
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRhenium coordination-induced conformational modulation in nitrogen-doped nanographene†
Eldhose V.
Varghese
a,
Yi-Hung
Liu
b,
Hsing-Yin
Chen
 a,
Chien-Hung
Li
a and
Chia-Hsiang
Chen
*ac
a,
Chien-Hung
Li
a and
Chia-Hsiang
Chen
*ac
aDepartment of Medicinal and Applied Chemistry, Kaohsiung Medical University, 80708 Kaohsiung, Taiwan. E-mail: chc@kmu.edu.tw
bDepartment of Chemistry, National Taiwan University, 10617 Taipei, Taiwan
cDepartment of Medical Research, Kaohsiung Medical University Hospital, 80708 Kaohsiung, Taiwan
First published on 6th May 2025
Abstract
The conformation of nanographene plays a pivotal role in determining its electronic, mechanical, and optical properties. Analyzing the conformation of nanographene is crucial because of its significant implications in various scientific and technological domains. In this work, we examined the changes in the structural conformation of nanographene upon coordination with a metal. We synthesized a series of nitrogen-doped nanographenes (2, 3, and 4) that differed by one C–C bond and their corresponding nanographene–Re complexes (2Re, 3Re, and 4Re). The conformations of the metal complexes were thoroughly analyzed using various spectroscopic techniques. A comparison of the X-ray crystal structures of nanographene before and after rhenium coordination revealed that rhenium coordination induced conformational changes in the nanographene moiety. Additionally, we assessed the catalytic activity of the nanographene–Re complexes toward the electrocatalytic hydrogen evolution reaction (HER). In the presence of a small amount of weak acid (1.5 mM acetic acid), the nanographene–Re complex exhibited hydrogen production at a low overpotential of 133 mV.
Introduction
Understanding the correlation between the conformational and electronic variables presents a significant challenge in chemistry.1,2 To fine-tune the characteristics of organic conjugated structures, steric control of conformation is frequently employed.3–6 The addition of bulky substituents at the ortho sites typically increases intramolecular steric repulsion, leading to a more twisted conformation. This twist can lead to interesting electronic, optical, chiroptical, and magnetic properties.6–11 This approach has been extensively utilized in the development of nonplanar polycyclic conjugated molecules with unique properties.11–14 The steric hindrances caused by these substituents impact not only the torsion angles and solubilities of the twisted compounds but also their electronic properties and molecular packing.6 Steric effects, along with other electronic effects such as inductive and resonance effects, work together to determine the shapes of π-conjugated skeletons. Based on numerous studies, by carefully manipulating the conformational effects, specific properties, such as unique reactivities, self-organization tendencies, interaction patterns with other molecules, and distinct physical characteristics, can be attained.15,16The most important feature of the twisted polyaromatic system is the appearance of intriguing helical chirality, and this system has great potential for utilization in nonlinear optics,17–21 chiroptical devices,22,23 asymmetric catalysis,24,25 organic electronics,26 molecular machines,27,28 and medicinal chemistry.29,30 The helical chirality observed in these systems is a result of strain-induced twisting; annulated π-systems with low stretching and conformational barriers can display dynamic chirality, enabling interconversion between enantiomers.31–33 The dynamic chirality present in certain cases can be utilized to achieve properties that can be switched based on chirality,34 along with facilitating accessibility to various conformers of a polyaromatic compound.35
Extended polycyclic aromatic hydrocarbons, also known as nanographenes, are finite segments of graphene composed of sp2-conjugated carbon atoms and have recently attracted significant attention due to their unique properties, such as extended π-conjugation, fine-tuned HOMO–LUMO gaps, long-wavelength absorption and emission, strong π–π interactions, increased mechanical strength, and potential applications in optoelectronic chemistry.36 The introduction of heteroatoms into the nanographene framework could facilitate nanographene-based ligands suitable for metal coordination.16,37,38 This subsequently facilitates the preparation of metal complexes with intriguing supramolecular properties. Despite recent advancements in the synthesis of extended nanographenes, their use as ligands remains largely unexplored. Only a few metal complexes with nanographene-containing ligands have been described thus far; however, they have shown great potential in the fields of supramolecular chemistry and homogeneous catalysis.38
In recent years, studies on nanographene–metal complexes have been limited and have focused mainly on the coordination ability of nanographenes with transition metals,39–43 while little attention has been given to conformational changes in the nanographene moiety upon coordination. However, recent studies have explored the effects of chemical reduction with alkali metals on the conformation of nanographenes. A geometric change in a nanographene derivative through multi-electron addition was recently reported; its conformation transformed from boat-shaped to chair-shaped.44 Moreover, the conformation of nanographene plays a pivotal role in determining its electronic, mechanical, and optical properties. To address this gap, a series of nitrogen-doped distorted nanographenes (2, 3, and 4) differing by one C–C bond and exhibiting helical chirality45 and their corresponding metal complexes were synthesized to investigate the conformational changes after coordination. In this study, a reaction scheme demonstrating chirality transformation in nanographene through metal coordination is presented. Unlike previous studies that achieved conformational changes through alkali metal-mediated chemical reduction,44 our method focuses on analyzing the conformational changes in nitrogen-doped nanographene via metal coordination.
Herein, we report a study on the chirality transformation in nanographenes upon coordination with rhenium. The influence of rhenium coordination on the structure of nanographenes was extensively validated through a range of spectroscopic techniques. This innovative approach to chirality transformation shows promise for the development of novel solid-state chiral and chiroptical materials. Following the confirmation of chirality transformation, we explored the potential of the use of the nanographene–metal complex in homogeneous catalysis and explicitly examined its catalytic activity in the electrocatalytic hydrogen evolution reaction. We also investigated the impact of C–C bond formation on catalytic performance, facilitating the sustainable utilization of carbon materials in environmental and energy applications.
Results and discussion
The procedures for synthesizing nanographenes 2, 3, and 4 and their corresponding nanographene–Re complexes are illustrated in Scheme 1. All synthesized complexes are air- and moisture-stable in the solid state. The structures of all complexes were confirmed using nuclear magnetic resonance (NMR), UV-vis, Fourier transform infrared (FT-IR) spectroscopy, high-resolution mass spectrometry, and single-crystal X-ray diffraction analyses.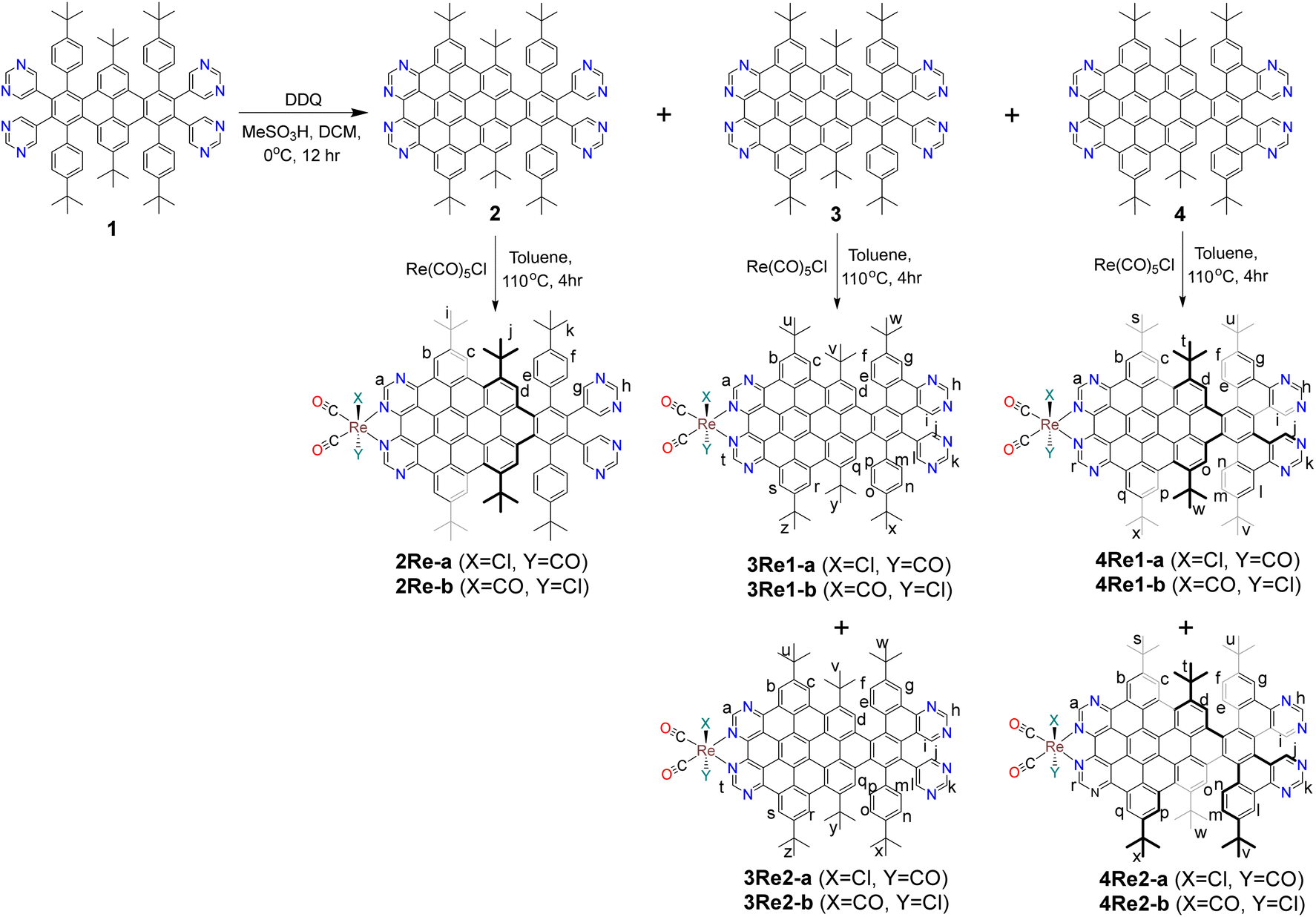 | ||
| Scheme 1 General scheme for the synthesis of nanographenes and nanographene–Re complexes. | ||
The nanographene–Re complex 2Re was synthesized by refluxing ligand 2 with one equivalent of the metal precursor Re(CO)5Cl in anhydrous toluene for 4 hours in a N2 atmosphere. One band appeared on the TLC plate; this band was caused by the stereoisomers 2Re-a and 2Re-b and could not be separated.
Upon refluxing six bond-fused nanographene 3 with Re(CO)5Cl, two distinct bands appeared on the TLC plate (Fig. S4†) and were subsequently isolated and named 3Re1 and 3Re2. NMR analysis revealed that both 3Re1 and 3Re2 had asymmetric structural patterns and underwent interconversion at room temperature (Fig. S18, S22, S37, and S38†). Details regarding the possible structures of compounds 3Re1 and 3Re2 are described in the ESI (Fig. S5).†
Complex 4Re was obtained by reacting ligand 4 with Re(CO)5Cl in hot toluene. Unlike nanographene 2, when nanographene 4 reacted with Re(CO)5Cl, two distinct bands (4Re1 and 4Re2) appeared on the TLC plate and were subsequently isolated (Fig. S1†). Single-crystal X-ray structure analysis revealed that the first band consisted of four different structures: 4Re1-a, 4Re1-a′, 4Re1-b, and 4Re1-b′ (Fig. 1). Compounds 4Re1-a and 4Re1-a′ are enantiomers, differing in the conformation of the nanographene ligand on the unfused pyrimidine side. Similarly, compounds 4Re1-b and 4Re1-b′ are also enantiomers. In contrast, 4Re1-a and 4Re1-b are stereoisomers and differ in the orientation of the Cl atom, and 4Re1-a′ and 4Re1-b′ are also stereoisomers. Interestingly, the stereoisomers and enantiomers had identical chemical shifts in the NMR spectrum, as depicted in Fig. 2.
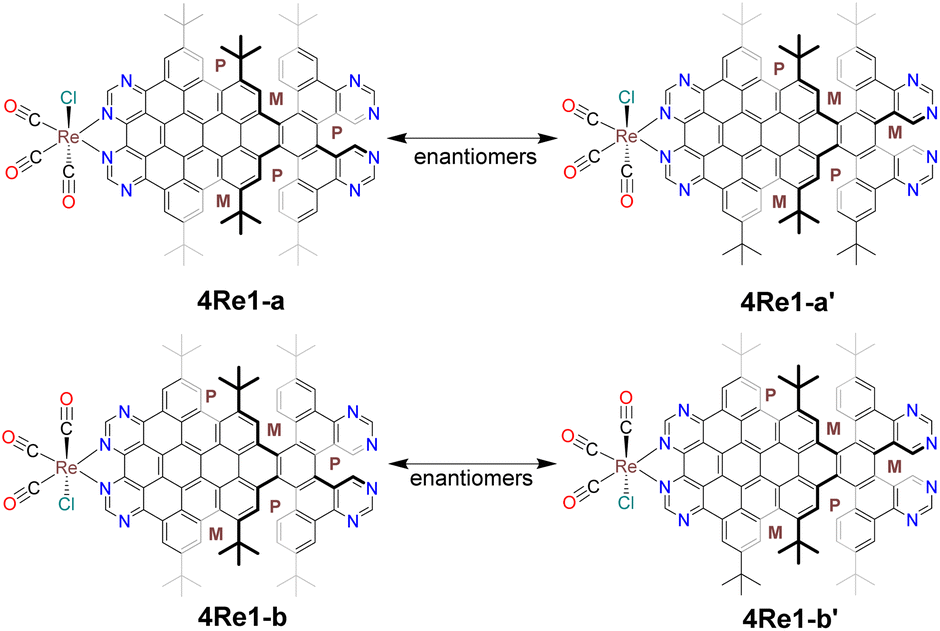 | ||
| Fig. 1 Four different structures of 4Re1 were obtained from single-crystal X-ray diffraction. 4Re1-a and 4Re1-a′ are enantiomers, and 4Re1-b and 4Re1-b′ are also enantiomers. | ||
 | ||
| Fig. 2 1H NMR spectra of compounds 2Re, 3Re1, 3Re2, 4Re1, and 4Re2 in CD2Cl2. The region between 7.0 and 10.5 ppm is enlarged for detail. The peaks of 4Re1 in the 4Re2 spectrum are marked with black dots, and the peaks of 4Re2 in the 4Re1 spectrum are marked with red dots. | ||
A comparison of the 1H NMR spectrum of the second band (4Re2) with that of (P,P,M,P,M)-4, revealed similar broad NMR signals and a matching NMR signal pattern (Scheme S1 and Fig. 2, S14, S28 and S31†). These results indicated that the second band potentially followed the (P,P,M,P,M) structural pattern in the nanographene moiety (Fig. S3†).
High-resolution mass spectrometry provided compelling evidence for the formation of the nanographene–metal complexes. The high-resolution mass spectrum of compound 2Re showed a main peak at m/z = 1515.5638 (Fig. S44†), which matched well with the calculated molecular mass of [M–Cl]+ at m/z = 1515.55982. The structural identities of both the six bond-fused Re complexes 3Re1 and 3Re2 and the seven bond-fused Re complexes 4Re1 and 4Re2 were initially verified using high-resolution mass spectrometry (HRMS). The HRMS results for 3Re1 and 3Re2 displayed peaks at m/z = 1513.5450 and 1513.5424, (Fig. S45 and S46†) respectively; these peaks closely corresponded to the calculated values of [M–Cl]+ at m/z = 1513.54417. Both the 4Re1 and 4Re2 complexes exhibited identical chemical formulae and mass signals, and their isotopic distribution patterns were in agreement with the experimental findings. In the HRMS analysis of 4Re1 and 4Re2, peaks at m/z values of 1511.5279 and 1511.5319 were observed (Fig. S47 and S48†) and consistent with the calculated value of 1511.52852.
The structures of the nanographene–Re complexes were further confirmed by NMR spectroscopy. The aromatic region of the 1H NMR spectra of 2Re, 3Re1, 3Re2, 4Re1, and 4Re2, recorded in CD2Cl2, is shown in Fig. 2. All proton signals were clearly assigned with the assistance of the 1H–1H COSY and 1H–1H ROESY NMR spectra. In the 1H NMR spectra of 2Re, 4Re1, and 4Re2, the signals of the protons corresponding to the pyrimidine ring on the fused side (Ha) shifted downfield relative to those of the free nanographene ligand; these proton signals appeared at 10.44, 10.46, and 10.47 ppm for compounds 2Re, 4Re1, and 4Re2, respectively, compared with 9.65 and 10.25 ppm for nanographenes 2 and 4, respectively; these results indicated the formation of Re–N coordination bonds due to the loss of electron density.
The formation of 2Re was confirmed using 1H NMR, as shown in Fig. 2. Similar to the parent nanographene 2, compound 2Re exhibited eight aromatic proton signals for a total of 22 aromatic protons in the 1H NMR spectrum; these were attributed to the nanographene moiety with a C2-axis. The aromatic protons in the fused part (Ha, Hb, Hc, and Hd) were observed in the more downfield region (δ = 10.45 to 9.37 ppm), whereas the aromatic protons of the non-fused side (He, Hf, Hg, and Hh) appeared between 8.84 and 7.25 ppm.
As expected, the 1H NMR spectra of 3Re1 and 3Re2 displayed an asymmetric structural pattern and showed few differences from that of 3, and the most appreciable differences were the downfield shifts of the Ha and Ht signals corresponding to the protons of the pyrimidine ring coordinated to the Re metal, with chemical shifts of δ = 10.45 and δ = 10.44 ppm, respectively. The majority of the protons in the non-fused aromatic rings were observed between 7 and 8.8 ppm. Owing to their asymmetrical structure, each tert-butyl group displayed six distinct peaks ranging from 1.4–1.9 ppm with equal intensities (Fig. S18 and S22†).
The 1H NMR spectra of nanographene 4 featured a set of signals indicative of a highly symmetric structure. In contrast, the 1H NMR spectra of 4Re1 displayed more signals than those of nanographene 4; these results were consistent with the loss of molecular C2 symmetry of the nanographene after coordination with the Re atom. Upon coordination with the Re atom, the aromatic protons exhibited distinct peaks in the NMR spectrum, especially the signals corresponding to the Hb/q, Hc/p, Hd/o, He/n, Hf/m and Hg/l protons. The peaks corresponding to the six tert-butyl groups also displayed an asymmetric pattern, with six different signals in the NMR spectrum (Fig. S26†). Compared with 4Re1, complex 4Re2 exhibited broadened signals in the NMR spectrum, suggesting fast interconversion between conformers on the NMR timescale. The 1H NMR spectrum of compound 4Re2 showed nine resonance signals in the region of 7.6–10.5 ppm; these signals corresponded to their aromatic protons Ha-r (Fig. 2 and Scheme 1), and the signals corresponding to the three tert-butyl groups were at 1.4–1.95 ppm. The number of these resonance signals corresponded to half of the protons of the molecules; these results indicated that the nanographene moiety of 4Re2 was C2 symmetric.
Compounds 4Re1 and 4Re2 caused difficulties during their isolation via chromatography because of their rapid interconversion at room temperature in solution. The interconversion of 4Re1 to 4Re2 was examined using 1H NMR, as shown in Fig. S35 and S36.† The conversion of compounds 4Re1 to 4Re2 reached a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio within 34 hours. Unlike their ligands, (P,P,M,M,M)- and (P,P,M,P,M)-4, the transformation of 4Re1 to 4Re2 continued over time, resulting in ratios of 40:60 for 4Re1 and 4Re2, respectively, at 74 hours, and subsequently remaining constant for an extended period. Conversely, the interconversion of 4Re2 to 4Re1 followed a different pathway and reached a ratio of 61:39 within 34 hours, with no further changes observed over an extended period. Fig. S39† shows the variable-temperature NMR spectra of mixtures of compounds 4Re1 and 4Re2. At room temperature, the 1H NMR spectrum of 4Re2 displayed broad signals for Hd/o, He/n, Hf/m, Hh/k, Hi/j, Hs/x, Ht/w, and Hu/v, indicating rapid interconversion between the conformers on the NMR timescale. However, upon decreasing the temperature to −35 °C, the signals of Hc/p, Hd/o, He/n, Hf/m, Hh/k, Hs/x, Ht/w and Hu/v of 4Re2 split into two distinct peaks. This observation indicated the existence of two sets of protons in different chemical environments; these peaks were attributed to the magnetic anisotropic effect caused by protons experiencing different ring currents in various twisted configurations. In contrast, the signals of 4Re1 were located at similar positions and showed minimal alterations. The variable-temperature NMR results revealed that 4Re2 existed in two identical conformers, which underwent a partial fusion of side flips across two blades. However, their conformations differed from those of 4Re1.
:1 ratio within 34 hours. Unlike their ligands, (P,P,M,M,M)- and (P,P,M,P,M)-4, the transformation of 4Re1 to 4Re2 continued over time, resulting in ratios of 40:60 for 4Re1 and 4Re2, respectively, at 74 hours, and subsequently remaining constant for an extended period. Conversely, the interconversion of 4Re2 to 4Re1 followed a different pathway and reached a ratio of 61:39 within 34 hours, with no further changes observed over an extended period. Fig. S39† shows the variable-temperature NMR spectra of mixtures of compounds 4Re1 and 4Re2. At room temperature, the 1H NMR spectrum of 4Re2 displayed broad signals for Hd/o, He/n, Hf/m, Hh/k, Hi/j, Hs/x, Ht/w, and Hu/v, indicating rapid interconversion between the conformers on the NMR timescale. However, upon decreasing the temperature to −35 °C, the signals of Hc/p, Hd/o, He/n, Hf/m, Hh/k, Hs/x, Ht/w and Hu/v of 4Re2 split into two distinct peaks. This observation indicated the existence of two sets of protons in different chemical environments; these peaks were attributed to the magnetic anisotropic effect caused by protons experiencing different ring currents in various twisted configurations. In contrast, the signals of 4Re1 were located at similar positions and showed minimal alterations. The variable-temperature NMR results revealed that 4Re2 existed in two identical conformers, which underwent a partial fusion of side flips across two blades. However, their conformations differed from those of 4Re1.
Upon defining the structures of the new nanographene–Re complexes, we proceeded to analyze their photophysical properties to establish the relationship between their structures and optical properties. UV-vis absorption and fluorescence spectra of compounds 2Re, 3Re1, 3Re2, 4Re1, and 4Re2 were measured in dichloromethane (Fig. 3), and their characteristics were compared to determine the effect of their molecular geometries and levels of conjugation on their electronic states. Compared with 3Re1, 3Re2, 4Re1, and 4Re2, compound 2Re showed a blueshifted absorption due to its fewer conjugated π-backbones.45–47 The maximum absorption for 2Re was observed at 233 and 394 nm, with molar extinction coefficients (ε) of 65400 M−1 cm−1 and 43200 M−1 cm−1, respectively, and these absorption peaks were attributed to π–π* transitions,42,45 additionally, the broader bands at 576 and 619 nm were assigned to the metal to ligand charge transfer.41,48 The absorption spectra of 3Re1 and 3Re2 shared similar profiles; with maximum absorption peaks (λmax) at 233 and 414 nm, and the metal to ligand charge transfer transitions were observed at 585 and 629 nm. Similarly, compounds 4Re1 and 4Re2 displayed a primary absorption band with two distinct peaks at λabs of 233 and 420 nm, along with the metal to ligand charge transfer transitions at 590 and 636 nm. The wavelengths of all absorbances and their corresponding absorption coefficients are listed in Table S1.† Upon dissolution in dichloromethane and exposure to UV light, all synthesized compounds exhibited orange fluorescence, and the emission peaks fell between 545 and 555 nm. A similar deduction was drawn from their fluorescence spectra; 4Re1 and 4Re2 exhibited redshifted emission compared with 2Re, 3Re1 and 3Re2 due to their extended π-system.
 | ||
| Fig. 3 UV-Vis absorption spectra (top) and normalized emission spectra (bottom) of compounds 2Re (green), 3Re1 (blue), 3Re2 (pink), 4Re1 (black), and 4Re2 (red) in CH2Cl2 (1 × 10−5 mol L−1). | ||
Single crystals of complexes 2Re and 4Re1 were obtained by layering n-pentane on top of a dichloromethane solution containing the respective compounds. Analysis of the single-crystal structures confirmed the chelation of the Re atom by the bipyrimidine moiety of the nanographene ligand.49 The X-ray structure revealed an octahedral configuration of the coordinated ligands surrounding the metal center, and the three carbonyls exhibited a facial form.
As shown in Fig. 4, the crystal structure of complex 2Re crystallized in a monoclinic crystal system with space group C2/c. The crystal structure indicated that the Re center adopted a distorted octahedral coordination geometry, as observed from the bond angles at the metal centers. The Re atom was coordinated to two nitrogen atoms of the pyrimidine ring, three carbonyl donors in a fac-orientation, and one chlorine atom. The bond lengths and angles around the Re atom were ordinary and comparable to those previously reported for other rhenium tricarbonyl complexes with bidentate ligands. For example, the average Re–carbonyl bond length was 1.95 Å, and the OC–Re–CO angles ranged from 90.29° to 92.65°. The Re–Cl bond length of 2.465 Å fell within the range observed in other rhenium tricarbonyl complexes.48,50,51 Within the unit cell, a pentane molecule was present, and the interplanar distance between the pentane molecule and the fused portion of 2Re was 3.515–3.567 Å.
 | ||
| Fig. 4 Crystal structure of compound 2Re with 50% thermal ellipsoids. Color code: carbon atoms, gray ellipsoids; chlorine atoms, green ellipsoids; nitrogen atoms, blue ellipsoids; oxygen atoms, red ellipsoids; ruthenium atoms, cyan ellipsoids. The hydrogen atoms are omitted for clarity. | ||
Considering the crystal structure of 4Re1, remarkably, four different structures, 4Re1-a, 4Re1-b, 4Re1-a′, and 4Re1-b′, coexisted in the crystal structure resulting from the slow solvent diffusion of pentane into its dichloromethane solution (Fig. 5). These compounds crystallized in a triclinic crystal system with space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . The crystal structure revealed a unit cell containing two pairs of enantiomers, and their nanographene moieties had conformations of (P,M,M,P,P) and (P,M,M,M,P). An analysis of the crystal structure of 4Re1 revealed that the enantiomers 4Re1-a and 4Re1-a′ differed in the conformation of the nanographene ligand between the pyrimidine ring on the noncoordinating side, whereas 4Re1-a and 4Re1-b differed in the orientation of the Cl atom. The Re atom was coordinated to two nitrogen atoms of the pyrimidine ring, with average Re–N bond lengths of 2.189 Å and 2.176 Å for compounds 4Re1-a and 4Re1-b, respectively. Furthermore, the average Re–C bond lengths were 1.915 Å and 1.972 Å, and the OC-Re-CO angles ranged from 87.05° to 92.48° and 79.36° to 90.49° for compounds 4Re1-a and 4Re1-b, respectively. The Re–Cl bond lengths were 2.494 Å for 4Re1-a and 2.487 Å for 4Re1-b; these values were on the higher side of the range observed in other rhenium tricarbonyl complexes.48,50,51 The structure of 4Re1-a consisted of three [5] helicene subunits with dihedral angles of 71.95° (C43–C44–C35–C34), 67.25° (C55–C56–C50–C49), and 69.63° (C60–C59–C38–C39). On the other hand, compound 4Re1-b had dihedral angles of 67.65° (C152–C150–C129–C130), 67.82° (C141–C142–C145–C146), and 72.56° (C134–C133–C126–C125) for the three [5] helicene moieties. The chelate part was nearly planar for both complexes 2Re and 4Re1, with only a slight deviation from the mean planes.
. The crystal structure revealed a unit cell containing two pairs of enantiomers, and their nanographene moieties had conformations of (P,M,M,P,P) and (P,M,M,M,P). An analysis of the crystal structure of 4Re1 revealed that the enantiomers 4Re1-a and 4Re1-a′ differed in the conformation of the nanographene ligand between the pyrimidine ring on the noncoordinating side, whereas 4Re1-a and 4Re1-b differed in the orientation of the Cl atom. The Re atom was coordinated to two nitrogen atoms of the pyrimidine ring, with average Re–N bond lengths of 2.189 Å and 2.176 Å for compounds 4Re1-a and 4Re1-b, respectively. Furthermore, the average Re–C bond lengths were 1.915 Å and 1.972 Å, and the OC-Re-CO angles ranged from 87.05° to 92.48° and 79.36° to 90.49° for compounds 4Re1-a and 4Re1-b, respectively. The Re–Cl bond lengths were 2.494 Å for 4Re1-a and 2.487 Å for 4Re1-b; these values were on the higher side of the range observed in other rhenium tricarbonyl complexes.48,50,51 The structure of 4Re1-a consisted of three [5] helicene subunits with dihedral angles of 71.95° (C43–C44–C35–C34), 67.25° (C55–C56–C50–C49), and 69.63° (C60–C59–C38–C39). On the other hand, compound 4Re1-b had dihedral angles of 67.65° (C152–C150–C129–C130), 67.82° (C141–C142–C145–C146), and 72.56° (C134–C133–C126–C125) for the three [5] helicene moieties. The chelate part was nearly planar for both complexes 2Re and 4Re1, with only a slight deviation from the mean planes.
 | ||
| Fig. 5 Crystal structures of compounds 4Re1-a (a), 4Re1-a′ (b), 4Re1-b (c), and 4Re1-b′ (d) with 50% thermal ellipsoids. Color code: carbon atoms, gray ellipsoids; chlorine atoms, green ellipsoids; nitrogen atoms, blue ellipsoids; oxygen atoms, red ellipsoids; ruthenium atoms, cyan ellipsoids. The hydrogen atoms are omitted for clarity. | ||
The formation of the single crystals of nanographene–metal complexes enabled the evaluation of structural deformation upon coordination. Upon coordination, complex 4Re1 exhibited structural deformation where the π-extended cores of the molecules bent inward, resulting in an average end-to-end distance of 12.182 Å between the two terminal carbon atoms situated in the pentacene unit; this value was slightly smaller than that of the nanographene ligand at 12.308 Å (Fig. S66†). Interestingly, the dihedral angles of the three [5] helicene moieties also decreased post-coordination compared with those of the free ligand.
The analysis of the deformation of nanographene 2 due to metal coordination involved the examination of selected angles and bond lengths. Compared with the original nanographene 2, the π-extended cores of 2Re bent inward, with a slight decrease in the average molecular length (from 12.272 Å to 12.038) between the pentacene units (Fig. S67†) and changes in the dihedral angles (Fig. 6). Compound 2Re displayed a similar trend to that of 4Re1; these results indicated an overall conformational deviation of the carbon scaffold after coordination with the Re metal.
 | ||
| Fig. 6 Crystal structure of nanographene 2 (a) and complex 2Re (b). Color code: carbon atoms, gray ellipsoids; chlorine atoms, green ellipsoids; nitrogen atoms, blue ellipsoids; oxygen atoms, red ellipsoids; ruthenium atoms, cyan ellipsoids. The hydrogen atoms are omitted for clarity. | ||
The crystal structure of nanographene 2 after coordination with Re showed a preference for a different structural pattern (Fig. 6). Upon careful analysis of the 2Re conformation, the conformation on the fused side resembled that of 4Re1. These findings from the available X-ray crystal structure supported the hypothesis regarding the modulation of the nanographene conformation through metal coordination. Therefore, coordination with rhenium induced a conformational change in the studied nanographene ligands 2 and 4.
To ascertain that the conformational change in nanographene is induced by rhenium coordination rather than thermal effects, nanographene 2 was reacted with Re(CO)5Cl at 40 °C for 24 hours while maintaining all other reaction conditions constant. The 1H NMR spectrum of the resulting compound was identical to that of 2Re (Fig. S32†), confirming that the applied temperature did not induce the conformational change in the nanographene. Likewise, when nanographene 4 was treated with Re(CO)5Cl under the same conditions, the 1H NMR spectrum of the product matched those of 4Re1 and 4Re2 (Fig. S33 and S34†). These findings collectively demonstrate that the conformational changes observed in 2Re and 4Re are attributable to rhenium coordination rather than thermal effects.
To evaluate the redox behavior of the complexes under noncatalytic conditions, cyclic voltammetry was conducted in N2-saturated anhydrous THF solutions containing a 0.1 mM complex and 0.1 M Bu4NPF6 as the supporting electrolyte. For every electrochemical analysis, the term 4Re refers to a mixture of 4Re1 and 4Re2 since these two species rapidly interconverted in solution. Similarly, 3Re denotes a mixture of 3Re1 and 3Re2 in all electrochemical measurements. Under a N2 atmosphere in THF, complexes 2Re, 3Re, and 4Re exhibited four reduction redox processes (Fig. 7). Variable scan rate studies of these complexes revealed that the first and second reductions were completely reversible and that the third and fourth reductions were irreversible (Fig. S54†).
 | ||
| Fig. 7 CV curves of compounds 2Re (a), 3Re (b) and 4Re (c). All measurements were recorded at 100 mV s−1 under N2 in THF with 0.1 M TBAPF6 as the supporting electrolyte. | ||
The cyclic voltammogram of compound 4Re in THF exhibited two reversible reduction waves corresponding to the nanographene ligand, with half-wave potentials (Ered1/2) of −0.919 and −1.515 V, and two irreversible reduction waves at Epc = −2.04 and −2.37 V. Compound 3Re also exhibited two reversible reduction waves with nearly the same Ered1/2 values as those of compound 4Re, with Ered1/2 of −0.925 and −1.52 V. Compound 3Re also showed two irreversible reduction waves at more negative potentials than compound 4Re, with Epc = −2.21 and −2.41 V. For compound 2Re, an additional negative shift was observed for all redox peaks compared with those of compound 4Re. Compound 2Re exhibited two reversible reduction waves with half-wave potentials (Ered1/2) of −0.94 and −1.53 V and two irreversible reduction waves at Epc = −2.15 and −2.42 V. Additionally, all complexes exhibited an irreversible oxidation wave, with oxidation peaks at Epa = 1.29, 1.24 and 1.21 V for complexes 4Re, 3Re, and 2Re, respectively (Fig. S49†). The results from the electrochemical characterization of all compounds are provided in Table 1.
| Compound | E red1 [V] | E red2 [V] | E red3 [V] | E red4 [V] | E ox1 [V] |
|---|---|---|---|---|---|
| a Measurement in THF at room temperature by cyclic voltammetry. The E1/2 values correspond to reversible processes. b E pc corresponds to irreversible reduction processes. c E pa corresponds to irreversible oxidation processes. | |||||
| 2Re | −0.95 | −1.53 | −2.15 | −2.42 | 1.21 |
| 3Re | −0.925 | −1.52 | −2.21 | −2.41 | 1.24 |
| 4Re | −0.919 | −1.515 | −2.04 | −2.37 | 1.29 |
For each nanographene–metal complex, the first reduction of nanographene occurred at Ered1/2 = −0.919 V, −0.925 V, and −0.95 V, whereas the second reduction occurred at E1/2red = −1.515 V, −1.52 V, and −1.53 V for complexes 4Re, 3Re, and 2Re, respectively. These reductions were significantly shifted anodically compared with the bpy-moiety reductions in Re(bpy)(CO)3Cl, which occurred at −1.80 V (Fig. S50†).52–55 This behavior occurred because the lowest π* orbital of nanographene was much lower in energy than the lowest π* orbital of bpy of Re(bpy)(CO)3Cl. This was a direct result of the extensive delocalization of the nanographene ligand.39,52,56,57 Furthermore, the CV spectrum showed a positive shift in the reduction potential from 2Re to 3Re to 4Re; this shift was attributed to the progressively extended conjugation from 2Re to 3Re to 4Re. Additionally, the irreversible ReI/0 redox process occurred at less negative potentials in the nanographene–metal complexes: −2.04 V, −2.21 V, and −2.15 V for compounds 4Re, 3Re, and 2Re, respectively, compared with −2.27 V for Re(bpy)(CO)3Cl (Fig. S50†). This shift was caused by the stabilization of the nanographene ligand within the nanographene–metal complex, which reduced the electron density around the Re metal center. This reduction in electron density likely caused the metal center to be more susceptible to reduction, facilitating an easier reduction in the nanographene–metal complexes than Re(bpy)(CO)3Cl. At more negative potentials, a fourth irreversible reduction process was observed at Epc = −2.37 V, −2.41 V, and −2.42 V for 4Re, 3Re, and 2Re, respectively. These values closely resemble the reduction potentials of the corresponding free nanographene ligands 4, 3, and 2 (Fig. S51–S53†), suggesting ligand-centered processes. Additionally, an irreversible oxidation wave assigned to the Re I/II redox process was observed at Epa = 1.2 V, 1.24 V, and 1.21 V for 4Re, 3Re, and 2Re, respectively (Fig. S49†).53,58
All synthesized complexes were subsequently tested for the hydrogen evolution reaction (HER) using acetic acid as the proton source. The electrocatalytic ability of each complex to generate hydrogen was evaluated using cyclic voltammetry. In the presence of an active catalyst, the introduction of protons from a weak acid leads to an irreversible reduction peak in the cyclic voltammogram (CV). Analyzing this catalytic wave provided valuable insights into the catalyst's activity and efficiency. Acetic acid was employed for acid addition studies with each complex. The obtained CVs showed that 2Re, 3Re and 4Re all exhibited catalytic waves upon the addition of acetic acid (Fig. S55†). Hence, the preliminary findings indicated that each of these complexes indeed functioned as an electrocatalyst for proton reduction.
The presence of acid impacted the third redox wave for all complexes, as depicted in Fig. S55.† In these instances, the acid (AcOH) caused an increase in the reduction current and a loss of reversibility for complexes 2Re, 3Re, and 4Re. The reduction wave was influenced solely by the acid concentration within the range of 0.1 to 1.5 mM. Fig. S57 and S58† show the ratio of the catalytic current in the presence of acetic acid (ic) to the peak current for reduction in the absence of acid (ip) as a function of acid concentration in the experiment.
The observed catalytic current exhibited a linear increase in acid concentration for concentrations less than 1.5 mM acetic acid. However, no significant increase in the catalytic current was observed beyond this acid concentration. Upon the addition of acid, an irreversible peak corresponding to proton reduction was observed at approximately −2.1 V and −2 V for compounds 2Re and 3Re, respectively. Complex 4Re displayed electrocatalytic performance when the concentration of AcOH was increased to 1.5 mM; here, three catalytic waves were observed at approximately −1.85, −2.03, and −2.25 V.
Interestingly, the catalytic peak current reached a point independent of the acid concentration above a concentration of 1.5 mM AcOH. In this regime of acid concentration independence, the rate constant for H2 evolution (kobs or TOFmax) was determined, and the TOFmax values for 2Re and 3Re were 4.28 s−1 and 2.8 s−1, respectively, as determined through CV analysis. For complex 4Re, the TOFmax values for the three new peaks were 0.77, 0.93, and 1.23 s−1 for peaks at −1.85, −2.03, and −2.25 V, respectively. The effects of current enhancement were subsequently investigated in relation to the catalyst concentration. At a fixed concentration of the proton source (1.5 mM AcOH), the catalyst current increased for all catalysts. As shown in Fig. S59,† increasing the catalyst concentration led to a linear increase in Icat. Thus, the reaction follows first-order kinetics with respect to the catalyst concentration for all complexes since Icat is proportional to [cat].59 While turnover frequencies are important for characterizing electrocatalysts, they are equally crucial to the electrochemical overpotential; this overpotential represents the activation energy required to achieve those rates. The overpotential is defined as the difference between the experimentally observed potential at which the catalyst operates and the calculated thermodynamic potential for the reaction. Therefore, a lower overpotential is desirable since it indicates a more energetically efficient catalytic system. The calculated overpotentials are listed in Table 2. The synthesized complexes exhibited overpotentials ranging from 133–193 mV; these values were relatively low compared with those of previously reported molecular catalysts.60–70 Based on the overpotentials, 4Re appeared to be an excellent energetically efficient catalyst, with an overpotential of 133 mV. This enhanced performance is attributed to the extended π-conjugation of the ligand in 4Re compared to 3Re and 2Re. The greater conjugation facilitates increased electron delocalization and storage upon reduction, which stabilizes the reduced metal center and shifts the redox potential to more positive values.71–73 As a result, metal complexes bearing extended conjugated ligands generate catalytically active redox species at less negative potentials, thereby enabling catalytic reduction at lower overpotentials.
| Compound | Solvent (proton source) | η | TOF (s−1) | Duration (h) | FE | TON |
|---|---|---|---|---|---|---|
| 2Re | THF (1.5 mM AcOH) | 193 mV | 4.28 | 6 | 44 | 6.05 |
| 3Re | THF (1.5 mM AcOH) | 163 mV | 2.8 | 6 | 22 | 3.4 |
| 4Re | THF (1.5 mM AcOH) | 133 mV | 0.93 | 6 | 28 | 3.3 |
To assess the electrochemical HER capability of the nanographene–Re complexes, controlled potential electrolysis (CPE) was conducted using a potential based on half of the maximum catalytic current observed in the CV measurements. Compared with CV, bulk electrolysis enables the study of catalysis on a larger scale and for a longer duration. Bulk electrolysis was performed in a single-compartment cell using working electrodes with a larger surface area to investigate the bulk changes in the solution. In the CPE study, the potential of the working electrode remained constant while the resulting current at the counter electrode was measured. Since the charge is defined as the integral of the current with respect to time, the build-up of charge over the course of the reaction can be tracked. Following the completion of the experiment, a gas chromatograph of the headspace was conducted to verify the presence of hydrogen gas.
To validate the electrochemical HER ability of 4Re observed in the CV measurements, controlled potential electrolysis (CPE) was conducted at different applied potentials. Initially, a six-hour CPE experiment was carried out at an applied potential Eapp of −1.83 V. Gas chromatography (GC) analysis revealed that a negligible amount of H2 was generated during the six-hour electrolysis. A six-hour CPE experiment was subsequently performed at an applied potential Eapp of −1.93 V. Gas chromatography (GC) analysis demonstrated that H2 was the primary product during the six-hour electrolysis, with a faradaic efficiency (FE) of 28% and a corresponding TON value of 3.3. Our results clearly indicated that 4Re could catalyze the electrochemical HER at a low overpotential. In the third catalytic wave (Eapp = −2.13 V), the main catalytic product was also H2, with an FE of 7% and an associated TON of 2.3. During the six-hour electrolysis, compound 3Re at an applied potential of −2.03 V generated only H2 gas as the product, with a faradaic efficiency of 22% and a TON value of 3.4. On the other hand, compound 2Re exhibited a higher faradaic efficiency of 44% for H2 formation at an applied potential of −2.13 V, with a TON value of 6.05 over the same duration. Comparatively, compound 2Re displayed better FE and TON values than compounds 4Re and 3Re during the six-hour electrolysis. When no catalyst or acetic acid was present, the GC results revealed negligible H2 formation during the six-hour electrolysis. This outcome indicated that the synthesized nanographene metal complex 4Re possessed the ability to perform the electrochemical HER at an extremely low overpotential of 133 mV, utilizing a very low concentration of AcOH.
Conclusion
In conclusion, we successfully synthesized and fully characterized a series of nanographene metal complexes (2Re, 3Re, and 4Re). Structural changes in the nanographene ligands upon metal coordination were investigated using various spectroscopic techniques. These analyses confirmed that metal coordination induced conformational alterations in the ligands, and X-ray crystal structure analysis provided robust evidence to support this observation. Specifically, the structure of nanographene 2 after coordination with Re showed a preference for a different structural pattern. Moreover, the X-ray crystal structures of complexes 2Re and 4Re1 revealed that coordination with rhenium induced a conformational shift in the nanographene ligands to adopt a similar structural pattern on the fused side. These findings highlight a possible strategy for modulating nanographene structures through metal coordination. Furthermore, the catalytic performance of these nanographene metal complexes for the electrochemical hydrogen evolution reaction (HER) was evaluated. These results indicated that complex 4Re could facilitate the HER at a low overpotential of 133 mV with a minimal AcOH concentration. Additionally, increasing the conjugation degree through C–C bond formation effectively reduced the catalytic overpotential. These results provide valuable insights into the design of efficient HER catalysts with reduced overpotentials.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Eldhose V. Varghese: methodology, investigation, formal analysis, writing – original draft. Yi-Hung Liu: investigation. Hsing-Yin Chen: investigation. Chien-Hung Li: investigation. Chia-Hsiang Chen: conceptualization, methodology, supervision, writing – original draft, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for the support of this work by funding from the National Science and Technology Council, Taiwan (NSTC 112-2113-M-037-013 and NSTC 113-2113-M-037-018), and Kaohsiung Medical University, Taiwan (KMU-TB114009).Notes and references
- P. Štacko, J. C. M. Kistemaker, T. van Leeuwen, M.-C. Chang, E. Otten and B. L. Feringa, Locked Synchronous Rotor Motion in a molecular motor, Science, 2017, 356, 964–968 CrossRef PubMed
.
- D. A. Leigh, Genesis of the Nanomachines: The 2016 Nobel Prize in Chemistry, Angew. Chem., Int. Ed., 2016, 55, 14506–14508 CrossRef CAS PubMed
- E. A. Braude, F. Sondheimer and W. F. Forbes, Steric Effects in the Electronic Spectra of Organic Compounds, Nature, 1954, 173, 117–119 CrossRef CAS
- G. Bott, L. D. Field and S. Sternhell, Steric Effects. A Study of a Rationally Designed System, J. Am. Chem. Soc., 1980, 102, 5618–5626 CrossRef CAS
- J. C. M. Kistemaker, P. Štacko, D. Roke, A. T. Wolters, G. H. Heideman, M.-C. Chang, P. van der Meulen, J. Visser, E. Otten and B. L. Feringa, Third-Generation Light-Driven Symmetric Molecular Motors, J. Am. Chem. Soc., 2017, 139, 9650–9661 CrossRef CAS PubMed
- W. Fan, T. Winands, N. L. Doltsinis, Y. Li and Z. Wang, A Decatwistacene with an Overall 170° Torsion, Angew. Chem., Int. Ed., 2017, 56, 15373–15377 CrossRef CAS PubMed
- S. H. Pun and Q. Miao, Toward Negatively Curved Carbons, Acc. Chem. Res., 2018, 51, 1630–1642 CrossRef CAS PubMed
- W. Jiang, Y. Li and Z. Wang, Tailor-Made Rylene Arrays for High Performance n-Channel Semiconductors, Acc. Chem. Res., 2014, 47, 3135–3147 CrossRef CAS PubMed
- G. Liu, T. Koch, Y. Li, N. L. Doltsinis and Z. Wang, Nanographene Imides Featuring Dual-Core Sixfold [5]Helicenes, Angew. Chem., Int. Ed., 2019, 58, 178–183 CrossRef CAS PubMed
- S. H. Pun, C. K. Chan, J. Luo, Z. Liu and Q. Miao, A Dipleiadiene-Embedded Aromatic Saddle Consisting of 86 Carbon Atoms, Angew. Chem., Int. Ed., 2018, 57, 1581–1586 CrossRef CAS PubMed
- M. Ball, Y. Zhong, Y. Wu, C. Schenck, F. Ng, M. Steigerwald, S. Xiao and C. Nuckolls, Contorted Polycyclic Aromatics, Acc. Chem. Res., 2015, 48, 267–276 CrossRef CAS PubMed
- J. B. Lin, T. K. Shah, A. E. Goetz, N. K. Garg and K. N. Houk, Conjugated Trimeric Scaffolds Accessible from Indolyne Cyclotrimerizations: Synthesis, Structures, and Electronic Properties, J. Am. Chem. Soc., 2017, 139, 10447–10455 CrossRef CAS PubMed
- R. A. Pascal and T. Acenes, Chem. Rev., 2006, 106, 4809–4819 Search PubMed
- S. Ito, S. Hiroto, S. Lee, M. Son, I. Hisaki, T. Yoshida, D. Kim, N. Kobayashi and H. Shinokubo, Synthesis of Highly Twisted and Fully π-Conjugated Porphyrinic Oligomers, J. Am. Chem. Soc., 2015, 137, 142–145 CrossRef CAS PubMed
- Z. Zhang, C.-L. Chen, Y.-A. Chen, Y.-C. Wei, J. Su, H. Tian and P.-T. Chou, Tuning the Conformation and Color of Conjugated Polyheterocyclic Skeletons by Installing ortho-Methyl Groups, Angew. Chem., Int. Ed., 2018, 57, 9880–9884 CrossRef CAS PubMed
- R. Kotani, H. Sotome, H. Okajima, S. Yokoyama, Y. Nakaike, A. Kashiwagi, C. Mori, Y. Nakada, S. Yamaguchi, A. Osuka, A. Sakamoto, H. Miyasaka and S. Saito, Flapping Viscosity Probe that Shows Polarity-Independent Ratiometric Fluorescence, J. Mater. Chem. C, 2017, 5, 5248–5256 RSC
- T. Verbiest, S. V. Elshocht, M. Kauranen, L. Hellemans, J. Snauwaert, C. Nuckolls, T. J. Katz and A. Persoons, Strong Enhancement of Nonlinear Optical Properties Through Supramolecular Chirality, Science, 1998, 282, 913–915 CrossRef CAS PubMed
- Y. Zhong, T. J. Sisto, B. Zhang, K. Miyata, X. Y. Zhu, M. L. Steigerwald, F. Ng and C. Nuckolls, Helical Nanoribbons for Ultra-Narrowband Photodetectors, J. Am. Chem. Soc., 2017, 139, 5644–5647 CrossRef CAS PubMed
- T. Otani, A. Tsuyuki, T. Iwachi, S. Someya, K. Tateno, H. Kawai, T. Saito, K. S. Kanyiva and T. Shibata, Facile Two-Step Synthesis of 1,10-Phenanthroline-Derived Polyaza[7]helicenes with High Fluorescence and CPL Efficiency, Angew. Chem., Int. Ed., 2017, 56, 3906–3910 CrossRef CAS PubMed
- N. J. Schuster, D. W. Paley, S. Jockusch, F. Ng, M. L. Steigerwald and C. Nuckolls, Electron Delocalization in Perylene Diimide Helicenes, Angew. Chem., Int. Ed., 2016, 55, 13519–13523 Search PubMed
- Y. Hu, X.-Y. Wang, P.-X. Peng, X.-C. Wang, X.-Y. Cao, X. Feng, K. Müllen and A. Narita, Benzo-Fused Double [7]Carbohelicene: Synthesis, Structures, and Physicochemical Properties, Angew. Chem., Int. Ed., 2017, 56, 3374–3378 CrossRef CAS PubMed
- T. Mori, Chiroptical Properties of Symmetric Double, Triple, and Multiple Helicenes, Chem. Rev., 2021, 121, 2373–2412 CrossRef CAS
- G. Albano, G. Pescitelli and L. Di Bari, Chiroptical Properties in Thin Films of π-Conjugated Systems, Chem. Rev., 2020, 120, 10145–10243 CrossRef CAS PubMed
- D. Sakamoto, I. Gay Sánchez, J. Rybáček, J. Vacek, L. Bednárová, M. Pazderková, R. Pohl, I. Císařová, I. G. Stará and I. Starý, Cycloiridated Helicenes as Chiral Catalysts in the Asymmetric Transfer Hydrogenation of Imines, ACS Catal., 2022, 12, 10793–10800 CrossRef CAS
- K. Yavari, P. Aillard, Y. Zhang, F. Nuter, P. Retailleau, A. Voituriez and A. Marinetti, Helicenes with Embedded Phosphole Units in Enantioselective Gold Catalysis, Angew. Chem., Int. Ed., 2014, 53, 861–865 CrossRef CAS PubMed
- S. R. Peurifoy, T. J. Sisto, F. Ng, M. L. Steigerwald, R. Chen and C. Nuckolls, Dimensional Control in Contorted Aromatic Materials, Chem. Rec., 2019, 19, 1050–1061 Search PubMed
- H. Isla and J. Crassous, Helicene-Based Chiroptical Switches, C. R. Chim., 2016, 19, 39–49 CrossRef CAS
- P. Ravat, T. Šolomek and M. Juríček, Helicenes as Chiroptical Photoswitches, ChemPhotoChe, 2019, 3, 180–186 CrossRef CAS
- J. L. Rushworth, A. R. Thawani, E. Fajardo-Ruiz, J. C. M. Meiring, C. Heise, A. J. P. White, A. Akhmanova, J. R. Brandt, O. Thorn-Seshold and M. J. Fuchter, [5]-Helistatins: Tubulin-Binding Helicenes with Antimitotic Activity, JACS Au, 2022, 2, 2561–2570 CrossRef CAS PubMed
- P. A. Summers, A. P. Thomas, T. Kench, J.-B. Vannier, M. K. Kuimova and R. Vilar, Cationic Helicenes as Selective G4 DNA Binders and Optical Probes for Cellular Imaging, Chem. Sci., 2021, 12, 14624–14634 RSC
- R. H. Janke, G. Haufe, E.-U. Würthwein and J. H. Borkent, Racemization Barriers of Helicenes: A Computational Study1, J. Am. Chem. Soc., 1996, 118, 6031–6035 CrossRef CAS
- J. Elm, J. Lykkebo, T. J. Sørensen, B. W. Laursen and K. V. Mikkelsen, Racemization Mechanisms and Electronic Circular Dichroism of [4]Heterohelicenium Dyes: A Theoretical Study, J. Phys. Chem. A, 2011, 115, 12025–12033 CrossRef CAS PubMed
- J.-J. Zhang, M.-C. Tang, Y. Fu, K.-H. Low, J. Ma, L. Yang, J. J. Weigand, J. Liu, V. W.-W. Yam and X. Feng, One-Pot Synthesis of Boron-Doped Polycyclic Aromatic Hydrocarbons via 1,4-Boron Migration, Angew. Chem., Int. Ed., 2021, 60, 2833–2838 CrossRef CAS PubMed
- C.-T. Chen, C.-C. Tsai, P.-K. Tsou, G.-T. Huang and C.-H. Yu, Enantiodivergent Steglich Rearrangement of O-carboxylazlactones Catalyzed by a Chirality Switchable Helicene Containing a 4-Aminopyridine Unit, Chem. Sci., 2017, 8, 524–529 RSC
- C. Weiss, D. I. Sharapa and A. Hirsch, Coronenohelicenes with Dynamic Chirality, Chem. - Eur. J., 2020, 26, 14100–14108 CrossRef CAS PubMed
- J. Merz, A. Steffen, J. Nitsch, J. Fink, C. B. Schürger, A. Friedrich, I. Krummenacher, H. Braunschweig, M. Moos, D. Mims, C. Lambert and T. B. Marder, Synthesis, Photophysical and Electronic Properties of Tetra-Donor- or Acceptor-Substituted Ortho-Perylenes Displaying Four Reversible Oxidations or Reductions, Chem. Sci., 2019, 10, 7516–7534 RSC
- X.-Y. Wang, X. Yao, A. Narita and K. Müllen, Heteroatom-Doped Nanographenes with Structural Precision, Acc. Chem. Res., 2019, 52, 2491–2505 CrossRef CAS PubMed
- X. Qiao, Q. Li, R. N. Schaugaard, B. W. Noffke, Y. Liu, D. Li, L. Liu, K. Raghavachari and L.-s. Li, Well-Defined Nanographene–Rhenium Complex as an Efficient Electrocatalyst and Photocatalyst for Selective CO2 Reduction, J. Am. Chem. Soc., 2017, 139, 3934–3937 CrossRef CAS PubMed
- S. M. Draper, D. J. Gregg, E. R. Schofield, W. R. Browne, M. Duati, J. G. Vos and P. Passaniti, Complexed Nitrogen Heterosuperbenzene: The Coordinating Properties of a Remarkable Ligand, J. Am. Chem. Soc., 2004, 126, 8694–8701 CrossRef CAS PubMed
- B. El Hamaoui, F. Laquai, S. Baluschev, J. Wu and K. Müllen, A Phosphorescent Hexa-peri-hexabenzocoronene Platinum Complex and its Time-Resolved Spectroscopy, Synth. Met., 2006, 156, 1182–1186 CrossRef CAS
- J. I. Mapley, J. N. Smith, G. E. Shillito, S. J. Fraser-Miller, N. T. Lucas and K. C. Gordon, Exploring the Excited States of a Hexa-peri-hexabenzocoronene-Substituted Dipyridophenazine Ligand and Its Metal Complexes, Inorg. Chem., 2023, 62, 11028–11036 CrossRef CAS PubMed
- D. J. Gregg, E. Bothe, P. Höfer, P. Passaniti and S. M. Draper, Extending the Nitrogen-Heterosuperbenzene Family: The Spectroscopic, Redox, and Photophysical Properties of “Half-Cyclized” N-1/2HSB and Its Ru(II) Complex, Inorg. Chem., 2005, 44, 5654–5660 CrossRef CAS PubMed
- H. He, J. Lee, Z. Zong, N. Liu, Y. Noh, V. M. Lynch, J. Oh, J. Kim, J. L. Sessler and X.-S. Ke, Precisely Metal Doped Nanographenes via A Carbaporphyrin Approach, Nat. Commun., 2025, 16, 1534 CrossRef CAS PubMed
- Y. Zhu, J. Borstelmann, O. Bertleff, J. Bergner, Z. Wei, C. Neiss, A. Görling, M. Kivala and M. A. Petrukhina, Unveiling the Multielectron Acceptor Properties of π-Expanded Pyracylene: Reversible Boat to Chair Conversion, J. Am. Chem. Soc., 2024, 146, 14715–14723 CrossRef CAS PubMed
- E. V. Varghese, C.-F. Gao, Y.-L. Chang, H.-Y. Chen and C.-H. Chen, Synthesis of Distorted Nitrogen-Doped Nanographenes by Partially Oxidative Cyclodehydrogenation Reaction, Chem.–Asian J., 2022, 17, e202200114 CrossRef CAS PubMed
- K. Y. Cheung, C. K. Chan, Z. Liu and Q. Miao, A Twisted Nanographene Consisting of 96 Carbon Atoms, Angew. Chem., Int. Ed., 2017, 56, 9003–9007 CrossRef CAS PubMed
- A. Graczyk, F. A. Murphy, D. Nolan, V. Fernández-Moreira, N. J. Lundin, C. M. Fitchett and S. M. Draper, Terpyridine-Fused Polyaromatic Hydrocarbons Generated via Cyclodehydrogenation and Used as Ligands in Ru(ii) Complexes, Dalton Trans., 2012, 41, 7746–7754 RSC
- A. B. S. Elliott, R. Horvath, X.-Z. Sun, M. G. Gardiner, K. Müllen, N. T. Lucas, M. W. George and K. C. Gordon, Long-Lived Charge Transfer Excited States in HBC-Polypyridyl Complex Hybrids, Inorg. Chem., 2016, 55, 4710–4719 CrossRef CAS PubMed
- Deposition numbers 2422960 (for 2Re) and 2422959 (for 4Re) contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
- A. Seridi, M. Wolff, A. Boulay, N. Saffon, Y. Coulais, C. Picard, B. Machura and E. Benoist, Rhenium(I) and Technetium(I) Complexes of a Novel Pyridyltriazole-Based Ligand Containing an Arylpiperazine Pharmacophore: Synthesis, Crystal Structures, Computational Studies and Radiochemistry, Inorg. Chem. Commun., 2011, 14, 238–242 CrossRef CAS
- M. Obata, A. Kitamura, A. Mori, C. Kameyama, J. A. Czaplewska, R. Tanaka, I. Kinoshita, T. Kusumoto, H. Hashimoto, M. Harada, Y. Mikata, T. Funabiki and S. Yano, Syntheses, Structural Characterization and Photophysical Properties of 4-(2-pyridyl)-1,2,3-triazole Rhenium(i) Complexes, Dalton Trans., 2008, 3292–3300 RSC
- S. K. Chandy, S. A. Bowers, M. Yin, L. Liu, K. Raghavachari and L.-s. Li, Proton-Coupled, Low-Energy Pathway for Electrocatalytic CO2 Reduction at Re(Diimine) Complexes with a Conjugated Pyrazinyl Moiety, Inorg. Chem., 2022, 61, 17505–17514 CrossRef CAS PubMed
- J. D. B. Koenig, Z. S. Dubrawski, K. R. Rao, J. Willkomm, B. S. Gelfand, C. Risko, W. E. Piers and G. C. Welch, Lowering Electrocatalytic CO2 Reduction Overpotential Using N-Annulated Perylene Diimide Rhenium Bipyridine Dyads with Variable Tether Length, J. Am. Chem. Soc., 2021, 143, 16849–16864 CrossRef CAS PubMed
- K. Talukdar, S. Sinha Roy, E. Amatya, E. A. Sleeper, P. Le Magueres and J. W. Jurss, Enhanced Electrochemical CO2 Reduction by a Series of Molecular Rhenium Catalysts Decorated with Second-Sphere Hydrogen-Bond Donors, Inorg. Chem., 2020, 59, 6087–6099 CrossRef CAS PubMed
- J. M. Smieja and C. P. Kubiak, Re(bipy-tBu)(CO)3Cl−improved Catalytic Activity for Reduction of Carbon Dioxide: IR-Spectroelectrochemical and Mechanistic Studies, Inorg. Chem., 2010, 49, 9283–9289 CrossRef CAS PubMed
- M. Yin, S. K. Chandy, S. A. Bowers, K. Raghavachari and L.-S. Li, Selective Electrocatalyzed Reduction of CO2 to Formate by Mononuclear Mo(Diimine)(CO)4 Complexes, ChemCatChem, 2023, 15, e202300523 CrossRef CAS
- W. Nie, D. E. Tarnopol and C. C. L. McCrory, Enhancing a Molecular Electrocatalyst's Activity for CO2 Reduction by Simultaneously Modulating Three Substituent Effects, J. Am. Chem. Soc., 2021, 143, 3764–3778 CrossRef CAS PubMed
- J. D. B. Koenig, W. E. Piers and G. C. Welch, Promoting Photocatalytic CO2 Reduction through Facile Electronic Modification of N-annulated Perylene Diimide Rhenium Bipyridine Dyads, Chem. Sci., 2022, 13, 1049–1059 RSC
- P. Karak, S. K. Mandal and J. Choudhury, Exploiting the NADP+/NADPH-like Hydride-Transfer Redox Cycle with Bis-Imidazolium-Embedded Heterohelicene for Electrocatalytic Hydrogen Evolution Reaction, J. Am. Chem. Soc., 2023, 145, 17321–17328 CrossRef CAS PubMed
- C.-L. Wang, H. Yang, J. Du and S.-Z. Zhan, Catalytic Performance of a Square Planar Nickel Complex for Electrochemical- and Photochemical-driven Hydrogen Evolution from Water, Inorg. Chem. Commun., 2021, 131, 108780 CrossRef CAS
- T. H. To, D. B. Tran, V. Thi Thu Ha and P. D. Tran, Electrocatalytic H2 Evolution Using Binuclear Cobalt Complexes As Catalysts, RSC Adv., 2022, 12, 26428–26434 RSC
- H.-B. Cui, J.-H. Li, X. Zhang, M. Zhou, Z.-Z. Huang, Y.-C. Lai, J.-X. Qiu, Y.-J. Ren and H.-X. Zhang, Electrocatalytic Hydrogen Evolution by Co(II) Complexes of Bistriazolylpyridines, Int. J. Hydrogen Energy, 2023, 48, 10891–10902 CrossRef CAS
- J. Liu, R.-Z. Liao, F. W. Heinemann, K. Meyer, R. P. Thummel, Y. Zhang and L. Tong, Electrocatalytic Hydrogen Evolution by Cobalt Complexes with a Redox Non-Innocent Polypyridine Ligand, Inorg. Chem., 2021, 60, 17976–17985 CrossRef CAS PubMed
- L. Chen, B. Xie, T. Li, C. Lai, J.-X. Cao, R.-W. Ji, M.-N. Liu, W. Li, D.-L. Zhang and J.-Y. He, Heteroleptic Nickel Complexes Bearing O-Methyldithiophosphate and Aminodiphosphine Monosulfide Ligands As Robust Molecular Electrocatalysts for Hydrogen Evolution, Appl. Organomet. Chem., 2022, 36, e6725 CrossRef CAS
- F. Kamatsos, M. Drosou and C. A. Mitsopoulou, Heteroleptic Thiolate Diamine Nickel Complexes: Noble-Free-Metal Catalysts in Electrocatalytic And Light-Driven Hydrogen Evolution Reaction, Int. J. Hydrogen Energy, 2021, 46, 19705–19716 CrossRef CAS
- Q.-C. Chen, S. Fite, N. Fridman, B. Tumanskii, A. Mahammed and Z. Gross, Hydrogen Evolution Catalyzed by Corrole-Chelated Nickel Complexes, Characterized in all Catalysis-Relevant Oxidation States, ACS Catal., 2022, 12, 4310–4317 CrossRef CAS
- A. Upadhyay, H. Meena, R. K. Jha, Kanika and S. Kumar, Isolation of Monomeric Copper(Ii) Phenolate Selenoether Complexes Using Chelating Ortho-Bisphenylselenide-Phenolate Ligands and their Electrocatalytic Hydrogen Gas Evolution Activity, Dalton Trans., 2022, 51, 7284–7293 RSC
- S. Kumar Padhi, E. Ahmad, S. Rai and B. Panda, Kinetics and Mechanistic Study of Electrocatalytic Hydrogen Evolution by [Co(Fc-Tpy)2]2+, Polyhedron, 2020, 187, 114677 CrossRef CAS
- H.-J. Yin, Z. Wang, Z.-Y. Zhao, X.-Y. Jiang, J.-Y. Yu, L.-M. Yang, Y.-M. Zhang, W. Liu and C.-L. Ni, Synthesis, Crystal Structure and Properties of Electro-Catalysis for Hydrogen Production of a Molecular Nickel Catalyst Based on Bis(1,2,5-Thiadiazole-3,4-Dithiolate) Ligand, J. Mol. Struct., 2023, 1274, 134501 CrossRef CAS
- K. Sudhakar and P. K. Panda, Tuning Proton Reduction Efficiencies of Copper Corrole in Electrocatalysis via Multiple β-Chloro Substitution, ACS Appl. Energy Mater., 2022, 5, 13492–13500 CrossRef CAS
- M. Drosou, F. Kamatsos and C. A. Mitsopoulou, Recent Advances in the Mechanisms of the Hydrogen Evolution Reaction by Non-Innocent Sulfur-Coordinating Metal Complexes, Inorg. Chem. Front., 2020, 7, 37–71 RSC
- H. M. Castro-Cruz and N. A. Macías-Ruvalcaba, Porphyrin-Catalyzed Electrochemical Hydrogen Evolution Reaction. Metal-Centered and Ligand-Centered Mechanisms, Coord. Chem. Rev., 2022, 458, 214430 CrossRef CAS
- H. Rao, Z.-Y. Wang, H.-Q. Zheng, X.-B. Wang, C.-M. Pan, Y.-T. Fan and H.-W. Hou, Photocatalytic Hydrogen Evolution from A Cobalt/Nickel Complex with Dithiolene Ligands Under Irradiation With Visible Light, Catal. Sci. Technol., 2015, 5, 2332–2339 RSC
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2422959 and 2422960. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc02827b |
| This journal is © The Royal Society of Chemistry 2025 |