
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnusually air-stable copper(I) complexes showing high selectivity for carbon monoxide†
Borna
Saeednia‡
 ,
Aria M.
Sragow‡
,
Yannan
Lin‡
,
Colton J.
Sheehan
,
Amy S.
Metlay
,
Michael R.
Gau
,
Samantha A.
Dye
,
Sarah P.
O'Konski
,
Thomas E.
Mallouk
and
Ivan J.
Dmochowski
*
,
Aria M.
Sragow‡
,
Yannan
Lin‡
,
Colton J.
Sheehan
,
Amy S.
Metlay
,
Michael R.
Gau
,
Samantha A.
Dye
,
Sarah P.
O'Konski
,
Thomas E.
Mallouk
and
Ivan J.
Dmochowski
*
Department of Chemistry, University of Pennsylvania, 231 S. 34th St., Philadelphia, PA 19104, USA. E-mail: ivandmo@sas.upenn.edu
First published on 19th February 2025
Abstract
We report two Cu(I)-tren host molecules with unusual air-stability, as revealed by strong preference for axial CO binding over bent O2. Spectroscopy, electrochemical, and X-ray crystal structure analyses indicate that the phenyl rotators of the capsule select for small axial ligands.
Structural features that regulate binding of diatomic molecules to metal centres are of central importance in many biochemical and chemical-industrial processes.1,2 As dioxygen and carbon monoxide can compete for binding at protein metal centres, probing the structural origins of selectivity between O2 and CO at metalloproteins is of interest.3–6 Selective CO binding holds importance for sensing applications7,8 and also for delivery, due to its pathophysiological and therapeutic roles.9 Here we highlight copper proteins that bind dioxygen and perform vital functions such as production of norepinephrine (dopamine-β-hydroxylase, DBH), mitochondrial function (cytochrome c oxidase, COX), and O2 transport in molluscs and arthropods (hemocyanin).10 These and many other copper proteins carefully regulate the Cu(I)/Cu(II) redox couple, for purposes such as electron transfer, oxidation of substrates, and O2 transport.11,12 Synthetic models for the study of cuproproteins are well-precedented13,14 and provide an opportunity to examine stereoelectronic features that control function.15 However, Cu(I) systems demonstrating CO/O2 selectivity are rare,16–18 and control of selectivity by steric factors alone has, to the best of our knowledge, never been demonstrated.
Metalloproteins can achieve ligand selectivity by varying the size and accessibility of the binding pocket.15 Many metalloproteins, including the hemocyanins (where a single dioxygen ligand coordinates two copper centres), possess deeply-buried active sites in which few guests bind.19 Inspired by this selectivity, we were interested in creating a model system with a defined metallo-cavity for ligands to enter.20 However, many classes of macromolecules possessing hydrophobic cavities such as cucurbiturils,21–23 calixarenes,24–27 resorcinarenes,28 and cyclodextrins29,30 generally lack defined metal-binding sites. Hemicryptophanes are a class of small-molecule cages31,32 that are excellent candidates for metalloprotein-inspired binding pockets, owing to their well-defined hydrophobic cavity created by a cyclotriveratrylene (CTV) north pole33–36 and the ease of attaching a south pole with a metal-binding site.31,37,38 Furthermore, hemicryptophanes and other CTV-based supramolecular structures have been used in copper-binding systems by the groups of Hardie and Dutasta.39–41 In designing the south pole, we were inspired by myoglobin and haemoglobin. While CO/O2 discrimination is an important feature of these iron-haem proteins, which has been mimicked in various small-molecule and protein models,42–45 such discrimination has not yet been observed in an analogous monocopper protein.
First synthesised by Ogawa,46L1 (Fig. 1) is a particularly versatile hemicryptophane. In addition to selective guest47 and anion48 binding with protonated L1, Zn(II)-coordinated L1 catalyses the hydrolysis of activated alkyl carbonates46,49 and Ru(III)-L1 can catalyse the oxidation of primary alcohols.50 Here, we sought to prepare Cu(I)-bound L1 and investigate its ligand-binding properties (Fig. 1). L1 provides an unusual example of a tripodal amine-substituted copper ligand. Many other tripodal amine- and amide-based Cu(I) systems are known, which use ligands such as tren, TMPA, and their derivatives51–55 to explore the effects of alkyl groups, chelate ring size, and steric bulk on substrate binding.56 However, with rare exceptions,57–60 most notably from the Reinaud group,61–65 these complexes lack a well-defined cavity at the metal site.
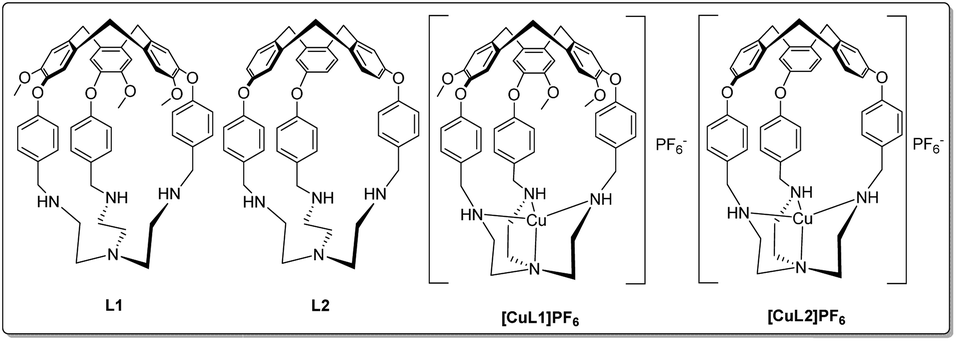 | ||
| Fig. 1 Structures of L1, L2, [CuL1]PF6, and [CuL2]PF6. | ||
In this work, we synthesised the copper(I) complex [CuL1]PF6 from the previously-reported L1.46,47 We observed air-stability and sought to elucidate the origin of this effect, as few examples of air-stable, 4-coordinate Cu(I) complexes with an open coordination site exist due to their typically high oxidation potential.66–70 In similar complexes, portal size and/or steric blockage have been observed to control the size of the guests that can access the cavity.47 Seeking to increase the portal size, we synthesised L2 (Fig. 1), a tren-hemicryptophane identical to the L1 ligand, but lacking the methoxy groups on the north pole (see ESI† for synthetic details). The resulting copper complex [CuL2]PF6 maintained an unusual degree of air-stability, despite its more accessible cavity.
Interestingly, no signs of oxidation in the solid state were observed for either [CuL1]PF6 or [CuL2]PF6 upon storage as the solid powder under air for several months (Fig. S2†). To assess stability in the solution phase, a solution of each Cu(I) compound was prepared in DMSO-d6 under air. Both solutions were monitored by 1H NMR spectroscopy for two weeks, at the end of which the solutions were bubbled with air for 1 h (Fig. S3 and S4†). In the case of [CuL1]PF6, no meaningful changes to the spectrum were observed after two weeks of air exposure (Fig. S3b†), nor was bubbling air observed to induce decomposition (Fig. S3c†). For [CuL2]PF6, slight decomposition was observable after air exposure for two weeks (Fig. S4b†). Subsequent bubbling with air did not appear to cause significant additional decomposition (Fig. S4c†). However, repetition of these experiments with the internal standard revealed a 26% loss of compound over 14 days (Fig. S5 and S6†). This is presumably due to the formation of a paramagnetically broadened Cu(II) species. While susceptible to oxidation in solution over several weeks, the compound demonstrates an unusual lack of air-sensitivity compared to other N3Cu(I) complexes, which generally undergo oxidation on the time scale of seconds to minutes.71,72 Both Cu(I) complexes are unusually air-stable in the solution phase, but [CuL1]PF6 appears to be marginally more stable than [CuL2]PF6. From this, it appears that the size of the portals is important, but cannot fully explain the diminished reactivity towards dioxygen in [CuL1]PF6.
We hypothesised that increased air stability results from a cavity that is too constrained to easily accommodate Cu(I)–O2 coordination. Binding to carbon monoxide—a ligand of similar size to dioxygen—was investigated for [CuL1]PF6 and [CuL2]PF6 using infrared (IR) spectroscopy in solution (Fig. 2). When purged with CO, in addition to the free CO peak at 2137 cm−1, a peak was observed at 2079 cm−1 for both [CuL1]PF6 and [CuL2]PF6, which we attribute to the formation of the Cu(I)–CO complex. Control experiments using fresh samples under dinitrogen showed neither IR peak. The 2079 cm−1 stretching frequency for the bound peak is not differentiable between [CuL1]PF6 and [CuL2]PF6, which is expected given the similar Cu(I)-tren ligand electronic environment. These vibrational frequencies are in line with other N3CuCO complexes reported in the literature, which possess a νCO of 2040–2100 cm−1.73,74 The experiment was repeated using 13C-labeled CO, demonstrating the expected isotope shift (Fig. S7†). Binding studies were continued by comparing 1H NMR spectra for both compounds under air, after degassing through freeze–pump–thaw cycles, and upon pressurization with CO to 20 psi (Fig. 3). The proton NMR spectra of both compounds showed changes indicative of binding behaviour in the presence of CO, eliciting peak broadening and changes in chemical shift. The degassed and under-air spectra show no differences. As expected, the most-affected NMR peaks belong to the south pole and linker regions of the molecule, nearest the copper centre. In order to remove CO and restore the ligand-free NMR spectrum for both compounds, it was necessary to degas rigorously using the freeze–pump–thaw method. The changes to both the 1H and 13C NMR spectra (Fig. S8–S14†) of [CuL1]PF6 and [CuL2]PF6 can be attributed to the presence of the CO as the guest.75,76 Notably, however, no signal corresponding to free or bound carbon monoxide was observed in the 13C NMR spectrum for either compound. Based on the evidence for a significant Cu(I)–CO bound population, we inferred that the CO off-rate is in an intermediate exchange regime resulting in extreme broadening of the CO-bound peak (Fig. S10 and S11†).77 This hypothesis was confirmed by applying 13C-labeled CO to the sample, upon which one broad 13C NMR peak corresponding to bound and free CO was observed (Fig. S12 and S13†).
