
Cooperating with additives: low-cost hole-transporting materials for improved stability of perovskite solar cells†
Paavo
Mäkinen‡
 a,
Daniele
Conelli‡
b,
G. Krishnamurthy
Grandhi
a,
Gian Paolo
Suranna
bc,
Paola
Vivo
*a and
Roberto
Grisorio
*b
a,
Daniele
Conelli‡
b,
G. Krishnamurthy
Grandhi
a,
Gian Paolo
Suranna
bc,
Paola
Vivo
*a and
Roberto
Grisorio
*b
aHybrid Solar Cells, Faculty of Engineering and Natural Sciences, Tampere University, P.O. Box 541, FI-33014, Finland. E-mail: paola.vivo@tuni.fi
bDipartimento di Ingegneria Civile, Ambientale, del Territorio, Edile e di Chimica (DICATECh), Politecnico di Bari, Via Orabona 4, 70125 Bari, Italy. E-mail: roberto.grisorio@poliba.it
cCNR-NANOTEC – Institute of Nanotechnology, c/o Campus Ecoteckne, Via Monteroni, 73100 Lecce, Italy
First published on 20th November 2024
Abstract
The widespread adoption of perovskite-based solar technologies is strictly related to the cost reduction of the hole-transporting component in the device, while maintaining compatibility with its absorbing active layer. To date, several organic systems have been developed to compete with the pioneering 2,2′,7,7′-tetrakis(N,N-di-4-methoxyphenylamino)-9,9′-spirobifluorene (Spiro-OMeTAD) used as the benchmarking hole-transporting material (HTM). However, an easily accessible platform to construct economically competitive HTM scaffolds as alternatives to Spiro-OMeTAD is still lacking. In this study, we propose a straightforward route (excluding organometallic cross-coupling reactions) to prepare nonconventional HTMs (BTF and BTC) based on a bithiophene core decorated with unsymmetrical triarylamine groups. The two HTMs are implemented in dopant-free n-i-p perovskite solar cells (PSCs) to evaluate their performance and long-term behaviour. Despite enhancing hole extraction and transport at the perovskite/HTM interface compared to the Spiro-OMeTAD benchmark, BTC does not perform exceptionally as an undoped HTM in PSCs (PCE = 14.0% vs. 16.5% of the doped Spiro-OMeTAD reference). Moreover, the efficiencies of unencapsulated devices rapidly degraded over time (T80: ∼57 days) due to weak HTM adhesion at the perovskite interface. Conversely, using tert-butylpyridine as the sole additive slightly increases performance (PCE = 14.8%) and remarkably improves device resilience to ambient exposure (PCE = 15.4% after 401 days), representing one of the longest shelf-stability experiments ever reported. Other dopant/additive formulations are unproductive in terms of both efficiencies and device resistance. These results indicate that focusing on the molecular design of low-cost HTMs and investigating the appropriate HTM/additive systems can be a promising strategy for developing efficient and stable PSCs.
Introduction
The pressing necessity of replacing fossil fuels with renewable energy sources to produce electricity has forced the research to explore low-cost solar technologies with a wide availability of raw materials and alternative to traditional silicon-based photovoltaic devices. Perovskite solar cells (PSCs) have exhibited considerable photovoltaic performances without requiring expensive manufacturing processing. In conventionally configured n-i-p PSCs, the perovskite active layer is sandwiched between an electron-transporting layer (ETL) and a hole-transporting material (HTM).1 To achieve high power conversion efficiencies (PCEs), both ETL and HTM must efficiently transport the photogenerated charges in the perovskite absorbing layer to the corresponding electrodes under illumination.2 In common PSCs, the ETLs are made of metal oxides (TiO2 or SnO2),3 while there is still room for improvement in HTM design to enhance hole mobility and thus reduce charge losses during transport. Moreover, HTM optimization is crucial for reducing production costs, which is essential for the large-scale commercialization of PSCs.To date, many organic HTMs have been proposed as alternatives to the expensive 2,2′,7,7′-tetrakis(N,N-di-4-methoxyphenylamino)-9,9′-spirobifluorene (Spiro-OMeTAD), which is the conventional benchmark for PSCs.4 High performances in Spiro-OMeTAD-based PSCs can be only achieved upon the addition of additives or dopants to increase its conductivity. These substances generally include 4-tert-butylpyridine (tBP), lithium bis-(trifluoromethylsulfonyl)imide (Li-TFSI), and a cobalt complex (FK209).5 However, their presence can promote the rapid device degradation by increasing hygroscopicity and forming nonconductive byproducts,6 which negatively impacts the long-term performance of PSCs. To address these instability issues, various HTMs have been designed for incorporation into PSCs without using dopants.7 However, synthesising dopant-free organic HTMs often involves complex preparation sequences, and there are relatively few reports effectively documenting low-cost, easily synthesizable HTMs that can be used in highly efficient dopant-free PSCs. In this context, common design strategies involve selecting a core building block (linear, star-shaped or 3D) and adorning it with suitable peripheral groups.8,9 Typical core scaffolds incorporate triphenylamine,10 thiophene-based derivatives,11,12 fluorene,13 and pyrene,14 only to cite a few. The peripheral groups generally include electron-donating diarylamine, triarylamine, and carbazole units,15 which provide the appropriate energy levels to function as hole extractors at the perovskite interface. At the same time, using unsymmetrical diaryl- or triaryl-amine groups to decorate the core is conceived to increase the hole mobility of the corresponding HTM.16 Additionally, unsymmetrical substitution at the nitrogen atom of the diarylamine group could lower the highest occupied molecular orbital (HOMO) energy level of the corresponding HTM, which might help increase the open-circuit voltage (VOC) and fill factor (FF) of the resulting PSCs.17 These structural variations inevitably increase the number of synthetic steps, impacting on the overall HTM cost.
In this study, we synthesised two low-cost largely π-extended HTMs (namely, BTF and BTC) to evaluate their performances in dopant-free PSCs and their long-term behaviour. We demonstrate that BTC can effectively enhance hole extraction and transport at the perovskite/HTM interface in comparison to the Spiro-OMeTAD benchmark. Notwithstanding these advantages, BTC did not perform exceptionally as an undoped HTM in PSCs, and their efficiencies declined more rapidly over time compared to the benchmark. On the contrary, the presence of tBP as the sole additive favourably impacted device stability, resulting in superior PCE after 401 days of aging compared to the initial value. Therefore, this work reveals that the molecular design of low-cost HTMs, in relation to additive/dopant incorporation, has promising potential for achieving efficient and stable PSCs.
Results and discussion
Molecular design and synthesis
The scaffold of the two HTMs was conceived with (i) a bithiophene core, (ii) two unsymmetrical electron-donating diarylamine peripheral groups and (iii) a benzene spacer in between them. The three components of the corresponding HTM were chosen to support the hole mobility (the bithiophene core) through the π-conjugation extension (benzene spacer), while increasing the HOMO energy level (due to the diarylamine group). This latter component was made unsymmetrical for the simultaneous presence of the 4-methoxybenzene unit (co-adjuvating the electron-donating character of the entire moiety) and a π-extended (2-fluorenyl or 3-carbazolyl) group. To preserve the chemical stability of the corresponding target molecules and ensure good solubility in nonpolar solvents used for their deposition in PSCs, these units were suitably functionalised with alkyl groups. In detail, the fluorene unit was protected at its C9 position (easily susceptible of oxidation) with a spiro-cyclohexane derivatisation (thus conceiving BTF), while the carbazole nitrogen atom was functionalised with a linear hexyl chain. The complete structures of BTF and BTC, along with the synthetic approach adopted for their preparation, are shown in Scheme 1.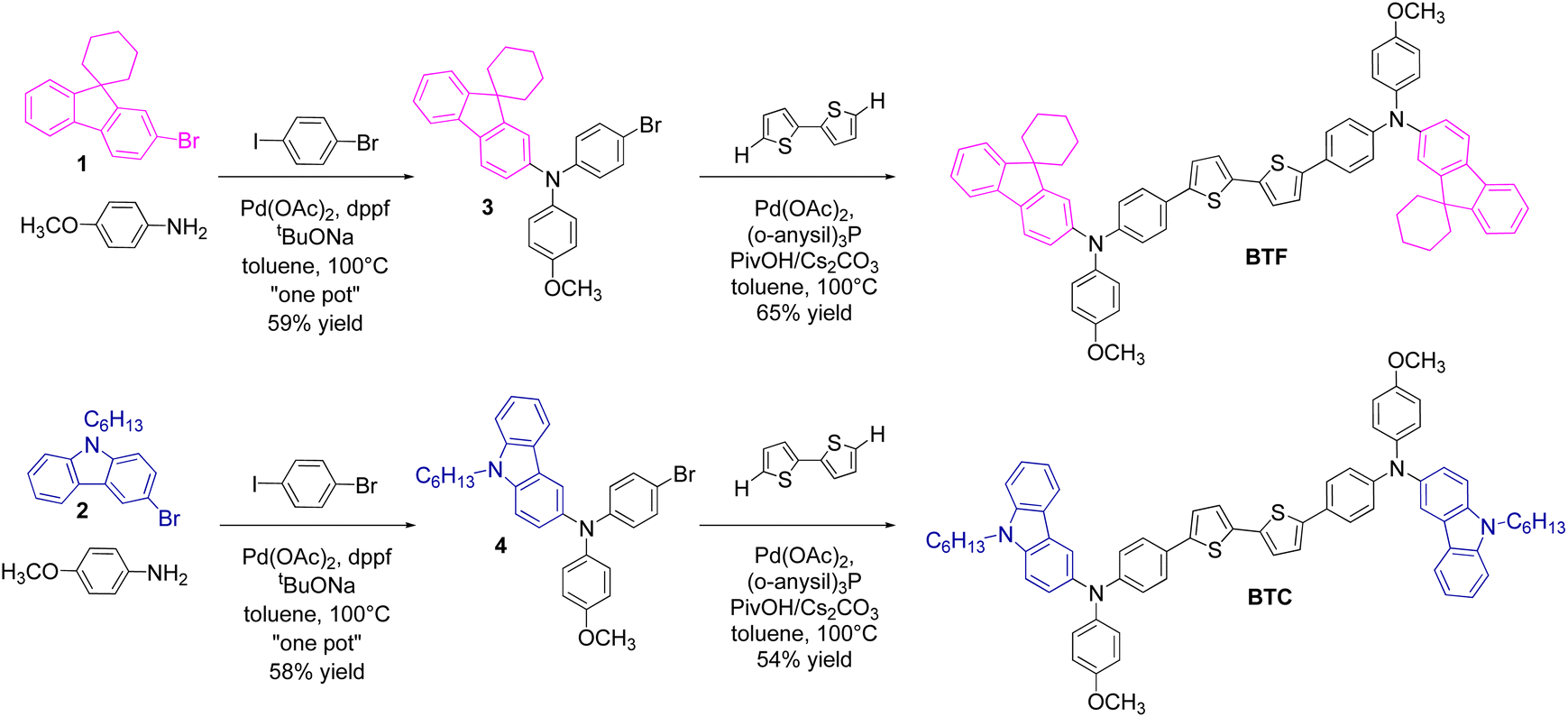 | ||
