
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAchieving highly efficient carbon radical-mediated cross-coupling reaction in a confined radical microenvironment within a metal–organic framework†
Ying-Lin
Li
a,
Ning
Li
*a,
Zhi-Bin
Mei
a,
Jun-Rong
Li
a,
Su-Juan
Yao
a,
Fei
Yu
 b,
Shun-Li
Li
a,
Jiao-Min
Lin
*a,
Jiang
Liu
*a and
Ya-Qian
Lan
a
b,
Shun-Li
Li
a,
Jiao-Min
Lin
*a,
Jiang
Liu
*a and
Ya-Qian
Lan
a
aNational and Local Joint Engineering Research Center of MPTES in High Energy and Safety LIBs, Engineering Research Center of MTEES (Ministry of Education), Key Lab. of ETESPG (GHEI), School of Chemistry, South China Normal University, Guangzhou, 510006, China. E-mail: ningl0314@163.com; linjm@m.scnu.edu.cn; liuj0828@m.scnu.edu.cn; Web: https://www.yqlangroup.com
bJiangsu Collaborative Innovation Centre of Biomedical Functional Materials, Jiangsu Key Laboratory of New Power Batteries, School of Chemistry and Materials Science, Nanjing Normal University, Nanjing, 210023, China
First published on 20th May 2025
Abstract
It has been well-demonstrated that the combination of photosensitive (PS), hydrogen atom transfer (HAT) and single electron transfer (SET) processes can achieve efficient radical-mediated organic synthesis, but such reaction systems are usually homogeneous, requiring additional HAT agents and can only activate one substrate. Here, we constructed two crystalline porous materials, Zr/Hf-NDI, which possess excellent light absorbing capacity and a confined radical microenvironment, making them able to integrate PS, HAT, and SET processes to simultaneously activate two substrates. Thus, as heterogeneous photocatalysts, they exhibited excellent catalytic performance for the carbon radical-mediated cross-coupling reaction between alcohols and o-phenylenediamine (OPD) to synthesize benzimidazoles (yield > 99%). More importantly, they displayed very good substrate compatibility, especially for OPD substrates with electron-withdrawing groups, even surpassing those of noble metal catalysts. In situ characterizations combined with theoretical calculations showed that the high activity of these catalysts arose from: (i) the metal-oxo clusters and radical NDI˙− ligands can form hydrogen bonding traction activation for the alcohol substrate, and thus facilitate it to generate key intermediate α-carbon radical through a HAT process; (ii) the OPD substrate, acting as an electron donor, forms strong D–A interaction with the NDI ligand and activates the NDI and itself into radicals NDI˙− and OPD˙+, respectively, via an SET process, further promoting the reaction. To the best of our knowledge, this is the best performing crystalline porous catalyst for photocatalytic carbon radical-induced benzimidazole synthesis.
Photocatalytic C–H bond activation to form carbon radicals for cross-coupling reactions has attracted tremendous attention due to its eco-friendly nature, low energy consumption, high atom-economy, and readily available starting materials.1 Nevertheless, activating C–H bond is challenging because of its high bond energy, especially the sp3 C–H bond.2 The marriage of homogeneous photocatalysts and HAT catalysts represents one of the most efficient and versatile reaction systems for C–H bond activation (Scheme 1a).3 In this system, the photocatalysts are transformed into their excited states through a photosensitization (PS) process, while the HAT catalysts are transformed into highly electrophilic heteroatom-centered radicals driven by the excited photocatalysts through a single electron transfer (SET) process and then abstract hydrogen atoms from the C–H bond to generate alkyl radicals for the subsequent cross-coupling reactions.4 Currently, a diversity of molecular photocatalysts5 (e.g. Ru-, Rh- and Ir-metal complexes) and HAT catalysts6 (e.g. quinone, peroxides and Ar-SH) have been developed to accommodate various types of C–H bond activation. Nevertheless, because the molecular photocatalysts and the additional HAT catalysts are separated and serve as homogeneous catalysts, they inevitably suffer from poor charge transfer, low recyclability and difficulty in product separation.7 Besides, in this system, the SET process is wasted to activate the HAT catalyst. In fact, the SET process can also be utilized to activate the reaction substrates to generate radicals.8 In this case, by combining the carbon radical formed by the HAT process, the cross-coupling reactions may be easier to carry out. Furthermore, if both reaction substrates can be activated, better substrate compatibility may be achieved for the cross-coupling reactions. In this context, the construction of heterogeneous photocatalysts with both HAT and SET functions to realize more efficient radical-mediated cross-coupling reactions is highly important and desired, yet it still remains a big challenge.9
 | ||
| Scheme 1 Schematic illustration of the photocatalytic C–H activation methods. (a) PS, SET and HAT processes occurring in a homogeneous system for C–H activation in previous work. (b) PS, SET and HAT processes occurring in a heterogeneous system for C–H activation in this work. | ||
In principle, heterogeneous photocatalysts that can integrate PS, HAT, and SET processes can not only avoid the use of additional HAT mediators, but also have better charge transfer effect and the ability to activate two types of reaction substrate through SET and HAT processes, respectively and thus have higher photocatalytic performance and substrate compatibility (Scheme 1b). Theoretically, such heterogeneous photocatalysts should have the following structural and functional properties: (i) containing a H atom binding site/group (e.g. quinone, sulfhydryl, or pyridine N-oxide groups) to fulfill the HAT catalytic function;10 (ii) having suitable redox potential and/or an electron-deficient or electron rich structure and thus being able to accept an electron from the substrates or give out electrons to the substrates through the SET process;11 (iii) possessing a unique porous structure and multiple adsorption sites to facilitate the adsorption, activation, and confined reaction of the substrates;12 (iv) having excellent chemical stability, light adsorption, and photoinduced electron–hole separation efficiencies for efficient solar energy utilization and catalytic recycling.13 In addition, a well-defined structure of the photocatalyst is also crucial as it can provide a platform for studying the reaction mechanisms and structure–activity relationships and for further optimizing and designing efficient photocatalysts.
Based on the above considerations, we designed and synthesized two new MOFs, Zr/Hf-NDI, which are constructed from a binuclear Zr/Hf-oxo cluster and naphthalenediimide (NDI) ligand. In these MOFs, the NDI ligand with multiple carbonyl groups and an electron-deficient structure can accept electrons (through an SET process) to form a free radical anion (NDI˙−) and simultaneously possess better HAT activity of the excited quinones.14 Therefore, by combining with host–substrate interactions and the pore confined effect, these MOFs have great potential to serve as photocatalysts for the carbon radical-mediated organic reactions. Based on this, we chose the cross-coupling reaction of alcohols and o-phenylenediamine (OPD) to form benzimidazoles as a model reaction, which is a carbon radical-mediated organic and value-added reaction that uses renewable biomass sources to prepare benzimidazole drugs in one step, but currently, it primarily relies on noble metal catalysts.15 In this reaction, the adsorption and activation of the alcohol molecule, as well as the abstraction of its α-C–H atom (through a HAT process) to form an α-carbon radical can be achieved by the synergy between the Zr/Hf-oxo cluster and NDI˙− ligand through hydrogen bonding traction activation. Besides, the OPD substrate forms donor–acceptor (D–A) interactions with the NDI ligand and serves as an electron donor to excite it. While doing so, OPD is converted into a radical species, further promoting this reaction (Fig. 1a). As expected, catalytic experiments revealed that these radical Zr/Hf-NDI˙− have excellent photocatalytic performance for this carbon radical-mediated reaction, giving more than 99% substrate conversion and 99% product selectivity within 12 h of irradiation. More importantly, they showed excellent compatibility with various substrates, including OPD derivatives bearing electron-withdrawing groups—substrates that are usually challenging for noble metal catalysts. To the best of our knowledge, this is the first report of an efficient non-noble metal-based MOF photocatalyst for the carbon radical-mediated cross-coupling reaction between alcohols and OPD to synthesize benzimidazoles. In situ characterizations and density functional calculations (DFT) revealed that the radical, SET, and HAT properties, combined with the D–A interactions and hydrogen bonding traction activation, endow these catalysts with excellent photocatalytic performance. This work represents the first example of the rational design of radical MOFs that can integrate PS, HAT, and SET processes for an efficient photocatalytic carbon radical-mediated organic reaction.
 | ||
| Fig. 1 Design concept and crystal structure of the photocatalyst. (a) Schematic illustration of the desired photocatalyst with a confined microenvironment and SET and HAT functions for activating OPD and alcohol substrates to form the corresponding radical species for the subsequent cross-coupling reaction. (b) The three-dimensional porous structure of Zr-NDI, which possesses large 1D rectangular channels along the c-axis. (c) The microenvironment of the 1D channel, which possess potential D–A interaction and hydrogen bond binding sites for the capture of electron rich and alcohol substrates. | ||
Single-crystal X-ray diffraction analysis revealed that Zr-NDI and Hf-NDI are isomorphic porous materials (Table S1†). Zr-NDI was taken as an example to describe their crystal structures. Zr-NDI was built up by 6-connected binuclear Zr2 units and 2-connected NDI2− ligands, featuring a three-dimensional (3D) porous structure and pcu topology (Fig. 1b and S1–S4†). Although some of its pores were occupied by uncoordinated H2NDI ligands that form strong hydrogen bonds with the framework (Fig. S5†), Zr-NDI possessed large 1D rectangular channels (2.4 × 10.4 Å) along the c-axis and a solvent accessible void of about 21.6% (Fig. 1b). It is interesting that in Zr-NDI, the aromatic surfaces of the electron-deficient NDI2−/H2NDI ligands are parallel to the channel, which makes them easily form D–A interactions with other electron rich guest/substrate molecules (Fig. 1c). Besides, there are a large number of uncoordinated carbonyl groups from the NDI2−/H2NDI ligands on the channel surface, which can also serve as hydrogen bonding sites for trapping guest/substrate molecules, e.g. alcohols (Fig. 1c). Thus, Zr-NDI possesses potential binding sites for both aromatic and alcohol substrates, which would benefit their adsorption, activation and coupling reaction. The structure of Hf-NDI is similar to that of Zr-NDI and is shown in Fig. S6 and S7.†
The powder X-ray diffraction (PXRD) patterns of the as-synthesized crystal samples of Zr-NDI and Hf-NDI were consistent with the simulated ones derived from the SCXRD data, demonstrating their high crystallinity (Fig. S8 and S9†). The Fourier transform infrared spectroscopy (FT-IR) spectra showed that these two compounds contain similar components and functional groups (Fig. S10 and S11†). Thermogravimetric analysis (TGA) revealed that their skeletons can be stabilized up to 400 °C (Fig. S12 and S13†). To test their chemical stability, samples of Zr-NDI and Hf-NDI were immersed in different solvents for 24 h, including N,N-dimethylformamide (DMF), N,N-diethylformamide (DEF), iPrOH, CH3OH, CH3CH2OH, CH3CN and H2O. The results revealed that both Zr-NDI and Hf-NDI have excellent chemical stability in these solvents (Fig. S14 and S15†). The Brunauer–Emmett–Teller (BET) surface area of Zr-NDI and Hf-NDI was estimated to be 992.6 and 949.6 m2 g−1 by N2 sorption isotherm at 77 K, respectively, which indicated that these MOFs have permanent porosity (Fig. S16 and S17†). The excellent chemical stability and high BET surface area laid the foundation for their application in photocatalytic reactions.
Given that NDI is a typical electron-deficient molecule16 capable of accepting electrons from electron rich molecules to form a free radical anion NDI˙−, we propose that Zr-NDI and Hf-NDI may also exhibit radical character. Taking Zr-NDI as a representative object, in the presence of triethylamine (TEA) vapor as an electron donor, the color of the Zr-NDI sample gradually deepened from red to black (Fig. 2a and S18†), suggesting the formation of free radical Zr-NDI˙−. Due to the visible color change, the solid-state ultraviolet-visible (UV-Vis) diffuse reflection spectrum was adopted to evaluate the photosensitivity. As shown in Fig. 2a, compared with the ligand H2NDI, which exhibited light absorption in the ultraviolet region, the light absorption of Zr-NDI expands to 600 nm. Moreover, for the radical Zr-NDI˙−, the light absorption can almost cover the entire visible light region, suggesting that the formation of radicals can endow the sample with excellent light absorption capacity. Such significant enhancement of light absorption of Zr-NDI˙− implies the charge transfer interaction between the electron-donating guest molecule (the TEA) and the electron-accepting NDI molecule. In addition, the appearance of a new absorption band at around 650 nm that corresponds to the characteristic absorption of NDI˙− also suggests the formation of radical Zr-NDI˙−. To further verify this, electron paramagnetic resonance (EPR) analysis was performed. As shown in Fig. 2b, a strong signal with the g-factor of 2.004 was detected for the sample of Zr-NDI˙−, confirming the formation of the radical anion.17 Moreover, we also found that other amines, such as ammonia, ethylamine, and ethylenediamine, can also be used to induce the generation of free radicals of Zr-NDI (Fig. S19–S22†). Nevertheless, the light adsorption of Zr-NDI activated by ammonia, ethylamine or ethylenediamine is weaker than that activated by TEA. Thus, we used TEA to activate Zr-NDI for the subsequent catalytic experiments. Notably, the PXRD diffraction patterns of Zr-NDI and Zr-NDI˙− were highly similar, demonstrating that the formation of radical anions did not affect the crystal structure (Fig. S8†). For Hf-NDI, similar radical properties can also be observed (Fig. S23 and S24†).
 | ||
| Fig. 2 Photophysical characterization of Zr-NDI and Zr-NDI˙−. (a) UV-Vis diffuse reflection spectrum of H2NDI, Zr-NDI and Zr-NDI˙−. (b) EPR spectra of solid radical Zr-NDI˙−. (c) Tauc plot of Zr-NDI and Zr-NDI˙− determined by the Kubelka–Munk formula from the original UV-Vis diffuse reflection spectrum. (d) Mott–Schottky plot measurement for Zr-NDI˙−. Inset: energy diagram of the HOMO and LUMO levels of Zr-NDI˙−. (e) Transient photocurrent responses of Zr-NDI and Zr-NDI˙− under Xe lamp irradiation. (f) EIS Nyquist plots of Zr-NDI and Zr-NDI˙−. | ||
According to the solid-state UV-Vis diffuse reflection spectra and the Kubelka–Munk function, the optical band gaps (Eg) of Zr-NDI˙−, Zr-NDI, Hf-NDI˙−, and Hf-NDI were estimated to be 1.53, 1.89, 1.54, and 1.85 eV (Fig. 2c and S25†), respectively. Mott–Schottky (MS) measurements were conducted to determine the lowest unoccupied molecular orbital (LUMO) energy level positions at frequencies of 500, 1000, and 1500 Hz. The results showed that the LUMOs of Zr-NDI˙−, Zr-NDI, Hf-NDI˙−, and Hf-NDI were −1.08, −1.16, −1.13, and −1.14 V (vs. NHE, pH = 7), respectively. Based on the band gap and the value of the LUMO, the highest orbital positions (HOMOs) of these compounds were calculated to be 0.45, 0.73, 0.41, and 0.71 V (Fig. 2d and S26–S28†), respectively. The transient photocurrent response tests showed that the activated MOFs, Zr-NDI˙− and Hf-NDI˙−, possessed a stronger transient photocurrent density under periodic light illumination, which is about 2 and 10 times higher than that of the original Zr-NDI and Hf-NDI, respectively. These results demonstrated that MOFs with free radicals can facilitate the separation and transfer of photogenerated carriers (Fig. 2e and S29†). Moreover, electrochemical impedance spectroscopy (EIS) revealed that Zr/Hf-NDI˙− exhibited smaller resistance, confirming that the introduction of delocalized electrons in the MOFs can contribute to a more rapid charge transfer process (Fig. 2f and S30†). On the whole, it seems that the stable free radical anion in the MOFs can significantly change their photophysical properties, including widening the light absorption, narrowing the band gaps, enhancing the photoresponsivity, reducing the electrical impedance, and promoting the photogenerated carrier separation. All of these properties may together contribute to the significant improvement of the photocatalytic performance.
Radical-induced cross-coupling of o-phenylenediamine (OPD) with alcohol is an attractive route for synthesizing benzimidazoles, owing to its high efficiency and high selectivity, but it usually requires noble metal (Pt, Pd, Au, etc.)-based catalysts.15b,18 Considering the excellent photophysical and radical properties and the potential SET and HAT effects, as well as the abundant adsorption and active sites of these MOFs, we thus investigated their photocatalytic performance for benzimidazole synthesis using OPD and ethanol as starting materials. The reaction was carried out using 10 mg Zr-NDI˙− as a catalyst, and 0.05 mmol OPD and 10 mL ethanol as raw materials, in an Ar atmosphere with Xe lamp (300 W) irradiation. After irradiation for 12 h, both the yield and selectivity of 2-methylbenzimidazole (2MBZ) can reach up to 99% (a small amount of hydrogen can be detected in the gas product), which suggests high catalytic activity of Zr-NDI˙− for this reaction (Fig. 3a). Screening experiments revealed that, without light or without Zr-NDI˙−, almost no 2MBZ or 11% yield of 2MBZ can be achieved, proving that both light irradiation and Zr-NDI˙− are indispensable for this reaction (Fig. 4a). In addition, when the Ar atmosphere was changed to air or oxygen, the yield of 2MBZ decreased to about 96% and 74%, respectively. In addition, reducing the amount of ethanol to 2 or 5 mL, the yield of 2MBZ decreased to 7% and 69%, respectively (Table S2†). Besides, the activation time of catalyst Zr-NDI˙− by TEA can also affect the catalytic activity. Specifically, the catalytic activity of Zr-NDI˙− increased with the increase of the activation time and the best performance was reached at 9 h, which was probably because the concentration of the free radical in the Zr-NDI˙− catalyst increased with the TEA activation time (Fig. S31 and S32†).
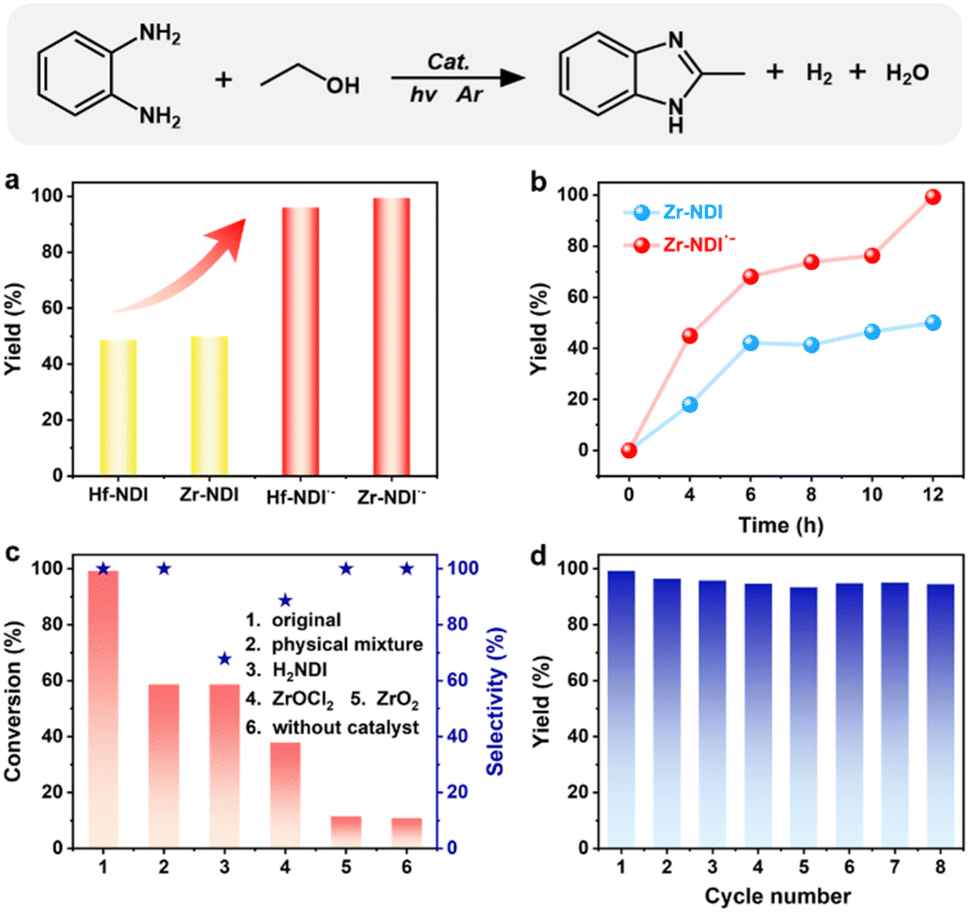 | ||
| Fig. 3 The photocatalytic performance of Zr-NDI and Zr-NDI˙− for the cross-coupling reaction of OPD and ethanol. (a) Yields of 2MBZ for Zr-NDI, Hf-NDI, Zr-NDI˙− and Hf-NDI˙− under the standard reaction conditions. (b) Time-dependent yields of 2MBZ for Zr-NDI and Zr-NDI˙−. (c) Conversion of OPD and selectivity of 2MBZ for various different catalysts under similar reaction conditions. (d) Cycling performance of Zr-NDI˙−. | ||
 | ||
| Fig. 4 The characterization of the reaction mechanism. (a) The photocatalytic performance of control experiments over Zr-NDI˙−. (b) EPR spectra of Zr-NDI˙− in the presence of DMPO as a trapping agent under light irradiation and in the dark. (c) HRMS spectra of the DMPO adduct to the ˙CH(CH3)OH intermediate under the standard conditions ([M + H+] C8H18O2N+: 160.2333, found: 160.1329). (d) In situ DRIFTS of the photocatalytic synthesis of 2MBZ using OPD and ethanol as reactants over Zr-NDI˙−. | ||
We then evaluated the photocatalytic performances of Hf-NDI˙−, Zr-NDI and Hf-NDI. The results revealed that when taking Hf-NDI˙− as a catalyst, the yield of 2MBZ can reach 96% in 12 h, which is similar to that of Zr-NDI˙−. Meanwhile, using Zr-NDI or Hf-NDI as catalysts, the yield of 2MBZ can only reach 50% and 49% (Fig. 3a), respectively, which is much lower than that of Zr-NDI˙− and Hf-NDI˙−. This suggests that MOFs with stable free radical properties show better photocatalytic performance under the analogical reaction conditions. To confirm this, the time-dependent 2MBZ yields of the Zr-NDI˙− and Zr-NDI catalysts were investigated. As shown in Fig. 3b, the 2MBZ yields of Zr-NDI˙− were about two times higher than that of Zr-NDI at different time points, around 4 to 12 h, confirming that radical Zr-NDI˙− indeed has higher catalytic activity than Zr-NDI. This is probably because the radical Zr-NDI˙− has better light adsorption and photoinduced electron–hole separation efficiencies, as well as a better HAT effect than that of the parent Zr-NDI. Considering that radical materials usually have a photothermal effect, and this may also be one of the factors responsible for the better photocatalytic performance, we thus investigated the photothermal effect of Zr-NDI and Zr-NDI˙−. As shown in Fig. S33 and S34,† the temperature difference (ΔT) of Zr-NDI˙− can reach 73.1 °C under irradiation for 500 s, while that of Zr-NDI can only reach 65.6 °C under the same conditions, suggesting that Zr-NDI˙− indeed has a better photothermal effect than Zr-NDI. In theory, the photothermal effect of the catalysts may also contribute to improving the photocatalytic performance, although the photocatalytic reaction is carried out at room temperature under circulating condensate water. Thus, we thought that the better photothermal effect of Zr-NDI˙− may also be one of the contributing factors for its better photocatalytic performance.
For comparison and to gain more insight into the catalytic mechanisms, the catalytic performances of the ligand H2NDI, ZrOCl2, ZrO2, and the physical mixture of ZrOCl2 and H2NDI were investigated. The results revealed that when using H2NDI as a catalyst, the conversion of OPD was only 59%, and the selectivity of the target product 2MBZ decreased to 68% due to the formation of the by-product, N-ethyl-2-methylbenzimidazole (NE2MBZ) (Fig. S35†). Meanwhile, for ZrOCl2 (as a homogeneous catalyst) and ZrO2, the conversion of OPD was 38% and 12% (Fig. 3c), respectively, although the selectivity for 2MBZ was high at 89% and 100%, respectively. These results suggest that both the ligand and metal contribute to this catalytic reaction, but neither alone can simultaneously achieve high substrate conversion and product selectivity. Conversely, when using a mixture of ZrOCl2 and H2NDI as a catalyst, the conversion of OPD was about 59% and the selectivity of 2MBZ was approaching 100%, which was better than that of ZrOCl2 and H2NDI alone, and comparable to that of Zr-NDI but still far inferior to that of Zr-NDI˙−. Note that for this mixture catalyst, the ZrOCl2 was dissolved in the solution during the photocatalytic reaction, suggesting that it should be a homogeneous catalyst. Nevertheless, it has better catalytic performance than that of ZrOCl2 and H2NDI alone, indicating that there was a synergistic effect between the metal centre and the ligand. Besides, the catalytic performance of Zr-NDI˙− was much better than that of this mixture catalyst, suggesting that the existence of free radicals indeed improved the photocatalytic performance and enhanced the reaction rate.
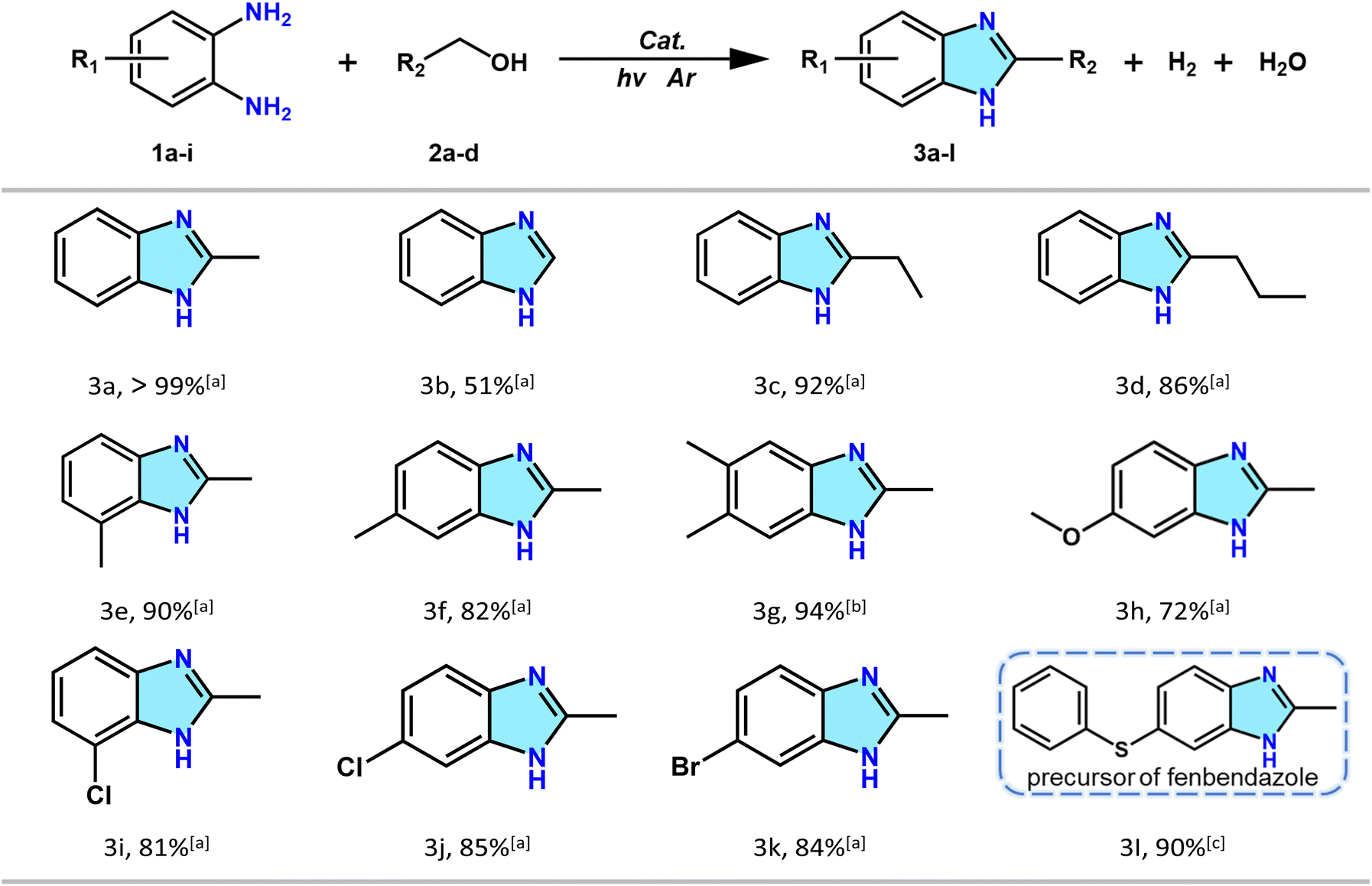
To explore the universality of catalyst Zr-NDI˙− for this reaction, different alcohol and diamine substrates were studied. As shown in Table 1, various alcohol substrates, including methanol, propanol, and butanol, can all react smoothly with OPD to give the corresponding benzimidazoles 3b, 3c and 3d with yields of 51%, 92%, and 86%, respectively. Note that for methanol, the yield of benzimidazole was relatively low. This was probably because the C–H bond of methanol is harder to activate, which was also observed in a previous report. We then investigated OPD substrates with electron-donating groups and electron withdrawing groups. When the OPD substrates contained electron-donating groups, such as –CH3 and –OCH3, considerable yields of the corresponding benzimidazoles were obtained (3e, 90%; 3f, 82%; 3g, 94%; 3h, 72%). Furthermore, when OPD substrates bearing electron-withdrawing groups, such as –Cl and –Br were used, the corresponding benzimidazole products were still obtained with yields up to 80% (3i, 81%; 3j, 85%; 3k, 84%), which surpasses previously reported results using noble metal catalysts (Tables S3 and S4†). Notably, it is difficult to obtain high benzimidazole yield for OPD substrates bearing electron-withdrawing groups in the presence of noble metal catalysts. This is probably because the noble metal catalysts are usually loaded on metal oxides, which, however, show weak adsorption capacity for OPD substrates bearing electron-withdrawing groups, resulting in low benzimidazole yields for these substrates. In contrast, in our system, the OPD substrates can form strong π–π stacking interactions with the rigid NDI ligands, making them easy to be adsorbed into the porous channels of the Zr-NDI˙− catalyst. Besides, the porous channels can provide a confined space to accelerate the collision between OPD and the alcohol substrates, thus resulting in a high benzimidazole yield for OPD substrates with both electron-donating and electron-withdrawing groups. To further demonstrate the potential application of this catalytic reaction, we tried to synthesize the precursor of fenbendazole, which is an effective and less toxic anthelmintic drug. The results revealed that the yield of the fenbendazole precursor (3l) can also reach 90% within 24 h, demonstrating the excellent photocatalytic performance of the Zr-NDI˙− catalyst and the great application potential of this catalytic system.
The PXRD patterns and FT-IR spectrum of Zr-NDI˙− before and after the photocatalytic reaction are nearly unchanged (Fig. S36–S38†), confirming its high integrity and heterogeneous catalytic nature. Besides, the UV-Vis diffuse reflection spectrum of Zr-NDI˙− after the reaction exhibited the characteristic absorption peak of NDI˙− at about 650 nm, which is consistent with that before the photocatalytic reaction, suggesting that its radical nature retained. This is also confirmed by the EPR spectrum of the Zr-NDI˙− sample after the reaction, which exhibited a characteristic EPR signal at g = 2.004. These results indicate that the Zr-NDI˙− catalyst is stable and its free radical nature can remain valid after the photocatalytic reaction (Fig. S39 and S40†). To further demonstrate the heterogeneous catalytic nature and catalytic stability of Zr-NDI˙−, recycling photocatalytic tests were carried out. The results showed that it maintained good catalytic efficiency with >95% yield throughout eight cycles (Fig. 3d), indicating that Zr-NDI˙− has good stability and reusability. To the best of our knowledge, this is the first time that MOFs with free radical activity have been used as photocatalysts for radical-induced efficient benzimidazole synthesis using OPD and alcohol as starting materials. Furthermore, their catalytic performances are even better than those of the noble metal catalysts, especially for OPD substrates bearing electron-withdrawing groups.
Encouraged by the excellent catalytic performance of Zr-NDI˙−, we further investigated the reaction mechanism of this photocatalytic reaction. According to the reports, there are two main mechanisms for this reaction. Generally, the alcohol was thought to be oxidized to an aldehyde intermediate first and then condensed with diamine to produce benzimidazole. Recently, a new radical route was proposed, which includes the formation of a C-centered radical through the dissociation of the α-C–H bond of the alcohol and then coupling it with diamine to produce benzimidazole.18,19 To study the reaction mechanism of our photocatalytic system, a series of controlled experiments were carried out (Fig. 4a). When TEOA was added as a hole scavenger or CCl4 as an electron scavenger to this catalytic reaction, the yield of 2MBZ decreased to 40% and 88%, respectively, and the latter almost ceased the production of hydrogen, indicating that photogenerated holes and electrons participated and contributed to this reaction. In this context, Zr-NDI˙− with better photogenerated charge carrier separation and transfer capacity displayed better photocatalytic performance than that of Zr-NDI. Conversely, when acetaldehyde replaced ethanol for this reaction, the yield of 2MBZ decreased to 21%, suggesting that this reaction follows the radical mechanism, rather than the traditional oxidation mechanism.
To demonstrate the participation of radicals in this reaction, a radical scavenger (2,6,6-tetramethylpiperidineoxy, TEMPO) was added to the reaction, and the results showed that it greatly suppressed the formation of 2MBZ (yield of 57%), implying that the reaction proceeded via a radical pathway. To confirm this, in situ EPR characterization was carried out. As displayed in Fig. 4b, using 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) as a spin-trapping agent, strong free radical signals of the carbon center were detected under full light illumination, while no EPR signal could be found in the dark, indicating the presence of free radicals upon light irradiation. To further identify the species of the free radicals produced in this reaction, high-resolution mass spectrometry (HRMS) was used to analyze the mixture after photocatalytic reaction. As shown in Fig. 4c, when DMPO was used as a trapping agent, the hydroxyethyl radical (˙CH(CH3)OH)-trapped adduct and its corresponding molecular fragments can be found. This evidence strongly proved that this reaction proceeds via radical pathway and the Zr-NDI˙− catalyst should be responsible for the cleavage of the α-C–H bond of ethanol to form the key carbon radical species. To gain an in-depth understanding of the reaction process of the photo-conversion of OPD and ethanol to 2MBZ, in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) characterization was performed. As displayed in Fig. 4d, in the dark, after achieving adsorption equilibrium for 30 min, no new IR band/peak was observed, indicating that the reaction cannot occur in dark conditions. In contrast, after light illumination, new peaks located at 1635, 1500, 1275, and 1056 cm−1 are present. The peak at 1500 cm−1 can be attributed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of the benzene ring, while the presence of the peak at 1635 cm−1 indicated that the two substrates had reacted to form a CN bond.20 Additionally, the peaks located at 1275 and 1056 cm−1 correspond to the C–N and C–H bonds, respectively, and their presence indicates that the imidazole ring has been closed and the final product 2MBZ was formed.
C bond of the benzene ring, while the presence of the peak at 1635 cm−1 indicated that the two substrates had reacted to form a CN bond.20 Additionally, the peaks located at 1275 and 1056 cm−1 correspond to the C–N and C–H bonds, respectively, and their presence indicates that the imidazole ring has been closed and the final product 2MBZ was formed.
To gain a deeper understanding of the catalyst's function and the detailed reaction mechanisms, density functional theory (DFT) calculations were performed. As illustrated by the optimized host-guest structure (Fig. 5a), a strong π–π interaction is observed between the uncoordinated ligand and the OPD molecule at a distance of approximately 3.58 Å. This interaction results in significant charge redistribution, making electrons more likely to flow from the –NH2 group on OPD to the carbonyl oxygen in the catalyst (Fig. 5b). This suggests that the OPD and the NDI ligand can serve as the electron donor (D) and electron acceptor (A), respectively, and they form strong D–A interactions. This coincides with the experimental phenomenon, i.e., when Zr-NDI was immersed in ethanol solution of OPD under light, its light absorption slightly expanded to the 600–700 nm region and exhibited the characteristic EPR peak of a radical anion (Fig. S41 and S42†). That is, reaction substrate OPD as an electron donor, underwent a single electron transfer (SET) process with Zr-NDI, resulting in the in situ generation of radical anion NDI˙−. Additionally, the presence of numerous oxygen and hydrogen atoms on the pore surface facilitates the adsorption of ethanol molecules in the catalyst channel through multiple hydrogen bonding interactions (distances of approximately 2.03–2.57 Å, Fig. 5c), enhancing their activation. The differential charge density map shows a significant reduction in charge density in the –CH2OH region of the ethanol molecule, particularly around the α-carbon hydrogen atom, indicating a tendency to lose electrons (Fig. 5d). This electron deficiency theoretically destabilizes the chemical bond, making it more prone to cleavage. Consequently, the hydrogen bonding traction activation between the ethanol molecule and the catalyst provides a basis for the activation of the ethanol molecule to form a carbon radical upon light irradiation.
 | ||
| Fig. 5 The DFT calculation and proposed reaction mechanism. (a) Optimized structure of OPD in the Zr-NDI pores. (b) Diagram of the charge density difference between OPD and Zr-NDI. (c) Optimized structure of ethanol in the Zr-NDI pores. (d) Diagram of the charge density difference between ethanol and Zr-NDI. (e) Free energy diagram for cross-coupling of OPD and ethanol to 2MBZ with or without catalyst. (f) Proposed reaction mechanism for photocatalytic 2MBZ synthesis over Zr-NDI. | ||
To further verify the essential role of the catalyst in substrate activation, we calculated the potential energy profile of the reaction pathway with and without the catalyst (Fig. 5e and S43, S44†). Compared to the blank catalyst (−0.28 eV), the absorption of the substrate with Zr-NDI is an exothermic process, showing an energy decrease of −1.80 eV, which indicates that the catalyst exhibits excellent adsorption capacity for the substrates. More importantly, the energy barrier for the dissociation of ethanol into ˙CH(CH3)OH in the presence of the catalyst is 0.95 eV, significantly lower than that in the absence of the catalyst (1.78 eV). This provides strong evidence that Zr-NDI contributes to the activation of the α-C–H bond of ethanol, synergizing with the HAT process to form the carbon radical reactive species ˙CH(CH3)OH. Subsequently, the activated substrates undergo a series of reactions to yield the final product 2MBZ.
Combining the experimental results and DFT calculation analysis, we proposed a plausible photocatalytic reaction mechanism for the synthesis of 2MBZ (Fig. 5f). Under light irradiation, Zr-NDI generates photogenerated carriers, and the two reactants are adsorbed in the pores of Zr-NDI through D–A and hydrogen bonding interactions, respectively. Following a SET process, Zr-NDI is excited to the radical anion Zr-NDI˙−, while OPD is converted into a cationic amine radical. Subsequently, through the hydrogen bonding traction activation formed with the Zr/Hf-oxo cluster and ligand, the ethanol molecule is activated and transformed into the intermediate species ˙CH(CH3)OH by a HAT process. These intermediates rapidly interact within the confined pores, leading to the formation of a Schiff base through dehydration and deprotonation, which subsequently undergoes dehydrocyclization to yield the final product, 2-methylbenzimidazole (2MBZ). Concurrently, the hydrogen protons extracted by the holes reacted with the photogenerated electrons to produce hydrogen. Based on the theoretical calculations, it is clear that the cleavage of the α-C–H bond of the ethanol molecule is an energy-consuming process. Thus, it should be the key step for this reaction. As the efficiency of this step depends on the HAT effect of the catalyst, the radical Zr-NDI˙− has better performance in this step than that of Zr-NDI. Therefore, based on the experimental results, the enhanced photocatalytic performance of the radical Zr-NDI˙− compared to the parent Zr-NDI is not only due to its better photoelectric properties but also due to its superior HAT capability.
In summary, we for the first time designed and constructed two heterogeneous MOFs, Zr-NDI and Hf-NDI. Due to their excellent light absorbing capacity and confined radical microenvironment, which contains multiple binding sites for the adsorption of OPD and alcohol substrates (through DA interaction and hydrogen bond interaction, respectively), these MOFs can integrate PS, HAT, and SET processes to simultaneously activate two substrates, achieving efficient photocatalytic carbon radical-mediated cross-coupling reaction to synthesize benzimidazole drugs. In particular, the radicals Zr-NDI˙− and Hf-NDI˙− can achieve a maximum approaching >99% substrate conversion and product selectivity. Furthermore, they have very good substrate compatibility. These results have surpassed those of the noble metal catalysts. To the best of our knowledge, this work presents the first example of the rational design of radical MOFs that integrate PS, HAT, and SET processes for efficient photocatalytic carbon radical-mediated organic reaction.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Y.-Q. L, J. L, and Y.-L. L conceived and designed the idea. Y.-L. L synthesized the catalysts and designed the experiments. Y.-L. L, Z.-B. M, J.-R. L and S.-J. Y assisted with experiments and related characterizations. Y.-Q. L, J. L, J.-M. L, N. L, F. Y, S.-L. L and Y.-L. L discussed the result and prepared the manuscript. All the authors reviewed and contributed to this paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R&D Program of China (2024YFA1510700, 2023YFA1507204 and 2023YFA1507201), the National Natural Science Foundation of China (22225109, 22271104, 22201046, 22301139 and U24A20550), Young Top Talents of Pearl River Talent Program of Guangdong Province (2021QN02L617), Guangdong Basic and Applied Basic Research Foundation (2023B1515120060 and 2023A1515030097), Natural Science Foundation of Jiangsu Province (BK 20230375) and the Open Foundation of the State Key Laboratory of Coordination Chemistry, Nanjing University (SKLCC2313).Notes and references
- (a) J. Twilton, C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, Nat. Rev. Chem., 2017, 1, 1–19 CrossRef; (b) H. Yi, G. Zhang, H. Wang, Z. Huang, J. Wang, A. K. Singh and A. Lei, Chem. Rev., 2017, 117, 9016–9085 CrossRef CAS PubMed; (c) G. Ji, L. Zhao, J. Wei, J. Cai, C. He, Z. Du, W. Cai and C. Duan, Angew. Chem., Int. Ed., 2021, 61, e202114490 CrossRef PubMed; (d) R.-R. Liang, Z. Han, P. Cai, Y. Yang, J. Rushlow, Z. Liu, K.-Y. Wang and H.-C. Zhou, J. Am. Chem. Soc., 2024, 146, 14174–14181 CrossRef CAS PubMed.
- (a) K. P. S. Cheung, S. Sarkar and V. Gevorgyan, Chem. Rev., 2021, 122, 1543–1625 CrossRef PubMed; (b) Z. Liu, M. Li, G. Deng, W. Wei, P. Feng, Q. Zi, T. Li, H. Zhang, X. Yang and P. J. Walsh, Chem. Sci., 2020, 11, 7619–7625 RSC.
- (a) K. Minami, K. Ohmatsu and T. Ooi, ACS Catal., 2022, 12, 1971–1976 CrossRef CAS; (b) W. Zhou, I. A. Dmitriev and P. Melchiorre, J. Am. Chem. Soc., 2023, 145, 25098–25102 CrossRef CAS PubMed; (c) Q. Liu and L.-Z. Wu, Natl. Sci. Rev., 2017, 4, 359–380 CrossRef CAS.
- (a) L. Capaldo and D. Ravelli, Eur. J. Org Chem., 2017, 2017, 2056–2071 CrossRef CAS PubMed; (b) L. Capaldo, L. L. Quadri and D. Ravelli, Green Chem., 2020, 22, 3376–3396 RSC.
- (a) X. Wang, J. He, Y.-N. Wang, Z. Zhao, K. Jiang, W. Yang, T. Zhang, S. Jia, K. Zhong, L. Niu and Y. Lan, Chem. Rev., 2024, 124, 10192–10280 CrossRef CAS PubMed; (b) K. Feng, E. R. Raguram, J. R. Howard, E. Peters, C. Liu, M. S. Sigman and S. L. Buchwald, J. Am. Chem. Soc., 2024, 146, 26609–26615 CrossRef CAS PubMed; (c) X. Zhao, X. Zhu, K. Wang, J. Lv, S. Chen, G. Yao, J. Lang, F. Lv, Y. Pu, R. Yang, B. Zhang, Z. Jiang and Y. Wan, Nat. Commun., 2022, 13, 4180 CrossRef CAS PubMed.
- (a) M. Schlegel, S. Qian and D. A. Nicewicz, ACS Catal., 2022, 12, 10499–10505 CrossRef CAS PubMed; (b) J.-K. Jin, K. Wu, X.-Y. Liu, G.-Q. Huang, Y.-L. Huang, D. Luo, M. Xie, Y. Zhao, W. Lu, X.-P. Zhou, J. He and D. Li, J. Am. Chem. Soc., 2021, 143, 21340–21349 CrossRef CAS PubMed; (c) J. Yan, H. Tang, E. J. R. Kuek, X. Shi, C. Liu, M. Zhang, J. L. Piper, S. Duan and J. Wu, Nat. Commun., 2021, 12, 7214 CrossRef CAS PubMed.
- (a) H. Pang, G. Liu, D. Huang, Y. Zhu, X. Zhao, W. Wang and Y. Xiang, Angew. Chem., Int. Ed., 2023, 62, e202313520 CrossRef CAS PubMed; (b) Y. Zhang, Y. Zhang, Y. Guo, S. Liu and X. Shen, Chem Catal., 2022, 2, 1380–1393 CrossRef CAS.
- (a) C. Mao, Y. Huang, H. Li, T. Xu, J. Chen and S. Liao, ACS Catal., 2024, 14, 15133–15139 CrossRef CAS; (b) S. Sil, A. Santha Bhaskaran, S. Chakraborty, B. Singh, R. Kuniyil and S. K. Mandal, J. Am. Chem. Soc., 2022, 144, 22611–22621 CrossRef CAS PubMed; (c) L. Zeng, T. Zhang, R. Liu, W. Tian, K. Wu, J. Zhu, Z. Wang, C. He, J. Feng, X. Guo, A. I. Douka and C. Duan, Nat. Commun., 2023, 14, 4002 CrossRef CAS PubMed; (d) I. Ghosh, T. Ghosh, J. I. Bardagi and B. König, Science, 2014, 346, 725–728 CrossRef CAS PubMed.
- E. P. Geunes, J. M. Meinhardt, E. J. Wu and R. R. Knowles, J. Am. Chem. Soc., 2023, 145, 21738–21744 CrossRef CAS PubMed.
- (a) P. La Manna, M. De Rosa, C. Talotta, A. Rescifina, G. Floresta, A. Soriente, C. Gaeta and P. Neri, Angew. Chem., Int. Ed., 2019, 59, 811–818 CrossRef PubMed; (b) Y. Xin, S. Wang, H. Yuan, T. Hou, W. Zhu, Y. Liu, Y. Yao, W. Zhang, S. Liang and L. Wang, Chem, 2021, 7, 2118–2136 CrossRef CAS; (c) N. Li, S. J. Yao, M. J. Wei, J. He, W. Chi and Y. Q. Lan, Small, 2022, 19, 2206724 CrossRef PubMed.
- (a) Y. Shang, W. Li, Y. Ma, B. Li, Q. Xu, Y. Du, Y. Peng, Y. Wang and Y. Zhu, Adv. Funct. Mater., 2024, 2406533 CrossRef CAS; (b) J. Jing, J. Li, Y. Su and Y. Zhu, Appl. Catal., B, 2023, 324, 122284 CrossRef CAS; (c) J. Li, S. Song, J. Meng, L. Tan, X. Liu, Y. Zheng, Z. Li, K. W. K. Yeung, Z. Cui, Y. Liang, S. Zhu, X. Zhang and S. Wu, J. Am. Chem. Soc., 2021, 143, 15427–15439 CrossRef CAS PubMed.
- X. Wang, M. Alzayer, A. J. Shih, S. Bose, H. Xie, S. M. Vornholt, C. D. Malliakas, H. Alhashem, F. Joodaki, S. Marzouk, G. Xiong, M. Del Campo, P. Le Magueres, F. Formalik, D. Sengupta, K. B. Idrees, K. Ma, Y. Chen, K. O. Kirlikovali, T. Islamoglu, K. W. Chapman, R. Q. Snurr and O. K. Farha, J. Am. Chem. Soc., 2024, 146, 3943–3954 CrossRef CAS PubMed.
- (a) H. Li, M. Eddaoudi, M. O'Keeffe and O. M. Yaghi, Nature, 1999, 402, 276–279 CrossRef CAS; (b) J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef PubMed; (c) H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed; (d) D. Birnthaler, R. Narobe, E. Lopez-Berguno, C. Haag and B. König, ACS Catal., 2023, 13, 1125–1132 CrossRef CAS.
- (a) Y. Shen, Y. Gu and R. Martin, J. Am. Chem. Soc., 2018, 140, 12200–12209 CrossRef CAS PubMed; (b) S. Halder, S. Mandal, A. Biswas and D. Adhikari, Green Chem., 2023, 25, 2840–2845 RSC.
- (a) Z.-S. Huang, Y.-F. Wang, M.-Y. Qi, M. Conte, Z.-R. Tang and Y.-J. Xu, Angew. Chem., Int. Ed., 2024, e202412707 CAS; (b) Y. F. Wang, M. Y. Qi, M. Conte, Z. R. Tang and Y. J. Xu, Angew. Chem., Int. Ed., 2023, 62, e202304306 CrossRef CAS PubMed.
- (a) D. Liu, J. Wang, X. Bai, R. Zong and Y. Zhu, Adv. Mater., 2016, 28, 7284–7290 CrossRef CAS PubMed; (b) H. Hu, Y. Y. Zhang, H. Ma, Y. Yang, S. Mei, J. Li, J. F. Xu and X. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202308513 CrossRef CAS PubMed.
- (a) Y. Yan, N.-N. Zhang, L. M. Tauche, K. Thangavel, A. Pöppl and H. Krautscheid, Inorg. Chem. Front., 2022, 9, 5016–5023 RSC; (b) S. Park, J. Lee, B. Kim, C.-Y. Jung, S.-E. Bae, J. Kang, D. Moon and J. Park, J. Am. Chem. Soc., 2024, 146, 9293–9301 CrossRef CAS PubMed.
- (a) Y. Shiraishi, Y. Sugano, S. Tanaka and T. Hirai, Angew. Chem., Int. Ed., 2010, 49, 1656–1660 CrossRef CAS PubMed; (b) Y. Qin, M. Hao, C. Xu and Z. Li, Green Chem., 2021, 23, 4161–4169 RSC.
- Z. Li, Z. Ye, L. Chen, J. Cui and J. Chen, ACS Appl. Nano Mater., 2020, 3, 10720–10731 CrossRef CAS.
- (a) G. Wu, Y. Liu, Y. He, J. Feng and D. Li, Appl. Catal., B, 2021, 291, 120061 CrossRef CAS; (b) W. Liu, Y. Wang, H. Huang, J. Wang, G. He, J. Feng, T. Yu, Z. Li and Z. Zou, J. Am. Chem. Soc., 2023, 145, 7181–7189 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2351583 and 2351584. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc01242b |
| This journal is © The Royal Society of Chemistry 2025 |