
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNon-organic solvent extraction of capsaicinoids from oil combined with fluorescent lateral flow immunoassay strips for on-site identification of illegally recycled waste cooking oil†
Yulian
Wang‡
a,
Yuxiang
Wu‡
b,
Deji
Gesang
a,
Zixuan
Dong
a,
Zhaoxu
Qin
a,
Qingchun
Li
a,
Jin
Li
a,
Qianxu
Zhou
a and
Guoqing
Shi
 *a
*a
aSchool of Chemistry and Biological Engineering, University of Science and Technology Beijing, Beijing 100083, China
bSchool of Agricultural Engineering and Food Science, Shandong University of Technology, Zibo, 255049, Shandong Province, China
First published on 20th December 2024
Abstract
The illicit use of recycled waste cooking oil poses a threat to food safety, yet there is currently a lack of on-site identification methods. This study targets a key component of recycled cooking oil, capsaicinoids, and establishes a rapid detection method for identifying illegally recycled waste cooking oil on-site. The method involves extracting capsaicinoids from the oil using a non-organic solvent extractant, and then detecting it using fluorescent lateral flow immunoassay (LFIA) strips. The preparation conditions of LFIA test strips were first optimized in this study. Subsequently, a 0.02 mol L−1 solution of dimethyl-β-cyclodextrin was optimized as the extractant for capsaicin. The optimized sample extraction conditions involve a sample-to-extractant volume ratio of 1:2 and an extraction time of 1 minute, with a total extraction and detection time not exceeding 15 minutes. This method demonstrates a limit of detection (LOD) for natural capsaicin in oil samples of 0.14 μg kg−1, with a detection range spanning 0.46 to 81 μg kg−1. The method yields recovery rates between 88.76% and 115.79%, with coefficient of variation (CV) values ranging from 1.80% to 13.37%. Cross-reactivity rates for dihydrocapsaicin and synthetic capsaicin exceed 90%, while the impact of common contaminants or additives in other edible oils on detection is minimal. In conclusion, this approach fulfills the technical criteria mandated by the China Food and Drug Administration for distinguishing capsaicin compounds in recycled oil, offering advantages such as simplicity, rapidity, and “green” operation, making it suitable for rapid on-site identification of illegally recycled waste cooking oil.
1. Introduction
Illegally recycled waste cooking oil, also known as gutter oil, carries a high risk of causing harm to human health.1 The differentiation between refined gutter oil and conventional edible oil is essential due to their nearly identical appearances. Therefore, the development of a low-cost and easy-to-use technique for identifying gutter oil is valuable for preventing its illegal use.At present, the identification of refined gutter oil primarily relies on chromatographic or spectroscopic techniques. These methods are crucial for detecting specific compounds present in recycled oil that resist removal during the refining process, including monoglycerides,2 diglycerides, free fatty acids,3 polycyclic aromatic hydrocarbons,4 triglycerides,5 primary and secondary oxidation products generated after heat treatment,6 long-chain fatty aldehydes,7 unsaturated fatty acids,8 and triacylglycerides.9 Capsaicin is a vanilloid alkaloid that provides the sensation of spiciness. It exhibits high chemical stability and is commonly found in spicy seasonings in the forms of natural capsaicin (CAP), synthetic capsaicin (S-CAP), and dihydrocapsaicin (D-CAP).10 The addition of spicy condiments during cooking results in a notable presence of capsaicin compounds in the used oil. The high boiling point, lipid solubility, and heat stability of capsaicin make its elimination challenging during the refining process of kitchen waste oil.11 Detecting the presence of capsaicin in edible oil serves as an effective method for distinguishing recycled oil.12 In accordance with the regulations stipulated in the “Determination of Capsaicin in Edible Oils” (BJS201801) by the China National Food and Drug Administration, an oil sample may be deemed irregular if the total capsaicin content (including CAP, S-CAP, and D-CAP) in the sample is 1.0 μg kg−1 or higher, indicating a potential use of recycled oil.
High-performance liquid chromatography-tandem mass spectrometry (HPLC-MS/MS) is a common method for detecting capsaicin in edible oils, with a detection limit of 0.03 μg kg−1 as specified in BJS201801. Additionally, Wu et al. reported a method using HPLC with a fluorescence detector to detect CAP and D-CAP in vegetable oils.13 Recent research has focused on extraction methods for capsaicin in edible oils, including solid-phase extraction,14 liquid–liquid extraction combined with solid-phase extraction,15 matrix solid-phase dispersion,16 magnetic solid-phase extraction using graphene oxide–Fe3O4 (GO–Fe3O4) nanocomposites,17 and magnetic molecularly imprinted polymers.18 Gel permeation chromatography and immunoaffinity extraction have also been utilized.19,20 These detection methods offer high sensitivity and accuracy, but their implementation for on-site rapid identification of recycled cooking oil is challenging due to the requirement of large instruments, skilled operators, long analysis times, and high costs.
In recent years, several studies have documented rapid detection methods developed targeting capsaicin. These methods are primarily divided into two categories, one of which is based on the spectroscopic or electrochemical characteristics of capsaicin and its derivatives for rapid detection. For example, Liu et al. demonstrated the use of chloroform and NaOH solution as extractants for the agitation and centrifugation of edible oils.21 Subsequent pH adjustment with H2SO4 solution enabled the direct detection of enhanced signals of capsaicin within 10 minutes using a portable Raman spectrometer with silver nanoparticle solution, reaching a detection limit of 2.9 μg L−1. In a separate study,22 a 2% NaOH solution was employed for the extraction of edible oils, and capsaicin derivatives were synthesized by the addition of NaNO2 solution. The identification of derivatives in gutter oils was achieved through surface-enhanced resonance Raman scattering spectroscopy, with a detection limit of 1.0 × 10−8 M. Overall, these spectroscopic approaches typically entail the use of relatively expensive detection instruments. Qin et al. conducted ultrasonic-assisted extraction of capsaicin from edible oil by adding a methanol–tetrahydrofuran solution and shaking for 30 minutes.23 They detected capsaicin in the oil using a dual-mode colorimetric sensor based on graphene oxide (GO) and gold nanoparticles (AuNPs), with a detection limit of 0.14 μg L−1. Wang et al. introduced methanol into edible oil, followed by vortex centrifugation for 20 minutes.24 They employed an electrochemical sensor based on multi-walled carbon nanotubes/molecularly imprinted polymer (MWCNTs–MIP) to detect capsaicin in the oil, achieving a detection limit of 0.02 μmol L−1. Fang and Duan utilized methanol for the extraction of capsaicin from sewer oil, followed by ultrasonic centrifugation and filtration.25 They detected capsaicin in the edible oil using a bimetallic MOF nanocage electrochemical sensor, with a detection limit of 0.4 μM. While these electrochemical methods do not require expensive equipment, their detection sensitivity often falls short of requirements.
The second category of rapid detection methods relies on capsaicin antibody-based immunoassays. These methods include enzyme-linked immunosorbent assay (ELISA),12 LFIA techniques utilizing colloidal gold, quantum dots, and time-resolved fluorescence microspheres as labeling agents,12,26–28 fluorescence polarization immunoassay (FPIA),29 electrochemical immunosensors,30 non-enzyme immunoassay based on DMSNs@PDA@Pt,10 and homogeneous fluorescence immunoassay based on AuNPs (AgNPs) quenching multicolor QDs@hydrogel beads.31 All these approaches necessitate intricate sample preparation steps involving methanol extraction, nitrogen drying, or rotary evaporation to eliminate methanol solvents, re-dissolve the extracted compounds in buffer solutions, and other complexities. Recently, Zhao et al. combined immunodetection methods with the amplification capability of the CRISPR cas12a system to establish a highly sensitive detection method for capsaicin in soybean oil.32 However, this method necessitates complex sample pretreatment steps including dichloromethane oil removal, petroleum ether extraction of capsaicin, nitrogen drying to eliminate petroleum ether, and resolubilization of the extract in a buffer solution. It is evident that current approaches are hindered by expensive instrumentation costs, intricate sample preparation procedures, and the use of toxic organic solvents, making it challenging to achieve cost-effective on-site rapid identification of gutter oil.
Cyclodextrins (CDs) are cyclic oligosaccharides comprising a cylindrical structure of at least six D-(+)-glucopyranose units linked by α-(1,4) glycosidic bonds.33 Due to their hydrophilic outer surface and lipophilic inner cavity, CDs are commonly employed as solubilizing agents for non-polar drugs.34 Recent findings indicate that cyclodextrin derivatives can enhance the transfer of lipophilic atrazine from the oil phase to the water phase, enabling the direct extraction of acetochlor from oil samples using cyclodextrin derivative solutions.35 This study aims to investigate the direct extraction of capsaicin from the oil phase using cyclodextrin derivative solutions and assess the feasibility of a field-deployable rapid detection technology based on this organic solvent-free extraction method and capsaicin lateral flow immunoassay test strips.
2. Experimental
2.1 Materials
Carboxyl functionalized europium-based fluorescent nanoparticles (EuNPs, average diameter 0.19 μm, solid content 1%, carboxyl concentration 139 ueq g−1) were purchased from Bangs laboratories. Carboxyl functionalized europium-based fluorescent nanoparticles (EuNPs, average diameter 0.07 μm, solid content 0.53%, carboxyl concentration 148 ueq g−1) were purchased from Shanghai Suyuan Biotechnology Co., Ltd. Ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC, 98%) and ethanolamine were purchased from Aladdin (Shanghai, China). N-Hydroxysulfoxide succinimide (sulfo-NHS, 98%) was purchased from MERYER (Shanghai, China). Morpholine ethanesulfonic acid (MES, 99%) was purchased from J&K Scientific (Beijing, China). Bovine serum albumin (BSA, 98%) was purchased from Sphere-MFCIS Biotechnology, Inc. Goat anti-mouse IgG (BR) was purchased from Hangzhou Sonice Biotechnology Co., Ltd. Hydroxyethyl-β-cyclodextrin (E-β-CD, 99%) was purchased from Shanghai Yuanye Biotechnology Co., Ltd. Dimethyl-β-cyclodextrin (DM-β-CD, 98%) was purchased from Zhejiang Lianshuo Biotechnology Co., Ltd. Sulfobutyl-β-cyclodextrin (S-β-CD, 99%) was purchased from Shanghai Yuanye Biotechnology Co., Ltd. Methyl-β-cyclodextrin (M-β-CD) and (2-hydroxypropyl)-β-cyclodextrin (HP-β-CD, 97%) were purchased from Beijing Bailingwei Technology Co., Ltd. Capsaicin (CAP, AR) was purchased from Beijing Yili Fine Chemicals Co., Ltd. Synthetic capsaicin (S-CAP, AR) and dihydro-capsaicin (D-CAP, AR) were purchased from Aladdin (Shanghai, China). Certified reference materials for capsaicin-containing oil (including CAP, S-CAP and D-CAP) were purchased from National Institute of Metrology (China).Capsaicin antibodies (AbCAP) and CAP-BSA were purchased from Shandong Landu Biotechnology Co., Ltd. Staphylococcus aureus Protein G (SPG,97.6%) was purchased from Dalian Meilun Biotechnology Co., Ltd. Disulfide bis(succinimide propionic acid ester) (DSP, 97%) was purchased from Shanghai Yuanye Biotechnology Co., Ltd. Polyvinyl chloride (PVC) sheets, glass fibre membranes (CB08) and absorbent pads (SX27) were obtained from Kinbio (Shanghai, China). Nitrocellulose (NC) membranes (Pall Vivid 120) were purchased from Pall Biotech (Maharashtra, India).
2.2 Instruments
The XYZ HM3010 dispensing platform was purchased from Jiening Biotechnology Co., Ltd. (Shanghai, China). A guillotine cutter was purchased from ANTOKUN Co., Ltd. (Hangzhou, China). A portable fluorescence detector (FIC-H1) was purchased from Jensen Biotechnology Co., Ltd. (Beijing, China). A high speed freezing centrifuge purchased from SCILOGEX (USA).2.3 Production of Ab@EuNP conjugates
Following the method described by Mao et al.,36 a protocol was adapted to immobilize CAP antibodies onto EuNPs using SPG. Briefly, EDC and sulfo-NHS were mixed with EuNPs at molar ratios of 0.32 mmol and 1.84 mmol per mg of EuNPs, and the mixture was shaken for 20 minutes. The solution was then centrifuged at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 15 minutes, the supernatant was removed, and the pellet was washed three times with 300 μL of phosphate-buffered saline (PBS, containing 10 mmol L−1 phosphate and 154 mmol L−1 NaCl, pH 7.2). After resuspending the pellet in 600 μL of PBS, 500 μL of 0.4 mg mL−1 SPG solution was added, followed by a 2 hour incubation. Subsequently, 100 μL of 10% BSA was added for blocking and incubated for 30 minutes. The sample was then centrifuged at 12000 rpm for 15 minutes, the supernatant was removed, and the pellet was resuspended in 250 μL of PBS–Tween buffer (PBST, containing 10 mmol L−1 phosphate, 154 mmol L−1 NaCl, and 0.5% Tween-20, pH 7.2). Next, 250 μL of CAP antibody solution was added and incubated for 1 hour, followed by the addition of 250 μL of 0.4 mol L−1 DSP solution for crosslinking. The sample was incubated for 30 minutes and the reaction was terminated by adding 37.5 μL of 1 mol L−1 ethanolamine. After centrifuging at 12000 rpm for 15 minutes and resuspending in 600 μL of PBS, the sample was stored at 4 °C for future use.
000 rpm for 15 minutes, the supernatant was removed, and the pellet was washed three times with 300 μL of phosphate-buffered saline (PBS, containing 10 mmol L−1 phosphate and 154 mmol L−1 NaCl, pH 7.2). After resuspending the pellet in 600 μL of PBS, 500 μL of 0.4 mg mL−1 SPG solution was added, followed by a 2 hour incubation. Subsequently, 100 μL of 10% BSA was added for blocking and incubated for 30 minutes. The sample was then centrifuged at 12000 rpm for 15 minutes, the supernatant was removed, and the pellet was resuspended in 250 μL of PBS–Tween buffer (PBST, containing 10 mmol L−1 phosphate, 154 mmol L−1 NaCl, and 0.5% Tween-20, pH 7.2). Next, 250 μL of CAP antibody solution was added and incubated for 1 hour, followed by the addition of 250 μL of 0.4 mol L−1 DSP solution for crosslinking. The sample was incubated for 30 minutes and the reaction was terminated by adding 37.5 μL of 1 mol L−1 ethanolamine. After centrifuging at 12000 rpm for 15 minutes and resuspending in 600 μL of PBS, the sample was stored at 4 °C for future use.
2.4 Preparation of the LFIA strip
The nitrocellulose (NC) membrane was humidified at 25 °C and 55% humidity for at least one hour. A solution of 0.4 mg mL−1 CAP–BSA (in PBS buffer, 10 mmol L−1, pH 7.2) and 0.2 mg mL−1 goat anti-mouse IgG (in PBS buffer, 10 mmol L−1, pH 7.2) was streaked onto the T and C lines of the NC membrane (at a speed of 0.5 μL cm−1), followed by drying in a 37 °C constant temperature incubator for 24 hours. The Ab@EuNP conjugate and the release solution (containing 10 mmol L−1 phosphate, 100 mmol L−1 sucrose, 5 mg mL−1 BSA, 2.5 mg mL−1 Tween-20, 0.03% Proclin 300) were mixed in a 1:1 volume ratio and sprayed onto a 30 × 1 cm glass fiber membrane at a speed of 9 μL cm−1 using a dispensing platform, then dried in a 37 °C constant temperature incubator for 24 hours to produce the conjugate pad. A 5 mL sample pad treatment solution (containing 20 mmol L−1 PB, 20 mg mL−1 trehalose, 10 mg mL−1 Tween-20) was pipetted onto a 30 × 2 cm glass fiber membrane for complete infiltration, followed by drying in a 37 °C constant temperature incubator for 24 hours to produce the sample pad. The sample pad, conjugate pad, NC membrane, and absorbent paper were sequentially adhered to a PVC plate, then cut into 4 mm-wide test strips using a strip cutter, and assembled into a cartridge, thus forming the CAP detection card. The CAP detection cards were stored in a desiccator for future use.
2.5 Detection procedure
The principle behind the competitive LFIA assay for CAP detection is depicted in Scheme 1a. Initially, the sample is applied onto the sample pad and capillary action drives the lateral flow of the liquid. The liquid first encounters the conjugate pad, causing the dissolution of the Ab@EuNP present on it. Subsequently, the liquid carries the Ab@EuNP flow along the NC membrane. Upon passing through the T line region, the CAP in the liquid competes with the immobilized CAP–BSA on the NC membrane to bind with the Ab@EuNP. As the liquid reaches the C line region, any unbound Ab@EuNP is captured by the immobilized anti-mouse antibody at that location. After scanning the test strip with a fluorescence reader, a fluorescence distribution spectrum is generated. The fluorescence peak at the T line is notably lower in positive samples compared to negative samples.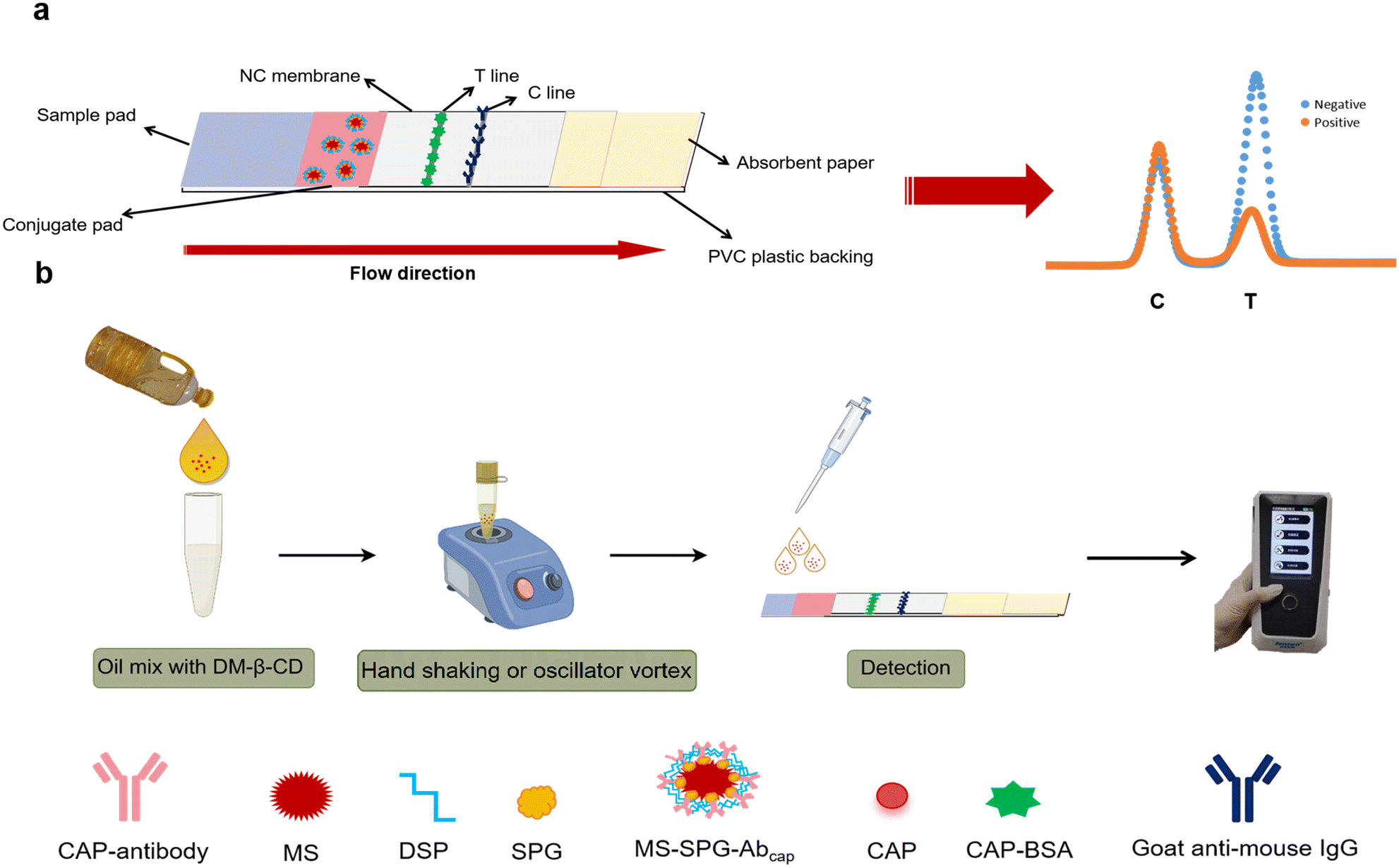 | ||
| Scheme 1 (a) The structure and detection principle of the LFIA strip. (b) On-site extraction and test process. By Figdraw. | ||
100 μL of the test solution was dropped into the sample well of the CAP detection card. It was then allowed to flow for 10 minutes. Subsequently, the detection card was inserted into the fluorescence immunoassay analyzer to measure the fluorescence intensity (FI) values of the T line and C line. The T/C ratio and inhibition rate (I%) were calculated according to the formula outlined below
 | (1) |
2.6 Extraction efficiency
Blank soybean oil samples were mixed with an extraction solvent solution in a 1:2 ratio, vigorously shaken for 1 minute, and then allowed to stand for 2 minutes. The lower aqueous phase was collected as the solvent to prepare standard solutions of CAP at concentrations of 0, 0.1, 0.4, 1.6, 3.1, 6.2, 25, and 100 ng mL−1. The strip was used to detect these solutions and calculate the corresponding I% for each CAP concentration. A plot of CAP concentration against I% was generated to create CAP detection standard curves for each extraction solvent solution.
