
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnantiomer-enriched π-extended helicenes with a perylene core from binaphthol: axial-to-helical chirality transfer with a stepwise Scholl reaction mechanism†
Zhi-Ao
Li
,
Ke-Lin
Zhu
,
Nai-Te
Yao
 ,
Jiaqi
Liang
,
Yi-Ling
Shang
,
Ye
Zhang
and
Han-Yuan
Gong
*
,
Jiaqi
Liang
,
Yi-Ling
Shang
,
Ye
Zhang
and
Han-Yuan
Gong
*
College of Chemistry, Beijing Normal University, No. 19, XinWai St, HaiDian District, Beijing, 100875, China. E-mail: hanyuangong@bnu.edu.cn
First published on 28th April 2025
Abstract
π-Extended helicenes, as chiral nanographene molecules, possess unique chiral structures and properties, making them highly valuable for applications in advanced chiral materials. However, their enantioselective synthesis typically requires complex resolution processes or the use of expensive catalysts and labor-intensive substrates. This study introduces a cost-effective strategy that utilizes binaphthol, an axially chiral precursor, in the Scholl reaction to generate a twisted perylene core. This approach leads to the formation of an enantiomer-enriched helical structure with an enantiomeric ratio of up to 80.1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :19.9, without the need for specialized ligands or substrates. The enantiomeric purity is further enhanced through crystal selection or thin-layer chromatography (TLC), eliminating the need for high-performance liquid chromatography (HPLC). The resulting helicenes exhibit a circular dichroism dissymmetry factor (|gabs|) of up to 0.01, accompanied by near-infrared (NIR) emission and a fluorescence quantum yield of 41.1%. These results underscore the practicality and efficiency of binaphthol in enantioselective synthesis, expanding the scope of the Scholl reaction to produce enantiomerically enriched helicenes with excellent optical properties.
:19.9, without the need for specialized ligands or substrates. The enantiomeric purity is further enhanced through crystal selection or thin-layer chromatography (TLC), eliminating the need for high-performance liquid chromatography (HPLC). The resulting helicenes exhibit a circular dichroism dissymmetry factor (|gabs|) of up to 0.01, accompanied by near-infrared (NIR) emission and a fluorescence quantum yield of 41.1%. These results underscore the practicality and efficiency of binaphthol in enantioselective synthesis, expanding the scope of the Scholl reaction to produce enantiomerically enriched helicenes with excellent optical properties.
Introduction
π-Extended helicenes, with their complex molecular structures, extensive π-electron distribution, liquid-phase processability, and stable chirality, are promising candidates for chiral functional materials.1–10 Advances in π-extended helicene synthesis methods have enabled the development of more sophisticated helical structures, including longer [n]helical cores,11–13 extended π-conjugation systems,14,15 multi-helical configurations,16–21 heteroatom incorporation,22–28 and fully fused multi-layered molecules.17,21 This diversity in π-electron distribution and asymmetry in molecular architecture enhances π-extended helicenes' chiral properties. However, most synthesized helicenes are racemates, requiring chiral resolution via high-performance liquid chromatography (HPLC) to obtain pure enantiomers. This limitation restricts the large-scale production of enantiomerically pure helicenes, impeding their practical applications and further constraining related research efforts. The enantioselective synthesis of helicene derivatives has received considerable attention due to their potential use in advanced functional materials. Several strategies have been developed, including asymmetric catalysis for helical core formation,29–31 diastereomer separation,32 cycloaddition reactions with chiral ligands,33 and the use of stereochemically pre-designed substrates.34–38 These methods depend on expensive transition metal catalysts29–31,33 or pre-synthesized, stereochemically constrained substrates,34–38 which preclude racemic mixtures. Alternatively, diastereomer synthesis often relies on introducing multiple chiral centers, necessitating tedious separation processes. These limitations underscore the urgent need for innovative, cost-effective, and scalable strategies for π-extended helicenes. A promising alternative lies in the use of cost-efficient axially chiral compounds to achieve chiral transmission, providing a practical and generalizable approach to overcoming these challenges.Chiral transfer methods using affordable chiral chemicals provide a practical approach to enantioselective synthesis. Binaphthol (BINOL), known for its axial chirality,39 is widely employed in the creation of chiral molecules,40–42 asymmetric catalysis, and chiral complexes with lanthanide metals.43–45 However, utilizing binaphthalene's axial chirality in the Scholl reaction to build a perylene core has faced significant challenges, primarily due to the extreme reaction conditions and harsh reagents required for the intramolecular oxidative coupling of binaphthalene.46,47 Studies have demonstrated that attaching electron-donating substituents to the binaphthalene backbone increases its susceptibility to intramolecular coupling reactions, thus facilitating the formation of the perylene core. Building on these findings, we identified alkylated BINOL as a more suitable synthetic precursor for this strategy. The alkylation enhances the electronic properties of BINOL, making it more reactive and enabling smoother coupling reactions, ultimately improving the efficiency of the perylene core synthesis.48
Herein, we report the synthesis of the enantiomer-enriched π-extended [7]helicene (1), featuring a perylene core. This was achieved by transferring axial chirality from BINOL derivatives to the helical chirality of the resulting helicenes. The enantiomer-enriched helicenes 1A and 1B served as key intermediates in the stepwise process. Using commercially available chiral-pure BINOL, we achieved an enantiomeric ratio (er) of up to 80.1:19.9 through a DDQ/CH3SO3H-promoted Scholl reaction at a suitable reaction starting temperature (253 K). This method, which requires neither specific substrate design nor chiral ligands, facilitated the synthesis of two additional intermediates of 1, namely 1A and 1B, through straightforward adjustments of reaction time and temperature. These results suggest a stepwise formation mechanism for the helical skeleton. Stereochemical analysis demonstrated that the enantiomeric ratio (er) of intermediate 1A remained consistent with that of the target molecule 1. This suggests that the loss of chirality occurs during the formation of intermediate (1A) from substrate (4), and does not affect the final enantiomeric ratio (Scheme 1).
 | ||
| Scheme 1 (a) Previous studies on the enantioselective synthesis of (π-extended) helicenes utilizing strategies, including transition metal catalysis with chiral ligands, pre-designed substrates, and diastereomer separation. (b) Enantioselective synthesis of helicenes with a perylene core achieved in this work via a chiral transmission strategy using cost-effective BINOL-derived substrates, along with an investigation of the optical properties of the target molecule and its intermediates. | ||
Compound 1 exhibited excellent crystallinity in a CH2Cl2/EtOH solvent system. Enantiomer-rich samples formed plate-like crystals, whereas racemic mixtures produced needle-like samples, enabling efficient chiral resolution via simple crystal selection. Notably, compound 1 could be successfully resolved into its enantiomers using thin-layer chromatography (TLC); however, this method proved ineffective for the separation of compounds 1A and 1B. These two methods allowed for the isolation of samples with enantiomeric ratios exceeding 99:1, eliminating the need for high-performance liquid chromatography (HPLC).
Photophysical characterization revealed that compounds 1A, 1B, and 1 exhibit fluorescence emission peaks at 515, 605, and 705 nm, respectively, with absolute quantum yield (ΦF) values of 83.6%, 73.6%, and 41.1%. Notably, compound 1 exhibited strong chiral optical activity, as indicated by a dissymmetry factor (|gabs| = 0.01), along with near-infrared emission at wavelengths exceeding 780 nm.
Results and discussion
Synthesis optimization
The white solid precursor of 1 (Rac, S-, or R-4) was synthesized with a yield of 60% via a Suzuki reaction between 2 (Rac, S-, or R-2) and 3 at 373 K for 12 h in toluene, using Pd(PPh3)4 as a catalyst and Cs2CO3 as a base (Scheme 2). In our initial attempt at the Scholl reaction without any chiral reagents or pre-chiral resolution, DDQ and CH3SO3H were used at 303 K. After 4 h, the primary product was 1B, a mauve solid with one remaining chemical bond to form 1. Extending the reaction time to 12 h resulted in the formation of 1 with an enantiomeric ratio (er) of approximately 67:33 and a yield of 50% as blue solid.
 | ||
| Scheme 2 Low-temperature strategy for the synthesis of enantiomer-enriched (M)-1 from (R)-2. Details of the final Scholl reaction, including acid selection and observed reaction phenomena, are summarized in the ESI.† | ||
To increase enantiomeric enrichment, the reaction temperature was lowered, and various alternative acids were explored to optimize the reaction conditions for better stereoselectivity. Below 253 K, the reaction could not be triggered. CF3SO3H, commonly used in Scholl reactions, resulted in substrate decomposition without product formation across various temperatures. CF3CO2H initiated the reaction at 253 K but was ineffective at lower temperatures. Although 98% H2SO4 successfully produced the intermediate compound at 253 K, prolonged reaction times led to decomposition. Ultimately, CH3SO3H was selected as the preferred acid, as it provided the best balance of reactivity and stability. Careful dispersion of CH3SO3H in CH2Cl2 (fivefold volume) was employed to prevent coagulation and ensure consistent reaction conditions (Table S1†).
At 253 K, the intermediate 1A was obtained as the main product, displaying a yellow-green colour and green fluorescence. Gradually increasing the temperature by 10 K allowed the reaction to progress, yielding 1B as the primary product after 12 h. Finally, maintaining the reaction at 303 K for an additional 12 h completed the conversion to 1. Using this stepwise, temperature-increasing method, the er value improved significantly, reaching 80.1:19.9 (Scheme 2). This result highlights the importance of controlled, incremental temperature adjustments for achieving higher enantioselectivity. The relationship between the enantiomeric ratios of the intermediates and the target product will be analyzed in the final section of this paper.
Mass spectrometric characterization of 1A, 1B and 1
The formation of intermediated products 1A and 1B and final product 1 from 4 was confirmed using matrix-assisted laser desorption ionization time-of-flight high-resolution mass spectrometry (MALDI-TOF HRMS). The observed isotope patterns closely matched the simulated data, with HRMS results indicating a stepwise decrease in molecular mass differences (4 or 2) attributable to progressive bond formation (Fig. S1–S6†).The crystal structures of 1A, 1B and 1
Single crystals of the intermediate products 1A·1.125CH2Cl2 and 1B·5EtOH were obtained by slow evaporation of a CH2Cl2 solution (10 mg/5 mL) into 10 mL of EtOH. In 1A·1.125CH2Cl2, the chiral nature of 1A arises from its fused-ring skeleton and the steric hindrance imposed by tert-butylphenyl groups, resulting in a C2-symmetric axis and a helical backbone. Analysis of the crystal structure of 1A reveals the formation of a perylene core, with outer chemical bonds expanding its π-conjugated system (Fig. 1a and S10a†). The packing mode of 1A exhibits alternate stacking of enantiomers with intermolecular carbon atomic distances ranging from 3.217 to 3.677 Å (Fig. S11 and S12†). | ||
| Fig. 1 Single-crystal structures, packing modes, and geometrical analysis of enantiomeric helicene derivatives. (a) Single crystal structure and packing mode of (M)-1A in 1A·1.125CH2Cl2. (b) Single crystal structure and packing mode of (M)-1B in 1B·5EtOH. (c) Single crystal structures of (M)-1 and (P)-1, as well as sums of torsional angles (ϕ), and dihedral angle (θ) between hexagon A and hexagon G shown in (Rac)-1·1.5CH2Cl2·H2O, (M)-1·2.5CH2Cl2 and (P)-1·0.75CH2Cl2·2.75H2O. (d) The distance between the centroids of the overlapped two hexagons in the terminal of [7]helicene kernel shown in (Rac)-1·1.5CH2Cl2·H2O, (M)-1·2.5CH2Cl2 and (P)-1·0.75CH2Cl2·2.75H2O. (e) Packing mode of 1 in (Rac)-1·1.5CH2Cl2·H2O, (M)-1·2.5CH2Cl2 and (P)-1·0.75CH2Cl2·2.75H2O. (P)-1 or (M)-1 are marked in yellow or blue, respectively. Note: tert-butyl groups and phenyl ring substituents are omitted for clarity in the molecular structure. | ||
Conversely, 1B·5EtOH displays C1 symmetry, characterized by a partially fused ring system, a rigid conformation, and a [5]helicene core formed by a new inner chemical bond (Fig. 1b and S10b†). The packing mode of 1B features a staggered, nearly parallel arrangement of enantiomers with intermolecular carbon atomic distances ranging from 3.045 to 3.828 Å (Fig. S13 and 14†).
Single crystals of (Rac)-1·1.5CH2Cl2·H2O, (M)-1·2.5CH2Cl2, and (P)-1·0.75CH2Cl2·2.75H2O were obtained through gradual evaporation of their solutions at 298 K. Each solution was prepared by dissolving 10 mg of the compound in 10 mL of CH2Cl2/EtOH (1:4, v/v), forming blue crystals in EtOH upon CH2Cl2 evaporation.
In the single crystal structure of (Rac)-1·1.5CH2Cl2·H2O, molecule 1 displays a C2 symmetric axis (Fig. 1c and S10c†) with a dihedral angle (θ) of 40.4° between the terminal hexagons of the [7]helicene core in (M)- and (P)-1. The total torsional angle (ϕ) is nearly identical for both enantiomers, measuring 131.5° for (M)-1 and 132.1° for (P)-1 (Fig. 1c). The terminal hexagon centroids are separated by 4.536 Å (Fig. 1d), and no significant π–π interactions are observed between adjacent molecules, with intermolecular carbon distances ranging from 3.641 to 4.107 Å. The enantiomers are arranged in a staggered, nearly parallel manner (Fig. S15 and S16†).
In the single crystals of (M)-1·2.5CH2Cl2 and (P)-1·0.75CH2Cl2·2.75H2O, the molecular architecture mirrors that in (Rac)-1·1.5CH2Cl2·H2O, with differences only in chirality and packing modes (Fig. 1e). In these enantiomer crystals, molecules exhibit nearly parallel stacking and folded stacking arrangements, with intermolecular distances between 3.390 and 3.489 Å ((M)-1) and 3.355 to 3.493 Å ((P)-1) (Fig. S17–S19†).
These distinct structural features align with the proposed stepwise mechanism for the formation of compound 1, wherein 1A represents an intermediate with perylene core and, and 1B progresses toward a partially completed helical structure. The crystallographic data provide direct evidence supporting the gradual transformation of intermediates into the final product.
The NMR spectroscopic characterization of 1A, 1B, 1 and related aromaticity analysis
NMR spectroscopic analysis of 1A, 1B, and 1 was performed in CDCl3 at 298 K (Fig. S26–S31†). The NMR spectral data of 1A and 1 exhibit signal degeneracy in both their 1H NMR and 13C NMR spectra, consistent with their C2 symmetries (Fig. 2a and c). These findings indicate that the molecular symmetry is preserved in solution, as the equivalent chemical environments lead to fewer distinct signals. In contrast, the 1H NMR and 13C NMR spectra of 1B display distinct signals corresponding to its C1 symmetry, reflecting the loss of degeneracy due to its lower molecular symmetry (Fig. 2b). These spectral differences provide further confirmation of the structural and symmetry variations among the intermediates and the final product. | ||
| Fig. 2 Molecular symmetry and aromaticity analysis of helicene derivatives via extended 1H NMR spectroscopy and NICS calculations. High-field regions of the extended 1H NMR spectra for 1A (a), 1B (b), and 1 (c), showing 5, 10, and 5 signals, respectively, corresponding to the protons on the tert-butyl groups. These results indicate the molecular symmetries of C2 for 1A, C1 for 1B, and C2 for 1 in solution. (d) Schematic representation of compound structural symmetry. (e) Extended 1H NMR spectrum of 1 and the corresponding absolute NICS(1)av (average sum of NICS(1) and NICS(−1)) values for hexagons II–VIII (darker blue indicating higher aromaticity) show varying degrees of aromaticity, while hexagon I (pink) exhibits anti-aromaticity. | ||
Proton signals Ha to Hl in compound 1 were successfully assigned using 2D NMR spectroscopy, although signals corresponding to aromatic substituents remained unassigned (Fig. S32–S41†). The simplified 1H and 13C NMR spectra of 1 indicate that it retains C2 symmetry in solution. Correlation with crystal structure data revealed that Ha exhibits the lowest chemical shift, attributed to significant shielding as it is positioned above the aromatic ring system. In the aromatic region, protons Hk, Hj, and Hl also displayed relatively low chemical shifts due to shielding effects from neighboring aromatic rings, whereas protons on the peripheral benzene units exhibited higher shifts (Fig. 2e). These findings highlight the influence of molecular geometry and electronic environment on the observed NMR chemical shifts.
To further investigate the electronic structure and aromaticity of compound 1, nucleus-independent chemical shift (NICS) calculations were performed at the GIAO-B3LYP/6-311+G(2d, p) level, alongside anisotropy of the induced current density (ACID) calculations at the CSGT-B3LYP/6-31G(d) level. The ACID analysis revealed a pronounced cyclic current along the molecular backbone, signifying strong aromaticity in the peripheral rings (Fig. S49†). NICS(1) and NICS(−1) calculations further identified hexatomic rings V, VII, and VIII on the helicene's outer edge as exhibiting the strongest shielding effects, consistent with 1H NMR and crystal structure observations (Fig. S48†). For example, protons Hj and Hk were located within the shielded zones of hexagon V, while proton Hl, situated at the junction of hexagons IV and V, displayed reduced shielding (Fig. 2e and S48†).
Additional NICS(0), NICS(−1), and NICS(1) analyses confirmed the anti-aromatic character of the perylene core (Fig. 2e and S48†), consistent with findings in perylene and related compounds.23 Combining data from chemical shifts, ACID and NICS calculations, and crystal structure analysis, it was concluded that peripheral hexagons, particularly II, V, VII, and VIII exhibit significant aromaticity and π-electron delocalization. In contrast, the internal hexagons demonstrate diminished aromaticity or exhibit anti-aromatic behavior. These findings provide a detailed picture of the electronic structure and aromatic properties of compound 1, supporting its unique structural features.
The study of the optoelectronic properties of 1A, 1B and 1
In CH2Cl2, the UV-vis absorption spectrum of compound 1 exhibits two maxima at 395 nm and 645 nm within the range of 350–750 nm. An absorption band spanning 500–750 nm gives the solution a characteristic blue appearance. Additional absorption bands are observed at 235 nm and 300 nm (Fig. 3a and S50†). Fluorescence measurements reveal that compound 1 emits at a maximum at a wavelength of 705 nm (Fig. 3a and S51†), with a fluorescence quantum yield (ΦF) of 41.1% and a fluorescence lifetime (τ) of 6.88 ns (Fig. S58†), confirming its near-infrared photoluminescence.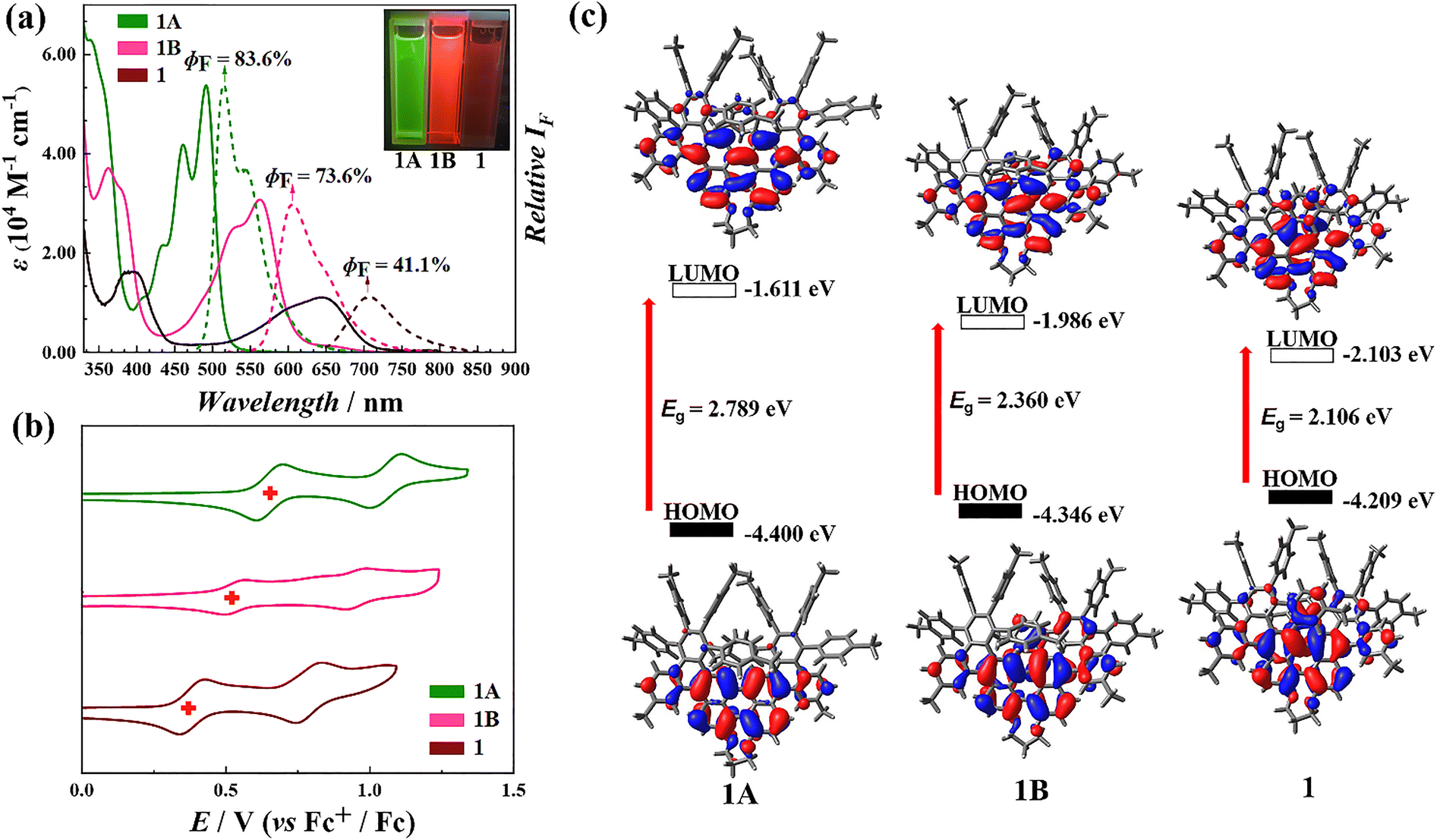 | ||
| Fig. 3 Optoelectronic properties and energy levels of helicene derivatives: UV-vis absorption, fluorescence spectra, cyclic voltammetry, and molecular orbital analysis. (a) Molar absorption (solid lines) and relative fluorescence spectra (dashed lines) of 1 (2.50 × 10−5 M, brown), 1A (1.25 × 10−5 M, green), and 1B (2.50 × 10−5 M, pink) in CH2Cl2. (b) Cyclic voltammetry (CV) curves showing the oxidation potentials of 1 (brown), 1A (green), and 1B (pink). Conditions: working electrode: Pt wire; reference electrode: Ag; scan rate: 100 mV s−1. (c) The calculated HOMO and LUMO orbitals, along with energy gaps for 1A (left), 1B (middle), and 1 (right). | ||
The optical properties of intermediates 1A and 1B reflect a progressive trend associated with increasing π-electron delocalization and expansion of the fused-ring system. As the molecular structure evolves, both absorption and emission wavelengths undergo a bathochromic shift, while fluorescence lifetime increases and quantum yield decreases. For 1A, the UV-vis spectrum shows absorption peaks at 405, 430, 460, and 490 nm, with fluorescence emission maxima at 515 and 545 nm (Fig. 3a, S52 and 53†). These emission peaks form a mirror image of the absorption spectrum, with a ΦF value of 83.6% and τ as 4.17 ns (Fig. S56†).
In the case of 1B, absorption maxima are observed at 530 and 560 nm, while fluorescence emission peaks occur at 605 and 650 nm. The optical spectra of 1B also exhibit a mirror-image relationship between absorption and emission (Fig. 3a, S54 and 55†), with a ΦF value of 73.6% and τ as 6.47 ns (Fig. S57†). These findings underscore the systematic evolution of optical properties as π-conjugation increases, highlighting the relationship between the molecular structure and photophysical behavior.
Electrochemical analysis of compounds 1, 1A, and 1B in CH2Cl2 using cyclic voltammetry (CV) and differential pulse voltammetry (DPV) identified two oxidation processes and one reduction process, with half-wave potentials (E1/2) at +0.772 V, +0.365 V, and −0.927 V for 1, +0.926 V, +0.500 V, and −0.941 V for 1B, +0.946 V, +0.629 V, and −0.986 V for 1A relative to the Fc+/Fc electrode (Fig. S59–S61†). The electrochemical results reveal a similar trend to previous findings: as the fused-ring system expands, the potential difference between the first oxidation and reduction potentials decreases, indicating a narrowing electrochemical gap. Additionally, the oxidation potential undergoes more pronounced shifts compared to the reduction potential, reflecting the system's increased electron delocalization (Fig. 3b).
To further elucidate the absorption properties, density functional theory (DFT) calculations were performed at the B3LYP/6-31G(d) level. For compound 1, the theoretical results closely match the experimental data, with absorption bands in the range of 550–700 nm attributed to the S0 → S1 transition. Bands in the 350–450 nm range are assigned to transitions from the ground state (S0) to higher excited states (Sn, n ≥ 2) (Fig. S62 and Table. S4†). Similarly, for the intermediates 1A and 1B, the theoretical results indicate that the longer-wavelength absorption bands correspond to the S0 → S1 transition, while the shorter-wavelength bands are associated with transitions from the ground state to higher excited states (Sn, n ≥ 2) (Fig. S63, 64, Tables S5 and S6†). Moreover, the energy gaps derived from theoretical calculations show the same trend as the optical and electronic data, with an extended π-conjugated system corresponding to a reduced energy gap (Fig. 3c and S65†). These findings underscore the relationship between the electronic structure and photophysical properties, as demonstrated by consistent experimental and computational results.
The chiral resolution, origins of chiral selectivity and chiral optical properties
The racemate samples exhibited a needle-like crystal morphology, while solutions containing an excess of each enantiomer ((M)- or (P)-1) produced two distinct forms: plate-shaped crystals for the enantiomer and needle-shaped crystals for the racemate (Fig. 4a and b). X-ray diffraction confirmed this difference in crystal morphology. Due to the clear distinction between the crystal forms and the ease of crystallization, enantiomers could be obtained through crystal selection rather than chiral HPLC (Fig. S67†). | ||
| Fig. 4 Chiral resolution, racemization mechanism, and optoelectronic properties of helicene derivatives: experimental and computational investigations. (a) Racemic crystals of 1. (b) Enantiomeric crystals of 1. (c) Thin-layer chromatography (TLC) results for 1 (n-hexane/CH2Cl2, 3:1, v/v). General chromatography (left) was achieved using sodium solyacrylate (PAAS-Na) as the adhesive (pH = 6.2–6.8). Chiral separation (right) was achieved using carboxymethylcellulose sodium (CMC-Na) as the adhesive (pH = 7–8), observed under UV light at 254 nm. Stationary (silica gel) phase granularity: 10–25 μm. (d) Proposed mechanism for partial racemization, with experimental evidence confirming chirality retention and transfer based on HPLC results. (e) Normalized circular dichroism (CD) spectra of 1, with the blue line representing (M)-1 and the red line representing (P)-1. (f) Relative transition state (TS) energies for 1A, 1B, and 1 calculated from TS energy calculations. (g) Calculated electric (μe, blue arrow) and magnetic (μm, red arrow) transition dipole moments for (M)-1 during the S0–S2 transition. For computational efficiency, tert-butyl groups were replaced with methyl groups. | ||
Additionally, chiral resolution was achieved using TLC (pH = 7–8) with CMC-Na (carboxymethylcellulose sodium) as an adhesive, with both methods yielding an enantiomeric ratio (er) greater than 99:1 (Fig. 4c). Subsequent chiral property tests were conducted on samples obtained via crystal-selection chiral resolution.
The failure to fully achieve high enantioselectivity in the synthesis of the target product 1, combined with the relatively rigid molecular skeleton of both the intermediate and the target molecule, suggests that racemization likely occurs during the conversion from substrate 4 to intermediate 1A. As a result, we hypothesize that the enantiomeric ratios of intermediate 1A and the final product 1, obtained under the same reaction conditions, should be similar. To test this hypothesis, we analyzed the enantiomeric ratios of intermediate 1A (enantiomer separation of 1B was not achieved by HPLC) obtained via the low-temperature strategy, as well as the target molecule 1. HPLC analysis revealed that both 1A and 1 produced from the low-temperature strategy maintained consistent enantiomeric ratios of approximately 80:20, supporting our hypothesis. Additionally, repeating the Scholl reaction with 1A as the substrate at 303 K (10.0 mg 1A; 12.6 mg DDQ; 10 mL CH2Cl2; 1 mL CH3SO3H) yielded compound 1 with the same enantiomeric ratio, further validating our findings (Fig. 4d and S68–S70†).
Notably, substrate 4 used in the Scholl reaction could not be successfully resolved into its chiral components. To investigate further, we performed a Suzuki-coupling reaction using bromobenzene and substrate 2 (Rac, S-, and R-2) under identical conditions to synthesize substrate 4* followed by HPLC analysis. The results indicated no evidence of racemization in 4* under this reaction condition, suggesting that substrate 4 likely retains its axial chirality during formation by the Suzuki-coupling reaction. These findings provide indirect evidence of the stability of substrate 4's chirality, supporting its role in the synthesis of enantiomer-enriched helicenes (Fig. S76†).
Our proposed mechanism involves a stepwise reaction pathway: intermediate 1A forms initially, characterized by the development of outer chemical bonds and the perylene core. This is followed by the formation of 1B, which incorporates an additional inner bond, ultimately leading to the final product 1 upon completion of the last inner bond. Racemization likely occurs during the conversion of substrate 4 to intermediate 1A, facilitated by the free rotation of the flexible σ-bond linking the BINOL and polybiphenyl analogs (Fig. 4d). Notably, lower reaction temperatures reduce both the reaction rate and σ-bond rotation, thereby minimizing racemization.
Circular dichroism (CD) spectra of (M)-1 and (P)-1 showed mirror images from 250 to 700 nm, with cotton effects at 252, 267, 312, 325, 343, 414, and 650 nm. The highest dissymmetry factor |gabs| of 0.01 occurred at 420 nm (Fig. 4e, S72 and S73†).
The dissymmetry factor (gabs) was calculated using g = 4|μm|·|μe|·cosθ/(|μm|2 + |μe|2), where μm is the magnetic transition dipole moment, μe is the electric transition dipole moment, and θ is the angle between μm and μe. For the S0–S2 transition of 1, θ is approximately 52.1°, |cosθ| is 0.61, and |μm|/|μe| is about 0.0192. The simulated |gabs| value of 0.011 closely aligns with the experimental value of 0.01 at 420 nm, suggesting that this maximum is due to the S0 → S2 transition (Fig. 4g and Table. S7†).
During the acquisition of the CD spectrum for intermediate 1A, we observed that obtaining a relatively symmetrical spectrum was challenging, likely due to its propensity for rapid racemization (Fig. S74†). To investigate this further, intermediates 1A, 1B, and product 1 were dissolved in chloroform and subjected to reflux heating for 24 hours, after which their CD signals were characterized. The results revealed that 1A underwent complete racemization, resulting in the loss of its CD signal. In contrast, the CD signals of 1B and 1 remained largely unchanged, indicating their significantly higher chiral stability (Fig. S77, 79 and 81†). To further investigate the racemization process, transition state (TS) calculations were performed to determine the energy barriers. The relative energy profiles and geometries were calculated using the semiempirical PM7 method within the MOPAC program. The results indicate that the racemization barrier for 1A is 25 kcal mol−1, whereas for 1B and 1, the barriers are substantially higher, at 75 and 73 kcal mol−1, respectively (Fig. 4f and Fig. S78, 80, 82†). These elevated TS energy values for 1B and 1 are attributed to their more rigid molecular backbones, which enhance structural stability, particularly at elevated temperatures.
These findings suggest that 1A is particularly prone to racemization during the formation of the helical skeleton, whereas 1B and 1 possess more stable stereostructures. Although the CD sample of 1B (Fig. S75†) was not enantiomerically pure (due to the limitations of HPLC for chiral analysis), the results imply that employing a low-temperature strategy could effectively mitigate the racemization of intermediate 1A. Once 1A is converted to 1B, racemization does not occur readily below 333 K.
These results, along with previous studies,35 indicate that the enantioselective synthesis of π-extended helicenes without chiral ligands or transition metals is enhanced by substrates with rigid structures and limited σ-bond rotation, which help to preserve molecular chirality.
Conclusions
In summary, we developed a refined strategy for constructing a perylene core from BINOL derivatives, enabling the synthesis of enantiomer-enriched π-extended helicenes (1). The optimized synthesis revealed that reaction temperatures above 253 K are necessary for the reaction to progress, while lower temperatures enhance enantioselectivity, achieving an enantiomeric ratio of up to 80.1:19.9. Partial racemization was observed, likely due to σ-bond rotation, as confirmed through the characterization of helicene intermediates (1A and 1B), detailed HPLC analysis, and supporting experimental evidence. Enantiomers of 1 were successfully resolved using crystal selection, exploiting the distinct crystal morphologies of racemates and enantiomers, or through facile thin-layer chromatography (TLC).
The π-extended [7]helicene (1) exhibits near-infrared emission and notable chiral optical properties, with a dissymmetry factor (|gabs|) of 0.01 at 420 nm. The chiral transfer process, based on readily available BINOL, provides an efficient route for synthesizing chiral fused-ring nanocarbon molecules. Building on this work, we are currently exploring more efficient and cost-effective strategies for producing enantioselective fused-ring nanocarbon compounds, which could further expand the potential applications of these unique molecules.
Data availability
All data supporting the findings of this study are available within the main text and the ESI,† or from the corresponding author upon reasonable request. Crystallographic data for compounds 1A, 1B, (Rac)-1, (M)-1 and (P)-1 have been deposited with the Cambridge Crystallographic Data Centre (CCDC) under deposition numbers 2411109 (1A), 2411107 (1B), 2411226 (Rac-1), 2411105 (M-1), and 2411106 (P-1). These data can be retrieved by accessing the following URL: https://www.ccdc.cam.ac.uk/solutions/csdcore/components/csd/.Author contributions
Z.-A. Li performed the experimental work and drafted the initial manuscript under the supervision of H.-Y. Gong. K.-L. Zhu, J. Liang, Y.-L. Shang, and Y. Zhang provided valuable assistance with compound synthesis. N.-T. Yao contributed to data collection and supported the analysis of theoretical calculations. The project was conceived and designed by Z.-A. Li and H.-Y. Gong, both of whom also contributed to the preparation of the final manuscript. Initial discoveries were made by Z.-A. Li and H.-Y. Gong. Crystallographic studies were supervised by H.-Y. Gong.Conflicts of interest
There are no conflicts to declare.Acknowledgements
H.-Y. Gong acknowledges financial support from the National Natural Science Foundation of China (92156009). H.-Y. Gong also thanks the staff at the BL17B1 beamline of the National Facility for Protein Science in Shanghai (NFPS), Shanghai Advanced Research Institute, CAS, as well as Dr Tongling Liang and Dr Xiang Hao at the Institute of Chemistry, Chinese Academy of Sciences, for technical support in X-ray diffraction data collection and analysis. Additional thanks go to Dr Junfeng Xiang, Dr Aijiao Guan, Dr Qian Li, and Dr Qiannan Bi for assistance with NMR data analysis, all from the Institute of Chemistry, Chinese Academy of Sciences.References
- M. Gingras, Chem. Soc. Rev., 2013, 42, 968–1006 RSC.
- M. Gingras, G. Félix and R. Peresutti, Chem. Soc. Rev., 2013, 42, 1007–1050 RSC.
- M. Gingras, Chem. Soc. Rev., 2013, 42, 1051–1095 RSC.
- Y. Zhu and J. Wang, Acc. Chem. Res., 2023, 56, 363–373 CrossRef CAS.
- Y.-J. Shen, N.-T. Yao, L.-N. Diao, Y. Yang, X.-L. Chen and H.-Y. Gong, Angew. Chem., Int. Ed., 2023, 62, e202300840 CrossRef CAS PubMed.
- Y.-J. Shen, L.-J. Peng, L.-N. Diao, N.-T. Yao, W.-K. Chen, Y. Yang, M. Qiu, W.-X. Zhu, X. Li, X.-Y. Wang and H.-Y. Gong, Org. Lett., 2024, 26, 7279–7284 CrossRef CAS.
- L.-J. Peng, X.-Y. Wang, Z.-A. Li and H.-Y. Gong, Asian J. Org. Chem., 2023, 12, e202300543 CrossRef CAS.
- T. Mori, Chem. Rev., 2021, 121, 2373–2412 CrossRef CAS PubMed.
- S. -H Pun, K.-M. Cheung, D. Yang, H. Chen, Y. Wang, S.-V. Kershaw and Q. Miao, Angew. Chem., Int. Ed., 2022, 61, e202113203 CrossRef.
- J.-K. Li, X.-Y. Chen, W.-L. Zhao, Y.-L. Guo, Y. Zhang, X.-C. Wang, A. C.-H. Sue, X.-Y. Cao, M. Li, C.-F. Chen and X.-Y. Wang, Angew. Chem., Int. Ed., 2022, 62, e202215367 CrossRef.
- G.-F. Huo, T.-M. Fukunaga, X. Hou, Y. Han, W. Fan, S. Wu, H. Isobe and J. Wu, Angew. Chem., Int. Ed., 2023, 62, e202218090 CrossRef CAS PubMed.
- K. Mori, T. Murase and M. Fujita, Angew. Chem., Int. Ed., 2015, 54, 6847–6851 CrossRef CAS PubMed.
- M. Toya, T. Omine, F. Ishiwari, A. Saeki, H. Ito and K. Itami, J. Am. Chem. Soc., 2023, 145, 11553–11565 CrossRef CAS.
- Y. Nakakuki, T. Hirose, H. Sotome, H. Miyasaka and K. Matsuda, J. Am. Chem. Soc., 2018, 140, 4317–4326 CrossRef CAS.
- Y. Nakakuki, T. Hirose, H. Sotome, M. Gao, D. Shimizu, R. Li, J.-Y. Hasegawa, H. Miyasaka and K. Matsuda, Nat. Commun., 2022, 13, 1475 CrossRef CAS PubMed.
- Y.-F. Wu, L. Zhang, Q. Zhang, S.-Y. Xie and L.-S. Zheng, Org. Chem. Front., 2022, 9, 4726–4743 RSC.
- W. Niu, Y. Fu, Z.-L. Qiu, C.-J. Schürmann, S. Obermann, F. Liu, A. A. Popov, H. Komber, J. Ma and X. Feng, J. Am. Chem. Soc., 2023, 145, 26824–26832 CrossRef CAS.
- H.-C. Huang, Y.-C. Hsieh, P.-L. Lee, C.-C. Lin, Y.-S. Ho, W.-K. Shao, C.-T. Hsieh, M.-J. Cheng and Y.-T. Wu, J. Am. Chem. Soc., 2023, 145, 10304–10313 CrossRef CAS.
- C. Li, Y. Yang and Q. Miao, Chem.–Asian J., 2018, 13, 884–894 CrossRef CAS PubMed.
- J. Tan, X. Xu, J. Liu, S. Vasylevskyi, Z. Lin, R. Kabe, Y. Zou, K. Müllen, A. Narita and Y. Hu, Angew. Chem., Int. Ed., 2023, 62, e202218494 CrossRef CAS PubMed.
- W. Niu, Y. Fu, Q. Deng, Z.-L. Qiu, F. Liu, A. A. Popov, H. Komber, J. Ma and X. Feng, Angew. Chem., Int. Ed., 2024, 63, e202319874 CrossRef CAS PubMed.
- K. Dhbaibi, L. Favereau and J. Crassous, Chem. Rev., 2019, 119, 8846–8953 CrossRef CAS PubMed.
- W.-W. Yang, Z.-H. Ren, J. Feng, Z.-B. Lv, X. Cheng, J. Zhang, D. Du, C. Chi and J.-J. Shen, Angew. Chem., Int. Ed., 2024, 63, e202412681 CrossRef CAS.
- D. Tan, J. Dong, T. Ma, Q. Feng, S. Wang and D.-T. Yang, Angew. Chem., Int. Ed., 2023, 62, e202304711 CrossRef CAS.
- G.-F. Huo, W.-T. Xu, Y. Han, J. Zhu, X. Hou, W. Fan, Y. Ni, S. Wu, H.-B. Yang and J. Wu, Angew. Chem., Int. Ed., 2024, 63, e202403149 CrossRef CAS.
- Y. Yu, C. Wang, F.-F. Hung, C. Chen, D. Pan, C.-M. Che and J. Liu, J. Am. Chem. Soc., 2024, 146, 22600–22611 CrossRef CAS PubMed.
- S. Qiu, A.-C. Valdivia, W. Zhuang, F.-F. Hung, C.-M. Che, J. Casado and J. Liu, J. Am. Chem. Soc., 2024, 146, 16161–16172 CrossRef CAS.
- J.-K. Li, X.-Y. Chen, Y.-L. Guo, X.-C. Wang, A. C.-H. Sue, X.-Y. Cao and X.-Y. Wang, J. Am. Chem. Soc., 2021, 143, 17958–17963 CrossRef CAS PubMed.
- S.-M. Guo, S. Huh, M. Coehlo, L. Shen, G. Pieters and O. Baudoin, Nat. Chem., 2023, 15, 872–880 CrossRef CAS PubMed.
- P. Redero, T. Hartung, J. Zhang, L. D. M. Nicholls, G. Zichen, M. Simon, C. Golz and M. Alcarazo, Angew. Chem., Int. Ed., 2020, 59, 23527–23531 CrossRef CAS.
- T. Hartung, R. Machleid, M. Simon, C. Golz and M. Alcarazo, Angew. Chem., Int. Ed., 2020, 132, 5709–5713 CrossRef.
- S. Chen, Z. Ge, Q. Jia, K.-P. Wang, L.-H. Gan and Z.-Q. Hu, Chem.–Asian J., 2019, 14, 1462–1466 CrossRef CAS.
- F. Morita, Y. Kishida, Y. Sato, H. Sugiyama, M. Abekura, J. Nogami, N. Toriumi, Y. Nagashima, T. Kinoshita, G. Fukuhara, M. Uchiyama, H. Uekusa and K. Tanaka, Nat. Synth., 2024, 3, 774–786 CrossRef CAS.
- M. Buendía, J. M. Fernández-García, J. Perles, S. Filippone and N. Martín, Nat. Synth., 2024, 3, 545–553 CrossRef.
- T. Ikai, K. Oki, S. Yamakawa and E. Yashima, Angew. Chem., Int. Ed., 2023, 62, e202301836 CrossRef CAS PubMed.
- A.-K. Swain, K. Kolanji, C. Stapper and P. Ravat, Org. Lett., 2021, 23, 1339–1343 CrossRef CAS PubMed.
- A.-K. Swain, K. Radacki, H. Braunschweig and P. Ravat, J. Org. Chem., 2022, 87, 993–1000 CrossRef CAS PubMed.
- P. Izquierdo-García, J. M. Fernández-García, J. Perles and N. Martín, J. Am. Chem. Soc., 2024, 146, 34943–34949 CrossRef PubMed.
- L. Pu, Chem. Rev., 2024, 124, 6643–6689 CrossRef CAS.
- R. Fu, Q.-Y. Zhao, H. Han, W.-L. Li, F.-Y. Chen, C. Tang, W. Zhang, S.-D. Guo, D.-Y. Li, W.-C. Geng, D.-S. Guo and K. Cai, Angew. Chem., Int. Ed., 2023, 62, e202315990 CrossRef CAS PubMed.
- R. Fu, D.-Y. Li, J.-H. Tian, Y.-L. Lin, Q.-Y. Zhao, W.-L. Li, F.-Y. Chen, D.-S. Guo and K. Cai, Angew. Chem., Int. Ed., 2024, 63, e202406233 CrossRef CAS PubMed.
- K. Zhang, J. Zhao, N. Zhang, J.-F. Chen, N. Wang, X. Yin, X. Zheng and P. Chen, J. Mater. Chem. C, 2022, 10, 1816–1824 RSC.
- J.-A. Adewuyi and G. Ung, J. Am. Chem. Soc., 2024, 146, 7097–7104 CrossRef CAS PubMed.
- N. F. M. Mukthar, N.-D. Schley and G. Ung, J. Am. Chem. Soc., 2022, 144, 6148–6153 CrossRef CAS PubMed.
- M. Deng, N.-D. Schley and G. Ung, Chem. Commun., 2020, 56, 14813–14816 RSC.
- K. Fujishiro, Y. Morinaka, Y. Ono, T. Tanaka, L.-T. Scott, H. Ito and K. Itami, J. Am. Chem. Soc., 2023, 145, 8163–8175 CrossRef CAS PubMed.
- Y. Uchida, T. Hirose, T. Nakashima, T. Kawai and K. Matsuda, Org. Lett., 2016, 18, 2118–2121 CrossRef CAS PubMed.
- Y. Hirao, T. Okuda, Y. Hamamoto and T. Kubo, ChemPlusChem, 2019, 85, 101–109 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2411105–2411107, 2411109, 2411226. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc01498k |
| This journal is © The Royal Society of Chemistry 2025 |