
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBorinine-FLP ring expansion: isolation of eight-membered B–P rings bridged by μ2 chalcogenide and chloronium ions†
Nathan C.
Frey‡
 a,
Samir Kumar
Sarkar‡
a,
Diane A.
Dickie
b,
Andrew
Molino
a and
Robert J.
Gilliard
Jr
*a
a,
Samir Kumar
Sarkar‡
a,
Diane A.
Dickie
b,
Andrew
Molino
a and
Robert J.
Gilliard
Jr
*a
aDepartment of Chemistry, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Building 18-596, Cambridge, MA 02139-4307, USA. E-mail: gilliard@mit.edu
bDepartment of Chemistry, University of Virginia, Charlottesville, Virginia 22904, USA
First published on 12th May 2025
Abstract
Boron–phosphorus (B–P) frustrated Lewis pairs (FLPs) are an important class of compounds for activating various small molecules. Utilizing the ring expansion reactivity of 9-chloro-9-borafluorene, a borinine-based FLP was synthesized. Various five-membered main-group element heterocycles were obtained via the reaction of the FLP with Me3NO, S8, and Se. Subsequent reduction of these species yielded the ring-expanded compounds, each featuring bridging B–E–B (E = O, S, Se) bonds. Similarly, halide abstraction from the FLP with AgNTf2 led to the formation of a cationic ring-expanded compound with a bridging B–Cl–B motif. This motif constitutes one of the first examples of a boron-stabilized chloronium ion, as verified using in-depth bonding analysis methods. Mechanistic pathways for the reduction- and halide abstraction-mediated ring expansion reactions are proposed with the aid of density functional theory. Electronic structure computations were performed to determine the best representation of bonding interactions in each compound, suggesting phosophorus(V)–chalcogen double bonding and chalcogen–boron(III) dative interactions within the heterocycles.
Introduction
Due to their partially filled d orbitals and broad range of oxidation states, transition metals have been used to activate a variety of chemical bonds. In recent years, however, there has been a surge in the use of main-group compounds to activate small molecules in a manner similar to transition metals. Collectively, this field has been recognized as “main-group metallomimetic chemistry”.1–9 At the forefront of these advances is boron, which by virtue of its vacant pz orbital, is inherently electron deficient.6,9 Nonetheless, recent developments in boron-based small molecule activation have shown that electron-rich boron systems, such as radicals,6,10–12 anions,13–20 and boron–boron multiply bonded systems,21–29 exhibit a broad array of reactivity toward various small molecules. One of the most commonly utilized methods of boron-based reactivity is through the use of Frustrated Lewis Pairs (FLPs).30–33Since the seminal report by Stephan,34 FLPs have been used to activate various small molecules, including H2, CO2, and NO.30–33,35 In typical FLPs, sterically bulky functional groups on the Lewis base (LB) and Lewis acid (LA) preclude the formation of typical acid–base adducts, resulting in a formal lone pair on the LB and an empty orbital on the LA. This configuration enables the FLP to act as both an electron donor and acceptor, resembling the frontier d orbitals of transition metals. Accordingly, FLPs have become a key component of main-group “metallomimetic” reactivity and catalysis for various hydrogenation, reduction, and small molecule capture reactions.30,32,33,36 While traditional FLPs consist of distinct Lewis acidic and basic fragments, recent studies have shown that species with highly strained rings with LB–LA moieties can also react like traditional FLPs.37–43 Accordingly, a theoretical study by Fernandez demonstrated that the ring-opening reactivity of these cyclic FLPs is dictated by ring strain.44 Electrochemical transformations of FLPs and their resulting products have been described to result in desired reactivity, including C–F functionalization and metal-free dehydrogenations.45–50 We hereby report the synthesis of a borinine-based FLP that activates chalcogen-containing small molecules to generate five-membered main-group element heterocycles, as well as the reactivity of these species with potassium graphite or silver(I) trifluoromethanesulfonamide to induce ring expansion.
Results and discussion
Expanding on our previous work to form bis(9-boraphenanthrene),51,52 and earlier reports by Fukushima,53 we sought to harness the ring expansion reactivity of 9-chloro-9-borafluorene to form reactive borinines. 9-Chloro-9-borafluorene was reacted with diphenyl(phenylethynyl)phosphane in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio in toluene under reflux conditions (Scheme 1), which led to the isolation of borylphosphinoethylene 1 as an off-white powder in 92% yield. The corresponding 11B{1H} resonance appeared at 0.3 ppm as a result of phosphorus-atom coordination to the boron center and is shifted upfield compared to 9-chloro-9-borafluorene (61.5 ppm), consistent with a tetracoordinate boron center.54 Single crystal X-ray diffraction (SC-XRD) analysis of compound 1 (Fig. 1) reveals that the six-membered boracycle ring is nonplanar due to phosphorus coordination, which forms a four-membered ring (B1–C13–C14–P1). The highly strained nature of the ring is indicated by the P1–C14–C13 angle (95.01(15)°), which is lower than the average for similar B–C–C–P motifs (96.504°, ranging from 94(1)° to 100.6(1)°),55 as well as the ideal C(sp2) bond angle of 120°. The B1–P1 bond length (2.018(2) Å) is also shorter than the average reported for similar systems with strained B–P bonds (2.062 Å, ranging from 1.985(1)° to 2.168(2)°).55
:1 ratio in toluene under reflux conditions (Scheme 1), which led to the isolation of borylphosphinoethylene 1 as an off-white powder in 92% yield. The corresponding 11B{1H} resonance appeared at 0.3 ppm as a result of phosphorus-atom coordination to the boron center and is shifted upfield compared to 9-chloro-9-borafluorene (61.5 ppm), consistent with a tetracoordinate boron center.54 Single crystal X-ray diffraction (SC-XRD) analysis of compound 1 (Fig. 1) reveals that the six-membered boracycle ring is nonplanar due to phosphorus coordination, which forms a four-membered ring (B1–C13–C14–P1). The highly strained nature of the ring is indicated by the P1–C14–C13 angle (95.01(15)°), which is lower than the average for similar B–C–C–P motifs (96.504°, ranging from 94(1)° to 100.6(1)°),55 as well as the ideal C(sp2) bond angle of 120°. The B1–P1 bond length (2.018(2) Å) is also shorter than the average reported for similar systems with strained B–P bonds (2.062 Å, ranging from 1.985(1)° to 2.168(2)°).55
 | ||
| Scheme 1 Synthesis of compound 1 from 9-chloro-9-borafluorene and diphenyl(phenylethynyl)phosphane. | ||
 | ||
| Fig. 1 Molecular structure of 1; hydrogen atoms are omitted for clarity. Anisotropic displacement parameters are depicted at 50% probability level. Selected distances (Å) and angles (°): B1–P1: 2.018(2); B1–Cl1: 1.885(3); C13–C14: 1.366(3); B1–C13–C14: 107.43(19); P1–C14–C13: 95.01(15)°. | ||
Because of the highly strained B–P ring, it was hypothesized that the four-membered ring may undergo ring expansion upon reaction with elemental chalcogens to form main-group element-doped five-membered rings, analogous to those previously reported.37,38,43 With compound 1 in hand, combination of the FLP with trimethylamine N-oxide (Me3N–O), elemental sulfur (S8), and grey selenium (Se) in dichloromethane (DCM) at room temperature led to the formation of compounds 2, 3, and 4, respectively (Scheme 2). Colorless, plate-shaped crystals of each species were grown in 90% (2), 93% (3), and 91% (4) yields. The 11B{1H} NMR spectra of compounds 2, 3, and 4 show signals at 10.9, 10.1, and 9.3 ppm, respectively, which are indicative of tetracoordinate boron centers. The 31P{1H} NMR peaks are observed at 72.9, 68.2, and 59.3 ppm, respectively, values that differ significantly from their E![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) PPh3 counterparts (OPPh3: 29.3 ppm, SPPh3: 43.2 ppm, SePPh3: 35.8 ppm).56 The identities of compounds 2–4 were verified via SC-XRD analysis (Fig. 2).
PPh3 counterparts (OPPh3: 29.3 ppm, SPPh3: 43.2 ppm, SePPh3: 35.8 ppm).56 The identities of compounds 2–4 were verified via SC-XRD analysis (Fig. 2).
 | ||
| Scheme 2 Ring expansion of compound 1 to form compounds 2–4. | ||
 | ||
| Fig. 2 Molecular structures of 2 (A), 3 (B), and 4 (C); hydrogen atoms are omitted for clarity. Anisotropic displacement parameters are depicted at 50% probability level. Selected distances (Å) and angles (°): 2: B1–P1: 2.527(3); B1–O1: 1.528(3); B1–Cl1: 1.915(3); P1–O1: 1.5433(17); C13–C14: 1.357(3); B1–C13–C14: 115.1(2); P1–C14–C13: 105.41(17); B1–O1–P1: 110.71(15) 3: B1–P1: 2.876(4); B1–S1: 1.968(3); B1–Cl1: 1.903(3); P1–S1: 2.0170(10); C13–C14: 1.352(5); B1–C13–C14: 121.0(3); P1–C14–C13: 112.3(2); B1–S1–P1: 92.39(4) 4: B1–P1: 2.965(5); B1–Se1: 2.110(4); B1–Cl1: 1.902(5); P1–S1: 2.1601(10); C13–C14: 1.349(5); B1–C13–C14: 123.2(3); P1–C14–C13: 113.7(3); B1–Se1–P1: 87.93(12). | ||
The solid-state structures of compounds 2–4 each feature B1–C13–C14–P1–E1 (E = O, S, Se) five-membered planar rings with an out-of-plane chlorine atom bound to the tetrahedral boron center. Relevant interatomic distances and angles are summarized in Fig. 2. As expected, the B1–P1 interatomic distance changes substantially from compound 1 (2.018(2) Å) after ring expansion to form compounds 2–4, reflecting the influence of the chalcogen size (2.527(3) Å for 2, 2.876(4) Å for 3, and 2.965(5) Å for 4). Because the phosphorus–chalcogen bond distances (1.5433(17) (2), 2.0170(10) (3), 2.1601(10) Å (4)) are all similar to the average C3PE bonds (1.495 Å (O), 1.978 Å (S), 2.127 Å (Se)), the PE interaction is most appropriately described as a double bond. Consequently, the boron–chalcogen interaction is best characterized as a dative interaction. The E→B distances in 2 (1.528(3) Å), 3 (1.968(3) Å), and 4 (2.110(4) Å) align with the average E→B distances reported in the literature (1.517 Å (O), 1.954 Å (S), 2.1670 Å (Se)).55 Notably, the B1–E1–P1 bond angle decreases as a function of chalcogen size, while the B1–C13–C14 and P1–C14–C13 bond angles each increase.
In an attempt to isolate the intramolecular phosphine-stabilized borinine anion via chemical reduction of compounds 2, 3, and 4 using 2 equivalents of potassium graphite (KC8), compounds 5, 6, and 7 were obtained from compounds 2, 3, and 4, respectively (Scheme 3, top). Colorless, plate-shaped crystals were obtained after several days in 88% yield for 5, 91% yield for 6, and 89% yield for 7.
 | ||
| Scheme 3 Synthesis of compounds 5, 6, 7 (top) and 8 (bottom). | ||
The 11B{1H} NMR spectra of compounds 5, 6, and 7 show signals at 0.7, −14.8, and −17.2 ppm, respectively, consistent with tetracoordinate boron centers. The 31P{1H} NMR peaks are observed at 18.1, 10.6, and −4.3 ppm for 5, 6, and 7, respectively. Both the 11B{1H} and 31P{1H} NMR peaks are shifted upfield compared to those of compounds 2, 3, and 4. When compound 1 was reacted with 1 equivalent of KC8 in THF, compound 5 was formed via THF activation. However, when THF was replaced with toluene, the reaction produced an analogue of compound 1 featuring a B–H bond, likely due to hydrogen abstraction from the solvent (Scheme S1†).
In attempts to form an intramolecular phosphine-stabilized borenium ion, compound 8 was synthesized by combining compound 1 with silver bis(trifluoromethanesulfonyl)imide (AgNTf2) in a 1:1 molar ratio in fluorobenzene at room temperature (Scheme 3, bottom). The 11B{1H}, 31P{1H}, and 19F NMR signals appear at 3.0, 21.3, and −77.6 ppm, respectively. Notably, the addition of excess AgNTf2 to compound 8 did not result in halide abstraction of the remaining chloride.
Compounds 5–7 were crystalized in a colorless, plate-like form from toluene at −37 °C. All three compounds (5–7) adopt a bowl-type geometry, where the phosphorus atoms of one borinine coordinate to the boron center of the other, forming an 8-membered ring with a central μ2-E (E = O, S, and Se) atom bound to the B atoms (Fig. 3A–C). The B–E–B (E = O, S, and Se) bond angles in 5–7 measure 121.7(3)°, 99.9(3)°, and 95.37(17)°, respectively. The average B–O, B–S, and B–Se bond lengths are 1.436(6), 1.895(10), and 2.041(6) Å, whereas the average B–P bond lengths in 5–7 are 2.078(4), 1.989(10), and 1.983(6) Å, respectively, each within the expected range. From these data, it is observed that when the B–E bond lengths increase, the B–P bond lengths decrease. Although similar dibora-bicyclononane structures with a μ2–O have been previously reported,57–65 the μ2–S (one example)66 and μ2–Se (no examples) bonds are exceedingly rare for this motif.
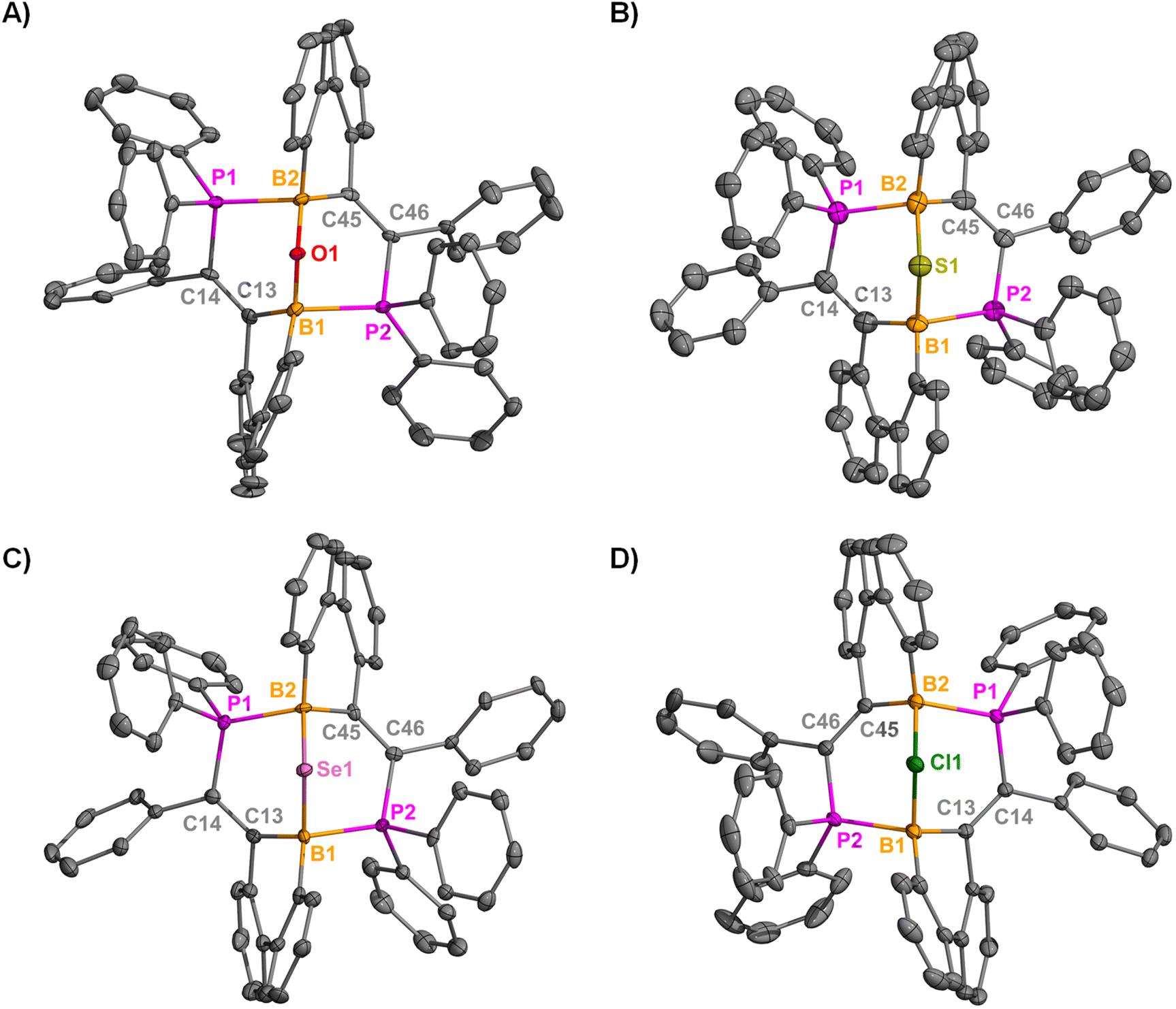 | ||
| Fig. 3 Molecular structures of 5 (A), 6 (B), 7 (C), and 8 (D); hydrogen atoms, solvents, and counteranions (8) are omitted for clarity. Anisotropic displacement parameters are depicted at 50% probability level. Selected distances (Å) and angles (°): 5: B1–P2: 2.080(4); B1–O1: 1.439(4); B2–P1: 2.075(4); B2–O1: 1.432(4); B1–O1–B2: 121.7(3) 6: B1–P2: 2.008(6); B1–S1: 1.893(6); B2–P1: 1.996(7); B2–S1: 1.902(7); B1–S1–B2: 99.9(3) 7: B1–P2: 1.976(4); B1–Se1: 2.035(5); B2–P1: 1.989(5); B2–Se1: 2.035(5); B1–Se1–B2: 95.37(17) 8: B1–P2: 1.9958(18); B1–Cl1: 1.9599(17); B2–P1: 1.9986(17); B2–Cl1: 1.9548(18); B1–Cl1–B2: 101.30(7). | ||
Compound 8 was obtained as yellow, block-like crystals from fluorobenzene at −37 °C, enabling its structural characterization via SC-XRD studies. Interestingly, compound 8 exhibits a bonding motif similar to 5–7, despite markedly different reaction conditions (Fig. 3D). The B1–Cl1 bond length (1.9599(17) Å) is nearly identical to the B2–Cl1 bond distance (1.9548(18) Å), and both exceed the average B–Cl bond length (1.812 Å, ranging from 1.41(4) Å to 2.360(2) Å) reported in the literature.55 Due to these similarities, compound 8 was subjected to theoretical analysis to understand its electronic structure (vide infra). Although other species containing a B–Cl–B bonding motif have been reported, they primarily exist as neutral or anionic species.67–72 The only known cationic B–Cl–B species were reported by Braunschweig73 and Chein and Chiu.74 It should be noted that neither Braunschweig nor Chein and Chiu characterized the bonding interactions between boron and chlorine within their reported species using theoretical methods. Braunschweig's [B2Cl3(IDip)2]+ displays significantly asymmetric B–Cl bonds (1.992(7) Å and 2.257(6) Å), which is the result of one B–Cl covalent interaction and one Cl→B dative interaction. However, the B–Cl bonds in [B2Cl3(IMes)2]+ (2.033(2) Å and 2.078(2) Å) are much more similar to one another. Furthermore, Chein and Chiu's masked borenium ion exhibits symmetric B–Cl bond lengths (1.991(1) Å and 1.990(1) Å), a feature also observed in compound 8.
With all compounds isolated, density functional theory (DFT) was employed to analyze the electronic structure of compounds 1–4. Analysis of the Wiberg Bond Indices (WBI) at the B3LYP75,76-D3(BJ)77/def2-TZVP78 level of theory indicates that the B–Cl in 1 can be regarded as a single bond (WBIB–Cl = 0.944). However, this bond weakens in the case of compound 2 (WBIB–Cl = 0.883), which can be rationalized by enhanced electron density at the boron center, resulting in weaker Cl→B π back-donation. As the chalcogen changes from S to Se, WBIB–Cl remains mostly unchanged (WBIB–Cl = 0.938 for 3, WBIB–Cl = 0.968 for 4) and reflects the reduced orbital overlap between B and the heavier chalcogens.
Model systems based on similar scaffolds by Rivard (A)40 and Nikonov (B),38 as well as (Ph)3P–BPh2Cl, were used as comparison points to the interaction between boron and phosphorus in compound 1. Representations of all model systems used for theoretical studies are available in Fig. S34.† Localized molecular orbital analysis (Intrinsic Bonding Orbitals, IBOs)79 and quantum theory of atoms in molecules (QTAIM) analysis describe bonding interactions between the boron and phosphorus atoms of compound 1 and model systems A, B, and (Ph)3P–BPh2Cl (Table S7†). The ρ(B–P) term denotes the electron density at the bond critical point (BCP) between boron and phosphorus, where a lower ρ value indicates a weaker covalent bond. The ρ(B–P) value for compound 1 is significantly lower than that of A and B, suggesting a more labile B–P bond in compound 1. Further evidence for the weak B–P bond in 1 is represented by the low H(B–P) value, which represents the total electron density at the BCP. Visualization of contour plots of the Laplacian of electron density for compounds 1, A, B, and (Ph)3P–BPh2Cl are available in Fig. S35–S38.† The curved nature of the bond paths between the boron and phosphorus atoms in 1, A, and B are a result of the strained B–C–C–P ring in each system, while the bond path in (Ph)3P–BPh2Cl is linear.
Localized molecular orbital analysis of compounds 2–4 identifies localized bonds displaying a σ bond between E and B, both s and p character between P and E, and a single lone pair on E (Fig. 4 and S39†). To further examine the nature of the E–B bond in compounds 2–4, energy decomposition analysis in conjunction with the natural orbitals of chemical valence method (EDA-NOCV) was performed to contrast electron-sharing (ES) and donor–acceptor (DA) bonding models. EDA-NOCV results indicate that the E–B bond in compounds 2–4 is best described as a donor–acceptor interaction, in which a lone pair on the chalcogen coordinates to the boron center (Fig. 4B and Table S8†). The strength of these E–B dative interactions decrease with the heavier chalcogens (E–B: O; −146 kcal mol−1 > S; −125 kcal mol−1 > Se; −121 kcal mol−1). Taken together with the experimental SC-XRD data, these computational findings suggest that the phosphorus–chalcogen interaction retains double-bond character, accompanied by a chalcogen–boron dative interaction.
 | ||
| Fig. 4 (A) Intrinsic bonding orbitals (IBOs) of the B–O and PO (σ and π) bonding interactions, as well as E lone pairs, in compound 2 computed at the B3LYP-D3BJ/def2TZVP level of theory. Orbital iso-surfaces enclose 70% of the integrated electron density of the orbital. (B) NOCV deformation density of the most significant orbital interaction for 2. Direction of charge migration is red to blue. Hydrogen atoms are omitted for clarity. | ||
Further insight into the electronic structure of compounds 1–8 was achieved by analyzing their frontier molecular orbitals, computed at the CPCM(DCM)-B3LYP-D3(BJ)//B3LYP-D3(BJ)/def2-TZVP level of theory. In each compound 1–4, the highest occupied molecular orbital (HOMO) lies primarily on the borinine unit (Fig. S40†). However, notable differences are observed in the lowest unoccupied molecular orbital (LUMO) of these systems. Whereas the LUMO of compound 1 exhibits significant density on the endocyclic alkene carbons, compounds 2–4 have LUMOs that are more localized on the endocyclic alkene carbons, as well as the –PPh2 motif. Comparison of the HOMO of compounds 5–8 indicates that the each HOMO of each system lies on the borinine core, with additional contribution from the bridging chalcogen (O, S, Se for 5, 6, 7, respectively) (Fig. S41†). The LUMO of these systems lies primarily on the endocyclic alkene carbons. Despite the HOMO of compounds 5–7 possessing significant chalcogen non-bonding orbital contribution, the HOMO of compound 8+ (counter-anion not considered) possesses no Cl lone pair characteristics. Instead, the HOMO of compound 8+ resembles that of compounds 1–4 by residing primarily on the borinine backbone.
Although compound 8 is formally a cationic species, the nature of the bridging B–Cl–B moiety remains ambiguous due to the potential for both donor–acceptor and electron-sharing bonding interactions. Owing to this ambiguity, a more comprehensive theoretical analysis was conducted to elucidate the nature of the B–Cl–B bond and to assess the possibility of a bridged chloronium species. The Hirshfeld CM-5 charge80 on the Cl atom in 8+ was calculated to be +0.210, while the charge on both B atoms is −0.190 (Table 1). For comparison, the CM-5 charges of relevant literature compounds, including the chloroborane borenium ion reported by Chein and Chiu (C+),74 the [B2Cl3(IMes)2] cation reported by Braunschweig73 (D+), Me2Cl+ reported by Stoyanov and Reed,81 and pyCl+ and py2Cl+ reported by Riedel82 were also computed (Table 1). All species, with the exception of D+ (−0.040) and py2Cl+ (+0.080), exhibit a positive Cl atom charge of approximately +0.200, suggesting a chloronium ion. IBO analysis of 8+ indicates a single covalent interaction between the central chlorine atom and each boron center, identifying two B–Cl σ bonds and two lone pairs (s and p character) on chlorine (Fig. S42†). As observed in diaryliodonium ions theoretically analyzed by Huber and Legault,83 the s and p lone pairs on chlorine remain localized due to the bent geometry at chlorine, preventing delocalization to either boron center. EDA-NOCV analysis was subsequently performed on 8+ to compare both donor–acceptor and electron-sharing models for the bridging B–Cl–B moiety. EDA–NOCV results confirm that the most appropriate bonding model is an electron-sharing interaction between two triplet fragments: a chlorine cation and the diboracycle (Fig. S45 and Table S9†). A qualitative molecular orbital diagram for the B–Cl–B bonding interactions, including the relevant localized bonding orbitals, is provided in Fig. S42.† These combined theoretical results unambiguously identify the chlorine in compound 8 as a chloronium ion, representing one of the earliest examples of such a species supported by two boron centers.
| Compound | r(B–Cl) (Å) | CM5(B) | CM5(Cl) |
|---|---|---|---|
| 8+ | 1.963 | −0.19 | +0.21 |
| C+ | 2.020 | +0.01 | +0.16 |
| D+ | 1.830 (exocyclic) | −0.15 | −0.04 (exocyclic) |
| 2.043 (endocyclic) | — | +0.07 (endocyclic) | |
| Me2Cl+ | — | — | +0.32 |
| pyCl+ | — | — | +0.22 |
| py2Cl+ | — | — | +0.08 |
The mechanism to form boracyclohexadienes from 9-chloro-9-borafluorene has been previously reported by Braunschweig and Lin.84 Drawing inspiration from this study, density functional theory was employed to investigate a possible mechanism for the formation of compound 1 at the B3LYP-D3(BJ)/def2-TZVP level of theory (Fig. 5). Starting from I, the C1 carbon of diphenyl(phenylethynyl)phosphane acts as a nucleophile and attacks the boron center (TS-II, ΔG‡ = +23.2 kcal mol−1), resulting in formation of int-III (ΔΔG = −20.0 kcal mol−1), in which a phosphirenium ion and borafluorenate are formed. Through TS-IV (ΔΔG‡ = +15.8 kcal mol−1), the phosphirenium ring opens, resulting in a 1,2-shift of the diphenylphosphino group to the C2 carbon, as well as ring expansion of the borafluorene to form borinine int-V (ΔΔG = −35.9 kcal mol−1). The diphenylphosphino moiety then rotates to form compound 1 (ΔΔG = −4.3 kcal mol−1).
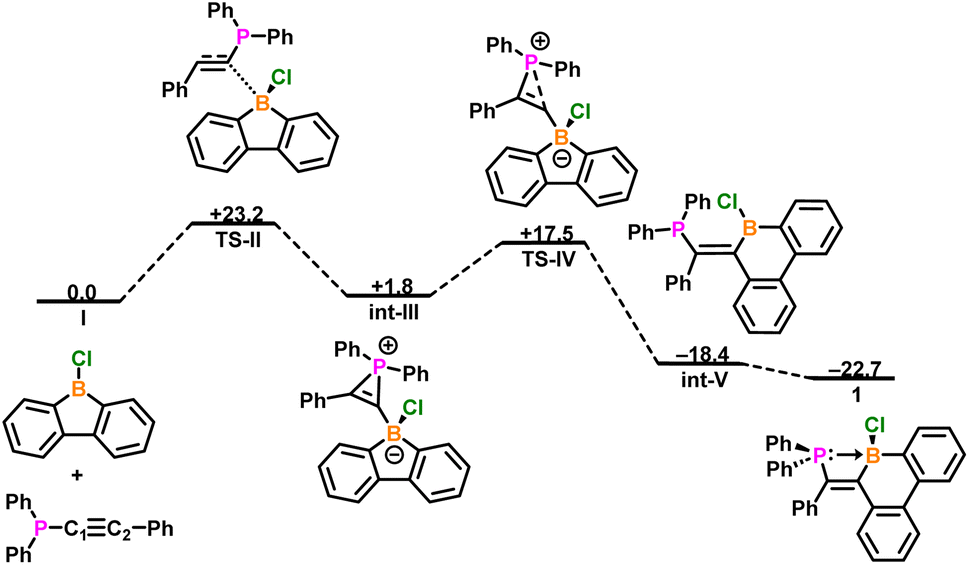 | ||
| Fig. 5 Calculated relative gas-phase free energies (ΔG, kcal mol−1) for the reaction between 9-chloro-9-borafluorene and diphenyl(phenylethynyl)phosphane to form compound 1 computed at the B3LYP-D3(BJ)/def2-TZVP level of theory. | ||
The second mechanism of interest involves the reductive ring expansion of compound 2 to form compound 5. A proposed mechanism for this process was computed at the B3LYP-D3(BJ)/def2-TZVP level of theory (Fig. 6). Addition of potassium metal to compound 2 results in the formation of int-A (ΔG = −22.6 kcal mol−1), in which the potassium atom is stabilized by the π-system of a neighboring phosphine ring and the central oxygen ring. From int-A, KCl leaves the coordination sphere of the reduced heterocycle (int-B, ΔΔG = −8.1 kcal mol−1), subsequently forming int-C (ΔΔG = +10.8 kcal mol−1), which suggests the formation of a phosphine-stabilized boryl radical. A second equivalent of int-C combines to produce the open-shell singlet int-D (ΔΔG = −7.9 kcal mol−1), in which the two int-C units are in close proximity to each other. A radical coupling transition state (TS-E, ΔΔG‡ = +27.7 kcal mol−1) from int-D was found to be the rate determining step of the mechanism. Once the bridging oxygen–boron bond is formed, the newly generated system becomes a closed-shell singlet (int-F, ΔΔG = −47.2 kcal mol−1). Subsequent addition of two equivalents of potassium metal results in the dipotassium species int-H (ΔΔG = −27.4 kcal mol−1). Loss of K2O from int-H leads to the ring-expanded compound 5 (ΔG = −87.1 kcal mol−1). To account for the formation of solid K2O, the computed ΔGrxn from int-H to compound 5 was adjusted by adding the experimental ΔGdeposition for KCl (−55.7 kcal mol−1) to the computed ΔGrxn. Despite our best efforts to model the potential energy surface for S and Se analogues, TS-E could not be located for either system. Nonetheless, we postulate that compounds 3 and 4 proceed through a similar mechanism as compound 2. The computed intermediates for formation of compounds 6 and 7 are available in Fig. S43 and S44.†
 | ||
| Fig. 6 Calculated relative gas-phase free energies (ΔG, kcal mol−1) for reduction and subsequent ring expansion of compound 2 to form compound 5 computed at the B3LYP-D3(BJ)/def2-TZVP level of theory. | ||
We then sought to propose a mechanism for the ring expansion of compound 1 to form compound 8 upon halide abstraction (Fig. 7). The mechanism begins with the removal of Cl− from 1, resulting in the phosphine-stabilized borenium ion int-1 (ΔG = +33.4 kcal mol−1). Subsequent addition of a second equivalent of 1 forms int-2 (ΔΔG = −0.3 kcal mol−1). TS-3 is the result of nucleophilic phosphine attack of P2 on B1 in an SN2 fashion (ΔΔG‡ = +31.4 kcal mol−1), resulting in dissociation of P1 from B1, forming int-4 (ΔG = −25.3 kcal mol−1). From int-4, nucleophilic attack of B2 by P1viaTS-5 (ΔΔG‡ = +0.7 kcal mol−1), results in the formation of 8+ (ΔΔG = −36.6 kcal mol−1). An alternative low-lying transition state (TS-3′), arising from chlorine nucleophilic attack at one of the boron sites (ΔΔG‡ = +5.5 kcal mol−1), was also found and results in the ring-closed int-4′ (ΔΔG = −7.5 kcal mol−1). However, this kinetic product readily reverts to int-2, whereas int-4 provides a significant thermodynamic sink that minimizes reversibility at room temperature.
 | ||
| Fig. 7 Calculated relative free energies (ΔG, kcal mol−1) for halide abstraction and subsequent ring expansion of compound 1 to form compound 8+ computed at the CPCM-B3LYP-D3-(BJ)/def2-TZVP level of theory. The dielectric constant (ε) was set to 5.42 to resemble fluorobenzene solvation. | ||
Conclusions
In summary, we report a new approach to expand the borole ring in 9-chloro-9-borafluorene using diphenyl(phenylethynyl)phosphane to yield a six-membered borinine ring that contains a four-membered B–P heterocycle (1). The computed mechanism for the ring expansion shows that the reaction proceeds through a 1,2-shift of the –PPh2 group. Compound 1 was then used to activate chalcogen sources (Me3N–O, S8, and Se) to form five-membered B–E–P heterocycles 2–4. Subsequent reduction of these species with KC8 results in K2E formation, thereby forming an eight-membered ring with a μ2-chalcogenide connecting two boron centers in compounds 5–7. A proposed mechanism shows that the rate-determining step of this reductive ring expansion is an open-shell singlet transition state, in which an intermolecular B–E bond is formed. Halide abstraction of compound 1 using silver bis(trifluoromethanesulfonyl)imide forms a proposed intermediate phosphine-stabilized borenium ion, which then ring expands to form compound 8. Using theoretical methods, we have assigned compound 8 as containing one of the first examples of a boron-stabilized chloronium ion.Data availability
Crystallographic data for 1–8 can be found in the Cambridge Crystallographic Database (CCDC: 2368500–2368507). All other data have been provided in the ESI,† including experimental details, NMR spectra, single-crystal X-ray diffraction data, and computational details.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the National Science Foundation Chemical Synthesis (CHE 2046544) and Major Research Instrumentation (CHE 2018870) programs for support of this work. We also thank the Alfred P. Sloan Foundation for a Sloan Research Fellowship. R. J. G. acknowledges additional laboratory support through a Beckman Young Investigator award from the Arnold & Mabel Beckman Foundation. N. C. F. acknowledges the National Science Foundation for a Graduate Research Fellowship (grant #1842490). N. C. F. thanks Professor Charles Edwin Webster for insightful conversations concerning the computational portions of the paper. The authors acknowledge the MIT SuperCloud and Lincoln Laboratory Supercomputing Center for providing resources that have contributed to the research results reported within this paper/report.Notes and references
- Z. Liu, Z. Wang, H. Mu, Y. Zhou, J. Zhou and Z. Dong, Nat. Commun., 2024, 15, 9849 CrossRef CAS.
- M. Shibuya, M. Matsuda and Y. Yamamoto, Chem.–Eur. J., 2021, 27, 8822–8831 CrossRef CAS.
- S. Bonfante, C. Lorber, J. M. Lynam, A. Simonneau and J. M. Slattery, J. Am. Chem. Soc., 2024, 146, 2005–2014 CrossRef CAS PubMed.
- A. Gärtner, U. S. Karaca, M. Rang, M. Heinz, P. D. Engel, I. Krummenacher, M. Arrowsmith, A. Hermann, A. Matler, A. Rempel, R. Witte, H. Braunschweig, M. C. Holthausen and M.-A. Légaré, J. Am. Chem. Soc., 2023, 145, 8231–8241 CrossRef.
- K. Chulsky, I. Malahov, D. Bawari and R. Dobrovetsky, J. Am. Chem. Soc., 2023, 145, 3786–3794 CrossRef CAS.
- M.-A. Légaré, C. Pranckevicius and H. Braunschweig, Chem. Rev., 2019, 119, 8231–8261 CrossRef.
- M.-A. Légaré, G. Bélanger-Chabot, R. D. Dewhurst, E. Welz, I. Krummenacher, B. Engels and H. Braunschweig, Science, 2018, 359, 896–900 CrossRef PubMed.
- H. Braunschweig, I. Krummenacher, M.-A. Légaré, A. Matler, K. Radacki and Q. Ye, J. Am. Chem. Soc., 2017, 139, 1802–1805 CrossRef CAS.
- Y. Su and R. Kinjo, Chem. Soc. Rev., 2019, 48, 3613–3659 RSC.
- Y. Su and R. Kinjo, Coord. Chem. Rev., 2017, 352, 346–378 CrossRef CAS.
- L. Kong and C. Cui, Synlett, 2021, 32, 1316–1322 CrossRef CAS.
- N. C. Frey, K. K. Hollister, P. Müller, D. A. Dickie, C. E. Webster and R. J. Gilliard Jr, Inorg. Chem., 2024, 63, 17639–17650 CrossRef CAS.
- M. Yamashita and K. Nozaki, Bull. Chem. Soc. Jpn., 2008, 81, 1377–1392 CrossRef CAS.
- M. Yamashita and K. Nozaki, in Synthesis and Application of Organoboron Compounds, ed. E. Fernández and A. Whiting, Springer International Publishing, Cham, 2015, pp. 1–37, DOI:10.1007/978-3-319-13054-5_1.
- M. Yamashita and K. Nozaki, Pure Appl. Chem., 2008, 80, 1187–1194 CrossRef CAS.
- K. E. Wentz, A. Molino, L. A. Freeman, D. A. Dickie, D. J. D. Wilson and R. J. Gilliard Jr, J. Am. Chem. Soc., 2022, 144, 16276–16281 CrossRef CAS.
- K. E. Wentz, A. Molino, L. A. Freeman, D. A. Dickie, D. J. D. Wilson and R. J. Gilliard Jr, Inorg. Chem., 2022, 61, 17049–17058 CrossRef CAS PubMed.
- K. E. Wentz, A. Molino, L. A. Freeman, D. A. Dickie, D. J. D. Wilson and R. J. Gilliard Jr, Angew. Chem., Int. Ed., 2023, 62, e202215772 CrossRef CAS PubMed.
- K. E. Wentz, A. Molino, S. L. Weisflog, A. Kaur, D. A. Dickie, D. J. D. Wilson and R. J. Gilliard Jr, Angew. Chem., Int. Ed., 2021, 60, 13065–13072 CrossRef CAS PubMed.
- K. E. Wentz, A. Molino, L. A. Freeman, D. A. Dickie, D. J. D. Wilson and R. J. Gilliard Jr, Inorg. Chem., 2021, 60, 13941–13949 CrossRef CAS PubMed.
- L. Englert, U. Schmidt, M. Dömling, M. Passargus, T. E. Stennett, A. Hermann, M. Arrowsmith, M. Härterich, J. Müssig, A. Phillipps, D. Prieschl, A. Rempel, F. Rohm, K. Radacki, F. Schorr, T. Thiess, J. O. C. Jiménez-Halla and H. Braunschweig, Chem. Sci., 2021, 12, 9506–9515 RSC.
- S. R. Wang, M. Arrowsmith, J. Böhnke, H. Braunschweig, T. Dellermann, R. D. Dewhurst, H. Kelch, I. Krummenacher, J. D. Mattock, J. H. Müssig, T. Thiess, A. Vargas and J. Zhang, Angew. Chem., Int. Ed., 2017, 56, 8009–8013 CrossRef CAS PubMed.
- T. E. Stennett, J. D. Mattock, L. Pentecost, A. Vargas and H. Braunschweig, Angew. Chem., Int. Ed., 2018, 57, 15276–15281 CrossRef CAS.
- M. Arrowsmith, J. Böhnke, H. Braunschweig and M. A. Celik, Angew. Chem., Int. Ed., 2017, 56, 14287–14292 CrossRef CAS PubMed.
- H. Braunschweig, T. Dellermann, W. C. Ewing, T. Kramer, C. Schneider and S. Ullrich, Angew. Chem., Int. Ed., 2015, 54, 10271–10275 CrossRef CAS.
- T. Brückner, M. Arrowsmith, M. Heß, K. Hammond, M. Müller and H. Braunschweig, Chem. Commun., 2019, 55, 6700–6703 RSC.
- J. Böhnke, T. Brückner, A. Hermann, O. F. González-Belman, M. Arrowsmith, J. O. C. Jiménez-Halla and H. Braunschweig, Chem. Sci., 2018, 9, 5354–5359 RSC.
- T. Matsubara, R. Yamasaki, T. Hori and M. Morikubo, Bull. Chem. Soc. Jpn., 2018, 91, 1683–1690 CrossRef CAS.
- H. Braunschweig, P. Constantinidis, T. Dellermann, W. C. Ewing, I. Fischer, M. Hess, F. R. Knight, A. Rempel, C. Schneider, S. Ullrich, A. Vargas and J. D. Woollins, Angew. Chem., Int. Ed., 2016, 55, 5606–5609 CrossRef CAS.
- D. W. Stephan, J. Am. Chem. Soc., 2015, 137, 10018–10032 CrossRef CAS PubMed.
- N. Li and W.-X. Zhang, Chin. J. Chem., 2020, 38, 1360–1370 CrossRef CAS.
- D. W. Stephan, Acc. Chem. Res., 2015, 48, 306–316 CrossRef CAS PubMed.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 6400–6441 CrossRef CAS PubMed.
- G. C. Welch, R. R. S. Juan, J. D. Masuda and D. W. Stephan, Science, 2006, 314, 1124–1126 CrossRef CAS.
- T. Wang, C. G. Daniliuc, G. Kehr and G. Erker, in Frustrated Lewis Pairs, 2021, ch. 3, pp. 87–112, DOI:10.1007/978-3-030-58888-5_3.
- B. L. Frenette and E. Rivard, Chem.–Eur. J., 2023, 29, e202302332 CrossRef CAS.
- A. S. Balueva, Y. Y. Efremov, V. M. Nekhoroshkov and O. A. Erastov, Bulletin of the Academy of Sciences of the USSR, Division of Chemical Science, 1989, vol. 38, pp. 2557–2560 Search PubMed.
- A. S. Balueva and G. N. Nikonov, Russ. Chem. Bull., 1993, 42, 341–343 CrossRef.
- S. Bontemps, M. Devillard, S. Mallet-Ladeira, G. Bouhadir, K. Miqueu and D. Bourissou, Inorg. Chem., 2013, 52, 4714–4720 CrossRef CAS.
- A. A. Omaña, R. K. Green, R. Kobayashi, Y. He, E. R. Antoniuk, M. J. Ferguson, Y. Zhou, J. G. C. Veinot, T. Iwamoto, A. Brown and E. Rivard, Angew. Chem., Int. Ed., 2021, 60, 228–231 CrossRef.
- V. K. Greenacre, M. B. Ansell, S. M. Roe and I. R. Crossley, Eur. J. Inorg. Chem., 2014, 2014, 5053–5062 CrossRef CAS.
- R. Liedtke, C. Eller, C. G. Daniliuc, K. Williams, T. H. Warren, M. Tesch, A. Studer, G. Kehr and G. Erker, Organometallics, 2016, 35, 55–61 CrossRef CAS.
- S. Porcel, G. Bouhadir, N. Saffon, L. Maron and D. Bourissou, Angew. Chem., Int. Ed., 2010, 49, 6186–6189 CrossRef CAS.
- S. Portela and I. Fernández, Z. Anorg. Allg. Chem., 2023, 649, e202200384 CrossRef CAS.
- E. J. Lawrence, V. S. Oganesyan, D. L. Hughes, A. E. Ashley and G. G. Wildgoose, J. Am. Chem. Soc., 2014, 136, 6031–6036 CrossRef CAS PubMed.
- E. J. Lawrence, E. R. Clark, L. D. Curless, J. M. Courtney, R. J. Blagg, M. J. Ingleson and G. G. Wildgoose, Chem. Sci., 2016, 7, 2537–2543 RSC.
- E. J. Lawrence, T. J. Herrington, A. E. Ashley and G. G. Wildgoose, Angew. Chem., Int. Ed., 2014, 53, 9922–9925 CrossRef CAS.
- G.-Q. Chen, G. Kehr, C. G. Daniliuc, M. Bursch, S. Grimme and G. Erker, Chem.–Eur. J., 2017, 23, 4723–4729 CrossRef CAS.
- D. Mandal, R. Gupta, A. K. Jaiswal and R. D. Young, J. Am. Chem. Soc., 2020, 142, 2572–2578 CrossRef CAS PubMed.
- L. J. C. van der Zee, J. Hofman, J. M. van Gaalen and J. C. Slootweg, Chem. Soc. Rev., 2024, 53, 4862–4876 RSC.
- K. K. Hollister, K. E. Wentz and R. J. Gilliard Jr, Acc. Chem. Res., 2024, 57, 1510–1522 CrossRef CAS.
- S. K. Sarkar, K. K. Hollister, A. Molino, A. D. Obi, C.-L. Deng, B. Y. E. Tra, B. M. Stewart, D. A. Dickie, D. J. D. Wilson and R. J. Gilliard Jr, J. Am. Chem. Soc., 2023, 145, 21475–21482 CrossRef CAS PubMed.
- Y. Shoji, N. Tanaka, S. Muranaka, N. Shigeno, H. Sugiyama, K. Takenouchi, F. Hajjaj and T. Fukushima, Nat. Commun., 2016, 7, 12704 CrossRef PubMed.
- H. Nöth and B. Wrackmeyer, Nuclear Magnetic Resonance Spectroscopy of Boron Compounds, Springer, Berlin, Heidelberg, 1978 Search PubMed.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B, 2016, 72, 171–179 CrossRef CAS.
- T. A. Albright, W. J. Freeman and E. E. Schweizer, J. Org. Chem., 1975, 40, 3437–3441 CrossRef CAS.
- Y. Zhang, M. Li, S. Chandrasekaran, X. Gao, X. Fang, H.-W. Lee, K. Hardcastle, J. Yang and B. Wang, Tetrahedron, 2007, 63, 3287–3292 CrossRef CAS PubMed.
- L. C. Wilkins, J. R. Lawson, P. Wieneke, F. Rominger, A. S. K. Hashmi, M. M. Hansmann and R. L. Melen, Chem.–Eur. J., 2016, 22, 14618–14624 CrossRef CAS.
- Q. Wu, M. Esteghamatian, N.-X. Hu, Z. Popovic, G. Enright, S. R. Breeze and S. Wang, Angew. Chem., Int. Ed., 1999, 38, 985–988 CrossRef CAS.
- V. Subramaniyan and G. Mani, Organometallics, 2018, 37, 127–135 CrossRef CAS.
- D. Kanichar, L. Roppiyakuda, E. Kosmowska, M. A. Faust, K. P. Tran, F. Chow, E. Buglo, M. P. Groziak, E. A. Sarina, M. M. Olmstead, I. Silva and H. H. Xu, Chem. Biodiversity, 2014, 11, 1381–1397 CrossRef CAS.
- R. Köster, A. Sporzyński, W. Schüßler, D. Bläser and R. Boese, Chem. Ber., 1994, 127, 1191–1199 CrossRef.
- A. Torres-Huerta, M. d. J. Velásquez-Hernández, L. G. Ramírez-Palma, F. Cortés-Guzmán, D. Martínez-Otero, U. Hernández-Balderas and V. Jancik, Inorg. Chem., 2017, 56, 10032–10043 CrossRef CAS.
- J.-H. Son, S. R. Tamang, J. C. Yarbrough and J. D. Hoefelmeyer, Z. Naturforsch., B, 2015, 70, 775–781 CrossRef CAS.
- D. Zhu, J. H. W. LaFortune, R. L. Melen and D. W. Stephan, Dalton Trans., 2019, 48, 2038–2045 RSC.
- N. Schulenberg, O. Ciobanu, E. Kaifer, H. Wadepohl and H.-J. Himmel, Eur. J. Inorg. Chem., 2010, 2010, 5201–5210 CrossRef.
- H. E. Katz, Organometallics, 1987, 6, 1134–1136 CrossRef CAS.
- S. P. Lewis, N. J. Taylor, W. E. Piers and S. Collins, J. Am. Chem. Soc., 2003, 125, 14686–14687 CrossRef CAS.
- K. Samigullin, I. Georg, M. Bolte, H.-W. Lerner and M. Wagner, Chem.–Eur. J., 2016, 22, 3478–3484 CrossRef CAS.
- A. Kobayashi, K. Suzuki, R. Kitamura and M. Yamashita, Organometallics, 2020, 39, 383–387 CrossRef CAS.
- M. J. Sgro, J. Dömer and D. W. Stephan, Chem. Commun., 2012, 48, 7253–7255 RSC.
- H. Braunschweig, A. Damme, R. D. Dewhurst, T. Kramer, T. Kupfer, K. Radacki, E. Siedler, A. Trumpp, K. Wagner and C. Werner, J. Am. Chem. Soc., 2013, 135, 8702–8707 CrossRef CAS.
- L. Englert, A. Stoy, M. Arrowsmith, J. H. Muessig, M. Thaler, A. Deißenberger, A. Häfner, J. Böhnke, F. Hupp, J. Seufert, J. Mies, A. Damme, T. Dellermann, K. Hammond, T. Kupfer, K. Radacki, T. Thiess and H. Braunschweig, Chem.–Eur. J., 2019, 25, 8612–8622 CrossRef CAS.
- D.-N. Shih, R. Boobalan, Y.-H. Liu, R.-J. Chein and C.-W. Chiu, Inorg. Chem., 2021, 60, 16266–16272 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785–789 CrossRef CAS PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- G. Knizia, J. Chem. Theory Comput., 2013, 9, 4834–4843 CrossRef CAS.
- A. V. Marenich, S. V. Jerome, C. J. Cramer and D. G. Truhlar, J. Chem. Theory Comput., 2012, 8, 527–541 CrossRef CAS PubMed.
- E. S. Stoyanov, I. V. Stoyanova, F. S. Tham and C. A. Reed, J. Am. Chem. Soc., 2010, 132, 4062–4063 CrossRef CAS.
- P. Pröhm, W. Berg, S. M. Rupf, C. Müller and S. Riedel, Chem. Sci., 2023, 14, 2325–2329 RSC.
- R. Robidas, D. L. Reinhard, S. M. Huber and C. Y. Legault, Chemphyschem, 2023, 24, e202200634 CrossRef CAS.
- Z. Wang, Y. Zhou, J.-X. Zhang, I. Krummenacher, H. Braunschweig and Z. Lin, Chem.–Eur. J., 2018, 24, 9612–9621 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, NMR spectra, single-crystal X-ray diffraction data, and computational details. CCDC 2368500–2368507. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc02000j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |