
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAccess to arynes from arenes via net dehydrogenation: scope, synthetic applications and mechanistic analysis†‡
Riley A.
Roberts§
 ,
Bryan E.
Metze§
,
Nicole
Javaly
,
Theresa M.
McCormick
* and
David R.
Stuart
*
,
Bryan E.
Metze§
,
Nicole
Javaly
,
Theresa M.
McCormick
* and
David R.
Stuart
*
Department of Chemistry, Portland State University, Portland, OR 97201, USA. E-mail: dstuart@pdx.edu
First published on 20th February 2025
Abstract
Arynes undergo a wide range of chemical transformations making them versatile reactive intermediates for organic synthesis. Access to arynes has long been dominated by pre-functionalised reagents, e.g., the venerable o-trimethylsilylaryl triflates. However, a move toward developing methods to access arynes that are both mild and efficient has prompted research into aryl “onium” aryne precursors. Here, we leverage aryl “onium” species as in situ or isolated intermediates in a net dehydrogenation of simple arenes as a novel and efficient way to access arynes. We describe a unified strategy in which two different tactics are employed to access diversely substituted arynes from simple arenes. (1) We developed a one-pot method that converts simple arenes into aryl thianthrenium salts and uses them in situ to generate arynes. (2) We developed a two-step process to convert arenes into aryl(Mes)iodonium salts and ultimately trapped arynes to expand the scope of compatible arenes. The net transformations from arenes to trapped arynes are complete with 2–4 hours. Mechanistic analysis through competition experiments, deuterium kinetic isotope effects (DKIE) and Density Functional Theory (DFT) provide key comparisons of the two approaches described in this work and yield a user's guide for selecting the appropriate “onium” leaving group based on the arene.
Introduction
The versatility of arynes as reactive intermediates continues to spur advances in organic synthesis including the discovery and development of asymmetric reactions and access to complex scaffolds.1–5 Although deprotonation/β-elimination was one of the earliest methods to generate arynes, the past several decades have been dominated by fluoride induced elimination of o-trimethylsilylaryl triflates because this has proven to be an exceptionally mild strategy (Scheme 1a).6 Notwithstanding the functional group compatibility of this approach,7 the primary limitation lies in the multi-step synthesis of o-trimethylsilylaryl triflates (Scheme 1a). The commercial availability of these reagents is therefore limited and the cost higher than other arylation reagents. Moreover, the synthetic sequence to access o-trimethylsilylaryl triflates requires strong bases and consequently the functional group compatibility of these aryne precursors is actually quite limited with respect to the aryl substituents (R-group in Scheme 1a). On balance, although the generation of arynes is mild, o-trimethylsilylaryl triflates do not provide efficient access to diversely substituted arynes and novel aryne precursors are needed to continue advancing synthetic capabilities with these reactive intermediates.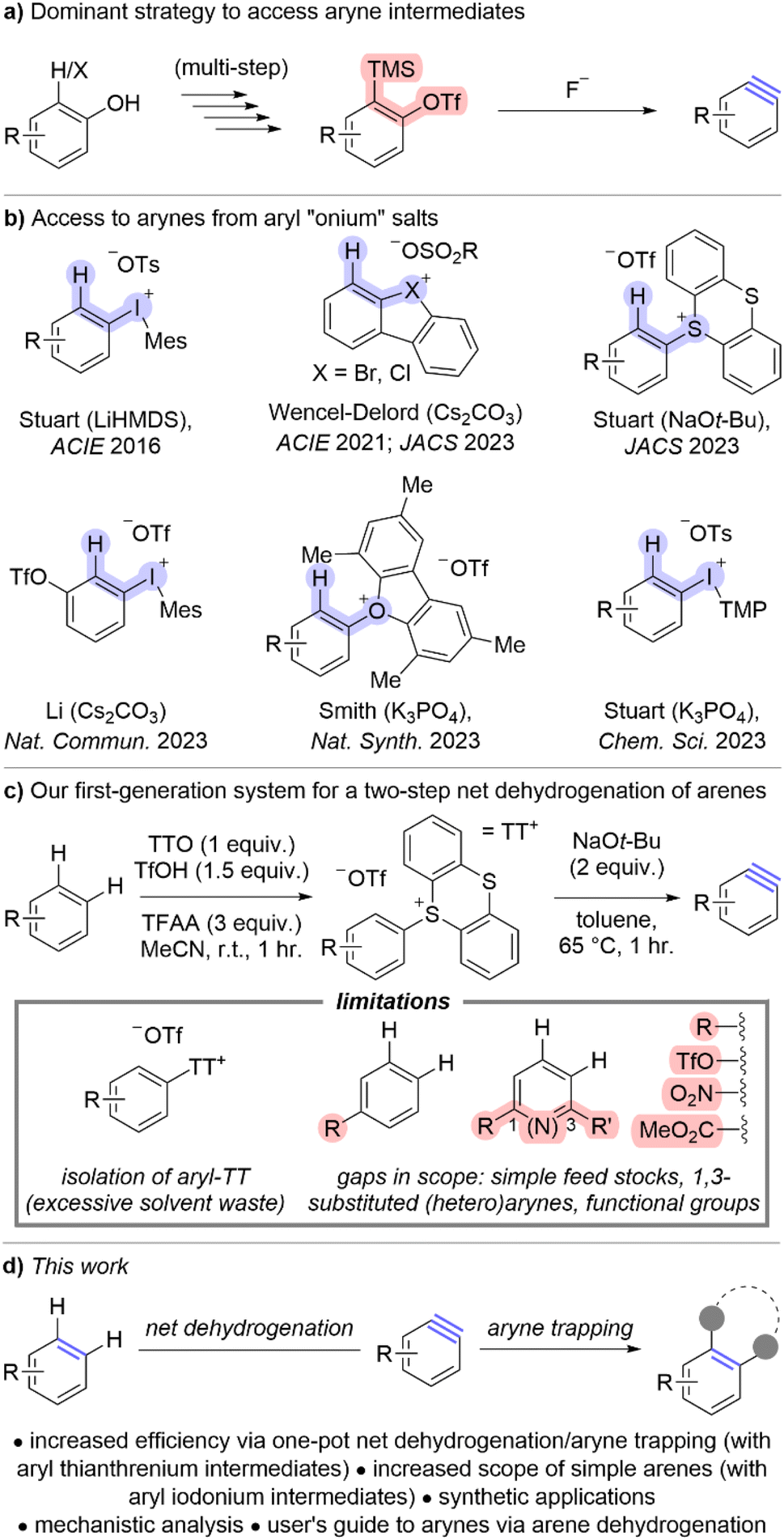 | ||
| Scheme 1 Overview of access to arynes. | ||
Aryl “onium” reagents have emerged as aryne precursors during a resurgence of methods based on ortho-deprotonation/β-elimination (Scheme 1b).7–13 These aryne precursors include those based on halogen leaving groups such as iodonium,7,8,12,14–18 bromonium,9,19 and chloronium,10,19 as well as chalcogen leaving groups such as sulfonium and oxonium (Scheme 1b).11,13,20,21 In several cases, these “onium” leaving groups permit the use of mild bases such as potassium and cesium carbonate and potassium phosphate to generate arynes,7,9,10,12,13,19,22 and these approaches rival or even exceed the functional group compatibility of fluoride-induced aryne generation of o-trimethylsilyl triflates.7 Although aryl “onium” reagents have provided a mild way to generate arynes via deprotonation, they have not universally improved the generality and efficiency of accessing arynes. For instance, cyclic diarylhalonium salts specifically deliver biaryl-derived arynes and therefore lack broad generality.9,10,19,23 Additionally, triaryloxonium salts require a substantial time and material investment to synthesize, similar to that of o-trimethylsilylaryl triflates.13 Unsymmetrical aryl(Mes)iodonium and aryl thianthrenium salts are uniquely positioned within this class of aryne precursors to effect both mild and efficient access to arynes (Scheme 1b).
Simple arenes are unconventional aryne precursors because they lack complimentary electrofugal and nucleofugal leaving groups for a redox neutral elimination event. Yet, the dehydrogenation of simple arenes represents an expansive source of latent aryne intermediates and even has the potential for late-stage functionalization of active pharmaceutical ingredients and agrochemicals. In 2023 we merged the C–H functionalization of arenes via thianthrenation and deprotonative aryne generation for a net dehydrogenation of simple arenes as an efficient way to access arynes (Scheme 1c).11 In our early optimisation, we found that aryl thianthrenium salts were more easily accessed than aryl(Mes)iodonium salts and we therefore developed a two-step sequence in which the aryl thianthrenium salts were isolated and subsequently used as aryne precursors (Scheme 1c). Although the isolation of aryl thianthrenium salts is relatively straightforward, only involving trituration and filtration, it does represent an additional, and potentially unnecessary, manipulation (Scheme 1c). Moreover, we found gaps in scope including simple feedstocks (i.e., benzene, toluene, anisole), polysubstituted arenes (i.e., 1,3-disubstituted arenes) including heteroarenes, and several useful functional groups such as triflate, nitro, and ester (Scheme 1c).
Herein, we describe a conceptual shift to access arynes from reagents that rely on pre-functionalization to those that only bear hydrogen substituents at the eventual aryne site (Scheme 1d). The methods we describe in this work use simple arenes to generate arynes, which substantially expands the breadth of arynes that are accessible. Moreover, we increase the efficiency of this approach so that a simple arene may be converted into a trapped aryne within hours and no isolation of aryl thianthrenium intermediate species is needed. Additionally, we show that tuning the C–H functionalization by installing iodonium leaving groups expands the scope of arenes that participate in these transformations. Finally, mechanistic analysis reveals the complementarity of sulfonium and iodonium leaving groups and this is specifically highlighted in a “user's guide” to employing simple arenes to access arynes.