CAP was spiked into blank oil samples to prepare a final concentration of 10 ng mL−1 (note: in certain standard documents, the concentration of compounds in oil is often expressed in μg kg−1. Given that the density of oil is approximately 0.9 kg L−1, converting the concentration unit from μg kg−1 to ng mL−1 requires multiplying the concentration value by 0.9. Conversely, to convert the concentration unit from ng mL−1 to μg kg−1, the concentration value needs to be divided by 0.9). The samples were extracted with a solvent solution, detected using a test strip, and the I% was calculated. The I% was then used in the standard curve to determine the concentration of CAP in the extraction liquid. The extraction efficiency was calculated using the formula below, where C is the CAP concentration in the extraction liquid, Vw is the volume of the extraction liquid, C0 is the initial CAP concentration in the oil sample, and Vo is the volume of the oil sample.
| Extraction efficiency % = (C × Vw/C0 × Vo) × 100% | (2) |
2.7 Detection of CAP in oil
The optimized extraction solution was mixed with the oil sample in a specific volume ratio. The mixture was vigorously shaken for 1 minute, followed by a 2 minute settling period to achieve phase separation between the oil and water. Subsequently, 100 μL of the aqueous phase was dropped onto an LFIA strip for the detection of CAP content (Scheme 1b).2.8 Standard curve
The procedure involved adding a standard solution of CAP to samples of soybean oil, ensuring thorough mixing to achieve a final CAP concentration of 81.00 μg kg−1. Subsequently, the spiked samples were diluted with soybean oil to achieve final CAP concentrations of 0, 0.04, 0.33, 1.00, 3.00, 9.00, 27.00, and 81.00 μg kg−1. Following an optimized extraction method, CAP was extracted from the samples, applied onto CAP detection cards, and the FI of the test (T) and control (C) lines was measured. B0 was defined as the T/C value corresponding to the blank solution. The T/C values of various CAP solutions were expressed relative to B0 as B/B0. A standard curve was generated by plotting B/B0 on the y-axis against the logarithm of CAP concentration on the x-axis.2.9 LOD, LOQ and working range
According to Eurachem guidelines,37 the LOD value of the method was initially estimated to be around 0.1 μg kg−1 through preliminary experiments. Subsequently, CAP was added to soybean oil to achieve a final concentration of 0.1 μg kg−1, serving as a low-concentration sample for determining the LOD and LOQ values. The sample was analyzed using the method in ten replicates, and the concentration of the sample was determined based on the standard curve to calculate the standard deviation (S0) of the measurement values, followed by the calculation of using the formula below:
using the formula below: | (3) |
Among them, with n = 10, the LOD and LOQ were determined as 3×  and 10×
and 10× 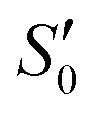 , respectively. The lower limit of the method's working range was defined by the LOQ, while the upper limit was established as the CAP concentration at which a noticeable decrease in the T/C value is visually observed.
, respectively. The lower limit of the method's working range was defined by the LOQ, while the upper limit was established as the CAP concentration at which a noticeable decrease in the T/C value is visually observed.
2.10 Selectivity
Different concentrations of D-CAP and S-CAP, ranging from 0 to 81.00 μg kg−1, were prepared in soybean oil. Standard curves were plotted using this method. The T/C values corresponding to blank oil samples were multiplied by 50% and then substituted into the three standard curves for CAP, D-CAP, and S-CAP. The IC50 values (50% inhibition concentration) for each compound were calculated. The cross-reactivity rate CR% was calculated using the following formula: | (4) |
Furthermore, two common additives in edible oils, tertiary butylhydroquinone (TBHQ) and vitamin E (VE), along with two prevalent contaminants, aflatoxin B1 (AFB1) and benzo[a]pyrene (Bap), and one frequently illegally added flavor, ethyl maltol, were selected for investigation. These compounds were individually spiked into soybean oil with 1 μg kg−1 of CAP at concentrations of TBHQ (400 μg kg−1), VE (400 μg kg−1), Bap (20 μg kg−1), and AFB1 (40 μg kg−1), respectively. These concentrations were twice the Maximum Residue Limit (MRL) specified in the Chinese National Standards for edible oils (GB2760-2014; GB2761-2017; GB2762-2022). Ethyl maltol was a potential unauthorized additive in edible oils, lacking an established MRL. The concentration used in this study was100 μg kg−1, approximately twice the highest concentration reported in oil samples from the Chinese market.38 The investigation was focused on the interference of these compounds in CAP detection.
2.11 Spiked recovery
In soybean oil, corn oil, peanut oil, and rapeseed oil, CAP standards were separately added to prepare samples with concentrations of 0.55, 1.11, and 5.55 μg kg−1. Additionally, a certain amount of soybean oil was taken and 0.5 μg kg−1 of capsaicin, dihydrocapsaicin, and synthetic capsaicin were added to achieve a total concentration of capsaicinoids of 1.5 μg kg−1 in the oil sample. The CAP content in the oil samples was determined using the method established in this study, and the recovery rate was calculated according to the following formula:| Recovery rate % = (Detected CAP/Spiked CAP) × 100% | (5) |
2.12 Accuracy
In this study, the total content of capsaicinoid compounds in the certified reference material (CRM) for edible oils (certification number: GBW(E)100503) was determined. The content of CAP, D-CAP, and S-CAP in this CRM was found to be 1.08 μg kg−1, 1.13 μg kg−1, and 1.14 μg kg−1, respectively. The bias value was calculated using the formula provided below, where X was represents the total content of capsaicinoid compounds detected using the method developed in this study, and X0 is the sum of the certified concentrations of the three compounds, with a value of 3.35 μg kg−1.| b% = (X − X0)/X0 × 100% | (6) |
2.13 Data processing
In this study, charts were created using Excel 2021, while standard curves were plotted using Origin 2018 software with curve fitting conducted using the Logistic model. Significance of differences was analyzed through one-way ANOVA, with a significance level set at P < 0.05 indicating statistical significance.3. Results and discussion
3.1 Optimization of the EuNP-LFIA strip for capsaicin
Two different sizes of EuNPs, 70 nm and 200 nm, available in the market were utilized as labeling materials to prepare Ab@EuNP. The response of LFIA test strips constructed with these two Ab@EuNPs towards CAP was compared. Both types of EuNPs were pre-modified with SPG according to the method described in Mao et al.,36 followed by the addition of CAP antibody at a concentration of 6 μg mL−1. The SPG facilitates the oriented immobilization of the antibody on the EuNP by binding to the Fc segment of the antibody. The NC membrane was coated with CAP–BSA at a concentration of 0.4 mg mL−1. LFIA test strips were prepared under the aforementioned conditions, and the FI values of the T line and the I% when detecting the blank solution and 5 ng mL−1 CAP solution are shown in Fig. 1a. Clearly, using 200 nm EuNPs resulted in a higher fluorescence signal and increased sensitivity for CAP detection. | ||
| Fig. 1 Optimization of preparation conditions for the LFIA strip for CAP. The effect on the FI values of the T line and the I% when detecting the blank solution and 5 ng mL−1 CAP solution (n = 3) by (a) different sizes of EuNPs; (b) different antibody concentrations; (c) different CAP–BSA concentrations; (d) different buffer pH values. | ||
Subsequently, the impact of varying quantities of antibodies labeled on EuNPs on the detection of CAP was investigated, with the results depicted in Fig. 1b. It is evident that as the concentration of CAP antibodies increases, the FI of the T line also rises. However, upon reaching a concentration of 7.6 μg mL−1, the FI plateaus, indicating saturation of the binding sites for CAP antibodies on the EuNPs. The trend in the change of I% diminishes gradually with increasing antibody concentration, leading to the selection of 7.6 μg mL−1 as the optimized antibody concentration, considering factors such as the cost of antibodies. The results indicate that in the three experimental groups with antibody concentrations of 1.9, 3.8, and 7.6 μg mL−1, the FI values of the test line (T line) increased with the antibody concentration, showing significant differences among them. However, the corresponding I% values in these three groups did not show significant variations. In the experimental groups with antibody concentrations of 7.6, 11.4, and 21.6 μg mL−1, the FI values of the T line did not increase significantly with the antibody concentration, whereas the I% values decreased significantly. The increase in FI values suggests an increase in the amount of antibodies bound to the surface of EuNPs. However, the plateauing of FI values does not necessarily indicate a saturation of antibody binding, as saturation may occur when the density of antibodies bound to EuNPs reaches the binding capacity limit of CAP–BSA on the T line. The competitive binding of CAP in the solution with immobilized CAP–BSA for Ab@EuNP occurs at the T line. The ratio of Ab@EuNP bound to CAP and CAP–BSA is largely unaffected by the quantity of antibodies on the surface of EuNPs when equilibrium is rapidly achieved in this reaction. Therefore, the I% remains relatively constant when the antibody concentration is between 1.9 and 7.6 μg mL−1. However, when the amount of antibodies bound to EuNPs is sufficiently high, even though a certain proportion of antibody binding sites may be occupied by CAP, the remaining sites can still ensure the capture of Ab@EuNP by CAP–BSA. This explains the phenomenon of the I% decreasing with an increase in antibody concentration between 7.6 and 21.6 μg mL−1.
The impact of CAP–BSA concentration on detection outcomes on the NC membrane was investigated under optimized antibody concentrations. The results are presented in Fig. 1c, showing a gradual increase in the FI value of the T line with increasing CAP–BSA concentration. Beyond 0.4 mg mL−1, the FI value plateaued, reaching its maximum inhibition rate at this point.
LFIA test strips were prepared at optimized antibody concentration and CAP-BSA coating concentration. The effect of different reaction buffer pH values on CAP detection was investigated, with results shown in Fig. 1d. When detecting the blank solution, the FI of the T line gradually decreased with increasing pH, indicating a decrease in the binding capacity between Ab@EuNP and CAP–BSA at higher pH levels. Conversely, when detecting the 5 ng mL−1 CAP solution, the I% initially increased with pH, reaching a peak at pH 7.5, and then decreased with further increase in pH. This suggests that at pH 7.5, the competitive binding advantage of free CAP in solution over CAP–BSA immobilized on the NC membrane with Ab@EuNP is maximized. Therefore, pH 7.5 was chosen as the optimized reaction pH.
An investigation was conducted to assess the impact of different detection times, as illustrated in Fig. S1.† When testing the blank solution, the fluorescence intensity (FI) of the test line increased progressively with longer detection periods. Upon reaching 10 minutes, the FI value leveled off, and there was no substantial decrease in the T/C ratio beyond this timeframe. Therefore, the optimized detection time was selected as 10 minutes.
3.2 Optimization of the extraction process
Five cyclodextrin derivatives, hydroxyethyl-β-cyclodextrin (E-β-CD), dimethyl-β-cyclodextrin (DM-β-CD), sulfobutylether-β-cyclodextrin (S-β-CD), methyl-β-cyclodextrin (M-β-CD), and (2-hydroxypropyl)-β-cyclodextrin (HP-β-CD), were selected and prepared in a phosphate buffer solution (pH 7.09) at a concentration of 0.03 mol L−1. Using these solutions, blank oil samples and spiked oil samples with 10 ng mL−1 of CAP were extracted separately. The LFIA method was employed to determine the corresponding I% for each extractant (Fig. S2†). The obtained I% values were then substituted into the standard curves for each extractant (Fig. S3†) to calculate the extraction efficiency of CAP in the oil samples for the different solutions (Fig. 2a). Analysis of Fig. 2a indicates that among the five cyclodextrin solutions tested, DM-β-CD exhibited the highest extraction efficiency for CAP in the oil phase at an equivalent molar concentration. Consequently, DM-β-CD was chosen as the extraction agent for subsequent experiments. | ||
| Fig. 2 Optimization of the extraction process. (a) The extraction efficiency of cyclodextrin derivative solutions for capsaicin in oil samples; (b) the extraction efficiency of different DM-β-CD concentrations for capsaicin; (c) the effect of pH on the extraction efficiency; (d) the comparation of experimental and theoretical Cw at different W/O ratios; (e) the effect of shaking time on the I%. | ||
The influence of various concentrations of DM-β-CD on the extraction efficiency of CAP was investigated. Various concentrations of DM-β-CD were prepared using phosphate buffer, as described earlier. The blank oil samples and spiked samples with 10 ng mL−1 of CAP were extracted, and the I% values were calculated (Fig. S4†). These values were then applied to their respective standard curves (Fig. S5†) to determine the extraction efficiency of different concentrations of DM-β-CD solutions (Fig. 2b). As depicted in Fig. 2b, the extraction efficiency of CAP exhibited an initial increase followed by a decrease with the rising concentration of DM-β-CD, peaking at a concentration of 0.02 mol L−1. Consequently, for subsequent experiments, the concentration of DM-β-CD was established at 0.02 mol L−1.
Due to the presence of dissociable amino groups in CAP, the pH of the solution can affect its dissociation state, potentially impacting the partition coefficient of CAP between oil and water phases. Therefore, we investigated the influence of the pH of the DM-β-CD solution on the extraction efficiency.
Solutions of 0.02 mol L−1 DM-β-CD were prepared using a phosphate buffer (10 mmol L−1) at pH 5.93 and 7.09, as well as a carbonate buffer (10 mmol L−1) at pH 8.71 and 9.77. The CAP in blank oil samples and spiked samples was extracted using the aforementioned solutions. The I% values were calculated (Fig. S6†), which were subsequently utilized in the standard curve (Fig. S7†) to ascertain the extraction efficiency of DM-β-CD solutions at various pH levels (Fig. 2c). The results indicate that the extraction efficiency of CAP is relatively low under neutral or slightly acidic conditions, but nearly doubles under weak alkaline conditions compared to neutral conditions. As the extraction recovery was highest in the buffer at pH 8.71, subsequent experiments utilized DM-β-CD solution prepared with a carbonate buffer at pH 8.71.
Subsequently, we examined the influence of the water-to-oil (W/O) volume ratio on the detection outcomes of CAP. The CAP in blank oil samples and spiked samples was extracted using a DM-β-CD solution at different W/O ratios. The I% values were calculated (Fig. S8†), which were subsequently utilized in the standard curve (Fig. S7†) to detect the corresponding CAP concentrations. The theorical concentration of CAP in the aqueous phase (Cw) can be determined through the equilibrium equation provided below:
| Kow = (Co0Vo − CwVw)/CwVo | (7) |
In this study, Co0 represents the initial concentration of CAP in the oil phase, where Kow is defined as Co/Cw, representing the equilibrium constant for the diffusion of CAP between the oil and water phases. Vw and Vo denote the volumes of the water and oil phases, respectively. Assuming Vw/Vo = n, eqn (7) yields Co0/Cw = Kow + n. Based on the experimental extraction efficiency of CAP obtained in the previous step, the values of Co and Cw at equilibrium were determined as 4.79 ng mL−1 and 2.60 ng mL−1, respectively, leading to a calculated Kow value of 1.84. Utilizing Kow and Co0, theoretical Cw values for different W/O ratios were computed. By comparing the experimentally measured  values under various W/O ratios with the theoretical values (as shown in Fig. 2d), it was observed that, except for the experiment with a W/O ratio of 1, the measured
values under various W/O ratios with the theoretical values (as shown in Fig. 2d), it was observed that, except for the experiment with a W/O ratio of 1, the measured  values closely matched the theoretical values. This discrepancy may be attributed to the limited water phase volume hindering the achievement of diffusion equilibrium or potential oil contamination during sampling. Additionally, an increase in the W/O ratio is observed to correspond with a gradual rise in the extraction efficiency (Fig. S9a†). Consequently, in subsequent experiments, a water-to-oil phase volume ratio of 2 was established.
values closely matched the theoretical values. This discrepancy may be attributed to the limited water phase volume hindering the achievement of diffusion equilibrium or potential oil contamination during sampling. Additionally, an increase in the W/O ratio is observed to correspond with a gradual rise in the extraction efficiency (Fig. S9a†). Consequently, in subsequent experiments, a water-to-oil phase volume ratio of 2 was established.
Based on the optimized conditions, the shaking time of the oil–water mixture system was ultimately optimized. As shown in Fig. 2e, an increase in shaking time leads to a rise in I%. However, once the shaking time reaches 1 minute, the corresponding I% stabilizes, showing no significant difference. Meanwhile, the corresponding changes in extraction efficiency also exhibit a similar trend (Fig. S9b†). At this point, the distribution of CAP between the oil and water phases may have reached equilibrium, hence 1 minute was selected as the shaking time for the oil–water system.
3.3 LOD and working range
In order to establish the LOD and LOQ, as well as the working range, a standard curve for CAP detection in edible oils was first generated (Fig. 3a), accompanied by the image of the LFIA strip in Fig. 3b. The standard curve for CAP is described by the regression equation y = −0.02 + 0.96/(1 + (x/6.10)^0.81), where x represents the concentration of CAP in edible oil samples (μg kg−1), and y denotes the B/B0 value. The method's LOD was determined to be 0.14 μg kg−1 following Eurachem guidelines, with the LOQ set at 0.46 μg kg−1 and a working range of 0.46–81 μg kg−1. According to the rapid testing method standard “Rapid Detection of Capsaicin in Edible Vegetable Oils – Fluorescence Immunoassay” (KJ202103) issued by the State Administration for Market Regulation of China, the LOD for capsaicin should be 0.4 μg kg−1. The LOD of this method is nearly three times lower than that stipulated in the standard method, thus fully meeting the requirements of the said standard. | ||
| Fig. 3 (a) Standard curves for three capsaicinoid compounds in edible oils; (b) LFIA strips under ultraviolet lamp irradiation. | ||
Fig. 3 also displays the standard curves of D-CAP and S-CAP. The curves for the three capsaicinoid compounds in the figure are essentially overlapping, indicating that the method developed in this study can simultaneously detect the total amount of these compounds. Given that the method's limit of quantification is below the specified detection concentration for identifying recycled cooking oil set by the China Food and Drug Administration (1.0 μg kg−1 for the total amount of CAP, D-CAP, and S-CAP), this approach is deemed suitable for distinguishing recycled cooking oil.
Table 1 presents a comparison between our method and other studies on capsaicin detection. The LOD of our method is similar to that of MMIP-HPLC-FLD for CAP detection. Compared to methods utilizing SERS, terahertz spectroscopy, and immunoassays, our LOD is lower but higher than the CRISPR–Cas12a iPOCT method. It is important to note that for practical food safety testing, lower detection limits are not always better.39 Both the LOD and LOQ values of our method are lower than the concentration of CAP used to determine recycled cooking oil, making it suitable for on-site identification due to its simple and environmentally friendly sample preparation.
| Detection method | Type of sample | LOD (μg kg−1) | Advantagea | Ref. |
|---|---|---|---|---|
| a Different letters represent various advantages for on-site detection: a – no need for heavy equipment, b – simple sample pre-treatment, c – total detection time, including sample processing, does not exceed 30 minutes, d – no requirement for organic solvents. | ||||
| Terahertz spectroscopy | Soybean oil | 1.25 | b | 40 |
| TRF-LFS | Gutter oil | 2.56 | abc | 28 |
| Electrochemical sensor | Gutter oil | 6.79 | a | 24 |
| Capture-SELEX | Edible oil | 0.16 | a | 23 |
| CRISPR-Cas12a iPOCT | Edible oil | 8.83 × 10−4 | a | 32 |
| SERS | Edible oil | 3.22 | ac | 21 |
| TRFICA | Vegetable oils | 0.6 | abc | 27 |
| MMIP-HPLC-FLD | Edible oil | 0.06 | b | 18 |
| EuNP-LFIA | Edible oil | 0.14 | abcd | This work |
In addition, various detection methods were compared for on-site detection based on their respective advantages (Table 1). It is evident that this method demonstrates significant advantages in on-site detection due to the utilization of environmentally friendly reagents, simple pre-treatment, and shorter detection times.
3.4 Selectivity
From Fig. 3, the IC50 values for CAP, D-CAP, and S-CAP were calculated as 5.02 μg kg−1, 5.41 μg kg−1, and 5.54 μg kg−1, respectively. Based on this, the cross-reactivity rates for detecting D-CAP and S-CAP using this method were determined to be 92.79% and 90.61%, respectively. Therefore, this method is capable of quantifying the total amount of the three capsaicinoid compounds mentioned. TBHQ, ethoxyquin (ethoxyquinoline), VE (vitamin E), AFB1 (aflatoxin B1), and ZEN (zearalenone) are commonly encountered artificial synthetic additives or natural contaminants in edible oils. This study investigated the impact of high concentrations of these interfering substances on the detection of CAP in edible oils (Fig. 4). The results indicate that in the presence of these interferents, there was no significant difference in the response (I%) of the test strips compared to when the interferents were absent, suggesting that these chemical substances do not interfere with the identification of recycled cooking oil. | ||
| Fig. 4 The impact of potential interferents in plant oils on capsaicin detection. | ||
3.5 Storage stability
The test cards were stored at room temperature in vacuum-sealed aluminum foil pouches for 6 months. Subsequently, the optimized sample extraction method was used to extract blank oil samples and the Certified Reference Material (CRM) containing CAP. The corresponding test solutions were then analyzed using the test cards, and the T/C values obtained were compared with those from the same samples tested six months prior. The results are presented in Fig. S10,† indicating no significant differences in the test results between the blank and CRM samples. This demonstrates the stability of the test cards for storage at room temperature for over 6 months.3.6 Trueness
In order to verify the accuracy of this method, this study initially conducted spiked recovery experiments on four different types of edible oils, namely soybean oil, corn oil, peanut oil, and rapeseed oil. As indicated in Table 2, the recovery rates of the three CAP concentrations in the four different types of edible oils ranged from 88.76% to 115.79%, with CV values ranging from 1.80% to 13.37%. To further validate the accuracy of this method, we applied it to detect a CRM sample, yielding a detection value of 3.53 μg kg−1 for the total content of capsaicinoid compounds, while the certified value was 3.35 μg kg−1, resulting in a bias percentage (b%) of 5.42%. These findings demonstrate the high accuracy of this method.| Sample | Spiked (μg kg−1) | Detected (μg kg−1) | Recovery (%) | CV (%) |
|---|---|---|---|---|
| Soybean oil | 0.55 | 0.64 | 115.79 | 10.79 |
| 1.11 | 1.26 | 113.72 | 11.61 | |
| 5.55 | 5.60 | 101.05 | 5.72 | |
| Corn oil | 0.55 | 0.51 | 92.49 | 10.05 |
| 1.11 | 1.23 | 111.07 | 13.37 | |
| 5.55 | 5.27 | 94.89 | 7.95 | |
| Peanut oil | 0.55 | 0.56 | 100.99 | 13.35 |
| 1.11 | 1.24 | 111.75 | 9.84 | |
| 5.55 | 4.93 | 88.76 | 8.38 | |
| Rapeseed oil | 0.55 | 0.52 | 94.19 | 10.97 |
| 1.11 | 1.15 | 103.56 | 7.45 | |
| 5.55 | 5.63 | 101.52 | 1.80 |
Conclusions
This study initially establishes a fluorescent LFIA method for detecting CAP by sequentially optimizing the particle size of EuNPs, the labeling concentration of antibodies, the coating concentration of CAP-BSA, and the pH of the reaction buffer. Following this, the DM-β-CD solution was chosen from a selection of five cyclodextrin derivatives for the purpose of extracting CAP from edible oil. The extraction conditions were then optimized, establishing the optimized concentration of the DM-β-CD solution at 20 mmol L−1 and the pH of the extraction solution at 8.7. The optimized extraction procedure involves mixing the extraction solution with the oil sample at a volume ratio of 2:1, vigorously shaking for 1 minute, followed by a 2 minute settling period to separate the oil and water phases. For analysis, 100 μL of the aqueous sample is dispensed onto the CAP detection card, and after 10 minutes, the FI of the detection card is measured using a handheld device to determine the CAP content in the sample. Due to the single-step sample preparation involved, the total detection time of this method is less than 15 minutes, highlighting its simplicity and speed. The LOD for CAP in oil samples using this method is 0.14 μg kg−1, with a working range of 0.46–81 μg kg−1. The cross-reactivity rates for D-CAP and S-CAP are both above 90%, enabling the total quantification of CAP, D-CAP, and S-CAP in oil samples. This method meets the technical requirements outlined in Chinese national standards for identifying recycled cooking oil by detecting the total amount of these three capsaicin compounds (1 μg kg−1). Further validation through spike recovery tests and CRM analysis confirms the accuracy and reliability of this method. It is important to emphasize that, in comparison to traditional detection methods, this method does not require the use of organic solvents for sample extraction. As a result, it enhances safety in the storage, transportation, usage, and waste disposal of reagents, eliminating the need for specialized laboratory facilities and allowing on-site application. This advancement can enable consumers and food safety authorities to conduct rapid on-site tests on edible oils present in agricultural markets, retail outlets, food processing facilities, restaurants, and other relevant settings. In summary, this study has established a field-applicable, simple, rapid, and accurate detection method for identifying gutter oils. Meanwhile, the research presented in this paper holds significant reference value for the development of rapid detection methods for other lipophilic contaminants in oils.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Author contributions
Y. Wang: methodology, validation, formal analysis, visualization, writing – original draft. Y. Wu: methodology, validation. D. G.: methodology. Z. D.: methodology. Z. Q.: methodology. Z. L.: methodology. J. L.: formal analysis. Q. Z.: formal analysis. G. S.: conceptualization, writing – review & editing, supervision, project administration.Conflicts of interest
The authors declare that they have no known competing interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The work was supported by the Fundamental Research Funds for the Central Universities (FRF-BR-23-02B). The authors thank Stork (https://www.storkapp.me/writeassistant/) for its linguistic assistance during the preparation of this manuscript.References
- J. Li, M. Zuo, W. Zhang, X. Zou and Z. Sun, Food Anal. Methods, 2022, 15, 3468–3478 CrossRef.
- G. Cao, C. Ding, D. Ruan, Z. Chen, H. Wu, Y. Hong and Z. Cai, Food Control, 2019, 96, 494–498 CrossRef CAS.
- H. Jin, L. Tu, Y. Wang, K. Zhang, B. Lv, Z. Zhu, D. Zhao and C. Li, Food Control, 2023, 145, 109448 CrossRef CAS.
- M. Su, Q. Jiang, J. Guo, Y. Zhu, S. Cheng, T. Yu, S. Du, Y. Jiang and H. Liu, LWT--Food Sci. Technol., 2021, 143, 111143 CrossRef CAS.
- G. Cao, Y. Hong, H. Wu, Z. Chen, M. Lu and Z. Cai, Food Control, 2021, 125, 107966 CrossRef CAS.
- H. Jin, H. Li, Z. Yin, Y. Zhu, A. Lu, D. Zhao and C. Li, Food Chem., 2021, 362, 130191 CrossRef CAS.
- W. Wang, Q. F. Yu, Y. Xiao and H. He, Chin. J. Anal. Chem., 2017, 45, 770–776 CAS.
- C. P. Lin, C. R. Liao, Y. F. Zhang, L. Xu, Y. Wang, C. L. Fu, K. M. Yang, J. Wang, J. He and Y. P. Wang, Lab Chip, 2018, 18, 595–600 RSC.
- T. T. Ng, S. Y. Li, C. C. A. Ng, P. K. So, T. F. Wong, Z. Y. Li, S. T. Chan and Z. P. Yao, Food Chem., 2018, 252, 335–342 CrossRef CAS PubMed.
- Z. Jin, W. Sheng, M. Sun, D. Bai, L. Ren, S. Wang, Z. Wang, X. Tang and T. Ya, J. Hazard. Mater., 2024, 466, 133670 CrossRef CAS PubMed.
- K. Tian, W. Wang, Y. Yao, X. Nie, A. Lu, Y. Wu and C. Han, J. Raman Spectrosc., 2018, 49, 472–481 CrossRef CAS.
- Y. Wu, J. Liu, J. Yu, J. Zhuang, F. Ma, J. Tan and Z. Shen, Talanta, 2022, 250, 123686 CrossRef CAS PubMed.
- J. Wu, J. Tang, Y. Xue, Z. Yan, Z. Hu, Y. Qi and W. Zhang, Zhongguo Youzhi, 2020, 45, 118–121 Search PubMed.
- X. Yang and S. Xiang, Zhongguo Youzhi, 2021, 46, 130–134 Search PubMed.
- C. Zhou, D. P. Ma, W. M. Cao, H. M. Shi and Y. R. Jiang, Food Addit. Contam.,:Part A, 2018, 35, 1447–1452 CrossRef CAS PubMed.
- X. Xu, S. Li, S. Chen, K. Fang, Z. Liu, L. Li, H. Zhou, Y. Zhang and C. Wang, Chin. J. Anal. Lab., 2019, 38, 191–195 CAS.
- Q. Lu, H. Guo, D. Li and Q. Zhao, J. Chromatogr., B, 2020, 1158, 122344 CrossRef CAS PubMed.
- Z. Liu, X. Wang, J. Chen, J. Gao, S. Yu and X. Wang, Microchem. J., 2020, 157, 105052 CrossRef CAS.
- J. Zhang, J. Li and X. Deng, Zhongguo Youzhi, 2021, 46, 82–86 Search PubMed.
- F. Ma, Q. Yang, B. Matthäus, P. Li, Q. Zhang and L. Zhang, J. Chromatogr., B, 2016, 1021, 137–144 CrossRef CAS.
- S.-H. Liu, X.-M. Lin, Z.-L. Yang, B.-Y. Wen, F.-L. Zhang, Y.-J. Zhang and J.-F. Li, Talanta, 2022, 245, 123488 CrossRef CAS PubMed.
- Z. G. Liu, S. H. Yu, S. P. Xu, B. Zhao and W. Q. Xu, ACS Omega, 2017, 2, 8401–8406 CrossRef CAS PubMed.
- M. Qin, I. M. Khan, N. Ding, Y. Sun, S. Qi, X. Dong, S. Niazi, Y. Zhang and Z. Wang, Sens. Actuators, B, 2023, 396, 134638 CrossRef CAS.
- M. Wang, B. Gao, Y. Xing and X. Xiong, Int. J. Electrochem. Sci., 2020, 15, 8437–8449 CrossRef.
- X. Fang and R. S. Duan, Front. Chem., 2022, 10, 822619 CrossRef CAS PubMed.
- J. Sun, L. Liu, S. Song, G. Cui and H. Kuang, Food Agric. Immunol., 2018, 29, 930–940 CrossRef CAS.
- D. Yuan, S. Li, L. Zhang, F. Ma, H. Wang, Q. Zhang and P. Li, Food Chem., 2023, 404, 134552 CrossRef CAS.
- Q. Wu, L. Yao, P. Qin, J. Xu, X. Sun, B. Yao, F. Ren and W. Chen, Food Chem., 2021, 357, 129739 CrossRef CAS.
- J. Zhang, M. Zhang, Q. Yang, L. Wei, B. Yuan, C. Pang, Y. Zhang, X. Sun and Y. Guo, Anal. Bioanal. Chem., 2022, 414, 6127–6137 CrossRef CAS.
- X. Sun, Q. X. Zhao, C. Y. Zha, J. L. Zhang, Z. R. Zhou, H. W. Dong, Q. Q. Yang, Y. M. Guo and S. C. Zhao, J. Electrochem. Soc., 2022, 169, 057502 CrossRef CAS.
- Z. Jin, W. Sheng, Z. Wang, X. Tang, T. Ya, S. Wang, Q. Ji, C. Fan and Y. Liu, Food Chem., 2025, 464, 141570 CrossRef CAS.
- Y. Zhao, W. Wu, X. Tang, Q. Zhang, J. Mao, L. Yu, P. Li and Z. Zhang, Biosens. Bioelectron., 2023, 225, 115102 CrossRef CAS.
- B. G. Poulson, Q. A. Alsulami, A. A. Sharfalddin, E. F. El Agammy, F. Mouffouk, A.-H. M. Emwas, L. Jaremko and M. Jaremko, Polysaccharides, 2022, 3(1), 1–31 CrossRef CAS.
- K. U. Khan, M. U. Minhas, S. F. Badshah, M. Suhail, A. Ahmad and S. Ijaz, Life Sci., 2022, 291, 120301 CrossRef CAS PubMed.
- Z. Li, Y. Wu, Z. Li, B. Yu, X. Mao and G. Shi, Analytical methods: advancing methods and applications, 2023 Search PubMed.
- X. Mao, Y. Wu, H. Chen, Y. Wang, B. Yu and G. Shi, Anal. Methods, 2020, 12, 5628–5634 RSC.
- Eurachem Guide: The Fitness for Purpose of Analytical Methods – A Laboratory Guide to Method Validation and Related Topics, ed. B. Magnusson and U. Örnemark, 2nd edn, 2014, ISBN 978-91-87461-59-0, Available from http://www.eurachem.org Search PubMed.
- M. Su, W. Mi, Y. Zhang, M. Lv and W. Shen, J. Food Compos. Anal., 2022, 114, 104822 CrossRef CAS.
- H. Moulahoum and F. Ghorbanizamani, Biosens. Bioelectron., 2024, 264, 116670 CrossRef CAS PubMed.
- Y. Xia, W. Liu, Y. Shi, S. Younas, C. Liu and L. Zheng, J. Food Sci., 2022, 87, 567–575 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sd00306c |
| ‡ These authors contribute equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |