
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Probing the ferredoxin:hydrogenase electron transfer complex by infrared difference spectroscopy†
Selmihan
Sahin
ab,
Johanna
Brazard
c,
Kilian
Zuchan
a,
Takuji B. M.
Adachi
 c,
Ulrich
Mühlenhoff
d,
Ross D.
Milton
*a and
Sven T.
Stripp
*e
c,
Ulrich
Mühlenhoff
d,
Ross D.
Milton
*a and
Sven T.
Stripp
*e
aUniversity of Geneva, Department of Inorganic and Analytical Chemistry, Sciences II, Quai Ernest-Ansermet 30, Geneva 4, 1211, Switzerland. E-mail: ross.milton@unige.ch
bSuleyman Demirel University, Department of Chemistry, Faculty of Engineering and Natural Sciences, Cunur, Isparta, 32260, Turkiye
cUniversity of Geneva, Department of Physical Chemistry, Sciences II, Quai Ernest-Ansermet 30, Geneva 4, 1211, Switzerland
dPhilipps-Universität Marburg, Institute of Cytobiology, Center for Synthetic Microbiology (SYNMIKRO), Karl-von-Frisch-Str. 14, Marburg, 35043, Germany
eUniversity of Potsdam, Institute of Chemistry, Spectroscopy and Biocatalysis, Karl-Liebknecht-Straße 24-25, Potsdam, 14476, Germany. E-mail: sven.stripp@uni-potsdam.de
First published on 30th April 2025
Abstract
Ferredoxins are small iron–sulfur proteins that engage in one-electron transfer with oxidoreductases across all domains of life. The catalyzed reactions often include multiple electrons, e.g., in the two-electron reduction of NADP+ during photosynthesis or the reduction of protons to H2 by the metalloenzyme hydrogenase. To date, the microscopic details of how ferredoxins facilitate multiple electron redox chemistry are unknown. Ferredoxins of the Allochromatium vinosum subfamily contain two [4Fe–4S] clusters, which allows for two one-electron transfer reactions. However, the iron–sulfur clusters of conventional 2[4Fe–4S]-type ferredoxins have very similar reduction potentials and conclusive evidence for the transfer of two electrons during a single protein–protein interaction (PPI) has not been reported. In this work, the electron transfer complexes between the clostridial 2[4Fe–4S] ferredoxin, CpFd, and [FeFe]-hydrogenases from both Clostridium pasteurianum (CpI) and Chlamydomonas reinhardtii (CrHydA), were investigated. Introducing a non-canonical amino acid near to one of the iron–sulfur clusters of CpFd permitted the quantification of electric field changes via the vibrational Stark effect by Fourier-transform infrared (FTIR) spectroscopy. Upon reduction, in situ FTIR difference spectroscopy reported on protein structural changes and microscale thermophoresis revealed that the affinity between ferredoxin and hydrogenase is modulated by redox-dependent PPIs. Prompted by these findings, we suggest a model how ferredoxin efficiently facilitates multiple electron redox chemistry based on individual one-electron transfer reactions.
Introduction
Protein–protein interactions (PPIs) are central to biochemical reactions,1 including electron transfer (ET) between metalloproteins.2–4 Small ET proteins such as flavodoxins, blue-copper proteins, and cytochromes5–7 as well as iron–sulfur proteins8 serve as redox partners in virtually all metabolic networks. This encompasses photosynthesis and the Calvin–Benson cycle, aerobic respiration, and the citric acid cycle, oxidative phosphorylation, and many other redox processes in the cell. Identifying PPIs is a considerable challenge as most of these interactions are transient and subtle.Ferredoxins (Fds) are small metalloproteins that engage in ET with various oxidoreductases.9 They contain either [2Fe–2S] or [4Fe–4S] clusters, depending on their occurrence in eukaryotes or prokaryotes. The primary and secondary coordination sphere of ferredoxin modulates their standard reduction potential (E0′) over hundreds of millivolts,10 which led to the distinction of “low-potential” and “high-potential” ferredoxins.11 The mesophilic bacterium Clostridium pasteurianum produces a low-potential ferredoxin (CpFd) that contains two [4Fe–4S] clusters (F and F′, Fig. 1A) with similar reduction potentials (Em = −400 ± 10 mV vs. SHE).12 In contrast, the 2[4Fe–4S] ferredoxin from Azotobacter vinelandii shows distinct Ems of −486 mV and −644 mV vs. SHE, as a result of two individual one-electron transfer steps at very low potential.13 Similar data was reported for the 2[4Fe–4S] ferredoxin from Allochromatium vinosum, where polar interactions with the clusters were identified to diversify the E0′s.14 In variance to these “alvin-type” ferredoxins, often discussed in the context of N2 fixation,15 Burgess and co-workers have argued that the homogeneity of E0′s in clostridial ferredoxins should allow for both one- and two-electron transfers at the same potential.13 To the best of our knowledge, however, two-electron reduction of clostridial ferredoxin has not been shown, and it is established that CpFd likely acts a one-electron carrier functioning at similar E0′s like eukaryotic ferredoxins.16
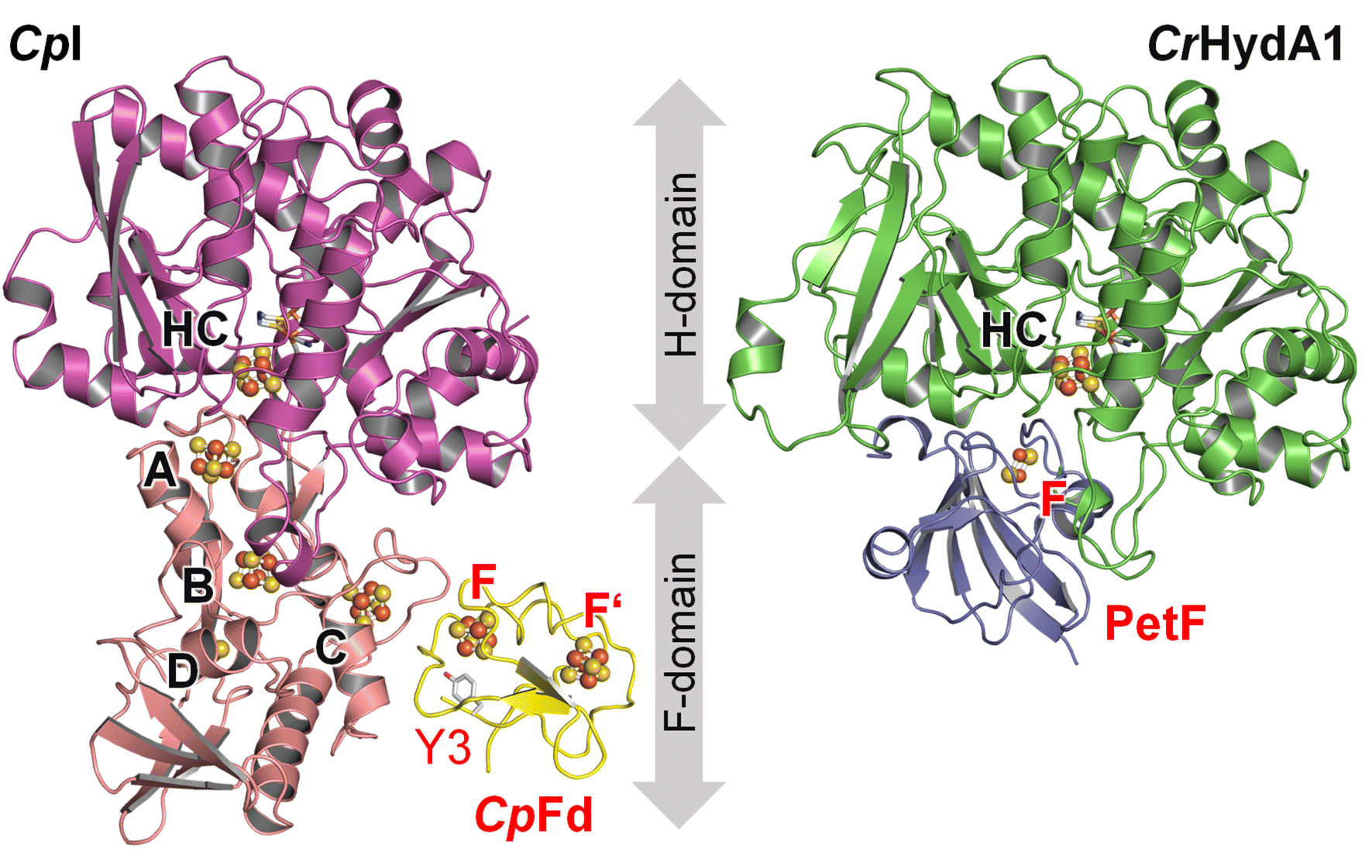 | ||
| Fig. 1 In silico models of the ferredoxin:hydrogenase complex. Left: [FeFe]-hydrogenase CpI (PDB ID 6N59) containing the H-cluster (HC) in the H-domain (magenta) as well as [4Fe–4S] clusters A–C and [2Fe–2S] cluster D in the F-domain (pink). Ferredoxin CpFd (yellow, PDB ID 1CLF) binds two [4Fe–4S] clusters F and F′, apart by ∼9.4 Å (shortest distance). The shortest distance between clusters C and F is ∼8.0 Å. In this study, tyrosine Y3 of CpFd (highlighted) is mutated to a cyano-phenylalanine. Right: [FeFe]-hydrogenase CrHydA1 (green, homology model) containing only the H-cluster (HC). An F-domain is missing. Ferredoxin PetF (blue, PDB ID 2MH7) binds a single [2Fe–2S] cluster F. The distance between clusters HC and F is ∼8.7 Å. Models generated with ClusPro 2.33 Coordinates for both complexes are supplied in ESI.† | ||
Ferredoxin CpFd is well-suited for ET with hydrogenase (Fig. 1). Hydrogenases are oxidoreductases that catalyze the reversible conversion of molecular hydrogen (H2), two protons (H+), and two electrons (hydrogen turnover).17,18 In the class of [FeFe]-hydrogenase, catalysis takes place at an unique iron–sulfur-based cofactor, the “hydrogen-forming” H-cluster, which is connected to the protein surface by a proton transfer pathway19 and an electron relay of various iron–sulfur clusters in the so-called F-domain.20–22 While the [FeFe]-hydrogenase from C. pasteurianum contains three [4F–4S] clusters and one [2Fe–2S] cluster (CpI, Fig. 1), only two [4Fe–4S] clusters are found in the [FeFe]-hydrogenase from Desulfovibrio desulfuricans, and no such “accessory” clusters are found in the [FeFe]-hydrogenase from Chlamydomonas reinhardtii (CrHydA1, Fig. 1).23 These enzymes are of interest to biotechnological H2 production, given their high proton reduction rates of up to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 s−1 close to the formal (or, biological standard) potential of hydrogen (E0′2H+/H2 = −414 mV vs. SHE).24–26In vivo, [FeFe]-hydrogenase competes for ferredoxin with other oxidoreductases, e.g., in NAD(P)H production, lipid maturation, and N2 fixation.27–29 Understanding electron flux in hydrogen turnover, it is important to characterize intermolecular ET between [FeFe]-hydrogenase and ferredoxin (Fig. 1), which relies on PPIs and, presumably, a successive exchange of single electrons.30–32
000 s−1 close to the formal (or, biological standard) potential of hydrogen (E0′2H+/H2 = −414 mV vs. SHE).24–26In vivo, [FeFe]-hydrogenase competes for ferredoxin with other oxidoreductases, e.g., in NAD(P)H production, lipid maturation, and N2 fixation.27–29 Understanding electron flux in hydrogen turnover, it is important to characterize intermolecular ET between [FeFe]-hydrogenase and ferredoxin (Fig. 1), which relies on PPIs and, presumably, a successive exchange of single electrons.30–32
Fourier-transform infrared (FTIR) spectroscopy is ideally suited to detect PPIs, mainly due to its sensitivity detecting small changes in the hydrogen-bonded C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and N–H groups of the peptide backbone that are informative of secondary structural changes.34 However, the spectral overlap with solvent (H2O) complicates the analysis, in particular when transient and/or subtle changes are addressed. These challenges can be overcome by FTIR difference spectroscopy where the activity of a sample is induced by a specific trigger (light, potential jumps, reactant titrations, etc.) that allow analyzing the spectral changes rather than the absolute spectra.35–39
O and N–H groups of the peptide backbone that are informative of secondary structural changes.34 However, the spectral overlap with solvent (H2O) complicates the analysis, in particular when transient and/or subtle changes are addressed. These challenges can be overcome by FTIR difference spectroscopy where the activity of a sample is induced by a specific trigger (light, potential jumps, reactant titrations, etc.) that allow analyzing the spectral changes rather than the absolute spectra.35–39
Introducing specific chromophores by protein engineering is a complementary strategy. Here, the reporter group shows favorable spectroscopic properties like a high extinction coefficient or a distinct absorption frequency. This can be achieved by amber codon suppression using an aminoacyl-tRNA synthetase/tRNA pair designed for the selective insertion of non-canonical amino acids40 like 4-cyano-L-phenylalanine (pCNF), which contains an IR-active nitrile group (–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) that can be probed to measure changes in the electric field via the vibrational Stark effect (VSE).41–43 The VSE originates from the small differences in dipole moment between the first vibrationally exited state ν1 compared to the ground state ν0. An external electric field projected onto the nitrile group by the protein environment interacts differently with the ν1 or ν0 dipole moment.44 The measured frequency shift Δν is then proportional to the electric field vector
N) that can be probed to measure changes in the electric field via the vibrational Stark effect (VSE).41–43 The VSE originates from the small differences in dipole moment between the first vibrationally exited state ν1 compared to the ground state ν0. An external electric field projected onto the nitrile group by the protein environment interacts differently with the ν1 or ν0 dipole moment.44 The measured frequency shift Δν is then proportional to the electric field vector  with the negative difference in dipole moments
with the negative difference in dipole moments  , the later which is also referred to as Stark tuning rate. The introduction of pCNF within a protein yields a local electric field probe.45
, the later which is also referred to as Stark tuning rate. The introduction of pCNF within a protein yields a local electric field probe.45
To investigate site-selective redox changes in ferredoxin CpFd under turnover conditions, we replaced tyrosine Y3 by the non-canonical amino acid pCNF near the [4Fe–4S] cluster F (Fig. 1). With the aim of minimizing any other changes to CpFd, we targeted the only tyrosine residue of the protein. Our CpFd:CpI model indicates that the pCNF residue is positioned ∼6.0 Å from the CpFd:CpI interface, and that the distal [4Fe–4S] cluster of CpI (C) is about 11.4 Å away (Fig. S1†). We used in situ attenuated total reflectance (ATR) FTIR difference spectroscopy to investigate CpFd in complex with [FeFe]-hydrogenases CpI or CrHydA1, triggering reduction and oxidation by changing the gas atmosphere between H2, N2, or O2. Only in the CpFd:CpI complex, a subtle yet significant shift of the nitrile band was observed, accompanied by clear changes in protein secondary structure.
Results and discussion
The production of holo-CpFd (wild type) and holo-Y3pCNF-CpFd in E. coli C321.ΔA.opt was confirmed by electronic spectroscopy, where one broad band at ∼400 nm reflects the presence of the oxidized [4Fe–4S] clusters (Fig. 2A).46 In comparison to wild-type CpFd, the total protein yield of Y3pCNF-CpFd decreased five-fold. This can be explained by translation stalling until the arrival of the pCNF-charged tRNA, or by a low cytoplasmic concentration of free pCNF. Importantly, the production of Y3pCNF-CpFd was also found to decrease by a further five-fold in the absence of pCNF in the culture medium, confirming the selectivity of the orthogonal aminoacyl-tRNA synthetase/tRNA pair for tRNA charging with pCNF. The incorporation of pCNF was initially verified by fluorescence spectroscopy (Fig. S2†). | ||
| Fig. 2 Electronic spectroscopy and protein film electrochemistry. (A) Electronic spectra of wild-type CpFd (red) and Y3pCNF-CpFd (black) recorded in aerobic MOPS/NaOH buffer (0.1 M, pH 7) at 25 °C. The broad band at 400 nm shows the presence of oxidized iron–sulfur clusters ([4Fe–4S]2+). (B) Representative cyclic voltammograms of wild-type CpFd (red) and Y3pCNF-CpFd (black) recorded in aerobic potassium phosphate/NaOH buffer (25 mM pH 7.5, 0.1 M NaCl, and 50 mM MgCl2) at 25 °C with a scan rate of 25 mV s−1. | ||
We next determined whether the presence of pCNF induces a significant change in E0′ of its neighboring [4Fe–4S] cluster using protein film voltammetry (Fig. 2B). Due to either (i) fast intermolecular ET between the [4Fe–4S] clusters of CpFd or (ii) electronically similar environments around the [4Fe–4S] clusters, a single pair of redox peaks at −390 ± 7 mV vs. SHE was observed, in good agreement with previous investigations.47,48 In the case of the Y3pCNF-CpFd, a single pair of redox peaks was observed at −378 ± 2 mV vs. SHE, suggesting that the presence of pCNF did not introduce a significant change in Em (Student's t-test, P = 0.0996).
The incorporation of pCNF was then verified by two independent vibrational spectroscopic approaches. Raman spectra reveal a single band at 2234 cm−1 that is exclusively observed in the Y3pCNF-CpFd sample and can be assigned to the nitrile stretching vibration of pCNF (Fig. S2†). The relatively high frequency and broad band shape suggests hydrogen-bonding with the solvent.45 Independent confirmation was achieved by FTIR spectroscopy (Fig. 3A). Interestingly, we found that the pCNF band position shifted by 4–5 cm−1 toward lower frequencies when water was removed from the protein film (Fig. S3†). Therefore, any comparison must be performed under unchanging hydration levels.
 | ||
| Fig. 3 Steady-state vibrational spectroscopy. (A) ATR FTIR spectra of wild-type CpFd (red) and Y3pCNF-CpFd (black) confirming the presence of the nitrile band of pCNF at ∼2234 cm−1 (inset). (B) Second derivate FTIR spectra of the Y3pCNF-CpFd:CpI complex (black) show five strong bands (a)–(e) between 2150 and 1750 cm−1 that have been assigned to the diatomic ligands of the H-cluster in the oxidized resting state Hox. These bands are missing in the Y3pCNF-CpFd spectrum (red); however, at higher frequencies both spectra show the nitrile band of pCNF (inset). In complex with CpI, this band is red-shifted by 3–4 cm−1 relative to CpFd. | ||
No meaningful nitrile shifts were detected upon electrochemical reduction or oxidation, in the presence of the chemical reductant dithionite, or by the oxidation of Y3pCNF-CpFd with O2 (Fig. S4†). Both of these approaches have been exploited to reduce or oxidize ferredoxins in the past.49–51 To probe the reduction and oxidation of Y3pCNF-CpFd under turnover conditions, we mixed the protein with its natural redox partner, the [FeFe]-hydrogenase CpI,52 in a ratio of 5:1 (1 mM CpFd and 200 μM CpI). The second derivative FTIR spectrum shows that the nitrile band is found at ∼2230 cm−1 (Fig. 3B), suggesting less solvent access around Y3pCNF in the presence of CpI. At lower frequencies, the spectrum reveals strong absorbance bands (a)–(e) that have been assigned to the cyanide (CN−) and carbon monoxide (CO) ligands of CpI.24–26 The IR signature is characteristic for the oxidized resting state of the H-cluster, Hox, and unaffected by the presence of CpFd.
We hypothesize that the reduction and oxidation of CpI would result in ET with Y3pCNF-CpFd.30–32 Therefore, a Y3pCNF-CpFd:CpI protein film was probed in the presence of N2, H2, or O2 employing our unique setup for in situ ATR FTIR spectroscopy.39 For this, 1 μL protein solution (200 μM) was concentrated on the ATR crystal under a stream of dry N2 and re-hydrated with N2 aerosol until the protein film adopted an equilibrium with the humidity in the gas phase. Five N2/H2 cycles followed by incubation with O2 were performed, where a treatment with O2 at the end of the experiment deactivates CpI – [FeFe]-hydrogenases are O2-sensitive53 – ultimately impeding H2 oxidation and subsequent ET toward Y3pCNF-CpFd. To ensure that the system adopted equilibrium conditions, each gas treatment was performed for 180 s before the spectra were compared. The apparent rate constants for reduction and auto-oxidation of CpI are k1 = 2.8 ± 0.1 s and k–1 = 51 ± 1.6 s, respectively (Fig. S8†). The data in Fig. 4A illustrate how Hox converts into redox states Hred and Hsred under 10% H2, auto-oxidation in the absence of H2 (100% N2), and oxygenic deactivation of CpI in the presence of 10% O2. Fig. 4B shows the respective second derivative spectra in the energy regime of the pCNF nitrile band (same data set as in panel A). We observe a distinct up-shift from 2229.5 ± 0.15 cm−1 to 2231.3 ± 0.15 cm−1 in the presence of 10% H2 (Δν = 1.8 cm−1) that is reversible when the gas atmosphere is switched back N2. Plotting the band position against gas treatment (Fig. 4C) reveals the reversibility of the shift and illustrates that once CpI is deactivated with 10% O2, subsequent treatment with H2 no longer impacts the nitrile band. Significant hydration changes that would affect the nitrile frequency were avoided (Fig. 4D and S3†).
 | ||
| Fig. 4 In situ analysis of the ferredoxin:hydrogenase complex. (A) ATR FTIR spectra of the H-cluster's CO ligands show the repeated reduction under 10% H2 (increase of Hred and Hsred at 1900 and 1894 cm−1, respectively) and oxidation under N2 (Hox at 1970 and 1947 cm−1). In the presence of 10% O2, all CO bands disappear due to disintegration of the H-cluster. (B) Second derivative ATR FTIR spectra of the Y3pCNF-CpFd:CpI complex in the frequency regime of the nitrile band under N2 (black), 10% H2 (red), or 10% O2 (blue). (C) The shift of the nitrile band as a function of gas composition. After deactivation of CpI with O2, a reaction with H2 is no longer observed. (D) Shift of the nitrile band as a function of the hydration level (measured at 3350 cm−1). The pCNF up-shift for increasingly humid protein films can be followed by linear regression (dashed line, data from Fig. S3†). In comparison, any changes in hydration level during the N2/H2/O2 titration are insignificant, which allows excluding any unspecific pCNF band shifts. | ||
With a Stark tuning rate of 0.71 cm−1 mV−1 cm−1 for pCNF,54 a 1.8 cm−1 shift is equivalent to a local electric field change of +2.53 mV cm−1, well in agreement with reports for other proteins.55–57 The observed up-shift indicates an accumulation of negative charge in front of the nitrile group's N-atom upon reduction, e.g., due to protein structural changes, the reduction of [4Fe–4S] cluster F of CpFd, or a reduction of [4Fe–4S] cluster C of CpI (Fig. 1). The later interpretation is particularly interesting as Artz et al. have argued that cluster C controls hydrogenase activity due to its low E0′ of approximately −450 mV vs. SHE, functioning as a “gate keeper” for catalytic ET.30 Whether the nitrile shift is a general feature of hydrogenase reduction or specific to the complex with CpI can be probed in presence of CrHydA1 as an alternative [FeFe]-hydrogenase. The so-called minimal [FeFe]-hydrogenase CrHydA1 lacks the iron–sulfur cluster containing F-domain of CpI (Fig. 1) but has been demonstrated to interact with both plant-type and bacterial ferredoxins.58–60 Surprisingly, however, a nitrile shift was not observed when a Y3pCNF-CpFd:CrHydA1 film was reacted with H2 (Fig. S5†). As this argues against a detection of reduced cofactors, we speculate that the Stark shift of the pCNF nitrile band is due to protein structural changes in the Y3pCNF-CpFd:CpI complex and PPI-coupled ET events specific for the interaction with the F-domain of CpI.
To investigate redox-dependent protein structural changes, FTIR difference spectra of Y3pCNF-CpFd in the presence of CpI or CrHydA1 were analyzed. Spectra under N2 were subtracted from spectra under 10% H2 yielding “H2–N2” difference spectra recorded in a time-dependent fashion. Fig. S6† shows the temporal evolution of H2–N2 difference spectra for Y3pCNF-CpFd:CpI, 5–25 s after initial contact with H2. These data depict the conversion of steady-state oxidized (equilibrium under N2) into steady-state reduced (equilibrium under H2) and as such are unrelated to the microscopic velocity of complex formation or catalysis. We emphasize that transient modulations of the ferredoxin:hydrogenase complex cannot be visualized by this approach. Fig. 5A shows H2–N2 difference spectra after 25 s for different samples. Between 2150 and 1750 cm−1, negative bands indicate depletion of the oxidized H-cluster (Hox, as accumulated under N2) while positive bands indicate evolution of the reduced H-cluster states Hred and Hred′ as well as Hsred and Hhyd in the presence of H2 (Table S2†).61 Below 1700 cm−1, the spectra show differential signals at 1670 and 1624 cm−1 as well as 1548 and 1535 cm−1. A negative band pattern becomes visible around 1515 cm−1. The prominent 1670/1624 feature can be assigned to secondary structural changes involving the amide I band, i.e., a formation of β-sheets over random coil structures.34 Importantly, as the amide I band is dominated by the CO stretching frequency of the protein backbone, the lack of spectral shifts in D2O is in agreement with this assignment (Fig. S6†). Moreover, the unchanging band position at 1670 cm−1 in D2O allows a decrease in the hydration level to be excluded (Fig. S6† additionally shows how spectral differences in the OH stretching regime of water were negligible). Smaller features below 1600 cm−1 might stem from amide II changes; however, no significant downshifts were observed upon deuteration. A reference experiment with Y3pCNF-CpFd and O2-deactivated CpI demonstrates that the formation of any difference feature below 1700 cm−1 is dependent on hydrogenase activity; the blue trace in Fig. 5A was calculated from the final H2 treatment in Fig. 4C and basically resembles a baseline.
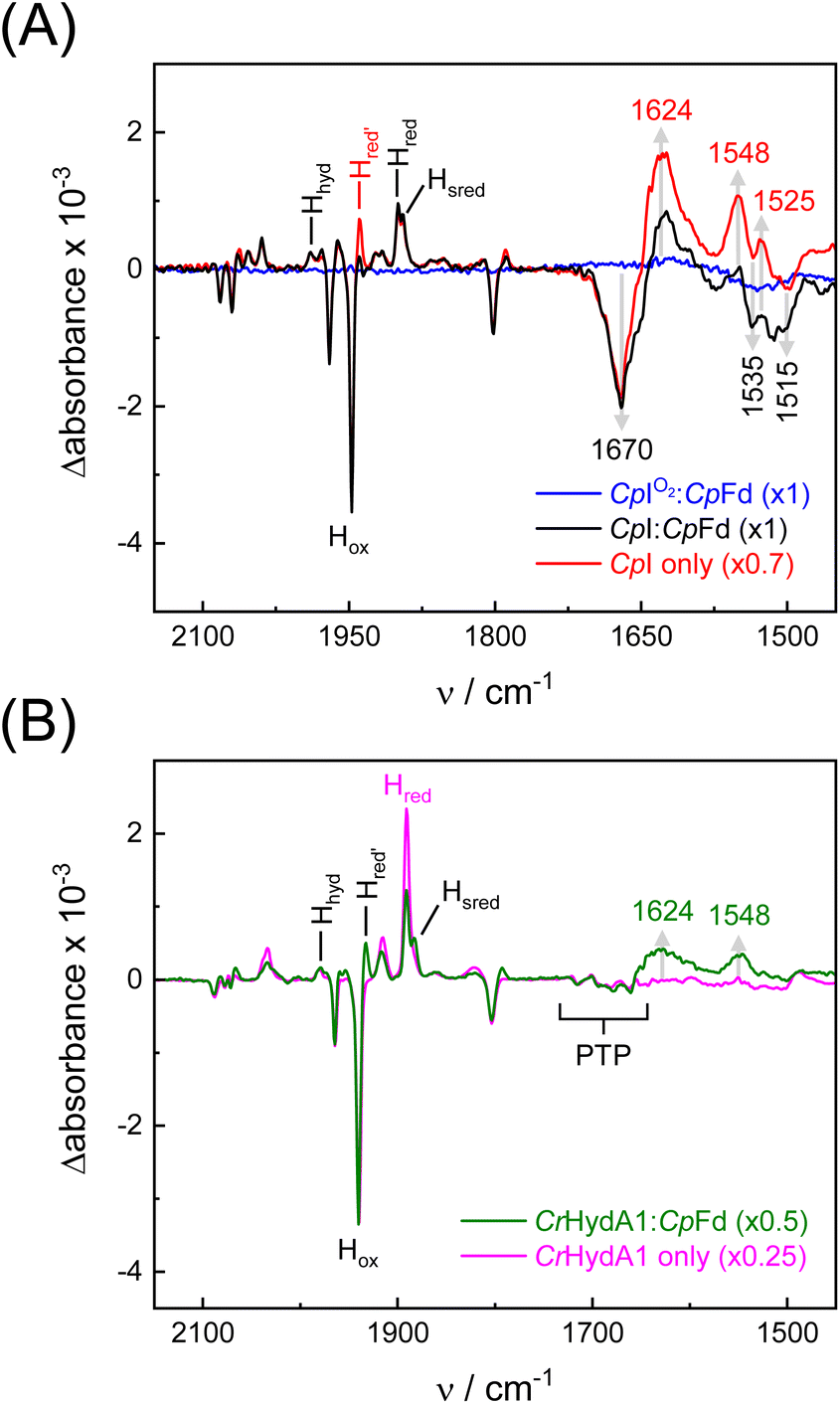 | ||
| Fig. 5 Infrared difference spectroscopy. (A) H2–N2 ATR FTIR difference spectra of Y3pCNF-CpFd:CpI (black) and CpI alone (red) after 25 s. Bands above 1750 cm−1 are assigned to H-cluster ligands in the Hox state (negative) and various reduced states (positive). At lower frequencies, the 1670/1624 cm−1 down-shift in the amide I regime indicates protein structural changes, alongside up-shifts at 1535/1548 cm−1 and 1515/1525 cm−1. The blue spectrum serves as a negative reference where CpI was inactivated with O2 ahead of the reaction with H2. (B) H2–N2 ATR FTIR difference spectra of Y3pCNF-CpFd:CrHydA1 (green) and CrHydA1 alone (magenta) after 50 s. Note the lack of the difference features in CrHydA1 in the amide I regime while only small changes are observed in the presence of Y3pCNF-CpFd. “PTP” refers to small difference features explored earlier.62 All spectra are normalized to the main CO band of Hox (1947 cm−1 in CpI, 1940 cm−1 in CrHydA1) with factors as indicated in the legends. | ||
Further exploring the origin of protein structural changes, we probed CpI in the absence of ferredoxin (Fig. 5A). Surprisingly, a very similar difference spectrum was observed, with slightly more pronounced difference features at 1548/1535 cm−1 and 1525/1515 cm−1 as well as a greater accumulation of the Hred′ state, indicative of a fully reduced composition of [4Fe–4S] clusters.63 This comparison permits the observed changes to be assigned to the hydrogenase rather than the ferredoxin. A similar interpretation was proposed by Voloshyn et al., detecting structural changes in the “sensory” [FeFe]-hydrogenase from Thermoanaerobacter mathranii upon reduction.64 As the Stark shift of Y3pCNF-CpFd is observed exclusively in complex with CpI (Fig. S4 and S5†), we speculate that the nitrile group near the [4Fe–4S] cluster of ferredoxin reports on changes in protein structure rather than redox state. Therefore, we analyzed the H2–N2 difference spectrum of a Y3pCNF-CpFd:CrHydA1 protein film in the next step. Band changes below 1700 cm−1 were nearly absent (the green traces in Fig. 5B only show small positive bands), and not detected at all when CrHydA1 was reduced in the absence of CpFd. These findings permit the origin of protein structural changes to be localized: assuming that the iron–sulfur clusters of the F-domain in CpI mediate ET between H-cluster and ferredoxin30–32 while the H-cluster in CrHydA1 interacts with ferredoxin directly,65–67 our data now strongly suggest that protein structural changes of the F-domain modulate PPIs and ET between hydrogenase and ferredoxin.
Whether reduction leads to a formation or dissociation of the ferredoxin:hydrogenase complex is unclear from the FTIR data. Therefore, we performed anaerobic microscale thermophoresis (MST) analyses to compare the dissociation constants (Kd) of CpI with CpFd or Y3pCNF-CpFd under reducing or oxidizing conditions. In the presence of hydrogenase, the characteristic dithionite-bleach68 of the Alexa Fluor 647 dye used for labeling ferredoxin was found to be reversible and facilitated a comparison of Kd for CpFd:CpI and Y3pCNF-CpFd:CpI under reducing conditions (Fig. 6 and S7†). This “protection” of the Alexa Fluor 647 label on ferredoxin was specific to CpI and not observed with other proteins (Fig. S7†). From a fit of the data, a Kd of 12.4 ± 0.97 μM was derived for wild-type CpFd and a similar Kd of 11.3 ± 0.71 μM was obtained for Y3pCNF-CpFd, suggesting that the mutation does not alter the interaction with CpI. These values are in excellent agreement with Kd values obtained for solution-based H2 evolution assays of CpI and dithionite-reduced CpFd (16.5 ± 5 μM).69 Quinkal and co-workers reported a maximal H2 evolution rate (Vmax) of 720 μmol H2 per min per mg, which underscores the kinetic relevance of the Kd values determined by MST here. Notably, no significant interaction could be detected between oxidized wild-type CpFd and CpI (Fig. S7†), cementing the importance of redox states in PPIs. The clostridial ferredoxin has been shown to support proton reduction with other [FeFe]-hydrogenases, e.g., CrHydA1.70 The low Kd of 2.41 ± 0.29 μM obtained from our MST experiments (Fig. S7†) supports this observation and justifies comparing CpFd:CpI and CpFd:CrHydA1 in the FTIR experiments. Note that CrHydA1 forms an ever stronger complex with its natural redox partner, PetF (Fig. S7†).
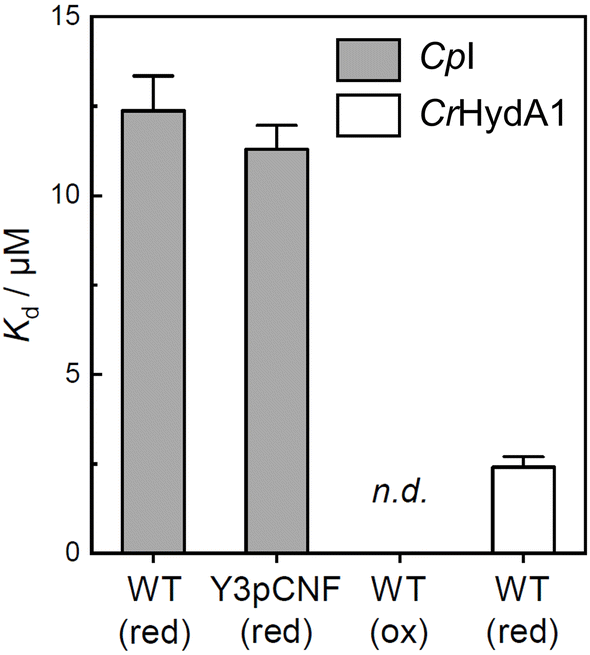 | ||
| Fig. 6 Microscale thermophoresis. Dissociation constants (Kd) of the CpFd:CpI (WT) and Y3pCNF-CpFd:CpI complex under reducing or oxidizing conditions (red/ox). Under oxidizing conditions in the absence of dithionite, an interaction between CpFd:CpI was not detected (n.d.). The open bar depicts the Kd of the CpFd:CrHydA1 complex under reducing conditions. See Fig. S7† for the titration curves and additional data. | ||
Oxidizing conditions in the MST experiment resemble the equilibrium under N2 in the FTIR experiment as most [FeFe]-hydrogenases convert into the oxidized state Hox in the absence of external reductants.71 Equally, reducing conditions in the MST experiment resemble the equilibrium under H2 in the FTIR experiment. Both dithionite and H2 are commonly used as reductants for hydrogenases, although H2 may be the preferable agent.72,73 Based on these conservative assumptions, the H2–N2 difference spectra in Fig. 5A can now be explained with complex formation upon reduction of CpI and CpFd in the presence of H2. The negative bands at 1670, 1535, and 1515 cm−1 are assigned to the F-domain in the “free”, oxidized form while positive bands at 1624, 1548, and 1525 cm−1 are assigned to the F-domain in the ferredoxin-bound, reduced form. As emphasized above, our data do not directly reflect the transient modulation of the ferredoxin:hydrogenase complex; however, an underlying mechanism presents itself. We speculate that redox-dependent protein structural changes facilitate complex formation and intermolecular ET in the presence of reducing equivalents. Once oxidized – e.g., upon H2 evolution – the complex reverts into the oxidized structure and releases the ferredoxin. The distal [4Fe–4S] cluster of the F-domain (C in Fig. 1) must play a crucial role as level indicator in this mechanism, as emphasized by Artz et al. earlier.30 Managing ET via the F-domain may explain the superior catalytic activity of bacterial [FeFe]-hydrogenases like CpI over algal [FeFe]-hydrogenases like CrHydA1.
Conclusions
In summary, we present a dynamic analysis of the electron transfer complex between ferredoxin CpFd and the structurally diverse [FeFe]-hydrogenases CpI and CrHydA1. The introduction of the non-canonical amino acid pCNF as a Stark probe permitted electric field changes upon reduction or oxidation of the complex to be determined, and FTIR difference spectroscopy uncovered pronounced protein structural changes within the F-domain of CpI. Such dynamic behavior is rarely picked up in structural biology, although the “sampling” of different conformations in cryogenic electron microscopy (cryo EM) may lead to respective findings.74–76 The MST affinity analysis of CpFd and CpI confirmed that the observed electric field and protein structural changes reflect a formation of the protein–protein complex under reducing conditions, prompted by redox-dependent PPIs. We speculate that the CpFd:CpI complex may be favored when redox states are complementary, i.e., oxidized CpI may “select” for reduced CpFd, and vice versa (most [FeFe]-hydrogenases are bidirectional77). Similar concepts of interprotein ET have been discussed early on.78Our observations now explain the typically superior performance of hydrogenases with ferredoxin over general reductants. On the one hand, the specificity of contact secures optimal ET distances between the iron–sulfur clusters, as sensed by the local Stark probe pCNF. On the other hand, global changes at the ET interface allow dissociation of the complex once electron transfer has taken place, i.e., upon oxidation. This can lead to an oscillation between “on and off” when the ferredoxin pool, an important metabolic marker,79–81 is sufficiently reduced. This way, the two-electron chemistry of hydrogenase is promoted by a one-electron redox partner like ferredoxin.
We note that two-electron redox chemistry with 2[4Fe–4S]-type ferredoxins (i.e., proceeding via two one-electron transfers) cannot be strictly excluded. A single interaction between CpI and CpFd may function to transfer a total of two electrons, given that the two [4Fe–4S] clusters are of similar potential and situated within physiologically relevant ET distance.82 However, evidence for redox-dependent PPIs in the interaction with ferredoxin can be found in a wide range of systems, hinting at a general phenomenon. For example, the [2Fe–2S]-type ferredoxin of Anabaena PCC7119 undergoes distinct changes upon reduction,83 and when Kurisu et al. crystallized the first ferredoxin-dependent protein complex in 2001,84 the authors highlighted structural changes in both ferredoxin and Fd:NADP+ reductase (FNR). The importance of such changes was rationalized by Xu et al. who used paramagnetic NMR spectroscopy to investigate the ternary complex between ferredoxin and Fd:Trx reductase (FTR) that “catalyzes” reduction of thioredoxin (Trx) eventually.85 Recent data from Steinhilper et al. suggests that similar principles may apply to other ferredoxin-driven redox processes as well.86
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
S. Sahin acknowledges funding from a Marie Curie-Skłodowska Individual Fellowship under the European Union's Horizon 2020 research and innovation program (101024443, “BERCO2”). Ross D. Milton acknowledges the Swiss National Science Foundation (200021_191985) for support. Sven T. Stripp acknowledges funding by the German Research Foundation DFG (STR1554/6-1 and STR1554/8-1). Thanks to Joachim Heberle for providing access to his laboratory and spectrometers at Freie Universität Berlin. Takuji B. M. Adachi acknowledges the Société Académique de Genève and Fondation Ernst et Lucie Schmidheiny for financial support. We thank Alexandre Jolly for assistance with pCNF-containing protein expression. We acknowledge Dr Jacek Kozuch, Prof. Esteban Vöhringer-Martinez, and Dr Rodrigo Recabarren for discussing the effect of electric field changes within proteins.References
- O. Keskin, A. Gursoy, B. Ma and R. Nussinov, Principles of protein−protein interactions: what are the preferred ways for proteins to interact?, Chem. Rev., 2008, 108(4), 1225–1244, DOI:10.1021/cr040409x.
- H. B. Gray and J. R. Winkler, Electron transfer in proteins, Annu. Rev. Biochem., 1996, 65, 537–561, DOI:10.1146/annurev.bi.65.070196.002541.
- H. B. Gray and J. R. Winkler, Long-Range electron transfer, Proc. Natl. Acad. Sci. U. S. A., 2005, 102(10), 3534–3539, DOI:10.1073/pnas.0408029102.
- J. L. Dempsey, J. R. Winkler and H. B. Gray, Proton-coupled electron flow in protein redox machines, Chem. Rev., 2010, 110(12), 7024–7039, DOI:10.1021/cr100182b.
- J. Sancho, Flavodoxins: sequence, folding, binding, function and beyond, Cell. Mol. Life Sci., 2006, 63(7–8), 855–864, DOI:10.1007/s00018-005-5514-4.
- F. De Rienzo, R. R. Gabdoulline, M. C. Menziani and R. C. Wade, Blue copper proteins: a comparative analysis of their molecular interaction properties, Protein Sci., 2000, 9(8), 1439–1454, DOI:10.1110/ps.9.8.1439.
- D. Alvarez-Paggi, L. Hannibal, M. A. Castro, S. Oviedo-Rouco, V. Demicheli, V. Tórtora, F. Tomasina, R. Radi and D. H. Murgida, Multifunctional cytochrome c: learning new tricks from an old dog, Chem. Rev., 2017, 117(21), 13382–13460, DOI:10.1021/acs.chemrev.7b00257.
- H. Beinert, Iron–sulfur proteins: ancient structures, still full of surprises, J. Biol. Inorg. Chem., 2000, 5(1), 2–15 CrossRef CAS PubMed.
- D. O. Hall, R. Cammack and K. K. Rao, Role for ferredoxins in the origin of life and biological evolution, Nature, 1971, 233(5315), 136–138, DOI:10.1038/233136a0.
- F. Capozzi, S. Ciurli and C. Luchinat, Coordination sphere versus protein environment as determinants of electronic and functional properties of iron–sulfur proteins, Struct. Bond., 1998, 90, 127–160 CrossRef CAS.
- P. Stephens, D. Jollie and A. Warshel, Protein control of redox potentials of iron–sulfur proteins, Chem. Rev., 1996, 96(7), 2491–2514 CrossRef CAS PubMed.
- R. C. Prince and M. W. Adams, Oxidation-reduction properties of the two Fe4S4 clusters in Clostridium pasteurianum ferredoxin, J. Biol. Chem., 1987, 262(11), 5125–5128, DOI:10.1016/S0021-9258(18)61163-9.
- H. S. Gao-Sheridan, H. R. Pershad, F. A. Armstrong and B. K. Burgess, Discovery of a novel ferredoxin from azotobacter vinelandii containing two [4Fe–4S] clusters with widely differing and very negative reduction potentials, J. Biol. Chem., 1998, 273(10), 5514–5519, DOI:10.1074/jbc.273.10.5514.
- E. Saridakis, P. Giastas, G. Efthymiou, V. Thoma, J. M. Moulis, P. Kyritsis and I. M. Mavridis, Insight into the protein and solvent contributions to the reduction potentials of [4Fe–4S]2+/+ clusters: crystal structures of the allochromatium vinosum ferredoxin variants C57A and V13G and the homologous Escherichia coli ferredoxin, J. Biol. Inorg. Chem., 2009, 14(5), 783–799, DOI:10.1007/s00775-009-0492-x.
- J. Meyer, Ferredoxins of the third kind, FEBS Lett., 2001, 509(1), 1–5, DOI:10.1016/S0014-5793(01)03049-6.
- K. TAGAWA and D. I. ARNON, Ferredoxins as electron carriers in photosynthesis and in the biological production and consumption of hydrogen gas, Nature, 1962, 195(4841), 537–543, DOI:10.1038/195537a0.
- W. Lubitz, H. Ogata, O. Rüdiger, E. Reijerse, O. Rudiger and E. H. Reijerse, Chem. Rev., 2014, 114(8), 4081–4148, DOI:10.1021/cr4005814.
- P. M. Vignais and B. Billoud, Occurrence, classification, and biological function of hydrogenases: an overview, Chem. Rev., 2007, 107(10), 4206–4272, DOI:10.1021/cr050196r.
- H. Tai, S. Hirota and S. T. Stripp, Proton transfer mechanisms in bimetallic hydrogenases, Acc. Chem. Res., 2021, 54(1), 232–241, DOI:10.1021/acs.accounts.0c00651.
- Y. Nicolet, C. Piras, P. Legrand, C. E. Hatchikian and J. C. Fontecilla-Camps, Desulfovibrio desulfuricans iron hydrogenase: the structure shows unusual coordination to an active site fe binuclear center, Structure, 1999, 7(1), 13–23, DOI:10.1016/S0969-2126(99)80005-7.
- J. W. Peters, W. N. Lanzilotta, B. J. Lemon and L. C. Seefeldt, X-ray crystal structure of the Fe-only hydrogenase (CpI) from Clostridium pasteurianum to 1.8 angstrom resolution, Science, 1998, 282(5395), 1853–1858, DOI:10.1126/science.282.5395.1853.
- J. Esselborn, N. Muraki, K. Klein, V. Engelbrecht, N. Metzler-Nolte, U.-P. Apfel, E. Hofmann, G. Kurisu and T. Happe, A structural view of synthetic cofactor integration into [FeFe]-hydrogenases, Chem. Sci., 2016, 7, 959–968, 10.1039/C5SC03397G.
- S. T. Stripp and T. Happe, How algae produce hydrogen—news from the photosynthetic hydrogenase, Dalton Trans., 2009,(45), 9960, 10.1039/b916246a.
- H. Land, M. Senger, G. Berggren and S. T. Stripp, Current state of [FeFe]-hydrogenase research – biodiversity and spectroscopic investigations, ACS Catal., 2020, 10(13), 7069–7086, DOI:10.1021/acscatal.0c01614.
- J. T. Kleinhaus, F. Wittkamp, S. Yadav, D. Siegmund and U.-P. Apfel, [FeFe]-hydrogenases: maturation and reactivity of enzymatic systems and overview of biomimetic models, Chem. Soc. Rev., 2021, 50(3), 1668–1784, 10.1039/D0CS01089H.
- J. A. Birrell, P. Rodríguez-Maciá, E. J. Reijerse, M. A. Martini and W. Lubitz, The catalytic cycle of [FeFe] hydrogenase: a tale of two sites, Coord. Chem. Rev., 2021, 449, 214191, DOI:10.1016/j.ccr.2021.214191.
- I. Yacoby, S. Pochekailov, H. Toporik, M. L. Ghirardi, P. W. King and S. Zhang, Photosynthetic electron partitioning between [FeFe]-hydrogenase and ferredoxin:NADP+ – oxidoreductase (FNR) enzymes in vitro, Proc. Natl. Acad. Sci. U. S. A., 2011, 108(23), 9396–9401, DOI:10.1073/pnas.1103659108.
- B. W. Burkhart, H. P. Febvre and T. J. Santangelo, Distinct physiological roles of the three ferredoxins encoded in the hyperthermophilic archaeon Thermococcus kodakarensis, mBio, 2019, 10(2) DOI:10.1128/mBio.02807-18.
- H. Addison, T. K. A. Glatter, G. Hochberg and J. G. Rebelein, Two distinct ferredoxins are essential for nitrogen fixation by the iron nitrogenase in Rhodobacter capsulatus, mBio, 2024, 15(3) DOI:10.1128/mbio.03314-23.
- J. H. Artz, D. W. Mulder, M. W. Ratzloff, C. E. Lubner, O. A. Zadvornyy, A. X. LeVan, S. G. Williams, M. W. W. Adams, A. K. Jones, P. W. King and J. W. Peters, Reduction potentials of [FeFe]-hydrogenase accessory iron–sulfur clusters provide insights into the energetics of proton reduction catalysis, J. Am. Chem. Soc., 2017, 139(28), 9544–9550, DOI:10.1021/jacs.7b02099.
- C. E. Lubner, J. H. Artz, D. W. Mulder, A. Oza, R. J. Ward, S. G. Williams, A. K. Jones, J. W. Peters, I. I. Smalyukh, V. S. Bharadwaj and P. W. King, A site-differentiated [4Fe–4S] cluster controls electron transfer reactivity of Clostridium acetobutylicum [FeFe]-hydrogenase I, Chem. Sci., 2022, 13(16), 4581–4588, 10.1039/D1SC07120C.
- C. Gauquelin, C. Baffert, P. Richaud, E. Kamionka, E. Etienne, D. Guieysse, L. Girbal, V. Fourmond, I. André, B. Guigliarelli, C. Léger, P. Soucaille and I. Meynial-Salles, Roles of the F-domain in [FeFe] hydrogenase, Biochim. Biophys. Acta, Bioenerg., 2018, 1859(2), 69–77, DOI:10.1016/j.bbabio.2017.08.010.
- D. Kozakov, D. R. Hall, B. Xia, K. A. Porter, D. Padhorny, C. Yueh, D. Beglov and S. Vajda, The cluspro web server for protein-protein docking, Nat. Protoc., 2017, 12(2), 255–278, DOI:10.1038/nprot.2016.169.
- A. Barth, Infrared spectroscopy of proteins, Biochim. Biophys. Acta, Bioenerg., 2007, 1767(9), 1073–1101, DOI:10.1016/j.bbabio.2007.06.004.
- W. Mäntele, Reaction-induced infrared difference spectroscopy for the study of protein function and reaction mechanisms, Trends Biochem. Sci., 1993, 18(6), 197–202, DOI:10.1016/0968-0004(93)90186-Q.
- C. Kötting and K. Gerwert, Proteins in action monitored by time-resolved FTIR spectroscopy, ChemPhysChem, 2005, 6(5), 881–888, DOI:10.1002/cphc.200400504.
- T. Kottke, V. A. Lórenz-Fonfría and J. Heberle, The grateful infrared: sequential protein structural changes resolved by infrared difference spectroscopy, J. Phys. Chem. B, 2017, 121(2), 335–350, DOI:10.1021/acs.jpcb.6b09222.
- V. A. Lórenz-Fonfria, Infrared difference spectroscopy of proteins: from bands to bonds, Chem. Rev., 2020, 120(7), 3466–3576, DOI:10.1021/acs.chemrev.9b00449.
- S. T. Stripp, In situ infrared spectroscopy for the analysis of gas-processing metalloenzymes, ACS Catal., 2021, 11(13), 7845–7862, DOI:10.1021/acscatal.1c00218.
- S. Sahin and R. D. Milton, Evolving enzymatic electrochemistry with rare or unnatural amino acids, Curr. Opin. Electrochem., 2022, 35, 101102, DOI:10.1016/j.coelec.2022.101102.
- H. Kim and M. Cho, Infrared probes for studying the structure and dynamics of biomolecules, Chem. Rev., 2013, 113(8), 5817–5847, DOI:10.1021/cr3005185.
- S. D. Fried and S. G. Boxer, Measuring electric fields and noncovalent interactions using the vibrational stark effect, Acc. Chem. Res., 2015, 48(4), 998–1006, DOI:10.1021/ar500464j.
- S. D. Fried and S. G. Boxer, Electric fields and enzyme catalysis, Annu. Rev. Biochem., 2017, 86(1), 387–415, DOI:10.1146/annurev-biochem-061516-044432.
- E. S. Park, S. S. Andrews, R. B. Hu and S. G. Boxer, Vibrational stark spectroscopy in proteins: a probe and calibration for electrostatic fields, J. Phys. Chem. B, 1999, 103(45), 9813–9817, DOI:10.1021/jp992329g.
- J. B. Weaver, J. Kozuch, J. M. Kirsh and S. G. Boxer, Nitrile infrared intensities characterize electric fields and hydrogen bonding in protic, aprotic, and protein environments, J. Am. Chem. Soc., 2022, 144(17), 7562–7567, DOI:10.1021/jacs.2c00675.
- J. C. M. Tsibris and R. W. Woody, Structural studies of iron–sulfur proteins, Coord. Chem. Rev., 1970, 5(4), 417–458, DOI:10.1016/S0010-8545(00)80100-9.
- F. A. Armstrong, R. Camba, H. A. Heering, J. Hirst, L. J. C. Jeuken, A. K. Jones, C. Le′ger and J. P. McEvoy, Fast voltammetric studies of the kinetics and energetics of coupled electron-transfer reactions in proteins, Faraday Discuss., 2000, 116, 191–203, 10.1039/b002290j.
- R. D. Milton, J. C. Ruth, J. S. Deutzmann and A. M. Spormann, Methanococcus maripaludis employs three functional heterodisulfide reductase complexes for flavin-based electron bifurcation using hydrogen and formate, Biochemistry, 2018, 57(32), 4848–4857, DOI:10.1021/acs.biochem.8b00662.
- F. A. Armstrong, H. A. O. Hill and N. J. Walton, Direct electrochemical reduction of ferredoxin promoted by Mg2+, FEBS Lett., 1982, 145(2), 241–244, DOI:10.1016/0014-5793(82)80175-0.
- S. G. Mayhew, D. Petering, G. Palmer and G. P. Foust, Spectrophotometric titration of ferredoxins and chromatium high potential iron protein with sodium dithionite, J. Biol. Chem., 1969, 244(11), 2830–2834, DOI:10.1016/S0021-9258(18)91702-3.
- B. Hosein and G. Palmer, The kinetics and mechanism of oxidation of reduced spinach ferredoxin by molecular oxygen and its reduced products, Biochim. Biophys. Acta, Bioenerg., 1983, 723(3), 383–390, DOI:10.1016/0005-2728(83)90045-2.
- J. B. Therien, J. H. Artz, S. Poudel, T. L. Hamilton, Z. Liu, S. M. Noone, M. W. W. Adams, P. W. King, D. A. Bryant, E. S. Boyd and J. W. Peters, The physiological functions and structural determinants of catalytic bias in the [FeFe]-hydrogenases CpI and CpII of clostridium pasteurianum strain W5, Front. Microbiol., 2017, 8, 1–11, DOI:10.3389/fmicb.2017.01305.
- S. T. Stripp, G. Goldet, C. Brandmayr, O. Sanganas, K. A. Vincent, M. Haumann, F. A. Armstrong and T. Happe, How oxygen attacks [FeFe] hydrogenases from photosynthetic organisms, Proc. Natl. Acad. Sci. U. S. A., 2009, 106(41), 17331–17336, DOI:10.1073/pnas.0905343106.
- I. T. Suydam and S. G. Boxer, Vibrational stark effects calibrate the sensitivity of vibrational probes for electric fields in proteins, Biochemistry, 2003, 42(41), 12050–12055, DOI:10.1021/bi0352926.
- A. T. Fafarman and S. G. Boxer, Nitrile bonds as infrared probes of electrostatics in ribonuclease S, J. Phys. Chem. B, 2010, 114(42), 13536–13544, DOI:10.1021/jp106406p.
- A. S. Adesina, K. Świderek, L. Y. P. Luk, V. Moliner and R. K. Allemann, Electric field measurements reveal the pivotal role of cofactor–substrate interaction in dihydrofolate reductase catalysis, ACS Catal., 2020, 10(14), 7907–7914, DOI:10.1021/acscatal.0c01856.
- I. T. Suydam, C. D. Snow, V. S. Pande and S. G. Boxer, Electric fields at the active site of an enzyme: direct comparison of experiment with theory, Science, 2006, 313(5784), 200–204, DOI:10.1126/science.1127159.
- S. T. Stripp and T. Happe, How algae produce hydrogen – news from the photosynthetic hydrogenase, Dalton Trans., 2009, 45, 9960–9969, 10.1039/b916246a.
- C. Gauquelin, C. Baffert, P. Richaud, E. Kamionka, E. Etienne, D. Guieysse, L. Girbal, V. Fourmond, I. André, B. Guigliarelli, C. Léger, P. Soucaille and I. Meynial-Salles, Roles of the F-domain in [FeFe] hydrogenase, Biochim. Biophys. Acta, Bioenerg., 2018, 1859(2), 69–77, DOI:10.1016/j.bbabio.2017.08.010.
- L. Girbal, G. Von Abendroth, M. Winkler, P. Benton, I. Meynial-Salles, C. Croux, J. W. Peters, T. Happe and P. Soucaille, Homologous and heterologous overexpression in clostridium acetobutylicum and characterization of purified clostridial and algal fe-only hydrogenases with high specific activities, Appl. Environ. Microbiol., 2005, 71(5), 2777–2781, DOI:10.1128/AEM.71.5.2777.
- J. Khushvakov, R. Nussbaum, C. Cadoux, J. Duan, S. T. Stripp and R. D. Milton, Following electroenzymatic hydrogen production by rotating ring disk electrochemistry and mass spectrometry, Angew. Chem., Int. Ed., 2021, 60(18), 10001–10006, DOI:10.1002/anie.202100863.
- M. Senger, V. Eichmann, K. Laun, J. Duan, F. Wittkamp, G. Knör, U.-P. Apfel, T. Happe, M. Winkler, J. Heberle and S. T. Stripp, How [FeFe]-hydrogenase facilitates bidirectional proton transfer, J. Am. Chem. Soc., 2019, 141(43), 17394–17403, DOI:10.1021/jacs.9b09225.
- P. Rodríguez-Maciá, N. Breuer, S. DeBeer and J. A. Birrell, Insight into the redox behavior of the [4Fe–4S] subcluster in [FeFe] hydrogenases, ACS Catal., 2020, 10(21), 13084–13095, DOI:10.1021/acscatal.0c02771.
- I. Voloshyn, C. Schumann, P. R. Cabotaje, A. Zamader, H. Land and M. Senger, Secondary structure changes as the potential H2 sensing mechanism of group D [FeFe]-hydrogenases, Chem. Commun., 2024, 60(78), 10914–10917, 10.1039/D4CC03098B.
- M. Winkler, S. Kuhlgert, M. Hippler and T. Happe, Characterization of the key step for light-driven hydrogen evolution in green algae, J. Biol. Chem., 2009, 284(52), 36620–36627, DOI:10.1074/jbc.M109.053496.
- K. Sybirna, P. Ezanno, C. Baffert, C. Léger and H. Bottin, Arginine171 of chlamydomonas reinhardtii [Fe–Fe] hydrogenase HydA1 plays a crucial role in electron transfer to its catalytic center, Int. J. Hydrogen Energy, 2013, 38(7), 2998–3002, DOI:10.1016/j.ijhydene.2012.12.078.
- S. Rumpel, J. F. Siebel, M. Diallo, C. Farès, E. J. Reijerse and W. Lubitz, Structural insight into the complex of ferredoxin and [FeFe] hydrogenase from Chlamydomonas reinhardtii, ChemBioChem, 2015, 16(11), 1663–1669, DOI:10.1002/cbic.201500130.
- B. Jagilinki, M. Willis, F. Mus, R. Sharma, L. Pellows, D. Mulder, Z. Yang, L. Seefeldt, P. King, G. Dukovic and J. Peters, Microscale thermophoresis (MST) as a tool to study binding interactions of oxygen-sensitive biohybrids, Bio Protoc., 2024, 14(1350) DOI:10.21769/BioProtoc.5041.
- I. Quinkal, V. Davasse, J. Gaillard and J.-M. Moulis, On the role of conserved proline residues in the structure and function of Clostridium pasteutianum 2[4Fe–4S] ferredoxin, Protein Eng., 1994, 7, 681–687 CrossRef CAS , https://academic.oup.com/peds/article/7/5/681/1576519.
- L. Girbal, G. von Abendroth, M. Winkler, P. M. C. Benton, I. Meynial-Salles, C. Croux, J. W. Peters, T. Happe and P. Soucaille, Homologous and heterologous overexpression in Clostridium acetobutylicum and characterization of purified clostridial and algal Fe-only hydrogenases with high specific activities, Appl. Environ. Microbiol., 2005, 71(5), 2777–2781, DOI:10.1128/AEM.71.5.2777-2781.2005.
- J. Duan, S. Mebs, K. Laun, F. Wittkamp, J. Heberle, T. Happe, E. Hofmann, U. P. Apfel, M. Winkler, M. Senger, M. Haumann and S. T. Stripp, Geometry of the catalytic active site in [FeFe]-hydrogenase is determined by hydrogen bonding and proton transfer, ACS Catal., 2019, 9(10), 9140–9149, DOI:10.1021/acscatal.9b02203.
- M. A. Martini, O. Rüdiger, N. Breuer, B. Nöring, S. Debeer, P. Rodríguez-Maciá and J. A. Birrell, The nonphysiological reductant sodium dithionite and [FeFe] hydrogenase: influence on the enzyme mechanism, J. Am. Chem. Soc., 2021, 143(43), 18159–18171, DOI:10.1021/jacs.1c07322.
- M. Senger, J. Duan, M. V. Pavliuk, U.-P. Apfel, M. Haumann and S. T. Stripp, Trapping an oxidized and protonated intermediate of the [FeFe]-hydrogenase cofactor under mildly reducing conditions, Inorg. Chem., 2022, 61(26), 10036–10042, DOI:10.1021/acs.inorgchem.2c00954.
- Y. Dong, S. Zhang, Z. Wu, X. Li, W. L. Wang, Y. Zhu, S. Stoilova-McPhie, Y. Lu, D. Finley and Y. Mao, Cryo-EM structures and dynamics of substrate-engaged human 26S proteasome, Nature, 2019, 565(7737), 49–55, DOI:10.1038/s41586-018-0736-4.
- D. Shvarev, D. Januliene and A. Moeller, Frozen motion: how cryo-EM changes the way we look at ABC transporters, Trends Biochem. Sci., 2022, 47(2), 136–148, DOI:10.1016/j.tibs.2021.11.008.
- H. L. Rutledge, B. D. Cook, H. P. M. Nguyen, M. A. Herzik and F. A. Tezcan, Structures of the nitrogenase complex prepared under catalytic turnover conditions, Science, 2022, 377(6608), 865–869, DOI:10.1126/science.abq7641.
- A. Fasano, H. Land, V. Fourmond, G. Berggren and C. Léger, Reversible or irreversible catalysis of H+/H2 conversion by FeFe hydrogenases, J. Am. Chem. Soc., 2021, 143(48), 20320–20325, DOI:10.1021/jacs.1c09554.
- G. McLendon and R. Hake, Interprotein electron transfer, Chem. Rev., 1992, 92(3), 481–490, DOI:10.1021/cr00011a007.
- E. A. Peden, M. Boehm, D. W. Mulder, R. Davis, W. M. Old, P. W. King, M. L. Ghirardi and A. Dubini, Identification of global ferredoxin interaction networks in Chlamydomonas reinhardtii, J. Biol. Chem., 2013, 288(49), 35192–35209, DOI:10.1074/jbc.M113.483727.
- J. T. Atkinson, I. Campbell, G. N. Bennett and J. J. Silberg, Cellular assays for ferredoxins: a strategy for understanding electron flow through protein carriers that link metabolic pathways, Biochemistry, 2016, 55(51), 7047–7064, DOI:10.1021/acs.biochem.6b00831.
- B. W. Burkhart, H. P. Febvre and T. J. Santangelo, Distinct physiological roles of the three ferredoxins encoded in the hyperthermophilic archaeon Thermococcus kodakarensis, mBio, 2019, 10(2) DOI:10.1128/mBio.02807-18.
- C. C. Page, C. C. Moser, X. Chen and P. L. Dutton, Natural engineering principles of electron tunnelling in biological oxidation-reduction, Nature, 1999, 402(6757), 47–52, DOI:10.1038/46972.
- R. Morales, M.-H. Charon, G. Hudry-Clergeon, Y. Pétillot, S. Norager, M. Medina and M. Frey, Refined X-ray structures of the oxidized, at 1.3 Å, and reduced, at 1.17 Å, [2Fe−2S] ferredoxin from the Cyanobacterium anabaena PCC7119 show redox-linked conformational changes, Biochemistry, 1999, 38(48), 15764–15773, DOI:10.1021/bi991578s.
- G. Kurisu, M. Kusunoki, E. Katoh, T. Yamazaki, K. Teshima, Y. Onda, Y. Kimata-Ariga and T. Hase, Structure of the electron transfer complex between ferredoxin and ferredoxin-NADP(+) reductase, Nat. Struct. Biol., 2001, 8(2), 117–121, DOI:10.1038/84097.
- X. Xu, P. Schürmann, J. S. Chung, M. A. S. Hass, S. K. Kim, M. Hirasawa, J. N. Tripathy, D. B. Knaff and M. Ubbink, Ternary protein complex of ferredoxin, ferredoxin:thioredoxin reductase, and thioredoxin studied by paramagnetic NMR spectroscopy, J. Am. Chem. Soc., 2009, 131(48), 17576–17582, DOI:10.1021/ja904205k.
- R. Steinhilper, L. Boß, S.-A. Freibert, V. Schulz, N. Krapoth, S. Kaltwasser, R. Lill and B. J. Murphy, Two-stage binding of mitochondrial ferredoxin-2 to the core iron–sulfur cluster assembly complex, Nat. Commun., 2024, 15(1), 10559, DOI:10.1038/s41467-024-54585-4.
Footnote |
| † Electronic supplementary information (ESI) available: Evaluation of the CpFd:CpI complex model, fluorescence and Raman spectra, additional infrared spectra, and MST data. See DOI: https://doi.org/10.1039/d5sc00550g |
| This journal is © The Royal Society of Chemistry 2025 |