| Scheme 1 Synthetic procedure for their obtainment and chemical structure of BTF and BTC. | ||
The fluorene- and carbazole-based precursors (1 and 2, respectively) were used to prepare the unsymmetrical triarylamine building blocks (3 and 4 including the fluorene and the carbazole groups, respectively) through an unprecedent “one-pot” Buchwald–Hartwig cross-coupling reaction involving p-anisidine in a first stage. The catalytic system was composed of palladium acetate (5% mol with respect to p-anisidine) as the catalyst precursor, 1,1′-bis(diphenylphosphino)ferrocene (dppf, 2.0 equivalents with respect to Pd) as the ligand and sodium t-butylate (3.0 equivalents with respect to p-anisidine) as the base. This peculiar catalyst/ligand combination was chosen in the light of its high selectivity in the formation of diarylamines, thus reducing the amount of side products in this step of the reaction. Once ascertained the complete conversion of the two substrates (upon 4 hours in toluene at 100 °C) forming the corresponding (not isolated) diarylamines, 1-iodo-4-bromobenzene was added into the reaction mixture for generating the corresponding triarylamines (3 and 4 in Scheme 1). The in situ cross-coupling between the formed diarylamine intermediate and 1-iodo-4-bromobenzene was promoted by the same catalytic system remained still active after the previous relatively short reaction time. To favour the triarylamine formation, a better leaving group (iodide) was inserted into the substrate, warranting the direct functionalization of the resulting products. The two triarylamine 3 and 4 were obtained in 59% and 58% yield, respectively, in comparison to the initial p-anisidine reagent. While affording a complex building block, this approach allowed to reduce the amount of catalysts, reagents, and solvents used for isolating the intermediate compounds, thus favourably impacting on the overall cost and waste burden of the final HTMs.
The synthesis of BTF and BTC was completed by a C–H direct arylation reaction between bithiophene and the triarylamine building block 3 or 4, respectively. The reaction was carried out in the presence of palladium acetate as the catalyst, tris(4-methoxyphenyl)phosphine as the ligand, pivalic acid as the proton scavenger, and cesium carbonate as the base in toluene at 100 °C. The two HTMs were obtained in 65% (BTF) and 54% (BTC) yield with a total production cost of 85.75 € per g and 101.60 € per g for BTF and BTC, respectively. These values (obtained considering the current commercial prices of all chemicals and solvents used for syntheses and purifications) are comparable with that of Spiro-OMeTAD (∼92 $ per g),18 but further cost reductions can be expected by optimising the reaction conditions of the single steps. Although energy consumption is also important when the synthetic sequence contains high temperature reactions, we cannot consider this parameter in the cost computation of the materials, because it is unrelated to the quantity of the reagents employed in a lab-scale reaction. In fact, energy consumption for maintaining a specific temperature is independent from the reaction volume and cannot be relativized to the product mass.
Theoretical studies
To gain insight into both the conformational aspects and the electronic density distribution of the frontier orbitals (HOMO and lowest unoccupied molecular orbital, LUMO) in BTF and BTC, we carried out theoretical calculations based on density functional theory (DFT) at the B3LYP/6-31G* level with the Gaussian 09 program suite.19 As shown in Fig. 1A, the optimised geometry of BTF exhibited a relatively small dihedral angle (14.2°) between the two thiophene rings, while a greater distortion was observed between the thiophene and benzene units (dihedral angle = 24.4°). Conversely, the conformational optimization in the case of BTC evidenced a perfect planarity of the bithiophene core, showing 0.1° between the two adjacent thiophene units, while a similar dihedral angle (24.3°) was verified at the connection between the bithiophene moiety and the benzene ring (Fig. 1A). Probably, the higher electron-donating properties of the carbazole unit favours the orbital overlap of the electron-withdrawing bithiophene unit inducing a more planar conformation of the BTC core. At the same time, the theoretical conformations of the two HTMs suggest the possibility of favourable intermolecular interactions in the solid state, propaedeutic to an efficient charge transport. Notwithstanding these conformational differences concerning the bithiophene core, the electronic densities of their frontier orbitals are similarly distributed (Fig. 1A). In detail, the LUMO distributions are mainly localised over the bithiophene core and the adjacent π-linkages. The LUMO energy levels are −1.53 eV (BTF) and −1.41 eV (BTC). Conversely, the HOMO electronic densities are uniformly distributed on the molecules, thus also involving the peripheral groups. The theoretical HOMO energy levels are −4.48 eV (BTF) and −4.31 eV (BTC) and this difference can be explained by the electron-donating character of the carbazole unit increasing the HOMO energy level of BTC with respect to BTF.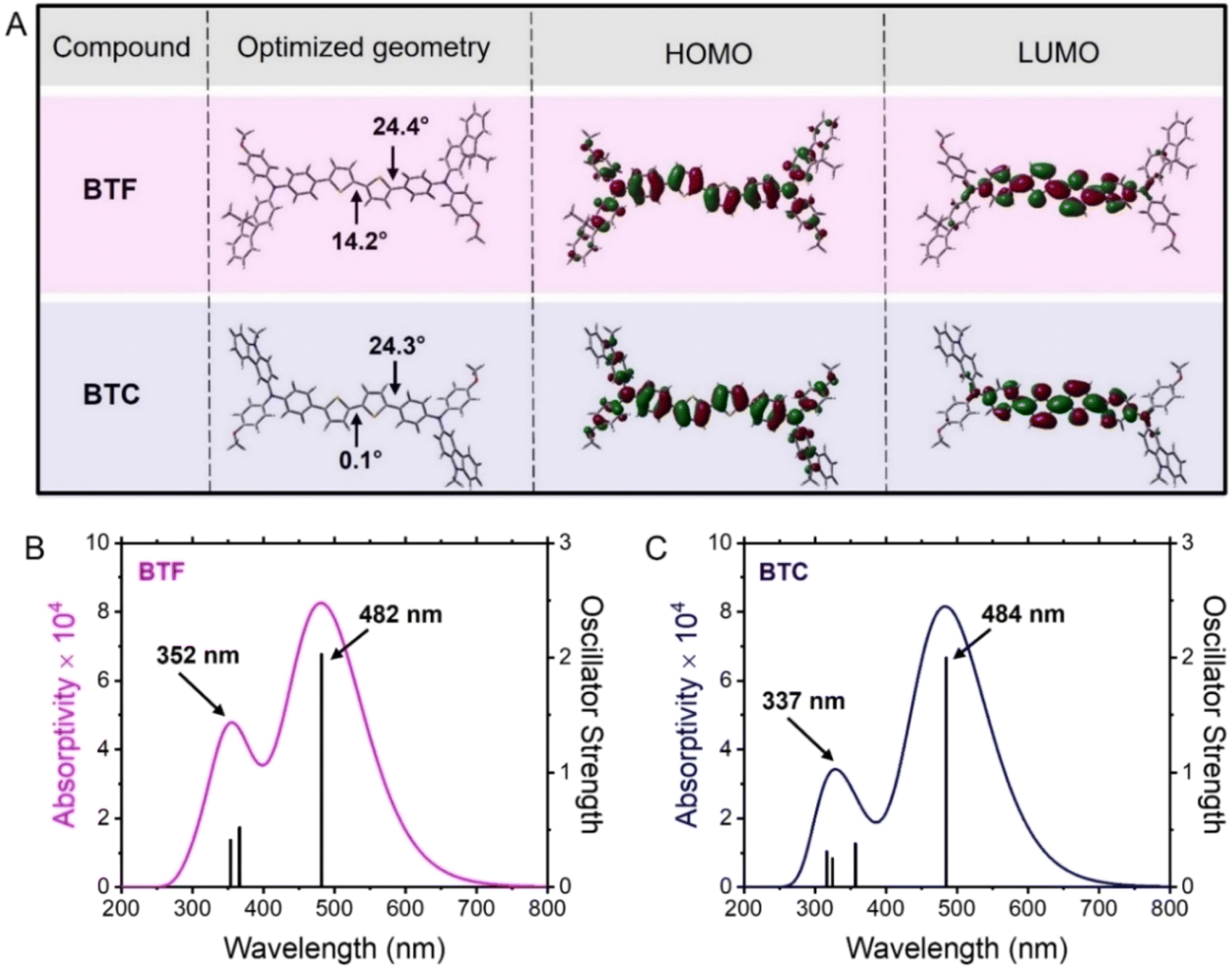 | ||
| Fig. 1 (A) Optimised molecular geometry, HOMO and LUMO isodensity surfaces (0.02e Å−3) of BTF and BTC. (B) Simulated absorption band of BTF including the main vertical electronic transitions (oscillator strength >0.2) determining the relevant profile. (C) Simulated absorption band of BTC including the main vertical electronic transitions (oscillator strength >0.2) determining the relevant profile. | ||
To gain insight into the optical properties of the two HTMs from the theoretical point of view, we carried out time-dependent DFT (TD-DFT) calculations at the same level of the theory (B3LYP/6-31G*). The simulated absorption profiles are reported in Fig. 1B and C along with the main (oscillator strength >0.2) electronic transitions. In both cases, the theoretical absorption profile is composed of two bands. The lower energy band resulted from the HOMO → LUMO transition and the corresponding electronic transitions are placed at 482 nm and 484 nm for BTF and BTC, respectively. Although a slight discrepancy was observed with respect to the experimental optical values (vide infra), it resulted a bathochromic shift of the absorption maximum in BTC in comparison to the counterpart. Excitation of an electron from the HOMO to LUMO level causes a slight electron density transfer from peripheral groups to the core, which can create an effective charge separation. In both cases, the second band at shorter wavelengths results from the convolution of less intense vertical electronic transitions involving different molecular orbitals, as reported in Table S1.† In detail, the theoretical absorption band of BTF at 352 nm (Fig. 2B) is composed of the fluorene- and bithiophene-localised π–π* transitions (Fig. S1†). The theoretical convolved absorption peak of BTC at 334 nm (Fig. 1C) is composed of the bithiophene- and p-anisyl-localised π–π* transitions (Fig. S1†). This latter fragment of the molecule is less π-extended in comparison to the fluorene unit thus determining the resulting hypsochromic shift.
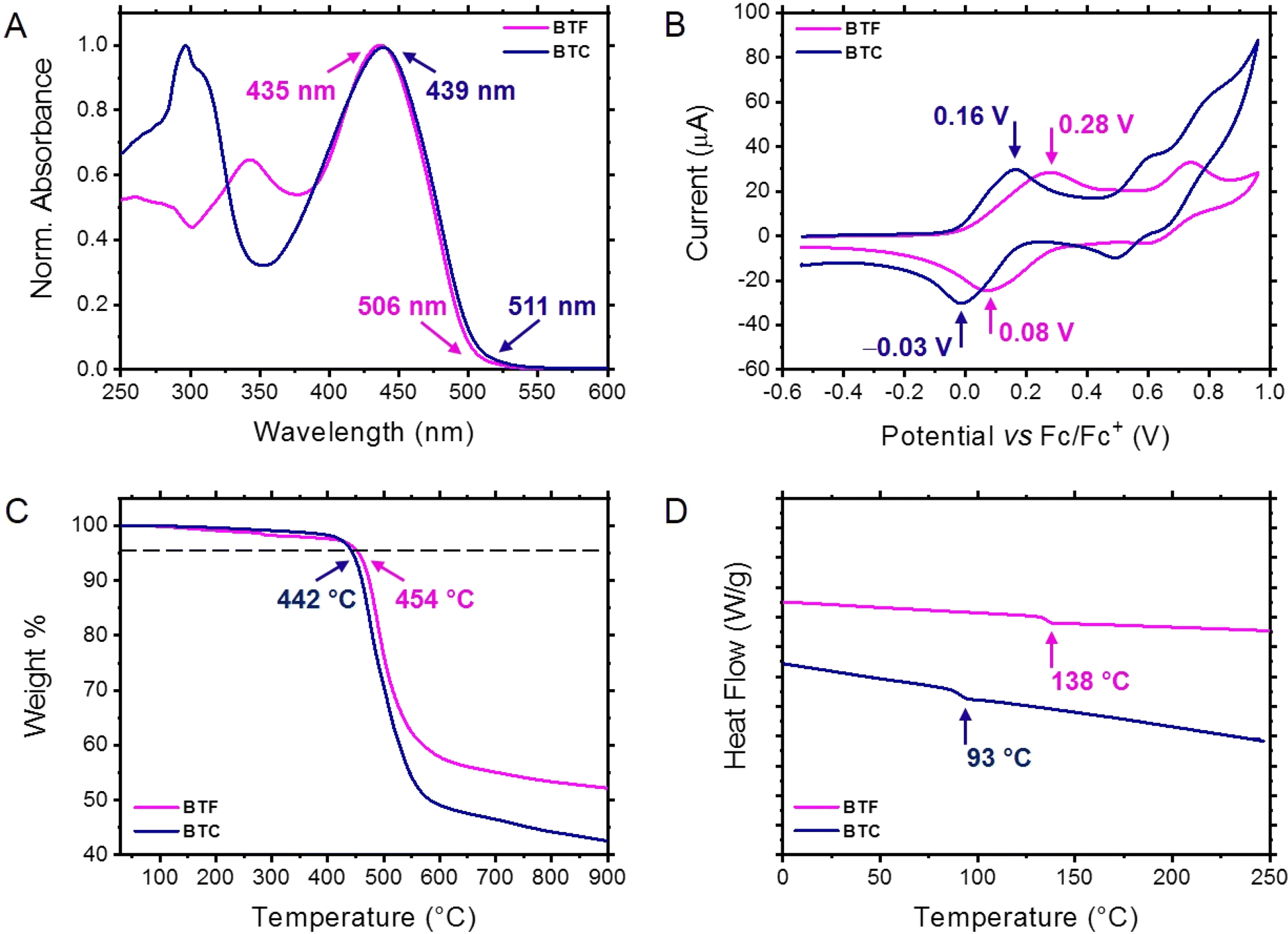 | ||
| Fig. 2 (A) UV-vis absorption spectra of BTF and BTC recorded in o-dichlorobenzene evidencing the numerical values of the absorption maxima and onsets. (B) Anodic CV measurements of BTF and BTC recorded in methylene chloride evidencing the potential values of the reversible oxidation peaks. (C) TGA plots of BTF and BTC highlighting the decomposition temperature at 5% weight loss. (D) DSC traces of BTF and BTC showing the glass transition temperatures. | ||
Optical, electrochemical, and thermal properties
The UV-vis absorption spectra of the two HTMs were recorded in methylene chloride (1.0 × 10−5 M). As displayed in Fig. 2A, both BTF and BTC showed similar absorption profiles with two distinct absorption bands: a higher energy absorption band (λmax = 342 nm and 296 nm for BTF and BTC, respectively) and a lower energy absorption band (λmax = 435 nm and 439 nm for BTF and BTC, respectively). According to our theoretical calculations, we can ascribe the higher energy absorption peaks to the localised π–π* electronic transitions of the aromatic components (bithiophene and peripheral units) and the lower energy absorption peaks to the intramolecular donor–acceptor charge transfer electronic transitions. The λmax redshift recorded for BTC with respect to BTF can be ascribed to the presence of electron-donating carbazole unit, increasing the charge transfer entity of the corresponding transition, as suggested by theoretical calculations (vide supra). Conversely, the fluorene-based chromophore induces the π-conjugation extension of the peripheral groups, promoting the bathochromic shift of the absorption shoulder with respect to the corresponding peak in BTC. The longer wavelength onsets of the relevant absorption spectra were used to calculate the optical band gaps of BTF and BTC (2.46 eV and 2.43 eV, respectively) calculated from the equation Egap = 1240/λ, where λ is taken as the onset of the absorption spectra (Fig. 2A).To evaluate the energy levels of the HTMs, we carried out electrochemical investigations by using cyclic voltammetry (CV). The cyclic voltammetries shown in Fig. 2B were recorded for HTM solutions (∼10−4 M in CH2Cl2) with 0.1 M tetrabutylammonium tetrafluoroborate as the supporting electrolyte at a scan rate of 0.1 V s−1. The two HTMs exhibited reversible oxidation events while no reduction peaks could be recorded in these conditions. This behaviour confirms that the designed molecules are more prone to hole transfer rather than to accept electrons. Upon calibration with ferrocene, the oxidation potential values (+0.18 V and +0.06 V for BTF and BTC, respectively) were established by using the half-wave potential of the anodic and cathodic peaks of the first oxidation event. From these values, the HOMO energy level of BTF and BTC were found to be −5.28 eV and −5.17 eV, respectively, resulting from the equation EHOMO = −(Eox + 5.10). The observed differences, in accordance with the theoretical calculations, can be ascribed to the electron-donating nature of the carbazole unit.
To test the thermal properties of the synthesised HTMs, we carried out thermogravimetric analyses (TGA) and differential scanning calorimetry (DSC) measurements under a nitrogen atmosphere. The TGA profiles revealed a remarkable thermal stability for the two compounds exhibiting decomposition temperatures (corresponding to a 5% weight loss) of 454 °C and 442 °C for BTF and BTC, respectively (Fig. 2C). Although the decomposition temperatures are reasonably suited for photovoltaic applications for both compounds, the higher thermal stability shown by BTF in comparison to BTC can be explained with the absence of linear aliphatic chains decorating its molecular structure.20
The existence of phase transitions within the thermal stability range of the two HTMs was evaluated by investigating the corresponding DSC traces. As reported in Fig. 2D, BTF and BTC showed glass transition events (138 °C and 93 °C, respectively), suggesting a more stable amorphous nature for the fluorene-based HTM. Again, the higher glass transition temperature (Tg) exhibited by BTF can be attributed to the cyclic functionality of the peripheral fluorene groups. These values are remarkably higher than that recorded for the analogous compound functionalised with the peripheral bis(4-methoxyphenyl)amino group (M102, Tg: 79 °C).21 Differently from Spiro-OMeTAD,22 the synthesised compounds are thus amorphous not exhibiting the characteristic transitions of the crystalline state. This behaviour can retard the long-term deterioration of the HTM film under the operating conditions.
Charge transfer processes and hole mobility
Fig. 3A shows the energy levels diagram including BTF, BTC and the benchmark Spiro-OMeTAD23 related to the other device components (TiO2, FTO, perovskite, and Au), evidencing the feasibility of the photogenerated charge pathway in multilayer photovoltaic devices. Based on the HOMO energy levels estimated by CV measurements, it can be deduced that these values are higher than the perovskite valence band (−5.65 eV) and both the hole extraction from the active layer and transport to Au electrode are feasible. The HOMO energy levels of BTF and BTC were lower than the gold work function, suggesting the rapid transfer of charge carriers from the perovskite to the relevant electrode. Moreover, the HOMO levels of BTF and BTC are more negative than Spiro-OMeTAD. Since the VOC values of PSCs mainly depend on the difference between the HOMO levels of the HTM and the quasi-Fermi level of the perovskite active layer, a higher VOC would be expected using BTF or BTC in comparison with the Spiro-OMeTAD reference as HTMs in PSCs.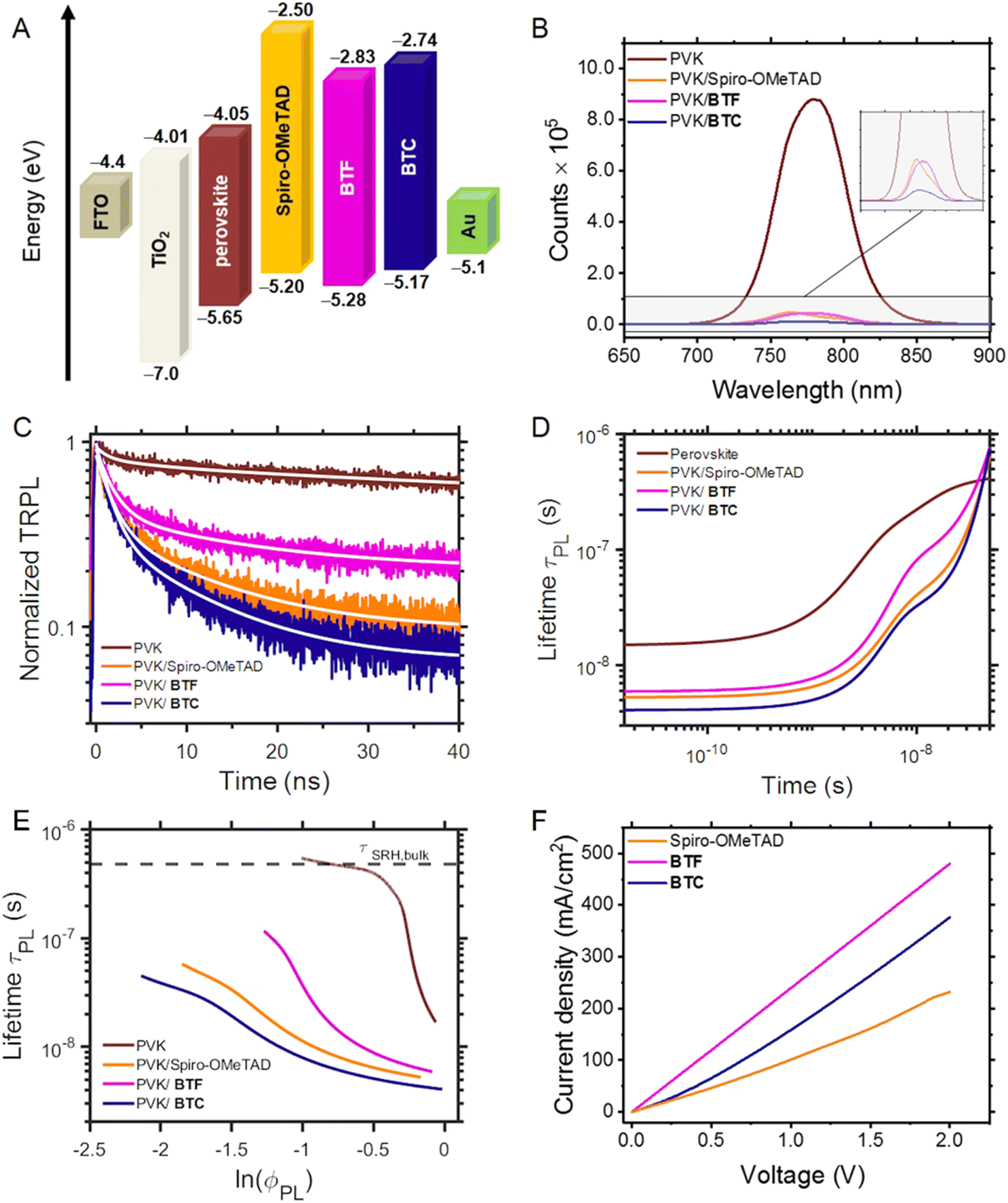 | ||
| Fig. 3 (A) Energy-level diagram for the tested hole transport material and other solar cell layers. (B) PL spectra of pure perovskite film on glass and glass/perovskite/HTM stacks, excited at 600 nm. (C) Time-resolved PL (TRPL) of decay curves of the samples excited 483 nm and monitored at 744 nm. The white lines represent exponential decay curves fitted to the data. (D) Differential lifetime τPL calculated from the fitted TRPL curves plotted as a function of time. (E) Differential lifetime as a function of the logarithm of TRPL intensity ln(ϕPL), serving as a relative measure for quasi-Fermi level splitting. The plateau of τPL for the pure perovskite sample was used to estimate bulk Shockley Read Hall lifetime τSRH,bulk. (F) J–V curves obtained in the space-charge limited current measurements. | ||
The LUMO energy levels of BTF and BTC were calculated by adding the value of their optical band gaps (vide supra) to the corresponding HOMO energy level. It was detected that these values are −2.83 eV and −2.74 eV for BTF and BTC, respectively, which are both remarkably higher than the energy level of the perovskite conduction band (−4.05 eV). As in the case of Spiro-OMeTAD, this energy configuration hampers the electron transfer from the perovskite conduction band to the HTM layer, thus optimising the charge separation at the perovskite/HTM interface.
We investigated the charge transfer process at the perovskite/HTM interface by analysing the entity of the photoluminescence (PL) quenching of the perovskite layer upon the HTM deposition atop. Fig. 3B shows steady-state PL of the perovskite films on which the two HTMs and the Spiro-OMeTAD benchmark are deposited. The PL intensity of both perovskite/BTF and perovskite/BTC bilayer showed a significant decrease in comparison with the pristine perovskite film, indicating the suppression of the radiative recombination between the photogenerated electrons and holes. In detail, the PL quenching efficiency was found to be 94.9% for BTF (and comparable to the value recorded for Spiro-OMeTAD), which resulted to be inferior with respect to that observed for BTC (98.6%). We attribute these interfacial properties to the more planar structure of BTF and BTC in comparison to that of Spiro-OMeTAD. The structural planarity, in fact, can favour the charge transfer at the perovskite/HTM interface owing to an approximately uniform distribution of electron density and the closer spatial proximity between the two entities. To explain the differences between BTF and BTC, we infer that defects caused by uncoordinated Pb2+ can be effectively passivated through coordination interactions by Lewis bases such as N and S atoms.24 Effectively, the additional heteroatoms of the peripheral groups in BTC are expected to reduce the interfacial potential barrier for the charge transfer providing passivation modes.25,26 However, these further surface interactions are insufficient to provide stability to the perovskite/HTM interface in devices (vide infra).
This interpretation was confirmed by analysing the time resolved PL (TRPL) of the proposed bilayers (Fig. 3C), which evidenced shorter excited state lifetimes for BTC in comparison with the other HTMs (BTF and Spiro-OMeTAD). As merely fitting the TRPL decay curves with one or multi exponential decay functions (narrow white lines in Fig. 3C) does not provide sufficient information on charge transfer kinetics, we analysed the decays in terms of differential lifetime with the following equation:
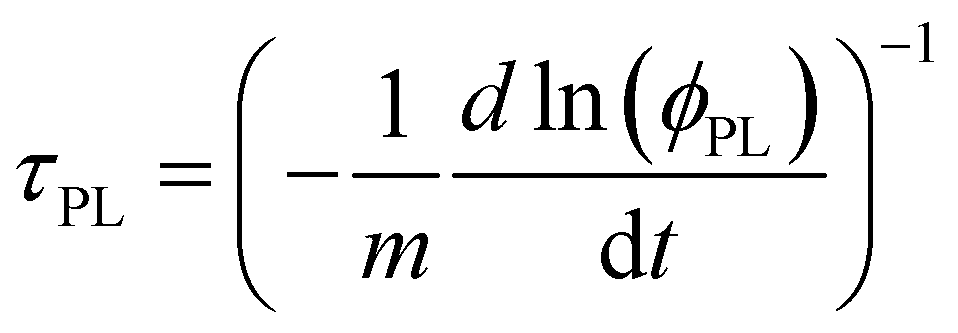 | (1) |
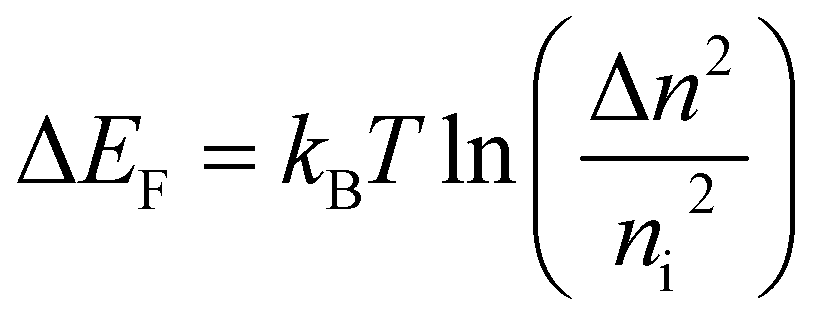 | (2) |
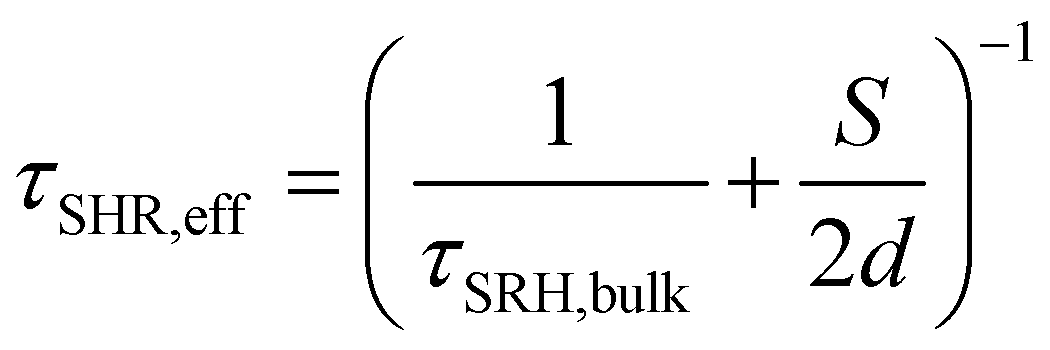 | (3) |
To determine the hole mobility of novel HTMs, hole-only devices were fabricated with the FTO/PEDOT:PSS/HTM/Au configuration. The space-charge-limited-current (SCLC) method was used for estimating the hole mobility. The mobility of the hole carriers was taken from the current density vs. voltage curve (Fig. 3F) and fitted to Mott–Gurney law, as described in ESI.† Here, the hole mobility of undoped Spiro-OMeTAD (4.28 × 10−4 cm2 V−1 s−1) was comparable to that of BTF (2.36 × 10−4 cm2 V−1 s−1) and BTC (1.21 × 10−4 cm2 V−1 s−1). However, doping with 4-tert-butylpyridine (tBP), as well as lithium bis(trifluoromethanesulfonyl)imide (Li-TFSI) and FK209 salts improved the hole mobility of Spiro-OMeTAD (1.02 × 10−3 cm2 V−1 s−1) compared to the two synthesised HTMs.
Photovoltaic device performances
We investigated the functionalisation effect of the bithiophene core on photovoltaic performances of BTF- and BTC-based PSCs fabricated with a planar n-i-p structure (FTO/TiO2/perovskite/HTM/Au) and an active area of 20 mm2 (Fig. S2†). The study on the concentration and spin rate optimization for the HTM deposition is reported in ESI (Fig. S3).† A comparison of the best J–V curves of PSCs based on Spiro-OMeTAD and undoped BTF and BTC along with the statistical distribution of the figures of merit concerning these devices is reported in Fig. 4A and B, respectively, while the corresponding photovoltaic parameters are summarised in Table 1.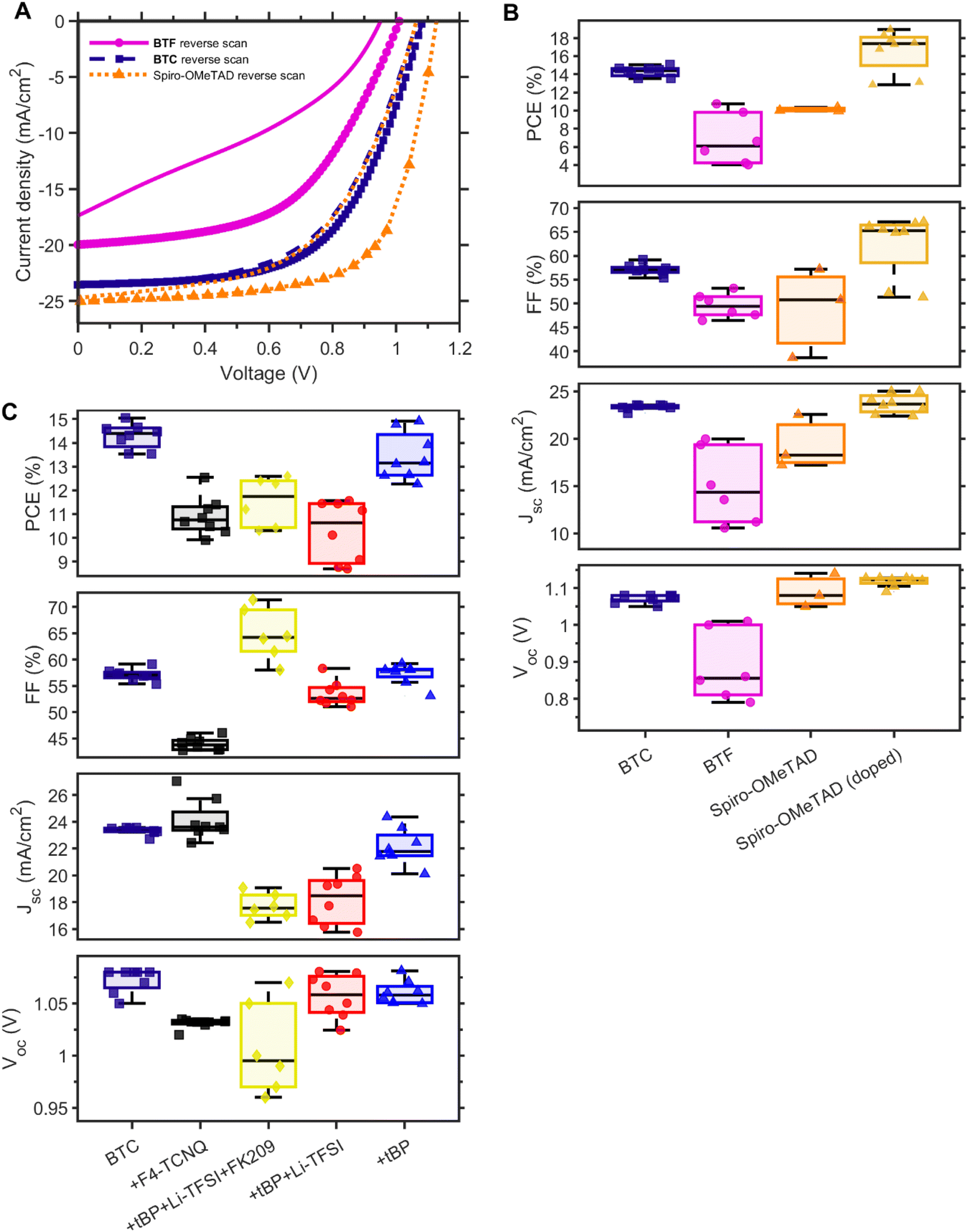 | ||
| Fig. 4 (A) Current density (J)–voltage (V) curves of champion devices employing BTF, BTC and doped Spiro-OMeTAD. Forward sweeps are plotted as lines without the markers. (B) Power conversion efficiency (PCE), fill factor (FF), short-circuit current density (JSC), and open-circuit voltage (VOC) of solar cell pixels employing BTF, BTC, Spiro-OMeTAD, and tBP:Li-TFSI:FK209-doped Spiro-OMeTAD. Measurements were conducted the day after fabrication. (C) Figures of merit of devices employing undoped BTC and BTC doped with F4-TCNQ as well as different combinations of tBP, Li-TFSI, and FK209. | ||
| HTM | Doping | V OC (mV) | J SC (mA cm−2) | FF (%) | PCE (%) at 1 day | PCE (%) at 401 days | |
|---|---|---|---|---|---|---|---|
| Undoped | Spiro-OMeTAD30 | 972 | 22.83 | 47.0 | 10.4 | ||
| BTF | 887 | 14.97 | 49.6 | 6.8 | |||
| BTC | 1073 | 23.32 | 57.1 | 14.3 | 7.9 | ||
| Doped | Spiro-OMeTAD | tBP:Li-TFSI:FK209 | 1073 | 23.13 | 66.7 | 16.5 | 12.8 |
| BTC | tBP:Li-TFSI:FK209 | 1007 | 17.70 | 64.8 | 11.5 | ||
| BTC | F4-TCNQ | 1043 | 26.54 | 44.1 | 10.5 | 7.1 | |
| BTC | tBP:Li-TFSI | 1008 | 22.78 | 42.1 | 8.1 | 11.8 | |
| BTC | tBP | 1060 | 25.67 | 57.3 | 13.4 | 13.8 |
In the case of undoped HTMs, the highest PCEs were obtained for BTC (14.3%), as the result of the higher JSC values (23.27 mA cm−2) and FF (56.5%), in comparison to both the BTF counterpart (6.8% PCE) and Spiro-OMeTAD reference (10.4% PCE). The high FFs observed in the BTC case can be attributed to the fast hole extraction, as evidenced by the highest surface recombination velocity observed for the perovskite/BTC sample (refer to Fig. 3E). The high JSC values for BTC-containing devices align well with the highest PL quenching efficiency of 98.6% achieved by BTC, compared to other HTMs, BTF and Spiro-OMeTAD (refer to Fig. 3b). This observation further suggests that the introduction of a linear alkyl chain in BTC does not disturb its charge collection properties. Furthermore, a better film quality can probably be achieved in comparison to BTF (Fig. S4†), which showed lower PCE, notwithstanding its superior hole mobility. As a support of this observation, we can observe that also the FF in the device with BTF is reduced to 49.6%, which can be attributed to a nonuniform coverage of the perovskite layer by the HTM and its low hole mobility. The non-uniform coverage might indicate sub-optimal interface formation between the perovskite and BTF layers. This could explain the significantly low VOC values compared to the other two types of devices, despite the suitable band alignment for hole transfer (BTF ≥ BTC > Spiro-OMeTAD). Furthermore, the main parameter leading to the lower performance of the undoped Spiro-OMeTAD reference in comparison to BTC is the FF, notwithstanding the good solubility of the benchmark in the solvent used for the deposition. The introduction of dopants did not impact on the overall device performance of BTC (Table 1 and Fig. 4C). The average PCE value (11.5%) obtained by using the same composition of dopants (tBP:Li-TFSI:FK209) implemented in Spiro-OMeTAD was found to be surprisingly remarkably lower than that recorded for the benchmark (16.5%). Probably, this complex formulation of dopants damages the ordered intermolecular packing of the HTM film (responsible for the efficient charge transport within the device) counterbalancing the ordinary positive effects of doping on the device performances. This result motivated us to explore simpler doping formulations, including F4-TCNQ, tBP:Li-TFSI, and the sole tBP. In these cases, however, the observed PCEs recorded at ‘day 1’ were found to be lower than the performance exhibited by the undoped BTC-based PSCs (Table 1). Nevertheless, the average efficiencies of tBP-doped solar cells (13.4%) approached those of the undoped counterparts, confirming that the incorporation of additives/dopants disturbs the ordered packing of the HTM film.
Device stability
Evaluating the long-term stability of PSCs is likely the most important factor in determining their value for large-scale applications.31 Therefore, we initially assessed the long-term stability of PSCs containing undoped BTF and BTC, using the behaviour of doped Spiro-OMeTAD as a reference under the same storage conditions (unencapsulated devices kept under 21 °C and 10–20% relative humidity). The evolution of their average figures of merit as a function of time is shown in Fig. 5, while their normalisation is reported in Fig. S5.† Considering that the studied HTMs (BTF, BTC and Spiro-OMeTAD) have a hydrophobic nature, they act as a protective layer on the perovskite material isolating it from humidity, which is believed to be mainly responsible for the performance degradation of PCSs over time, damaging the perovskite crystalline structure. Therefore, given these premises, the evolution of efficiency over time must be ascribed to alterations of the perovskite/HTM interface and/or to the use of hygroscopic dopants. Focusing exclusively on performance, we observed that the average PCE of the undoped BTC-based PSCs slightly increased after 11 days (PCE = 14.2%) and reached the maximum value (PCE = 15.4%) after 6 days. This efficiency effectively rivals that of the doped Spiro-OMeTAD-based PSCs (PCE = 15.5%), which slightly deteriorated after the same storage period (Fig. 5A). It is reasonable to suppose that the gradual oxygenation of the undoped HTM layers over time can promote the photo-oxidation of the material with the formation of the corresponding radical ions.32 These species (BTC˙+/O2˙−) are expected to “dope” the HTM layer and to effectively improve its charge transport properties,32 thus raising the overall device performances in the initial stages of the long-term stability evaluation. By contrast, this initial PCE improvement was not observed in the case of BTF, due to its higher oxidation potential in comparison to the BTC counterpart (vide supra), probably inhibiting the formation of the corresponding radical cations.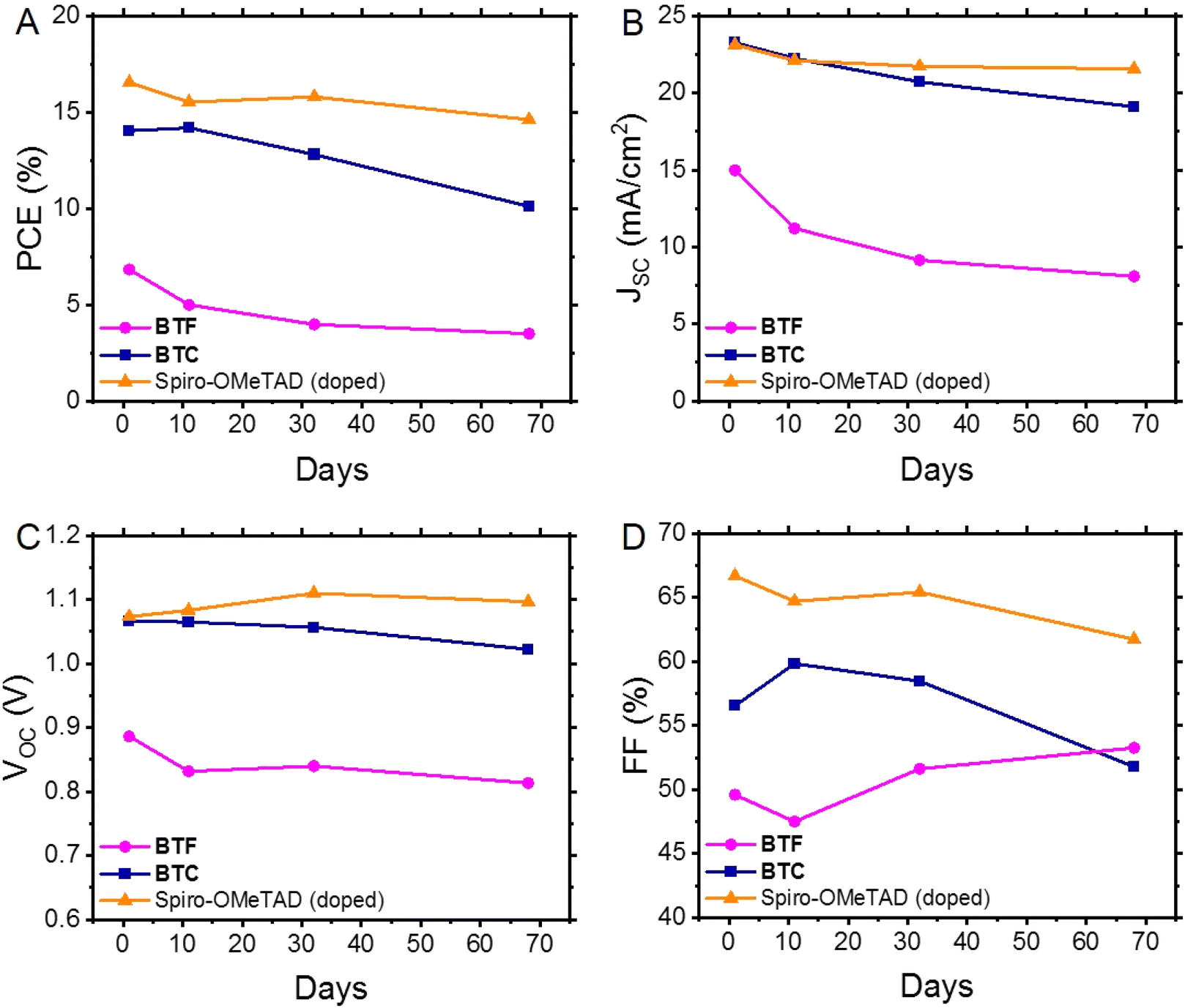 | ||
| Fig. 5 Evolution over time of the figures of merit in undoped BTF- and BTC-based PSCs in comparison to doped Spiro-OMeTAD benchmark: (A) power conversion efficiency, (B) short-circuit current density, (C) open circuit voltage, and (D) fill factor. | ||
At the end of our long-term investigations of such configured devices, the PSCs incorporated with doped Spiro-OMeTAD retained most of the initial PCE (88.5%) after 68 days, while the corresponding parameter of undoped BTC and BTF significantly deteriorated (72.1% and 51.5%, respectively) after the same period. A deep analysis on the behaviour of the individual figures of merit over time (Fig. 5B–D) revealed that the PCE drop observed for BTC is mainly caused by the JSC and FF deterioration. This effect was even more pronounced than that observed in the case of doped Spiro-OMeTAD. The well-known hygroscopicity of the additive/dopant formulation reasonably explains the instability of doped Spiro-OMeTAD-based devices, while we verified the PCE deterioration of the BTC-based devices, notwithstanding the progressive oxygen incorporation into the HTM layer guaranteeing the doping supply. This behaviour can be attributed to the reduced number of methoxy groups that can efficiently passivate the coordinatively unsaturated lead ions at the perovskite/HTM interface,13,33–35 reducing the adhesion capacities of the organic compound to the perovskite surface during the device functioning. In fact, favouring the charge recombination at the perovskite/HTM interface negatively impacts the JSC values.36 To support this interpretation, we exposed a BTC solution to a 300 W Xe-lamp, thus verifying the initial formation of the corresponding radical cations (Fig. S6†) during the simple irradiation required to acquire the basic device figures of merit. At the same time, a more prolonged treatment probably caused a deterioration of the material, that could negatively impact the device stability containing undoped BTC films under real application conditions. The same interpretation can be used to explain to instability of BTF-based devices, which is amplified by the low resilience of the fluorene units toward the C-9 oxidation.37 Next, we exclusively analysed the long-term behaviour of the differently doped BTC-based PSCs owing to their better performances in undoped devices (Fig. 6 and S7†). Since the PCE exhibited by the BTC-based PSCs doped with the conventional formulation used for Spiro-OMeTAD was much lower in comparison to the benchmark, we decided not to investigate the long-term stability of these devices. Furthermore, the use of the conventional formulation of dopants (tBP:Li-TFSI:FK209) in Spiro-OMeTAD determined a rapid deterioration (T80 < 100 days) of the corresponding devices under the applied storage conditions. Despite the low average initial PCE value (10.5%) observed for F4-TCNQ-doped PSCs, the study on the long-term stability of these devices is particularly attractive, because the chemical doping agent (F4-TCNQ) is not hygroscopic unlike the Li-TFSI additive. Therefore, by monitoring its trend, we can exclude the role of moisture captured by the dopant in humid conditions on the long-term performances of the corresponding devices. We observed an increase of the PCE values in the first stage of the temporal investigations reaching the value of 12.0%, after that a drastic drop of the efficiencies was recorded (7.1%), as evident in Fig. 6A. The same tendency was detected for the champion device (Table 2). We analysed these results by breaking down the role of each figure of merit on the resulting efficiency. While a remarkable decrease in the average JSC was observed, especially in the initial stages (Fig. 6B), the average VOC remained relatively stable over time (Fig. 6C). A similar trend was observed for the tBP:Li-TFSI-doped devices, where a significant increase in PCE was evident in the initial stages of aging (Fig. 6A), primarily due to an enhancement in VOC and FF (Fig. 6C and D), while JSC remained substantially constant over time (Fig. 6B). However, these same figures of merit (VOC and FF) can also be considered responsible for the deterioration of long-term performance. The observed average PCE passed from 15.6% recorded at the 28th day of aging to 11.8% after 401 days. The same trend was observed for the tBP:Li-TFSI-doped champion device (Table 2). In the case of BTC-based PSCs solely employing tBP as the additive, the effects of self-doping are obviously attenuated with respect to the case including the presence of lithium salts. In fact, a limited PCE improvement was observed in the initial stages of aging, which, however, remained substantially constant up to 401 days. Apart from the initial drop of JSC counterbalance by an improvement of VOC, it is important to note the preservation of the FF values over time not observed for the other devices (Fig. 6B–D). Furthermore, these results are impressive in comparison to the behaviour exhibited by similarly engineered HTMs.35
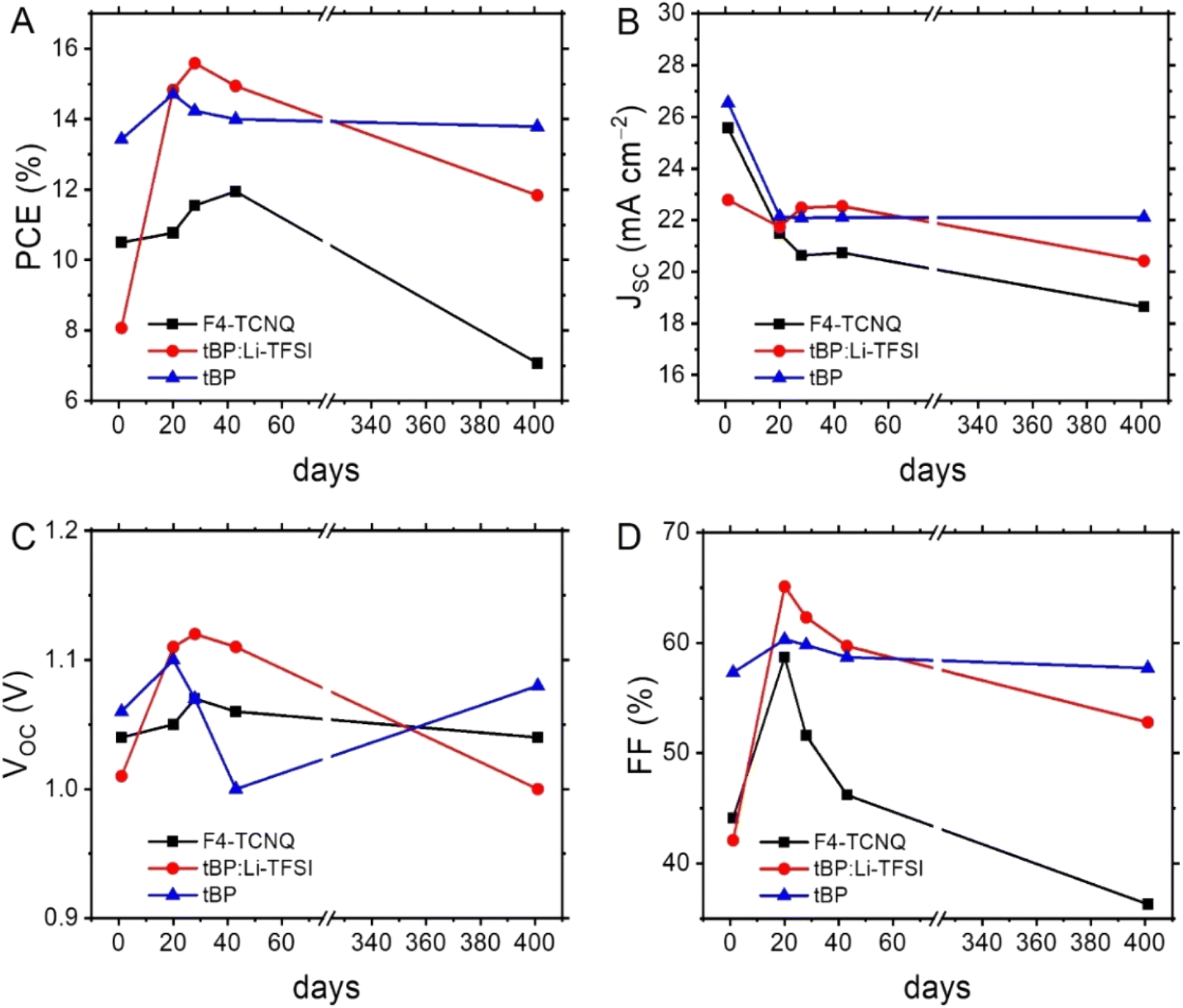 | ||
| Fig. 6 Evolution over time of the figures of merit in differently doped BTC-based PSCs: (A) PCE, (B) JSC, (C) VOC, and (D) FF. | ||
| HTM (champion) | Doping | Day 1 PCE (%) | Day 20 PCE (%) | Day 43 PCE (%) | Day 401 PCE (%) |
|---|---|---|---|---|---|
| BTC | F4-TCNQ | 12.5 | 13.8 | 14.3 | 8.3 |
| BTC | tBP:Li-TFSI | 10.1 | 17.7 | 17.1 | 14.7 |
| BTC | tBP | 14.8 | 16.6 | 15.5 | 15.4 |
Generally, tBP is introduced in the additive formulation for the HTM deposition due to the very low solubility of Li-TFSI in chlorobenzene, but its presence seems to be detrimental for the device stability.38,39 At the same time, the main role of Li-TFSI in the HTM solution is to provide the counterions for the oxidised HTM to form stable complexes. We infer that in the case of tBP-modified BTC-based devices, the additive can efficiently passivate the surface defects of the perovskite layer40–44 favouring the durable adhesion of the HTM propaedeutic to the long-term stability of the corresponding devices. Moreover, tBP is the least expensive among the additives generally employed in PSCs, thus supporting the large-scale development of this technology in terms of sustainability.
To assess the impact on operational stability, we conducted accelerated aging tests for devices employing undoped BTC and BTC with tBP-additive, as well as doped Spiro-OMeTAD reference. The maximum power point of the samples was tracked under continuous 1-sun illumination and 65 °C heating in an intrinsic nitrogen atmosphere, corresponding to the ISOS-L-2I protocol.45 The normalised results obtained from 8 pixels of each type of sample are presented in Fig. S8.† All samples exhibited rapid initial degradation, but we observe superior stability for BTC-based samples and even further improvement with the addition of tBP.
Conclusions
A straightforward route (not involving organometallic cross-coupling reactions) was applied to prepare nonconventional HTMs (BTF and BTC) based on a bithiophene core decorated with unsymmetrical triarylamine groups partially lacking methoxy groups. The impact of different peripheral substitution was analysed using theoretical, optical, electrochemical, and photophysical characterizations. The two HTMs were initially tested in dopant-free n-i-p PSCs to evaluate their performance and long-term behaviour. Despite enhancing hole extraction and transport at the perovskite/HTM interface in comparison to the Spiro-OMeTAD reference, BTC (which performed better than the BTF counterpart) did not achieve exceptional results as an undoped HTM in PSCs (PCE = 14.0% vs. 16.5% of the doped Spiro-OMeTAD reference). Moreover, their efficiencies rapidly degraded over time (T80: ∼57 days), likely due to the deterioration of the perovskite/HTM interface. The introduction of additives was systematically investigated for their impact on the photovoltaic performance and stability of BTC-based PSCs. It was observed that HTM photo-oxidation is responsible for the remarkable improvement of the efficiencies in the initial aging stages (PCE up to 17.7%). While the coexistence of tBP and Li-TFSI was detrimental to long-term device stability, the unique structure of BTC could durably tolerate the presence of tBP. This not only slightly increased performance (PCE = 14.8%) but also remarkably improved device resilience to ambient exposure (PCE = 15.4% after 401 days). These results indicate that suitably engineered HTMs can compete with the expensive Spiro-OMeTAD benchmark in both performance and production costs. Additionally, selecting the proper HTM/additive system can be a promising strategy to preserve long-term device stability.Data availability
The authors confirm that the data supporting the findings of this study are available within the article [and/or its ESI†].Conflicts of interest
There are no conflicts to declare.Acknowledgements
P. M. thanks the financial support of the Finnish Cultural Foundation (grant 00240880, Kalle & Dagmar Välimaa fund). G. K. G. acknowledges Tampere Institute for Advanced Study for postdoctoral funding. P. V. thanks the Research Council of Finland, Decision No. 347772 and Jane and Aatos Erkko foundation (SOL-TECH project). The work is part of the Research Council of Finland Flagship Programme, Photonics Research and Innovation (PREIN), decision number 346511. PRIN project SUPERNANO (code 2022C7Z2RA) funded by the Italian Ministry for Universities and Research (MUR), PRIN project BOMBCAT (code P202253ANE) funded by the Italian Ministry for Universities under the National Recovery and Resilience Plan (NRRP), and the Project “Network 4 Energy Sustainable Transition – NEST” code PE0000021, funded under NRRP, Mission 4 Component 2 Investment 1.3 – Concession Decree No. 1561 of 11.10.2022 of MUR by the European Union – Next Generation EU are gratefully acknowledged.References
- M. M. Lee, J. Teuscher, T. Miyasaka, T. N. Murakami and H. J. Snaith, Science, 2012, 338, 643 CrossRef CAS PubMed
.
- L. Tian, F. Wen, W. Zhang, H. Zhang, H. Yu, P. Lin, X. Liu, S. Zhou, X. Zhou, Y. Jiang, T. Chen, Z. Ma, M. Zhang and Y. Huang, ACS Appl. Mater. Interfaces, 2020, 12, 49648 CrossRef CAS PubMed
- W. Hu, W. Zhou, X. Lei, P. Zhou, M. Zhang, T. Chen, H. Zeng, J. Zhu, S. Dai, S. Yang and S. Yang, Adv. Mater., 2019, 31, 1806095 CrossRef PubMed
- P. Murugan, T. Hu, X. Hu and Y. Chen, J. Mater. Chem. A, 2022, 10, 5044 RSC
- L. Nakka, Y. Cheng, A. G. Aberle and F. Lin, Adv. Energy Sustainability Res., 2022, 3, 2200045 CrossRef CAS
- E. Kasparavicius, M. Franckevicius, V. Malinauskiene, K. Genevicius, V. Getautis and T. Malinauskas, ACS Appl. Energy Mater., 2021, 4, 13696 CrossRef CAS
- W. Wang, J. Zhou and W. Tang, J. Mater. Chem. A, 2022, 10, 1150 RSC
- J. Urieta-Mora, I. García-Benito, A. Molina-Ontoria and N. Martín, Chem. Soc. Rev., 2018, 47, 8541 RSC
- P. Mahajan, B. Padha, S. Verma, V. Gupta, R. Datt, W. C. Tsoi, S. Satapathi and S. Arya, J. Energy Chem., 2022, 68, 330 CrossRef CAS
- A. Farokhi, H. Shahroosvand, G. D. Monache, M. Pilkington and M. K. Nazeeruddin, Chem. Soc. Rev., 2022, 51, 5974 RSC
- M. Sasikumar, G. Maddala, M. Ambapuram, M. Subburu, J. R. Vaidya, S. N. Babu, P. Chetti, R. Mitty and S. Pola, Sustainable Energy Fuels, 2020, 4, 4754 RSC
- M. Yan, Y. Wang, C. Chen, X. Ding, M. Zhai, Z. Xia, C. Liu, W. Zhang and M. Cheng, J. Phys. Chem. Lett., 2024, 15, 11119 CrossRef CAS PubMed
- P. Mäkinen, F. Fasulo, M. Liu, G. K. Grandhi, D. Conelli, B. Al-Anesi, H. Ali-Löytty, K. Lahtonen, S. Toikkonen, G. P. Suranna, A. B. Muñoz-García, M. Pavone, R. Grisorio and P. Vivo, Chem. Mater., 2023, 35, 2975 CrossRef
- X. Zhang, X. Liu, F. F. Tirani, B. Ding, J. Chen, G. Rahim, M. Han, K. Zhang, Y. Zhou, H. Quan, B. Li, W. Du, K. G. Brooks, S. Dai, Z. Fei, A. M. Asiri, P. J. Dyson, M. K. Nazeeruddin and Y. Ding, Angew. Chem., 2024, 136, e202320152 CrossRef
- K. Radhakrishna, S. B. Manjunath, D. Devadiga, R. Chetri and A. T. Nagaraja, ACS Appl. Energy Mater., 2023, 6, 3635 CrossRef CAS
- N. J. Jeon, H. Na, E. H. Jung, T.-Y. Yang, Y. G. Lee, G. Kim, H.-W. Shin, S. Il Seok, J. Lee and J. Seo, Nat. Energy, 2018, 3, 682 CrossRef CAS
- F. Wu, J. Liu, G. Wang, Q. Song and L. Zhu, Chem.–Eur. J., 2016, 22, 16636 CrossRef CAS PubMed
- M. L. Petrus, T. Bein, T. J. Dingemans and P. Docampo, J. Mater. Chem. A, 2015, 3, 12159 RSC
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed
- R. Grisorio, C. Piliego, P. Cosma, P. Fini, P. Mastrorilli, G. Gigli, G. P. Suranna and C. F. Nobile, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 2093 CrossRef CAS
- Y. Wu, Z. Wang, M. Liang, H. Cheng, M. Li, L. Liu, B. Wang, J. Wu, R. Prasad Ghimire, X. Wang, Z. Sun, S. Xue and Q. Qiao, ACS Appl. Mater. Interfaces, 2018, 10, 17883 CrossRef CAS
- T. Malinauskas, D. Tomkute-Luksiene, R. Sens, M. Daskeviciene, R. Send, H. Wonneberger, V. Jankauskas, I. Bruder and V. Getautis, ACS Appl. Mater. Interfaces, 2015, 7, 11107 CrossRef CAS
- L. Nakka, Y. Cheng, A. G. Aberle and F. Lin, Adv. Energy Sustainability Res., 2022, 3, 2200045 CrossRef CAS
- Z. Li, X. Sun, Z. Li, X. Yu, X. Wu, C. Zhong, D. Liu, D. Lei, A. K. Y. Jen and Z. Zhu, Angew. Chem., Int. Ed., 2021, 60, 7227 CrossRef
- B. X. Zhao, C. Yao, K. Gu, T. Liu, Y. Xia and Y.-L. Loo, Energy Environ. Sci., 2020, 13, 4334 RSC
- Z. J. Li, B. H. Jo, S. J. Hwang, T. H. Kim, S. Somasundaram, E. Kamaraj, J. Bang, T. K. Ahn, S. Park and H. J. Park, Adv. Sci., 2019, 6, 1802163 CrossRef
- L. Krückemeier, B. Krogmeier, Z. Liu, U. Rau and T. Kirchartz, Adv. Energy Mater., 2021, 11, 2003489 CrossRef
- M. Liu, S. Dahlström, C. Ahläng, S. Wilken, A. Degterev, A. Matuhina, M. Hadadian, M. Markkanen, K. Aitola, A. Kamppinen, J. Deska, O. Mangs, M. Nyman, P. D. Lund, J.-H. Smått, R. Österbacka and P. Vivo, J. Mater. Chem. A, 2022, 10, 11721 RSC
- M. Liu, M. Endo, A. Shimazaki, A. Wakamiya and Y. Tachibana, ACS Appl. Energy Mater., 2018, 1, 3722 CrossRef CAS
- T. Braukyla, R. Xia, T. Malinauskas, M. Daskeviciene, A. Magomedov, E. Kamarauskas, V. Jankauskas, Z. Fei, C. Roldán-Carmona, C. Momblona, M. K. Nazeeruddin, P. J. Dyson and V. Getautis, Sol. RRL, 2019, 3, 1900224 CrossRef
- C. C. Boyd, R. Cheacharoen, T. Leijtens and M. D. McGehee, Chem. Rev., 2019, 119, 3418 CrossRef CAS PubMed
- S. Wang, W. Yuan and Y. S. Meng, ACS Appl. Mater. Interfaces, 2015, 7, 24791 CrossRef CAS
- Y. Guo, L. He, J. Guo, Y. Guo, F. Zhang, L. Wang, H. Yang, C. Xiao, Y. Liu, Y. Chen, Z. Yao and L. Sun, Angew. Chem., Int. Ed., 2022, 61, e202114341 CrossRef CAS
-
(a) L. Wan, W. Zhang, S. Fu, L. Chen, Y. Wang, Z. Xue, Y. Tao, W. Zhang, W. Song and J. Fang, J. Mater. Chem. A, 2020, 8, 6517 RSC
- H. Yao, T. Wu, B. Wu, H. Zhang, Z. Wang, Z. Sun, S. Xue, Y. Hua and M. Liang, J. Mater. Chem. A, 2021, 9, 8598 RSC
- B. Yang, J. Suo, F. Di Giacomo, S. Olthof, D. Bogachuk, Y. Kim, X. Sun, L. Wagner, F. Fu, S. M. Zakeeruddin, A. Hinsch, M. Grätzel, A. Di Carlo and A. Hagfeldt, ACS Energy Lett., 2021, 6, 3916 CrossRef CAS
- R. Grisorio, G. Allegretta, P. Mastrorilli and G. P. Suranna, Macromolecules, 2011, 20, 7977 CrossRef
- X. Liang, Y. Ming, S.-H. Lee, G. Fu, S.-U. Lee, T.-I. Kim, H. Zhang and N.-G. Park, ACS Appl. Mater. Interfaces, 2024, 16, 32147 CrossRef CAS
- J. Zhang, T. Zhang, L. Jiang, U. Bach and Y.-B. Cheng, ACS Energy Lett., 2018, 3, 1677 CrossRef CAS
- S. Wang, M. Sina, P. Parikh, T. Uekert, B. Shahbazian, A. Devaraj and Y. S. Meng, Nano Lett., 2016, 16, 5594 CrossRef CAS
- S. N. Habisreutinger, N. K. Noel, H. J. Snaith and R. J. Nicholas, Adv. Energy Mater., 2017, 7, 1601079 CrossRef
- N. K. Noel, A. Abate, S. D. Stranks, E. S. Parrott, V. M. Burlakov, A. Goriely and H. J. Snaith, ACS Nano, 2014, 10, 9815 CrossRef PubMed
- S. Song, E. Young Park, B. Soo Ma, D. J. Kim and H. H. Park, Adv. Energy Mater., 2021, 11, 2003382 CrossRef CAS
- Y. Y. Kim, S. S. Shin, N. J. Jeon, T.-S. Kim and J. Seo, Adv. Energy Mater., 2021, 11, 2003382 CrossRef
- M. V. Khenkin, E. A. Katz, A. Abate, G. Bardizza, J. J. Berry, C. Brabec, F. Brunetti, V. Bulović, Q. Burlingame, A. Di Carlo, R. Cheacharoen, Y.-B. Cheng, A. Colsmann, S. Cros, K. Domanski, M. Dusza, C. J. Fell, S. R. Forrest, Y. Galagan, D. Di Girolamo, M. Grätzel, A. Hagfeldt, E. von Hauff, H. Hoppe, J. Kettle, H. Köbler, M. S. Leite, S. Liu, Y.-L. Loo, J. M. Luther, C.-Q. Ma, M. Madsen, M. Manceau, M. Matheron, M. McGehee, R. Meitzner, M. K. Nazeeruddin, A. F. Nogueira, Ç. Odabaşı, A. Osherov, N.-G. Park, M. O. Reese, F. De Rossi, M. Saliba, U. S. Schubert, H. J. Snaith, S. D. Stranks, W. Tress, P. A. Troshin, V. Turkovic, S. Veenstra, I. Visoly-Fisher, A. Walsh, T. Watson, H. Xie, R. Yıldırım, S. M. Zakeeruddin, K. Zhu and M. Lira-Cantu, Nat. Energy, 2020, 5, 35 CrossRef
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4se01356e |
| ‡ These authors contribute equally. |
| This journal is © The Royal Society of Chemistry 2025 |