
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRegulating CRISPR/Cas12a trans-cleavage on the hairpin DNA–MB nanointerface for enhanced multiplexed sensing application†
Chenxi
Zhao
a,
Lijie
Du
a,
Dike
Jiang
b,
Jing
Hu
 *a and
Xiandeng
Hou
*ac
*a and
Xiandeng
Hou
*ac
aAnalytical & Testing Centre, Sichuan University, Chengdu, Sichuan 610064, China. E-mail: houxd@ scu.edu.cn; hujing2019@scu.edu.cn
bKey Laboratory of Animal Diseases and Human Health of Sichuan Province, College of Veterinary Medicine, Sichuan Agricultural University, Chengdu, 611130, China
cKey Lab of Green Chem & Tech (MOE) at College of Chemistry, Sichuan University, Chengdu, Sichuan 610064, China
First published on 19th May 2025
Abstract
RNA-programmed clustered regularly interspaced short palindromic repeats (CRISPR) and CRISPR-associated systems (Cas) have garnered considerable attention in gene editing and molecular diagnostics. However, the precise control over the catalytic activity and cleavage site of Cas12a, a typical subtype of the Cas family, as well as its combination with downstream dynamic DNA technology remains challenging. Here, it was demonstrated that the cleavage activity and site can be modulated by adjusting the root, loop and stem of hairpin DNA reporters attached onto magnetic beads (MBs). The accelerated and controlled Cas12a trans-cleavage on the MB-hairpin DNA nanointerfaces is leveraged to trigger downstream hybridization chain reaction (HCR). Capitalizing on fluorescence (FL) labeling or double-stranded DNA (dsDNA) intercalators (SYBR green I or Ru-dppz), multiplexed detection (label or label-free, single-mode FL or dual-mode FL/ICP-MS) can be realized. Taking porcine pseudorabies virus (PRV) DNA as a model target, the potential application of this approach was demonstrated in clinical diagnosis, which achieves 100% accuracy that rivals the gold standard PCR. This methodology holds promise in a wide array of biological applications.
Introduction
The efficient trans-cleavage nuclease activity of the RNA-programmed CRISPR-Cas12a system has spurred intense research interest from gene editing to imaging and molecular diagnostics.1–3 Upon the base pairing of a nucleic acid target to the complementary crRNA guide, the trans-cleavage activity of Cas12a nuclease is activated, randomly cleaving single-stranded DNA (ssDNA) fragments.4 Typically, in the presence of a reporter ssDNA with a fluorophore and a quencher labelled at both ends (FQ-ssDNA), the fluorescence will witness a gradual increase with the degradation of the ssDNA reporter, entailing the signal transduction of molecular recognition events and diagnostics.5Although CRISPR/Cas12a-based technology has demonstrated great potential in molecular diagnostics,6 limitations become evident when low levels of specific nucleic acid targets in complex real samples are to be rapidly detected. For example, the trans-cleavage activity is highly dependent on the random diffusion and collision between the activated Cas12a protein and the DNA reporter.7 In the case of free FQ-ssDNA reporters, the binding and interplay with activated Cas12a protein is far from sufficient.8 Besides, the inefficient quenching of the fluorophore by the quencher leads to a relatively high fluorescence background and compromises the detection sensitivity. Therefore, a reporter ssDNA with a length of less than 30 nt has been usually adopted to ensure proximity and efficient Förster resonance energy transfer (FRET) between the fluorophore and quencher.9 The use of short free FQ-ssDNA reporters, however, brings about a new dilemma. That is, the resultant DNA fragments from the arbitrary site of trans-cleavage cannot be leveraged for downstream dynamic DNA nanotechnology (such as hybridization chain reaction or strand displacement amplification), which has considerably limited the flexibility of sensor design and analytical performance. Probably because of this, current signal amplification strategies mainly concentrate on upstream pre-amplification including polymerase chain reaction (PCR) and isothermal techniques (e.g. recombinase polymerization amplification, RPA and loop-mediated isothermal amplification, LAMP). Most noteworthy are those that are well-known such as DETECTR,10 SHERLOCK,11 HOLMES,12 Cas12aVDet13 and so forth. To address these limitations and fulfil the capability of CRISPR-Cas systems, it is imperative to control the kinetics, activity and site of CRISPR-Cas12a cleavage, and bridge the gap with downstream dynamic DNA technology.
Regarding kinetics, a feasible resolution may be the use of simple three-dimensional scaffolds that colocalize DNA reactants in a confined space, such as loading abundant DNA onto gold nanoparticles (AuNPs) or magnetic beads (MBs).14–16 Accelerated reaction kinetics and enhanced performance have been widely observed in the literature and our previous work.17,18 Featuring cost-effective and facile magnetic separation, trans-cleavage on the MB-DNA nanointerfaces enables simpler, faster, and improved diagnosis of possible HBV infections. In the pursuit of higher Cas12a trans-cleavage activities, other DNA secondary structures (e.g. hairpin,19 triplex,20 and G-quadruplexes21,22) have been embraced by researchers. The special structural arrangement of these DNA secondary structures has, for some unknown reasons, contributed to faster reaction kinetics and enhanced activities. Molecular dynamics studies have demonstrated that the stem-loop DNA has a higher affinity to the Cas12a catalytic pocket than ssDNA, and hairpins with different loop lengths make a big difference.23 This has significant implications in the design of CRISPR-Cas systems with controllable activity and site of cleavage, potentially compatible with dynamic DNA nanotechnology and holding promise for an array of biological applications. In terms of signal output, alternative methods, such as electrochemical methodology,24 visual detection25,26 and inductively coupled plasma mass spectrometry (ICP-MS),27,28 have been well established, among which ICP-MS has the advantage of low background, high sensitivity and wide dynamic range. Nonetheless, the developed detection platforms are mostly single-mode methods. The combination of two or more approaches, namely dual-mode or multi-mode detection, could potentially enhance the accuracy and reliability of bioassays by the cross validation between/among different methods.29–31 However, to the best of our knowledge, the rational control of CRISPR-Cas12a trans-cleavage on the MB-hairpin DNA nanointerfaces has not been reported, nor its marriage with downstream dynamic DNA technology, nor have the ICP-MS based dual-mode or multiplexed detection platforms been developed for molecular diagnostic purposes.
Herein, we proposed the construction of various hairpin DNA-modified MBs and investigated the trans-cleavage behavior on these different 3D nanointerfaces. The use of MB-hairpin DNA probes promises to accelerate the trans-cleavage kinetics, simplify the separation and purification process, and serve as a platform for the implementation of downstream reactions. After a critical evaluation of the impact of the spacer (or root), stem and loop domain, the controlled CRISPR-Cas12a trans-cleavage with desirable catalytic activity and cleavage site was attained. On top of that, the hybridization chain reaction (HCR) was further conducted with the hairpin DNA–MB residues after controlled Cas12a cleavage. In addition to fluorescent labelling, capitalizing on the intercalation of SYBR green I or Ru-dipyridophenazine (dppz) to the long dsDNA concatemers from the HCR, label-free fluorescence and/or inductively coupled plasma mass spectrometry (ICP-MS) detection were also described. Taking porcine pseudorabies virus (PRV) DNA as a model target, the potential application of the FL/ICP-MS dual-mode CRISPR-Cas12a detection platform in clinical diagnosis was explored, achieving 100% accuracy that rivals the gold standard PCR. This study attempts to provide a deeper understanding and better control of the Cas12a cleavage that can be used to assist the development of improved diagnostic platforms and holds promise in a wide array of biological applications.
Results and discussion
Feasibility of trans-cleavage on the hairpin DNA–MB nanointerface
The feasibility of trans-cleavage on the hairpin DNA–MB interface was investigated in the first place employing fluorescence for signal output, and compared to those using a free ssDNA and a free hairpin DNA as substrates (Fig. 1A). To this end, the two terminals of the free ssDNA and hairpin DNA strands were labeled with a fluorophore (F) and a quencher (Q), respectively, while those of the hairpin DNAs to be assembled on MBs were labeled with biotin (B) and a fluorophore (F), yielding hairpin DNA–MB nanointerfaces via specific streptavidin–biotin interaction. The sequences of the ssDNA-FQ, the hairpin DNA-FQ and the hairpin DNA-FB were listed in Table S1.† Unless otherwise specified, a conserved sequence of PRV DNA containing a 5′ TTTG protospacer-adjacent motif (PAM) was selected as a model target and used to activate the CRISPR-Cas12a system in a feasibility test and experimental conditions exploration. Once activated, the ssDNA-FQ, the hairpin DNA-FQ, and the hairpin DNA-FB-MBs were degraded with the fluorescence of the labeled fluorophores recovered. The fluorescence emission of 6-FAM at 520 nm (Ex = 480 nm) was recorded at cleavage time of 3, 5, 10, 15 and 20 min for each system. As shown in Fig. 1B and S1,† in the presence of target PRV DNA, the fluorescence intensities gradually increased with cleavage time, indicating that CRISPR-Cas12a worked well on not only the free ssDNA and the hairpin DNA, but also the hairpin DNA–MB nanointerfaces. Note that an H1M microplate reader was used throughout this work for fluorescence detection, and the fluorescence intensities recorded at a specific wavelength (as in Fig. 1B) were different from those read from the fluorescence spectra (Fig. S1†). Compared with the free ssDNA-FQ, the hairpin-FQ and the hairpin-FB-MB substrates featured a significantly lower fluorescence background due to the close contact between the fluorophore and the quencher, coinciding with previous publications,32,33 with further advantages such as higher affinity of CRISPR-Cas12a towards hairpin DNA structures and faster rate of trans-cleavage. More importantly, the localized MBs with high hairpin DNA density showed an even faster trans-cleavage rate and higher signal-to-background ratios (Fig. S2†), and this is of great importance for practical applications allowing for the easy separation and purification of the MBs simply using a magnet.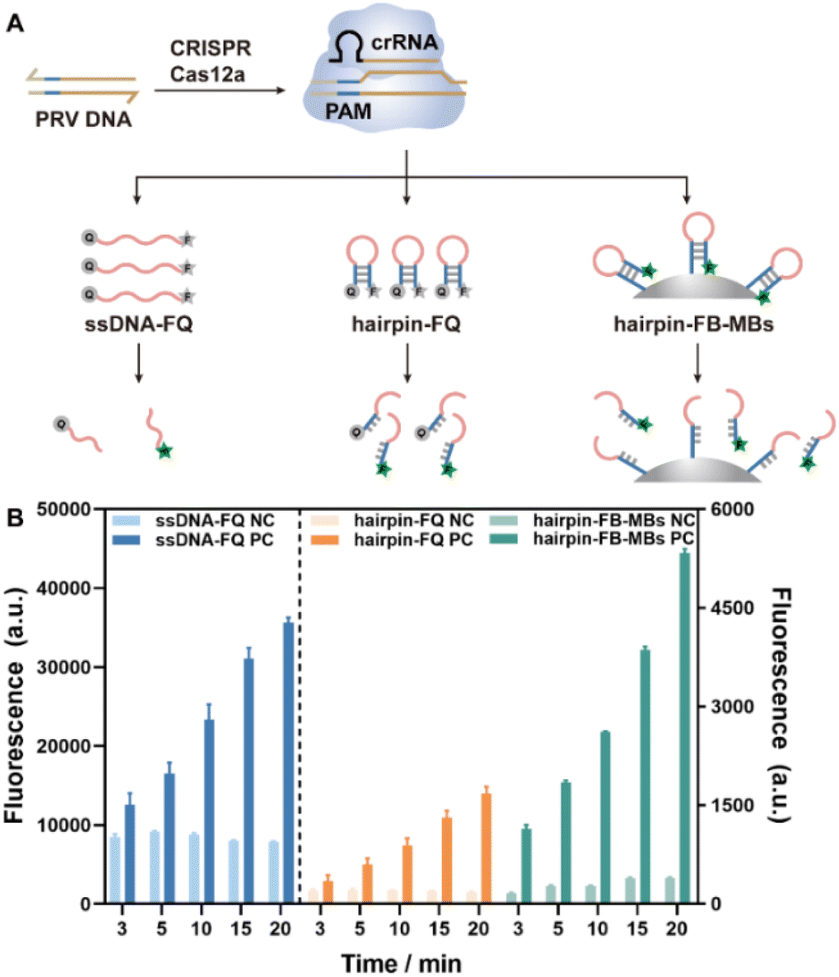 | ||
| Fig. 1 (A) Schematic illustration of the CRISPR-Cas12a trans-cleavage of the ssDNA-FQ, the hairpin DNA-FQ and the hairpin DNA-FB-MBs; and (B) the fluorescence intensities at different cleavage time using the ssDNA-FQ, the hairpin DNA-FQ and the hairpin DNA-FB-MBs as substrates, respectively. | ||
Exploring controlled cleavage on the hairpin DNA–MB nanointerface
Hairpin DNA, also known as a stem-loop structure, is one of the specific DNA secondary structures consisting of an unpaired loop and a double-stranded stem occasionally with elongated sticky ends (or called root).34 Since activated Cas12a indistinguishably cleaves single strands, both the loop and root region of the hairpin structure can be cleaved. Therefore, the controlled trans-cleavage of the hairpin DNA on MBs is crucial for biosensor development.To evaluate the influence of the root region on the trans-cleavage of CRISPR-Cas12a, a series of hairpin DNA sequences with the same loop but different root length (0, 3, 6, 10 nt) were separately assembled onto the MBs (Fig. 2A). All the hairpin DNA sequences were labeled with a FAM fluorophore at the 5′ end. When activated Cas12a cleaved the root, the hairpin DNA will detach from the surface of MBs into the supernatant, generating a fluorescence emission signal; while activated Cas12a cleaved the loop portion, the stem structures remained stable at room temperature due to the high melting temperatures (Tm, Fig. S3†), thereby no fluorescence was detected in the supernatant. As can be clearly seen from Fig. 2B and S4,† with the increase of the root length, more intense fluorescence was detected in the supernatant, implying higher probability of cleavage occurring at the root region. The proportions of hairpin DNA–MBs cleaved at the root region were roughly estimated, which reached approximately 50% when the root length was 10 nt (Fig. 2C). Presumably, the hairpin DNA–MBs with longer roots featured lower steric hindrance and more accessible cleavage sites, thus facilitating the recognition and cleavage of Cas12a to release more fluorophores. In contrast, when the root length was 0 nt, the trans-cleavage towards the root could be largely inhibited and cleavage was confined to the loop section, achieving precise control of the cleavage site.
 | ||
| Fig. 2 (A) Schematic illustration of the CRISPR-Cas12a trans-cleavage of hairpin DNA–MBs with different root lengths; (B) the fluorescence intensities of the supernatant solutions from the cleavage of hairpin DNA–MBs with different root lengths; and (C) the estimated proportions of cleavage occurring at the root region. | ||
Since the RuvC domain is responsible for the trans-cleavage activity of Cas12, and it has a higher binding affinity towards the hairpin structures than the ssDNA, hairpin sequences with the same stem structure but different loop lengths (0C, 5C, 10C, 15C and 20C, C represents cytosine) were attached to the MBs to investigate the impact of the loop structure. Here, the root and stem lengths were kept at 0 nt and 24 nt, respectively, and the hairpin DNA sequences were all labeled with a FAM fluorophore at the 5′ end. As expected, in the presence of activated CRISPR-Cas12a, only the loop domain will be cleaved. However, the hairpin DNA with cleaved loops would not dissociate from the MBs as the stem part was rather stable (Tm = 69.2 °C, Fig. S5†). To visualize the proportions of hairpin DNA with Cas12a-cleaved loops, two means were adopted to denature the stem dsDNA and release the FAM-labelled ssDNA (Fig. 3A), heating at 75 °C for 10 min or Exonuclease III (Exo III) digestion. In this manner, the detected fluorescence intensities of FAM could reveal the impact of loop length on the trans-cleavage activity of CRISPR-Cas12a. As demonstrated in Fig. 3B and C, irrespective of the denaturing method adopted (heating or Exo III), the supernatant fluorescence increased with the loop length, reaching the maximum at a loop length of 15 nt and then levelled off. This suggests a stronger binding of hairpin DNA with greater loop lengths to the Cas12a catalytic pocket, substantiating that the loop length can be used to regulate the trans-cleavage activity.
 | ||
| Fig. 3 (A) Schematic illustration of the CRISPR-Cas12a trans-cleavage of hairpin DNA–MBs with different loop lengths. The loop-cleaved hairpin DNA–MBs were denatured by either heating at 75 °C for 10 min or digesting with exonuclease III to release the fluorophores; (B) the fluorescence intensities of the supernatant solutions from the cleavage of hairpin DNA–MBs with different loop lengths (loop-cleaved hairpin DNA–MBs denatured by heating); and (C) the fluorescence intensities of the supernatant solutions from the cleavage of hairpin DNA–MBs with different loop lengths (loop-cleaved hairpin DNA–MBs denatured by Exo III). | ||
Stem length has a direct impact on the stability of the hairpin structure. Hairpin sequences with the same loop structure (10 nt) but different stem lengths (6, 8, 10, 12 and 14 bp) were then assembled on the MBs (Fig. S6†). Hereafter, the hybridization chain reaction (HCR) was incorporated to facilitate the combination of CRISPR-Cas12a with downstream dynamic DNA technology, and in turn, to evaluate the impact of stem length on the trans-cleavage (Fig. 4A). Specifically, the ssDNA fragment between the loop and the MBs served as an initiator (H0) to trigger the chain hybridization reaction with H1-FAM and H2 strands. The HCR among H0, H1 and H2 in solution was verified by the polyacrylamide gel electrophoresis (PAGE) analysis of the HCR products (Fig. S7†). Changes in the Gibbs free energy during the HCR simulated by NUPACK software also proved the hybridization of H0, H1 and H2 (Fig. S8†). In addition, the H0-modified MBs became more negative after H1 and H2 were introduced (Fig. S9†), demonstrating the feasibility of the HCR among H0, H1 and H2 on MBs. Since a free poly-C tail may be generated when the hairpin was cleaved by Cas12a, H0 with different lengths of poly-C tail (0, 5C, 10C) were used to trigger the HCR either in solution or on the MBs (Fig. S10†). PAGE analysis demonstrated that long-chain fragments exceptionally occurred in the lanes loaded with the HCR products from H0-xC, H1 and H2 (Fig. S10B†). Moreover, the fluorescence intensities originating from the HCR of MBs-H0-xC with H1-FAM and H2 were less affected by the length of the poly-C tail (Fig. S10C†), implying that the poly-C tail would not interfere with the HCR reaction.
 | ||
| Fig. 4 (A) Schematic illustration of the CRISPR-Cas12a trans-cleavage of hairpin DNA–MBs with different stem lengths. The loop-cleaved hairpin DNA–MBs were separated, heated or otherwise, and hybridized with H1-FAM and H2; (B) the fluorescence intensities from the HCR of loop-cleaved hairpin DNA–MBs (no heating) with H1-FAM and H2; and (C) the fluorescence intensities from the HCR of loop-cleaved hairpin DNA–MBs (heating) with H1-FAM and H2. | ||
The Cas12a trans-cleavage of hairpin DNA–MBs with different stem length was studied and the results are presented in Fig. 4 (here the H0 sequence was kept the same for all stem lengths of hairpin DNA–MBs). With stems of length 6 or 8 bp, the H0 fragment within the hairpin structure easily hybridized with H1-FAM and H2, yielding intense fluorescence whether in the absence of PRV DNA (inactivated CRISPR-Cas12a, NC, Fig. 4B) or in the presence of PRV DNA (activated CRISPR-Cas12a, PC, Fig. 4B); whereas with stems of length 10 bp and longer, the hairpin structure on MBs was so stable that the HCR became rather difficult, generating negligible fluorescence whether the loop was cleaved or not (NC/PC, Fig. 4B); however, if heating denaturing was implemented on the loop-cleaved hairpin DNA–MBs prior to the HCR, significant fluorescence can be observed for those with stem length longer than 10 bp (PC, Fig. 4C). To better visualize this process, FAM-labeled hairpin DNAs (root length 10 nt, stem length 14 bp, loop length 10 nt) were assembled onto the MBs (Fig. S11A†). In the presence of activated Cas12a, both the loop and the root domain would be cleaved, releasing root-cleaved FAM-hairpin into the solution and loop-cleaved DNA hairpins were retained on MBs. After heating, the loop-cleaved hairpin DNA–MBs were denatured and the FAM-labeled ssDNA fragments were further released into the solution. Following the introduction of ROX-labeled H1 (H1-ROX) and H2 strands, the HCR would be initiated on the MBs. The emergence of 6-FAM fluorescence in the solutions from both the root-cleaved hairpin DNA and the heating-denatured loop-cleaved hairpin DNA–MBs, as well as the ROX fluorescence after the HCR on MBs, altogether verified the whole process described above (Fig. S11B†). MBs-HP with longer stem lengths allowed further implementation of downstream HCR on the MBs after trans-cleavage and heating denaturing. Of note, the fluorescence from the loop-cleaved hairpin undergoing heating denaturing was higher than that from the root cleaved, which, again, proved the higher binding affinity of Cas12a to the loop domain than the root.
Leveraging the controlled trans-cleavage on hairpin DNA–MB nanointerfaces and the HCR for multiplexed detection of PRV DNA
From the above discussion, it can be concluded that the stem length and root length would exert a significant impact on the stability of the hairpin structure and the cleavage site, while the cleavage activity was partially affected by the loop domain (higher binding affinity of Cas12a to the loop domain than ssDNA) and undoubtedly, the concentration of activated Cas12a. The controlled trans-cleavage on the hairpin DNA–MB nanointerfaces was leveraged for the detection of the model target PRV DNA, combining downstream isothermal HCR amplification and various methods for multiplexed signal readout.To this end, hairpin DNA with the same loop sequence (10C) but different stem/root lengths (6 bp/18 nt, 12 bp/12 nt, 14 bp/10 nt, 24 bp/0 nt) was modified on MBs (denoted as MBs-HP-6, MBs-HP-12, MBs-HP-14 and MBs-HP-12, respectively), and subjected to trans-cleavage by the CRISPR-Cas12a activated with different concentrations of PRV dsDNA (Fig. 5A). The MBs were collected after cleavage, heating denatured or otherwise, followed by the introduction of H1-FAM and H2 strands to initiate possible HCR assembly. In this fashion, the ultimate fluorescence intensities were closely correlated to the concentration of PRV DNA. Since Cas12a specifically cleaves ssDNA instead of RNA, DNA-RNA chimera hairpin structures have been recently used to control the cleavage site of Cas12a and expose the ssRNA sequence for subsequent assays.35–37 Here, DNA-RNA chimera hairpin assembled MBs (denoted as MBs-HP-D-R, 0 nt of root, 24 bp of stem, 10 nt of loop) were prepared in parallel, following the same procedures as DNA hairpin-MBs. Theoretically, the activated Cas12a merely cleaves ssDNA in the DNA-RNA chimera hairpin, generating MBs-ssRNA to trigger the HCR assembly with H1 and H2 strands. The feasibility to use the Cas12a-cleaved DNA-RNA chimera hairpin to initiate the HCR assembly with H1 and H2, both in solution and on MBs, was experimentally verified as shown in Fig. S12 and S13.†
 | ||
| Fig. 5 (A) Schematic illustration of CRISPR-Cas12a-mediated detection of PRV DNA employing different MBs-HP as cleaving substrates and the HCR for amplification. The relationship between the fluorescence intensities and the concentrations of PRV DNA using: (B) MBs-HP-6 as trans-cleavage substrates without heating denaturing; (C) MBs-HP-12 as trans-cleavage substrates without heating denaturing; (D) MBs-HP-12 as trans-cleavage substrates with heating denaturing; (E) MBs-HP-14 as trans-cleavage substrates with heating denaturing; (F) MBs-HP-24 as trans-cleavage substrates with heating denaturing; or (G) MBs-HP-D-R as trans-cleavage substrates without heating denaturing. | ||
Parameters affecting the Cas12a trans-cleavage were carefully evaluated, taking MBs-HP-D-R as an exemplified substrate (Fig. S14†). After that, the fluorescence intensities (Fig. 5) and corresponding fluorescence spectra (Fig. S15†) were recorded for Cas12a activated with different concentrations of PRV DNA using MBs-HP-6, MBs-HP-12, MBs-HP-14, MBs-HP-24 and MBs-HP-D-R as cleaving substrates, respectively. The results are presented in Fig. 5B–G. In the case of HP-6 with a root length of 18 nt and a stem length of 6 bp, the hairpin was ready to initiate the HCR with H1-FAM and H2 in the absence of PRV DNA and without heating denaturing. Note that the CRISPR-Cas12a system was inactivated when no target PRV DNA was added and the ultimate fluorescence in Fig. 5 referred to the portion retained on the MBs. With the increase in the concentration of PRV DNA, the fluorescence from the HCR gradually decreased, likely because the activated Cas12a easily cleaved the root of HP-6 on this occasion, and thus part of the HP-6 was detached from the MBs and subsequent HCR assembly was inhibited. When MBs-HP-12 (stem length 12 bp, root length 12 nt) served as the trans-cleavage substrate, the hairpin structure was stabler due to the greater stem length, and thus the HCR hardly occurred among HP-12, H1-FAM and H2, and much lower fluorescence (<2000 a.u.) was observed when no PRV DNA was added. The fluorescence intensities augmented at low concentrations of PRV DNA but declined at higher concentrations of PRV DNA, regardless of whether heating denaturing was applied or not (Fig. 5B and C). Presumably, at low concentrations of PRV DNA, the activated Cas12a preferentially cleaved the loop section due to the higher binding affinity, hence favouring the following HCR process and yielding stronger fluorescence; however, when the concentration of PRV DNA further increased, the probability of HP-12 to be cleaved at the root section soared so that less HP-12 was retained on MBs for the ensuing HCR. Note that heating denaturing seemed to facilitate the HCR at low concentrations of PRV DNA in the case of HP-12 and HP-14 (Fig. 5D and E), probably by exposing more single strands containing the H0 sequence. Furthermore, when MBs-HP-24 with the maximum stem length and minimum root length were to be cleaved, the trend became totally different, and the fluorescence intensities keeping increasing with the concentration of PRV DNA from 0 nM to 20 nM (Fig. 5F). This clearly indicates that controlled Cas12a trans-cleavage can be attained via hairpin DNA structure design and leveraged for biosensing purposes. It should be noted that when the MBs-DNA-RNA chimeric hairpin was used as the cleaving substrates, the HCR assembly could be triggered after Cas12a trans-cleavage of the DNA fragments without heating denaturing, likely due to the instability of DNA-RNA duplexes. The changes in fluorescence intensities versus the concentration of PRV DNA resembled that of MBs-HP-14 with heating denaturing (Fig. 5G). Despite that, DNA-RNA chimera hairpins were high in acquisition costs and low in stability and HCR efficiency, thus highlighting the advantage of using hairpin DNA-assembled MB substrates over their DNA-RNA counterparts in this study.
Apart from fluorescence labeling, label-free detection methods can be developed by taking advantage of the binding of intercalators (such as DNA-staining dye, SYBR green I, and Ru-dppz complexes) with the long dsDNA concatemers derived from the HCR. Moreover, the fluorescence of Ru-dppz and the considerable Ru atoms embedded into the dsDNA may be further exploited for an FL/ICP-MS dual-mode signal output (Fig. S16†). In this way, the label-free and dual-mode detection of PRV DNA can be realized. As manifested in Fig. S17,† capitalizing on SYBR green I or Ru-dppz for dsDNA staining, the fluorescence intensities of SYBR (single-mode) as well as the fluorescence intensities of Ru-dppz and the ICP-MS intensities of 101Ru (dual-mode) increased linearly with the concentration of PRV DNA from 0.5 to 10 nM following the procedures described earlier.
Clinical sample analysis
To date, various methods have been developed for the diagnosis of PRV DNA,38–40 mostly relying on real-time PCR (RT-PCR). Herein, a dual-mode FL/ICP-MS protocol was employed for the clinical diagnosis of possible PRV infections in swine. Twelve blood samples were collected from healthy and diseased swine, and the PRV-relevant DNA was extracted from the blood samples and identified by RT-qPCR as six PRV-negative and six PRV-positive samples (Table S4†). Meanwhile, the PCR amplicons were analyzed by the dual-mode method established in this work, combining controlled Cas12a trans-cleavage of MBs-HP-24, HCR assembly and Ru-dppz staining (Fig. 6A). The fluorescence and ICP-MS signals were recorded for these samples and visualized by heatmaps (Fig. 6B and E) and box plots (Fig. 6C and F). Evidently, there were significant differences in the FL and ICP-MS intensities between the group of negative and positive samples. Diagnoses by the developed dual-mode approach were in good agreement with those by RT-qPCR (Table S4†). Besides, the receiver operating characteristic (ROC) curve demonstrated that the Area Under the Curve (AUC) of both fluorescence and mass spectrometry methods was 1 (Fig. 6D and G), suggesting that this method should have important implications in clinical diagnosis with utmost sensitivity, specificity and accuracy of 100% that rivals the gold standard PCR. | ||
| Fig. 6 (A) Schematic illustration of the workflow for the clinical diagnosis of PRV infection using the developed protocol, combining controlled Cas12a trans-cleavage of MBs-HP-24, HCR assembly, Ru-dppz staining and FL/ICP-MS dual-mode detection. (B–D) Heatmaps, box plots and ROC curves of the PRV-negative and PRV-positive samples by fluorescence detection; and (E–G) heatmaps, box plots and ROC curves of the PRV-negative and PRV-positive samples by ICP-MS detection. Data presented as mean ± SD (n = 3) and compared with a two-tailed paired Student's t-test, **P < 0.01. | ||
Conclusions
In summary, the site, activity and kinetics of CRISPR-Cas12a trans-cleavage were regulated by conducting cleavage on the hairpin DNA–MB nanointerfaces. The results in this work demonstrated that the root length and stem length determined the cleavage site and stability of the hairpin structure (both uncleaved and loop-cleaved), while the cleavage activity was mainly affected by the loop domain (higher binding affinity of Cas12a to the loop domain than ssDNA) and the concentration of activated Cas12a. The controlled trans-cleavage on the hairpin DNA–MB nanointerfaces was leveraged for biosensing and molecular diagnostic combining isothermal HCR assembly, bridging the gap between the CRISPR-Cas system with downstream dynamic DNA technology. Apart from fluorescence labelling, label-free methods adopting dsDNA intercalators such as SYBR Green I and Ru-dppz were also exploited for single-mode or FL/ICP-MS dual-mode signal readout. Label-free detection, together with the cross-validation capability endowed by dual-mode signal readout, permitted simple, reliable and accurate clinical diagnosis of PRV infections that rivalled the gold-standard PCR. Above all, this study provides a deeper understanding and better control of the Cas12a cleavage that can be used to assist the development of improved diagnostic platforms, with significant implications in a wide array of biological applications.Data availability
Experimental details and additional data can be found in the attached ESI.†Author contributions
Chenxi Zhao, Jing Hu and Xiandeng Hou designed the research; Chenxi Zhao performed the research; Chenxi Zhao, Lijie Du and Jing Hu analysed the data; Dike Jiang collected the clinical samples; Chenxi Zhao drafted the manuscript, and Jing Hu and Xiandeng Hou edited it.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by the National Natural Science Foundation of China (Grant No. 22476136) and the “111 Centre” (B17030). We would like to thank Dr Chenghui Li of the Analytical & Testing Centre of Sichuan University for helpful assistance with zeta potential tests.References
- Y. Tang, L. Gao, W. Feng, C. Guo, Q. Yang, F. Li and X. C. Le, The CRISPR–Cas toolbox for analytical and diagnostic assay development, Chem. Soc. Rev., 2021, 50, 11844–11869 RSC.
- M. M. Kaminski, O. O. Abudayyeh, J. S. Gootenberg, F. Zhang and J. J. Collins, CRISPR-based diagnostics, Nat. Biomed. Eng., 2021, 5, 643–656 CrossRef CAS PubMed.
- P. Liu, Y. Lin, X. Zhuo, J. Zeng, B. Chen, Z. Zou, G. Liu, E. Xiong and R. Yang, Universal crRNA acylation strategy for robust photo-initiated one-pot CRISPR–Cas12a nucleic acid diagnostics, Angew. Chem., Int. Ed., 2024, 63, e202401486 CrossRef CAS PubMed.
- Z. Hu, A. Sun, J. Yang, G. Naz, G. Sun, Z. Li, J. Gogo Liu, S. Zhang and X. Zhang, Regulation of the CRISPR-Cas12a system by methylation and demethylation of guide RNA, Chem. Sci., 2023, 14, 5945–5955 RSC.
- J. S. Chen, E. Ma, L. B. Harrington, M. Da Costa, X. Tian, J. M. Palefsky and J. A. Doudna, CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity, Science, 2018, 360, 436–439 Search PubMed.
- R. Sun, Y. Zhao, W. Wang, J. G. Liu and C. Chen, Nonspecific interactions between Cas12a and dsDNA located downstream of the PAM mediate target search and assist AsCas12a for DNA cleavage, Chem. Sci., 2023, 14, 3839–3851 Search PubMed.
- S. Chen, R. Wang, S. Peng, S. Xie, C. Lei, Y. Huang and Z. Nie, PAM-less conditional DNA substrates leverage trans-cleavage of CRISPR-Cas12a for versatile live-cell biosensing, Chem. Sci., 2022, 13, 2011–2020 RSC.
- F. Li, J. Li, W. Yang, S. Yang, C. Chen, L. Du, J. Mei, Q. Tang, X. Chen, C. Yao, D. Yang, X. Zuo and P. Liu, Framework-hotspot enhanced trans cleavage of CRISPR-Cas12a for clinical samples detection, Angew. Chem., Int. Ed., 2023, 62, e202305536 CrossRef CAS PubMed.
- H. Hevekerl, T. Spielmann, A. Chmyrov and J. Widengren, Förster resonance energy transfer beyond 10 nm: Exploiting the triplet state kinetics of organic fluorophores, J. Phys. Chem. B, 2011, 115, 13360–13370 CrossRef CAS PubMed.
- J. S. Chen, E. Ma, L. B. Harrington, M. Da Costa, X. Tian, J. M. Palefsky and J. A. Doudna, CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity, Science, 2018, 360, 436–439 CrossRef CAS PubMed.
- M. J. Kellner, J. G. Koob, J. S. Gootenberg, O. O. Abudayyeh and F. Zhang, SHERLOCK: nucleic acid detection with CRISPR nucleases, Nat. Protoc., 2019, 14, 2986–3012 CrossRef CAS PubMed.
- S. Li, Q. Cheng, J. Wang, X. Li, Z. Zhang, S. Gao, R. Cao, G. Zhao and J. Wang, CRISPR-Cas12a-assisted nucleic acid detection, Cell Discovery, 2018, 4, 20 CrossRef PubMed.
- B. Wang, R. Wang, D. Wang, J. Wu, J. Li, J. Wang, H. Liu and Y. Wang, Cas12aVDet: A CRISPR/Cas12a-based platform for rapid and visual nucleic acid detection, Anal. Chem., 2019, 91, 12156–12161 CrossRef CAS PubMed.
- J. I. Cutler, E. Auyeung and C. A. Mirkin, Spherical nucleic acids, J. Am. Chem. Soc., 2012, 134, 1376–1391 CrossRef CAS PubMed.
- A. E. Prigodich, A. H. Alhasan and C. A. Mirkin, Selective enhancement of nucleases by polyvalent DNA-functionalized gold nanoparticles, J. Am. Chem. Soc., 2011, 133, 2120–2123 CrossRef CAS PubMed.
- J. I. Cutler, D. Zheng, X. Xu, D. A. Giljohann and C. A. Mirkin, Polyvalent oligonucleotide iron oxide nanoparticle “Click” conjugates, Nano Lett., 2010, 10, 1477–1480 CrossRef CAS PubMed.
- X. Fu, Y. Shi, F. Peng, M. Zhou, Y. Yin, Y. Tan, M. Chen, X. Yin, G. Ke and X. Zhang, Exploring the trans-cleavage activity of CRISPR/Cas12a on gold nanoparticles for stable and sensitive biosensing, Anal. Chem., 2021, 93, 4967–4974 CrossRef CAS PubMed.
- C. Zhao, L. Du, J. Hu and X. Hou, Recombinase polymerase amplification and target-triggered CRISPR/Cas12a assay for sensitive and selective Hepatitis B virus DNA analysis based on lanthanide tagging and inductively coupled plasma mass spectrometric detection, Anal. Chem., 2024, 96, 15059–15065 CrossRef CAS PubMed.
- N. Bagheri, A. Chamorro, A. Idili and A. Porchetta, PAM-engineered toehold switches as input-responsive activators of CRISPR-Cas12a for sensing applications, Angew. Chem., Int. Ed., 2024, 63, e202319677 CrossRef CAS PubMed.
- T. Li, R. Hu, J. Xia, Z. Xu, D. Chen, J. Xi, B. Liu, J. Zhu, Y. Li, Y. Yang and M. Liu, G.-triplex: A new type of CRISPR-Cas12a reporter enabling highly sensitive nucleic acid detection, Biosens. Bioelectron., 2021, 187, 113292 CrossRef CAS PubMed.
- Y. Li, T. Li, B. Liu, R. Hu, J. Zhu, T. He, X. Zhou, C. Li, Y. Yang and M. Liu, CRISPR-Cas12a trans-cleaves DNA G-quadruplexes, Chem. Commun., 2020, 56, 12526–12529 RSC.
- T. Li, D. Chen, X. He, Z. Li, Z. Xu, R. Li, B. Zheng, R. Hu, J. Zhu, Y. Li and Y. Yang, Leveraging Cas13a's trans-cleavage on RNA G-quadruplexes for amplification-free RNA detection, Chem. Commun., 2024, 60, 3166–3169 RSC.
- M. Rossetti, R. Merlo, N. Bagheri, D. Moscone, A. Valenti, A. Saha, P. R. Arantes, R. Ippodrino, F. Ricci, I. Treglia, E. Delibato, J. van der Oost, G. Palermo, G. Perugino and A. Porchetta, Enhancement of CRISPR/Cas12a trans-cleavage activity using hairpin DNA reporters, Nucleic Acids Res., 2022, 50, 8377–8391 CrossRef CAS PubMed.
- Y. Dai, R. A. Somoza, L. Wang, J. F. Welter, Y. Li, A. I. Caplan and C. C. Liu, Exploring the trans-cleavage activity of CRISPR-Cas12a (cpf1) for the development of a universal electrochemical biosensor, Angew. Chem., Int. Ed., 2019, 58, 17399–17405 CrossRef CAS PubMed.
- L. Ma, L. Peng, L. Yin, G. Liu and S. Man, CRISPR-Cas12a-powered dual-mode biosensor for ultrasensitive and cross-validating detection of pathogenic bacteria, ACS Sens., 2021, 6, 2920–2927 CrossRef CAS PubMed.
- Y. Jiang, M. Hu, A. Liu, Y. Lin, L. Liu, B. Yu, X. Zhou and D. Pang, Detection of SARS-CoV-2 by CRISPR/Cas12a-enhanced colorimetry, ACS Sens., 2021, 6, 1086–1093 CrossRef CAS PubMed.
- Y. Xu, B. Chen, M. He, G. Yuan and B. Hu, Dual-amplification single-particle ICP-MS strategy based on strand displacement amplification CRISPR/Cas12a amplification for homogeneous detection of miRNA, Anal. Chem., 2025, 97, 811–817 CrossRef CAS PubMed.
- Z. Deng, J. Hu, R. Liu and Y. Lv, Elemental probe-based CRISPR/Cas12a biosensing for sensitive tobramycin detection, At. Spectrosc., 2022, 43, 201–206 CAS.
- T. Zhang, C. Huang, Y. Jiao, L. Shao, D. Jiang, F. Li, W. Li and X. Gao, ICP-MS and fluorescence dual-mode detection of ZIKV-RNA based on quantum dot labeling with hybridization chain reaction, Talanta, 2024, 269, 125463 CrossRef CAS PubMed.
- G. Xiao, B. Chen, M. He and B. Hu, Dual-mode detection of avian influenza virions (H9N2) by ICP-MS and fluorescence after quantum dot labeling with immuno-rolling circle amplification, Anal. Chim. Acta, 2020, 1096, 18–25 CrossRef CAS PubMed.
- B. Chen, G. Xiao, M. He and B. Hu, Elemental mass spectrometry and fluorescence dual-mode strategy for ultrasensitive label-free detection of HBV DNA, Anal. Chem., 2021, 93, 9454–9461 CrossRef CAS PubMed.
- X. Wu, N. Pi, F. Deng, S.-Y. Tang, C. Zhang, L. Zhu, F. Sun, X. He, H. Li, S. Zhao, R. Xiang and Y. Li, Circular DNA enhanced amplification-free CRISPR/Cas12a assays for end-user friendly ultra-sensitive Porphyromonas gingivalis diagnosis, Microchem. J., 2024, 207, 111983 CrossRef CAS.
- E. Paialunga, N. Bagheri, M. Rossetti, L. Fabiani, L. Micheli, A. Chamorro-Garcia and A. Porchetta, Leveraging synthetic antibody–DNA conjugates to expand the CRISPR-Cas12a biosensing toolbox, ACS Synth. Biol., 2025, 14, 171–178 CrossRef CAS PubMed.
- J. Huang, X. Yang, X. He, K. Wang, J. Liu, H. Shi, Q. Wang, Q. Guo and D. He, Design and bioanalytical applications of DNA hairpin-based fluorescent probes, TrAC, Trends Anal. Chem., 2014, 53, 11–20 CrossRef CAS.
- K. Shi, S. Xie, R. Tian, S. Wang, Q. Lu, D. Gao, C. Lei, H. Zhu and Z. Nie, A CRISPR-Cas autocatalysis-driven feedback amplification network for supersensitive DNA diagnostics, Sci. Adv., 2021, 7, eabc7802 CrossRef CAS PubMed.
- C. Zhao, Z. Yang, T. Hu, J. Liu, Y. Zhao, D. Leng, K. Yang and G. An, CRISPR-Cas12a based target recognition initiated duplex-specific nuclease enhanced fluorescence and colorimetric analysis of cell-free DNA (cfDNA), Talanta, 2024, 271, 125717 CrossRef CAS PubMed.
- X. Zhao, X. Tian, Y. Wang, L. Li, Y. Yu, S. Zhao and J. Zhang, CRISPR-Cas12a-activated palindrome-catalytic hairpin assembly for ultrasensitive fluorescence detection of HIV-1 DNA, Anal. Chim. Acta, 2022, 1227, 340303 CrossRef CAS PubMed.
- H. Kang, X. Yang, R. Jiang, P. Gao, Y. Zhang, L. Zhou, X. Ge, J. Han, X. Guo and H. Yang, Ultrasensitive and visual detection of pseudorabies virus based on CRISPR-Cas12b system, Microb. Pathog., 2025, 203, 107447 CrossRef CAS PubMed.
- K. C. Shi, X. Hu, Y. W. Yin, Y. W. Shi, Y. Pan, F. Long, S. P. Feng and Z. Q. Li, Development of a triplex crystal digital RT-PCR for the detection of PHEV, PRV, and CSFV, Front. Vet. Sci., 2024, 11, 26–39 Search PubMed.
- L. Cao, W. Lv, A. Li, L. Yang, F. Zhou, F. Wen, S. Yuan, S. Huang, Z. Li and J. Guo, A SYBR green I-based multiplex real-time PCR for simultaneous detection of pseudorabies virus, porcine circovirus 3 and porcine parvovirus, BMC Vet. Res., 2025, 21, 10–19 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc01759a |
| This journal is © The Royal Society of Chemistry 2025 |