
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImproving the electrocatalytic activity of Pd nanoparticles through electronic coupling interaction with a Ni2P–MoS2 hybrid support for ethanol electro-oxidation in an alkaline medium†
Thabo
Matthews
 *abc,
Makhaokane Paulina
Chabalala
bc,
Siyabonga Patrick
Mbokazi
bc,
Memory
Zikhali
bc,
Tarekegn Heliso
Dolla
d,
Anatolijs
Šarakovskis
e,
Guntars
Vaivars
efg,
Tunde Lewis
Yusuf
h,
Rhiyaad
Mohamed
a and
Nobanathi Wendy
Maxakato
*bc
*abc,
Makhaokane Paulina
Chabalala
bc,
Siyabonga Patrick
Mbokazi
bc,
Memory
Zikhali
bc,
Tarekegn Heliso
Dolla
d,
Anatolijs
Šarakovskis
e,
Guntars
Vaivars
efg,
Tunde Lewis
Yusuf
h,
Rhiyaad
Mohamed
a and
Nobanathi Wendy
Maxakato
*bc
aHySA/Catalysis Centre of Competence, Catalysis Institute, Department of Chemical Engineering, University of Cape Town, Cape Town 7701, South Africa. E-mail: matthews.thabo@uct.ac.za/; matthewsThabo@gmail.com
bDepartment of Chemical Sciences, University of Johannesburg, Doornfontein, 2028, South Africa. E-mail: nmaxakato@uj.ac.za
cCenter for Nanomaterials Research, University of Johannesburg, South Africa
dInstitute for Catalysis and Energy Solutions (ICES), College of Science, Engineering and Technology, University of South Africa (UNISA), Private Bag X6, Florida, 1710, South Africa
eInstitute of Solid State Physics, University of Latvia, Kengaraga Iela 8, LV-1063 Riga, Latvia
fFaculty of Chemistry, University of Latvia, Jelgavas Iela 1, LV-1004 Riga, Latvia
gInstitute of Chemical Physics, University of Latvia, Jelgavas Iela 1, LV-1004 Riga, Latvia
hDepartment of Chemistry, Faculty of Natural and Agricultural Sciences, University of Pretoria, Private Bag X20, Hatfield 0028, Pretoria, South Africa
First published on 13th February 2025
Abstract
To improve the performance of direct ethanol fuel cells (DEFCs), which are hindered by traditional catalysts, having matters pertaining to stability, activity, and selectivity in reaction environments, various electrocatalysts such as Pd/Ni2P, Pd/MoS2, and Pd/Ni2P–MoS2 were synthesized using the microwave-assisted NaBH4–ethylene glycol reduction method. The research findings suggest that the Pd/Ni2P–MoS2 catalyst we developed had the highest activity (1579 mA mgPd−1), approximately 21 times greater than that of commercial Pd/C. The stability of the electrocatalysts were examined using chronoamperometry (CA) and cyclic voltammetry (CV) measurements, which indicated that the Pd/Ni2P–MoS2 electrocatalyst had good stability towards the ethanol oxidation reaction (EOR) in alkaline electrolyte. Electrochemical impedance spectroscopy (EIS) analysis showed that the Pd/Ni2P–MoS2 electrocatalyst had lower charge transfer resistance, indicating better electrochemical kinetics. According to XRD, HR-TEM, XPS, and electrochemical analysis, the enhanced electrocatalytic activity, long-term stability of the Pd/Ni2P–MoS2 electrocatalyst were attributable to the interface synergism as well as electronic and strain effects between the Pd, Ni2P, and MoS2 interactions. This resulted in a downshift in the d-band center of the Pd/Ni2P–MoS2 electrocatalyst, weakening intermediate adsorption and the adsorbate metal interaction.
1 Introduction
The widespread use of CO2 emitting fossil fuels has made meeting the world's energy demand a significant challenge. This has put a strain on the planet's natural resources. Therefore, developing renewable energy resources globally, is essential to ensure sustainable growth, especially in developing countries. Direct alcohol fuel cells are promising energy conversion and storage technologies, suitable for portable and mobile devices. Both monohydric and polyhydric alcohols have appreciable high energy densities, high boiling points, and low volatilities, which makes them easy to store. Direct ethanol fuel cell (DEFCs have several advantages.1,2 It has an existing infrastructure for production, high specific energy, and abundance. However, ethanol usage is limited due to the slow kinetics of the ethanol oxidation reaction (EOR) and the requirement to cleave the C–C bond, which has presented significant challenges.1–5Electrocatalyst design is essential in developing DEFC electrocatalysts with improved performance. It is possible to modify electrocatalysts by changing their shape, composition, support materials, electronic state, structural effects, and preparatory methods. Various synthesis methods have been used to control the compositions and structures of bimetallic electrocatalysts, aiming to enhance the electrocatalyst activity, CO selectivity, and durability of Pd-based electrocatalysts. For example, in their study, Shu et al.6 demonstrated that the PdCuCo electrocatalyst, showed a significantly higher peak current density for the electro-oxidation of fuel under alkaline conditions. The performance was ten times greater than that of the Pd/C catalyst. It is well known that the use of appropriate metals and supports can adjust the electronic structure and affect the intrinsic activity of electrocatalysts. Doping and alloying protocols have been shown to impact the electronic structure of Pd and improve the performance of the electro-oxidation reaction. Junfeng Liu et al.7 evaluated these catalyst desgin strategies, reporting that a P-doped PdMo bimetallene exhibits superior performance in the electro-oxidation of various alcohols due to the altered Pd electronic structures.
For enhanced Pd electrocatalytic activity, Wang et al.8 synthesized a Pd–Ni–P ternary catalyst (by introducing Ni and P into Pd) that showed high EOR electrocatalytic performance. In another study, Ye et al.9 prepared stable 3D hierarchical Pd@CoP nanosheets to promote the oxidation or removal of CO and CH3CO species. Similarly, Chen et al.10 improved the EOR performance in alkaline media by introducing P into a CuPdNi alloy, forming a CuPdNiP catalyst. From these studies, we concluded that P promotes bifunctional effects, shortens the interatomic distance, and promotes metal support interactions (MSIs). Improving the activity and stability of Pd, with lower Pd loading, promotes the prospects for commercialization.
Both experimental and theoretical evaluations have shown that Ni2P exhibits higher electrocatalytic activity and stability for various reactions, such as the hydrogen evolution reaction (HER) and formic acid oxidation reaction (FAOR). These improved electrocatalytic or photochemical catalytic properties are due to the electronic charge transfer from the interaction of Ni and P and active metal site exposure effects. Chang et al.11 synthesized Pt–Ni2P/C electrocatalysts, wherein the 30 wt% loading of Ni2P exhibited higher performances relative to Pt/C, Ni-promoted Pt/C, and P-promoted Pt/C catalysts, as assessed by electrochemical evaluation. On the other hand, metal dichalcogenides, such as MoS2, WS2, etc., have attracted attention for catalytic applications due to their plausible electrical and electrochemical properties. In particular, MoS2 is interesting because it is easy to synthesize and cost-effective and has longer active edges. All these features make MoS2 an efficient component for preparing a hybrid electrocatalyst. For example, Tang et al.12 synthesized Pt-decorated porous coral reef-like MoS2/nitrogen-doped biocarbon, which showed superior CO tolerance and was an effective methanol oxidation reaction (MOR) catalyst. In a different study, Meng Li et al.13 reported the use of ultrafine Pt/MoP-NC for the MOR, and attributed the improved performance to a strong synergistic interface effect between platinum (Pt) and molybdenum phosphide (MoP) which was strengthened by the electronic donation properties of MoP and in turn enhanced both the catalytic activity and stability. The same phenomenon was put forward by Yubin K. et al.14 where they alluded to Pt doping and interfacial engineering with MoSe2 which accelerated MOR kinetics.
In light of the aforementioned efforts and drawing inspiration from previous findings as the foundation, we report for the first time the promotion of Pd by Ni2P–MoS2 for the ethanol oxidation reaction. We show that the Ni2P–MoS2 promotion effect on Pd was significant. In this study, we detail the synthesis of the Pd/Ni2P–MoS2 electrocatalyst using an ethylene glycol/NaBH4 microwave-assisted reduction method and its characterization as well as electrocatalytic properties. Through the interface of MoS2 with Ni2P and Pd nanoparticles, we achieved a synergy between Pd and Ni2P–MoS2. The hybrid Pd/Ni2P–MoS2 electrocatalyst demonstrated superior electrocatalytic activity with a mass activity of 1579 mA mgPd−1 and an onset potential of 0.48 (V vs. RHE) while also exhibiting remarkable durability after 500 cycles, retaining 88.5% of the initial current.
2 Experimental
2.1 Chemicals
The following chemicals were all purchased from Sigma-Aldrich. Molybdenum(IV) sulphide (MoS2) powder (<2 μm, 99%), nickel chloride hexahydrate, NiCl2·6H2O, red phosphorus, ethanol, 99.9%, palladium(II) chloride PdCl2 (98.0%), Pd/C commercial 10 wt%, dimethyl ether (DME), ethylene glycol (EG), 99.5.0%, potassium hydroxide (KOH) pellets, ferrocene C10H10Fe, and hydrogen peroxide H2O2.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of Ni2P–MoS2 was used in the experiment. First, 800 mg of Ni2P and 800 mg of MoS2 were mixed using a pestle and mortar for 15 minutes. The resulting mixture was then dissolved in 50 mL of a solution containing equal parts of ethanol and distilled water. This solution was placed in a glass beaker and stirred continuously for 1 hour while bubbling N2/Ar gas to prevent oxide formation. The solution was then transferred to a stainless-steel autoclave and heated to 150 °C for 3 hours. The contents were then centrifuged and dried at 80 °C for 6 hours. Finally, the resulting powder was named Ni2P–MoS2.
:1) ethanol and double distilled water. The black solid was dried in a vacuum oven at 60 °C for 24 h and labeled as Pd/Ni2P–MoS2. The other electrocatalysts were synthesized using the same method with appropriate chemicals, specifically Pd/Ni2P without the addition of MoS2 and Pd/MoS2 without the addition of Ni2P.
:1 ratio of Ni2P–MoS2 was used in the experiment. First, 800 mg of Ni2P and 800 mg of MoS2 were mixed using a pestle and mortar for 15 minutes. The resulting mixture was then dissolved in 50 mL of a solution containing equal parts of ethanol and distilled water. This solution was placed in a glass beaker and stirred continuously for 1 hour while bubbling N2/Ar gas to prevent oxide formation. The solution was then transferred to a stainless-steel autoclave and heated to 150 °C for 3 hours. The contents were then centrifuged and dried at 80 °C for 6 hours. Finally, the resulting powder was named Ni2P–MoS2.
:1) ethanol and double distilled water. The black solid was dried in a vacuum oven at 60 °C for 24 h and labeled as Pd/Ni2P–MoS2. The other electrocatalysts were synthesized using the same method with appropriate chemicals, specifically Pd/Ni2P without the addition of MoS2 and Pd/MoS2 without the addition of Ni2P.
2.2 Physicochemical instrumentation
The samples were analysed for their crystalline nature using an X'Pert PRO PANalytical diffractometer at room temperature, with CuKα radiation (λ = 1.5406 Å), covering the 2θ range of 10–90°. Other tests performed included FTIR spectroscopy using a PerkinElmer FTIR spectrophotometer and RAMAN spectroscopy using a WITec alpha300R confocal Raman spectrometer with a laser wavelength of 532 nm and 600 grooves per mm, using a 50X objective and a 100 μm fiber. Images were recorded using scanning transmission electron microscopy (STEM) with a JEOL JEM-ARM200F at 200 kV. Pd concentrations were measured using inductively coupled plasma-optical emission spectrometry (ICP-OES), while X-ray photoemission spectroscopy (XPS) measurements were performed in an ultra-high vacuum (UHV) chamber equipped with a SPECS PHOIBOS 150 hemispherical electron energy analyser, with an Al Kα excitation line (hν = 1486.71 eV).2.3 Electrochemical characterization
A Dropsens ustat 4000 and a standard three-electrode setup were used for the electrochemical measurements. The glassy carbon working electrode (WE) had an area of 0.07065 cm2, while the counter was made of a Pt sheet and Ag/AgCl (3 M KCl) was used as the reference electrode. To prepare the electrocatalyst ink, 10 mg of the electrocatalyst was dispersed in 50/50 (v/v) ultrapure H2O/CH3CH(OH)CH3 solution containing 30 μL of DME. The catalyst ink was then ultrasonicated at room temperature for 3 hours. 5 μL of the electrocatalyst ink was drop-coated on the active area of the GCE and dried at 80 °C in an oven. A freshly prepared N2 gas-saturated mixture of 0.5 M CH3CH2OH and 1 M KOH was used as the electrolyte. The CV measurements were performed between a potential window of 0.0 to 1.1 V (V vs. RHE) at 50 mV s−1 (scan direction from left to right) at room temperature, and the third scans were recorded. Electrochemical impedance spectra (EIS) at 0.59 V (vs. RHE) in the frequency range of 10 Hz to 100000 kHz were also recorded. Chronoamperometry measurements were conducted in N2-saturated 1 M KOH + 0.5 M ethanol at 0.59 (V vs. RHE) using a Gamry interface 1010E 27143. To convert all potentials to the RHE scale, the Nernst equation ERHE = EAg/AgCl + 0.059 × pH + 0.197 was used. A 10 wt% commercial Pd/C electrocatalyst was used as the benchmark for all electrochemical tests under similar reaction conditions.
3 Results and discussion
3.1 Synthesis chemistry of Pd/Ni2P–MoS2
Initially, Ni2P and MoS2 were mixed in water and ethanol.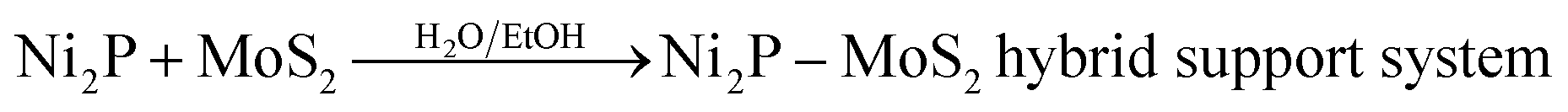 | (1) |
For the deposition of Pd nanoparticels (NPs) onto the Ni2P–MoS2 electrocatalyst (assuming the anchoring of Pd NPs onto the catalyst surface), we postulate that on anchoring the Pd NPs they should mostly be in the zero state via the reduction of PdCl2. This reduction process is carried out in a mixture of NaHB4/EG and is facilitated by microwave irradiation. We propose that Na+ from NaBH4/NaOH reacts with Cl from PdCl2 to form NaCl, which is then washed off. The representative equation is shown below:
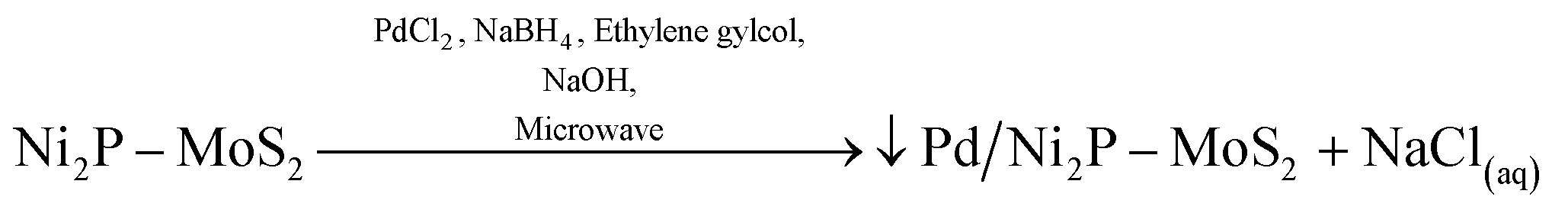 | (2) |
During our study, we assumed that the precursor reduction to a metal results in the complete oxidation of EG into CO2 and H2O, indicating the maximum reducing power of the alcohol.
| CH2OH − CH2OH + 3PdCl2 → ↓3Pd + ↑2CO2 + 6HCl | (3) |
3.2 Physicochemical characterization
The prepared electrocatalysts' arrangement and composition were determined using XRD, FTIR, and Raman spectroscopy. Fig. 1(a) presents the XRD patterns for the electrocatalytic materials. The presence of sharp and intense diffraction peaks in all samples attests to their high crystallinity. Fig. 1(a) illustrates MoS2 characteristic diffraction peaks at 14.1°, 29.0°, 33.68°, 44.0°, 59.56°, and 60.2°, indexed to (002), (004), (100), (006), (110), and (008) planes, respectively, of the 2H-phase of MoS2 (JCPDS 37-1492).15 Additionally, there is an extra peak at around 2θ ≈ 28.8° corresponding to the (004) peak of 1T-phase MoS2, suggesting the presence of a minor amount of 1T-phase molybdenum disulfide (MoS2) alongside the predominant 2H-phase. Molybdenum disulfide has different phases, with the natural 2H-phase being semiconducting and thermodynamically favored. This phase consists of two S–Mo–S layers formed from edge-sharing MoS6 trigonal prisms, whereas the metallic 1T phase features a single S–Mo–S layer of edge-sharing MoS6 octahedra. Recent studies show that adding metallic atoms to MoS2 nanosheets can induce the transformation from the 2H phase into the 1T phase, leading to lower charge transfer resistance and improved catalytic performance.16–18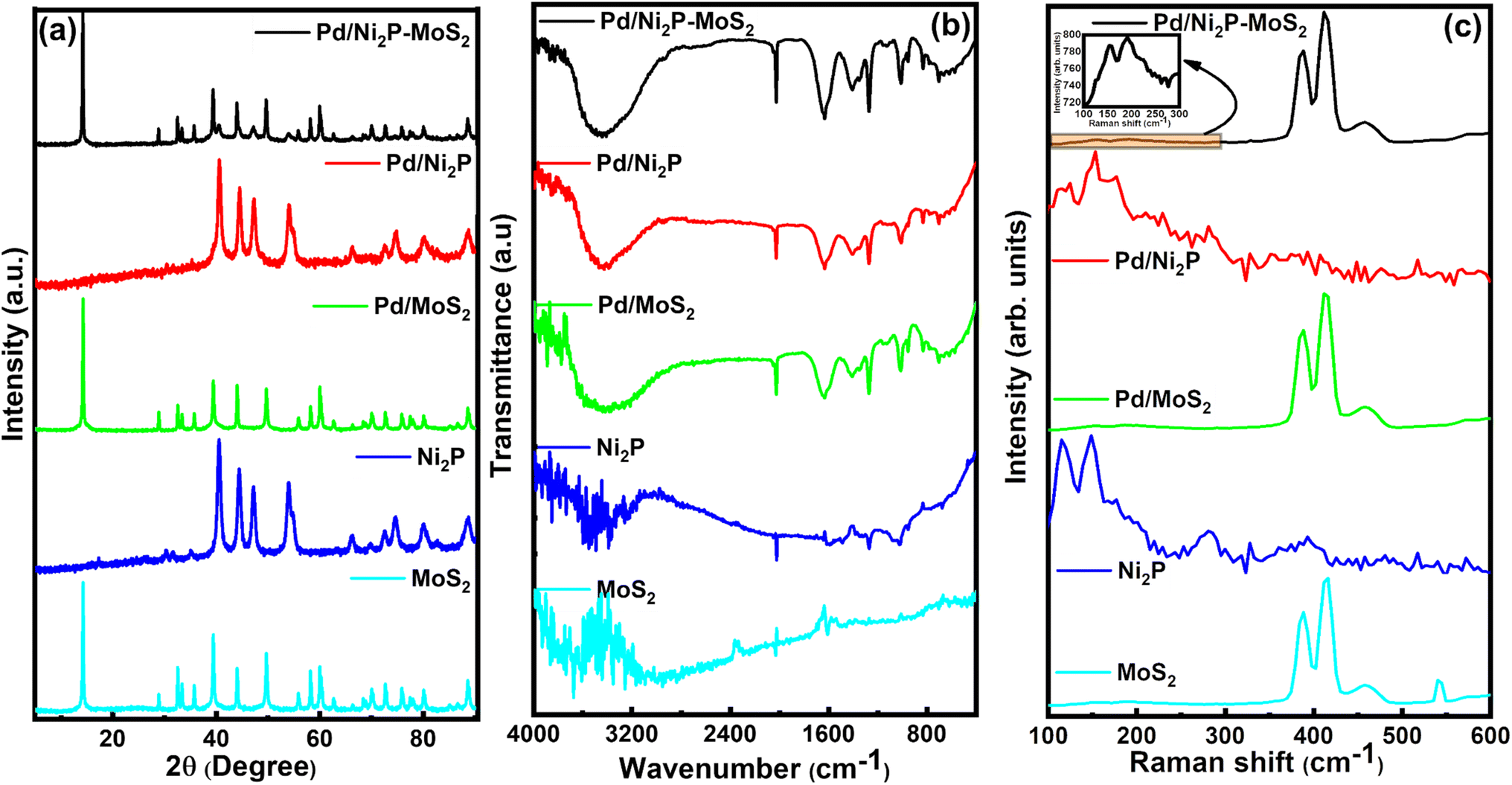 | ||
| Fig. 1 (a) XRD patterns, (b) FTIR spectra, and (c) Raman spectra of the synthesised electrocatalysts. | ||
The (002) peak predominance indicates a preferential orientation along the (001) direction in the prepared materials. For Ni2P, the diffraction peaks at 40.6°, 44.7°, 47.3°, 54.2°, and 54.7° are indexed to (111), (021), (210), (300), and (002) planes, respectively, and relate to the hexagonal phase of Ni2P (JCPDS Card No. 65-1989).19,20 The Pd diffaction lines are invisible on Pd/MoS2, Pd/Ni2P, and Pd/Ni2P–MoS2 samples due to overlap, low loading, smaller sizes, and good dispersion of Pd nanoparticles; as seen from the HR-TEM. This is seen by the broadening of the peak base where Pd appears. Thus, the XRD pattern of Pd/Ni2P–MoS2 confirms the successful formation of the hybrid catalyst.
The surface chemical states of the samples were analyzed using Fourier transform infrared (FTIR) spectroscopy. The FTIR spectra of Ni2P, MoS2, Pd/Ni2P, Pd/MoS2, and Pd/Ni2P–MoS2 are shown in Fig. 1(b). The peak at 3419 cm−1 is a strong signal from the adsorbed –OH groups. The peaks from 1300 cm−1 to 1700 cm−1 indicate the presence of water and the O–H group bending vibration.21–24 MoS2 exhibits characteristic Mo–S and S–S vibrations at 600 and 905 cm−1. The two bands at 1042 cm−1 and 918 cm−1 are attributed to species with phosphorus.21,25
Raman spectra shown in Fig. 1(c) were used to assess the structural properties of the electrocatalysts. The distinct peaks at 381 and 410 cm−1 in MoS2, Pd/MoS2, and Pd/Ni2P–MoS2 samples are assigned to the E2g1 and A1g of Mo–S phonon mode belonging to the 2H crystalline phase of MoS2.26,27 Previous research studies have shown similar findings to the ones presented here.28 This further proves that the composite structure of MoS2 and Ni2P has successfully formed. The deposition of Pd atoms onto Ni2P, MoS2, and Ni2P–MoS2 leads to a shift of approximately 2.0 cm−1 in the A1g peak. This indicates that Pd interacts with support materials and can considerably modify the electronic structure, as confirmed from XPS analysis in Fig. 3 and 4. Similarly, the E2g1 and A1g peaks of Ni2P–MoS2 exhibit a slight red shift compared to MoS2 due to interfacial stress resulting from the interaction between Ni2P and MoS2. This shift suggests that the formation of the MoS2–Ni2P heterojunction induces changes in the Raman spectra of MoS2, confirming the successful preparation of MoS2–Ni2P. Furthermore, the peak separations (A1g − E12g) in the bare supports are 29.7, 25.4, and 23.2 cm−1, with Pd-loaded supports showing a slight decrease in the peak separation attributable to introducing S vacancies during chemical synthesis; this peak separation further proves successful Pd anchoring and support hybridization and that MoS2 is a good candidate for Pd anchoring.28–30
The morphology of the electrocatalysts was studied using HR-TEM, as seen in Fig. 2. Ni2P and MoS2 have a sheet-like morphology, as shown in Fig. 2(a) and (b), respectively, with SAED pattern insets. Fig. 2(a) shows lattice fringes, confirming that the desired Ni2P phase was successfully formed. The distinct lattice fringes with a d-spacing of 0.20 nm were observed for the Ni2P crystal, corresponding to the (201) lattice plane of the hexagonal Ni2P phase.31 The SAED pattern of Ni2P has three diffraction rings representing the (111), (300), and (321) planes of Ni2P, from the inner to the outer ring. Fig. 2(c) and (d) show Pd NPs anchored onto the surface of Ni2P and MoS2, respectively, with 2 and 3.4 nm sizes. The different sizes of the Pd NPs indicate that the supports interact with the NPs differently. The HR-TEM image in Fig. 2(e) confirms the microstructure of the Ni2P–MoS2 nanosheet hybrids with Pd NPs on the surface, demonstrating the feasibility of achieving the hybrid electrocatalyst using an NABH4/EG microwave-assisted reduction method. The Pd NPs' particle size on the Ni2P–MoS2 is 3.4 nm, as that for Pd/MoS2, which proves that MoS2 plays a major role in the anchoring of Pd NPs, as explained based on the shift and separation from Raman spectra (Fig. 1(c)). The synthesized materials are highly crystalline, as seen from the SAED patterns shown in Fig. 2 (a and b insets) and (b–d) (iii), as well as the XRD patterns.
 | ||
| Fig. 2 HR-TEM images of (a) Ni2P, (b) MoS2, (c) (i) Pd/Ni2P, (d) (i) Pd/MoS2, and (e) (i) Pd/Ni2P–MoS2, (ii) particle size distributions, and (iii) SAED patterns. | ||
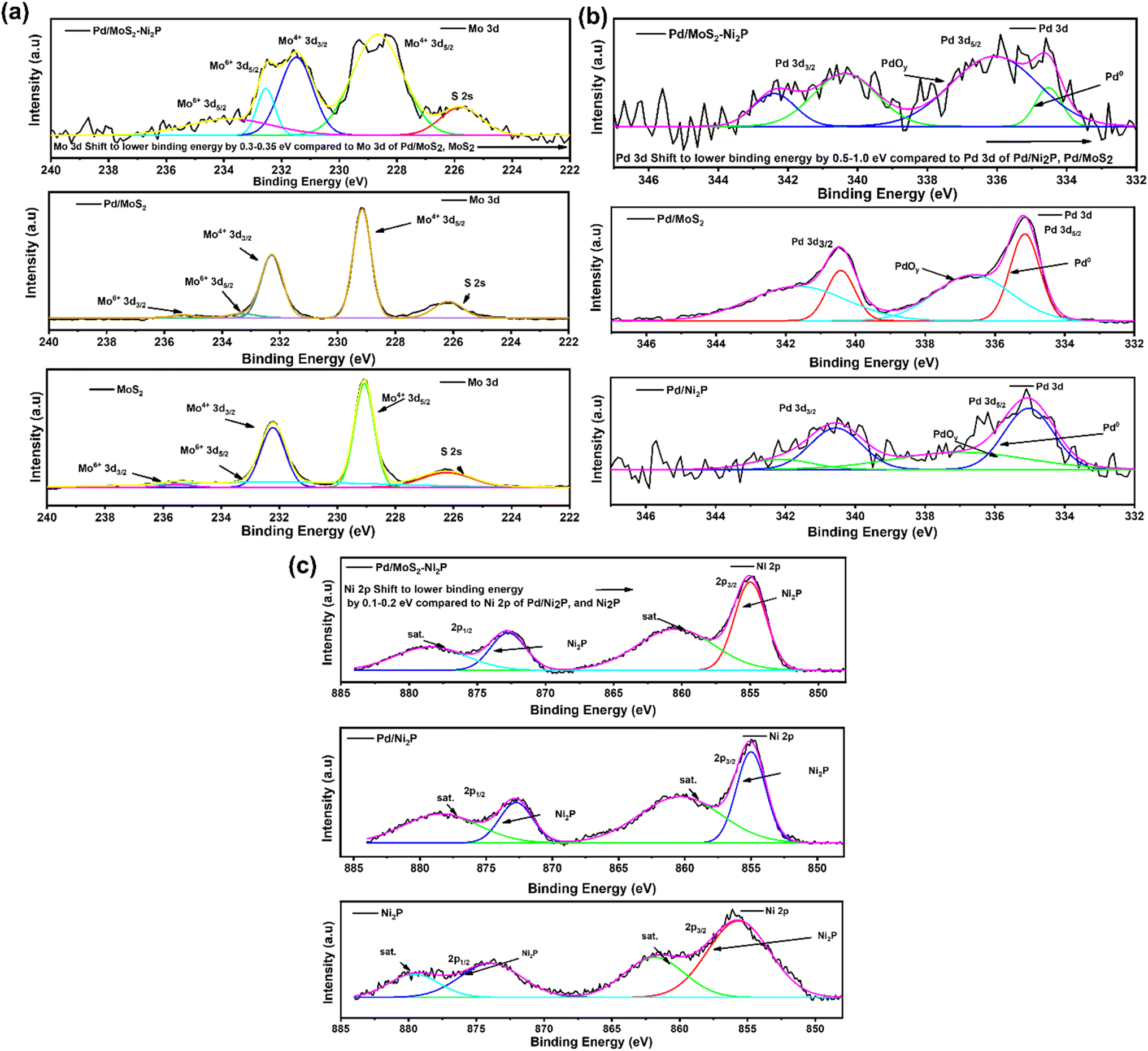
XPS was used to examine the surface chemical composition of Pd/MoS2–Ni2P, Pd/MoS2, Pd/Ni2P, Ni2P, and MoS2 hybrid nanomaterials. In comparison, the survey spectra in Fig. 3(a) demonstrate the presence of C, O, P, Pd, S, Mo, and Ni for the main composite and subsequent elements for the other nanomaterials. Furthermore, the presence of C and O elements can be seen in the survey spectra and Fig. S4 in the ESI,† which could be a result of absorbed gaseous molecules. The deconvoluted spectra of S 2p exhibit twin peaks of S 2p1/2 and S 2p3/2 at the corresponding BEs of 163.8 and 162.6 eV, which is attributed to S2− in MoS2 of Pd/MoS2–Ni2P, Pd/MoS2, and MoS2 nanomaterials, Fig. 3(b). Pd/MoS2–Ni2P and Pd/MoS2 have slightly different binding energy (BE) values than pure MoS2 (S 2p3/2 = 162.4 eV), indicating charge transfer from Pd to MoS2.32 The distinctive peak of phosphate species is centered at 133.67 eV for P 2p, as shown in Fig. 3(c) for the nanocomposites with the nickel phosphide constituent.33 The P 2p is shifted to a higher BE relative to Ni2P; we postulate that this represents the interaction of Pd with Ni2P to form species such as Ni–P–Pd or Pd–P.
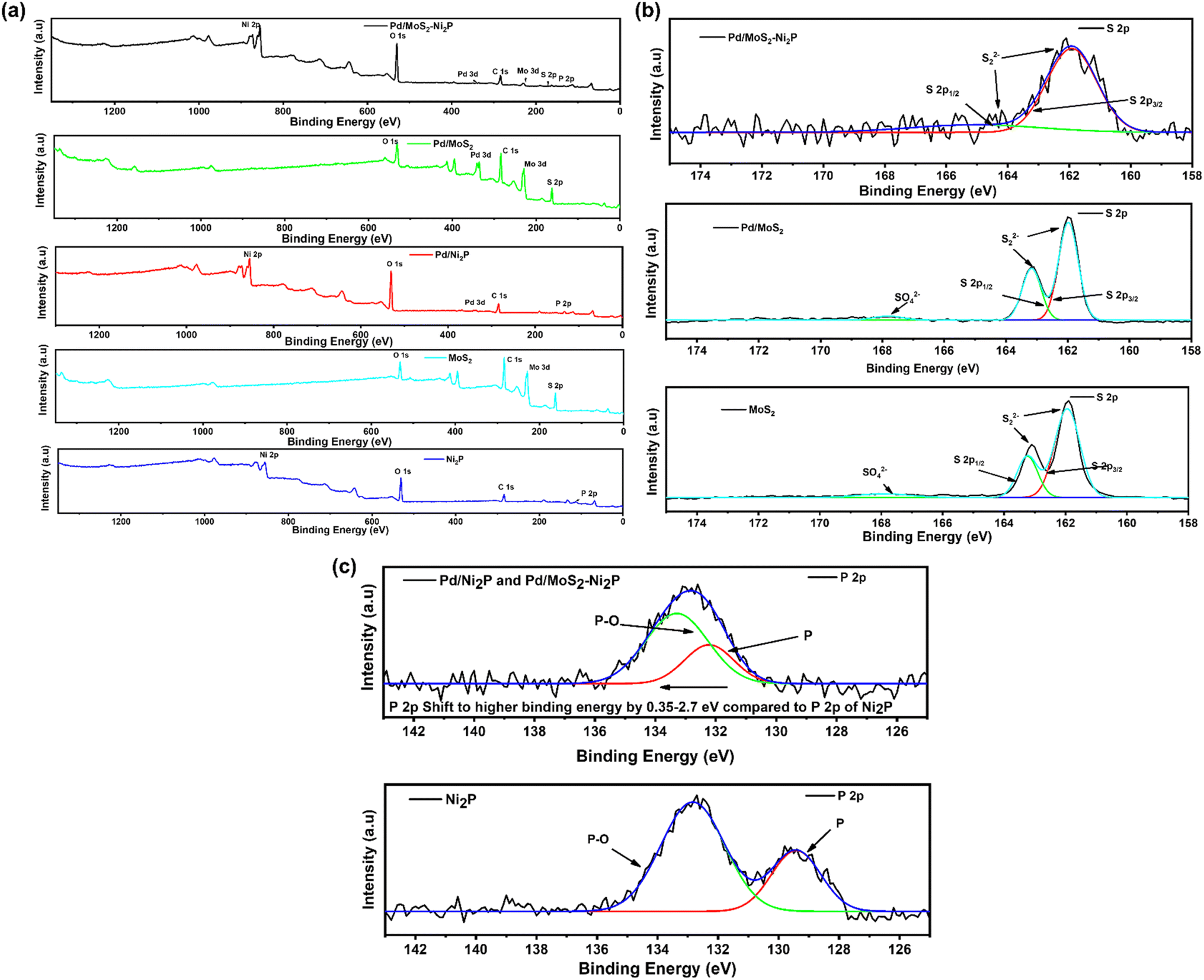 | ||
| Fig. 3 XPS spectra. (a) Survey scans, (b) S 2p and (c) P 2p. | ||
Fig. 4(a) shows high-resolution spectra of the Mo 3d for Pd/MoS2–Ni2P, Pd/MoS2, and MoS2 electrocatalysts. The doublets of the Mo4+ bands at 229.5 eV and 232.7 eV in the Mo 3d region are ascribed to Mo 3d5/2 and Mo 3d3/2 orbitals, respectively, and correspond well with the literature.34 When Pd/MoS2–Ni2P is formed, the major composite species undergoes changes in binding energy (BE). The core-level Mo peaks in Pd/MoS2–Ni2P indicate a shift to lower BEs relative to pristine MoS2 and Pd/MoS2. This change is attributable to a decrease in the Fermi level (EF) caused by p-type doping, as previously seen in another research study. This demonstrates successful Mo substitution with Pd dopants, and the covalent bonds within the lattice are reported to be responsible for the stabilization of Pd atoms.35
 | ||
| Fig. 4 XPS spectra. (a) Survey, (b) Pd 3d, (c) O 1S, and (d) C 1S for 1% Pd at 10%, 20%, and 30% V2O5–C, 3% Pd at 10%, 20% and 30% V2O5–C, and 5% Pd/at 10%, 20% and 30% V2O5–C. | ||
The high-resolution spectrum's Pd 3d regions display four peaks, as seen in Fig. 4(b). Pd 3d5/2 peaks at 335.8 eV and 336.8 eV, respectively, correspond to Pd0/2+.36 It is interesting to note that the Pd BEs in Pd/Ni2P and Pd/MoS2 occur at the same BE, and there is a notable shift in Pd/MoS2–Ni2P, indicating great electronic coupling interaction. Thus, there is a shift in BEs for Pd0 of Pd/MoS2–Ni2P compared to metallic Pd (3d5/2 = 335.1 eV) on Pd/Ni2P and Pd/MoS2, which implies charge transfer from Pd to double support MoS2–Ni2P.37 Therefore, substantial Pd and MoS2 contacts occur for Pd/MoS2–Ni2P, implying that Pd catalysts for ethanol electro-oxidation have enhanced catalytic characteristics. The shift in Pd binding energy for Pd/MoS2Ni2P indicated that the insertion of Pd particles resulted in a substantial electronic alteration with Ni2P due to the strain effect.38
The Ni 2p deconvoluted spectra, Fig. 4(c), contain peaks at 855.15 (Ni 2p3/2) and 872.83 eV (Ni 2p1/2) for Ni2+ presence. Satellite peaks were seen at 860.57 and 878.57 eV. The change in the electronic environment of the nickel atoms during composite creation causes the shift in BE for Ni 2p for Pd/MoS2–Ni2P, indicating successful hybridization of the nanocomposite, which had a great effect as seen in the shifting of 3d Pd BE in Fig. 4(b).
3.3 Electrochemical characterization
Cyclic voltammetry was utilized to evaluate the performance of the synthesized electrocatalysts in an alkaline medium. The commercial Pd/C, Pd/Ni2P–MoS2, Pd/Ni2P, and Pd/MoS2 electrocatalysts were assessed in 1 M KOH, Fig. 5(a). The PdO reduction peak in the potential range of 0.6–0.8 (V vs. RHE) for all catalysts indicates efficient alkaline electrocatalysis, with Pd/Ni2P–MoS2 having the highest catalytic activity for electrochemical reactions in an alkaline medium, as observed in the negative scan. The current density of the electrocatalysts in the negative scan is arranged in ascending order as follows: Pd/MoS2 < Pd/C < Pd/Ni2P < Pd/Ni2P–MoS2.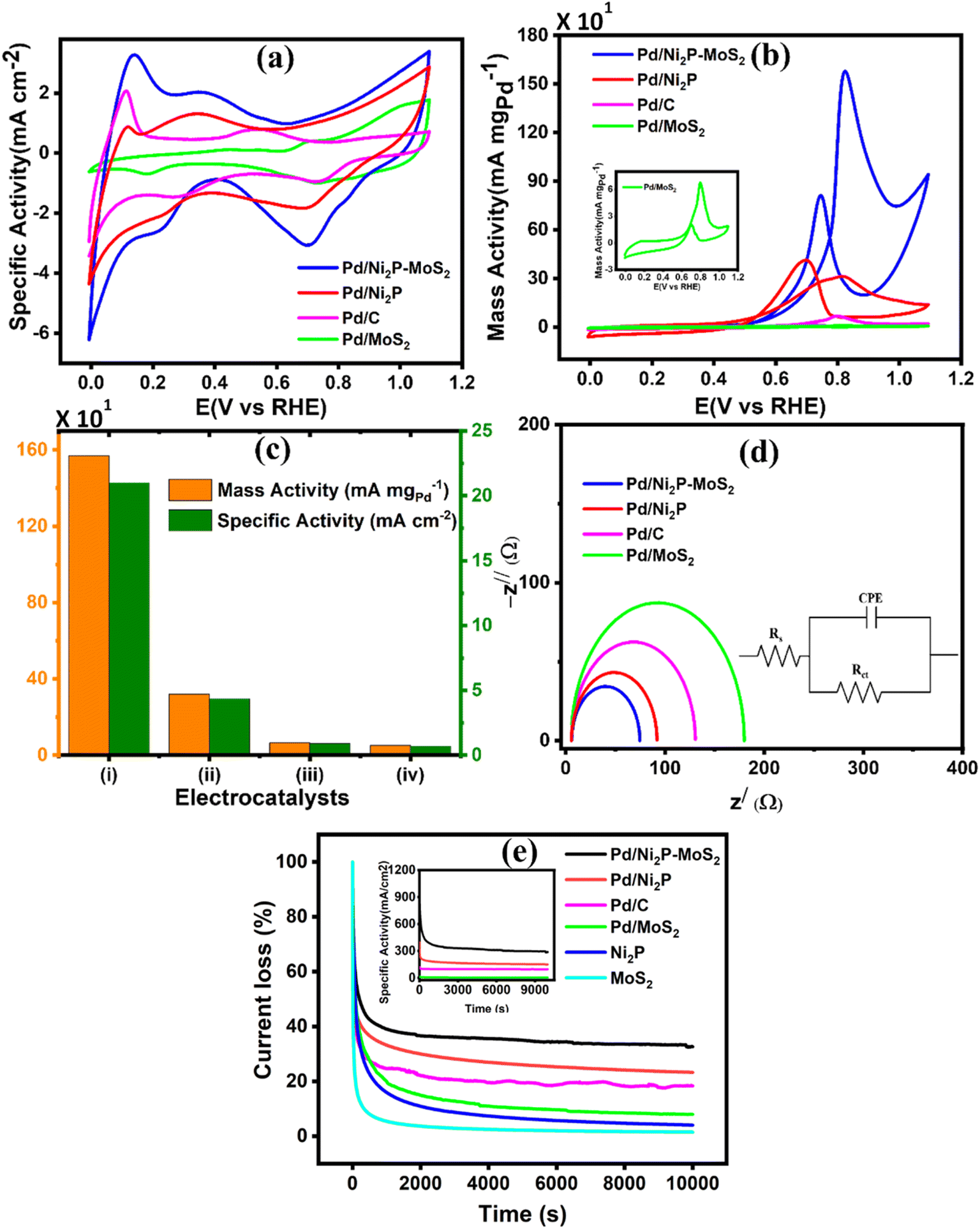 | ||
| Fig. 5 (a) Cyclic voltammograms in 1 M KOH, (b) ethanol oxidation at a scan rate of 50 mV s−1, (c) the maximum mass activity and specific activity for (i) Pd/Ni2P–MoS2, (ii) Pd/Ni2P, (iii) Pd/C and (iv) Pd/MoS2, (d) Nyquist plots at 0.55 V in 1 M KOH + 0.5 M CH3CH2OH, and (e) chronoamperometric curves measured at 0.59 (V vs. RHE). | ||
The electrochemically active surface area (ECSA) (eqn (4)) for the electrocatalysts was determined using the PdO reduction peak based on the literature as depicted in Fig. 5(a)
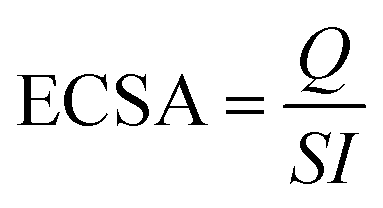 | (4) |
The CV cycles of the multi-metallic electrocatalysts Pd/Ni2P–MoS2, Pd/Ni2P, and Pd/MoS2 and commercial Pd/C were studied in 0.5 M ethanol and 1 M KOH; their cyclic voltammograms are displayed in Fig. 5(b). All the corresponding catalysts show distinct anodic scans, as expected. The observed forward anodic scan is ascribed to the electro-oxidation of fresh CH3CH2OHads on electrocatalyst, whilst the reverse scan is attributed to the removal of intermediates species such as COad, COHad, HCOad, and HCOOad that accumulate on the electrocatalyst surface resulting from the EOR. The ternary metal-based electrocatalyst Pd/Ni2P–MoS2 recorded the highest current density of 1579 mA mgPd−1 with the lowest onset potential, meaning better kinetics and low activation energy, which appears to be 5, 22, and 23 times more than that of of Pd/Ni2P (314.8 mA mgPd−1), Pd/C (71.9 mA mgPd−1) and Pd/MoS2 (69.2 mA mgPd−1) respectively. The current densities are plausible relative to others reported in the literature, as shown in Table S2.† The onset potential (Eonset) of the catalysts, as shown in Table 1, followed the descending order as follows: Pd/MoS2 (0.64 V vs. RHE) > Pd/C (0.54 V vs. RHE) > Pd/Ni2P (0.52 V vs. RHE) > Pd/Ni2P–MoS2 (0.48 V vs. RHE).
| Catalysts | ICP-OES | E onset (V vs. RHE) | Forward scan | Reverse scan | ECSAb (m2 g−1) | EISc | Chronoamperometry | Cycle studies | |
|---|---|---|---|---|---|---|---|---|---|
| i f | I b | R ct (Ω) | R s (Ω) | Retained currentd (%) |
|
||||
a
i
f and Ib (mA mg−1Pd).
b Electrochemical surface area (based on the PdO peak).
c Electrochemical Impedance parameters: Rct charge transfer resistance values from EIS fitting (Nyquist) and Rs solution resistance values from EIS fitting (Nyquist).
d @ 1000 s from CA.
e
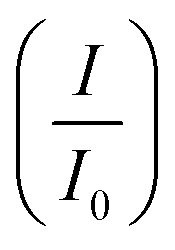 electrocatalyst deactivation rate, from cyclic studies. electrocatalyst deactivation rate, from cyclic studies.
|
|||||||||
| Pd/Ni2P–MoS2 | 5.24 | 0.480 | 1579 | 812 | 113.4 | 68.36 | 5.68 | 33.19 | 0.885 |
| Pd/Ni2P | 4.98 | 0.520 | 314.8 | 413.2 | 78.30 | 86.62 | 5.79 | 23.97 | 0.868 |
| Pd/C | 10.00 | 0.540 | 71.9 | 25.7 | 37.88 | 125.13 | 5.98 | 17.37 | 0.674 |
| Pd/MoS2 | 5.77 | 0.640 | 69.2 | 22.2 | 6.79 | 174.01 | 6.04 | 8.47 | 0.654 |
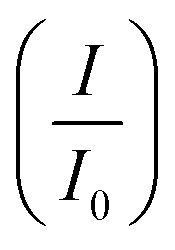
Pd/Ni2P–MoS2 recorded the most negative Eonset compared to the other electrocatalysts; this observation can be due to high reaction kinetics and low overpotential, hence its high catalytic activity. Furthermore, the good active characteristics of Pd/Ni2P–MoS2 may be due to the increased number of active sites as a result of combining nickel phosphide and molybdenum disulfide. The two inorganic support materials in Pd/Ni2P–MoS2 have demonstrated a remarkable electrocatalytic improvement for the ethanol electrooxidation reaction, incorporating their well-reported electrocatalytic characteristics, such as high conductivity and mass transport of Ni2P, and the unique properties of MoS2, such as large surface area and high electrochemical accessibility, and, most importantly, their bifunctional effect promotes electron transfer between the electrode and the reactants, leading to enhanced electrocatalytic performance.
As shown in Fig. 5(b), Pd/C appears to outperform Pd/MoS2; this observation might be due to the poor conductivity of MoS2. Fig. 5(c) shows mass activity and specific activity of (i)-Pd/Ni2P–MoS2, (ii)-Pd/Ni2P, (iii)-Pd/C, and (iv)-Pd/MoS2. Pd/Ni2P–MoS2 recorded the largest mass activity (1579 mA mgPd−1) and specific activity (216.1 mA cm2) when compared to other reference electrocatalysts; this indicates its improved efficiency for the EOR under alkaline conditions. The remarkable activity of Pd/Ni2P–MoS2 is attributed to interface synergism and electronic and strain effects among Pd, Ni2P, and MoS2. Also, the ultimate electron coupling within the nanocomposite boosted the Pd NPs' electrocatalytic activity. The Pd/Ni2P–MoS2 electrocatalyst exhibits metal support interactions that enhances its durability, stability, electrocatalytic activity, and selectivity. The electronic effect at the interface promotes electron transfer towards the direct C1-12e-ethanol electro-oxidation route for DEFCs. The proposed Pd/Ni2P–MoS2 with interface synergism and overall nano-structural cooperation gives maximum electrocatalytic power and operates for more than 10000 s, surpassing the performance of other electrocatalysts.
Electrochemical impedance spectroscopy was used to further investigate the remarkable reaction kinetics of the electrocatalysts by determining their charge transfer (Rct) and solution resistance (Rs) in relation to their Nyquist arch diameter. The obtained Nyquist plots, together with the corresponding circuit, are shown in Fig. 5(d). The arch diameter of the Nyquist plots was used to measure electrocatalyst reaction kinetics by determining their Rct at the electrode–electrolyte interface. The arch diameter presented decreased as follows: Pd/MoS2 > Pd/C > Pd/Ni2P > Pd/Ni2P–MoS2. This was due to the extent of electronic coupling interaction with Ni2P, MoS2, and Ni2P–MoS2. Thus, Pd/Ni2P–MoS2 has the smallest Rct and Rs, as indicated in Table 1; this further proves the composite catalyst's great kinetics reactions due to increased reaction active sites, leading to exceptional catalyst performance seen in Fig. 5(b). The excellent catalytic reaction kinetics can further be ascribed to the synergistic effect of the Pd–Ni–Mo alloy, that is, there is easy movement of electrons within the Pd/Ni2P–MoS2 and the electrode–electrolyte interface. The Pd/MoS2 electrocatalyst recorded the highest Rct and Rs values with a large Nyquist arch as demonstrated; this shows sluggish reaction kinetics and high overpotential, and the least catalytic activity was obtained in CV. This can be ascribed to the layered structure of MoS2, which hinders electron transfer between the catalyst surface and reactants.
The vulnerability of the catalysts to CO poisoning is one of the few drawbacks limiting fuel cell commercialization. Thus, potentiostatic chronoamperometry was used to evaluate electrocatalyst stability at a constant potential of 0.59 (V vs. RHE) for 10000 s in 0.5 M CH3CH2OH and 1 M KOH electrolyte solution as shown in Fig. 5(e). The inset in Fig. 5(e) shows the CA curve as a function of current density and time, but for a better comparison, the CA curve was plotted as a function of % current loss and time. A high initial current density was observed due to a transient potential jump that induced a significant oxidation current. However, this current density gradually decreased because of the diffusion of reactants and the accumulation of poisoning species, such as adsorbed carbon monoxide (COads), on the electrode surface, which deactivate the active sites of the electrocatalyst.39,40 The electrocatalysts initially experienced a sharp decline in current density within the first 1000 seconds before stabilizing at a constant current level from 2500 s. The constant current density region represents the current density each electrocatalyst retained after CO poisoning. The retained current density increased in the order: Pd/MoS2 > Pd/C > Pd/Ni2P > Pd/Ni2P–MoS2 as shown in Table 1. Pd/Ni2P–MoS2 retained the highest current density and is more stable than commercial Pd/C. The strong metal–phosphorus interaction can explain this enhanced anti-poisoning ability, which prevents leaching and corrosion.41 The modulation of the electronic structure may also influence this effect14 from the Ni2P–MoS2 hybrid substrate. This modulation promotes the electro-oxidation and removal of adsorbed CO-like species on the Pd surface. These findings further suggest that using Ni2P and MoS2 inorganic materials as hybrid support materials improves the electrocatalysts' stability for the EOR in alkaline media. According to a recent argument by Hofstead-Duffy et al.,42 assessing electrocatalysts using If/Ib is still debatable. They proposed that If and Ib share the same chemical origin, which means that the empirical criterion of If/Ib is inadequate for assessing the resistance of electrocatalysts to poisoning. As we concur with their findings, we complemented the above findings in this study by calculating the poisoning rate, σ (%s−1) (eqn (5)). From CA, the linear decay current density portion was used to calculate the long-term poisoning rate of adsorbed species:
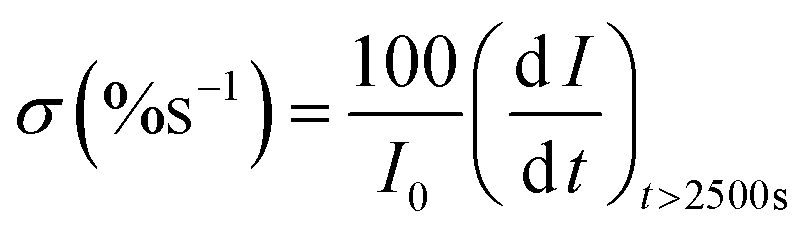 | (5) |
 t = time in seconds and I0 = extrapolated current density % at the point
t = time in seconds and I0 = extrapolated current density % at the point 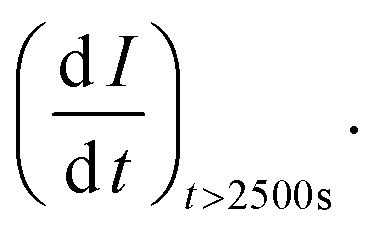 Jiang et al.43 proposed the procedure for the MOR on PtRu electrocatalysts. The same procedure was used by Guo et al.32 for the MOR and Chen et al.10 for glucose oxidation. From these studies, we have postulated that the slope should be calculated from t = ts where ts is the time when the CA curve starts to stabilize. The σ (%s−1) values followed the order: Pd/Ni2P–MoS2 < Pd/Ni2P < Pd/C < Pd/MoS2, as shown in Table 1. Among the synthesized electrocatalysts and commercial Pd/C, Pd/Ni2P–MoS2 exhibited the highest tolerance to poisonous intermediates and the lowest poisoning rate during the EOR. The σ (%s−1) values give a clear view of the contribution of the hybrid support relative to the reduction in poisoning rates for the Pd/Ni2P–MoS2 electrocatalyst. The low poisoning rate for Pd/Ni2P–MoS2 is due to synergistic effects, electronic coupling between Pd and Ni2P/MoS2, and MSI.44,45 These findings also suggest that Pd/Ni2P–MoS2 has high chemical stability despite the initial drop in current density. Thus, the nanocomposite utilizes the synergy between Ni2P and MoS2 to slow the decay and promote active site regeneration by removing poison. The Pd/MoS2 electrocatalyst has the highest σ (%s−1) value, indicating that it is the most unstable and susceptible to intermediate poisoning due to a lack of Pd NP protection. This is evident from the significant migration and coalescence/Ostwald ripening shown in Fig. 7(a); this causes a reduction of the electrochemical active centers, which is also apparent in Fig. 6(a).
Jiang et al.43 proposed the procedure for the MOR on PtRu electrocatalysts. The same procedure was used by Guo et al.32 for the MOR and Chen et al.10 for glucose oxidation. From these studies, we have postulated that the slope should be calculated from t = ts where ts is the time when the CA curve starts to stabilize. The σ (%s−1) values followed the order: Pd/Ni2P–MoS2 < Pd/Ni2P < Pd/C < Pd/MoS2, as shown in Table 1. Among the synthesized electrocatalysts and commercial Pd/C, Pd/Ni2P–MoS2 exhibited the highest tolerance to poisonous intermediates and the lowest poisoning rate during the EOR. The σ (%s−1) values give a clear view of the contribution of the hybrid support relative to the reduction in poisoning rates for the Pd/Ni2P–MoS2 electrocatalyst. The low poisoning rate for Pd/Ni2P–MoS2 is due to synergistic effects, electronic coupling between Pd and Ni2P/MoS2, and MSI.44,45 These findings also suggest that Pd/Ni2P–MoS2 has high chemical stability despite the initial drop in current density. Thus, the nanocomposite utilizes the synergy between Ni2P and MoS2 to slow the decay and promote active site regeneration by removing poison. The Pd/MoS2 electrocatalyst has the highest σ (%s−1) value, indicating that it is the most unstable and susceptible to intermediate poisoning due to a lack of Pd NP protection. This is evident from the significant migration and coalescence/Ostwald ripening shown in Fig. 7(a); this causes a reduction of the electrochemical active centers, which is also apparent in Fig. 6(a).
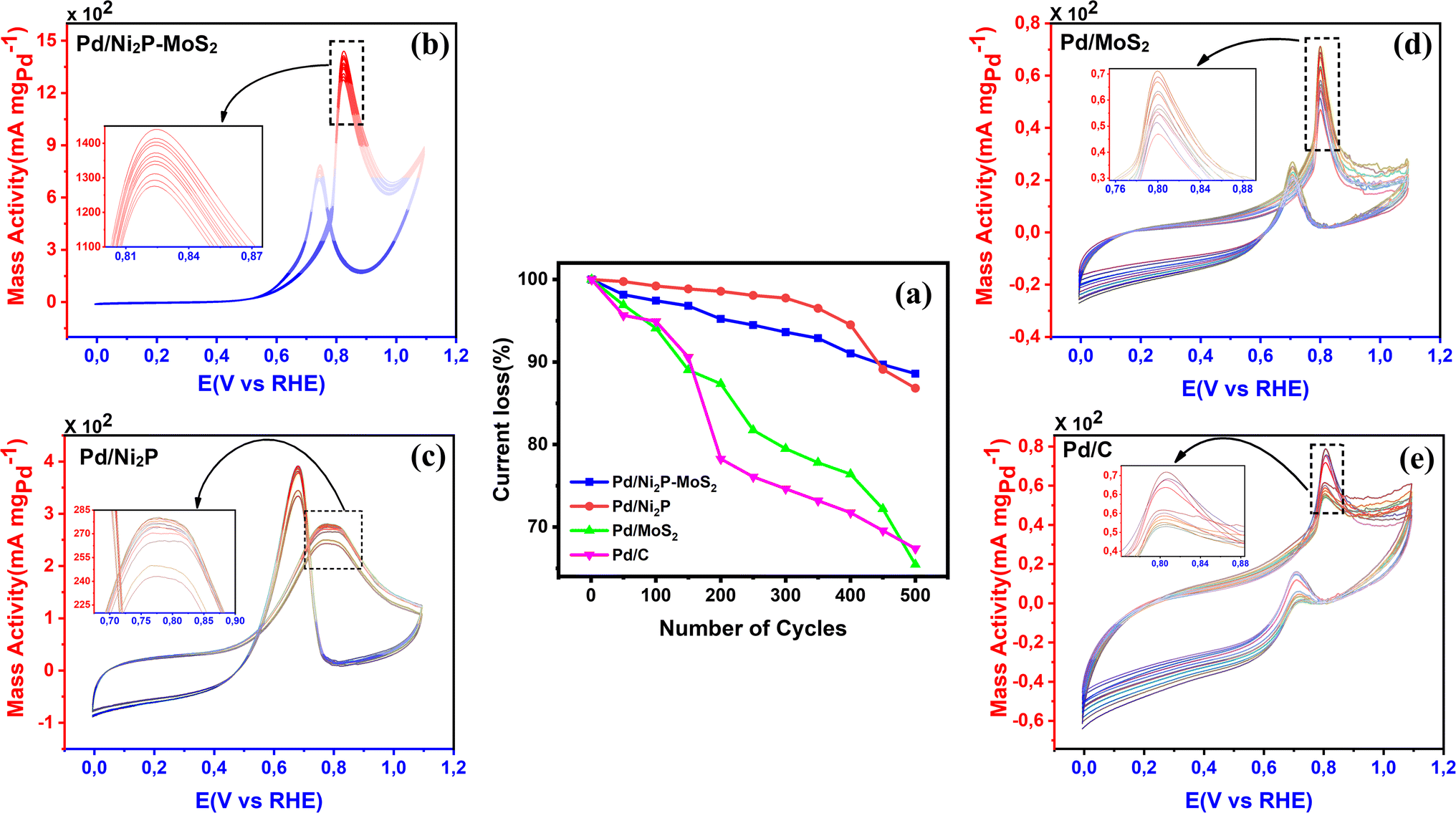 | ||
| Fig. 6 (a) Line profile of CV cycles and (b–e) the durability tests performed by CV continuous cyclization in 0.5 M ethanol and 1 M KOH solution for 500 cycles of Pd/Ni2P–MoS2, Pd/Ni2P, Pd/MoS2, and Pd/C. | ||
CV was further used to assess the durability of the synthesized electrocatalysts. Fig. 6(a) shows that Pd/Ni2P–MoS2 had the highest cycle durability relative to other electrocatalysts and commercial Pd/C. Fig. 6(b–e) show the actual CV cycles with the respective insets for forward oxidation peaks. The insets clearly show the current decay profile as pseudo-contours. From the inset of Pd/Ni2P–MoS2 (Fig. 6(b)) we see a gradual drop of the CV oxidation peak with an increasing cycle number, which we postulated MSI and better dispersion on the hybrid support system as compared to randomized and non-uniform for Fig. 6(c–e) based on the CV cycle profiles. From the cycle studies, we complemented the findings by calculating the electrocatalyst deactivation rate, where I (current density) was normalized by I0 (initial current)46 and the 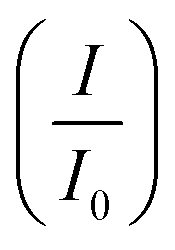 ratios are shown in Table 1. For the Pd supported on Ni2P–MoS2 containing the electrocatalyst had a
ratios are shown in Table 1. For the Pd supported on Ni2P–MoS2 containing the electrocatalyst had a  of 0.885 > Pd/C (0.674) and all the electrocatalysts indicating excellent cycling durability, which is summarized in Table S1.† On the other hand, Pd-supported electrocatalysts on mono-supports had a low
of 0.885 > Pd/C (0.674) and all the electrocatalysts indicating excellent cycling durability, which is summarized in Table S1.† On the other hand, Pd-supported electrocatalysts on mono-supports had a low 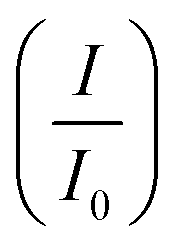 rate, which proves that the synergy and collaboration effect in a hybrid support system is fundamental in ensuring durability. The
rate, which proves that the synergy and collaboration effect in a hybrid support system is fundamental in ensuring durability. The 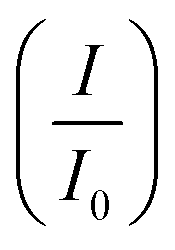 ratios match CA curve findings, confirming the significance of hybrid support formation.
ratios match CA curve findings, confirming the significance of hybrid support formation.
To further confirm the structural and morphological preservation of the electrocatalysts. Fig. 7(a and b) show HR-TEM images after the 10000 s CA stability test and 500 CV cycles, respectively. The Pd NP distribution is not greatly affected, with minimal morphological changes. Of note is the small increase in the size of Pd NPs in Pd/MoS2 assumed to be caused by the coalescence of the Pd NPs as they retained the spherical-like morphology. The major change seen was a reduction in the inter Pd NP distance. Thus, there was very little Pd NP agglomeration after the CA and cycle test, showing the electrocatalyst robustness, thus confirming the existence of MSI with the electrocatalyst matrix.
 | ||
| Fig. 7 HR-TEM images after durability and stability testing for (a) Pd/Ni2P, (b) Pd/MoS2, and (c) Pd/Ni2P–MoS2. | ||
3.4 Complementary stability test (1 year cycle)
From the above-discussed results, we chose the most active electrocatalyst, Pd/Ni2P–MoS2, for further electrochemical evaluation to corroborate stability. The Pd/Ni2P–MoS2 stability was investigated using a modified GCE termed “GCE_1” that was kept for a period of one year in an open environment. The Pd/Ni2P–MoS2 modified GCE was tested using cyclic voltammetry and chronoamperometry every 3 months. As seen from Fig. 8, the current density of GCE_1 fluctuates between the nominal value in Fig. 5(c); we postulate that active site regeneration is possible. The presented bars in Fig. 8 are an average of current density obtained after every 3 months. The GCE_1 was kept in an open environment, and the results clearly indicated that external weather factors minimally affected the electrocatalytic activity. This result means that the electrocatalytically active sites can be maintained over a period of one year, as per this study. As for the CA current retained, there was not much loss of current, which supports the results of cycle studies in Fig. 6(b). As shown in Fig. 8, the %CA current loss was from ∼45% → ∼38.9%. This plausible minimal current loss can be explained by MSI as proved by BE shifts in XPS, confirming the interaction of materials in the catalyst matrix and probable active site regeneration during the CA run.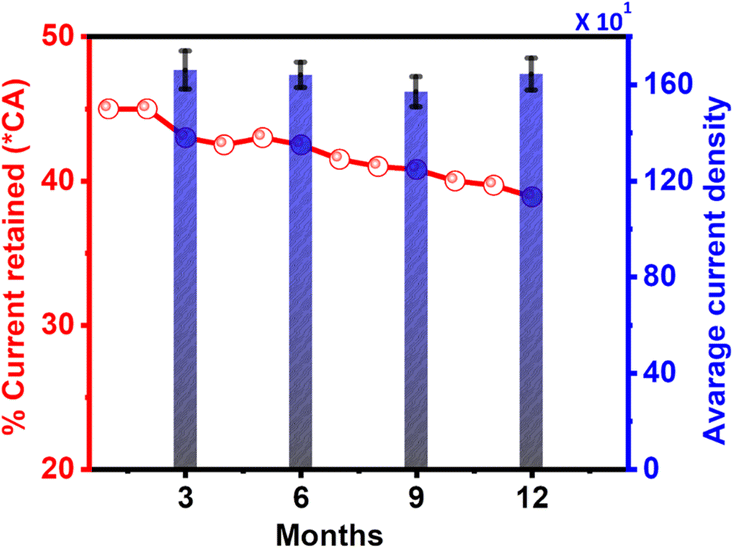 | ||
| Fig. 8 Average current density for every three months of storage and the % retained current from chronoamperometry (*CA) for the Pd/Ni2P–MoS2 electrocatalysts in 1 M KOH + 0.5 M ethanol. | ||
4 Conclusion
A structural design has been developed to enhance the efficiency and lifespan of direct ethanol fuel cells (DEFCs) by engineering the synergistic interface of Pd/Ni2P–MoS2. Initial findings indicate that Pd/Ni2P–MoS2 exhibits greater electrocatalytic activity and improved steady-state performance for the ethanol oxidation reaction (EOR). The incorporation of the hybrid Ni2P–MoS2 support significantly boosts the electrocatalytic activity of Pd for ethanol electro-oxidation through interface synergism and electronic coupling interaction. The Pd/Ni2P–MoS2 electrocatalysts demonstrate the highest electrochemical catalytic activity for the EOR, with a mass activity of 1579 mA mgPd−1, a more negative onset potential of 0.48 V vs. RHE, and excellent durability, retaining 89% of the initial current after 500 cycles. This study illustrates that the Ni2P–MoS2 hybrid support enhances electrocatalytic performance in comparison to the Pd/C electrocatalyst for the EOR in an alkaline medium. Nevertheless, it is essential to further assess other binary or ternary Pd-based electrocatalysts supported on Ni2P–MoS2@carbon to increase the reaction rate and achieve high selectivity towards CO2 formation while breaking the C–C bond.Data availability
All data and materials used in this article are available upon reasonable request.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This research was funded by the University Research Council, International Postgraduate Scholarships (2021–2023), the Centre for Nanomaterials Science Research, University of Johannesburg, South Africa, University of Cape Town 2025–2026 URC Postdoctoral Fellowship, Nurturing Emerging Scholars Programme, National Research Foundation of South Africa: Grant Numbers 118148, and 138083 and the Centre for Nanomaterials Science Research, University of Johannesburg, South Africa. We acknowledge the University of Johannesburg (UJ) FRC for financial support and Faculty of Science, Department of Chemical Sciences at the University of Johannesburg, South Africa. Funding was also provided by the Department of Science and Innovation (DSI), through the Hydrogen South Africa (HySA) Catalysis Centre of Competence Programme.References
- L. An, T. S. Zhao and Y. S. Li, Carbon-neutral sustainable energy technology: Direct ethanol fuel cells, Renewable Sustainable Energy Rev., 2015, 50, 1462–1468, DOI:10.1016/j.rser.2015.05.074.
- J. Guo, R. Chen, F.-C. Zhu, S.-G. Sun and H. M. Villullas, New understandings of ethanol oxidation reaction mechanism on Pd/C and Pd2Ru/C catalysts in alkaline direct ethanol fuel cells, Appl. Catal., B, 2018, 224, 602–611, DOI:10.1016/j.apcatb.2017.10.037.
- B. Cermenek, J. Ranninger and V. Hacker, Alkaline direct ethanol fuel cell, Elsevier, 2019, vol. 1904, DOI:10.1016/B978-0-12-811458-2.00015-8.
- Y. Zheng, X. Wan, X. Cheng, K. Cheng, Z. Liu and Z. Dai, Advanced catalytic materials for ethanol oxidation in direct ethanol fuel cells, Catalysts, 2020, 10(2), 166, DOI:10.3390/catal10020166.
- M. Z. F. Kamarudin, S. K. Kamarudin, M. S. Masdar and W. R. W. Daud, Review: Direct ethanol fuel cells, Int. J. Hydrogen Energy, 2013, 38(22), 9438–9453, DOI:10.1016/j.ijhydene.2012.07.059.
- Y. Shu, et al., Hollow Echinus-like PdCuCo Alloy for Superior Efficient Catalysis of Ethanol, ACS Appl. Mater. Interfaces, 2018, 10(5), 4743–4749, DOI:10.1021/acsami.7b17731.
- J. Liu, et al., Phosphorus doped PdMo bimetallene as a superior bifunctional fuel cell electrocatalyst, Chem. Eng. J., 2024, 486, 150258, DOI:10.1016/j.cej.2024.150258.
- Y. Wang, F.-F. Shi, Y.-Y. Yang and W.-B. Cai, Carbon supported Pd–Ni–P nanoalloy as an efficient catalyst for ethanol electro-oxidation in alkaline media, J. Power Sources, 2013, 243, 369–373, DOI:10.1016/j.jpowsour.2013.06.021.
- S.-H. Ye, J.-X. Feng and G.-R. Li, Pd Nanoparticle/CoP Nanosheet Hybrids: Highly Electroactive and Durable Catalysts for Ethanol Electrooxidation, ACS Catal., 2016, 6(11), 7962–7969, DOI:10.1021/acscatal.6b02263.
- D. Chen, et al., Enhancing Ethanol Oxidation Reaction Activity of P-Doped CuPdNi Nanocatalyst by Optimizing Surface-Atom Distribution, ACS Appl. Energy Mater., 2019, 2(8), 5525–5533, DOI:10.1021/acsaem.9b00715.
- J. Chang, L. Feng, C. Liu, W. Xing and X. Hu, Ni2P enhances the activity and durability of the Pt anode catalyst in direct methanol fuel cells, Energy Environ. Sci., 2014, 7(5), 1628–1632, 10.1039/c4ee00100a.
- B. Tang, et al., Porous coral reefs-like MoS2/nitrogen-doped bio-carbon as an excellent Pt support/co-catalyst with promising catalytic activity and CO-tolerance for methanol oxidation reaction, Electrochim. Acta, 2017, 246, 517–527, DOI:10.1016/j.electacta.2017.06.052.
- M. Li, F. Yang, J. Chang, A. Schechter and L. Feng, MoP-NC Nanosphere Supported Pt Nanoparticles for Efficient Methanol Electrolysis, Acta Phys.-Chim. Sin., 2023, 2301005, DOI:10.3866/PKU.WHXB202301005.
- Y. Kuang, W. Qiao, S. Wang, F. Yang and L. Feng, Doping and Interfacial Engineering of MoSe2 Nanosheets by NH3 Plasma Promoted Pt for Methanol Electrolysis, ACS Mater. Lett., 2024, 6(5), 1722–1731, DOI:10.1021/acsmaterialslett.4c00244.
- M. Liu, et al., Interfacial electronic structure engineering on molybdenum sulfide for robust dual-pH hydrogen evolution, Nat. Commun., 2021, 12(1), 5260 CrossRef PubMed.
- D. Tsikritzis, N. Tsud, T. Skála and L. Sygellou, Unravelling the phase transition of 2H-MoS2 to 1T-MoS2 induced by the chemical interaction of Pd with molybdenum disulfide–graphene hybrids, Appl. Surf. Sci., 2022, 599, 153896, DOI:10.1016/j.apsusc.2022.153896.
- Y. Yan, X. Ge, Z. Liu, J.-Y. Wang, J.-M. Lee and X. Wang, Facile synthesis of low crystalline MoS2 nanosheet-coated CNTs for enhanced hydrogen evolution reaction, Nanoscale, 2013, 5(17), 7768, 10.1039/c3nr02994h.
- Y. Li, H. Wang, L. Xie, Y. Liang, G. Hong and H. Dai, MoS2 Nanoparticles Grown on Graphene: An Advanced Catalyst for the Hydrogen Evolution Reaction, J. Am. Chem. Soc., 2011, 133(19), 7296–7299, DOI:10.1021/ja201269b.
- E. Ramírez-Mondragón, O. E. Contreras, U. J. Tamayo-Pérez and M. T. Oropeza-Guzmán, Synthesis and characterization of Ni2P and MoS2 on MWCNT as an innovative catalytic material for hydrogen generation, Appl. Surf. Sci., 2020, 503, 144163 CrossRef.
- H. Sun, et al., Porous multishelled Ni2P hollow microspheres as an active electrocatalyst for hydrogen and oxygen evolution, Chem. Mater., 2017, 29(19), 8539–8547 CrossRef.
- M. Asim, et al., Synergetic effect of Au nanoparticles and transition metal phosphides for enhanced hydrogen evolution from ammonia-borane, J. Colloid Interface Sci., 2023, 638, 14–25, DOI:10.1016/j.jcis.2023.01.122.
- Y. Xiao, et al., Ball-milled Ni2P/g-C3N4 for improved photocatalytic hydrogen production, Int. J. Hydrogen Energy, 2023, 48(41), 15460–15472, DOI:10.1016/j.ijhydene.2023.01.086.
- S. Carenco, C. Boissière, L. Nicole, C. Sanchez, P. Le Floch and N. Mézailles, Controlled Design of Size-Tunable Monodisperse Nickel Nanoparticles, Chem. Mater., 2010, 22(4), 1340–1349, DOI:10.1021/cm902007g.
- W. Wang, et al., Earth-abundant Ni2P/g-C3N4 lamellar nanohydrids for enhanced photocatalytic hydrogen evolution and bacterial inactivation under visible light irradiation, Appl. Catal., B, 2017, 217, 570–580, DOI:10.1016/j.apcatb.2017.06.027.
- Y. Xu, R. Wang, Z. Liu, L. Gao, T. Jiao and Z. Liu, Ni2P/MoS2 interfacial structures loading on N-doped carbon matrix for highly efficient hydrogen evolution, Green Energy Environ., 2022, 7(4), 829–839, DOI:10.1016/j.gee.2020.12.008.
- D. Vikraman, et al., Theoretical evaluation and experimental investigation of layered 2H/1T-phase MoS2 and its reduced graphene-oxide hybrids for hydrogen evolution reactions, J. Alloys Compd., 2021, 868, 159272 CrossRef.
- X. Yu, Y. Feng, Y. Jeon, B. Guan, X. W. Lou and U. Paik, Formation of Ni–Co–MoS2 nanoboxes with enhanced electrocatalytic activity for hydrogen evolution, Adv. Mater., 2016, 28(40), 9006–9011 CrossRef PubMed.
- F. Sultana, et al., An insight to catalytic synergic effect of Pd-MoS2 nanorods for highly efficient hydrogen evolution reaction, Arabian Journal of Chemistry, 2022, 15(5), 103735, DOI:10.1016/j.arabjc.2022.103735.
- D. M. Sim, et al., Controlled Doping of Vacancy-Containing Few-Layer MoS2via Highly Stable Thiol-Based Molecular Chemisorption, ACS Nano, 2015, 9(12), 12115–12123, DOI:10.1021/acsnano.5b05173.
- H. Lv, et al., Pd decorated MoS2 nanoflowers as photothermal catalyst for enhanced NIR-induced 4-nitrophenol reduction, J. Environ. Chem. Eng., 2023, 11(5), 110375, DOI:10.1016/j.jece.2023.110375.
- F.-C. Pan, H. He, Z.-X. Yang, Q. Zheng, D. Lin and Y. Huo, Rationally designed Ni2P/WS2/Co9S8@C multi-interfacial electrocatalyst for efficient overall water splitting, Chem. Eng. J., 2022, 446, 136961, DOI:10.1016/j.cej.2022.136961.
- Y. Guo, et al., Preparation of MoS2 nanosheets to support Pd species for selective steerable hydrogenation of acetylene, J. Mater. Sci., 2021, 56, 2129–2137 CrossRef.
- R. Bernasconi, et al., Electrocatalytic layers for hydrogen evolution reaction based on nickel phosphides: cost-effective fabrication and XPS characterization, J. Mater. Sci., 2022, 57(20), 9370–9388 CrossRef.
- M. Acerce, D. Voiry and M. Chhowalla, Metallic 1T phase MoS2 nanosheets as supercapacitor electrode materials, Nat. Nanotechnol., 2015, 10(4), 313–318 CrossRef PubMed.
- L. Z. Hao, et al., Self-powered broadband, high-detectivity and ultrafast photodetectors based on Pd-MoS 2/Si heterojunctions, Phys. Chem. Chem. Phys., 2016, 18(2), 1131–1139 RSC.
- A. Drelinkiewicz, M. Hasik and M. Kloc, Pd/polyaniline as the catalysts for 2-ethylanthraquinone hydrogenation. The effect of palladium dispersion, Catal. Lett., 2000, 64, 41–47 CrossRef CAS.
- H. Li, K. Yu, X. Lei, B. Guo, H. Fu and Z. Zhu, Hydrothermal synthesis of novel MoS2/BiVO4 hetero-nanoflowers with enhanced photocatalytic activity and a mechanism investigation, J. Phys. Chem. C, 2015, 119(39), 22681–22689 CrossRef CAS.
- R. Kottayintavida and N. K. Gopalan, Nickel phosphate modified carbon supported Pd catalyst for enhanced alcohol electro oxidation, Int. J. Hydrogen Energy, 2020, 45(19), 11116–11126 CrossRef.
- Y. Zhou, Q. Wang, X. Tian, J. Chang and L. Feng, Electron-enriched Pt induced by CoSe2 promoted bifunctional catalytic ability for low carbon alcohol fuel electro-reforming of hydrogen evolution, J. Energy Chem., 2022, 75, 46–54, DOI:10.1016/j.jechem.2022.08.009.
- W. Qiao, L. Yu, J. Chang, F. Yang and L. Feng, Efficient bi-functional catalysis of coupled MoSe2 nanosheet/Pt nanoparticles for methanol-assisted water splitting, Chin. J. Catal., 2023, 51, 113–123, DOI:10.1016/S1872-2067(23)64469-9.
- Y. Shi and B. Zhang, Recent advances in transition metal phosphide nanomaterials: Synthesis and applications in hydrogen evolution reaction, Chem. Soc. Rev., 2016, 45, 1529–1541, 10.1039/c5cs00434a.
- A. M. Hofstead-Duffy, D.-J. Chen, S.-G. Sun and Y. J. Tong, Origin of the current peak of negative scan in the cyclic voltammetry of methanol electro-oxidation on Pt-based electrocatalysts: a revisit to the current ratio criterion, J. Mater. Chem., 2012, 22(11), 5205, 10.1039/c2jm15426a.
- J. Jiang and A. Kucernak, Electrooxidation of small organic molecules on mesoporous precious metal catalysts. II: CO and methanol on platinum–ruthenium alloy, J. Electroanal. Chem., 2003, 543, 187–199, DOI:10.1016/S0022-0728(03)00046-9.
- H. An, et al., Synthesis and performance of palladium-based catalysts for methanol and ethanol oxidation in alkaline fuel cells, Electrochim. Acta, 2013, 102, 79–87, DOI:10.1016/j.electacta.2013.03.142.
- J. Goel and S. Basu, Effect of support materials on the performance of direct ethanol fuel cell anode catalyst, Int. J. Hydrogen Energy, 2014, 39(28), 15956–15966, DOI:10.1016/j.ijhydene.2014.01.203.
- J. Han, et al., Role of Au-TiO2 interfacial sites in enhancing the electrocatalytic glycerol oxidation performance, Electrochem. Commun., 2018, 96, 16–21, DOI:10.1016/j.elecom.2018.09.004.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4se01223b |
| This journal is © The Royal Society of Chemistry 2025 |