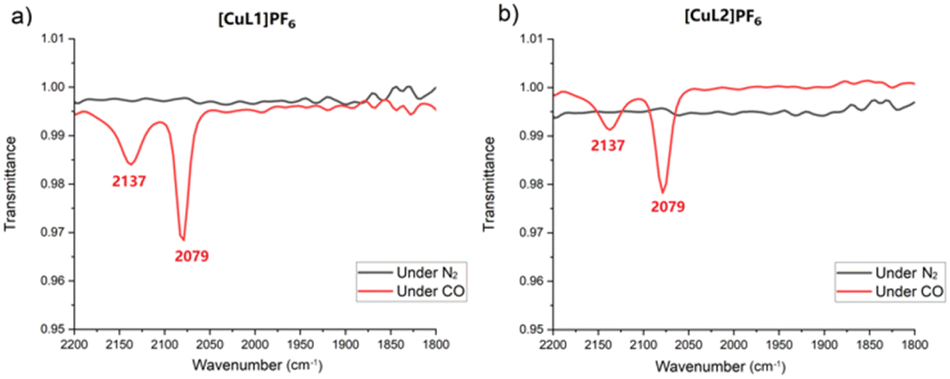 | ||
| Fig. 2 IR spectra of (a) [CuL1]PF6 under N2 (grey) and CO (red) and (b) [CuL2]PF6 under N2 (grey) and CO (red). | ||
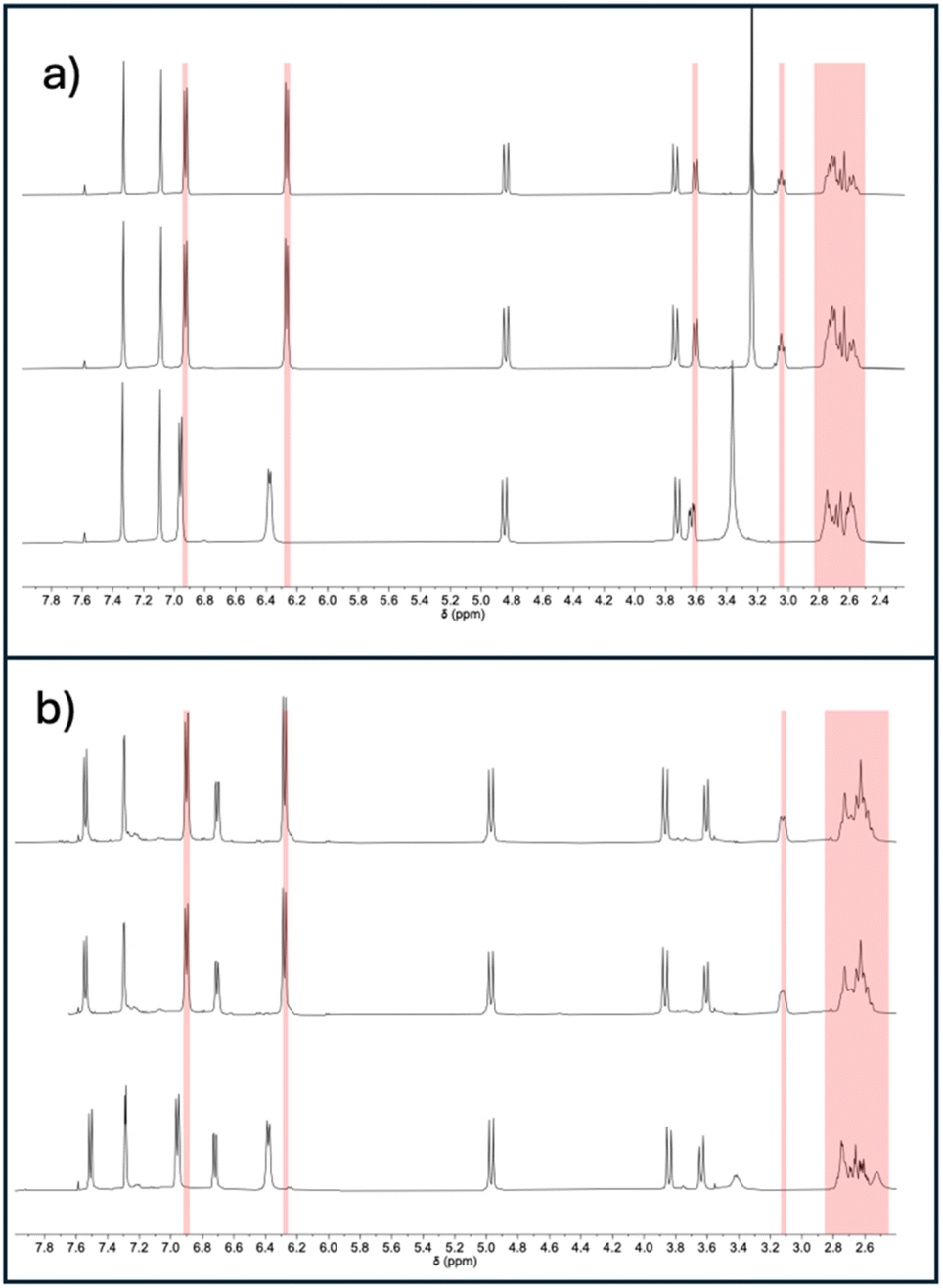 | ||
| Fig. 3 (a) 1H NMR spectra (500 MHz, CD3CN) of [CuL1]PF6 under air (top), after degassing (middle) and under a 20 psi pressure of carbon monoxide (bottom). (b) 1H NMR spectra (500 MHz, CD3CN) of [CuL2]PF6 under air (top), after degassing (middle) and under a 20 psi pressure of carbon monoxide (bottom). | ||
To examine carbon monoxide binding further, cyclic voltammetry (CV) experiments were performed in the absence and presence of carbon monoxide for [CuL1]PF6 and [CuL2]PF6 (Fig. 4). Both compounds displayed highly irreversible electron-transfer kinetics in ligand-free and ligand-bound states. We propose that the irreversible nature of the CVs is attributable to several factors, including the rigidity of the tren ligand within the capsule. In similar copper complexes, ligand rearrangement is necessary to facilitate redox changes at the metal.78,79 Considerable evidence supports the role of a “rack” or entatic state in copper electron-transfer proteins, in which small movements in one or more axial ligands assist the complex in shifting between preferred coordination geometries, i.e., tetrahedral Cu(I) and square planar/higher-coordinate Cu(II).11,80 Our system may demonstrate the opposite: the movement of ligands is constrained by the macrocycle, hindering electron transfer. This phenomenon has been seen in similar TREN-based systems displaying irreversible electron-transfer kinetics, in which the constrained ligand enforces a particular coordination geometry upon the metal centre, thus favouring the corresponding oxidation state.58 The very sluggish ET kinetics observed for these Cu model systems helps to explain their notable Cu(I) air-stability. We note that the extremely slow ET kinetics observed for both Cu(I)–CO species (Fig. 4) may be attributable to the high inner-sphere reorganisation energy associated with rapid CO ligand exchange.81
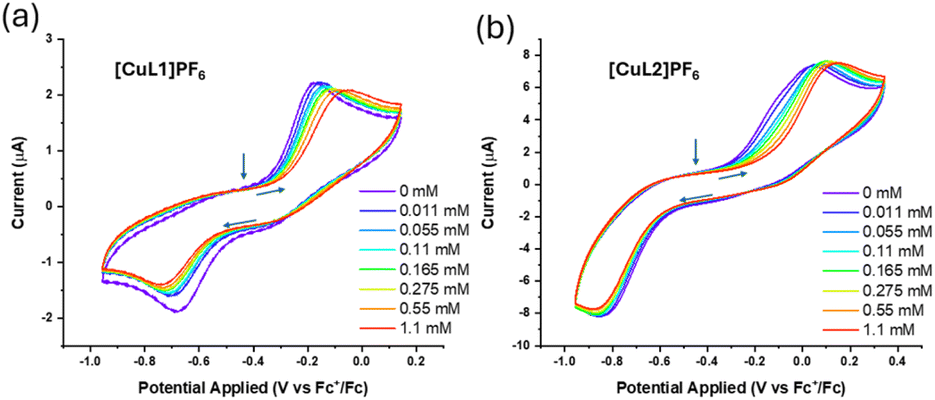 | ||
| Fig. 4 Cyclic voltammograms for (a) [CuL1]PF6 and (b) [CuL2]PF6 in the presence of varying concentrations of carbon monoxide. All experiments were performed in anhydrous DMSO with 250 mM TBAPF6 supporting electrolyte, glassy carbon working electrode, Pt mesh counter electrode, and Ag/AgNO3 reference electrode calibrated to Fc+/Fc at a scan rate of 100 mV s−1. Arrows show the start point of the scan (−0.457 V vs. Fc+/Fc) and the scan direction. | ||
Titration of CO revealed a correlation between the CO concentration and reduction potential of both compounds. As more CO was titrated during the CV measurement, both apparent E1/2 values shifted anodically, providing additional evidence that CO is coordinating to the metal sites in [CuL1]PF6 and [CuL2]PF6. Consistent with observations from 1H NMR, purging the sample with nitrogen did not revert the CV to its original state in either case. Finally, the Cu(I) oxidation potentials measured in both [CuL1]PF6 and [CuL2]PF6 indicate that oxidation by dioxygen should be thermodynamically favourable. This makes the relative Cu(I) air-stability all the more striking, particularly with such prominent CO binding. The larger portals in [CuL2]PF6 barely affect CO binding or air stability.
To shed light on the preference for CO binding and against O2 binding, we sought to obtain various crystal structures to visualize the cavity. Crystals suitable for single crystal X-ray diffraction were obtained for [CuL1]PF6 by vapor diffusion of ether into acetonitrile (Fig. 5a). Aware of the “induced fit” model observed for related supramolecular complexes,82 we suspected that a structure of the empty capsule L1 would not accurately predict the size and shape of the cavity when a ligand is present. Therefore, we also sought to crystallise Cu(I)–ligand and Cu(II)–ligand complexes that should more closely resemble the putative Cu(II)–O2 complex. Thus, L1 was crystallised in the presence of Cu(OTf)2.† The resulting X-ray structure contained a molecule of acetonitrile residing in the cavity and bound axially to the copper (Fig. 5b), similar to the expected binding for CO. Crystallisation of L1 was also attempted in the presence of Cu(OTf)2 and n-Bu4NN3 in hopes of capturing a ligand coordinated to the Cu centre in a bent fashion, similar to the expected dioxygen coordination geometry.53,83,84 It was revealed that under these conditions an azide anion was bound to the copper(II) centre (Fig. 5c).
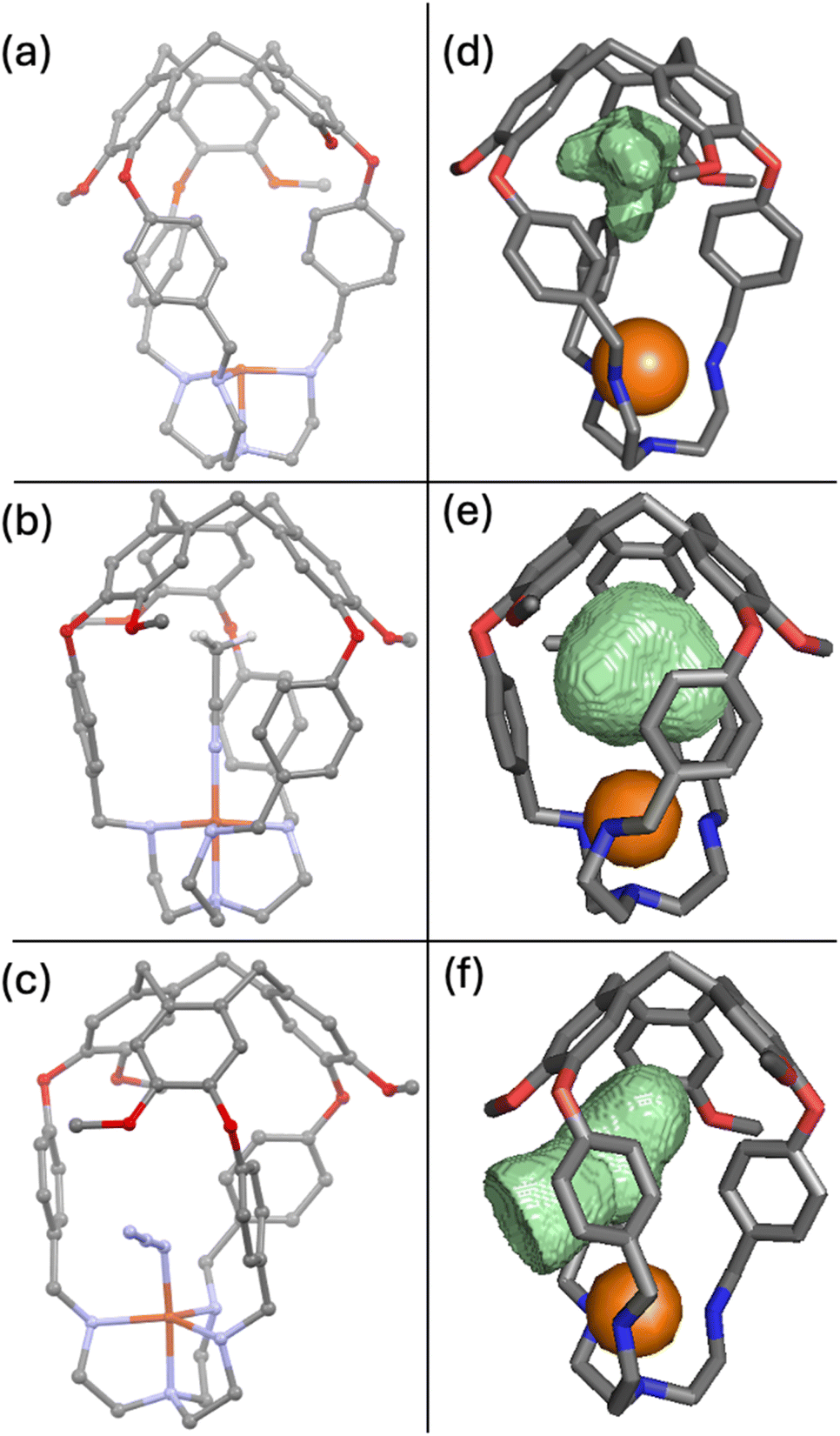 | ||
| Fig. 5 Crystal structures of (a) [CuL1]PF6, (b) [CuL1(MeCN)](OTf)2 and (c) [CuL1(N3)](OTf)2 (counter ions have been removed for clarity). Mapped out cavities for (d) [CuL1]PF6, (e) [CuL1(MeCN)](OTf)2 and (f) [CuL1(N3)](OTf)2 overlaid with the corresponding crystal structure. Guests and counter ions have been removed for clarity. | ||
We mapped the surface of the ligand-accessible cavity for each of these crystal structures using Molovol.85 Cavity surfaces overlaid with the crystal structure (not including the ligands) are presented in Fig. 5d–f. For the empty structure, we observed the cavity to be elongated and narrow near the copper centre, perfectly suited to accommodate acetonitrile (Fig. 5b) or carbon monoxide when bound axially. In fact, the cavity's oblong shape does not permit a bent ligand to bind entirely inside the host molecule. Instead, upon binding to azide at a bent angle, the aryl linkers rotate, allowing the azide ligand to thread between them such that it is only partially contained within the host cavity (Fig. 5c and f). While making space for the ligand, the linker conformational change comes at an entropic cost as it restricts the rotation of the linkers. Azide binding suggests that such binding angles are attainable, but the loss of entropy needs to be compensated for by enthalpic gains. As such, we propose that O2 binding in [CuL1]PF6 and [CuL2]PF6 lacks sufficient enthalpy to fully compensate for the loss of entropy in these host molecules at rt.
A related phenomenon has been observed previously: in the Karlin group's seminal studies of Cu(I)–CO and –O2 binding, the CO adduct formation rate constant is higher than that of the O2 adduct,86 which was attributed to a slower kOff rate of CO for entropic reasons pertaining to ligand re-organisation. In comparison, the intermediate exchange that we observe in the 13CO NMR may be due to the more constrained nature of the ligand, in which a smaller entropic penalty would be paid for the re-ligation of the TREN ligand than for a pendant pyridyl ligand.73
These observations carry implications for the study of cuproproteins and Cu-enzyme mimetics as a whole. Several model systems in the literature use CO as a redox-inactive ligand with similar binding behaviour to O2.15,87,88 Our model system, while unusual, highlights the possibility that some monocopper proteins may bind CO and discriminate against O2, as is well known to occur in haemoglobin. Arthropod and molluscan hemocyanins display differing selectivities for CO and O2,89 but this effect is presumably mediated by the cooperativity seen in anywhere from 6 to 160 dicopper sites.90 The current study suggests the possibility of achieving discrimination in a monocopper protein based upon binding angle and cavity shape.
In conclusion, we have synthesised two unusually air-stable Cu(I) tren-hemicryptophane host molecules. We propose that our system is the first CO-binding Cu(I) complex for which O2 binding/reaction is demonstrated to be electronically favourable, but is disfavoured due to steric factors alone. IR, NMR, and CV investigations yield strong evidence of discrimination for CO over O2. These findings shed insight on an alternate approach for stabilizing Cu(I) species, in which the coordination of a fifth ligand, as preferred by Cu(II), is made entropically unfavourable. This concept may be possible to adapt for small-molecule catalyst development and directed evolution of copper enzymes in the future, granting unusual ligand selectivity and reactivity.
Data availability
Our laboratory maintains all laboratory notebooks in a secure site, and maintains electronic copies of all spectra on secure servers. We have provided the most critical data in the MS and ESI,† and we will be happy to provide any additional information relevant to this paper upon request from reviewers or the broader public, after publication.Author contributions
Conceptualization, Y. L., B. S., A. M. S., C. J. S., A. S. M., and I. J. D.; methodology, Y. L., B. S., A. M. S., S. A. D., and S. P. O.; investigation, Y. L., B. S., A. M. S., C. J. S., A. S. M., M. R. G., P. J. C.; formal analysis, B. S.; writing – original draft, A. M. S. and B. S.; writing – review and editing, A. M. S., B. S., C. J. S., A. S. M., and I. J. D.; supervision, I. J. D. and T. E. M.; project administration, I. J. D. and T. E. M.; funding acquisition, I. J. D. and T. E. M.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by NIH Grant R35-GM-131907 to I. J. D. NMR spectrometer use was supported by the NSF Major Research Instrumentation Program (NSF CHE-1827457), NIH supplement awards 3R01GM118510-03S1 and 3R01GM087605-06S1, and Vagelos Institute for Energy Science and Technology. We thank Dr Jun Gu and Dr Chad W. Lawrence for assistance with NMR spectroscopy experiments and Dr Charles W. Ross III for mass spectrometry expertise. The authors thank current members of the Dmochowski lab, and also Prof. Kang Du and Dr Walter Johnsen for fruitful discussions. I. J. D. fondly remembers being an undergraduate attending a lecture by George Whitesides in 1992 where GW described his recent publication in Science, “How To Make Water Run Uphill”.91 The trap was set, and I. J. D. was caught! I. J. D. received wonderful training in the GW laboratory, and is still fascinated by studying complexity in molecular systems more than 30 years later.Notes and references
- E. A. Lewis and W. B. Tolman, Chem. Rev., 2004, 104, 1047–1076 CrossRef CAS PubMed.
- L. Gonzaga de França Lopes, F. S. Gouveia Júnior, A. Karine Medeiros Holanda, I. Maria Moreira de Carvalho, E. Longhinotti, T. F. Paulo, D. S. Abreu, P. V. Bernhardt, M.-A. Gilles-Gonzalez, I. Cirino Nogueira Diógenes and E. Henrique Silva Sousa, Coord. Chem. Rev., 2021, 445, 214096 CrossRef.
- A. C. Rosenzweig and M. H. Sazinsky, Curr. Opin. Struct. Biol., 2006, 16, 729–735 CrossRef CAS PubMed.
- L. M. Blomberg, M. R. A. Blomberg and P. E. M. Siegbahn, J. Inorg. Biochem., 2005, 99, 949–958 CrossRef CAS PubMed.
- T. Shimizu, D. Huang, F. Yan, M. Stranava, M. Bartosova, V. Fojtíková and M. Martínková, Chem. Rev., 2015, 115, 6491–6533 CrossRef CAS PubMed.
- T. G. Spiro, M. Z. Zgierski and P. M. Kozlowski, Coord. Chem. Rev., 2001, 219–221, 923–936 CrossRef CAS.
- J. Ohata, K. J. Bruemmer and C. J. Chang, Acc. Chem. Res., 2019, 52, 2841–2848 CrossRef CAS PubMed.
- X. Liu, N. Li, M. Li, H. Chen, N. Zhang, Y. Wang and K. Zheng, Coord. Chem. Rev., 2020, 404, 213109 CrossRef CAS.
- N. Bauer, X. Yang, Z. Yuan and B. Wang, Chem. Sci., 2023, 14, 3215–3228 RSC.
- K. I. Tishchenko, E. K. Beloglazkina, A. G. Mazhuga and N. V. Zyk, Rev. J. Chem., 2016, 6, 49–82 CrossRef CAS.
- E. I. Solomon, D. E. Heppner, E. M. Johnston, J. W. Ginsbach, J. Cirera, M. Qayyum, M. T. Kieber-Emmons, C. H. Kjaergaard, R. G. Hadt and L. Tian, Chem. Rev., 2014, 114, 3659–3853 CrossRef CAS PubMed.
- P. K. Hota, A. Jose, S. Panda, E. M. Dunietz, A. E. Herzog, L. Wojcik, N. Le Poul, C. Belle, E. I. Solomon and K. D. Karlin, J. Am. Chem. Soc., 2024, 146, 13066–13082 CrossRef CAS PubMed.
- R. H. Holm and E. I. Solomon, Chem. Rev., 2004, 104, 347–348 CrossRef CAS PubMed.
- V. Mahadevan, R. K. Gebbink and T. D. P. Stack, Curr. Opin. Chem. Biol., 2000, 4, 228–234 CrossRef CAS.
- H. R. Lucas and K. D. Karlin, in 9 Copper-Carbon Bonds in Mechanistic and Structural Probing of Proteins as well as in Situations where Copper is a Catalytic or Receptor Site, De Gruyter, 2015, pp. 295–362 Search PubMed.
- O. Green, B. A. Gandhi and J. N. Burstyn, Inorg. Chem., 2009, 48, 5704–5714 CrossRef CAS PubMed.
- G. Thiabaud, G. Guillemot, I. Schmitz-Afonso, B. Colasson and O. Reinaud, Angew. Chem., Int. Ed., 2009, 48, 7383–7386 CrossRef CAS PubMed.
- L. Le Clainche, Y. Rondelez, O. Sénèque, S. Blanchard, M. Campion, M. Giorgi, A. F. Duprat, Y. Le Mest and O. Reinaud, Comptes Rendus Acad. Sci. – Ser. IIC Chem., 2000, 3, 811–819 CAS.
- K. A. Magnus, H. Ton-That and J. E. Carpenter, Chem. Rev., 1994, 94, 727–735 CrossRef CAS.
- M. J. Wiester, P. A. Ulmann and C. A. Mirkin, Angew. Chem., Int. Ed., 2011, 50, 114–137 CrossRef CAS.
- J. W. Lee, S. Samal, N. Selvapalam, H.-J. Kim and K. Kim, Acc. Chem. Res., 2003, 36, 621–630 CrossRef CAS PubMed.
- K. I. Assaf and W. M. Nau, Chem. Soc. Rev., 2014, 44, 394–418 RSC.
- B. Tang, J. Zhao, J.-F. Xu and X. Zhang, Chem.–Eur. J., 2020, 26, 15446–15460 CrossRef CAS PubMed.
- G. Sachdeva, D. Vaya, C. M. Srivastava, A. Kumar, V. Rawat, M. Singh, M. Verma, P. Rawat and G. K. Rao, Coord. Chem. Rev., 2022, 472, 214791 CrossRef CAS.
- F. Sansone, L. Baldini, A. Casnati and R. Ungaro, New J. Chem., 2010, 34, 2715–2728 RSC.
- O. Santoro and C. Redshaw, Coord. Chem. Rev., 2021, 448, 214173 CrossRef CAS.
- M. Durmaz, E. Halay and S. Bozkurt, Beilstein J. Org. Chem., 2018, 14, 1389–1412 CrossRef CAS PubMed.
- M. J. McIldowie, M. Mocerino and M. I. Ogden, Supramol. Chem., 2010, 22, 13–39 CrossRef CAS.
- G. Crini, Chem. Rev., 2014, 114, 10940–10975 CrossRef CAS PubMed.
- E. Engeldinger, D. Armspach and D. Matt, Chem. Rev., 2003, 103, 4147–4174 CrossRef CAS PubMed.
- D. Zhang, A. Martinez and J.-P. Dutasta, Chem. Rev., 2017, 117, 4900–4942 CrossRef CAS PubMed.
- M. J. Hardie, in Supramolecular Chemistry, John Wiley & Sons, Ltd, 2012 Search PubMed.
- M. Yoshizawa, J. K. Klosterman and M. Fujita, Angew. Chem., Int. Ed., 2009, 48, 3418–3438 CrossRef CAS PubMed.
- A. J. Kirby, Angew Chem., Int. Ed. Engl., 1996, 35, 706–724 CrossRef.
- B. Ginovska, O. Y. Gutiérrez, A. Karkamkar, M.-S. Lee, J. A. Lercher, Y. Liu, S. Raugei, R. Rousseau and W. J. Shaw, ACS Catal., 2023, 13, 11883–11901 CrossRef CAS.
- P. Bhandari and P. S. Mukherjee, ACS Catal., 2023, 13, 6126–6143 CrossRef CAS.
- Y. Makita, T. Danno, K. Ikeda, H.-H. Lee, T. Abe, K. Sogawa, A. Nomoto, S. Fujiwara and A. Ogawa, Tetrahedron Lett., 2017, 58, 4507–4509 CrossRef CAS.
- S. A. Ikbal, C. Colomban, D. Zhang, M. Delecluse, T. Brotin, V. Dufaud, J.-P. Dutasta, A. B. Sorokin and A. Martinez, Inorg. Chem., 2019, 58, 7220–7228 CrossRef CAS PubMed.
- T. K. Ronson, H. Nowell, A. Westcott and M. J. Hardie, Chem. Commun., 2010, 47, 176–178 RSC.
- A. Schmitt, S. Collin, C. Bucher, V. Maurel, J.-P. Dutasta and A. Martinez, Org. Biomol. Chem., 2015, 13, 2157–2161 RSC.
- O. Perraud, J.-B. Tommasino, V. Robert, B. Albela, L. Khrouz, L. Bonneviot, J.-P. Dutasta and A. Martinez, Dalton Trans., 2013, 42, 1530–1535 RSC.
- C. K. Chang and T. G. Traylor, Proc. Natl. Acad. Sci. U. S. A., 1975, 72, 1166–1170 CrossRef CAS.
- D. H. Busch, L. L. Zimmer, J. J. Grzybowski, D. J. Olszanski, S. C. Jackels, R. C. Callahan and G. G. Christoph, Proc. Natl. Acad. Sci. U. S. A., 1981, 78, 5919–5923 CrossRef CAS.
- J. P. Collman, A. Dey, R. A. Decreau, Y. Yang, A. Hosseini, E. I. Solomon and T. A. Eberspacher, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 9892–9896 CrossRef CAS PubMed.
- J. P. Collman, A. Dey, C. J. Barile, S. Ghosh and R. A. Decréau, Inorg. Chem., 2009, 48, 10528–10534 Search PubMed.
- Y. Makita, K. Sugimoto, K. Furuyoshi, K. Ikeda, S. Fujiwara, T. Shin-ike and A. Ogawa, Inorg. Chem., 2010, 49, 7220–7222 CrossRef CAS PubMed.
- Y. Lin, M. R. Gau, P. J. Carroll and I. J. Dmochowski, J. Org. Chem., 2022, 87, 5158–5165 CrossRef CAS PubMed.
- Y. Lin, K. Du, M. R. Gau and I. J. Dmochowski, Chem. Sci., 2023, 14, 291–297 RSC.
- Y. Makita, K. Ikeda, K. Sugimoto, T. Fujita, T. Danno, K. Bobuatong, M. Ehara, S. Fujiwara and A. Ogawa, J. Organomet. Chem., 2012, 706–707, 26–29 CrossRef CAS.
- Y. Makita, T. Fujita, T. Danno, M. Inoue, M. Ueshima, S. Fujiwara and A. Ogawa, Supramol. Catal., 2012, 1, 11–13 Search PubMed.
- M. P. Lanci, V. V. Smirnov, C. J. Cramer, E. V. Gauchenova, J. Sundermeyer and J. P. Roth, J. Am. Chem. Soc., 2007, 129, 14697–14709 CrossRef CAS PubMed.
- A. Poater and L. Cavallo, Inorg. Chem., 2009, 48, 4062–4066 CrossRef CAS PubMed.
- C. Würtele, E. Gaoutchenova, K. Harms, M. C. Holthausen, J. Sundermeyer and S. Schindler, Angew. Chem., Int. Ed., 2006, 45, 3867–3869 CrossRef PubMed.
- A. K. Mishra and S. Verma, Inorg. Chem., 2010, 49, 3691–3693 CrossRef CAS PubMed.
- L. Zhou, D. Powell and K. M. Nicholas, Inorg. Chem., 2007, 46, 2316–2321 CrossRef CAS PubMed.
- T. Brückmann, J. Becker, C. Würtele, M. T. Seuffert, D. Heuler, K. Müller-Buschbaum, M. Weiß and S. Schindler, J. Inorg. Biochem., 2021, 223, 111544 CrossRef PubMed.
- A. Mondal, K. P. Reddy, S. Som, D. Chopra and S. Kundu, Inorg. Chem., 2022, 61, 20337–20345 CrossRef CAS PubMed.
- D. K. Chand and P. K. Bharadwaj, Inorg. Chem., 1998, 37, 5050–5055 CrossRef CAS.
- L. Chaloner and X. Ottenwaelder, Inorg. Chim. Acta, 2018, 481, 106–112 CrossRef CAS.
- L. Chaloner, A. Khomutovskaya, F. Thomas and X. Ottenwaelder, Dalton Trans., 2016, 45, 11109–11119 RSC.
- S. Paria, Y. Morimoto, T. Ohta, S. Okabe, H. Sugimoto, T. Ogura and S. Itoh, Eur. J. Inorg. Chem., 2018, 2018, 1976–1983 CrossRef CAS.
- G. Izzet, B. Douziech, T. Prangé, A. Tomas, I. Jabin, Y. Le Mest and O. Reinaud, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 6831–6836 CrossRef CAS PubMed.
- G. De Leener, D. Over, C. Smet, D. Cornut, A. G. Porras-Gutierrez, I. López, B. Douziech, N. Le Poul, F. Topić, K. Rissanen, Y. Le Mest, I. Jabin and O. Reinaud, Inorg. Chem., 2017, 56, 10971–10983 CrossRef CAS PubMed.
- G. Izzet, J. Zeitouny, H. Akdas-Killig, Y. Frapart, S. Ménage, B. Douziech, I. Jabin, Y. Le Mest and O. Reinaud, J. Am. Chem. Soc., 2008, 130, 9514–9523 CrossRef CAS PubMed.
- G. Izzet, M.-N. Rager and O. Reinaud, Dalton Trans., 2007, 771–780 RSC.
- D. Sengupta, P. Melix, S. Bose, J. Duncan, X. Wang, M. R. Mian, K. O. Kirlikovali, F. Joodaki, T. Islamoglu, T. Yildirim, R. Q. Snurr and O. K. Farha, J. Am. Chem. Soc., 2023, 145, 20492–20502 CrossRef CAS PubMed.
- M. P. Tang, L. Zhu, Y. Deng, Y. Shi, S. K.-M. Lai, X. Mo, X.-Y. Pang, C. Liu, W. Jiang, E. C. M. Tse and H. Y. Au-Yeung, Angew. Chem., Int. Ed., 2024, 63, e202405971 CrossRef CAS PubMed.
- S. Ostrowska, P. Arnaut, D. J. Liptrot, C. S. J. Cazin and S. P. Nolan, Chem. Commun., 2023, 59, 9126–9129 RSC.
- L. Le Clainche, M. Giorgi and O. Reinaud, Eur. J. Inorg. Chem., 2000, 2000, 1931–1933 CrossRef.
- C. Chuang, K. Lim, Q. Chen, J. Zubieta and J. W. Canary, Inorg. Chem., 1995, 34, 2562–2568 CrossRef CAS.
- S. Schindler, Eur. J. Inorg. Chem., 2000, 2000, 2311–2326 CrossRef.
- K. Komiyama, H. Furutachi, S. Nagatomo, A. Hashimoto, H. Hayashi, S. Fujinami, M. Suzuki and T. Kitagawa, Bull. Chem. Soc. Jpn., 2004, 77, 59–72 CrossRef CAS.
- H. C. Fry, H. R. Lucas, A. A. Narducci Sarjeant, K. D. Karlin and G. J. Meyer, Inorg. Chem., 2008, 47, 241–256 CrossRef CAS PubMed.
- Y. Rondelez, O. Sénèque, M.-N. Rager, A. F. Duprat and O. Reinaud, Chem.–Eur. J., 2000, 6, 4218–4226 CrossRef CAS PubMed.
- K. E. Chaffee, H. A. Fogarty, T. Brotin, B. M. Goodson and J.-P. Dutasta, J. Phys. Chem. A, 2009, 113, 13675–13684 CrossRef CAS PubMed.
- L. Garel, J.-P. Dutasta and A. Collet, Angew Chem., Int. Ed. Engl., 1993, 32, 1169–1171 CrossRef.
- L. Mureddu and G. W. Vuister, FEBS J., 2019, 286, 2035–2042 CrossRef CAS PubMed.
- P. J. Griffin, B. J. Charette, J. H. Burke, J. Vura-Weis, R. D. Schaller, D. J. Gosztola and L. Olshansky, J. Am. Chem. Soc., 2022, 144, 12116–12126 CrossRef CAS PubMed.
- G. Coullerez, E. Malmström and M. Jonsson, J. Phys. Chem. A, 2006, 110, 10355–10360 CrossRef CAS PubMed.
- B. Dicke, A. Hoffmann, J. Stanek, M. S. Rampp, B. Grimm-Lebsanft, F. Biebl, D. Rukser, B. Maerz, D. Göries, M. Naumova, M. Biednov, G. Neuber, A. Wetzel, S. M. Hofmann, P. Roedig, A. Meents, J. Bielecki, J. Andreasson, K. R. Beyerlein, H. N. Chapman, C. Bressler, W. Zinth, M. Rübhausen and S. Herres-Pawlis, Nat. Chem., 2018, 10, 355–362 CrossRef CAS PubMed.
- C. Zong, C. J. Wilson, T. Shen, P. Wittung-Stafshede, S. L. Mayo and P. G. Wolynes, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 3159–3164 CrossRef CAS PubMed.
- O. Taratula, P. A. Hill, N. S. Khan, P. J. Carroll and I. J. Dmochowski, Nat. Commun., 2010, 1, 148 CrossRef PubMed.
- Y. Kobayashi, K. Ohkubo, T. Nomura, M. Kubo, N. Fujieda, H. Sugimoto, S. Fukuzumi, K. Goto, T. Ogura and S. Itoh, Eur. J. Inorg. Chem., 2012, 2012, 4574–4578 Search PubMed.
- S. Kim, J. Y. Lee, R. E. Cowley, J. W. Ginsbach, M. A. Siegler, E. I. Solomon and K. D. Karlin, J. Am. Chem. Soc., 2015, 137, 2796–2799 Search PubMed.
- J. B. Maglic and R. Lavendomme, J. Appl. Crystallogr., 2022, 55, 1033–1044 Search PubMed.
- H. C. Fry, D. V. Scaltrito, K. D. Karlin and G. J. Meyer, J. Am. Chem. Soc., 2003, 125, 11866–11871 CrossRef CAS PubMed.
- C. D. Kline and N. J. Blackburn, Biochemistry, 2016, 55, 6652–6661 CrossRef CAS PubMed.
- E. E. Chufán, S. T. Prigge, X. Siebert, B. A. Eipper, R. E. Mains and L. M. Amzel, J. Am. Chem. Soc., 2010, 132, 15565–15572 CrossRef PubMed.
- H. Decker, P. R. Connelly, C. H. Robert and S. J. Gill, Biochemistry, 1988, 27, 6901–6908 CrossRef CAS PubMed.
- H. Decker, N. Hellmann, E. Jaenicke, B. Lieb, U. Meissner and J. Markl, Integr. Comp. Biol., 2007, 47, 631–644 CrossRef CAS PubMed.
- M. K. Chaudhury and G. M. Whitesides, Science, 1992, 256, 1539–1541 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2359098–2359100. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc00237k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |