hydrogen bonding, confirmed by single-crystal X-ray analysis, and transforms thiosemicarbazones into bistable P-type photoswitches. Additionally, thiosemicarbazones dimerize or form aggregates through hydrogen bonding—a process that can be turned off or on with light. Overall, thiosemicarbazones offer tunable photochromic and supramolecular properties, rendering them a promising photoswitch for creating stimuli-responsive systems.
Introduction
Photoswitches refer to molecules capable of undergoing reversible conversion from one state, or isomer, to another upon exposure to light of a certain wavelength.1 The switching process has the potential to cause a substantial geometric shift through isomerization around a double bond, as demonstrated in stiff-stilbene,2,3 (hemi)indigo,4–6 azobenzene,7–10 and diazocine systems.11,12 This transformative event can be strategically harnessed to exert control over the interplay of molecules, in particular via supramolecular interactions.13–15 A notable degree of precision has been achieved in steering ion transport,16 supramolecular assemblies,17–24 non-equilibrium systems,25–27 photopharmacology,28,29 and catalysis,30–32 among various other functions, with light. Photoswitches thus prove instrumental for breaking the static boundaries of chemical processes and infusing them with newfound adaptability.Yet, tethering the moiety accountable for the non-covalent interactions to the photochromic scaffold may entail elaborate synthesis procedures33 and impair the switching performance relative to the standalone photoswitch.27 From the viewpoint of molecular design, rendering the supramolecular unit an integral component of the photoswitch structure-rather than attaching it later on-would simplify the process. This led us to explore thiosemicarbazones (TSCs), an organic compound class formally consisting of a thiourea unit linked to a CN double bond, whose photoswitching capabilities are yet to be fully unveiled (Fig. 1). On the one hand, thiourea groups are widely recognized as versatile hydrogen bonding partners. On the other hand, photoswitches featuring CN double bonds have attracted considerable attention as potential alternatives to traditional olefin or azo analogs, owing to their comparable or even superior properties.34–37 They offer distinct advantages, such as tunable properties, high photoconversion efficiency, ease of derivatization, and scalability in synthesis.
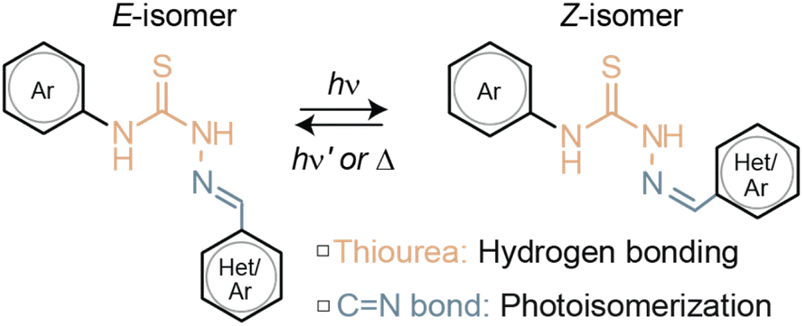 | ||
| Fig. 1 Photoisomerization of thiosemicarbazones. | ||
Hydrazones, extensively studied by Aprahamian et al., are one notable member of this photoswitch class.38 Besides their facile synthesis and high photostationary states (PSS), hydrazones demonstrate bistability, exhibiting photochromism in both directions (P-type) along with remarkable thermal stability of the metastable isomer lasting up to thousands of years. Additionally, some hydrazone derivatives can exhibit very short thermal half-lives, leading to T-type behavior in specific cases.39,40 Hecht et al. introduced acylhydrazones as another versatile photoswitch class within this category.41 In addition to displaying the desired photoswitch properties seen in hydrazones, acylhydrazones also present a range of thermal stabilities for their Z-isomer, ranging from short-lived to thermally stable derivatives. By suitable substitution, acylhydrazones display not only P-type but also T-type photochromism, wherein back-isomerization occurs via the ground state rather than the excited state. Unlike (acyl)hydrazones, the thiourea moiety in TSCs provides a strong binding motif for supramolecular interactions that may be adjustable with light. However, the photochromic properties of TSCs remain largely unexplored. Scattered reports exist in literature, in which light-induced proton transfer42–44 as well as E→Z isomerization of a single compound,45 both in the solid state, are described. Instead, thiosemicarbazones are primarily acknowledged for their biochemical activity (e.g., antibacterial, antifungal, and antitumor)46,47 as well as their coordination affinity to a broad range of metal ions.48
In this work, we comprehensively explore the photochromic properties of TSCs and introduce them as a promising class of photoswitches. The simple yet high-yielding synthesis allowed us to prepare a library of TSCs, including 23 compounds, and systematically investigate their structure–property relationships. In-depth optical and NMR spectroscopy measurements demonstrate the pronounced tuneability of the photoisomerization around the CN double bond. The library not only encompasses T-type photoswitches, featuring thermal half-lives of the metastable Z-isomer ranging from minutes to hours, along with stable Z-isomers, but also P-type photoswitches exhibiting photoisomerization in both switching directions. Moreover, the E-isomer of TSCs tends to form weakly associated dimers through hydrogen bonds in solution. The generation of the Z-isomer completely shuts down this interaction by sterically shielding the thiourea group, thereby providing the means to toggle between assembled and disassembled states on-demand with light. Overall, the easily accessible synthesis of TSCs, combined with their diverse photochromic properties and adjustable supramolecular interactions, positions them as a promising newcomer to the photoswitch family.
Results and discussion
Synthesis and molecular design
Thiosemicarbazones can be readily prepared in two simple synthetic steps with excellent yields and straight-forward purification. First, an aromatic isothiocyanate derivative was treated with hydrazine monohydrate in dry CH2Cl2 at 0 °C to yield the corresponding thiosemicarbazide within 30 min (Fig. 2A, see the ESI† for details). The condensation of the thiosemicarbazide with an aromatic carbonyl compound in ethanol and the presence of trace amounts of trifluoroacetic acid as a catalyst resulted in the formation of the final thiosemicarbazone. After both reaction steps, the products conveniently precipitated from the mixtures. Consequently, a simple filtration and washing process is all that was required to obtain highly pure molecules. A diverse range of thiosemicarbazides is also commercially available, which generally simplifies the synthesis further. To demonstrate the feasibility of the synthesis protocol, we performed the reactions on a gram-scale (Fig. 2A, right). Based on this rapid and efficient synthesis, we prepared a library of 23 differently substituted TSCs to explore structure–property relationships (Fig. 2B). The as-synthesized TSCs remained stable and showed no signs of hydrolysis of the CN double bond for at least one year, both in the solid state and when dissolved in CDCl3 and DMSO-d6. The compounds were ambiguously characterized using variety of analytical methods (i.e., 1H, 13C, and 19F NMR spectroscopy, FT-IR spectroscopy, and mass spectrometry; Fig. S1–S58 in the ESI†).
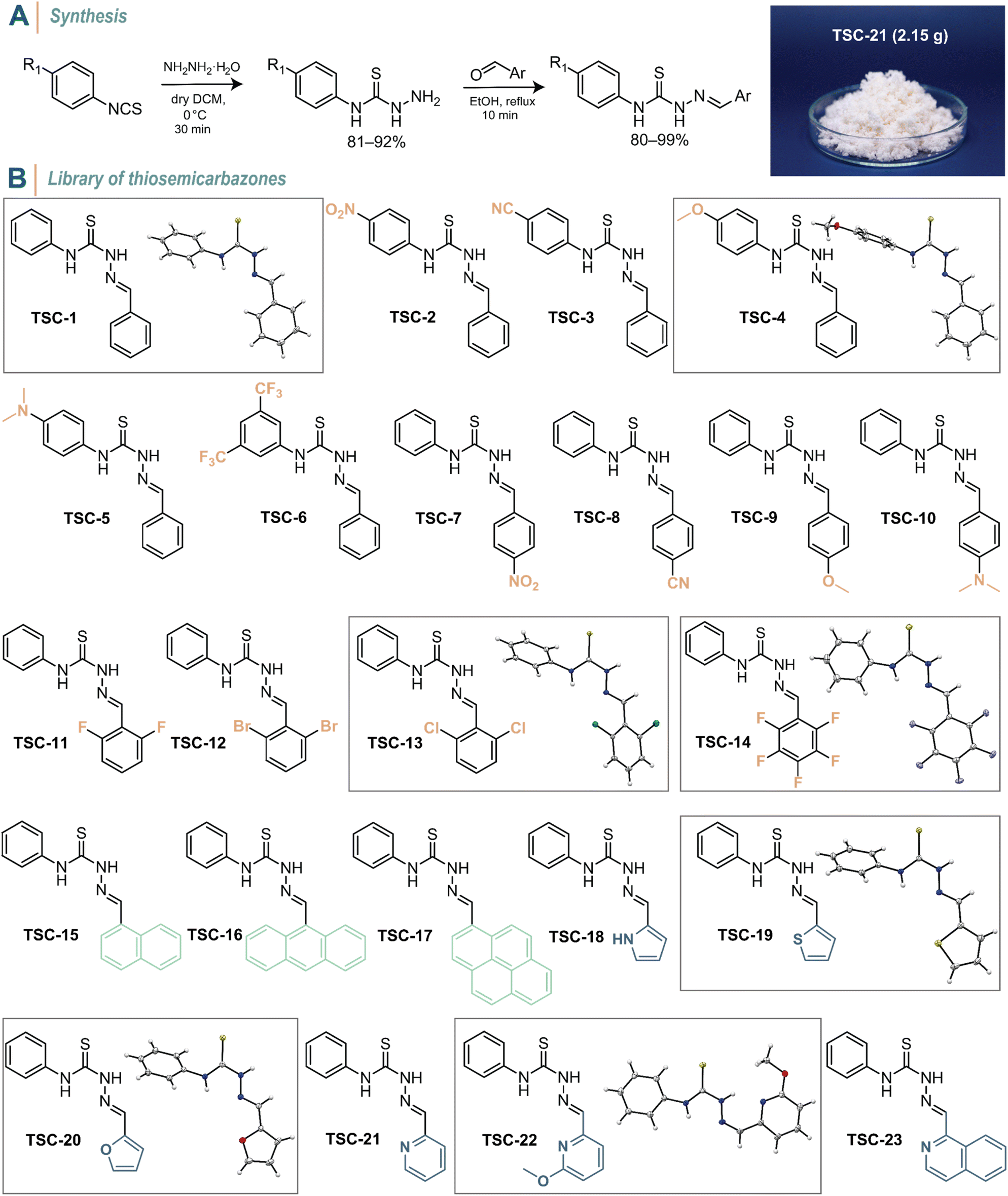 | ||
| Fig. 2 Library of TSCs. (A) General two-step synthesis scheme to prepare functionalized TSCs. A photograph of compound TSC-21 showcases the scalability of the synthesis. (B) Chemical structures of all synthesized TSCs. Single-crystal X-ray structures of TSC-1, TSC-13, TSC-14, TSC-19, TSC-20, and TSC-22 are depicted with thermal ellipsoids representing 50% probability (40% and 17% disorders of the furanyl and thienyl rings, respectively, have been omitted for clarity). C: grey, H: off-white, N: blue, S: yellow, O: red, Cl: green, and F: purple. | ||
Our molecular design process was driven by the following considerations: TSC-1 with phenyl groups attached to both the thiourea and CN double bond serves as the reference compound. In para-position of the N-phenyl group, we introduced –NO2 (TSC-2) and –CN (TSC-3) as electron-withdrawing (EW) groups as well as –OMe (TSC-4) and –NMe2 (TSC-5) as electron-donating (ED) groups. TSC-6 features two trifluoromethyl substituents positioned meta to the thiourea, as –CF3 groups hold a privileged position not only in drug and catalyst design (e.g., Schreiner's catalyst), but also in enhancing the fatigue resistance of photoswitches.49 We applied the same para-substitution pattern to the C-phenyl moiety (TSC-7–TSC-10) and incorporated various halides (TSC-11–TSC-14). The latter was motivated by the potential of ortho-halogenation in azobenzene to generate photoswitches with long-lived Z-isomers and near-quantitative PSS.50–52 The CN moiety was further functionalized with various extended π-systems (TSC-15–TSC-17) to shift the absorption profile of the TSCs towards the visible region of the electromagnetic spectrum. Lastly, we investigated heterocyclic derivatives (TSC-18–TSC-23), as heterocycles are commonly found in molecular photoswitches and can be crucial to their function.6,7,49
To gain deeper insight into the molecular geometry of TSCs, we analysed their molecular structure by single-crystal X-ray diffraction. Single crystals were obtained at room temperature by slow solvent evaporation. All analysed TSCs exhibited a trans–cis conformation on their thiourea core, corroborated by 2D NMR analysis (Fig. S59 in the ESI†). TSC-1, TSC-4, TSC-13, TSC-14, TSC-19, and TSC-20 crystallized in the E-configuration, while TSC-22 adopted the Z-configuration (Fig. S60–S66 in the ESI†). Analysis of TSC-22 indicated the presence of an intramolecular H-bond between the thiourea and pyridinyl functionalities (N–H⋯N: 1.990 Å) examination of the X-ray data also revealed the rotational freedom of the N-phenyl group, which exhibits dihedral angles in the range from 8° to 98°. In contrast, the C-phenyl group is oriented in the same plane as the CN and thiourea moieties unless a bulky substituent such as chlorine is incorporated, causing deviation from planarity by up to 35°. The detailed structural information obtained from single-crystal X-ray diffraction provided a foundational understanding of the molecular geometry of TSCs.
Optical properties of TSC-1
To explore the behaviour of thiosemicarbazones under light illumination, we first conducted UV-vis spectroscopy studies on TSC-1 in an acetonitrile solution (c = 3.8 × 10−5 M, 20 °C, Fig. 3A, S67–S69 in the ESI†). The dissolved compound showed an absorption maximum in the UV region (λmax = 319 nm) and a high extinction coefficient at this wavelength (ε = 35![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 M−1 cm−1, Table 1). Upon irradiation with an LED (λirr = 340 nm), the absorption at the absorption maximum decreased and a new absorption band at 302 nm built up, until reaching the PSS. Due to this hypsochromic shift (Δλmax = −17 nm), TSC-1 classifies as a negative photochrome. The clear isosbestic point at 295 nm suggests a clean conversion from TSC-1E to TSC-1Z upon irradiation. Complete back-isomerization to the thermodynamically stable E-isomer occurred thermally over time after the light source was turned off (Fig. 3B). The thermal half-life of the metastable Z-isomer was determined as t1/2 = 199 min. Illumination with light of other wavelengths did not induce back-isomerization, thus TSC-1 demonstrates pure T-type photoswitch behaviour. We further quantified the composition of both isomers at the PSS with 1H NMR spectroscopy, showing 44% of Z-isomer in the equilibrium state (Fig. 3C).
400 M−1 cm−1, Table 1). Upon irradiation with an LED (λirr = 340 nm), the absorption at the absorption maximum decreased and a new absorption band at 302 nm built up, until reaching the PSS. Due to this hypsochromic shift (Δλmax = −17 nm), TSC-1 classifies as a negative photochrome. The clear isosbestic point at 295 nm suggests a clean conversion from TSC-1E to TSC-1Z upon irradiation. Complete back-isomerization to the thermodynamically stable E-isomer occurred thermally over time after the light source was turned off (Fig. 3B). The thermal half-life of the metastable Z-isomer was determined as t1/2 = 199 min. Illumination with light of other wavelengths did not induce back-isomerization, thus TSC-1 demonstrates pure T-type photoswitch behaviour. We further quantified the composition of both isomers at the PSS with 1H NMR spectroscopy, showing 44% of Z-isomer in the equilibrium state (Fig. 3C).
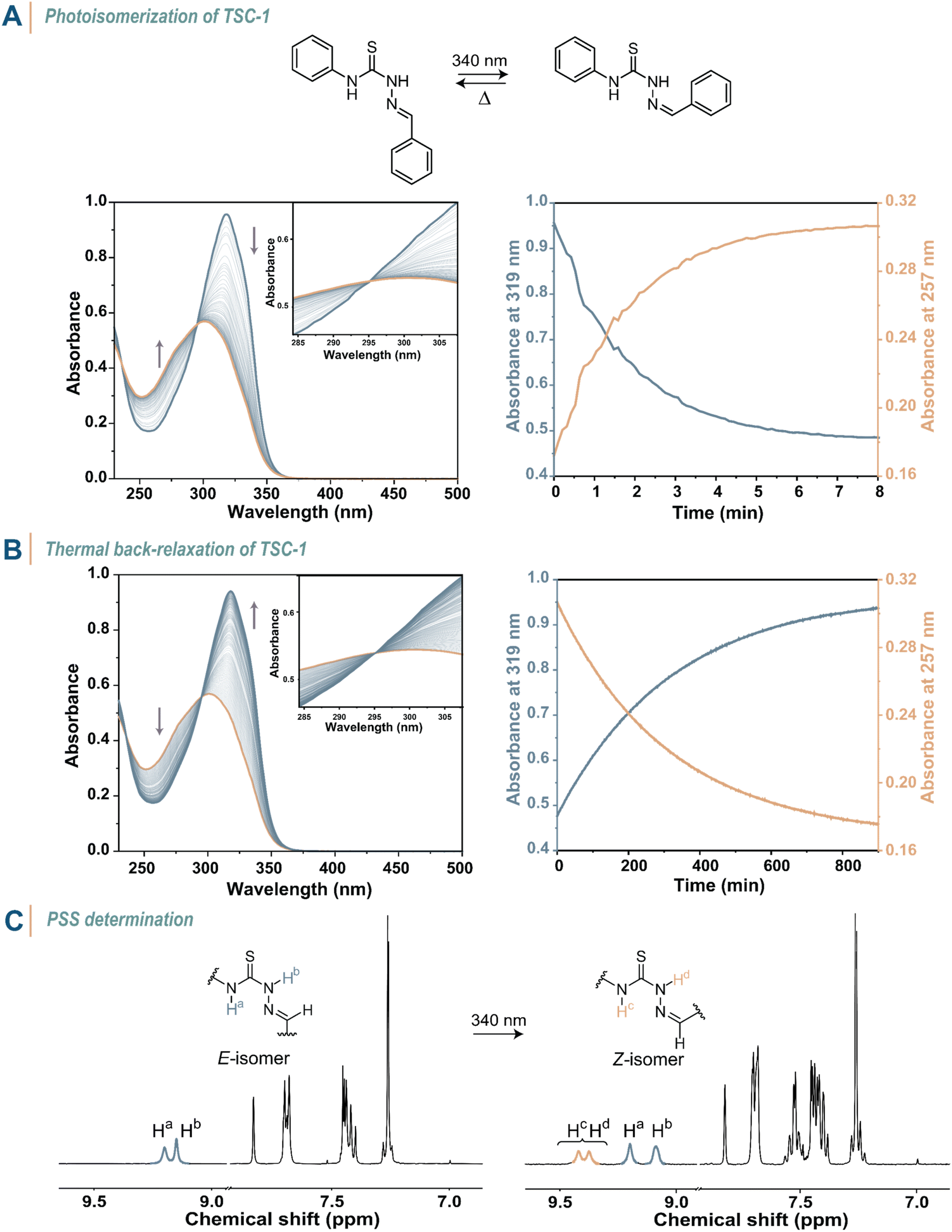 | ||
| Fig. 3 Photoisomerization studies on TSC-1. (A) Conversion of TSC-1E (blue line) in MeCN (c = 3.77 × 10−5 M) to TSC-1PSS (orange line) under constant light irradiation (λirr = 340 nm) followed over time (left) and absorbance at λ = 319 nm and 257 nm followed over time (right). (B) Thermal back-relaxation of TSC-1PSS (left) and absorbance at λ = 319 nm and 257 nm followed over time (right). (C) 1H NMR spectra (400 MHz, 25 °C, CDCl3) of TSC-1E before and after illumination with 340 nm until reaching the PSS of 44%. | ||
| Photoswitch | ε(E) [M−1 cm−1] | λ max(E) [nm] | λ max(PSS)a [nm] | Δλmaxb [nm] | t 1/2 |
E:Z ratio at PSS (λirr) |
|---|---|---|---|---|---|---|
| a Defined as the most red-shifted absorption band. b Defined as Δλmax = λmax(PSS) − λmax(E). c Measured at 20 °C; photoswitches were labeled as thermally stable when no thermal Z → E isomerization was observed for at least 60 min.41 d Determined based on 1H NMR experiments in MeCN-d3via LED illumination. e Determined based on 1H NMR experiments in CDCl3via LED illumination. In cases of short-lived Z-isomers, the PSS was not determined. f λ max for dark equilibrium state. | ||||||
| TSC-1 | 35400 |
319 | 302 | −17 | 199 min | 56:44 (365 nm)d |
| TSC-2 | 39700 |
328 | 321 | −7 | 32 min | n.d. |
| TSC-3 | 39000 |
321 | 304 | −17 | 1.35 min | n.d. |
| TSC-4 | 35000 |
317 | 307 | −10 | 20 min | n.d. |
| TSC-5 | 37200 |
316 | 312 | −4 | 118 min | n.d. |
| TSC-6 | 33100 |
321 | 318 | −3 | 1 min | n.d. |
| TSC-7 | 28300 |
361 | 351 | −10 | 258 min | 33:67 (365 nm)e |
| TSC-8 | 32500 |
337 | 328 | −9 | 213 min | 53:47 (365 nm)e |
| TSC-9 | 35200 |
328 | 317 | −11 | 37 min | n.d. |
| TSC-10 | 47500 |
367 | 437 | 70 | 11 min | n.d. |
| TSC-11 | 35200 |
318 | 293 | −25 | 10.7 h | 24:76 (365 nm)e |
| TSC-12 | 21300 |
309 | 284 | −25 | 1 day | 10:90 (365 nm)e |
| TSC-13 | 23400 |
314 | 283 | −31 | Stable | 8:92 (365 nm)e |
| TSC-14 | 37600 |
318 | 293 | −25 | 53 min | 25:75 (365 nm)e |
| TSC-15 | 28800 |
349 | 276 | −73 | 76 min | 18:82 (365 nm)e |
| TSC-16 | 12800 |
389 | 364 | −25 | Stable | 59:41 (365 nm)e |
| TSC-17 | 41600 |
406 | 405 | −1 | 66 min | n.d. |
| TSC-18 | 31900 |
338 | 402 | 64 | 6 min | n.d. |
| TSC-19 | 31300 |
337 | 407 | 70 | 46 min | n.d. |
| TSC-20 | 37200 |
328 | 323 | −5 | Stable | 22:78 (365 nm)d |
| TSC-21 | n.d. | 320f | 325 | 5 | Stable | 82:18 (405 nm)d |
| 67:33 (340 nm)d |
||||||
| TSC-22 | n.d. | 333f | 355 | 22 | Stable | 85:15 (365 nm)d |
| 36:64 (340 nm)d |
||||||
| TSC-23 | n.d. | 340f | 376 | 36 | 192 min | 22:78 (365 nm)d |
We then carried out a solvent screening study to assess if there is a correlation between polarity or hydrogen bonding ability of the solvent and the photoswitching performance of TSC-1 (Fig. S70–S74 in the ESI†). In highly polar solvents (e.g., DMF, DMSO) and protic media (e.g., MeOH, EtOH) barely any spectral changes were observed under UV light illumination, presumably because these solvents stabilize the E-isomer through hydrogen bonding,53 leading to rapid thermal back-isomerization of the Z-isomer. In contrast, TSC-1 responded well to light in less polar and aprotic solvents. For instance, the thermal half-lives observed in EtOAc and THF were measured at t1/2 = 66 and 83 min, respectively. Solvents with low hydrogen bonding capability, such as CHCl3, toluene, and acetonitrile, yielded a prolonged thermal stability of the Z-isomer with t1/2 = 120, 181, and 199 min, respectively. While a detailed mechanistic study is required to fully understand these effects, the observed trends are summarized in Table S2 of the ESI.†
To investigate the influence of sulfur on the optical properties of thiosemicarbazones, we compared TSC-1 with its oxygen analog, semicarbazone SC-1 (Fig. S75 in the ESI†). From the absorption profile in acetonitrile, it is apparent that SC-1 requires higher energy light for photoisomerization (λmax = 293 nm) and has a lower molar absorption coefficient at the absorption maximum (ε = 25000 M−1 cm−1) compared to TSC-1. These findings suggest that sulfur enhances light absorption efficiency and reduces the energy required for activation, making it advantageous for photoswitch design. This is consistent with studies demonstrating that the steric size of sulfur elongates the CX bond, which results in an energy drop of the 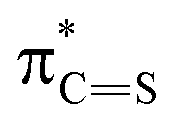 relative to
relative to 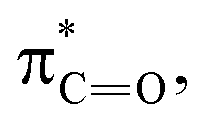 thereby reducing the HOMO–LUMO gap.54,55
thereby reducing the HOMO–LUMO gap.54,55
Interestingly, the steric repulsion of sulfur also increases the electronegativity of the CX bond, despite sulfur being the more electropositive chalcogen atom.56 This is supported by NMR spectroscopy, which shows a downfield shift of the N–H proton signals (Fig. S76 in the ESI†).
Photoswitching studies on TSC library
Satisfied with the pronounced photoisomerization of TSC-1, we continued with analyzing the optical properties of the other TSC compounds in acetonitrile solutions (Fig. S77–S115 in the ESI†). Aside from the molar extinction coefficients of the E-isomers ranging from 12800 to 47500 M−1 cm−1 at their λmax (Table 1), the key discoveries are summarized as follows. Anchoring EW and ED groups in para-position of the N-phenyl group had only marginal effect on the λmax values of the E-isomers of TSC-2–TSC-5, while the t1/2 values of the Z-isomers generally shortened. TSC-6 with two –CF3 groups in meta-position showed the lowest t1/2 of 1 min among all library members. The stability of the Z-isomer of these photoswitches shows no apparent correlation with EW or ED groups. This is likely due to the rotational flexibility of the N-phenyl group around the C–N bond and its considerable distance from the CN bond, which make electronic effects difficult to evaluate. Substituting the para-position of the C-phenyl moiety (TSC-7–TSC-10) resulted, however, in a clear trend, as ED groups decrease and EW groups increase the thermal half-lives of the Z-isomers relative to the parent TSC-1. Interestingly, the absorption maxima of TSC-7–TSC-9 shifted hypsochromically upon UV light illumination with Δλmax in the range of −9 to −11 nm, while TSC-10 functionalized with –NMe2 revealed a new absorption band in the visible region at 437 nm (Δλmax = 70 nm). TSC-10 thus classifies as a positive photochrome, with the benefit of distinct absorption band separation. Next in line, we studied the ortho-halogenated chromophores and observed a significant elevation in the thermal stability of their Z-isomers. While TSC-11 and TSC-12 feature fluorides and bromides, respectively, with half-lives of 10.7 h and 1 day, TSC-13 containing chlorides demonstrates bistability, at least under the operating conditions. TSC-14 bearing a pentafluorophenyl group, however, showed a significantly shorter half-life of 53 min, highlighting the exclusive role of ortho-halogens in stabilizing the Z-isomers. This trend is also evident in azobenzenes, indicating that ortho-halogenation contributes to the prolonged thermal stability of the Z-isomer.50–52 Furthermore, the high photoconversion yields observed in TSC-11–TSC-14 compared to other TSCs can be attributed to the greater separation between the π–π* spectral bands of the E- and Z-isomers, which facilitates a more selective and efficient light absorption of the E-isomer at the irradiation wavelength (Fig. S95–S101 in the ESI†).57 While TSC-11 and TSC-14 show partial overlap between these bands, resulting in PSS values of approximately 75%, TSC-12 and TSC-13 achieve exceptional PSS values of ≥90% (Fig. S116–S124 in the ESI†). By extending the π-conjugation through larger aromatic moieties, such as transitioning from phenyl to naphthyl (TSC-15), anthracenyl (TSC-16), and pyrenyl (TSC-17), the λmax of the E-isomers expectedly shifted towards the blue edge of the visible spectrum. To verify that anthracene does not form dimers as a side reaction under light irradiation, we exposed a CDCl3 solution of TSC-16 to 365 nm light and analyzed it using 1H NMR spectroscopy (Fig. S123 in the ESI†). No signals were detected around 4.5 ppm, which would indicate anthracene dimerization.58 Instead, the results confirmed the successful formation of 41% Z-isomer at the PSS.
Lastly, heterocyclic TSCs were explored, revealing a significant impact on the photoswitching performance. Similar to TSC-10, both pyrrolyl TSC-18 and thienyl TSC-19 demonstrate positive photochromism, characterized by a new red-shifted absorption band emerging at approximately 405 nm.41 The new absorption band observed in TSC-10, TSC-18, and TSC-19 may be attributed to an intramolecular hydrogen transfer occurring in the excited state, forming a tautomeric species. Similar behaviour has been reported for acylhydrazones,41 where theoretical calculations indicate that the Z-tautomer formed via intramolecular hydrogen transfer leads to a red shift in the absorption spectrum.59 This spectral feature was absent in furanyl TSC-20. Instead, the Z-isomer of TSC-20 displayed remarkable thermal stability, likely due to the presence of an intramolecular hydrogen bond between the thiourea N–H and the oxygen atom on the furan ring.
Intrigued by this finding, we anticipated a comparable stabilization of the Z-isomer in pyridinyl derivatives TSC-21–TSC-23. To explore this further, we first conducted a thorough analysis on TSC-21 using both UV-vis and NMR spectroscopy techniques. Illuminating a solution of TSC-21 with blue light (λirr = 405 nm) resulted in the formation of a thermally stable equilibrium state, similar to furanyl TSC-20. However, 1H NMR measurements unveiled that the initial mixture of TSC-21 in MeCN-d3 was composed of an almost equal E:Z ratio of 47:53 (Fig. S125 in the ESI†). This composition can be attributed to pyridine's stronger propensity as a hydrogen bond acceptor compared to furan. Upon exposure to blue light, the E:Z ratio converted to 82:18. More importantly, when the solution was then illuminated with 340 nm, the E:Z ratio slightly adjusted to 67:33. This observation marks the first evidence of P-type photochromism in TSCs. To enhance the stability of the Z-isomer even further, we introduced an electron-donating –OMe group in TSC-22 (Fig. 4A). Due to the boost in electron density around the nitrogen atom of the pyridinyl group, TSC-22 showed an enriched initial E:Z ratio of 2:98 in MeCN-d3 (Fig. S126 in the ESI†). The intramolecular H-bond and the presence of a pseudo six-membered aromatic ring contribute to the stability of TSC-21Z, as evidenced by its single-crystal X-ray structure. Photoisomerization (λirr = 365 nm) to the energetically less preferred E-isomer led to a thermally stable PSS (Fig. 4B), which was comprised of a E:Z ratio of 85:15. Subsequent back-isomerization with 340 nm light yielded an E:Z ratio of 36:64, consistent with the spectral changes observed in UV-vis spectroscopy (Fig. 4C).
 | ||
| Fig. 4 (A) Bidirectional photoisomerization of TSC-22. (B) Conversion of TSC-22 (orange line) in MeCN (c = 2.68 × 10−5 M) to TSC-22PSS (blue line) followed over time under constant irradiation (λirr = 365 nm). (C) Conversion of TSC-22PSS (blue line) to TSC-22 (orange line) followed over time under constant irradiation (λirr = 340 nm). The insets show changes in absorbance at 343 and 355 nm followed over time during illumination. | ||
Lastly, we were interested in the potential impact of an electron-deficient quinolinyl moiety on the photoswitching behaviour. As anticipated, 1H NMR analysis revealed that the initial mixture of TSC-23 in MeCN-d3 was composed of an E:Z ratio of 78:22 due to a weaker intramolecular hydrogen bond. Upon 365 nm light exposure, the E:Z ratio flipped to 22:78 (Fig. S127 in the ESI†). Interestingly, it was not possible to back-isomerize TSC-23Z with light. Instead this isomer thermally relaxed to the initial composition over time with t1/2 = 192 min. This underlines the crucial role of electron-rich pyridinyl groups in rendering TSCs bistable P-type photoswitches.
To maximize the utility of photoswitches, they should undergo photoisomerization with minimal fatigue over numerous switching cycles. Hence, we explored the light stability of several compounds. TSC-6 was selected as a fast-relaxing T-type photoswitch and subjected to repetitive photoisomerization with 365 nm light until reaching the PSS and then allowed to relax back thermally before initiating the next cycle (Fig. 5A). A similar experiment was performed on TSC-22, only with the difference that back-isomerization was induced using 340 nm light instead of thermal energy (Fig. 5B). Both chromophores demonstrated stability, showing no significant signs of fatigue over at least 10 switching cycles. Prolonged UV light irradiation for 5 h did also not induce a spectral change that indicate light-induced degradation, as evidenced by UV-vis spectroscopy experiments on TSC-4 and TSC-13 (Fig. 5C and D). These experiments highlight the reliability of thiosemicarbazones as photoswitches.
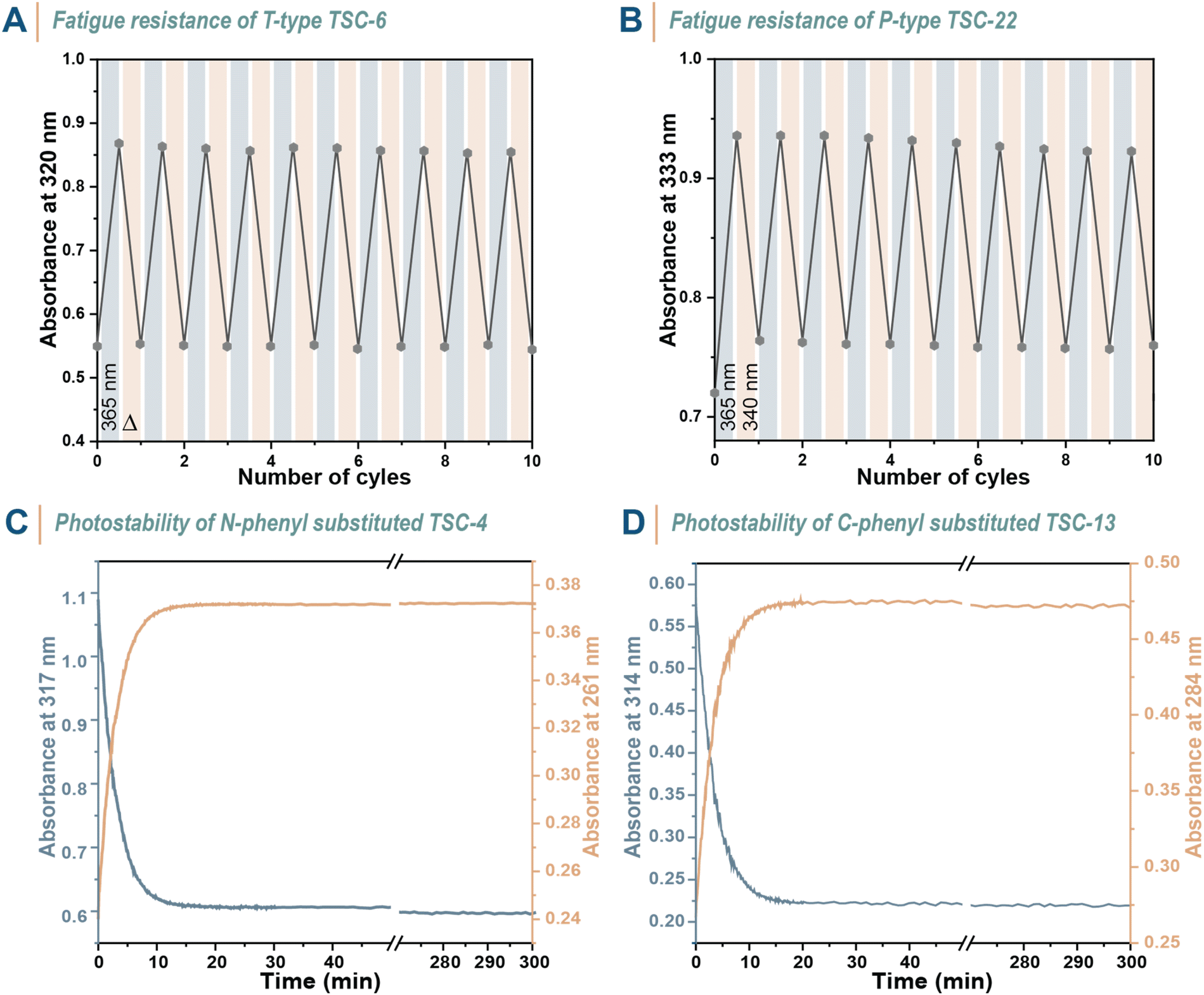 | ||
| Fig. 5 Photostability of TSCs. (A) Change in absorbance at λmax(E) during photoswitching cycles of TSC-6 in MeCN (c = 2.90 × 10−5 M) based on alternating irradiation with 365 nm LED until reaching the PSS and complete thermal back-relaxation. (B) Change in absorbance at λmax(E) during photoswitching cycles of TSC-22 in MeCN (c = 2.68 × 10−5 M) based on alternating irradiation with 365 and 340 nm LEDs until reaching the PSS in both directions. (C) Long-term irradiation of TSC-4 in MeCN (c = 3 × 10−5 M) and (D) of TSC-13 in MeCN (c = 3.14 × 10−5 M). | ||
To further evaluate the efficiency of the photoisomerization, quantum yields were determined for selected compounds at 365 nm. The halogenated TSCs were chosen due to their high thermal half-lives, with the following values: TSC-11 (ΦE→Z = 0.85), TSC-12 (ΦE→Z = 0.33), TSC-13 (ΦE→Z = 0.58), and TSC-14 (ΦE→Z = 0.83, Table S3 and Fig. S128 in the ESI†). These values are comparable or superior to those of (acyl)hydrazone photoswitches.41,60
Supramolecular properties
After investigating the structure–property relationship of thiosemicarbazones in terms of their photoswitching capability, we were inspired by the potential of modulating the supramolecular interactions between these molecules in solution using light. First, we assessed whether the E-isomer of thiosemicarbazones dimerizes in solution through hydrogen bonding between the sulfur atom of the thiourea group and hydrogen atoms Hb and Hc, as the single crystal structure analysis of TSC-13 suggests (Fig. 6A). When switching to the Z-isomer, this interaction is anticipated to be completely turned off by steric shielding of the binding site, thereby providing the opportunity to toggle between dimeric and monomeric states. We thus selected TSC-13E as a model compound due to its pronounced thermal stability and prepared a series of solutions with concentrations ranging from 9.82 to 0.57 mM in CDCl3. In 1H NMR spectra we observed a characteristic shift in the Hb and Hc signals towards high field as the concentration decreased (Fig. 6B and S129 in the ESI†). The chemical shift of Ha remained constant, as this proton is not directly involved in the dimerization process. Correlating the shift in Hb signal with the varying concentration allowed us to determine the binding constant which was obtained from two analyses with minimal standard deviation, yielding Kdim = 6.44 ± 0.01 M−1. Instead of adjusting the concentration of TSC-13E to determine its binding affinity—which is experimentally more elaborate—we also explored the option of ‘diluting’ the photoswitch in solution with light. We thus irradiated a CDCl3 solution containing TSC-13E (c = 9.83 mM) with 365 nm light and recorded 1H NMR spectra at various stages of the process (Fig. 6C and S130 in the ESI†). Through photoisomerization we gradually reduced the quantity of binding TSC-13E in solution and thus effectively diluted the compound with respect to the constant volume of solvent. Accordingly, we observed a consistent trend in the chemical shift of the Hb and Hc signals of TSC-13E, which yielded a comparable dimerization constant of 6.18 ± 0.34 M−1. At the same time, we confirmed that the formed TSC-13z did not participate in binding events due to steric constraints, since the new signals assigned to TSC-13Z, such as He at 8.6 ppm, remained constant throughout the experiment. The same experiments were conducted on TSC-11 yielding comparable values (Fig. S131 and S132 in the ESI†). These findings showcase that the non-covalent interactions between thiosemicarbazones can be turned off at will by means of light.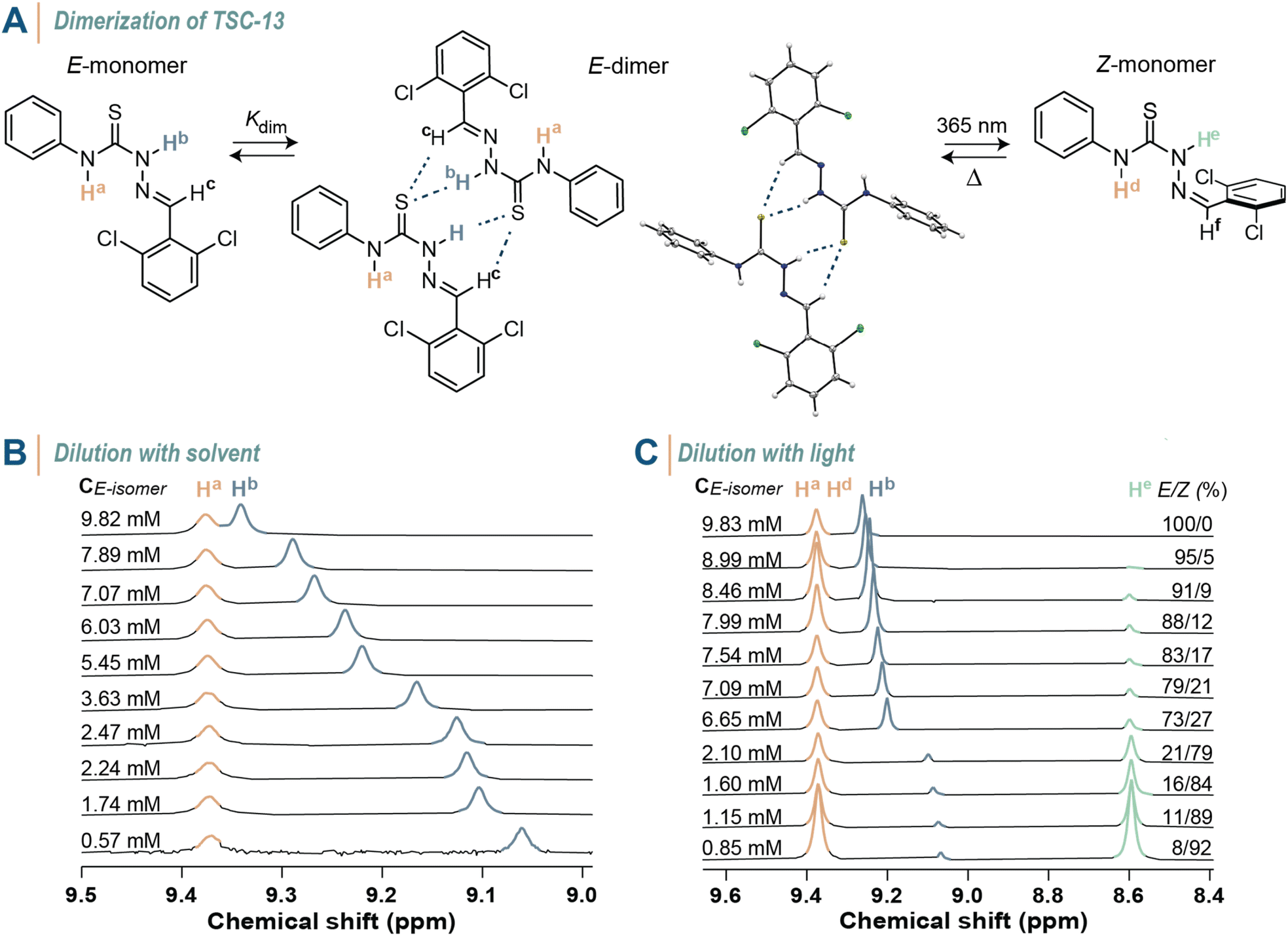 | ||
| Fig. 6 Light-controlled supramolecular interactions. (A) Dimerization of TSC-13E through H-bonding (as confirmed by single-crystal X-ray analysis), which can be turned off when switched to TSC-13Z. (B) 1H NMR spectra (400 MHz, 25 °C, CDCl3) of TSC-13 solutions with varying concentrations. (C) 1H NMR spectra (400 MHz, 25 °C, CDCl3) at various stages of irradiation with 365 nm highlighting the emergence of new signals attributed to TSC-13Z. | ||
Motivated to enhance the supramolecular interactions among thiosemicarbazones, we examined the behaviour of pyridine-substituted TSC-21 in a concentrated toluene solution. While the Z-isomer is inherently more stable in an apolar solvent due to intramolecular hydrogen bonds, we anticipated that photoisomerization to the E-isomer would unleash both binding sites—the thiourea and pyridine moiety—resulting in the formation of larger supramolecular aggregates (Fig. 7). When we exposed TSC-21 solution with an initial E:Z ratio of 20:80 to 365 nm light, we observed that 91% of the photoswitch precipitated from the solution. To recover the initial state, we simply heated the mixture, facilitating the back-isomerization to the soluble Z-isomer. A subsequent cycle of light-induced precipitation and heat-induced dissolution demonstrated the reproducibility of the process with the same E:Z ratio maintained and 90% of the E-isomer precipitated (Fig. S133 in the ESI†). To further study the aggregation behaviour, we performed SEM analysis on the precipitate, which forms a more complex morphology compared to TSC-21 obtained from toluene prior to light illumination (Fig. S134 in the ESI†).
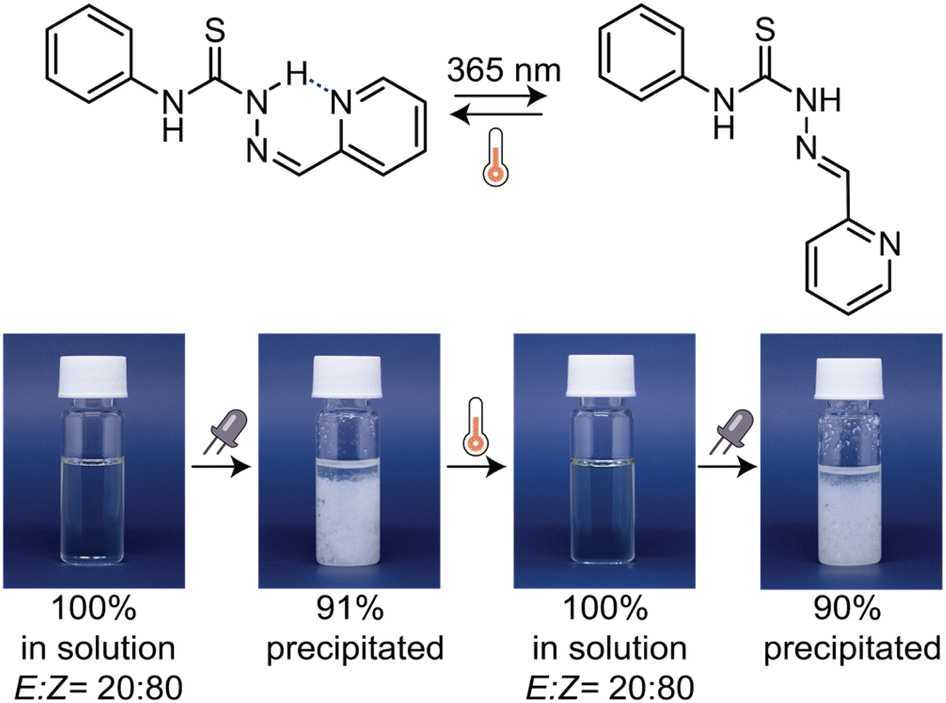 | ||
| Fig. 7 Photographs showing two cycles of the light-induced aggregation and precipitation of TSC-21 from a 15 mM solution in toluene with 365 nm LED, followed by the heat-induced dissolution due to thermal back-relaxation. | ||
Conclusions
This comprehensive study demonstrates the photoisomerization capabilities of thiosemicarbazones and their light-responsive supramolecular interactions. By systematically decorating thiosemicarbazones with various substituents, we uncovered a broad spectrum of characteristics, encompassing shifts in absorption maxima spanning 91 nm, PSS values reaching 92%, negative and positive photochromism, alongside T- and P-type photoswitching. The thermal stability of the less stable isomer showed tunability, with thermal half-lives ranging from as short as 1 min to as long as 1 day, and some remaining stable within the timeframe of observation. Additionally, thiosemicarbazones display dimerization or aggregation facilitated by intermolecular hydrogen bonding, which can be (de)activated by light on demand. This wide range of features combined with its ease of synthesis renders thiosemicarbazones an attractive and versatile newcomer in the photoswitch family. Given their potential biological activity and notable binding affinity to metal ions, thiosemicarbazones could find promising application in photopharmacology61,62 or light-controlled recognition of charged species.63 Relatively weak supramolecular interactions, as showcased in this study, have also shown to be essential in substrate activation,64,65 thereby suggesting tunable catalysis as another potential research direction. Overall, these findings pave the way for designing and developing novel light-responsive molecular systems with precisely adjustable functionalities.Data availability
The X-ray crystallographic coordinates for structures reported in this study have been deposited at the Cambridge Crystallographic Data Centre (CCDC) under deposition numbers 2358493 (TSC-1), 2358067 (TSC-4), 2358068 (TSC-13), 2358069 (TSC-14), 2358070 (TSC-19), 2358071 (TSC-20), and 2358072 (TSC-22).† Copies of the data can be obtained free of charge viahttps://www.ccdc.cam.ac.uk/structures/. All other data supplementary the findings of this study, including experimental procedures and compound characterization, NMR spectroscopy, single-crystal X-ray diffraction, UV/vis spectroscopy, are available within the article and its ESI.† All data are available from the corresponding author upon request.Author contributions
B. S. conducted syntheses, performed UV/vis and NMR spectroscopy experiments, analysed and interpreted data. B. B. solved the single crystal X-ray structures. B. S. and F. E. conceived the idea, designed the study, and wrote the manuscript. All authors discussed the results and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to Prof. Željko Tomović for fruitful scientific discussions and acknowledge Luc Rijnders for his contributions to the synthesis of TSCs. We also thank Dr Christos Pantazidis for the SEM analyses and Roegaya Sabera for the photographs. F.E. acknowledges the financial support by the Dutch Research Council NWO (VENI grant no. VI.Veni.212.215).Notes and references
- A. Goulet-Hanssens, F. Eisenreich and S. Hecht, Adv. Mater., 2020, 32, 1905966 CrossRef CAS PubMed.
- D. Villarón and S. J. Wezenberg, Angew. Chem., Int. Ed., 2020, 59, 13192–13202 CrossRef PubMed.
- F. Xu, J. Sheng, C. N. Stindt, S. Crespi, W. Danowski, M. F. Hilbers, W. J. Buma and B. L. Feringa, Chem. Sci., 2024, 15, 6763–6769 RSC.
- K. Kuntze, J. Viljakka, M. Virkki, C. Y. Huang, S. Hecht and A. Priimagi, Chem. Sci., 2023, 14, 2482–2488 RSC.
- C. Y. Huang and S. Hecht, Chem.–Eur. J., 2023, 29, e202300981 CrossRef CAS PubMed.
- V. Josef, F. Hampel, H. Dube, V. Josef, F. Hampel and H. Dube Friedrich-Alexander, Angew. Chem., Int. Ed., 2022, 61, 202210855–202210856 CrossRef PubMed.
- S. Crespi, N. A. Simeth and B. König, Nat. Rev. Chem., 2019, 3, 133–146 CrossRef CAS.
- J. Gemen, J. R. Church, T.-P. Ruoko, N. Durandin, M. J. Białek, M. Weißenfels, M. Feller, M. Kazes, M. Odaybat, V. A. Borin, R. Kalepu, Y. Diskin-Posner, D. Oron, M. J. Fuchter, A. Priimagi, I. Schapiro and R. Klajn, Nat. Rev. Chem., 2019, 3, 133–146 CrossRef.
- J. Isokuortti, K. Kuntze, M. Virkki, Z. Ahmed, E. Vuorimaa-Laukkanen, M. A. Filatov, A. Turshatov, T. Laaksonen, A. Priimagi and N. A. Durandin, Chem. Sci., 2021, 12, 7504–7509 RSC.
- S. A. M. Steinmüller, M. Odaybat, G. Galli, D. Prischich, M. J. Fuchter and M. Decker, Chem. Sci., 2024, 15, 5360–5367 RSC.
- H. Lee, J. Tessarolo, D. Langbehn, A. Baksi, R. Herges and G. H. Clever, J. Am. Chem. Soc., 2022, 144, 3099–3105 CrossRef CAS PubMed.
- J. Ewert, L. Heintze, M. Jordà-Redondo, J. S. Von Glasenapp, S. Nonell, G. Bucher, C. Peifer and R. Herges, J. Am. Chem. Soc., 2022, 144, 15059–15071 CrossRef CAS.
- Z. Yang, Z. Liu and L. Yuan, Asian J. Org. Chem., 2021, 10, 74–90 CrossRef CAS.
- F. Xu and B. L. Feringa, Adv. Mater., 2023, 35, 2204413 CrossRef CAS.
- E. Nieland, J. Voss and B. M. Schmidt, Synlett, 2022, 34, 975–982 Search PubMed.
- S. J. Wezenberg, L. J. Chen, J. E. Bos, B. L. Feringa, E. N. W. Howe, X. Wu, M. A. Siegler and P. A. Gale, J. Am. Chem. Soc., 2022, 144, 331–338 CrossRef CAS PubMed.
- J. de Jong, M. A. Siegler and S. J. Wezenberg, Angew. Chem., Int. Ed., 2024, 63, e202316628 CrossRef CAS PubMed.
- G. Tyagi, J. L. Greenfield, B. E. Jones, W. N. Sharratt, K. Khan, D. Seddon, L. A. Malone, N. Cowieson, R. C. Evans, M. J. Fuchter and J. T. Cabral, JACS Au, 2022, 2, 2670–2677 CrossRef CAS.
- D. Hugenbusch, M. Lehr, J. S. von Glasenapp, A. J. McConnell and R. Herges, Angew. Chem., Int. Ed., 2023, 62, e202212571 CrossRef CAS PubMed.
- E. Nieland, J. Voss and B. M. Schmidt, ChemPlusChem, 2023, 88, e202300353 CrossRef CAS PubMed.
- E. Weyandt, G. M. Ter Huurne, G. Vantomme, A. J. Markvoort, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2020, 142, 6295–6303 CrossRef CAS.
- L. Wei, S. T. Han, T. T. Jin, T. G. Zhan, L. J. Liu, J. Cui and K. Da Zhang, Chem. Sci., 2021, 12, 1762–1771 RSC.
- R. G. Dinardi, A. O. Douglas, R. Tian, J. R. Price, M. Tajik, W. A. Donald and J. E. Beves, Angew. Chem., Int. Ed., 2022, 61, e202205701 CrossRef CAS.
- A. D. W. Kennedy, R. G. Dinardi, L. L. Fillbrook, W. A. Donald and J. E. Beves, Angew. Chem., Int. Ed., 2022, 61, e202205701 CrossRef.
- M. Canton, J. Groppi, L. Casimiro, S. Corra, M. Baroncini, S. Silvi and A. Credi, J. Am. Chem. Soc., 2021, 143, 10890–10894 CrossRef CAS PubMed.
- M. Kathan, S. Crespi, N. O. Thiel, D. L. Stares, D. Morsa, J. de Boer, G. Pacella, T. van den Enk, P. Kobauri, G. Portale, C. A. Schalley and B. L. Feringa, Nat. Nanotechnol., 2022, 17, 159–165 CrossRef CAS PubMed.
- J. J. B. van der Tol, T. A. P. Engels, R. Cardinaels, G. Vantomme, E. W. Meijer and F. Eisenreich, Adv. Funct. Mater., 2023, 33, 202301246 CrossRef.
- P. Kobauri, F. J. Dekker, W. Szymanski and B. L. Feringa, Angew. Chem., Int. Ed., 2023, 62, e202300681 CrossRef CAS.
- K. Hüll, J. Morstein and D. Trauner, Chem. Rev., 2018, 118, 10710–10747 CrossRef.
- K. Grill and H. Dube, J. Am. Chem. Soc., 2020, 142, 19300–19307 CrossRef CAS PubMed.
- G. C. Thaggard, J. Haimerl, R. A. Fischer, K. C. Park and N. B. Shustova, Angew. Chem., Int. Ed., 2023, 62, e202302859 CrossRef CAS PubMed.
- Q. Qiu, Z. Sun, D. Joubran, X. Li, J. Wan, K. Schmidt-Rohr and G. G. D. Han, Angew. Chem., Int. Ed., 2023, 62, e202300723 CrossRef CAS PubMed.
- B. Tang, M. Pauls, C. Bannwarth and S. Hecht, J. Am. Chem. Soc., 2024, 146, 45–50 CrossRef CAS PubMed.
- L. Greb, G. Vantomme and J. M. Lehn, in Molecular Photoswitches: Chemistry, Properties, and Applications, 2 Volume Set, Wiley-VCH Verlag, 2022, vol. 1–2, pp. 325–349 Search PubMed.
- J. Wu, L. Kreimendahl, S. Tao, O. Anhalt and J. L. Greenfield, Chem. Sci., 2024, 15, 3872–3878 RSC.
- L. D. Thai, T. R. Guimaraes, L. C. Chambers, J. A. Kammerer, D. Golberg, H. Mutlu and C. Barner-Kowollik, J. Am. Chem. Soc., 2023, 145, 14748–14755 CrossRef CAS.
- D. Veselý, J. Jančík, M. Weiter, D. Blasi, N. Ivanova, J. Krajčovič and A. Georgiev, J. Photochem. Photobiol., A, 2022, 430, 113994 CrossRef.
- B. Shao and I. Aprahamian, Chem, 2020, 6, 2162–2173 CAS.
- Q. Li, H. Qian, B. Shao, R. P. Hughes and I. Aprahamian, J. Am. Chem. Soc., 2018, 140, 11829–11835 CrossRef CAS PubMed.
- H. Qian, S. Pramanik and I. Aprahamian, J. Am. Chem. Soc., 2017, 139, 9140–9143 CrossRef CAS PubMed.
- D. J. Van Dijken, P. Kovaříček, S. P. Ihrig and S. Hecht, J. Am. Chem. Soc., 2015, 137, 14982–14991 CrossRef CAS PubMed.
- Y. Che, L. Liu, Y. Tang, Y. Qiao, X. Zhao, S. Gao, S. Ding and D. Jia, New J. Chem., 2017, 41, 15229–15235 RSC.
- J. Hu, L. Liu, D. Jia, J. Guo, X. Xie, D. Wu and R. Sheng, J. Photochem. Photobiol., A, 2011, 217, 117–124 CrossRef CAS.
- X. Xie, L. Liu, D. Jia, J. Guo, D. Wu and X. Xie, New J. Chem., 2009, 33, 2232–2240 RSC.
- Y. Che, L. Liu, J. Zhao, Y. Yu and X. Zhao, Dyes Pigm., 2019, 169, 105–110 CrossRef CAS.
- Y. Tsai, N. Aira, D. Ensinck, C. Banchio, H. Gramajo, A. G. Suárez, R. A. Spanevello and A. M. Sarotti, ChemistrySelect, 2023, 8, e202303776 CrossRef CAS.
- E. Palma, P. Raposinho, M. P. C. Campello, D. Belo, J. F. Guerreiro, V. Alves, A. Fonseca, A. J. Abrunhosa, A. Paulo and F. Mendes, Eur. J. Inorg. Chem., 2021, 2021, 1337–1348 CrossRef CAS.
- A. I. Matesanz, A. B. Caballero, C. Lorenzo, A. Espargaró, R. Sabaté, A. G. Quiroga and P. Gamez, Inorg. Chem., 2020, 59, 6978–6987 CrossRef CAS PubMed.
- M. Herder, B. M. Schmidt, L. Grubert, M. Pätzel, J. Schwarz and S. Hecht, J. Am. Chem. Soc., 2015, 137, 2738–2747 CrossRef CAS PubMed.
- L. N. Lameijer, S. Budzak, N. A. Simeth, M. J. Hansen, B. L. Feringa, D. Jacquemin and W. Szymanski, Angew. Chem., Int. Ed., 2020, 59, 21663–21670 CrossRef CAS PubMed.
- K. Hema, A. B. Grommet, M. J. Białek, J. Wang, L. Schneider, C. Drechsler, O. Yanshyna, Y. Diskin-Posner, G. H. Clever and R. Klajn, J. Am. Chem. Soc., 2023, 24755–24764 CAS.
- D. Bléger, J. Schwarz, A. M. Brouwer and S. Hecht, J. Am. Chem. Soc., 2012, 134, 20597–20600 CrossRef PubMed.
- C. Laurence, S. Mansour, D. Vuluga, A. Planchat and J. Legros, J. Org. Chem., 2021, 86, 4143–4158 CrossRef CAS PubMed.
- C. Nieuwland and C. Fonseca Guerra, Chem.–Eur. J., 2024, 30, e202304361 CrossRef CAS PubMed.
- Y. L. Wu and A. I. Wright, Phys. Chem. Chem. Phys., 2023, 25, 1342–1348 RSC.
- C. Nieuwland, R. Verdijk, C. Fonseca Guerra and F. M. Bickelhaupt, Chem.–Eur. J., 2024, 30, e202304161 CrossRef CAS PubMed.
- R. Lin, P. K. Hashim, S. Sahu, A. S. Amrutha, N. M. Cheruthu, S. Thazhathethil, K. Takahashi, T. Nakamura, T. Kikukawa and N. Tamaoki, J. Am. Chem. Soc., 2023, 145, 9072–9080 CrossRef CAS PubMed.
- G. W. Breton and X. Vang, J. Chem. Educ., 1998, 75, 81 CrossRef CAS.
- M. Moreno, R. Gelabert and J. M. Lluch, Phys. Chem. Chem. Phys., 2019, 21, 16075–16082 RSC.
- B. Shao, H. Qian, Q. Li and I. Aprahamian, J. Am. Chem. Soc., 2020, 141, 8364–8371 CrossRef PubMed.
- S. Samanta, A. A. Beharry, O. Sadovski, T. M. McCormick, A. Babalhavaeji, V. Tropepe and G. A. Woolley, J. Am. Chem. Soc., 2013, 135, 9777–9784 CrossRef CAS PubMed.
- M. J. Fuchter, J. Am. Chem. Soc., 2020, 63, 11436–11447 CAS.
- M. Natali and S. Giordani, Chem. Soc. Rev., 2012, 41, 4010–4029 RSC.
- D. Larsen, L. M. Langhorn, O. M. Akselsen, B. E. Nielsen and M. Pittelkow, Chem. Sci., 2017, 8, 7978–7982 RSC.
- C. Thomas, F. Peruch, A. Deffieux, A. Milet, J. P. Desvergne and B. Bibal, Adv. Synth. Catal., 2011, 353, 1049–1054 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods, experimental procedures, NMR spectroscopy, single-crystal X-ray diffraction, UV/vis spectra, PSS determination, quantum yield, dimerization constant determination studies, light-induced aggregation, additional references. CCDC 2358067, 2358068, 2358069, 2358070, 2358071, 2358072 and 2358493. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc08530b |
| This journal is © The Royal Society of Chemistry 2025 |