Results and discussion
Increased efficiency
The overarching goal of this line of research is to convert simple arenes into arynes. We partially achieved this goal by developing a two-step procedure to install a thianthrenium leaving group followed by ortho-deprotonation and β-elimination (Scheme 1c).11 To further advance this goal, we re-evaluated this process to develop a one-pot procedure in which a simple arene can be selected from a chemical inventory and converted into a trapped aryne within several hours and without isolation of any intermediate species (Scheme 2). There were two major hurdles to merging the two-pot process into a one-pot procedure: (1) two different solvents were used and (2) the acidic and basic conditions of the two steps are incompatible. We selected 4-chloroanisole 1a as the simple arene for study and the conditions presented below in entry 1 of Table 1 are the result of a large number of experiments to rectify the challenges described above. Under these conditions 1a and TTO are dissolved in MeCN followed by the addition of triflic anhydride (Tf2O) at 0 °C to yield a homogenous light purple solution (Table 1). After stirring for 30 minutes at room temperature, solid potassium phosphate is added to the vial, which results in a loss of the purple color and the reaction mixture is stirred for an additional five minutes. Furan 2a and solid sodium tert-butoxide are added to the vial and it is placed in a pre-heated (65 °C) aluminum block for 60 minutes. The trapped aryne product 3aa is observed in 86% 1H-NMR yield from 1a; 3aa is obtained in 73% isolated yield after purification by column chromatography when the reaction is conducted on 0.5 mmol scale of 1a (Table 1). A summary of experiments to demonstrate the relevance of each component in the reaction sequence is shown in Table 1 (entries 2–8). Toluene was used in our prior work in the aryne forming step,11 however when the one-pot sequence is performed in toluene only trace (<5%) amounts of 3aa were observed in the crude 1H NMR spectrum (Table 1, entry 2). Triflic acid is a by-product of the C–H thianthrenation step and therefore leaving out the neutralization step (K3PO4) results in a greatly diminished yield of 3aa even with excess base in the second step (23%; Table 1, entry 3). Two other methods were tested to activate the TTO reagent under the one-pot conditions (Table 1, entries 4 and 5). The strongly acidic conditions involving a combination of TFAA and TfOH resulted in low yield (10%) of 3aa whereas conditions with the mixed anhydride TFAOTf resulted in comparable yield (83%) to those with Tf2O (Table 1, entries 4 and 5). The mixed anhydride TFAOTf, however, is substantially more expensive than Tf2O. Two other sulfonium leaving groups were also tested in this reaction (Table 1, entries 6 and 7). The tetrafluorinated analog of TTO, TFTO, resulted in low yield of 3aa (26%), though the dibenzothiophene analog, DBTO, resulted in a similar, but slightly lower, yield to that obtained with TTO (75%; Table 1, entries 6 and 7). Finally, LiHMDS as base instead of NaOt-Bu in step 2 resulted in substantially lower yield (34%; Table 1, entry 8).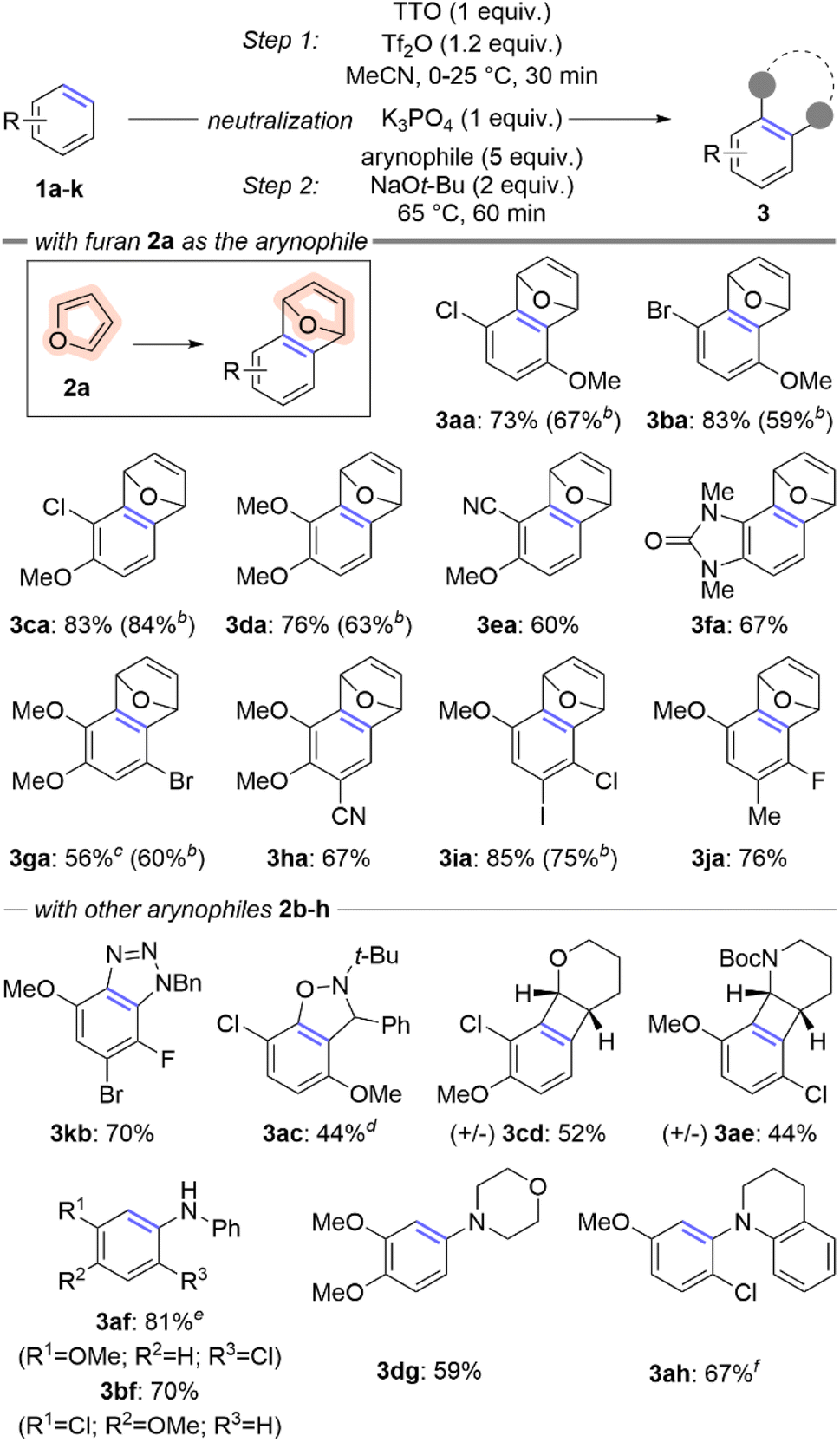 | ||
Scheme 2 Scope of simple arenes used to access arynes.a aConditions: 1 (0.5 mmol, 1 equiv.), TTO (0.5 mmol, 1 equiv.), Tf2O (0.6 mmol, 1.2 equiv.), MeCN (2 mL), 0–25 °C, 30 minutes; K3PO4 (0.5 mmol, 1 equiv.), r.t., 5 minutes; 2 (0.5–2.5 mmol, 1–5 equiv., see ESI‡); NaOt-Bu (1 mmol, 2 equiv.), 65 °C, 60 minutes. bYield obtained from 2-pot procedure.11cIsolated with 5% TTO as a contaminant. dMinor regioisomer isolated in 29% yield. eConducted on 7 mmol scale of 2a and isolated as a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of regioisomers (major isomer shown). fIsolated as an 8.1:1 mixture of regioisomers (major isomer shown). :1 mixture of regioisomers (major isomer shown). fIsolated as an 8.1:1 mixture of regioisomers (major isomer shown). | ||
|
|
||
|---|---|---|
| Entry | Deviation from standard conditions | NMR yield |
| a Conditions: 1a (0.1 mmol, 1 equiv.), TTO (0.1 mmol, 1 equiv.), Tf2O (0.12 mmol, 1.2 equiv.), MeCN (0.4 mL), 0 °C – r.t., 30 min; K3PO4 (0.1 mmol, 1 equiv.), r.t., 5 min; 2a (0.5 mmol, 5 equiv.), NaOt-Bu (0.2 mmol, 2 equiv.), 65 °C, 60 min; yields determined by 1H NMR spectroscopic analysis of crude reaction mixtures with 1,3,5-trimethoxybenzene as internal standard. b Isolated yield on 0.5 mmol scale of 1a. | ||
| 1 | Nonea | 86%, b73% |
| 2 | Toluene instead of acetonitrile as solvent | <5% |
| 3 | No K3PO4 added after step 1 | 23% |
| 4 | TFAA + TfOH instead of Tf2O | 10% |
| 5 | TFAOTf instead of Tf2O | 83% |
| 6 | TFTO instead of TTO | 26% |
| 7 | DBTO instead of TTO | 75% |
| 8 | LiHMDS instead of NaOt-Bu | 34% |
The scope of the one-pot method to convert simple arenes into trapped arynes was evaluated with ten different arenes and nine different arynophiles (Scheme 2). Six of the arenes used in the one-pot method had also been previously used in our two-pot method (1a–d, 1g, 1i; Scheme 2). In four of the cases, the yield is higher in the one-pot method than the two-pot method by 6–24% (3aa, 3ba, 3da, and 3ia; Scheme 2). In the two cases in which the yield is not higher for the one-pot method, it is within 5% and the greater efficiency and reduced waste of the one-pot method make it preferable (3ca, 3ga; Scheme 2). Several new arenes were also evaluated in the one-pot method including 2-cyanoanisole (1e), a benzoimidazolone derivative (1f), and trisubstituted arenes (1h and 1j; Scheme 2). A range of different arynophiles were also compatible in the one-pot procedure, including those that undergo [3 + 2] cycloaddition such as benzyl azide20 (2b) and a nitrone (2c); as well as those that participate in [2 + 2] cycloaddition such as cyclic enol ether (2d) and cyclic enamine (2e; Scheme 2). The later reaction converts simple arenes into densly functionalized racemic benzocyclobutanes (3cd and 3ae; Scheme 2). Amines are one of the strongest arynophiles known,11,21,24,25 and are compatible in this one-pot procedure. Both aromatic and aliphatic (2f–h) amines trap arynes derived from simple arenes to generate new aryl amine products (3af, 3bf, 3dg, and 3ah; Scheme 2).
Increased scope
During evaluation of the scope of the one-pot method we found that several feedstock arenes resulted in low yield (Fig. 1). Specifically, when we attempted to use benzene, toluene, anisole, or 1,3-dimethoxybenzene in this reaction we only observed trace (<5%) yield of trapped aryne product (Fig. 1). However, C–H thianthrenation of these arenes is well-established;26–31 moreover, we have independently synthesized the corresponding aryl thianthrenium salts and observed trace yield when we attempted to generate and trap the corresponding arynes.32 | ||
| Fig. 1 Limitations of arene scope in generating arynes with aryl thianthrenium intermediates. | ||
In order to address the observed gaps in the scope of arenes used to access arynes via aryl thianthrenium intermediates, we considered alternative “onium” leaving groups. Our prior work in the area of generating arynes from aryl “onium” reagents suggested that iodonium leaving groups might be a viable intermediate to dovetail these missing arenes and their corresponding arynes.8 Although, several aryliodonium leaving groups are known in the context of generating arynes, including phenyl,33,34 mesityl,8,14–17,22 and 2,4,6-trimethoxyphenyliodonium,7,35 we hypothesized that the mesityliodonium is uniquely suited to this role for two key reasons. First, the 2,6-dimethyl groups of mesityl block deprotonation on the mesityl ring and lead to selective aryne formation on the other ring of the diaryliodonium salts; this scenario is not necessarily predictable with the phenyliodonium leaving group. Second, the mesityliodonium group may be installed by C–H functionalization of arenes using commercially available mesityliodine diacetate;36,37 this scenario is not generally possible for installation of the 2,4,6-trimethoxyphenyliodonium group. We modified Dohi and Kita's conditions36 to synthesize and isolate aryl(Mes)iodonium salts and then used these as reagents to generate arynes and trap them with furan; in all cases the reported yields are for the two-step procedure from the simple arene (Scheme 3). We were unable to develop a one-pot procedure, as in the case of aryl thianthrenium intermediates, because we found that both the synthesis of and use of diaryliodoniums as aryne precursors is substantially more sensitive to solvent effects and low yields were observed in acetonitrile as the sole solvent.16 In the case of benzene we used commercially available phenyliodine diacetate (PIDA) because, consistent with Dohi and Kita's prior observations,36 benzene is unreactive with mesityliodine diacetate, even under forcing acidicconditions. The symmetrical diphenyliodonium triflate was used to generate benzyne which was trapped with furan in 59% overall yield from benzene 1l (3la; Scheme 3). Toluene, anisole, and diphenyl ether were all compatible under the standard conditions and generated trapped arynes in high overall yields (3ma, 3na, 3pa; Scheme 3). Several 1,2-disubstituted arenes were also compatible including those bearing functional groups that were not compatible with a thianthrenium leaving group, including triflate (1q), nitro (1s), phenyl (1t), and ester (1w) groups (3qa, 3sa, 3ta, 3wa; Scheme 3). We did not observe competitive loss of the triflate group in the case of 3qa (Scheme 3). In the case of a trifluoromethyl substituent, 1r-TT resulted in trace aryne adduct in our hands, though the isomer 4-trifluoromethyl anisole has been successfully used by others.21 Additionally, aryl(Mes)iodonium reagent 1r-IMes yielded an approximate 10:1 mixture of inseparable regioisomers due to competitive deprotonation at the 3- and 5-position (Scheme 3). When we replaced furan with a nitrone 2c to trap the aryne, a similar mixture of regioisomers were formed, yet were separable by chromatography and the major one isolated in 76% overall yield from 1r (3rc, Scheme 3). We have noted previously that substitution patterns impact the reactivity of aryl “onium” aryne precursors, and those derived from 1,3-substituted arenes are the least reactive.35 We found that using the aryl(Mes)iodonium intermediates and LiHMDS as base successfully generated arynes from these substrates which were trapped with furan (3ua, 3oa and 3va; Scheme 3). In the case of arene 1u, an 8:1 mixture of 1u-IMes isomers is obtained from iodination at the 4- and 6-positions, respectively, and when this mixture was carried forward a 10:1 mixture of isomers was obtained for the aryne adduct. The major isomer 3ua shown results from aryne formation at the 4,5-position and the minor isomer results from formation at the 5,6-position (Scheme 3). Similar isomer ratios were obtained for the synthesis and use of 1u-TT as an intermediate, but the yield of aryne adduct from 1u-TT is notably lower (3ua, 56%; Scheme 3). The use of aryl(Mes)iodonium salts has also provided access to a trapped pyridyne (3va) and to the best of our knowledge this constitutes the first example of a pyridine to pyridyne process (Scheme 3).
 | ||
| Scheme 3 Increased scope of arenes via two-step C–H functionalisation/aryne generation with aryl(Mes)iodonium intermediates.a aConditions: step 1: 1 (1 mmol, 1 equiv.), MesI(OAc)2 (1 mmol, 1 equiv.), TfOH (1 mmol, 1 equiv.), MeCN (1 mL), r.t., 3 hours; step 2: 1-IMes (0.5 mmol), 2a (2.5 mmol, 5 equiv.), LiHMDS (1 M in toluene, 0.55 mmol, 1.1 equiv.), toluene (2.5 mL total), r.t., 1 hour. bPhI(OAc)2 (0.5 mmol) instead of MesI(OAc)2 in step 1. cYield obtained from 1-TT salt under first-generation conditions with furan 2a as the aryne trap.11dNitrone 2c (0.5 mmol, 1 equiv.) used instead of 2a; minor regioisomer isolated in 8% yield. eUsed TMSOTf (1 mmol, 1 equiv.) instead of TfOH in step 1. fIsolated as a 10:1 mixture of regioisomers; the major isomer is shown, the minor isomer resulted from the aryne at the 5,6-position. gYield using 1u-TT as isolated intermediate. hYield using 1o-TT as in situ intermediate. | ||
Synthetic applications
The ability to efficiently convert arenes into arynes is aptly suited to late-stage functionalization. We have demonstrated this concept on O-methyl triclosan 1x, the methylated derivative of an anti-bacterial agent found in many consumer products (Scheme 4a). In this case we subjected 1x to our standard one-pot conditions via the in situ generated aryl thianthrenium and the trapped aryne 3xa was isolated in 74% yield (Scheme 4a). We have also shown that aryne intermediates can be used to access several naphthalene derivatives, namely naphthoquinones, naphthalenes, and naphthols (Scheme 4b). Arene 1a was used as the starting material in all three cases. First, a one-pot method to transform 1a into the corresponding naphthoquinone is achieved in 66% yield when 2j is used in the aryne trapping step and the crude reaction mixture is stirred with trifluoroacetic acid (4; Scheme 4b). Second, the aryne adduct 3aa can be converted into a naphthalene via “oxygen deletion”; the two-pot process for generating naphthalene 5 from arene 1a proceeds in 43% overall yield (Scheme 4b).38,39 Finally, the oxabicyclic ring of 3aa can be ring opened with HCl to generate the functionalized 1-naphthol 6; this process can be conducted in a single-pot and 6 can be isolated in 60% overall yield from 1a (Scheme 4b). Alternatively, 6 can be carried forward in the same flask to generate a derivative of propranolol (highlighted in grey on 7) in 34% overall yield from 1a (Scheme 4b).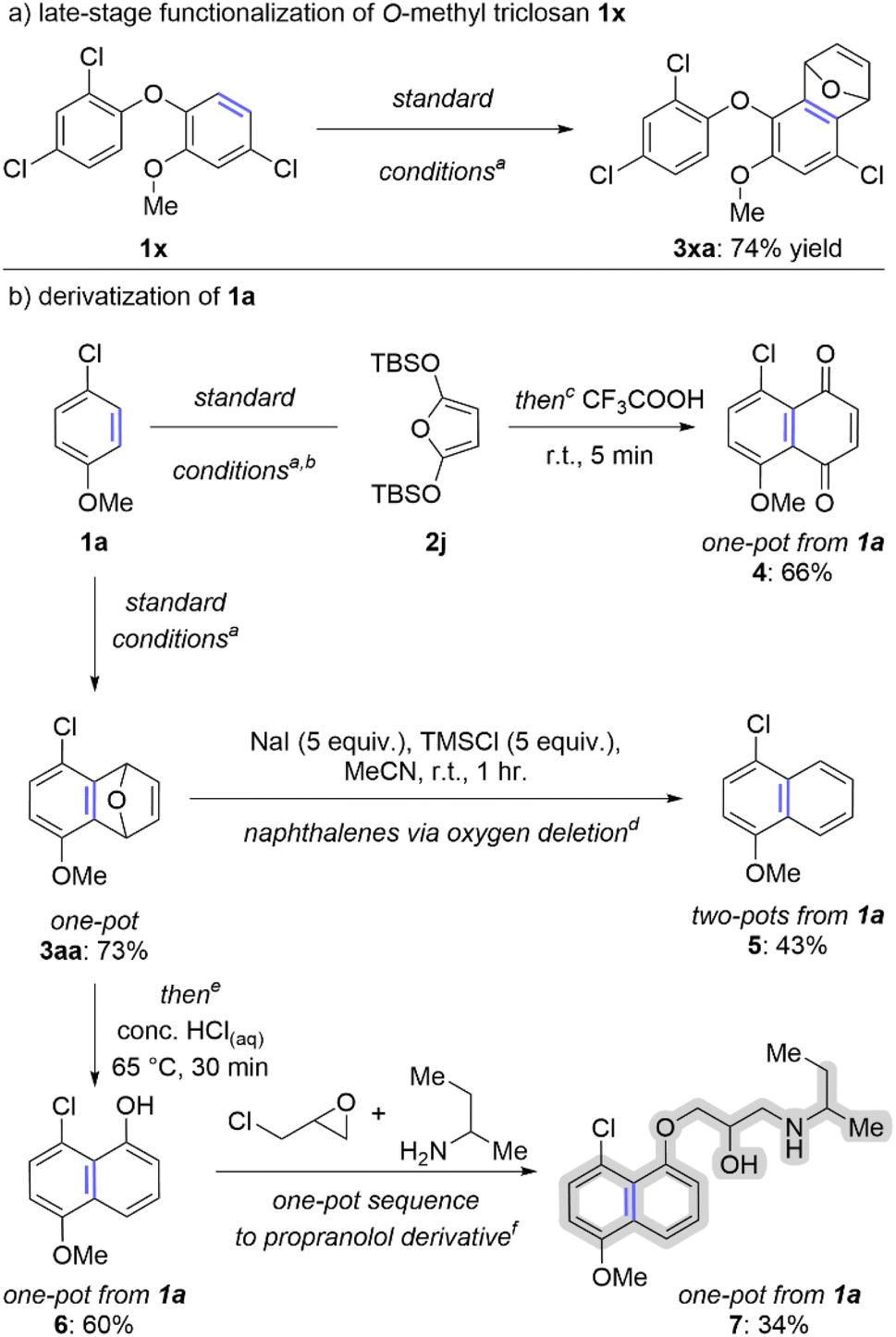 | ||
| Scheme 4 Synthetic applications of accessing arynes from simple arenes.a aO-Methyltriclosan (0.5 mmol, 1 equiv.), TTO (0.5 mmol, 1 equiv.), Tf2O (0.6 mmol, 1.2 equiv.), MeCN (2 mL), 0–25 °C, 30 minutes; K3PO4 (0.5 mmol, 1 equiv.), r.t., 5 minutes; 2a (2.5 mmol, 5 equiv., see ESI‡); NaOt-Bu (1 mmol, 2 equiv.), 65 °C, 60 minutes. bUsed 2j (2.5 mmol, 5 equiv.) instead of 2a as aryne trap. cAfter step 2 of the “standard conditions” described in (a) CF3COOH (1 mL, 26 equiv.), r.t., 5 min. d3aa (0.5 mmol, 1 equiv.), NaI (2.5 mmol, 5 equiv.), TMSCl (2.5 mmol, 5 equiv.), MeCN (1 mL), r.t. 1 hour. eStandard conditions conducted on 0.1 mmol scale of 1a; after step 2 of the “standard conditions” described in (a) conc. HClaq (1 mL, 11 equiv.), 65 °C, 30 min. fSee ESI‡ for conditions of multi-step, one-pot sequence. | ||
Mechanistic analysis
Our synthetic studies (vide supra) suggest that the two strategies described here for net arene dehydrogenation, one using aryl thianthrenium intermediates and the other using aryl(Mes)iodonium intermediates, have complementary arene scope. We have performed experiments to probe the mechanism of each step, C–H functionalization and aryne formation, and we have compared and contrasted the results for each tactic. We used deuterium kinetic isotope effects (DKIE), competition experiments, and Density Functional Theory (DFT), and our mechanistic analysis corroborates our synthetic studies; each step of the reaction has mechanistic subtleties that contribute to complementary overall scope based on the two aryl “onium” intermediates employed.The first step of the net dehydrogenation is the installation of the “onium” leaving group, and given that this occurs at an aromatic C–H bond we used DKIE analysis to probe this step. A mixture of toluene 1m and deuterated toluene 1m-d8 was subjected to the first step of the standard conditions for each approach (Scheme 5). Thianthrenation of toluene via activation by Tf2O in acetonitrile yielded a DKIE of 1.6 (Scheme 5a). This result is consistent with Wang's DKIE of 1.4 obtained for the same system albeit conducted in DCM,28 but stands in contrast to the DKIE of 2.7 obtained by Ritter when TTO is activated by a combination of TFAA and TfOH.40 Ritter has suggested that the stability of TT+˙ as a persistent radical and, therefore, the fast and reversible formation and cleavage of the C–S bond relative to slow deprotonation of the Wheland intermediate is the cause of the primary DKIE of 2.7. However, the substantially lower DKIE obtained by us and Wang, 1.6 and 1.4, respectively,28 suggest that the mode of activation may also impact the kinetic relevance of the C–H bond cleavage step. That is, the mode in which TTO is activated may generate distinct thianthrenium electrophiles and impact the relative barriers for C–S bond formation and C–H bond cleavage; the latter appears to be less kinetically relevant when TTO is activated by Tf2O (Scheme 5a).28 We also performed a DKIE analysis on the formation of tolyl(Mes)iodonium triflate, 1m-IMes, by the same method (Scheme 5b). In this case, we obtained a DKIE of 1.5, a value similar to the DKIE obtained for arene thianthrenation (Scheme 5b). These data suggest that the kinetic relevance of the C–H cleavage step is similar for thianthrenation and iodination steps and points toward the existence of a Wheland intermediate in both cases that undergoes relatively fast deprotonation.
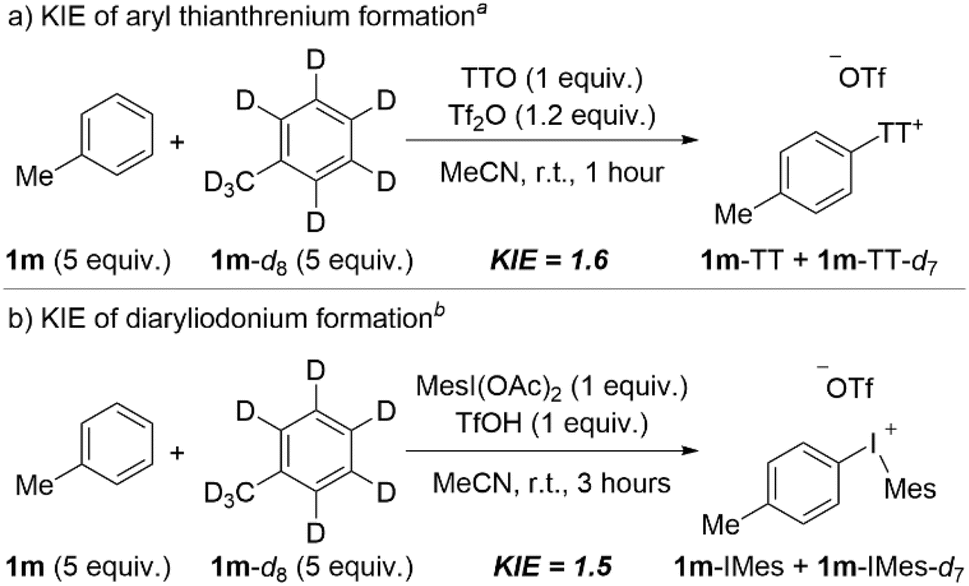 | ||
| Scheme 5 DKIE analysis for the formation of aryl “onium” species. aConditions: 1m (0.5 mmol, 5 equiv.), 1m-d8 (0.5 mmol, 5 equiv.), TTO (0.1 mmol, 1 equiv.), Tf2O (0.12 mmol, 1.2 equiv.), MeCN (0.4 mL), 0–25 °C, 1 hour. bConditions: 1m (0.5 mmol, 5 equiv.), 1m-d8 (0.5 mmol, 5 equiv.), MesI(OAc)2 (0.1 mmol, 1 equiv.), TfOH (0.1 mmol, 1 equiv.), MeCN (0.1 mL), r.t., 3 hours. | ||
To further probe the existence of a Wheland intermediate, we considered a Hammett analysis via one-pot competition experiments for both the formation of aryl thianthrenium and aryl(Mes)iodonium salts. However, our initial attempts to construct a Hammett plot in both cases were unsuccessful because electron-rich arenes, such as anisole and toluene completely out competed benzene in the case of thianthrenation. Moreover, we, and others,36 have found that benzene is unreactive with iodomesitylene diacetate under the conditions we used to install the iodonium leaving groups. Therefore, we conducted a series of competition experiments to construct a qualitative scale of relative arene reactivity for the formation of the two different aryl “onium” species (Scheme 6). We used an equal excess of the two competing arenes relative to TTO or MesI(OAc)2 (Scheme 6). Although the scales for the two processes appear similar, there are some important distinctions that warrant further comment. The order of arene relative reactivity for general classes of arenes is consistent for the two processes: benzene, toluene, chloroanisoles, anisole, dimethoxybenzenes is the order from least to most reactive. Additionally, thianthrenation has a broader scope of compatible arenes than iodination (Scheme 6). Although Hammett plots provide a ρ-value which can be a useful metric to describe the electronic properties of the arene during the rate determining step,40 individual plots must be constructed for a series of substituents at a given position (meta or para). In our relative reactivity scale, we have included polysubstituted arenes which demonstrates how the position and electronic effects of multiple substituents impacts reactivity. For instance, 2-, 3- and 4-chloroanisoles, 1c, 1u and 1a, respectively, have relative reactivity between toluene 1m and anisole 1n (Scheme 6). Therefore, although chloro is a deactivating substituent, the combination of chloro (deactivating) and methoxy (activating) is more reactive than a methyl (activating) substituent. Additionally, the relative position of the substituents has an impact on reactivity, which was not necessarily consistent between thianthrenation and iodination (Scheme 6). Again, considering the 2-, 3- and 4-chloroanisoles, the 1,4-substitution pattern was the least reactive in both thianthrenation and iodination reactions, in fact 4-chloroanisole 1a was not a viable substrate for iodination (Scheme 6). In the case of thianthrenation, 2-chloroanisole 1c was slightly more reactive than 3-chloroanisole 1u; though this order was reversed for iodination (Scheme 6). The trends observed for dimethoxybenzenes shared some similarities with those of chloroanisoles. For example, 1,4-dimethoxybeznene 1y was not a viable substrate for thianthrenation or iodination. Moreover, 1,2-dimethoxybenzene 1d was only compatible with thianthrenation but not iodination. In the case of thianthrenation, the 1,3-substitution pattern 1o is more reactive than 1,2-substitution pattern 1d (Scheme 6).
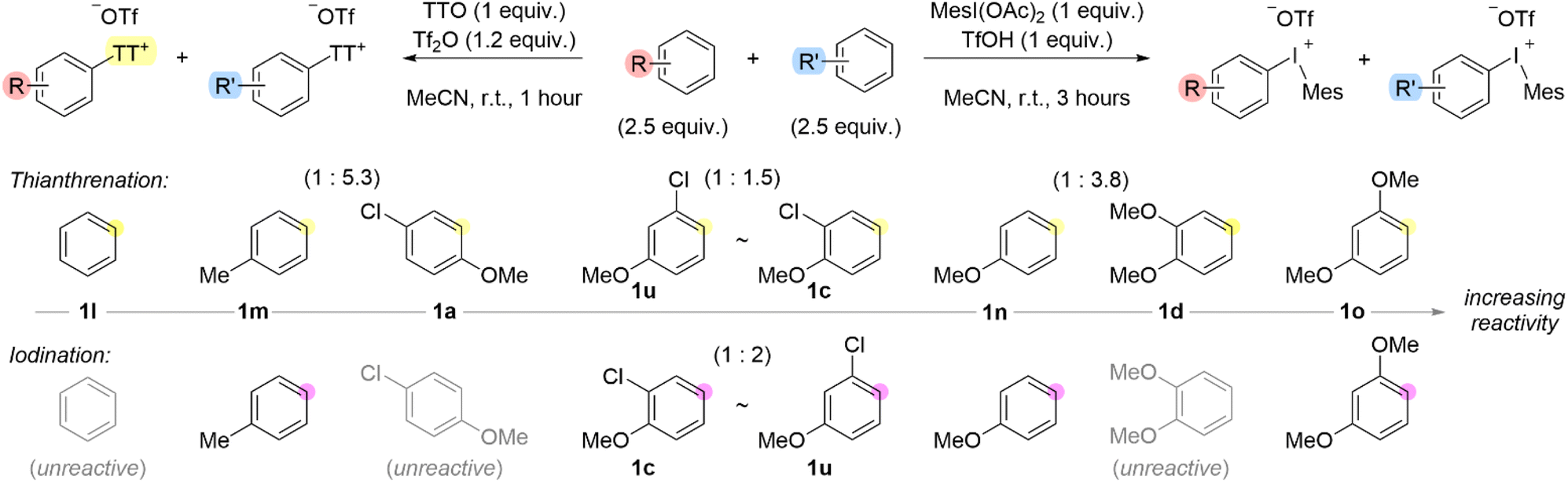 | ||
| Scheme 6 Arene relative reactivity scale for the formation of aryl thianthrenium and aryl(Mes)iodonium salts.a,b aConditions: 1 (1 mmol, 1 equiv.), MesI(OAc)2 (MIDA) (1 mmol, 1 equiv.), TfOH (1 mmol, 1 equiv.), MeCN (1 mL), r.t., 3 hours. bConditions: 1 (0.5 mmol, 1 equiv.), TTO (0.5 mmol, 1 equiv.), Tf2O (0.6 mmol, 1.2 equiv.), MeCN (2 mL), 0–25 °C, 30 minutes. | ||
The relative reactivity of 1,3- and 1,2-dimethoxybenzene 1o and 1d in thianthrenation and the lack of reactivity of 1,4-dimethoxybenzene 1y in both reactions prompted us to take a closer look at these substrates (Scheme 7). Electron-rich arenes, such as these, are known to be reductants via single electron transfer (SET), and arene radical cations have been suggested as intermediates in the formation of both aryl thianthrenium and diaryliodonium salts.40,41 1,3-Dimethoxybenzene 1o has a reduction potential of 1.50 vs. SCE,42 and readily forms 1o-IMes and 1o-TT in high yields (Scheme 7a). 1,2-Dimethoxybenzene 1d is a stronger reductant with a reduction potential of 1.43 vs. SCE, and is only compatible in the thianthrenation reaction (Scheme 7b). In the case of iodination of 1d, both 1d and MesI(OAc)2 are completely consumed in the reaction, but no product is formed (Scheme 7b). Moreover, a competition experiment between 1d and 1o in the formation of aryl(Mes)iodonium salts results in neither 1d-IMes or 1o-IMes. This data suggests that 1d inhibits the formation of 1o-IMes by decomposing MesI(OAc)2via SET. A similar scenario is observed for the attempted formation of an aryl(Mes)iodonium salt from 1,4-dimethoxybenzene 1y; both the arene and MesI(OAc)2 are consumed, but no product is observed (Scheme 7c). 1,4-Dimethoxybenzene 1y also inhibits the formation of 1o-IMes in a competition experiment. Thianthrenation of 1y is somewhat different from iodination. Although thianthrenation of 1y is unsuccessful and 1y is completely consumed, it does not inhibit the formation of 1o-TT in a separate competition experiment. Collectively, the data presented in Scheme 7 suggest that there is a key difference in the mechanisms for forming aryl thianthrenium and aryl(Mes)iodonium salts. Although both reactions are somewhat sensitive to the reduction potentials of arene substrates, thianthrenation reactions are not inhibited by strongly reducing arenes. This may suggest that arene radical cations are not productive intermediates in the formation of diaryliodonium salts under our conditions (Scheme 6 and 7).
 | ||
| Scheme 7 Attempted C–H functionalization of dimethoxybenzene isomers. aConditions: 1 (2 mmol, 1 equiv.), MesI(OAc)2 (MIDA) (2 mmol, 1 equiv.), TsOH (2 mmol, 1 equiv.), MeCN (2 mL), r.t., 3 hours. bConditions: 1 (0.5 mmol, 1 equiv.), TTO (0.5 mmol, 1 equiv.), Tf2O (0.6 mmol, 1.2 equiv.), MeCN (2 mL), 0–25 °C, 30 minutes. | ||
The aryne forming step involves deprotonation of an aromatic C–H bond and elimination of the “onium” leaving group. We used competition and DKIE experiments as well as DFT to analyze and compare this process for aryl thianthrenium and aryl(Mes)iodonium salts. Our synthetic and mechanistic analysis of the formation of aryl “onium” species revealed that aryl thianthrenation is broader in arene scope than aryl iodination (vide supra). Our experimental evidence suggests that the formation of arynes from aryl(Mes)iodonium salts is broader than from aryl thianthrenium salts. In order to gain direct evidence for the relative reactivity of aryl thianthrenium and aryl(Mes)iodonium salts as aryne precursors, we performed a competition experiment with deuterium labeled substrates (Scheme 8). 2-Fluorophenol was trideuteromethylated with DMSO-d6 to generate 1z-d3.43 The deuterated and non-deuterated analogs of 1z-TT and 1z-IMes were synthesized and used in competition experiments to generate and trap the corresponding aryne, ultimately leading to 3za and 3za-d3 (highlighted in pink, Scheme 8). Both scenarios of the deuterium label on the aryl thianthrenium or the aryl(Mes)iodonium salts were performed and in both cases the product 3za derived from the aryl(Mes)iodonium salt was the major product by ∼2.5:1 when LiHMDS is used (Scheme 8). We also noted a base effect in these competition experiments. The use of tert-butoxide resulted in exclusive formation of aryne from the aryl(Mes)iodonium salt which may be due to preferential complexation of the alkoxide with the iodonium relative to the thianthrenium (ESI pg S10‡). These data provide direct evidence that aryl(Mes)iodonium salts are more reactive aryne precursors than the corresponding aryl thianthrenium salts.
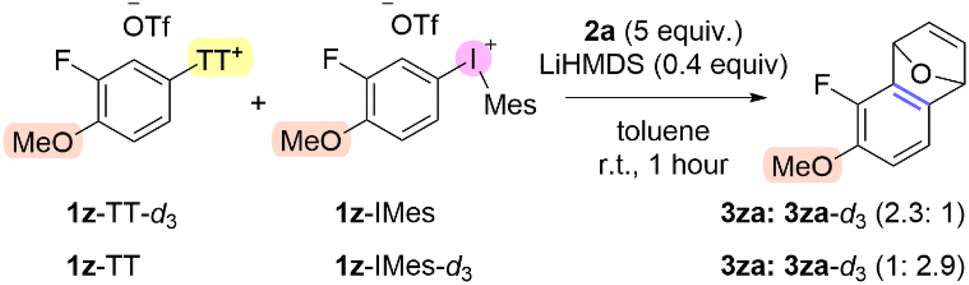 | ||
| Scheme 8 Competition of aryl thianthrenium and aryl(Mes)iodonium aryne precursors.a aConditions: 1z-TT-(d3) (0.05 mmol, 1 equiv.), 1z-IMes-(d3) (0.05 mmol, 1 equiv.), 2a (0.5 mmol, 5 equiv. relative to total 1z-TT + 1z-IMes), LiHMDS (0.04 mmol, 0.4 equiv. relative to total 1z-TT + 1z-IMes), toluene (0.5 mL), r.t., 1 hour; ratios determined from 1H NMR spectra of isolated product. | ||
Chalcogen leaving groups are relatively rare in aryne forming reactions,6 but “onium” groups based on these have received increased attention in recent years with C–H deprotonation/β-elimination sequences.11,13,20,21,39 As such there is relatively little known about the mechanism of this process,13 and nothing known when aryl thianthrenium salts are used. We measured the DKIE by one-pot competition experiments to glean an understanding of the kinetic relevance of the C–H deprotonation step in the formation of arynes from aryl thianthrenium and aryl(Mes)iodonium salts (Scheme 9). We synthesized a deuterated analog of 1b-TT and 1m-IMes to obtain the DKIE from aryl thianthrenium and aryl(Mes)iodonium salts, respectively (Scheme 9). In the case of 1b-TT and its deuterated analog, we observed a small DKIE of 1.4 (Scheme 9a), which is similar to the value obtained by Smith and co-workers for aryne formation from triaryloxonium ions.13 On the other hand, when we performed a competition experiment between 1m-IMes and its deuterated analog we observed a much larger DKIE of 4.9 when NaOt-Bu was used as base and 5.2 when LiHMDS was used (Scheme 9b). These data demonstrate that the C–H deprotonation step is more kinetically relevant in aryne formation from aryl(Mes)iodonium salts than from aryl thianthrenium salts.
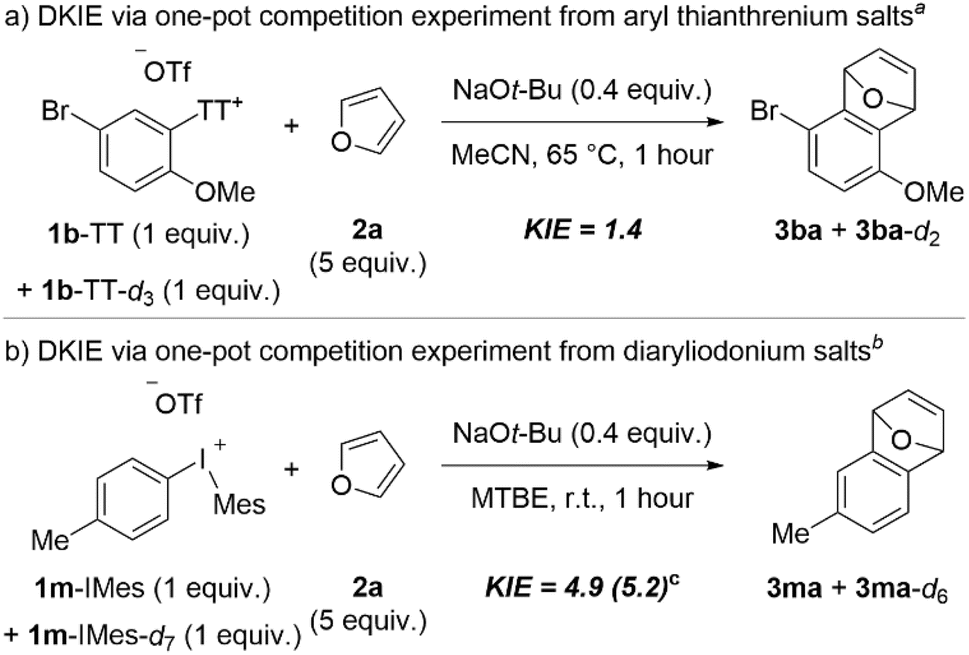 | ||
| Scheme 9 DKIE experiments for aryne formation from aryl thianthrenium or aryl(Mes)iodonium salts. aConditions: 1b-TT (0.25 mmol, 1 equiv.), 1b-TT-d3 (0.25 mmol, 1 equiv.), 2a (0.5 mmol, 5 equiv. relative to total [1b-TT]), NaOt-Bu (0.2 mmol, 0.4 equiv. relative to total [1b-TT]), MeCN (2.5 mL), 65 °C, 1 hour. bConditions: 1m-IMes (0.5 mmol, 1 equiv.), 1m-IMes-d3 (0.5 mmol, 1 equiv.), 2a (5 mmol, 5 equiv. relative to total [1m-IMes]), NaOt-Bu (0.4 mmol, 0.4 equiv. relative to total [1m-IMes]), MTBE (5 mL), r.t., 1 hour. cConducted on 0.05 mmol scale of 1m-IMes; LiHMDS (0.04 mmol, 0.4 equiv.), toluene (0.5 mL) used instead of NaOt-Bu and MTBE. | ||
To gain further insight into the C–H deprotonation/β-elimination sequence from aryl thianthrenium and aryl(Mes)iodonium salts, we performed DFT calculations on several different substrates (Scheme 10 and Fig. S1–S6‡). All intermediates and transition state structures were optimized with the M06-2x functional and the Def2-tzvpp basis set with SMD solvation (toluene) using Gaussian09.44–47 Given the range of solvents used in the experimental methods to access arynes from aryl thianthrenium and aryl(Mes)iodonium salts we surveyed solvation models (Table S1‡), and ultimately selected toluene as a single and representative solvent for DFT analysis. Our DFT analysis revealed that aryne formation from aryl thianthrenium and aryl(Mes)iodonium salts occur through distinct reaction pathways (Scheme 10). All aryl thianthrenium substrates analyzed by DFT (1b-TT, 1l-TT and 4-chlorophenyl thianthrenium) were found to generate a stable zwitterion (A, Scheme 10) upon deprotonation with tert-butoxide and passing through TS1 (Scheme 10 and Fig. S5‡). In the case of A, derived from 1b-TT, protonation (the reverse of the initial step) is lower in energy than elimination of the thianthrenium group through TS2 by 2.5 kcal mol−1 (Scheme 10, left). Therefore, the C–H deprotonation is reversible and TS2 is the highest energy point on the reaction pathway (Scheme 10, left). Conversely, the computed reaction pathway for all aryl(Mes)iodonium salts analyzed (1m-IMes, 1l-IMes and 4-chlorophenyl(Mes)iodonium) proceed through a single transition state (Scheme 10 and Fig. S2, S4, S6‡). In the transition state, epitomized by TS3, C–H and C–I bond cleavage is concerted but asynchronous; the C–H bond is elongated by 30% whereas the C–I bond is elongated by only 5%. Nevertheless, no zwitterionic intermediate could be found as TS3 collapses to the aryne dedihydro-1m as well as tert-butanol and mesityl iodide (Scheme 10, right). With these pathways established, the DKIE were calculated from the optimized geometries of the reactants and transition states for each reaction step.48,49 The overall DKIE for the two step aryne formation from 1b-TT was calculated (see eqn (SF1)‡),13,50 and both values were found to be in relatively good agreement (Scheme 10). The DKIE calculated from the DFT data for 1b-TT was 1.0 compared to the DKIE of 1.4 obtained from the wet lab experiment (Scheme 9 and 10, left). The relatively small DKIE reflects the reversible C–H deprotonation followed by rate-limiting thianthrene elimination, and thus the separation of the C–H deprotonation and rate-limiting steps. The DKIE calculated from the DFT data for 1m-IMes was 4.5 compared to the experimental value of 4.9 (Scheme 9 and 10, right). This much larger DKIE reflects C–H bond cleavage in the rate-limiting step of the reaction.
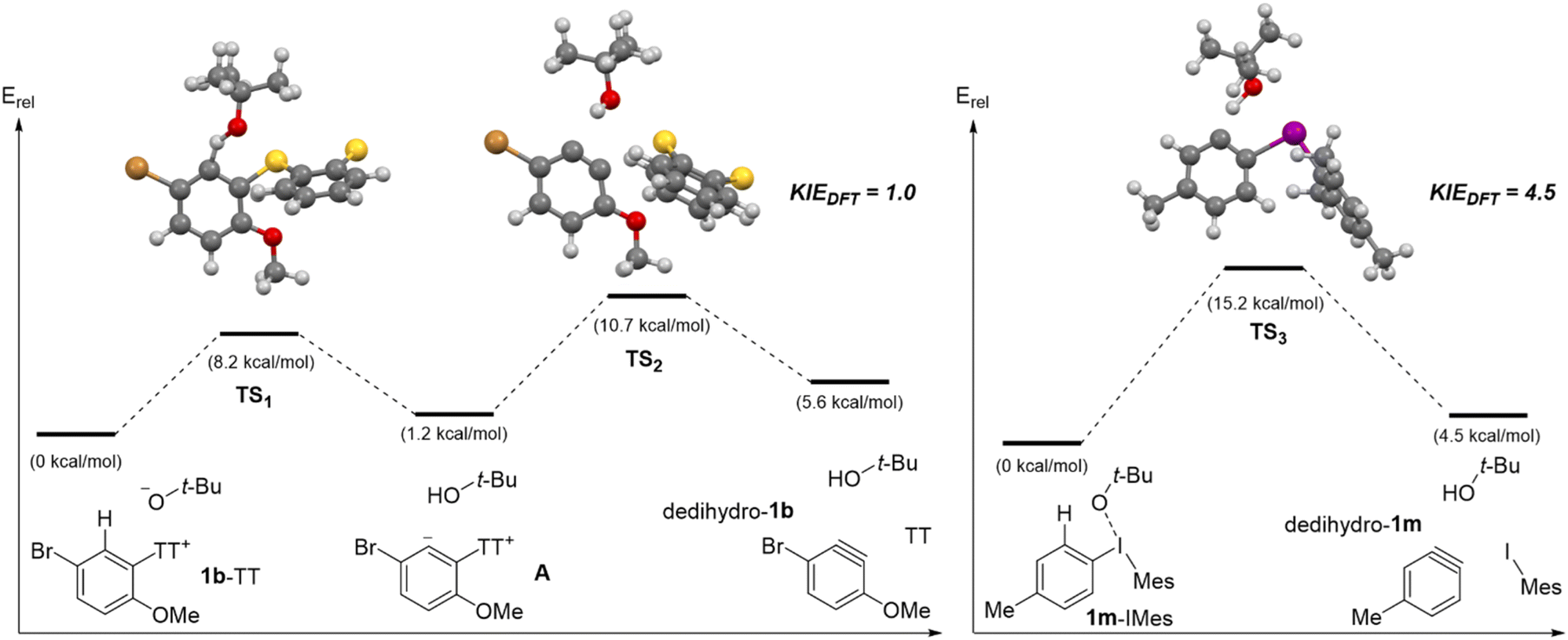 | ||
| Scheme 10 Calculated reaction landscape for 1b-TT and 1m-IMes.a aIntermediates and transition states were optimized at M06-2x/Def2-tzvpp with SMD solvation in toluene using Gaussian09.44–47 | ||
User's guide to accessing arynes from arenes
Collectively, our observations over the course of these studies can be distilled down to a user's guide for generating and trapping an aryne derived from a simple arene. We have demonstrated that the two tactics, using an aryl thianthrenium or an aryl(Mes)iodonium salt as an intermediate, are essentially complimentary with respect to the arene scope. For several classes of arene only the aryl thianthrenium salt can be synthesized obviating the use of aryl(Mes)iodonium salts for these substrates; in other cases the aryne cannot be accessed from the aryl thianthrenium and therefore the aryl(Mes)iodonium salts critically expand the arene scope. Our general strategy, shown in Scheme 11a, involves either a one-pot (for aryl thianthrenium) or two-pot (for diaryliodonium) process for net dehydrogenation and functionalization of arenes. The site of aryne formation largely depends on the substitution pattern of the arene and in most cases high selectivity (10:1 or greater) is observed (Scheme 11b). In mono-substituted arenes the aryne is formed exclusively at the 3,4-position (highlighted blue) because both thianthrenium and iodonium groups are installed with very high para-selectivity (4-position) and deprotonation occurs exclusively ortho to the leaving group (3-position; Scheme 11b). In mono-substituted arenes, no aryne formation is observed at the 2,3-position. In 1,2-disubstituted arenes the major site of aryne formation is at the 3,4-position. This selectivity is again initiated by high para-selectivity (4-position) for installation of thianthrenium and iodonium groups; this is followed by generally high selectivity for deprotonation at the 3-position (Scheme 11b).14 In a few cases, we have observed a minor product from deprotonation at the 5-position (i.e., 1r-IMes and 1t-IMes, Scheme 3), but an aryne is not formed at the 5,6-position for 1,2-substituted arenes; this numbering scheme is predicated on the strongest para-director at the 1-position. In 1,3-substituted arenes, the site of iodination may be impacted by the relative steric effects of the EDG and the R-group at the 3-position (Scheme 11b). When the R-group is a chloride as in 1u we observed preference (8:1) for installation of the iodonium at the 4-position over the 6-position (Scheme 11b). Subsequently, because the 3-position is blocked, deprotonation occurs at the 5-position. The minor aryne adduct arises from iodination at the 6-position and deprotonation at the 5-position (Scheme 11b). There is only one site for aryne formation to occur with 1,4-substituted arynes and it is at the 2,3-position (Scheme 11b). However, in this case it is important to point out that the aryne is flanked by two substituents and in these cases we have shown that the nature of the two substituents impacts the selectivity of nucleophilic addition to the aryne; that is, the site of thianthrenation is not necessarily the site of nucleophilic addition.11,21 It should also be noted that higher substitution patterns can be treated as an extension of disubstituted arenes. That is, 1,2,3-substituted arene react similarly to 1,2-substituted arenes (cf. 3ha and 3da, Scheme 2) and 1,2,4-substituted arenes react similarly to 1,4-substituted arenes (cf.3ia and 3aa, Scheme 2).
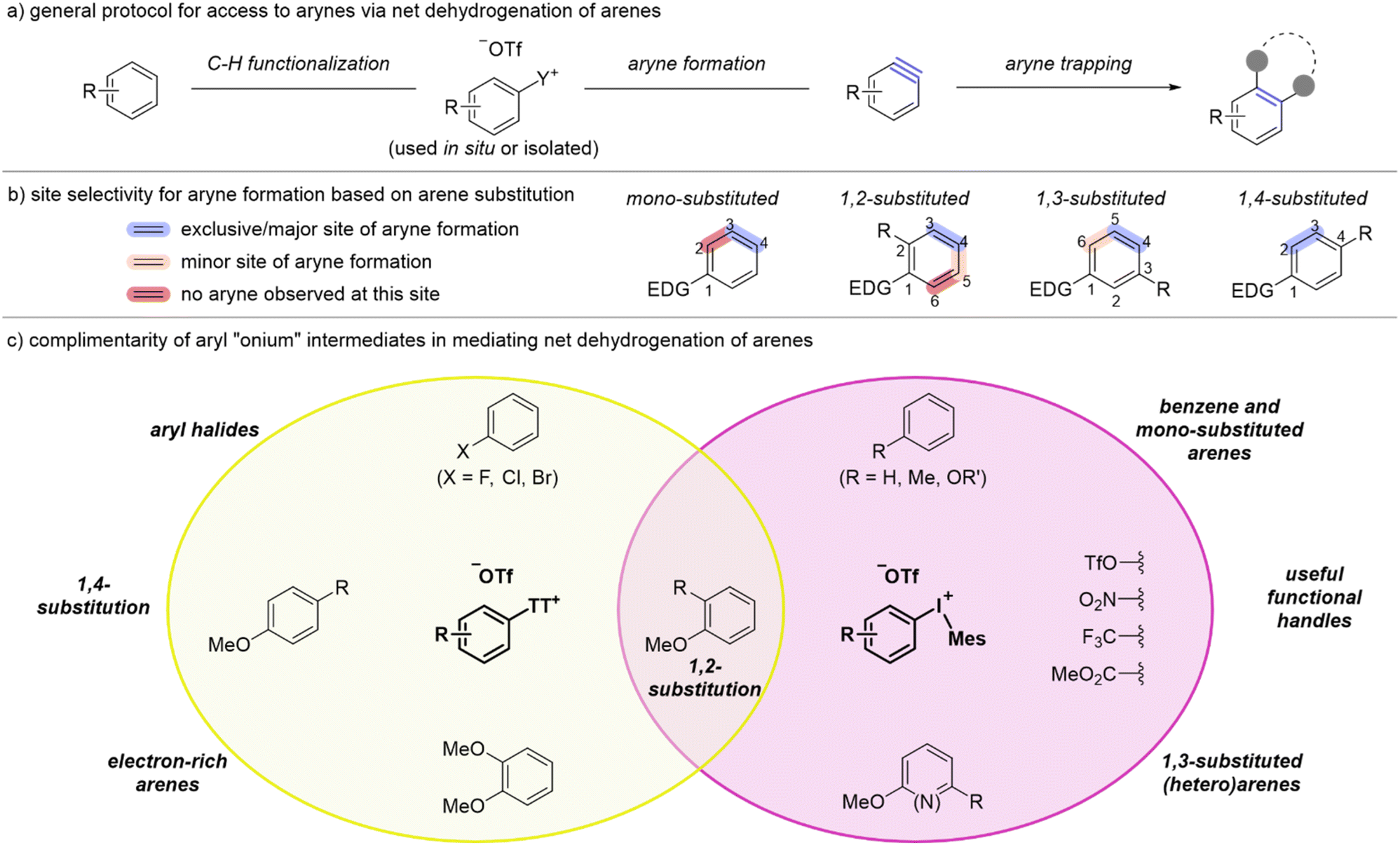 | ||
| Scheme 11 User's guide to accessing arynes from simple arenes (EDG = electron-donating group). | ||
Broadly, there are relatively few arenes for which there is overlap in the use of aryl thianthrenium and aryl(Mes)iodonium salts as intermediates to access arynes from simple arenes (Scheme 11c). 1,2-Disubstituted arenes hold this unique position and we recommend using the one-pot method via aryl thianthrenium salts (Table 1 and Scheme 2) as it is the most efficient approach to convert arenes into arynes. Other substrates for which aryl thianthrenium salts are the preferred mediator are aryl halides, and in this case we recommend using our first-generation two-pot method as the aryl halide is used as the solvent in the C–H thianthrenation step.11 Additionally, 1,4-substituted arenes and very electron-rich arenes, such as 1,2-dimethoxybenzene 1d, require the use of the corresponding aryl thianthrenium salt as an intermediate (Scheme 11c, yellow oval). The use of aryl(Mes)iodonium salts as an intermediate is recommended for the formation of arynes from benzene or mono-substituted feedstocks, such as toluene and anisole, 1,3-substituted arenes, including pyridines, and useful functional handles such as triflate, nitro and ester (Scheme 11c, purple oval).
Conclusions
Herein, we described a general strategy for accessing arynes from arenes. Two distinct approaches were discussed that depend on the arene substitution pattern and electronic effects. In the first case, a one-pot procedure, which passes through in situ generated aryl thianthrenium salts, was used. This is especially useful for generating arynes at remote sites on aryl halides and on 1,2- and 1,4-substituted arenes, as well as those arenes that are strong reducing agents. In the other case, a two-pot process, which passes through an isolated aryl(Mes)iodonium salt, was used. This is especially useful for generating arynes from mono-substituted arenes, 1,3-substituted arenes, and a substituted pyridine. Collectively, these two methods provide mild and efficient access to arynes within several hours from the unfunctionalized arene thereby substantively expanding the scope of arynes that are accessible. Moreover, these tactics provide opportunities for late-stage functionalization (triclosan) and access to a range of benzenoid-based scaffolds such as naphthoquinones, 1-naphthols, and naphthalenes; including the streamlined synthesis of pharmaceutical derivatives (propranolol). The net dehydrogenation of simple arenes is based on two distinct steps: C–H functionalization and C–H deprotonation/β-elimination. Mechanistic analysis via competition and DKIE experiments, as well as DFT reveal that there are both similarities and differences depending on the intermediate (thianthrenium or iodonium) enroute to the aryne. C–H functionalization via thianthrenation is broader in scope than iodination; however, mesityliodonium are more reactive leaving groups than the corresponding thianthrenium. C–H functionalization by both thianthrenation and iodination occur with very high para-selectivity and have small observed DKIE consistent with reversible formation of a Wheland intermediate and fast deprotonation. However, the iodination reaction is especially sensitive to SET events and the formation of aryl radical cations, whereas thianthrenation appears to be largely insensitive to this feature. Aryne formation from aryl thianthrenium salts and aryl(Mes)iodonium salts have distinct reaction pathways. Aryl thianthrenium salts undergo reversible deprotonation to generate a stable zwitterion that proceeds through rate-limiting thianthrenium elimination to produce the aryne. Aryl(Mes)iodonium salts undergo a concerted but asynchronous deprotonation/elimination event to generate the aryne. In both cases, experimental and computational determined DKIE values are consistent with these pathways. We envision that these platforms for generating arynes will engender new synthetic applications of arynes and increase the breadth of arynes that are accessible to the broader synthetic chemistry community.Data availability
The datasets supporting this article have been uploaded as part of the ESI.‡Author contributions
DRS, RAR, and BEM conceptualised the project. RAR and BEM conducted the laboratory experiments and curated the data. NJ performed the DFT experiments, supervised by TMM. DRS wrote the manuscript with input from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the National Science Foundation under grant no. 2247802 (DRS). The National Science Foundation provided funding for the BioAnalytical Mass Spectrometry Facility at PSU under grant no. 1828753. The National Science Foundation provided funding for the high-performance computing cluster at PSU under grant no. 1624776. The project described was supported, in part, by the Oregon State University Research Office. The content is solely the responsibility of the authors and does not necessarily represent the official views of the OSU Mass Spectrometry Center. The authors acknowledge the OSU Mass Spectrometry Center at Oregon State University and specific institutional instrument grants. Orbitrap Fusion Lumos – NIH #1S10OD020111-01, Waters Ion Mobility ToF Mass Spectrometer – NIH #1S10RR025628-01, Applied Biosystems 4000Qtrap – NIH #1S10RR022589-01, ABSciex Triple ToF 5600 – NIH #1S10RR027878-01.Notes and references
- H. Takikawa, A. Nishii, T. Sakai and K. Suzuki, Chem. Soc. Rev., 2018, 47, 8030–8056 RSC.
- D. B. Werz and A. T. Biju, Angew. Chem., Int. Ed., 2020, 59, 3385–3398 CrossRef CAS PubMed.
- S. M. Anthony, L. G. Wonilowicz, M. S. McVeigh and N. K. Garg, JACS Au, 2021, 1, 897–912 CrossRef CAS PubMed.
- K. Kamikawa, Nat. Rev. Chem, 2023, 7, 496–510 CrossRef PubMed.
- N. Kim, M. Choi, S.-E. Suh and D. M. Chenoweth, Chem. Rev., 2024, 124, 11435–11522 CrossRef CAS PubMed.
- J. Shi, L. Li and Y. Li, Chem. Rev., 2021, 121, 3892–4044 CrossRef CAS PubMed.
- B. E. Metze, R. A. Roberts, A. Nilova and D. R. Stuart, Chem. Sci., 2023, 14, 13885–13892 RSC.
- S. K. Sundalam, A. Nilova, T. L. Seidl and D. R. Stuart, Angew. Chem., Int. Ed., 2016, 55, 8431–8434 CrossRef CAS PubMed.
- M. Lanzi, Q. Dherbassy and J. Wencel-Delord, Angew. Chem., Int. Ed., 2021, 60, 14852–14857 CrossRef CAS PubMed.
- M. Lanzi, T. Rogge, T. S. Truong, K. N. Houk and J. Wencel-Delord, J. Am. Chem. Soc., 2023, 145, 345–358 CrossRef CAS PubMed.
- R. A. Roberts, B. E. Metze, A. Nilova and D. R. Stuart, J. Am. Chem. Soc., 2023, 145, 3306–3311 CrossRef CAS PubMed.
- H. Yuan, W. Yin, J. Hu and Y. Li, Nat. Commun., 2023, 14, 1841 CrossRef CAS PubMed.
- O. Smith, M. J. Hindson, A. Sreenithya, V. Tataru, R. S. Paton, J. W. Burton and M. D. Smith, Nat. Synth., 2023, 3, 58–66 CrossRef.
- A. Nilova, P. A. Sibbald, E. J. Valente, G. A. González-Montiel, H. C. Richardson, K. S. Brown, P. H. Cheong and D. R. Stuart, Chem.–Eur. J., 2021, 27, 7168–7175 CrossRef CAS PubMed.
- M. Wang and Z. Huang, Org. Biomol. Chem., 2016, 14, 10185–10188 RSC.
- S. S. Karandikar, B. E. Metze, R. A. Roberts and D. R. Stuart, Org. Lett., 2023, 25, 6374–6379 CrossRef CAS PubMed.
- H. Chen, J. Han and L. Wang, Beilstein J. Org. Chem., 2018, 14, 354–363 CrossRef CAS PubMed.
- Z. Zhang, X. Wu, J. Han, W. Wu and L. Wang, Tetrahedron Lett., 2018, 59, 1737–1741 CrossRef CAS.
- D. Carter Martos, M. De Abreu, P. Hauk, P. Fackler and J. Wencel-Delord, Chem. Sci., 2024, 15, 6770–6776 RSC.
- X.-W. Gu, Y.-H. Zhao and X.-F. Wu, Green Chem., 2023, 25, 6282–6286 RSC.
- J. Lyu, X. Zhang, Z. Luo, Y. Jiang, K. Xing, G. Zhang and C. Ding, Adv. Synth. Catal., 2024, 367, e202400889 CrossRef.
- X. Li, H. Chen, X. Liu, L. Wang and J. Han, ChemistrySelect, 2023, 8, e202301890 CrossRef CAS.
- M. Liu, H. Jiang, J. Tang, Z. Ye, F. Zhang and Y. Wu, Org. Lett., 2023, 25, 2777–2781 CrossRef CAS PubMed.
- N. F. Fine Nathel, L. A. Morrill, H. Mayr and N. K. Garg, J. Am. Chem. Soc., 2016, 138, 10402–10405 CrossRef CAS PubMed.
- B. E. Metze, A. Bhattacharjee, T. M. McCormick and D. R. Stuart, Synthesis, 2022, 54, 4989–4996 CrossRef CAS.
- F. Berger, M. B. Plutschack, J. Riegger, W. Yu, S. Speicher, M. Ho, N. Frank and T. Ritter, Nature, 2019, 567, 223–228 CrossRef CAS PubMed.
- X.-Y. Chen, X.-X. Nie, Y. Wu and P. Wang, Chem. Commun., 2020, 56, 5058–5061 RSC.
- J. Wu, Z. Wang, X.-Y. Chen, Y. Wu, D. Wang, Q. Peng and P. Wang, Sci. China Chem., 2020, 63, 336–340 CrossRef CAS.
- Y. Cai, S. Chatterjee and T. Ritter, J. Am. Chem. Soc., 2023, 145, 13542–13548 CrossRef CAS PubMed.
- B. Lansbergen, P. Granatino and T. Ritter, J. Am. Chem. Soc., 2021, 143, 7909–7914 CrossRef CAS PubMed.
- X.-X. Nie, Y.-H. Huang and P. Wang, Org. Lett., 2020, 22, 7716–7720 CrossRef CAS PubMed.
- L. Deng, B. Xiao, M. Zhou, H. Cao, T.-Y. Sun and Z. Dong, ChemRxiv, 2024, preprint, DOI:10.26434/chemrxiv-2024-b2wss.
- T. Kitamura, M. Yamane, K. Inoue, M. Todaka, N. Fukatsu, Z. Meng and Y. Fujiwara, J. Am. Chem. Soc., 1999, 121, 11674–11679 CrossRef CAS.
- A. Yoshimura, J. M. Fuchs, K. R. Middleton, A. V. Maskaev, G. T. Rohde, A. Saito, P. S. Postnikov, M. S. Yusubov, V. N. Nemykin and V. V. Zhdankin, Chem.–Eur. J., 2017, 23, 16738–16742 CrossRef CAS PubMed.
- A. Nilova, B. Metze and D. R. Stuart, Org. Lett., 2021, 23, 4813–4817 CrossRef CAS PubMed.
- T. Dohi, T. Hayashi, S. Ueda, T. Shoji, K. Komiyama, H. Takeuchi and Y. Kita, Tetrahedron, 2019, 75, 3617–3627 CrossRef CAS.
- D. R. Stuart, Diaryliodonium Salts, American Chemical Society, 2024 Search PubMed.
- M. Murai, T. Ogita and K. Takai, Chem. Commun., 2019, 55, 2332–2335 RSC.
- W. Xiao, C. Li, J. Lv, S. Xu, W. Shi, X. Su, J. Xue, B. Huang, Y. Zou, M. Yan and X. Zhang, Angew. Chem., Int. Ed., 2024, 63, e202411166 CAS.
- F. Juliá, Q. Shao, M. Duan, M. B. Plutschack, F. Berger, J. Mateos, C. Lu, X.-S. Xue, K. N. Houk and T. Ritter, J. Am. Chem. Soc., 2021, 143, 16041–16054 CrossRef PubMed.
- T. Dohi, N. Yamaoka and Y. Kita, Tetrahedron, 2010, 66, 5775–5785 CrossRef CAS.
- H. Roth, N. Romero and D. Nicewicz, Synlett, 2016, 27, 714–723 CAS.
- Z. Shen, S. Zhang, H. Geng, J. Wang, X. Zhang, A. Zhou, C. Yao, X. Chen and W. Wang, Org. Lett., 2019, 21, 448–452 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Accounts, 2008, 120, 215–241 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01 Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- R. S. Paton, Kinisot.py, version 2.0.2, 2016 Search PubMed.
- N. M. O’boyle, A. L. Tenderholt and K. M. Langner, J. Comput. Chem., 2008, 29, 839–845 CrossRef PubMed.
- R. E. Plata and D. A. Singleton, J. Am. Chem. Soc., 2015, 137, 3811–3826 CrossRef CAS PubMed.
Footnotes |
| † An earlier version of this work was posted as a pre-print: R. A. Roberts, B. E. Metze, N. Javaly, T. M. McCormick, D. R. Stuart, ChemRxiv, 2024, DOI: https://10.26434/chemrxiv-2024-04r0l (posted Nov. 13). |
| ‡ Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc00054h |
| § These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |