
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntramolecular asymmetric propargylation of esters and imides: C–H functionalisation enables stereocontrolled access to UCS1025A†
James C.
Corcoran
,
Jin
Zhu‡
,
Mason A.
Semenick
,
Anna L.
Welser§
and
Yi-Ming
Wang
 *
*
Department of Chemistry, University of Pittsburgh, Pittsburgh, Pennsylvania 15260, USA. E-mail: ym.wang@pitt.edu
First published on 4th June 2025
Abstract
We report an iridium-catalysed stereoselective cyclisation of aryl alkynes with tethered esters and imides to give silyl-protected O,O- and N,O-acetals. The optimised conditions afford chiral 5- and 6-membered heterocycles in excellent ee and moderate to high dr and can be isolated chromatographically as single diastereomers. A variety of esters, including acetates, formates, benzoates, and pivalates, can be employed to give functionalised tetrahydrofuran derivatives. Additionally, phthalimide- and maleimide-containing substrates give stereodefined nitrogen-containing bi- and tricyclic fused-ring products with potential applications in natural product synthesis, as illustrated by the synthesis of a known intermediate en route to furopyrrolizidine alkaloid UCS1025A.
Introduction
Esters are accessible and versatile functional groups that participate in a variety of practical C–C bond forming reactions as a stable, mildly electrophilic coupling partner. Despite these favourable properties and their prochiral nature, they are generally not used to establish stereochemistry in asymmetric synthesis. Relatively reactive carbanion equivalents (e.g., Grignard and alkyllithium reagents) are generally needed to allow for uncatalysed nucleophilic addition to esters; however, these reagents almost always undergo a second addition upon collapse of the initial tetrahedral intermediate to generate achiral bis-addition products in the alcohol oxidation state. Whilst controlled monoaddition is possible using specialised starting materials or Ni or Pd-catalysed cross-coupling technology, these reactions lead to achiral ketone products (Scheme 1A).1,2 | ||
| Scheme 1 Context of reported work in relation to ester addition and enantioselective acetal synthesis. | ||
In some cases, formation of a stereocentre is possible through the addition of a single equivalent of nucleophile and trapping of the tetrahedral intermediate to afford stable silyl O,O-acetals as products (Scheme 1A).3 However, this process has mostly been applied to nucleophiles of attenuated reactivity stabilised by α-heteroatomic substituents (e.g., F, Cl, P). Moreover, to the best of our knowledge, this strategy has not been applied to the context of stereocontrolled synthesis or asymmetric catalysis.
Compared to previous approaches, the direct, enantioselective addition of a carbon nucleophile into an ester constitutes an unexplored retrosynthetic disconnection for the assembly of acetals. Monocyclic O,O- and N,O-acetals have previously been prepared using asymmetric organocatalysis and transition metal catalysis.4 For instance, in early work, List and coworkers reported an intramolecular transacetalisation protocol using a chiral phosphoric acid catalyst (Scheme 1B).5 In addition, spirocyclic and fused bicyclic O,O- and N,O-acetals have previously been prepared using a number of strategies that employ transition metal-catalysed cascade processes (Scheme 1C).6 For example, Slaughter and coworkers reported an early example in which alkyne activation by cationic Au was leveraged to generate an oxocarbenium species, which underwent subsequent stereoselective alcohol addition, while Ding reported a tandem hydrogenation/Brønsted acid mediated cyclisation employing an Ir catalyst that mediated both processes.7 In contrast to previous strategies, in which stereoselective C–heteroatom bond formation plays a crucial role, we report herein the asymmetric addition of tethered carbanion nucleophiles to esters to generate acetals through the stereoselective construction of C–C bonds (Scheme 1D).
Given the important role of chiral N- and O-containing heterocycles in drug development and natural product synthesis,8 and their potential utility as precursors to electrophiles that engage in cross coupling,9 we undertook the exploration of cyclisation reactions of alkynes containing tethered electrophilic coupling partners. Based on recent work in Au catalysis report by Zhang and coworkers on intramolecular aldehyde addition,10 as well as recent work in Fe- and Ir-catalysed C–H functionalisation chemistry reported by our group,11 we considered the possibility of using simple esters as underexplored electrophiles for reaction with catalytically generated organoiridium nucleophiles to afford O,O-acetals. In particular, we hypothesised that ester-tethered alkyne 1 could undergo complexation-assisted deprotonation at the propargylic position upon coordination to iridium complex II (Scheme 2),11 and that the ester group in the resultant allenyliridium species IV would then be susceptible to intramolecular nucleophilic attack upon activation of the carbonyl by a suitable Lewis acid (e.g., R3SiOTf) to form cyclic silyl acetal 2. Although conceptually similar to previously investigated reactivity, the weak inherent electrophilicity of the ester, combined with the mild reaction conditions required for the survival of the acetal product, renders the implementation of the proposed reactivity a significant synthetic challenge.
 | ||
| Scheme 2 Proposed catalytic cycle for the iridium catalysed cyclisation of ester-tethered alkynes to give acetals (TMPH = 2,2,6,6-tetramethylpiperidine). | ||
We suspected that, given a similar degree of electrophilicity, imides could represent another carbonyl based functional group that would take part in analogous chemistry. While many examples of 1,2-addition to imides that give N,O-acetals have been reported, few are enantioselective.12 Moreover, enantioselective additions performed under catalytic conditions are largely, but not exclusively,12e,g limited to the reduction of meso imides using hydride reagents. Given their accessibility and underutilisation in enantioselective carbonyl addition chemistry, new strategies could make esters and imides attractive substrates for the assembly of chiral oxygen and nitrogen containing heterocycles.
Results and discussion
At the outset, we examined model acetate substrate 1a using various electrophilic silyl sources under conditions previously developed by our group for iridium-catalysed enantioselective propargylic functionalisation (5 mol% [Ir(cod)Cl]2, 20 mol% Carreira's ligand (R)-L, and 2,2,6,6-tetramethylpiperidine (TMPH) as the base, Table 1). Among the Lewis acids examined, the bulky triisopropylsilyl triflate (TIPSOTf) proved to be the most effective, while smaller silyl triflates gave other silylation products as side products and were therefore less effective (entry 1 vs. 12 and 13). Decreasing the number of equivalents of TMPH and TIPSOTf simultaneously (entry 4), or increasing the excess of TMPH relative to TIPSOTf (entry 5) were both marginally detrimental to yield and enantiomeric excess (ee). Conducting the reaction at half of the standard catalyst loading resulted in lower, but still useful, yield (entry 6). The outcome (both enantioselectivity and reactivity) exhibited a marked dependence on the reaction temperature (entries 1–3), and in the exploration of scope, further adjustments to this parameter were made on an individual basis, depending on the steric and electronic properties of the substrate. Finally, 1,2-dichloroethane (DCE) was slightly preferable to CH2Cl2 as the solvent; however, both were far superior to a range of aromatic solvents investigated (entries 7–11).|
|
|||
|---|---|---|---|
| Entry | Change | Yieldb (%) | eec (%) |
a The dr of 2a was determined by 1H NMR spectroscopy to be >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in all cases.
b Yields were determined for the major diastereomer by 1H NMR spectroscopy using 1,1,2,2-tetrachloroethane as an internal standard.
c Enantiomeric excesses were determined by chiral HPLC.
d Isolated yield of major diastereomer.
e Enantiomeric excess not determined.
f The tert-butyldimethylsilyl (TBS) or triethylsilyl (TES) protected acetal products were observed in place of 2a. :1 in all cases.
b Yields were determined for the major diastereomer by 1H NMR spectroscopy using 1,1,2,2-tetrachloroethane as an internal standard.
c Enantiomeric excesses were determined by chiral HPLC.
d Isolated yield of major diastereomer.
e Enantiomeric excess not determined.
f The tert-butyldimethylsilyl (TBS) or triethylsilyl (TES) protected acetal products were observed in place of 2a.
|
|||
| 1 | None | 87 (80d) | 99 |
| 2 | At r.t. (21 °C) | 73 | 98 |
| 3 | At 40 °C | 86 | 96 |
| 4 | 1.5 eq. TIPSOTf, 2 eq. TMPH | 82 | 97 |
| 5 | 2 eq. TIPSOTf, 3 eq. TMPH | 83 | 96 |
| 6 | 2.5% [Ir(cod)Cl]2 | 70 | 97 |
| 7 | CH2Cl2 as solvent | 83 | 96 |
| 8 | o-Dichlorobenzene “ | 55 | 92 |
| 9 | Chlorobenzene “ | 12 | NDe |
| 10 | PhCF3 “ | 5 | NDe |
| 11 | Toluene “ | Trace | NDe |
| 12 | TBSOTf in place of TIPSOTf | 59f | NDe |
| 13 | TESOTf “ | 17f | NDe |
| 14 | TIPSCl “ | 0 | ND |

For five membered ring formation, this strategy proved applicable to a variety of esters, with 50 °C giving optimal results in most cases (Scheme 3). In addition to the original acetate substrate, a formate ester was used successfully to form unsubstituted acetal 2b. Similarly, a benzoate ester (2d) and 4-methoxybenzoate ester (2j) were also successful under these conditions. Furthermore, homologous substrates could be employed to provide the corresponding tetrahydropyrans 2c, 2g and 2h.
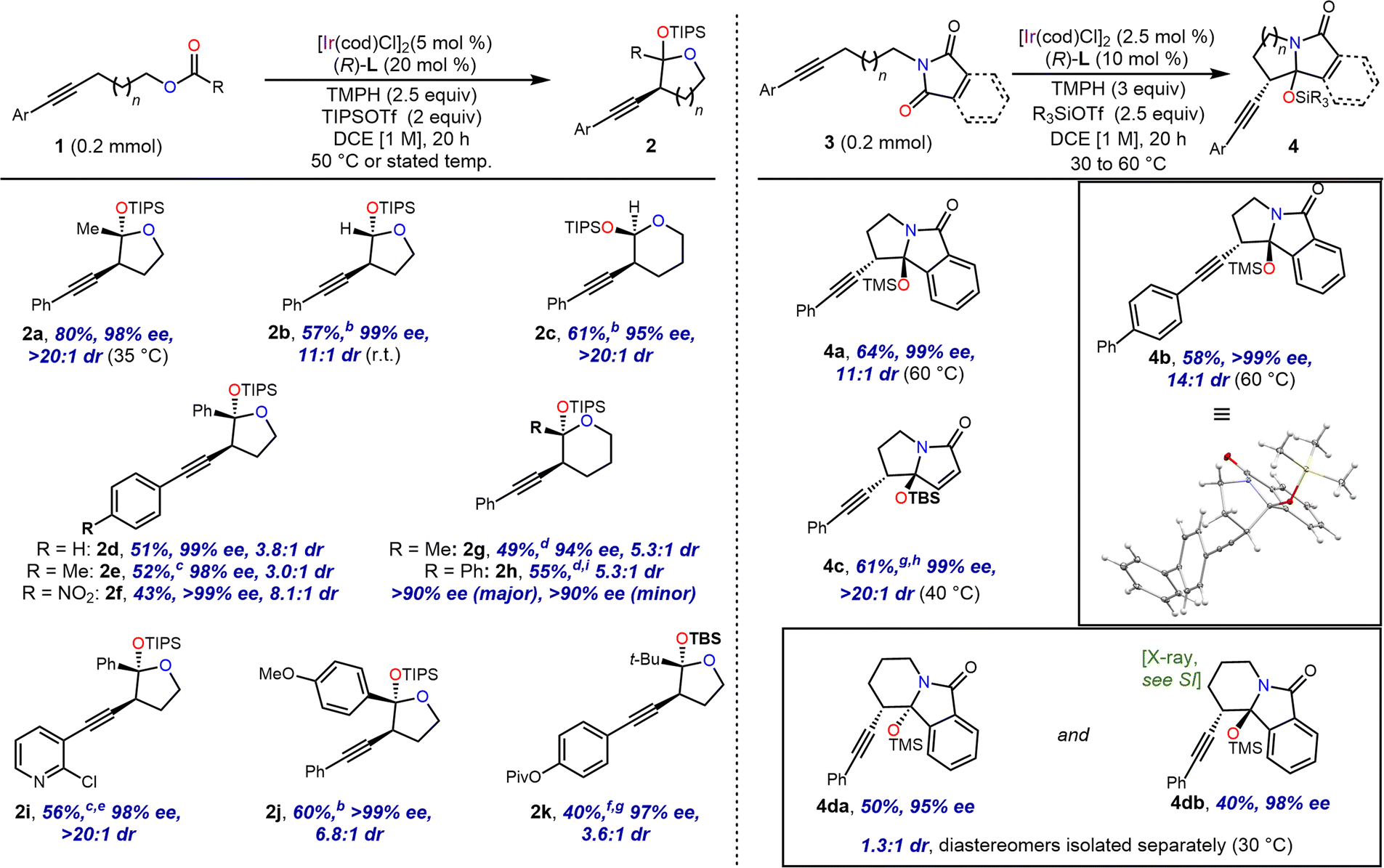 | ||
| Scheme 3 Substrate scope for ester and imide cyclisation. aIsolated yields and enantiomeric excesses (ee) are reported for the major diastereomer of each product. Diastereomeric ratios (dr) were determined by 1H NMR spectroscopy of the crude material before purification. b2.5 mol% [Ir(cod)Cl]2 and 10 mol% L. cStirred for 3 days. d10 mol% lithium bistriflimide (LiNTf2) used as an additive. e10 mol% [Ir(cod)Cl]2 and 40 mol% L. fStirred for 36 h. gt-Butyldimethylsilyl triflate (TBSOTf) used as silyl triflate. h5 mol% [Ir(cod)Cl]2 and 20 mol% L. iCombined yield of unseparated diastereomers. | ||
Interestingly, based on analysis of 1H NMR coupling constants, the relative configuration of the alkynyl and silyloxy groups of 2c was assigned as syn, rather than the anti-configuration assigned to 2g and 2h, as well as 2b and all analogous five-membered O,O-acetal products (see the ESI† for details for the stereochemical assignments). Benzoate ester 2e bearing a moderate electron donor on the alkynyl aryl group was tolerated, but cyclised slowly and needed a reaction time of 3 days. Moderate to strong electron withdrawing groups were also tolerated on this ring (2f, 2i, and 2k), though in the case of 2i, optimal yield required an extended reaction time and additional catalyst. A tethered pivalate underwent addition to give t-butyl substituted product 2k. In this example tert-butyldimethylsilyl triflate (TBSOTf) was used in place of TIPSOTf, as the latter gave low conversion of the starting material. This was presumed to be the result of crowding between the bulky acyl group and the silyl protecting group.
These transformations proceeded with excellent enantioselectivity. While diastereoselectivities ranged from moderate (3:1) to excellent (>20:1), the diastereomers formed were separable by silica gel chromatography, and the major diastereomer of each product could be isolated in diastereomerically pure form, except in the case of 2h, whose diastereomers were inseparable and were characterised as a mixture.
We then turned to nitrogen-tethered substrates for the generation of N,O-acetals. We found that while amides appeared insufficiently electrophilic to participate in the reaction, substrates bearing imides could be cyclised to give silyl-protected hemiaminals. Phthalimide- and maleimide-tethered alkynes gave rise to fused bi- and tricyclic products, which were also isolable as single diastereomers (Scheme 3). These cyclisation reactions exhibited optimal yield when the smaller TMS and TBS groups were used in place of TIPS, while maintaining excellent ee and dr. However, a substrate bearing a longer chain resulted in the formation of a six-membered ring in 4da and 4db with good ee but poor dr. In this case, the diastereomers were separable chromatographically, and each was isolated in useful yields.
We noted the close structural similarity of maleimide-derived 4c to key intermediates 6 and 7 used in previously reported total syntheses of pyrrolizidine natural product UCS1025A, a fungal alkaloid with anti-tumor and antibiotic activity.13 We sought to prepare these intermediates in a stereochemically pure form through a streamlined route using our intramolecular imide addition, thereby achieving a formal total synthesis of this natural product.
Although several completed and formal total syntheses have been published for UCS1025A, catalytic enantioselective catalysis has not been applied toward the synthesis of the key furopyrrolizidine ring system. Instead, previous approaches have relied on chiral pool and chiral resolution strategies to establish the stereochemistry.13 In this context, the current approach allows for the use of simple and accessible starting materials without the need to discard an unwanted stereoisomer. Moreover, we foresee the application of this approach to the synthesis of related furopyrrolizidine alkaloids exhibiting a range of biological activities (Scheme 4).14
 | ||
| Scheme 4 Application toward the synthesis of UCS1025A. | ||
Maleimide-tethered alkyne 3c was prepared in 96% yield using modified Mitsunobu conditions first reported by Walker.15 In particular, the addition of neopentyl alcohol as an additive was critical (96% yield vs. <10% yield without the additive). Alkyne 3c was subjected to iridium-catalysed cyclisation conditions on a 3 mmol scale to give 4c in 62% yield. To remove the extraneous aryl substituent, alkyne 4c was first subjected to semi-hydrogenation conditions to afford cis-configured alkene 5 in 88% yield. Ozonolysis followed by Pinnick oxidation of the resulting aldehyde then gave carboxylic acid 6 in 48% yield over 2 steps. Finally, iodolactonisation provided iodide 7 in 91% yield (23% over 6 steps) without erosion of stereochemical purity (99% ee). This compound, whose spectral data were in accord with those previously reported by Danishefsky and Lambert, had previously been converted to UCS1025A in three additional steps.13a Since the requisite aldehyde coupling partner for the Danishefsky synthesis can be prepared in enantioenriched form in six steps from commercial materials, the preparation of 7 as described above constitutes a nine-step (longest linear sequence) formal total synthesis of this natural product.16
Conclusions
In summary, we have developed an efficient method for the synthesis of cyclic silyl O,O- and N,O-acetals by applying a propargylic C–H functionalisation strategy to alkynes bearing ester and imide tethers as electrophiles. Products were generally isolated in moderate to good yield and excellent ee as single diastereomers after chromatographic purification. A formal synthesis of UCS1025A was achieved using our new method to perform the key cyclisation step to establish the stereochemistry of the pyrrolizidine fragment of the natural product. Further investigation of this strategy for the functionalisation of esters and imides is underway, including expansions to other weakly electrophilic functional groups and intermolecular settings.Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for 4b and 4db has been deposited at the CCDC under deposition numbers 2440355 and 2440354 respectively, and can be obtained from https://www.ccdc.cam.ac.uk/.Author contributions
James C. Corcoran: conceptualisation, investigation, data curation, writing—original draft preparation. Jin Zhu: conceptualisation, investigation. Mason A. Semenick: investigation. Anna L. Welser: investigation. Yi-Ming Wang: supervision, writing—review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge support from the National Science Foundation (CHE-2400082). Support for the single crystal X-ray diffractometer was provided by a grant from the National Science Foundation (CHE-2216178). We thank Dr Steven J. Geib (University of Pittsburgh) for collection of X-ray crystallographic data, Professor Paul Floreancig (University of Pittsburgh) for helpful discussions on the formal synthesis of UCS1025A, and Ruihan Wang and Aaron Charlack (Y.-M. W. group) for proofreading and revisions of the manuscript and ESI.†References
- (a) P. S. Fier, R. A. Roberts and R. T. Larson, Org. Lett., 2023, 25, 3131 CrossRef CAS PubMed; (b) X.-W. Han, Y. He, C. Gui, X.-Q. Chu, X.-F. Zhao, X.-H. Hu, X. Zhou, W. Rao and Z.-L. Shen, J. Org. Chem., 2024, 89, 13661 CrossRef CAS PubMed.
- (a) T. Ben Halima, W. Zhang, I. Yalaoui, X. Hong, Y.-F. Yang, K. N. Houk and S. G. Newman, J. Am. Chem. Soc., 2017, 139, 1311 CrossRef CAS PubMed; (b) J. Masson-Makdissi, J. K. Vandavasi and S. G. Newman, Org. Lett., 2018, 20, 4094 CrossRef CAS PubMed; (c) Y.-L. Zheng, P.-P. Xie, O. Daneshfar, K. N. Houk, X. Hong and S. G. Newman, Angew. Chem., Int. Ed., 2021, 60, 13476 CrossRef CAS PubMed.
- (a) H. Hauptmann and M. Mader, Synthesis, 1978, 1978, 307 CrossRef; (b) R. Krishnamurti, D. R. Bellew and G. K. S. Prakash, J. Org. Chem., 1991, 56, 984 CrossRef CAS; (c) R. P. Singh, G. Cao, R. L. Kirchmeier and J. n. M. Shreeve, J. Org. Chem., 1999, 64, 2873 CrossRef CAS PubMed; (d) I. Shiina, Y. Imai, M. Suzuki, M. Yanagisawa and T. Mukaiyama, Chem. Lett., 2004, 29, 1062 CrossRef; (e) C. Clarke, D. J. Fox, D. S. Pedersen and S. Warren, Org. Biomol. Chem., 2009, 7, 1329 RSC.
- (a) S. Vellalath, I. Čorić and B. List, Angew. Chem., Int. Ed., 2010, 49, 9749 CrossRef CAS PubMed; (b) J. H. Kim, I. Čorić, S. Vellalath and B. List, Angew. Chem., Int. Ed., 2013, 52, 4474 CrossRef CAS PubMed; (c) Z. Chen and J. Sun, Angew. Chem., Int. Ed., 2013, 52, 13593 CrossRef CAS PubMed; (d) W. Lim, J. Kim and Y. H. Rhee, J. Am. Chem. Soc., 2014, 136, 13618 CrossRef CAS PubMed; (e) L. Qiu, X. Guo, C. Ma, H. Qiu, S. Liu, L. Yang and W. Hu, Chem. Commun., 2014, 50, 2196 RSC; (f) A. Matsumoto, K. Asano and S. Matsubara, Chem. Commun., 2015, 51, 11693 RSC; (g) B. Mondal, K. Mondal, P. Satpati and S. C. Pan, Eur. J. Org Chem., 2017, 2017, 7101 CrossRef CAS; (h) S. Li, J. Lv and S. Luo, Org. Chem. Front., 2018, 5, 1787 RSC; (i) B. Ye, J. Zhao, K. Zhao, J. M. McKenna and F. D. Toste, J. Am. Chem. Soc., 2018, 140, 8350 CrossRef CAS PubMed; (j) N. Mistry and S. P. Fletcher, Chem. Sci., 2018, 9, 6307 RSC; (k) M. Li, Y. Liu and Y. J. Zhang, Org. Lett., 2022, 24, 6716 CrossRef CAS PubMed.
- I. Čorić, S. Vellalath and B. List, J. Am. Chem. Soc., 2010, 132, 8536 CrossRef PubMed.
- (a) I. Čorić and B. List, Nature, 2012, 483, 315 CrossRef PubMed; (b) Z. Sun, G. A. Winschel, A. Borovika and P. Nagorny, J. Am. Chem. Soc., 2012, 134, 8074 CrossRef CAS PubMed; (c) F. Wang, F. Chen, M. Qu, T. Li, Y. Liu and M. Shi, Chem. Commun., 2013, 49, 3360 RSC; (d) X. Wang, S. Dong, Z. Yao, L. Feng, P. Daka, H. Wang and Z. Xu, Org. Lett., 2014, 16, 22 CrossRef CAS PubMed; (e) Z. H. Du Peng, S. Guanshuo and K. Zou, Chin. J. Org. Chem., 2015, 35, 1641 CrossRef; (f) J. Li, L. Lin, B. Hu, X. Lian, G. Wang, X. Liu and X. Feng, Angew. Chem., Int. Ed., 2016, 55, 6075 CrossRef CAS PubMed; (g) A. Midya, S. Maity and P. Ghorai, Chem. Eur J., 2017, 23, 11216 CrossRef CAS PubMed; (h) J. Gong, Q. Wan and Q. Kang, Adv. Synth. Catal., 2018, 360, 4031 CrossRef CAS; (i) S. Ge, W. Cao, T. Kang, B. Hu, H. Zhang, Z. Su, X. Liu and X. Feng, Angew. Chem., Int. Ed., 2019, 58, 4017 CrossRef CAS PubMed; (j) M. Balha, C. Soni and S. C. Pan, Eur. J. Org Chem., 2019, 2019, 2552 CrossRef CAS; (k) C.-C. Xie, R. Tan and Y.-K. Liu, Org. Chem. Front., 2019, 6, 919 RSC.
- (a) S. Handa and L. M. Slaughter, Angew. Chem., Int. Ed., 2012, 51, 2912 CrossRef CAS PubMed; (b) X. Wang, Z. Han, Z. Wang and K. Ding, Angew. Chem., Int. Ed., 2012, 51, 936 CrossRef CAS PubMed.
- (a) S. J. Collier, M. A. K. Vogel, B. J. Wong and N. K. Modukuru, Chapter 1: Biocatalytic Approaches to Chiral Heterocycles, in Prog. Heterocycl. Chem., ed. G. W. Gribble and J. A. Joule, Elsevier, 2009, vol. 21, pp 1 Search PubMed; (b) Y. Krishna, K. Shilpa and F. Tanaka, Org. Lett., 2019, 21, 8444 CrossRef CAS PubMed; (c) M. Li, Y. Liu and Y. J. Zhang, Org. Lett., 2022, 24, 6716 CrossRef CAS PubMed; (d) T. Gupta, D. Rani, L. M. Nainwal and R. Badhwar, Chirality, 2024, 36, e23637 CrossRef CAS PubMed.
- (a) P. Maity, H. D. Srinivas and M. P. Watson, J. Am. Chem. Soc., 2011, 133, 17142 CrossRef CAS PubMed; (b) H. D. Srinivas, P. Maity, G. P. A. Yap and M. P. Watson, J. Org. Chem., 2015, 80, 4003 CrossRef CAS PubMed; (c) T. Haidzinskaya, H. A. Kerchner, J. Liu and M. P. Watson, Org. Lett., 2015, 17, 3857 CrossRef CAS PubMed.
- T. Li, X. Cheng, P. Qian and L. Zhang, Nat. Catal., 2021, 4, 164 CrossRef CAS PubMed.
- (a) J. Zhu, Y. Wang, A. D. Charlack and Y.-M. Wang, J. Am. Chem. Soc., 2022, 144, 15480 CrossRef CAS PubMed; (b) R. Ding, Y. Wang and Y.-M. Wang, Synthesis, 2023, 55, 733 CrossRef CAS PubMed; (c) J. Zhu, H. Xiang, H. Chang, J. C. Corcoran, R. Ding, Y. Xia, P. Liu and Y.-M. Wang, Angew. Chem., Int. Ed., 2024, 63, e202318040 CrossRef CAS PubMed; (d) J. Yu, Y. Xia, S. Dey, J. Zhu, K. S. Cheung, S. J. Geib and Y.-M. Wang, J. Am. Chem. Soc., 2024, 146, 27998 CAS.
- (a) K. Matsuki, H. Inoue, A. Ishida, M. Takeda, M. Nakagawa and T. Hino, Chem. Pharm. Bull., 1994, 42, 9 CrossRef CAS; (b) R. Romagnoli, E. C. Roos, H. Hiemstra, M. J. Moolenaar, W. Nico Speckamp, B. Kaptein and H. E. Schoemaker, Tetrahedron Lett., 1994, 35, 1087 CrossRef CAS; (c) J. Kang, J. W. Lee, J. I. Kim and C. Pyun, Tetrahedron Lett., 1995, 36, 4265 CrossRef CAS; (d) F.-E. Chen, H.-F. Dai, Y.-Y. Kuang and H.-Q. Jia, Tetrahedron: Asymmetry, 2003, 14, 3667 CrossRef CAS; (e) A. Whyte, A. Torelli, B. Mirabi and M. Lautens, Org. Lett., 2019, 21, 8373 CrossRef CAS PubMed; (f) T. Sengoku, A. Miyoshi, T. Tsuda, T. Inuzuka, M. Sakamoto, M. Takahashi and H. Yoda, Tetrahedron, 2020, 76, 131252 CrossRef CAS; (g) A. Selmani and S. Darses, Org. Lett., 2020, 22, 2681 CrossRef CAS PubMed.
- (a) T. H. Lambert and S. J. Danishefsky, J. Am. Chem. Soc., 2006, 128, 426 CrossRef CAS PubMed; (b) T. R. Hoye and V. Dvornikovs, J. Am. Chem. Soc., 2006, 128, 2550 CrossRef CAS PubMed; (c) R. M. de Figueiredo, R. Fröhlich and M. Christmann, Angew. Chem., Int. Ed., 2007, 46, 2883 CrossRef CAS PubMed; (d) K. Uchida, T. Ogawa, Y. Yasuda, H. Mimura, T. Fujimoto, T. Fukuyama, T. Wakimoto, T. Asakawa, Y. Hamashima and T. Kan, Angew. Chem., Int. Ed., 2012, 51, 12850 CrossRef CAS PubMed; (e) R. Sano, R. Kosuge, T. Tsubogo and H. Uchiro, Tetrahedron Lett., 2018, 59, 704 CrossRef CAS; (f) C. Cui and M. Dai, Chin. J. Chem., 2023, 41, 3019 CrossRef CAS.
- (a) T. A. Agatsuma, S. Nara, S. Matsumiya, R. Nakai, H. Ogawa, S. Otaki, S.-i. Ikeda, Y. Saitoh and Y. Kanda, Org. Lett., 2002, 4, 4387 CrossRef CAS PubMed; (b) K. C. Nicolaou, A. A. Shah, H. Korman, T. Khan, L. Shi, W. Worawalai and E. A. Theodorakis, Angew. Chem., Int. Ed., 2015, 54, 9203 CrossRef CAS PubMed; (c) T. Nogawa, M. Kawatani, M. Uramoto, A. Okano, H. Aono, Y. Futamura, H. Koshino, S. Takahashi and H. Osada, J. Antibiot., 2013, 66, 621 CrossRef CAS PubMed.
- M. A. Walker, J. Org. Chem., 1995, 60, 5352 CrossRef CAS.
- (a) R. M. de Figueiredo, M. Voith, R. Fröhlich and M. Christmann, Synlett, 2007, 2007, 0391 CrossRef; (b) J. He, J. E. Baldwin and V. Lee, Synlett, 2018, 29, 1117 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2440354 and 2440355. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc02523k |
| ‡ Current address: Massachusetts Institute of Technology, Cambridge, Massachusetts 02139 USA. |
| § Current address: PPG Research and Development Center, Allison Park, Pennsylvania 15101 USA. |
| This journal is © The Royal Society of Chemistry 2025 |