
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePursuing the holy grail of thermally activated delayed fluorescence emitters: a molecular strategy for reducing the energy gap and enhancing spin–orbit coupling†
Vladyslav
Ievtukhov
ab,
Antonio
Prlj
c,
Olga
Ciupak
d,
Michał
Mońka
a and
Illia E.
Serdiuk
 *a
*a
aFaculty of Mathematics, Physics and Informatics, University of Gdańsk, Wita Stwosza 57, 80-308 Gdańsk, Poland. E-mail: illia.serdiuk@ug.edu.pl; Tel: + 48 58 523 22 44
bFaculty of Chemistry, University of Gdańsk, Wita Stwosza 63, 80-308 Gdańsk, Poland
cDivision of Physical Chemistry, Institut Ruđer Bošković, Bijenička Cesta 54, 10000 Zagreb, Croatia
dDepartment of Organic Chemistry, Gdańsk University of Technology, Gabriela Narutowicza 11/12, 80-233 Gdańsk, Poland
First published on 9th May 2025
Abstract
The beauty and complexity of organic materials stem from the intricate interplay of their structural effects, where the same substituent can prove highly beneficial in one position yet entirely redundant in another. In this study, we demonstrate how a single halogen atom, strategically positioned, can significantly enhance the emissive properties of thermally activated delayed fluorescence (TADF) emitters. To address the inherent “low singlet–triplet energy gap (ΔEST) equals low spin–orbit coupling (SOC)” tradeoff in donor–acceptor (DA) emitters, we propose leveraging heavy-atom substitution to achieve two simultaneous effects: (1) reducing rotational heterogeneity and thereby lowering ΔEST at the macroscopic level due to the substituent's steric bulk, and (2) increasing SOC through the substituent's high atomic number (Z). In pursuit of efficient blue emitters, our investigation focused on DMAC-DPS derivatives with halogens positioned in the linker fragment at the ortho position to the donor. Fluorine substitution stabilized the charge-transfer state in polar media, resulting in a remarkably low activation energy of 20 meV. Bromine substitution enhanced SOC by over 20-fold in a nonpolar medium, while chlorine, striking a balance between the two, emerged as a “golden mean,” offering both low activation energy and sufficiently high SOC. Molecular dynamics analysis revealed that molecular vibrations which disrupt the linker benzene ring symmetry promote excited-state mixing. The resulting hybrid nature of the T1 state, involving electron density on the halogen, is particularly important for SOC enhancement in the chlorine derivative. These findings highlight that combining a mild heavy-atom effect (HAE) with the control of selective molecular vibrations offers a promising strategy for the design of efficient blue TADF emitters. Our results provide valuable insights for advancing the development of high-performance organic light-emitting materials.
Introduction
Thermally activated delayed fluorescence (TADF) has gained much attention as a promising solution to the problems of heavy-metal-free organic light emitting diode (OLED) technology targeted for eco-friendly display and lighting applications. Organic TADF materials are lightweight, flexible, and capable of being manufactured on large surfaces at low cost. One of the key challenges of OLED technology is the efficiency of light emission, which depends on the ability of the emissive component to effectively utilize both singlet and triplet excitons. TADF enables qualitative use of triplet excitons by converting them into singlet excitons, thereby enhancing emission efficiency. Efficiency and operational stability are specifically problematic issues for blue OLEDs; thus substantial efforts have been made recently to develop appropriate blue emitters.Organic TADF emitters typically involve donor–acceptor systems, where structural and electronic parameters affecting charge transfer (CT) between the donor (D) and acceptor (A) components play a crucial role in the key photophysical properties and electroluminescent performance in a device. The energy gap between the lowest singlet and triplet states (ΔEST) influences the efficiency of triplet harvesting. A smaller energy gap allows for faster reverse intersystem crossing (rISC), where triplet excitons are converted into singlet excitons, thus avoiding nonradiative deactivation which also has an impact on the stability of a device. The performance of TADF emitters is not solely governed by the energy gap. Spin–orbit coupling (SOC), the interaction between the electron's spin and its orbital motion, is equally crucial in facilitating rISC. Enhanced SOC helps to promote the transition of triplet excitons to singlet states, further improving the efficiency of light emission.
Most molecular design approaches developed thus far tend to compromise either the ΔEST or SOC. Minimizing ΔEST typically requires increasing the charge-transfer (CT) character in both the S1 and T1 states, which unfortunately reduces SOC. This happens because when the same CT nature dominates in S1 and T1, their electron and hole wavefunctions are spatially separated, diminishing the necessary orbital angular momentum interactions that drive SOC, thereby suppressing rISC. A third important parameter, the high radiative rate constant (kr), is also compromised when CT is stabilized. Conversely, increasing SOC usually involves enhancing the triplet character different from CT, which then sacrifices ΔEST. This trade-off is clearly illustrated by orthogonal DA emitters – characterized by low ΔEST, SOC, and kr – contrasting with planar “multiresonance” (MR) systems,1,2 which demonstrate significantly larger values for each of these parameters. For these reasons, within dominating molecular design strategies, achieving a balance between reducing ΔEST and increasing SOC is believed to be critical for designing high-performance OLEDs that can operate at lower voltages and higher efficiency, especially in the blue region.
In spite of recent progress in MR systems3–5 and discovery of inversed singlet–triplet emitters,6,7 this challenge for blue emitters seems to be far from an industrially acceptable solution. One could solve the “low ΔEST = low SOC” tradeoff described above by developing a molecular design for improving both the energy gap and SOC simultaneously. Among all-organic materials, DA emitters remain probably the best scaffold for such a purpose offering fast and efficient triplet harvesting, reasonable kr, good stability of both hole and electron charge carriers, and energies of frontier molecular orbitals suitable for the best known OLED architectures. In theory, orthogonal alignment of D and A can afford ΔEST as low as a few meV. However, this limit has not been probably achieved yet in a real macroscopic emitting system. The reported values of experimental activation energy (Ea), which is a function of ΔEST and reorganization energy (λ), usually exceed 50 meV, especially for blue emitters.8–11 Within the two-state rISC model which we introduced previously for orthogonal DA emitters with a strong CT character, such deviations are explained by molecular vibrations, specifically torsions, causing fluctuations of the dihedral angle θ between D and A (Fig. 1A). ΔEST and SOC are functions of θ: The closer θ is to 90° the lower the ΔEST and SOC. Following such considerations, to reach the theoretical limit of ΔEST and Ea one should minimize θ-rotational isomerism by retaining the orthogonal DA structure and strong CT character of S1 and T1 states. To tackle the “low ΔEST = low SOC” tradeoff, as a concept of this work, we suggest using heavy-atom substitution to simultaneously (1) decrease θ distribution and thus ΔEST in the macroscopic system due to the large size of the substituent; (2) increase SOC due to its high atomic number (Z).
 | ||
| Fig. 1 (A) The illustration of the two-state T1(3CT)–S1(1CT) model for rISC: dihedral angle θ in the example of the TMCz-BO emitter;12 ΔEST, SOC, and rotamer occurrence as a function of θ. (B) Some of the previously studied ortho-substituted TADF emitters. (C) Systems currently under investigation. | ||
The ortho position of the linker fragment (Fig. 1B) appears to play a crucial role in determining the ΔEST. The ortho-alignment of multiple donors or acceptors can be considered a distinctive molecular design approach. One of the earliest and most extensively studied groups of organic TADF materials comprises benzonitrile acceptors paired with several carbazole donors positioned in close proximity to one another.13–15 In these emitters and analogs,16 such structural arrangements promote a nearly orthogonal alignment of D and A, restrict rotational freedom, and achieve exceptionally high rISC rates in the blue region. In 5Cz-TRZ (Fig. 1B), such a design leads to minimal efficiency roll-off (<2.3%) and satisfactory operational stability (T90 of 600 h) at a luminance of 1000 cd m−2.17 In simpler, more atom-economic, archetypal blue DA emitters like DMAC-TRZ, DMAC-DPS, etc., which feature hydrogen atoms in the ortho positions, computational models predict a θ angle of 90° at the optimal ground and excited state geometries.18,19 However, the low barrier for θ-torsion results in deviations of up to ±30°. Substituting the ortho position with a methyl group, as demonstrated in TRZ emitters, significantly improves OLED performance.20 For instance, a yellowish-green emitter featuring a phenoxazine donor (TrzPXZ-Me, Fig. 1B) exhibited an extremely low EQE roll-off of 8% at 1000 cd m−2, attributed to an increased rISC rate.
In the pursuit of deep-blue TADF emitters, an elegant analysis of the effect of methyl substitution on the linker-donor fragment in the carbazole (Cbz)-TRZ scaffold system was presented in ref. 21. This study demonstrated a direct relationship between steric hindrance, energy gap modulation, and rISC rate enhancement – boosting the rate 600-fold to 1.8 × 105 s−1 when two methyl groups appear at ortho positions (Cbz-TRZ4, Fig. 1B) – and its subsequent impact on OLED performance. A similar reduction in ΔEST was observed in a Cbz-TRZ analog upon fluorine substitution (TCTZ-2F, Fig. 1B), resulting in a twofold increase in the rISC rate, reaching 1.9 × 105 s−1.22 Fluorine substitution was further shown to enhance the rISC rate and shorten the delayed fluorescence (DF) lifetime by more than half in DMAC–quinoxaline-based red emitters (FDMAC-CNQ, Fig. 1B), both in solution and various solid media.23 Experimentally, ΔEST was identified as a critical parameter for such TADF enhancement, decreasing from 90 meV to 40 meV upon fluorine substitution in solution-phase studies.
The improvements observed in ortho-substituted derivatives of emitters can largely be attributed to the restriction of θ-torsion, which enforces an orthogonal alignment and stabilizes the CT character. It is hypothesized that further strengthening of the donor–acceptor (DA) interaction, and thus the CT character, could enhance rISC rates. While this approach can be effective for green, yellow, and red systems, however, in the blue region, additional stabilization of the CT states poses a significant challenge, as it often compromises color purity. Consequently, balancing the CT character to achieve both fast rISC and high color purity remains a persistent problem in the development of blue TADF emitters.
Diphenylsulfone (DPS) derivatives are of particular interest for this study. Due to the strong electron-withdrawing nature and high triplet energy of the DPS acceptor fragment, these emitters exhibit a pronounced CT character in their S1 and T1 states, with minimal contribution from locally excited (LE) states. Combined with a high-energy LUMO, this results in the advantageous combination of a low ΔEST and sufficiently high S1–S0 transition energy, making DPS derivatives suitable for blue emission. Previous studies have also reported excellent TADF properties in (di)DMAC-DPS derivatives featuring ortho substituents. For instance, the introduction of methyl groups in the ortho position of the dimethylthioxanthene-S,S-dioxide acceptor (DMAC-TXO2-Me, Fig. 1B) enhances the rISC rate and EQE by several percentage points.24 However, in non-cyclic DPS structures, due to its electron-releasing character, methyl groups lead to destabilization of the CT character and an increase in ΔEST.25
Here, to implement the above mentioned concept, we explore the effect of various halogens (R) in the ortho position of the linker of the DMAC-DPS emitter (Fig. 1C). We prove the possibility of the simultaneous reduction of the ΔEST gap and increase in SOC in two ways. Firstly, the experimental investigations show that the rISC rate increases in the sequence R = H > F > Cl > Br due to a significant decrease in Ea and, in a selected medium, an increase in SOC. Secondly, the DFT calculations and molecular modeling predict reduction of θ-rotational freedom resulting in the narrowed ΔEST distribution. Calculations indicate that the heavy-atom effect (HAE) on SOC and the rISC rate is realized by the 3LE–1CT channel, but not the pure 3CT–1CT one. According to the results obtained, chlorine seems to be the best choice for an ortho-substituent affording efficient triplet harvesting and maintaining high PLQY.
Experimental and computational methods
Reagents and materials
Reagents for synthesis, solvents of respective grades, Zeonex ® polymer (ZNX) and the bis[2-(diphenylphosphino)phenyl]ether oxide (DPEPO, purified by sublimation) host for spectroscopic measurements were purchased and used without further purification.Synthesis
Emitters were synthesized according to the two-stage scheme as described below (Scheme 1). | ||
| Scheme 1 Synthesis of target emitters. | ||
F-H-DPStB obtained from 4-fluorobenzenesulfonyl chloride as a pale yellow solid in 63% yield. 1H NMR (500 MHz, CDCl3, δ): 8.00–7.96 (m, 1H), 7.86 (d, J = 8.8 Hz, 2H), 7.53 (d, J = 8.8 Hz, 2H), 7.21–7.17 (m, 2H), 1.32 (s, 9H). MALDITOF-MS, m/z: calc. for C16H17FO2S 292.09, found 293.20 [M + H]+.
F-F-DPStB obtained from 3,4-difluorobenzenesulfonyl chloride as a pale yellow solid in 51% yield. 1H NMR (500 MHz, CDCl3, δ): 7.86 (d, J = 8.8 Hz, 2H), 7.78–7.74 (m, 1H), 7.55 (d, J = 8.8 Hz, 2H), 7.32–7.29 (m, 1H), 1.34 (s, 9H). MALDITOF-MS, m/z: calc. for C16H16F2O2S 310.08, found 311.24 [M + H]+.
F-Cl-DPStB obtained from 3-chloro-4-fluorobenzenesulfonyl chloride as a colorless solid in 65% yield. 1H NMR (500 MHz, CDCl3, δ): 8.03 (dd, J = 8.0 Hz, J = 2.2 Hz, 1H), 7.88–7.85 (m, 3H), 7.57 (d, J = 8.9 Hz, 2H), 7.25–7.30 (m, 1H), 1.34 (s, 9H). MALDITOF-MS, m/z: calc. for C16H16ClFO2S 326.05, found 327.17 [M + H]+.
F-Br-DPStB obtained from 3-bromo-4-fluorobenzenesulfonyl chloride as a colorless solid in 85% yield. 1H NMR (500 MHz, CDCl3, δ): 8.34 (dd, J = 8.1 Hz, J = 4.5 Hz, 1H), 7.78 (d, J = 8.6 Hz, 2H), 7.44 (d, J = 8.6 Hz, 2H), 7.31 (dd, J = 7.8 Hz, J = 2.2 Hz, 1H), 7.17–7.13 (m, 1H), 1.34 (s, 9H). MALDITOF-MS, m/z: calc. for C16H16BrFO2S 370.00, found 371.09 and 373.09 isotope peaks [M + H]+.
DMAC-H-DPStB obtained from F-H-DPStB as a white crystalline solid in 89% yield. 1H NMR (500 MHz, C6D6, δ): 8.03 (d, J = 8.3 Hz, 2H), 7.92 (d, J = 8.3 Hz, 2H), 7.25 (d, J = 7.8 Hz, 2H), 7.02 (d, J = 8.3 Hz, 2H), 6.86 (d, J = 8.3 Hz, 2H), 6.82 (t, J = 7.5 Hz, 2H), 6.70 (t, J = 7.7 Hz, 2H), 6.03 (d, J = 8.2 Hz, 2H), 1.51 (s, 3H), 0.93 (s, 9H). 13C NMR (125 MHz, CDCl3, δ): 157.5, 146.2, 141.0, 140.2, 138.1, 131.3, 131.1, 130.3, 127.8, 126.5, 126.4, 125.4, 121.5, 114.7, 36.1, 35.3, 31.1, 30.9. HRMS, m/z: calc. for C31H31NO2S 481.207, found 482.219 [M + H]+.
DMAC-F-DPStB obtained from F-F-DPStB as a pale yellow crystalline solid in 83% yield. 1H NMR (500 MHz, C6D6, δ): 7.92 (d, J = 8.5 Hz, 1H), 7.85 (d, J = 8.3 Hz, 2H), 7.75 (d, J = 8.1 Hz, 1H), 7.24 (d, J = 7.3 Hz, 2H), 7.01 (d, J = 8.3 Hz, 2H), 6.82 (t, J = 7.5 Hz, 3H), 6.70 (t, J = 7.7 Hz, 2H), 6.02 (d, J = 8.2 Hz, 2H), 1.49 (s, 6H), 0.93 (s, 9H). 13C NMR (125 MHz, CDCl3, δ): 159.4, 158.0, 144.0, 143.9, 139.3, 137.3, 134.8, 133.1, 133.0, 130.7, 128.0, 126.7, 126.6, 125.5, 125.1, 125.0, 121.7, 117.6, 117.4, 113.2, 36.0, 35.4, 31.1, 30.9. HRMS, m/z: calc. for C31H30FNO2S 499.198, found 500.207 [M + H]+.
DMAC-Cl-DPStB obtained from F-Cl-DPStB as a white crystalline solid in 91% yield. 1H NMR (500 MHz, C6D6, δ): 8.42 (d, J = 1.8 Hz, 1H), 7.84 (d, J = 8.6 Hz, 2H), 7.82–7.77 (m, 1H), 7.26 (d, J = 7.7 Hz, 2H), 6.99 (d, J = 8.6 Hz, 2H), 6.87–6.76 (m, 3H), 6.68 (t, J = 7.7 Hz, 2H), 5.87 (d, J = 8.2 Hz, 2H), 1.52 (bs, 6H), 0.90 (s, 9H). 13C NMR (125 MHz, CDCl3, δ): 158.1, 143.7, 142.9, 138.7, 137.6, 137.4, 134.7, 131.0, 130.1, 128.0, 126.7, 126.6, 125.8, 121.5, 113.0, 36.0, 35.4, 31.1, 29.7. HRMS, m/z: calc. for C31H30ClNO2S 515.169, found 515.170 [M]+.
DMAC-Br-DPStB obtained from F-Br-DPStB as a white crystalline solid in 90% yield. 1H NMR (500 MHz, C6D6, δ): 8.61 (bs, 1H), 7.83 (d, J = 8.2 Hz, 2H), 7.27 (d, J = 7.6 Hz, 2H), 6.98 (d, J = 8.5 Hz, 2H), 6.82 (t, J = 7.5 Hz, 2H), 6.75 (d, J = 8.2 Hz, 1H), 6.67 (t, J = 7.7 Hz, 2H), 5.84 (d, J = 8.2 Hz, 2H), 1.60 (s, 3H), 1.53 (s, 3H), 0.90 (s, 9H). 13C NMR (125 MHz, CDCl3, δ): 158.0, 144.4, 143.5, 138.5, 137.4, 134.7, 134.2, 130.0, 128.8, 128.0, 127.8, 126.7, 126.6, 126.0, 121.4, 113.1, 68.0, 35.9, 31.1, 25.6. HRMS, m/z: calc. for C31H30BrNO2S 559.118 and 561.118, found 559.123 and 561.124 [M]+, 560.129 and 562.128 [M + H]+ corresponding to ions containing different isotopes of Br.
Sample preparation
Thin films were fabricated on quartz glass using a solution-processing approach, specifically the spin-coating method, with CHCl3 solutions containing the emitter and suitable host. To enhance compound solubility prior to spin-coating, an ULTRON ultrasonic bath (TME Electronic Components, Poland) was employed.Instrumentation
UV-vis absorption spectra were obtained using a Shimadzu UV-1900 spectrophotometer. Steady-state photoluminescence (PL) spectra were acquired with an FS5 spectrofluorometer (Edinburgh Instruments, UK) in front-face excitation geometry, achieving 1 nm resolution. Absolute PL quantum yields (PLQYs) of films were measured using a Quantaurus-QY spectrometer, while solution PLQYs utilized an FS5 system. Time-resolved PL spectra and PL intensity decay curves were measured using a YAG:Nd laser (EKSPLA PL2251A) and optical parametric generator (PG 401/SH) as excitation sources, combined with a Hamamatsu streak camera and Bruker grating spectrometer for detection.26 Optical filters (e.g., 325/50BP and 375LP) were used to minimize artefacts during excitation. Measurements of emission decays and spectra at different temperatures and of phosphorescence spectra at 10 K were enabled by using a helium cryostat (APD DE-202) and temperature controller (LakeShore 336). Rate constants (kr, kISC, and krISC) were derived from established equations. For data processing and visualization Fityk27 and Origin28 software were used.Quantum chemical calculations
Computational investigations were conducted using two approaches. For the first (1) stationary approach, to analyze the effect of θ-rotation as a key molecular vibration, we performed DFT/TDDFT29 geometry optimizations using the Gaussian 16 program package.30 Unconstrained geometry optimizations for the ground and excited S1, T1, and T2 states were conducted using the B3LYP functional31 and LANL2DZ basis set. Convergence of all geometry optimizations was verified by the absence of imaginary frequencies in obtained molecular vibrations. Next, using S0, S1, T1, and T2 states' optimal geometries, several input files were generated with the θ dihedral angle ranging from 60° to 120°. These geometries were used for stationary point calculations of molecular orbitals, electronic energies, singlet–triplet energy gaps, reorganization energies, and spin–orbit coupling matrix elements (SOCMEs) using the ORCA 4.2 software package32,33 with the B3LYP functional and Def2-SVP basis set with an included relativistic zero-order regular approximation (ZORA). In spite of the underestimation of long-range interactions and delocalization errors,34 TD-DFT B3LYP calculations including θ-torsions provide good correlation with the experimental data in polar media where the CT character dominates, and are specifically useful for analysis of pure CT and LE states without mixing of characters.18,35 Such a stationary approach allows for simplified analysis of individual CT–CT and CT–LE transitions and is useful for understanding how each rISC channel is affected by structural modifications.We further attempted to analyze the global effect of molecular vibrations and excited-state mixing within a more elaborate (2) molecular modeling approach. The efficiency of the rISC process in DMAC-DPS derivatives was estimated by calculating rates using the nuclear ensemble approach.36 Following de Sousa and de Silva, rISC rates can be calculated using the expression:
 | (1) |
 | (2) |
To calculate the rISC rate, nuclear geometries were sampled from the initially populated T1 state using quantum thermostat (QT) molecular dynamics simulations.37 QT dynamics incorporate the effect of zero-point vibrational motion that conventional Boltzmann sampling lacks. This method performs well for harmonic and moderately anharmonic systems and offers a computationally efficient alternative to the path-integral molecular dynamics approach.38
To analyze the distribution of θ-values for nuclear conformations, we sampled ensembles from molecular dynamics simulations. Since the simulations remained locked on one side of the torsional space due to the energetic barrier associated with reorienting the bulky tert-butyl-phenyl-sulfone tail, direct sampling of both torsional directions was not feasible within the simulation timescale. To account for this, we obtained distributions for θ-angles above 90° and reflected them to generate the corresponding values below 90°, ensuring a complete representation of chemically equivalent structures with opposite torsion directions.
ΔEST and SOCME were evaluated for each geometry sampled from the T1 nuclear distributions (see Generation of nuclear ensembles). Reorganization energies, on the other hand, were determined based on the optimized excited state structures. While DFT-PBE0 dynamics is suitable for exploring the nuclear density distribution of the T1 state, calculation of reorganization energies requires balanced optimizations of both T1 and S1 states. Therefore, geometry optimizations were performed with TDDFT. The TDDFT minimum energy structures were optimized with the M06-2X functional, the Def2-SVP basis set and a M4 grid, using Turbomole. For the bent structures, D3 dispersion correction was also included. The Tamm–Dancoff approximation was utilized, and the additional keyword $currswitchdisengage was necessary for successful optimizations. Energies of the optimized geometries were recalculated using ORCA, and these values served as the final input for determining the reorganization energy. Some further details on the calculation procedures can be found in the ESI.†
Results and discussion
In their absorption spectra, the compounds exhibit a typically low molar extinction coefficient in the long-wavelength S0–S1 band of CT character (Fig. 2A). Substitution with halogens causes a red shift in the band maximum, moving from 355 nm (R = H) to 363 nm (R = F), 372 nm (R = Cl), and 369 nm (R = Br). This shift indicates a significant electronic influence of the halogen on CT absorption via the acceptor fragment, with each halogen modifying its strength in accordance with the Hammett ortho constant42 (Fig. S1, ESI†). Meanwhile, a second, more complex band undergoes the opposite changes, shifting hypsochromically across the same halogen series. For example, the shoulder at ∼325 nm shifts to shorter wavelengths in the halogen derivatives. The transitions responsible for these bands are attributed to localized 1LE transitions, involving the 9,9-dimethyl-9,10-dihydroacridine (DMAC)-linker fragment.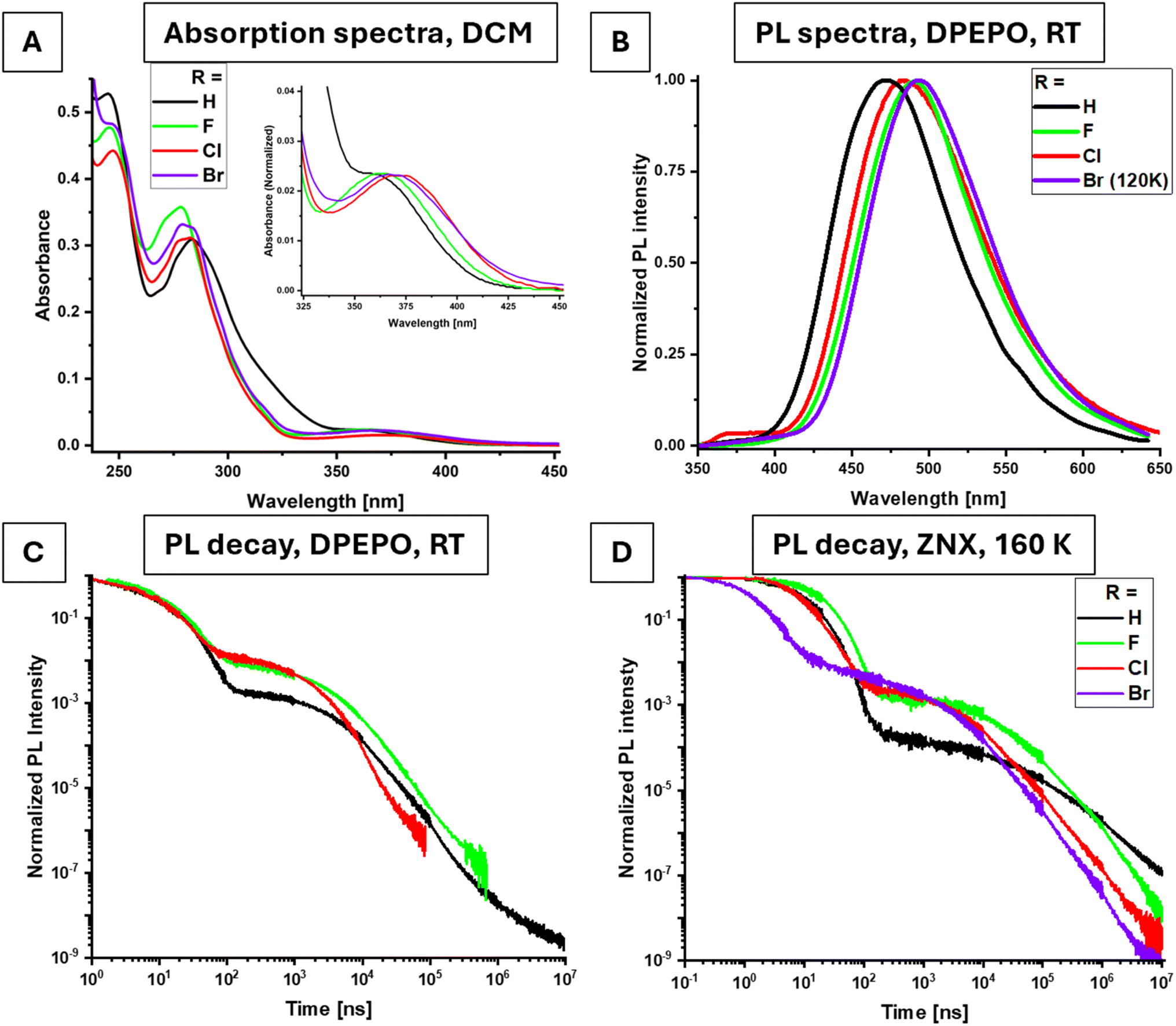 | ||
| Fig. 2 Absorption spectra in DCM solutions, c = 40 μM (A). PL spectra in the DPEPO film (10% w/w) at RT (B). PL decay curves in DPEPO films (10% w/w), RT (C) and ZNX films (0.5% w/w), 160 K (D). | ||
In films, all studied emitters exhibit blue TADF (Fig. 2B, S2A, S3 in ESI,† and Table 1) with distinct regions of prompt fluorescence (PF) and delayed fluorescence (DF) (Fig. 2C), resembling the behavior of previously studied DMAC-DPS analogs.19 In ZNX films, the DMAC-H-DPStB compound exhibits the most deep-blue photoluminescence (PL) with a peak emission (PLmax) at 428 nm. In DPEPO films, DMAC-F-DPStB shows the most red-shifted emission with a PLmax of 490 nm. This behavior indicates the important role of fluorine's electron-withdrawing nature, which enhances the acceptor strength and most effectively stabilizes the excited 1CT state in a polar (DPEPO) medium, but not in a nonpolar (ZNX) one or in the ground-state geometry as suggested by the absorption spectra discussed above. DMAC-Br-DPStB is not emissive at room temperature, but below 200 K its stationary PL spectra are similar to those of the F and Cl derivatives.
| Compound | T [K] | PLmax [nm] | PLQY [%] | τ PF [ns] | τ DF [μμs] | k r [106 s−1] | k nr [106 s−1] | k ISC [106 s−1] | k rISC [106 s−1] |
|---|---|---|---|---|---|---|---|---|---|
| ZNX film, 0.5% w/w | |||||||||
| DMAC-H-DPStB | 290 | 428 | 71 | 10.6 | 37.2 | 24 | 10 | 52 | 0.067 |
| 160 | — | 12.5 | 1.0 × 103 | — | — | 42 | 2.0 × 10−3 | ||
| DMAC-F-DPStB | 290 | 447 | 74 | 19.0 | 19.0 | 12 | 4.4 | 36 | 0.17 |
| 160 | — | 19.8 | 3.0 × 102 | — | — | 33 | 1.0 × 10−2 | ||
| DMAC-Cl-DPStB | 290 | 450 | 86 | 11.7 | 9.3 | 15 | 2.5 | 68 | 0.52 |
| 160 | — | 12.9 | 56.4 | — | — | 51 | 5.1 × 10−2 | ||
| DMAC-Br-DPStB | 160 | 451 | — | 2.0 | 14.2 | — | — | 4.3 × 102 | 0.52 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||||||||
| DPEPO film, 10% w/w | |||||||||
| DMAC-H-DPStB | 290 | 471 | 71 | 15.0 | 29 | 26 | 11 | 28 | 6.0 × 10−2 |
| 120 | — | 14.3 | 2.1 × 102 | — | — | 39 | 1.1 × 10−2 | ||
| DMAC-F-DPStB | 290 | 490 | 100 | 20.2 | 8.3 | 19 | 0 | 32 | 0.41 |
| 120 | — | 20.4 | 19.8 | — | — | 35 | 0.16 | ||
| DMAC-Cl-DPStB | 290 | 483 | 67 | 11.7 | 1.8 | 24 | 12 | 53 | 1.5 |
| 120 | — | 14.1 | 22.4 | — | — | 54 | 0.19 | ||
| DMAC-Br-DPStB | 120 | 493 | — | 3.0 | 33.4 | — | — | 3.1 × 102 | 0.46 |
Exclusively, DMAC-Cl-DPStB in ZNX exhibits a second, low-intensity band centered at 375 nm with a distinguishing vibronic structure (Fig. S2A, ESI†). This band almost vanishes in the DPEPO film (Fig. 2B) and is undetectable in other emitters at room temperature. When cooled down to 10 K, it appears in time-resoled emission spectra (TRES) in the fast regime of < 5 ns in ZNX films doped with DMAC-H-DPStB and DMAC-F-DPStB, and DPEPO films with DMAC-Br-DPStB (Fig. S4, ESI†). According to the excitation spectrum, which is different from that of the dominating 460 nm band (Fig. S2B, ESI†), and based on the previous reports,43 such a high-energy emission corresponds to the conformer with a bent DMAC fragment which is TADF inactive (see the discussion below, and in the ESI†).
Across the series R = H < F < Cl, the DF lifetime (τDF) decreases significantly, driven by a gradual increase in the rISC rate (Table 1 and Fig. 2C). For instance, in DPEPO films, the Cl derivative shows the shortest τDF of 1.8 μs, representing a 15-fold decrease compared to the unsubstituted compound (R = H). The corresponding rISC rate increases dramatically from 6.0 × 104 s−1 (R = H) to 1.5 × 106 s−1 (R = Cl), while maintaining a photoluminescence quantum yield (PLQY) near 70%. A similar trend is observed in the less polar ZNX films. The rate constant for direct intersystem crossing (kISC) shows only moderate sensitivity to halogen substitution, increasing by less than twofold in DMAC-Cl-DPStB. This indicates a selective enhancement of rISC, as highlighted by the ratio krISC/kISC [×10−3] which increases from 2.1 (R = H) to 8.7 (R = F) and 28 (R = Cl) in DPEPO films.
Interestingly, the fluorine derivative shows the lowest radiative rate constant kr (Table 1), while achieving the highest PLmax. This suggests that fluorine most effectively stabilizes the 1CT state due to its strong electron-withdrawing effect. The chlorine derivative, meanwhile, exhibits intermediate kr and PLmax values, coupled with outstanding triplet harvesting parameters, making it the optimal choice for enhancing TADF efficiency in the blue emission region.
The bromine-substituted derivative, perhaps the most intriguing in this study, emits light in the same spectral region as DMAC-F-DPStB when cooled below 200 K, where thermal nonradiative deactivation of excited states is suppressed. PL intensity decay curves at selected temperatures (Fig. 2D, S5 and S6 ESI†) reveal that, even at lower temperatures, the DF intensity remains remarkably high due to the fastest rISC rates. For instance, in ZNX films at 160 K, krISC exceeds 0.5 × 106 s−1, a remarkable 260-fold increase compared to DMAC-H-DPStB under identical conditions. In fact, ln(krISC) exhibits strong dependence on the atomic number (Z) of the halogen substituent, consistent with the HAE enhancing SOC (Fig. 3A). While the trend is more complex in frozen DPEPO films, the overall pattern persists. The ISC rate also shows sensitivity to bromine, increasing more than 10-fold compared to that of the unsubstituted emitter.
 | ||
| Fig. 3 (A) Dependence of ln(krISC) on the atomic number of the substituent (Z). (B and C) Dependence of ln(krISC) on 1/T and linear fit parameters in ZNX and DPEPO films, respectively. (D and E) Determined rISC activation energies for different emitters and as a function of van der Waals radius (Rw), respectively. (F and G) Determined SOC values for different emitters and in the function of Z of the substituent, respectively. (H) Statistically weighted SOCME for 3CT–1CT and 3LE–1CT transitions calculated on the B3LYP/Def2-SVP level of theory taking into account θ-rotamers. | ||
To evaluate whether the proposed molecular design strategy resolves the “low ΔEST = low SOC” tradeoff, temperature-dependent experiments were conducted. As a qualitative analysis, we examined TRES at 10 K (Fig. S4, ESI†). As described in detail in the ESI,† the TRES results reveal a diminishing energy difference between the S1 and T1 states with the introduction of halogen substituents. Furthermore, a significant amount of delayed fluorescence is observed in the microsecond domain for both DMAC-Cl-DPStB and DMAC-Br-DPStB, most likely indicating enhanced SOC that facilitates rISC even at low temperatures.
For a more quantitative analysis, we extracted key electronic parameters – Ea and SOC – by examining the temperature dependence of krISC on 1/T (Fig. 3B and C) using Arrhenius and Marcus–Hush equations as described previously35 and in detail in the ESI.† According to the Arrhenius equation (eqn (S1), ESI†), the slope of the linear dependence of ln(krISC) on 1/T is a function of Ea, while the intercept reflects the pre-exponential factor in the Marcus–Hush equation (eqn (S2), ESI†). Assuming Ea = ΔEST, the calculated values (Fig. 3D, F and Table S1, ESI†) allow for comparisons within the same medium, because reorganization energy dependent on the medium polarity is not explicitly included. Experimental data for DMAC-Br-DPStB were unavailable due to strong nonradiative deactivation with the variation in temperature. We compare thus determined Ea and SOC with the respective parameters from B3LYP analysis, which enables separate analysis of 3CT → 1CT and 3LE → 1CT transitions without substantial excited-state mixing.
E a and ΔE
In ZNX films, Ea decreases across the series R = H (82 meV) > F (70 meV) > Cl (61 meV) (Fig. 3D), correlating strongly with the van der Waals radius of the halogen substituent (Fig. 3E). In DPEPO films, Ea for the fluorine derivative drops to 22 meV – nearly four times lower than that of the unsubstituted emitter – but increases slightly to 31 meV for the chlorine derivative. These results suggest that in nonpolar ZNX films, the size of the substituent primarily governs Ea. We assume that in a more polar medium, the electronic effect of the substituent also becomes important, and thus the highly electronegative F substituent affords the lowest ΔEST and Ea in DPEPO films via the most efficient CT stabilization. Interestingly, as predicted by B3LYP calculations (Fig. S7, ESI†), halogen substituents have negligible influence on the energy gap between S1 and T1 states of strong CT character (ΔE3CT–1CT) at their respective energetic minima. The observed decrease in experimental Ea values is thus mostly caused by a higher barrier of θ-rotation: the analysis of rotamer probability (Fig. 4A) indicates gradual narrowing of the θ-distribution in the sequence (R) H < F < Cl. A bigger contribution of orthogonal rotamers results in a decrease in the statically weighted ΔE3CT–1CT value: 41 meV (R = H) > 33 meV (R = F) > 30 meV (R = Cl). A bromine atom appears to be too large, and thus orthogonal geometry is no longer optimal for CT states: the statically weighted ΔE3CT–1CT value increases up to 34 meV as the θ value moves to 84° in the T1(3CT) minimum. | ||
| Fig. 4 Dependences of rotamer population (A), T1–S1 (3CT–1CT, (C)) and T2–S1 (3LE–1CT, (D)) on the θ angle calculated using the B3LYP functional. A graphical visualization of electronic density changes during the T2–S1 transition in DMAC-Cl-DPStB based on molecular orbitals depicted in Fig. S8, ESI† (B); red circles indicate electronic density on the Cl atom. | ||
SOC
Fortunately, SOC increases consistently across the series R = H (0.13 cm−1) < F (0.18 cm−1) < Cl (0.28 cm−1) in ZNX films (Fig. 3F), showing a strong dependence on the atomic number of the substituent (Fig. 3G). DFT-predicted SOC matrix elements (SOCMEs) for the T2–S1 transition (Fig. 3H) further confirm this trend, underscoring the pivotal role of electronic density of the linker fragment in enabling the active rISC channel. The halogens' electronic density is involved in the transitions of such a 3LE state localized on the DMAC-linker fragment (Fig. 4B), thus affecting the change in the angular orbital momentum (ΔL). In the presence of Br, TDDFT calculations predict exceptionally high SOC for such a transition, exceeding 10 cm−1. In spite of the unavailability of the experimental estimation of SOC for DMAC-Br-DPStB, such a prediction of the strongest coupling of 3LE and 1CT states correlates perfectly with the highest rISC rate in ZNX films at 160 K (Fig. 3A).In more polar DPEPO films, DMAC-F-DPStB exhibits the lowest SOC (0.07 cm−1), which doubles for the Cl derivative (Fig. 3F). Such a decrease for DMAC-F-DPStB suggests that the main rISC channel is 3CT → 1CT, which is expected to dominate in polar media. Calculations indicate that the rISC mechanism is more complex. TDDFT-predicted SOCMEs for the 3CT → 1CT transitions decrease across the series R = H > F > Cl (Fig. 3H), consistent with experimental trends for R![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) H and F but not for Cl. The experimental evidence for the SOC increase in DMAC-Cl-DPStB can be explained by some contribution of the T2 state or the 3LE character to the T1 state, realized even at low temperatures via the previously suggested static excited-state mixing (SESM).12 Even small contributions of 3LE character involving electronic density on the Cl atom to T1 can provide a more effective rISC channel than the pure 3CT → 1CT one. Regarding DMAC-Br-DPStB, in DPEPO at 120 K, its rISC rate is the highest among other derivatives indicating the highest SOC, but it does not increase significantly as compared to ZNX. For this reason, the dependence of krISC on the atomic number in DPEPO is no longer exponential (Fig. 3A). Taking into account an over 50 times higher SOC predicted for 3LE → 1CT than for 3CT → 1CT (Fig. 3H), this supports the assumption that the HAE depends on the 3LE portion in T1, which decreases in a polar medium.
H and F but not for Cl. The experimental evidence for the SOC increase in DMAC-Cl-DPStB can be explained by some contribution of the T2 state or the 3LE character to the T1 state, realized even at low temperatures via the previously suggested static excited-state mixing (SESM).12 Even small contributions of 3LE character involving electronic density on the Cl atom to T1 can provide a more effective rISC channel than the pure 3CT → 1CT one. Regarding DMAC-Br-DPStB, in DPEPO at 120 K, its rISC rate is the highest among other derivatives indicating the highest SOC, but it does not increase significantly as compared to ZNX. For this reason, the dependence of krISC on the atomic number in DPEPO is no longer exponential (Fig. 3A). Taking into account an over 50 times higher SOC predicted for 3LE → 1CT than for 3CT → 1CT (Fig. 3H), this supports the assumption that the HAE depends on the 3LE portion in T1, which decreases in a polar medium.
These analyses demonstrate that in various media, Ea and ΔEST can be reduced and SOC can be increased through the introduction of heavy atoms at the ortho position of the linker. When comparing the F and Cl derivatives, DMAC-Cl-DPStB exhibits a more substantial SOC increase than Ea reduction, resulting in an over 3.5-fold higher rISC rate. In a polar medium, fluorine substitution seem to stabilize the CT states most effectively, beneficial for achieving low ΔEST and Ea; however, without a sufficiently high Z number, this stabilization is detrimental to SOC.
Solutions
To ensure that the observations for the emitters in films are not substantially driven by intermolecular effects, we investigated TADF and spin-flip dynamics in diluted solutions (Fig. S12–S14, Tables S3 and S4 in ESI†). By varying the solvent polarity, we obtained the dependence of krISC on the S1-state energy (ES1). For all three derivatives DMAC-H-DPStB, DMAC-F-DPStB, and DMAC-Cl-DPStB a linear dependence of ln(krISC) on ES1 was observed (Fig. 5), supporting a direct T1 → S1 mechanism for rISC. Along with krISC, kr also decreases with ES1 (Fig. S14A, ESI†) indicating stabilization of the CT character of S1 with increasing polarity. According to the current understanding,18,44 the polarity effect, manifested through a decrease in ES1, stabilizes both singlet and especially triplet CT states. This leads to a greater separation of the HOMO and LUMO, reducing the oscillator strength and ΔEST, thereby facilitating thermally activated rISC. The observed dependence also suggests that the stabilization of the CT state enhances the effectiveness of the 3CT → 1CT transition, consistent with previous studies. Within the same DA electronic system, a decrease in ΔEST with increasing polarity is thus proportional to ES1.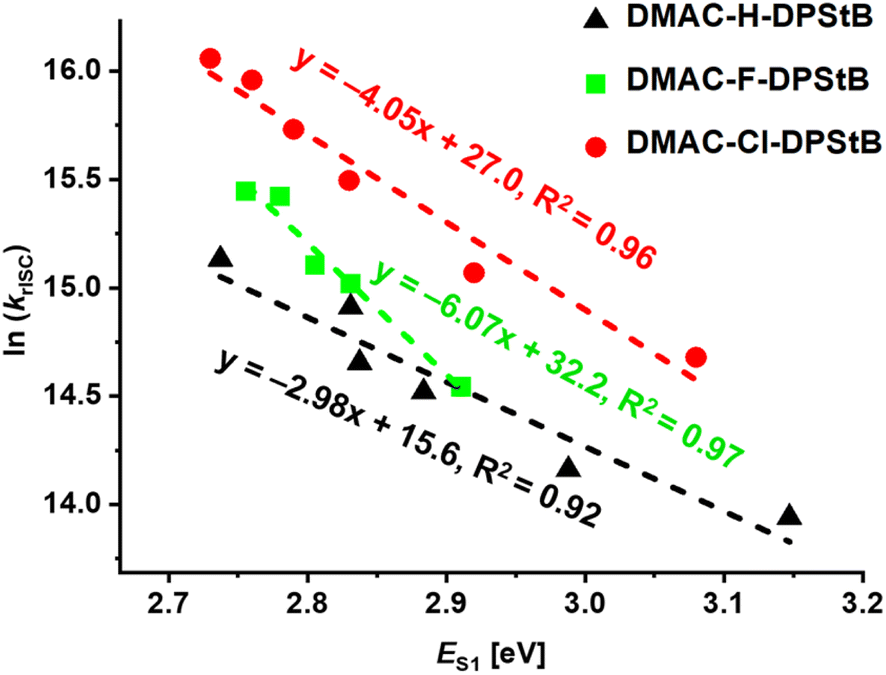 | ||
| Fig. 5 Dependence of the rISC rate constant on the energy of the S1 state in liquid solutions. | ||
However, for all compounds, ln(kISC) also increases with ES1 (Fig. S14B, ESI†) contrasting with the expected ISC for the 1CT → 3CT transition, which should decrease as its driving force (ΔEST) vanishes. Moreover, across all polarities, krISC is consistently the highest for Cl and lowest for H, indicating a similar trend for SOC (R= Cl > F > H). This contradicts the theoretical predictions of SOC for T1 → S1, which suggest the opposite trend (R = H > F > Cl). The observed discrepancies imply that similar to films, the T2 (3LE) state is influencing the rISC process even in polar solutions, despite the fact that the T2–T1 energy gap should be significantly increased in a polar medium due to the stabilization of T1 (3CT). We hypothesize that two lowest exited states are effectively coupled via dynamic excited-state mixing. This interaction is specifically important for Cl with the highest SOC of T2 → S1 resulting in the highest rISC. A similar effect was previously observed for the TMCz-BO emitter,12 where dynamic mixing between triplet states was shown to increase the efficiency of the rISC process.
Interestingly, the sensitivity of krISC to solvent polarity described by the slopes of fitted curves (Fig. 5) increases in the order: R = H(−3.0) < Cl(−4.0) < F(−6.1). We assume that this behavior can be rationalized by considering the polarizability and charge distribution in the H, F, and Cl derivatives. The rISC rate of DMAC-F-DPStB is the most sensitive to polarity, which is attributed to the unique combination of the highest electronegativity, relatively small size of the fluorine substituent and electronic properties of fluorine. F pulls electron density toward itself, causing stronger intrinsic polarization of the emitter. This results in a greater stabilization of the charge transfer state by polar solvents and as a consequence, a more efficient decrease in ΔEST. Such considerations also explain the lowest Ea of DMAC-F-DPStB in DPEPO films.
Molecular modeling
The experimental findings discussed above indicate that despite the confirmed T1–S1 mechanism of rISC, the HAE is mediated via the T2–S1 (3LE–1CT) channel, even in polar media where internal conversion should be inefficient. To explain this apparent inconsistency, we previously hypothesized the occurrence of SESM or dynamic excited-state mixing (DESM), wherein T1 and T2 states mix through vibrational modes, forming a hybrid CT–LE state. This mixing can occur dynamically in solutions or statically due to conformational trapping in films.In an attempt to model such a hybrid T1 state and the effect of molecular vibrations on rISC, we utilized the nuclear ensemble approach with electronic properties calculated using the M06-2X functional. The latter is better suited for the prediction of CT states compared to B3LYP due to its improved treatment of long-range interactions, and enables the modeling of mixed CT–LE systems. Regarding the ability to reproduce experimental conditions, this approach is applied for an isolated system in a vacuum, which allows for modeling scenarios of low polarity, where CT states are not strongly stabilized. First we take a look at the distribution of the θ-values for the ensembles of nuclear conformations sampled from molecular dynamics (Fig. 6A). The broadest distribution is observed for R = H, which has medium maxima at 112°/68°. For R = F, the distribution narrows only slightly, with medium maxima at 107°/73°. Due to the optimal steric effect of the substituent, the Cl derivative exhibits the narrowest θ-distribution (considering the width at half maximum) centered at around 90°. In contrast, the bromine atom appears too large to support the orthogonal geometry of the T1 state, leading to a θ-distribution in DMAC-Br-DPStB that resembles that of DMAC-H-DPStB, but with the contribution of the 90 ± 10° rotamers effectively diminished.
 | ||
| Fig. 6 Results of molecular modeling calculations: distribution of the dihedral angle θ (A), ΔEST (B), and SOCME (C) of the S1–T1 transitions; calculated for T1 geometry at the M06-2X level of theory. | ||
Such pronounced differences in the θ-distribution significantly impact the electronic properties, particularly the character of the T1 states. We studied the evolution of the CT character within the obtained ensemble of geometries using the charge-transfer number analysis introduced by Plasser.45 The S1 state exhibits >70% and >80% of CT character in most of the population in DMAC-H-DPStB and halogenated derivatives, respectively. The results summarized in Fig. S9 (ESI)† indicate that the T1 state contains a much higher contribution of the LE character. For example, in DMAC-H-DPStB, the CT character of T1 remains below 60% for the majority of conformers. Interestingly, while halogen substitution generally increases the CT character of T1, even in DMAC-Cl-DPStB, which demonstrates the strongest CT nature, the contribution of LE character remains considerable. Specifically, over 90% of the population exhibits CT numbers ranging between 0.9 and 0.5 (Fig. S9, ESI†). In the remaining species, the LE character dominates in T1. This further proves the hybrid nature of the triplet state, which plays a crucial role in the spin-flip dynamics.
The described geometry-induced changes in the electronic structure have the most pronounced effect on ΔEST (Fig. 6B), which determines the rISC activation energy. Due to the predominance of the orthogonal structure, the ΔEST distribution is the narrowest and closest to zero for DMAC-Cl-DPStB, followed by DMAC-Br-DPStB and DMAC-F-DPStB, and finally DMAC-H-DPStB. While only 11% of DMAC-H-DPStB species achieve ΔEST < 0.2 eV, almost 70% of the DMAC-Cl-DPStB population fulfills this criterion. As was mentioned above, this is attributed to the steric constraints imposed by the larger chlorine substituent, which restricts rotational freedom and stabilizes the orthogonal geometry. The average calculated ΔEST values are 0.38 eV (R = H), 0.32 eV (R = F), 0.19 eV (R = Cl), and 0.29 eV (R = Br), which are larger than the experimental Ea values obtained in ZNX films (Fig. 3A). This is primarily due to the absence of medium effects in the calculations. In real systems, even a nonpolar ZNX medium stabilizes CT states, leading to smaller ΔEST and Ea.
When it comes to SOC, the observed trend is not straightforward. The introduction of fluorine to DMAC-H-DPStB shifts the mode of the SOC distribution curve from 0.52 cm−1 to 0.33 cm−1 (Fig. 6C), which aligns well with the increased CT contribution in the T1 state. Replacing fluorine with chlorine results in a pronounced broadening of the SOC distribution: 99% of the population displays values in the range of 0.1–1.3 cm−1. Although the mode of distribution is at 0.3 cm−1, over 25% of the population shows SOC >0.5 cm−1. Notably, this increase in the population of species with enhanced SOC occurs despite the T1 state exhibiting the highest CT number and thus should favor the 3CT–1CT rISC channel with decreased SOC (Fig. 4C). The bromine derivative further supports this observation, displaying an even more pronounced HAE on the T1–S1 transition: SOC distribution becomes the broadest, ranging from 0.2 to 9.5 cm−1. Such SOC enhancement arises from the aforementioned contribution of LE character to the triplet state, which along with the HAE enables chlorine- and bromine-enhanced SOC for the 3LE–1CT transition. Regarding the effect of molecular vibrations, this transition itself shows relatively weak dependence on structural changes (Fig. 4D). However, as discussed above, the contribution of the LE character to T1 is strongly influenced by molecular vibrations.
To understand the molecular origins of triplet state mixing and the HAE on SOC, we analyzed different molecular conformations from the molecular dynamics ensemble of DMAC-Cl-DPStB, focusing on the evolution of natural transition orbitals (NTOs). Whilst the nature of S1 remains dominantly CT, the triplet state character specifically depends on the geometry (Fig. 7A, S11 and Table S2, ESI†). The analyzed molecular conformations can be categorized into three groups: the (1) normal group, where T1 is the CT state, and T2 is the LE state; (2) mixed group, where NTOs of T1 and T2 look nearly identical, exhibiting a mixed nature with delocalization over the DMAC donor and the linker; and (3) inverted group, a small fraction of conformations, where T1 is the LE state, and T2 is the CT one. The revealed differences in NTOs are induced by specific molecular vibrations. Most importantly, the mixing of LE and CT character (mixed group), is driven by vibrations of the linker aryl ring. Such vibrations break the substituted benzene ring symmetry and planarity, which manifests the deviations of the valence angles from 120° and/or a non-planar linker ring structure (Fig. 7B). We thus conclude that the structural fluctuations of the linker are the main reason for the mixed CT–LE nature of T1 in the studied emitters. These conclusions support and explain the mechanism of our previous hypotheses on SESM and DESM, coinciding with the other approaches on the TADF mechanism based on the existence of hybridized local charge-transfer (HLCT) states46 and T1–T2 vibronic coupling.47 Importantly, such mixed NTOs of T1 involve the electronic density localized on the chlorine atom, enabling the HAE on the T1–S1 transition.
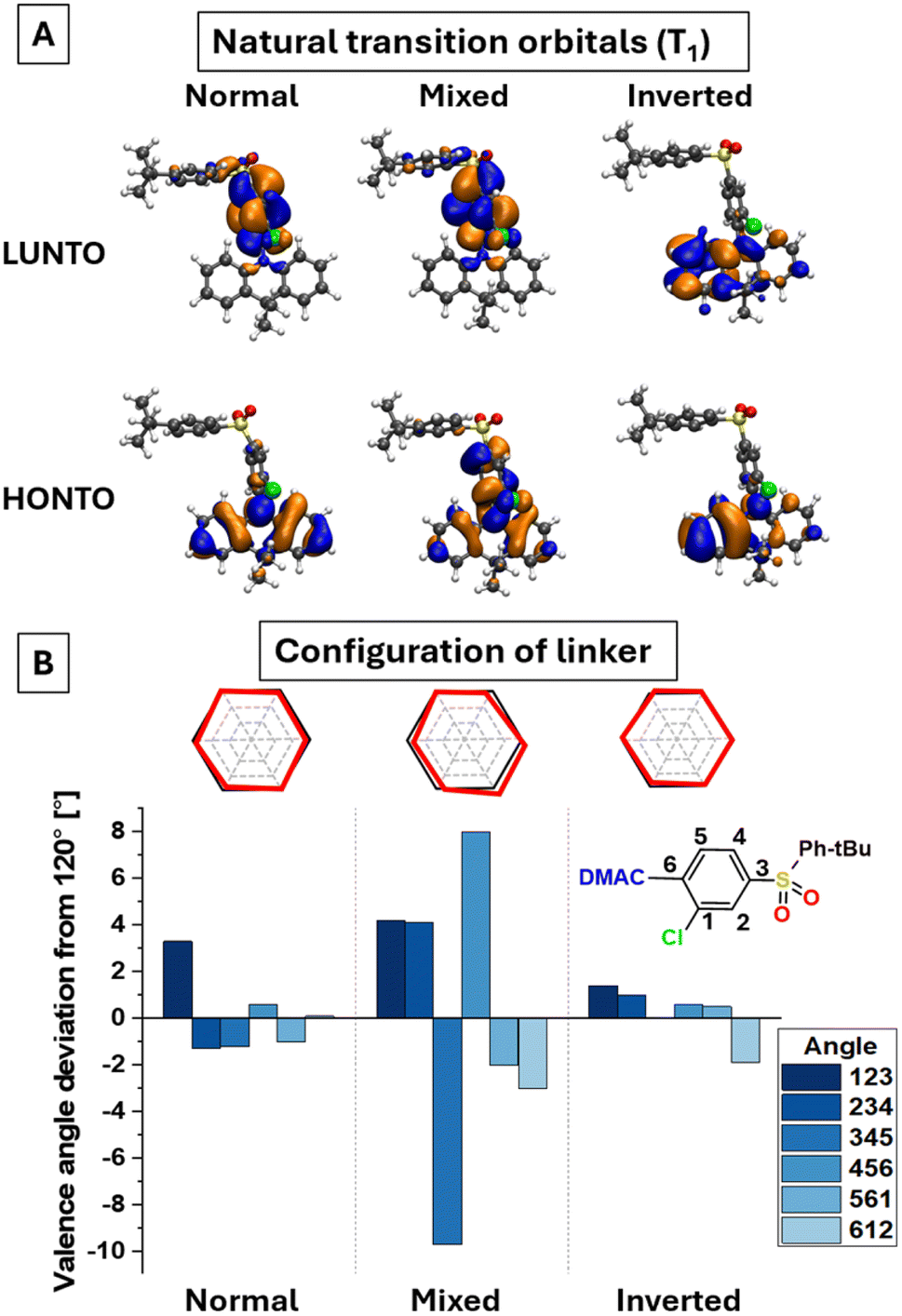 | ||
| Fig. 7 Excited-state mixing: correlation between electronic and geometric parameters in DMAC-Cl-DPStB. Changes in NTO (A), valence angles and conformation of the linker ring (B) among representative species in the modelled molecular ensemble. | ||
These combined factors result in a distinct trend in the rISC rates calculated according to eqn (1): 4.2 × 104 s−1 (R = H) 6.4 × 104 s−1 (R = F), 9.8 × 105 s−1 (R = Cl), and 7.2 × 106 s−1 (R = Br). While the absolute values are influenced by the model assumptions – such as the choice of approximate electronic structure methods, neglected environmental effects, and inherent model approximations – the observed trend strongly aligns with experimental data (Table 1). These results clearly emphasize the dominant role of the aforementioned structural and electronic parameters that narrow the distribution of ΔEST, which impacts the rISC rate exponentially. The role of SOC via quadratic dependence (eqn (1)) cannot be overlooked. In light of experimental results, the broader SOC distribution in DMAC-Cl-DPStB emerged as a key factor, allowing it to outperform DMAC-F-DPStB in ZNX and even DPEPO films, despite a lower activation energy in the latter (see above). Finally, we note that calculated reorganization energies, being 0.267 eV, 0.269 eV, 0.217 eV, and 0.224 eV for R = H, F, Cl, and Br, respectively, did not appear to be the dominant factor driving the rISC rate trends.
In addition to supporting the experimental findings, molecular modeling highlights that among other halogen substituents, Cl achieves a ‘golden mean’ for rISC enhancement by optimally balancing the reduction of ΔEST and noticeable SOC increase. In DMAC-Cl-DPStB, a moderate HAE enhances the interaction between triplet and singlet states through the 3LE character. Our analysis suggests that the relatively low rates calculated using eqn (1) are augmented by this interaction, as the 3LE state facilitates stronger spin-vibronic coupling, thereby improving rISC efficiency.
The low PLQY of DMAC-Br-DPStB at room temperature, despite its high SOC and relatively small ΔEST, hinders its suitability for light-emitting applications. This limitation is likely a result of efficient nonradiative deactivation, driven by strong coupling between the triplet and ground states. This assumption is supported by the M06-2X-predicted SOC values for T1 → S0, which are significantly higher for the Br derivative (42.6 cm−1) compared to Cl (9.21 cm−1), F (3.56 cm−1), and H (2.44 cm−1).
Finally, it should be noted that calculations reveal the presence of an additional bent conformer alongside the twisted conformer. The bent conformer exhibits significantly lower rISC rates due to larger ΔEST values, rendering it TADF inactive. While the bent structure is nearly isoenergetic with the twisted conformer in a vacuum, this difference is within the accuracy margin of computational methods. As a result, both conformers may coexist under experimental conditions, though as discussed above, the emission from the bent form is present only in trace amounts. Its limited contribution to TADF further supports the dominance of the twisted conformer in the observed photophysical behavior. For detailed discussion on the calculation results for the bent isomer, see the ESI.†
Conclusions
This study highlights the nuanced interplay between structural design and photophysical properties in developing efficient blue TADF emitters. By systematically introducing halogen substituents at the ortho position of the linker in DMAC-DPS derivatives, we demonstrate how judicious molecular modifications can address the inherent “low ΔEST = low SOC” tradeoff in donor–acceptor systems. Precisely targeted heavy-atom substitution emerges as a pivotal factor for optimizing TADF performance, enabling simultaneous reduction of activation energy and enhancement of spin–orbit coupling.Fluorine substitution stabilizes charge-transfer states, achieving the lowest ΔEST and Ea values among the derivatives, particularly in polar media. However, this stabilization, while beneficial for reducing nonradiative decay, limits the enhancement of SOC and thus the rISC rate. Chlorine substitution, in contrast, achieves a balance between reducing Ea and increasing SOC, making DMAC-Cl-DPStB the most efficient emitter in terms of rISC rates and overall TADF performance. Bromine substitution, while offering the highest SOC due to its atomic number, is accompanied by increased nonradiative deactivation, limiting its practical application under standard conditions.
Temperature-dependent studies revealed the crucial role of the medium's polarity and rigidity in modulating the interplay between CT and localized excited LE states. In a nonpolar environment, substituent size predominantly dictates Ea trends, while in polar media, halogen electronegativity further differentiates CT stabilization. Importantly, the participation of the 3LE state, as influenced by halogen electronic density, was shown to be a key driver of SOC and rISC efficiency, particularly in the chlorine and bromine derivatives.
The combination of experimental and computational insights underscores the potential of heavy-atom ortho-substitution as a strategy for improving TADF emitters. By modulating both steric and electronic effects, halogen atoms not only reduce ΔESTvia reduced θ-rotational freedom but also enhance SOC through 3LE state contributions enabled by static excited-state mixing. This dual benefit enables efficient triplet harvesting while maintaining blue emission with high photoluminescence quantum yields.
A detailed molecular dynamics analysis of DMAC-Cl-DPStB revealed that linker aryl ring vibrations induce structural fluctuations, which in turn activate the mixing of CT and LE states. This mixing plays a crucial role in enabling SOC and the HAE, shedding light on the role of vibronic effects in these phenomena.
The findings of this study provide a comprehensive framework for overcoming the intrinsic trade-offs in blue TADF emitter design. Hopefully, the demonstrated success of DMAC-Cl-DPStB in achieving high rISC rates with stable CT and LE state interplay offers a promising direction for future developments in blue TADF materials, bringing us closer to the realization of efficient and stable all-organic OLED technologies. As a key drawback, DMAC-Cl-DPStB exhibits a small but detectable presence of the bent-DMAC isomer, which is TADF-inactive due to an excessively large ΔEST. Therefore, future molecular design strategies should consider molecular flexibility to maximize SOC while minimizing nonradiative losses and also aiming to eliminate the inactive isomer for advanced OLED applications.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
I. E. S. – conceptualization, supervision, synthesis, methodology, writing. V. I. – investigation (spectroscopic analysis and QM calculations), data curation, formal analysis, visualization, writing (original draft). A. P. – investigation (QM and MM calculations), data curation, formal analysis, visualization, writing. O. C. – investigation (synthesis), data curation. M. M. – methodology, visualization, writing (review & editing).Conflicts of interest
The patent application P.448129, submitted to the Polish Patent Office, concerns the structure, synthesis, and photophysical properties of the compounds discussed herein.Acknowledgements
This work was supported by the National Science Centre, Poland, under the Sonata 16 project no. UMO-2020/39/D/ST5/03094. Quantum chemical calculations were performed on the computers of the Wroclaw Centre for Networking and Supercomputing (WCSS), Poland. A. P. acknowledges support from the Croatian Science Foundation (HRZZ), grant no. IP-2020-02-7262.References
- K. Stavrou, A. Danos, T. Hama, T. Hatakeyama and A. Monkman, Hot Vibrational States in a High-Performance Multiple Resonance Emitter and the Effect of Excimer Quenching on Organic Light-Emitting Diodes, ACS Appl. Mater. Interfaces, 2021, 13(7), 8643–8655 CrossRef CAS PubMed.
- T. Hatakeyama, K. Shiren, K. Nakajima, S. Nomura, S. Nakatsuka, K. Kinoshita, J. Ni, Y. Ono and T. Ikuta, Ultrapure Blue Thermally Activated Delayed Fluorescence Molecules: Efficient HOMO-LUMO Separation by the Multiple Resonance Effect, Adv. Mater., 2016, 28, 2777–2781 CrossRef CAS PubMed.
- X.-F. Luo, X. Xiao and Y.-X. Zheng, Recent progress in multi-resonance thermally activated delayed fluorescence emitters with an efficient reverse intersystem crossing process, Chem. Commun., 2024, 60, 1089–1099 RSC.
- X. Wu, B.-K. Su, D.-G. Chen, D. Liu, C.-C. Wu, Z.-X. Huang, T.-C. Lin, C.-H. Wu, M. Zhu, E. Y. Li, W.-Y. Hung, W. Zhu and P.-T. Chou, The role of host–guest interactions in organic emitters employing MR-TADF, Nat. Photonics, 2021, 15, 780–786 CrossRef CAS.
- J. Wang, P. Zou, L. Chen, Z. Bai, H. Liu, W.-C. Chen, Y. Hou, B. Z. Tang and Z. Zhao, Promising interlayer sensitization strategy for the construction of high-performance blue hyperfluorescence OLEDs, Light Sci. Appl., 2024, 13, 139 CrossRef CAS PubMed.
- N. Aizawa, Y.-J. Pu, Y. Harabuchi, A. Nihonyanagi, R. Ibuka, H. Inuzuka, B. Dhara, Y. Koyama, K.-I. Nakayama, S. Maeda, F. Araoka and D. Miyajima, Delayed fluorescence from inverted singlet and triplet excited states, Nature, 2022, 609, 502–506 CrossRef CAS PubMed.
- J. Ehrmaier, E. J. Rabe, S. R. Pristash, K. L. Corp, C. W. Schlenker, A. L. Sobolewski and W. Domcke, Singlet-triplet inversion in heptazine and in polymeric carbon nitrides, J. Phys. Chem. A, 2019, 123, 38 Search PubMed.
- P. Data, P. Pander, M. Okazaki, Y. Takeda, S. Minakata and A. P. Monkman, Dibenzo[a,j]phenazine-Cored Donor–Acceptor–Donor Compounds as Green-to-Red/NIR Thermally Activated Delayed Fluorescence Organic Light Emitters, Angew. Chem., Int. Ed., 2016, 55, 5739–5744 CrossRef CAS PubMed.
- X. Li, G. Baryshnikov, C. Deng, X. Bao, B. Wu, Y. Zhou, H. Ågren and L. Zhu, A three-dimensional ratiometric sensing strategy on unimolecular fluorescence–thermally activated delayed fluorescence dual emission, Nat. Commun., 2019, 10, 731 CrossRef CAS PubMed.
- F. B. Dias, Kinetics of thermal-assisted delayed fluorescence in blue organic emitters with large singlet-triplet energy gap, Philos. Trans. R. Soc., A, 2015, 373, 20140447 CrossRef PubMed.
- B. Yurash, H. Nakanotani, Y. Olivier, D. Beljonne, C. Adachi and T.-Q. Nguyen, Photoluminescence Quenching Probes Spin Conversion and Exciton Dynamics in Thermally Activated Delayed Fluorescence Materials, Adv. Mater., 2019, 10, 3646 Search PubMed.
- V. Ievtukhov, M. Mońka, O. Ciupak, I. Bylińska, P. Bojarski, K. Krzymiński and I. E. Serdiuk, Experimental Evidence of the Excited-State Mixing in the Blue Emitter for Organic Light-Emitting Diodes, J. Mater. Chem. C, 2023, 13, 68–80 RSC.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Highly Efficient Organic Light-Emitting Diodes from Delayed Fluorescence, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- T. Huang, Q. Wang, H. Zhang, Y. Xin, Y. Zhang, X. Chen, D. Zhang and L. Duan, Delocalizing Electron Distribution in Thermally Activated Delayed Fluorophors for High-Efficiency and Long-Lifetime Blue Electroluminescence, Nat. Mater., 2024, 23, 1523–1530 CrossRef CAS PubMed.
- C.-Y. Chan, M. Tanaka, H. Nakanotani and C. Adachi, Efficient and Stable Sky-Blue Delayed Fluorescence Organic Light-Emitting Diodes with CIEy Below 0.4, Nat. Commun., 2018, 9, 5036 CrossRef PubMed.
- J. Luo, S. Gong, T. Zhang, C. Zhong, G. Xie, Z.-H. Lu and C. Yang, Rational Design of Isophthalonitrile-Based Thermally Activated Delayed Fluorescence Emitters for OLEDs with High Efficiency and Slow Efficiency Roll-Off, Dyes Pigm., 2017, 147, 350–356 CrossRef CAS.
- L.-S. Cui, A. J. Gillett, S.-F. Zhang, H. Ye, Y. Liu, X.-K. Chen, Z.-S. Lin, E. W. Evans, W. K. Myers, T. K. Ronson, H. Nakanotani, S. Reineke, J.-L. Bredas, C. Adachi and R. H. Friend, Fast spin-flip enables efficient and stable organic electroluminescence from charge-transfer states, Nat. Photonics, 2020, 14, 636–642 CrossRef CAS.
- I. E. Serdiuk, M. Mońka, K. Kozakiewicz, B. Liberek, P. Bojarski and S. Y. Park, Vibrationally Assisted Direct Intersystem Crossing between the Same Charge-Transfer States for Thermally Activated Delayed Fluorescence: Analysis by Marcus–Hush Theory Including Reorganization Energy, J. Phys. Chem. B, 2021, 125(10), 2696–2706 CrossRef CAS PubMed.
- C. H. Ryoo, J. Han, J.-H. Yang, K. Yang, I. Cho, S. Jung, S. Kim, H. Jeong, C. Lee, J. E. Kwon, I. E. Serdiuk and S. Y. Park, Systematic Substituent Control in Blue Thermally Activated Delayed Fluorescence (TADF) Emitters: Unraveling the Role of Direct Intersystem Crossing between the Same Charge-Transfer States, Adv. Opt. Mater., 2022, 10, 2201622 CrossRef CAS.
- H. Li, J. Li, D. Liu and Y. Mei, Mechanism Evolution from Normal Fluorescence to Thermally Activated Delayed Fluorescence and Color Tuning over Visible Light Range: Effect of Intramolecular Charge Transfer Strength, Dyes Pigm., 2020, 183, 108732 Search PubMed.
- L.-S. Cui, H. Nomura, Y. Geng, J. U. Kim, H. Nakanotani and C. Adachi, Controlling Singlet-Triplet Energy Splitting for Deep-Blue Thermally Activated Delayed Fluorescence Emitters, Angew. Chem., Int. Ed., 2017, 56(6), 1571–1575 Search PubMed.
- Y. Li, J.-J. Liang, H.-C. Li, L.-S. Cui, M.-K. Fung, S. Barlow, S. R. Marder, C. Adachi, Z.-Q. Jiang and L.-S. Liao, The Role of Fluorine-Substitution on the π-Bridge in Constructing Effective Thermally Activated Delayed Fluorescence Molecules, J. Mater. Chem. C, 2018, 6(20), 5561–5568 Search PubMed.
- K. Zheng, F. Ni, Z. Chen, C. Zhong and C. Yang, Polymorph-Dependent Thermally Activated Delayed Fluorescence Emitters: Understanding TADF from a Perspective of Aggregation State, Angew. Chem., Int. Ed., 2020, 59(25), 9972–9976 CrossRef CAS PubMed.
- P. Stachelek, J. S. Ward, P. L. dos Santos, A. Danos, M. Colella, N. Haase, S. J. Raynes, A. S. Batsanov, M. R. Bryce and A. P. Monkman, Molecular Design Strategies for Color Tuning of Blue TADF Emitters, ACS Appl. Mater. Interfaces, 2019, 11(30), 27126–27136 CrossRef PubMed.
- H. Xia, Y. Tang, Y. Zhang, F. Ni, Y. Qiu, C.-W. Huang, C.-C. Wu and C. Yang, Highly Efficient Blue Electroluminescence Based on TADF Emitters with Spiroacridine Donors: Methyl Group Effect on Photophysical Properties, J. Mater. Chem. C, 2022, 10(12), 4478–4488 RSC.
- A. A. Kubicki, P. Bojarski, M. Grinberg, M. Sadownik and B. Kukliński, Time-resolved streak camera system with solid state laser and optical parametric generator in different spectroscopic applications, Opt. Commun., 2006, 269, 275–280 CrossRef.
- M. Wojdyr, A General-Purpose Peak Fitting Program, J. Appl. Crystallogr., 2010, 43, 1126–1128 CrossRef CAS.
- Origin, Origin 7.5, OriginLab Corp., Northampton, MA, 2003.
- J. K. Labanowski and J. W. Andzelm, Density Functional Methods in Chemistry, New York, 1991 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, et al., Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- A. D. Becke, A new mixing of Hartree-Fock and local density-functional theories, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- F. Neese, The ORCA Program System, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- B. de Souza, G. Farias, F. Neese and R. Izsák, Predicting Phosphorescence Rates of Light Organic Molecules Using Time-Dependent Density Functional Theory and the Path Integral Approach to Dynamics, J. Chem. Theory Comput., 2019, 15(3), 1896–1904 CrossRef CAS PubMed.
- P. Dev, S. Agrawal and N. J. English, Determining the Appropriate Exchange-Correlation Functional for Time-Dependent Density Functional Theory Studies of Charge-Transfer Excitations in Organic Dyes, J. Chem. Phys., 2012, 136(22), 224301 CrossRef PubMed.
- M. Mońka, I. E. Serdiuk, K. Kozakiewicz, E. Hoffman, J. Szumilas, A. Kubicki, S. Y. Park and P. Bojarski, Understanding the Internal Heavy-Atom Effect on Thermally Activated Delayed Fluorescence: Application of Arrhenius and Marcus Theories for Spin–Orbit Coupling Analysis, J. Mater. Chem. C, 2022, 10(23), 7925–7934 RSC.
- L. E. de Sousa and P. de Silva, Unified Framework for Photophysical Rate Calculations in TADF Molecules, J. Chem. Theory Comput., 2021, 17(9), 5815–5823 CrossRef PubMed.
- M. Ceriotti, M. Parrinello, T. E. Markland and D. E. Manolopoulos, Efficient stochastic thermostatting of path integral molecular dynamics, Phys. Rev. Lett., 2009, 103(3), 030603 CrossRef PubMed.
- A. Prlj, D. Hollas and B. F. E. Curchod, Deciphering the Influence of Ground-State Distributions on the Calculation of Photolysis Observables, J. Phys. Chem. A, 2023, 127(35), 7056–7068 CrossRef PubMed.
- GLE4MD, Generalized Langevin Equation for Molecular Dynamics, Accessed December 26, 2024, https://gle4md.org/.
- PHOTOX, ABIN: A Computational Tool for the Calculation of Photophysical Properties, Accessed December 26, 2024, https://github.com/PHOTOX/ABIN.
- Y. J. Franzke, C. Holzer, J. H. Andersen, T. Begušić, F. Bruder, S. Coriani, F. Della Sala, E. Fabiano, D. A. Fedotov and S. Fürst, et al., TURBOMOLE: Today and Tomorrow, J. Chem. Theory Comput., 2023, 19(20), 7202–7220 CrossRef PubMed.
- M. Charton, Nature of the ortho effect. V. Ortho-substituent constants, J. Am. Chem. Soc., 1969, 91(24), 6649–6654 CrossRef CAS.
- K. Stavrou, L. G. Franca, T. Böhmer, L. M. Duben, C. M. Marian and A. P. Monkman, Unexpected Quasi-Axial Conformer in Thermally Activated Delayed Fluorescence DMAC-TRZ, Pushing Green OLEDs to Blue, Adv. Funct. Mater., 2023, 33, 2300910 CrossRef CAS.
- D. K. A. P. Huu, S. Saseendran, R. Dhali, L. G. Franca, K. Stavrou, A. Monkman and A. Painelli, Thermally Activated Delayed Fluorescence: Polarity, Rigidity, and Disorder in Condensed Phases, J. Am. Chem. Soc., 2022, 144(33), 15211–15222 CrossRef PubMed.
- F. Plasser, TheoDORE: A Toolbox for a Detailed and Automated Analysis of Electronic Excited State Computations, J. Chem. Phys., 2020, 152(8), 084108 CrossRef CAS PubMed.
- W. Li, Y. Pan, R. Xiao, Q. Peng, S. Zhang, D. Ma, F. Li, F. Shen, Y. Wang, B. Yang and Y. Ma, Employing ∼100% Excitons in OLEDs by Utilizing a Fluorescent Molecule with Hybridized Local and Charge-Transfer Excited State, Adv. Funct. Mater., 2014, 24(11), 1609–1614 CrossRef CAS.
- J. Gibson, A. P. Monkman and T. J. Penfold, Understanding the Mechanism of Thermally Activated Delayed Fluorescence, ChemPhysChem, 2016, 17(19), 2956–2961 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional procedure details, results of analyses, experimental and computational investigations, and discussions on the properties of emitters in solutions and on the role of bent isomers. See DOI: https://doi.org/10.1039/d5sc00954e |
| This journal is © The Royal Society of Chemistry 2025 |