
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLayered ammonium metal phosphate based heterostructure with phosphate–sulfide interfacial synergy for efficient oxygen evolution and urea oxidation reactions†
Deepak Rajaram
Patil
a,
Santosh
Patil
 b,
Harish S.
Chavan
a,
Ah-yeong
Lee
a and
Kiyoung
Lee
*a
b,
Harish S.
Chavan
a,
Ah-yeong
Lee
a and
Kiyoung
Lee
*a
aDepartment of Chemistry and Chemical Engineering, Inha University, 100 Inha-ro, Michuhol-gu, 22212 Incheon, Republic of Korea. E-mail: kiyoung@inha.ac.kr
bDr Vishwanath Karad MIT World Peace University (MIT-WPU), Kothrud, Pune, 411038, India
First published on 14th February 2025
Abstract
This study unveils a highly efficient electrocatalyst based on hydrated ammonium metal phosphates (NH4MPO4·H2O) with a layered crystal structure and expanded interlayer spacing, facilitating rapid electron and ion transport for advanced oxygen evolution reaction (OER) applications. Addressing inherent limitations in conductivity and electroactive surface area, we engineered a heterostructured electrocatalyst by combining NH4NiPO4·H2O with CdIn2S4 and in situ formed Ni3S2 on nickel foam (NF) through a two-step hydrothermal process. The resulting NH4NiPO4·H2O/CdIn2S4/Ni3S2 (NPO/CINS) system leverages phosphate–sulfide interfacial interactions, significantly enhancing catalytic performance. Electrochemical tests reveal impressive OER and urea oxidation reaction (UOR) activities, achieving low overpotentials of 245 mV and 1.26 V at 10 mA cm−2, respectively. The obtained exceptional UOR efficiency exceeds that of previously reported oxide and sulfide-based heterostructure electrocatalysts. The NPO/CINS heterostructure demonstrates remarkable stability towards OER, with only 2% degradation over 65 hours of continuous operation, affirming its durability for high-performance applications. This work emphasizes the power of synergistic interfacial bonding, optimized electron transfer, and strategic structural design, positioning the NPO/CINS heterostructure as a pioneering catalyst for scalable energy solutions.
1. Introduction
Globally, hydrogen (H2) stands as a clean, sustainable fuel with transformative potential to address the pressing challenges of climate change and the urgent transition from finite fossil fuels to renewable energy sources.1 To date, materials such as platinum (Pt), iridium oxide (IrO2), and ruthenium oxide (RuO2) have set benchmarks as superior electrocatalysts for the hydrogen evolution reaction (HER) and the oxygen evolution reaction (OER), respectively.2–4 However, water electrolysis faces two significant challenges: (i) the high costs of these advanced catalysts and (ii) the inherently slow reaction kinetics of OER at the anode, which contribute to notable efficiency losses.5,6 In response, the pursuit of alternative anodic oxidation reactions with lower thermodynamic potentials has gained considerable attention as a route toward energy-efficient hydrogen production. A promising candidate is the urea oxidation reaction (UOR), which, with a notably lower thermodynamic potential (0.37 V vs. RHE) than OER (1.23 V vs. RHE), positions UOR-coupled electrolysis as a groundbreaking method to enhance hydrogen production efficiency.7,8 Despite this, UOR's six-electron transfer process (CO(NH2)2 + 6OH− → N2 + 5H2O + CO2 + 6e−) is hindered by slow kinetics, posing a critical challenge. To overcome these limitations, the development of bifunctional electrocatalysts that can drive both OER and UOR efficiently is essential for achieving low-energy, sustainable water splitting and advancing hydrogen production toward industrial feasibility.An extensive research effort has been directed towards the development of inexpensive and efficient electrocatalysts. Transition metal catalysts particularly Ni-based phosphides, oxides, sulfides and selenides hold promises for electrocatalysts owing to their low cost, intrinsic catalytic activity and chemical stability.9–16 Metal phosphates have emerged as a widely studied alternative to conventional oxygen evolution reaction (OER) catalysts, addressing the high cost and scarcity of benchmark catalysts.17,18 Their open framework structure facilitates both water oxidation and adsorption, while also acting as a proton acceptor during the oxidation of metal atoms. Of particular interest are hydrated ammonium metal phosphates (NH4MPO4·H2O), which have garnered considerable attention in catalysis and energy storage due to their exceptional structural features that promote rapid electron and ion transport.19,20 NH4MPO4·H2O, with its layered crystal structure and relatively large interlayer spacing, has demonstrated significant potential for enhancing OER activity. This distinct architecture creates efficient pathways for ion diffusion, accelerating electrochemical reactions and making these materials particularly advantageous for high-performance applications such as energy storage and electrocatalytic urea decomposition. However, despite these promising characteristics, ammonium metal phosphates still exhibit low intrinsic OER/UOR activity, largely due to poor electrical conductivity, limited electroactive surface area, and weak interfacial charge transfer.19,20 Enhancing these properties is key to unlocking the full catalytic potential of these materials, driving them toward superior performance in advanced energy applications.
One of the effective way of boosting the catalytic efficiency of electrocatalysts is to create heterostructures with additional catalysts.21,22 The heterostructure formation leads to leveraging synergistic effects, tuning electronic structures, increasing multiple active sites, improving mass transport properties, and enhancing stability.23,24 Specifically, transition metal sulfides (TMSs) display inherent metallic characteristics attributed to the uninterrupted network of M–M bonds in their structures.23,24 CdIn2S4 is known to be one of the best TMSs for photoelectrochemical water splitting due to its exceptional catalytic prowess in both the HER and OER, thereby enabling highly efficient water splitting.25 Furthermore, when integrated with supplementary catalysts or semiconductor materials, CdIn2S4 demonstrates synergistic interactions that amplify the overall efficiency of the water-splitting mechanism.26 Moreover, it exhibits robust stability under electrocatalytic environments, ensuring sustained performance over prolonged periods.
Herein, we report a facile two-step hydrothermal synthesis to grow binder-free NH4NiPO4·H2O/CdIn2S4/NF electrocatalyst and its assessment to electrocatalytic HER and OER reactions. Firstly, Ni-foam (NF) itself was utilized as the source of nickel, enabling uniform growth of NH4NiPO4·H2O nanostructures. The direct transformation of the nickel foam surface into a catalyst material enhances electrical conductivity, minimises ionic diffusion resistance, facilitates efficient electrolyte penetration, and maximizes the electroactive surface area. A unique combination of NH4NiPO4·H2O and CdIn2S4 is intended to leverage the strengths of both materials. The hydrothermal synthesis of NH4NiPO4·H2O/CdIn2S4 electrocatalyst resulted in in situ formation of new catalytic material Ni3S2 which further improved catalytic OER activity through a hybrid nanostructure. Ni3S2 has emerged as an appealing bi-functional catalyst that facilitates both the HER and OER in alkaline electrolysis, demonstrating its potential for practical and scalable use in electrochemical devices.22 While several previous studies have demonstrated the electrocatalytic activity of Ni-based NH4MPO4·H2O and/or sulfides as electrocatalysts, a critical aspect that has remained unexplored is the effect of phosphate–sulfide interfacial interactions. Therefore, by systematically investigating the ternary NH4NiPO4·H2O/CdIn2S4/Ni3S2 electrocatalyst, we highlight the benefits of unexplored phosphate/sulphite interface phenomena and their significance to advancing electrocatalytic activities.
2. Experimental
Nickel foam, ammonium phosphate monobasic (NH4H2PO4), cadmium nitrate tetrahydrate (CdNO3)2·4H2O and urea (CH4N2O), thioacetamide (C2H5NS), indium nitrate hydrate (In(NO3)3·H2O) were all of Sigma-Aldrich and used as received. NH4NiPO4·H2O was synthesized via a hydrothermal approach. First, nickel foam was pre-treated in 3 M HCl and then rinsed with water, each for 15 minutes, before being dried at 70 °C. In a typical synthesis, 0.1 M NH4H2PO4 and 0.1 M urea were dissolved in deionized water. This solution, along with the nickel foam substrate, was placed into a 50 mL autoclave and heated to 180 °C for 12 hours. Upon cooling, the substrate was removed, thoroughly rinsed and dried once more at 70 °C. The resulting Ni-foam substrate was then used for further characterization and synthesis steps.The CdIn2S4/Ni3S2 and NH4NiPO4·H2O/CdIn2S4/Ni3S2 electrodes were synthesized using a straightforward two-step hydrothermal method. First, 0.09 g of Cd(NO3)2·4H2O, 0.18 g of In(NO3)2·H2O, and 0.35 g of C2H5NS were dissolved in 35 mL of deionized water. The resulting solution was then transferred to a 50 mL Teflon-lined autoclave, containing both the pre-synthesized NH4NiPO4·H2O/NF substrate and bare Ni foam substrate, and heated at 160 °C for 10 hours. After the reaction, the autoclave was left to cool naturally overnight. The precipitate was collected, thoroughly washed with water and ethanol several times, and dried at 70 °C. The synthesized materials are labelled as follows: NH4NiPO4·H2O as NPO, CdIn2S4/Ni3S2 as CINS, and the composite NH4NiPO4·H2O/CdIn2S4/Ni3S2 as NPO/CINS.
The morphologies of the as-prepared electrocatalysts were examined using field-emission scanning electron microscopy (FESEM; S-4300, Hitachi) and field-emission transmission electron microscopy (FE-TEM, JEM-2100F, JEOL). Their elemental composition and distribution were determined via energy-dispersive X-ray spectrometry (EDX; EX-250, Horiba). High-resolution X-ray diffraction (XRD; X'Pert PRO MRD, Phillips) was employed to identify the crystalline phases, and X-ray photoelectron spectroscopy (XPS; K-Alpha, Thermos Scientific) was utilized to analyze the surface elemental composition. The Raman spectra were recorded in air using a Raman spectrometer (LabRAM Revolution, HORIBA) equipped with a 532 nm laser and 1800 lines per mm grating, with spectra collected at a resolution of 0.5 cm−1 spanning from 50 to 2000 cm−1. The analysis was conducted at the Inha University Core Facility Center for Sustainable Energy.
All electrochemical measurements were carried out using a biologic potentiostat workstation with a standard three-electrode configuration. The prepared binder-free electrodes served as the working electrodes, while a Hg/HgO electrode and Pt mesh were utilized as the reference and counter electrodes, respectively. Electrochemical measurements were conducted in two distinct electrolytes: 1.0 M KOH and 1.0 M KOH with 0.33 M urea. A potentiostat (VSP, Bio-Logics) electrochemical workstation was employed for these measurements. Electrode activation was achieved via cyclic voltammetry (CV) until a stable overpotential was reached, followed by linear sweep voltammetry (LSV) within a potential window of 0 to 0.6 V vs. SCE at a scan rate of 5 mV s−1. Electrode stability was assessed using chronopotentiometry (CP) at a constant current density of 10 mA cm−2. The Tafel equation (η = b![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) log|j| + a) was utilized to derive Tafel curves from the LSV data, relating the overpotential (η) to the current density (j) and the Tafel slope (b). Electrochemical impedance spectroscopy (EIS) measured interface charge transfer resistance across a frequency range of 1 Hz to 10 kHz.
log|j| + a) was utilized to derive Tafel curves from the LSV data, relating the overpotential (η) to the current density (j) and the Tafel slope (b). Electrochemical impedance spectroscopy (EIS) measured interface charge transfer resistance across a frequency range of 1 Hz to 10 kHz.
3. Results and discussion
The stepwise synthesis of the NPO, CINS, and the NPO/CINS heterostructure on Ni foam via a hydrothermal method is depicted in schematic I in ESI.† This schematic outlines the sequential deposition and integration processes that yield a robust and synergistic heterostructure. In step-I, the Ni foam acts as a reactive substrate for the in situ formation of NPO through a chemical dissolution process. The process begins with the ionization and hydrolysis of (NH4)2HPO4 in the hydrothermal environment, facilitated by the presence of urea. Urea plays a crucial role as a slow-releasing source of ammonia (NH3) and hydroxide ions (OH−), which help to regulate the pH and provide the necessary conditions for the reaction. The dissolved phosphate ions react with nickel ions released from the Ni foam surface during its partial dissolution. This reaction, driven by the consumption of oxygen, leads to the nucleation and growth of NPO directly on the Ni foam substrate. The resulting NPO exhibits strong adhesion due to its in situ formation, ensuring robust contact and electrical connectivity with the underlying Ni foam. The subsequent step involves the hydrothermal synthesis of CdIn2S4 in the presence of the NPO-coated Ni foam. During this process, sulfur precursors react with nickel ions from the foam, forming Ni3S2in situ as an intermediate layer. This dual process ensures intimate contact between the Ni3S2 and the growing CdIn2S4 particles. The coexistence of these phases enhances the heterostructure's electronic and catalytic properties. In the final step, the integration of NPO and CINS occurs through a synergistic assembly process, facilitated by the sequential reactions outlined in steps I and II. The heterostructure combines the unique properties of NPO, CdIn2S4, and Ni3S2, resulting in a highly interconnected and multifunctional material. The NPO provides a stable and conductive scaffold, while the CINS composite introduces catalytic active sites and enhanced charge transfer capabilities. The strong interfacial bonding between layers ensures mechanical stability and durability.The XRD patterns of NPO and CINS and NPO/CINS heterostructure are shown in Fig. S1.† In Fig. S1a,† the peaks can be attributed to a pure orthorhombic phase of NPO (JCPDF no. 86-1866), along with two sharp reflections from the Ni foam, marked by the asterisks (*). The XRD pattern of the CINS film exhibits two distinct sets of peaks, corresponding to CdIn2S4 and Ni3S4 (Fig. S1b†). Like NPO, CdIn2S4 crystallized in the cubic spinel structure, consistent with the JCPDS no. 27-060, while the Ni3S2 phase formed in a rhombohedral structure, corresponding to JCPDS no. 03-0863. The X-ray diffraction (XRD) pattern of NPO/CINS (Fig. S1c†) exhibits prominent diffraction peaks corresponding to the characteristic crystal planes of all three constituent phases, with no detectable impurity peaks, thereby confirming the successful synthesis of the heterostructure between NPO and CINS. This structural integrity and phase purity were further validated through FESEM.
Fig. 1a and S2a† present the FESEM images of NPO, showcasing a highly textured and layered morphology characterized by three-dimensional, flake-like formations. These flakes overlap and stack upon one another, creating a complex architecture that offers a vast surface area, ideal for enhancing electrocatalytic performance. The FESEM images of CINS (Fig. 1b and S2b†) reveal a porous, interconnected network structure, resembling a sponge-like framework. This material features rough surfaces with numerous interconnected voids or pores, giving rise to a continuous and convoluted texture formed by thin, wrinkled sheets or layers. The high surface area and porous nature make this structure highly advantageous for electrocatalytic applications, promoting efficient mass transport and active site exposure. The morphology of the NPO/CINS heterostructure (Fig. 1c and S2c†) reveals an integration of both phases, where the randomly oriented flake-like NPO structures are uniformly enveloped by the CINS, forming a net-like network. This synergistic combination of high surface area flakes and interconnected porous networks significantly enhances the material's overall performance by optimizing mass transport, increasing active site availability, and improving structural stability. Such a heterostructure holds great potential for high-performance applications in electrocatalysis. The detailed elemental composition of all the elements in NPO/CINS heterostructure from EDX spectrum is tabulated in Fig. 1d, indicating the presence of all the elements in the NPO/CINS heterostructure. The element nitrogen (N) may not be detected due to its presence potentially falling below the detection threshold of the EDX system.
 | ||
| Fig. 1 FESEM images of (a) NH4NiPO4·H2O, (b) CdIn2S4/Ni3S2 and (c) NH4NiPO4·H2O/CdIn2S4/Ni3S2 heterostructure. (d) Elemental composition in NH4NiPO4·H2O/CdIn2S4/Ni3S2 heterostructure determined from EDS. (e) TEM image of NPO flakes. The inset shows the elemental mapping for Ni, P, N, and O. (f) TEM image of CdIn2S4/Ni3S2. The inset shows elemental mapping for Cd, In, Ni, and S. | ||
The TEM images and corresponding elemental mapping in Fig. 1e and f further corroborate the structural and compositional characteristics of the NPO/CINS heterostructure. In Fig. 1e, the TEM image reveals the presence of thin, layered structures consistent with the flake-like morphology of NPO. The magnified inset shows a uniform distribution of Ni, P, N, and O elements across these flakes, highlighting the homogeneous composition of the NPO phase. Fig. 1f demonstrates the integration of CINS into the heterostructure. The TEM image displays agglomerated, porous regions, indicative of the sponge-like CINS framework consistent with FESEM image (Fig. 1b). The elemental mapping shows a well-dispersed distribution of Cd, In, Ni, and S, confirming the successful formation of the heterostructure. The overlap of these elements highlights the synergistic combination of NPO's flake-like architecture and CINS's porous network. This combination ensures enhanced active site availability, and increased structural stability, making the heterostructure highly favorable for electrocatalytic applications.
The X-ray photoelectron spectroscopy (XPS) analysis was employed to probe the surface composition and electronic structure of the NPO/CINS electrocatalysts, as illustrated in Fig. 2. In Fig. 2a, the high-resolution Ni 2p spectrum reveals two prominent sets of spin–orbit doublets, with binding energies at 855.7 eV and 873.2 eV, corresponding to Ni 2p3/2 and Ni 2p1/2, respectively. These peaks are further deconvoluted into Ni2+ and Ni3+ states. Specifically, the Ni2+ states are centered at 855.5 eV and 872.9 eV, while the Ni3+ states are centered at 856.1 eV and 874.3 eV.7,27 Additionally, satellite peaks are observed at 861.4 eV and 879.6 eV, confirming the presence of Ni in both Ni2+ and Ni3+ oxidation states.
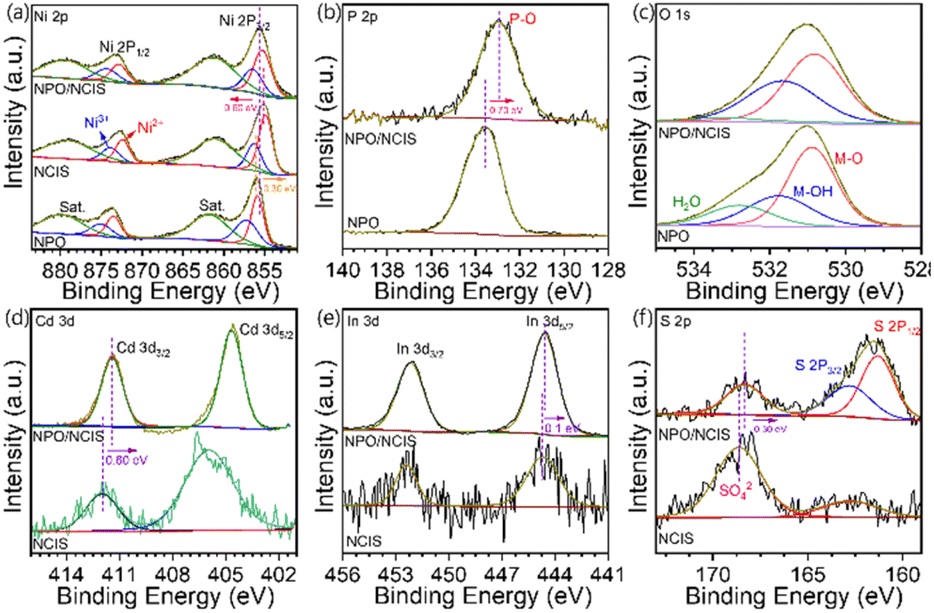 | ||
| Fig. 2 High-resolution XPS spectra of (a) Ni 2p, (b) P 2p, (c) O 1s, (d) Cd 3d, (e) In 3d and (f) S 2p in NH4NiPO4·H2O, CdIn2S4/Ni3S2, and NH4NiPO4·H2O/CdIn2S4/Ni3S2 heterostructure. | ||
To further explore the impact of the heterostructure between NPO and CINS, XPS measurements were also conducted on NPO and CINS as shown in Fig. 2a. Notably, the Ni 2p3/2 and Ni 2p1/2 peaks in the NPO/CINS heterostructure shift toward lower binding energies compared to NPO and shift toward higher binding energy compared to CNIS. This observation reveals a pronounced electron transfer from CINS to NPO mediated through Ni ions within the NPO/CINS heterostructure, substantially enhancing electron density around these Ni sites. Consequently, the intensity of Ni3+ oxidation states in the NPO/CINS heterostructure markedly increased compared to that of NPO and CINS, indicating a stabilized high-valence Ni environment facilitated by interfacial charge transfer. The deconvoluted P 2p spectra of NPO and the NPO/CINS heterostructure are illustrated in Fig. 2b. For NPO, the P 2p spectrum reveals a peak at 133.6 eV corresponding to P–O bonding. In the case of the heterostructure, the peak was shifted to lower binding energy (133 eV) compared to pristine NPO. As shown in Fig. 2c, the O 1s spectrum of NPO can be deconvoluted into two peaks at 530.9 eV, 531.8 eV, and 532.8 eV attributed to M–O, M–OH and adsorbed water species, respectively. Upon formation of the NPO/CINS heterostructure, the peak intensity of absorbed water decreased while that of OH increased, signifying an enhanced stabilization of OH groups at the heterostructure interface due to interfacial bonding and electron transfer effects. The Cd 3d spectrum of CINS displays two prominent peaks at 406 eV and 412.0 eV, corresponding to Cd 3d5/2 and Cd 3d3/2, respectively (Fig. 2d).25 Upon coupling with NPO, these peaks shifted to lower binding energies, indicating subtle electronic changes. The In 3d spectrum of CINS, as depicted in Fig. 2e, exhibits two distinct peaks at 444.7 eV and 452.4 eV, attributed to In 3d5/2 and In 3d3/2, respectively.25 Notably, these binding energies remain unchanged after coupling with NPO. For the S 2p spectrum in Fig. 2f, the peaks at 162.8 eV and 168.6 eV correspond to S 2p, associated with typical metal–sulfur bond and surface SO42− species exposed to air, respectively.28 However, in the NPO/CINS heterostructure, these peaks experience a negative shift, reflecting altered electronic environments. The variations in binding energies of Ni, O, Cd, and S can be ascribed to surface charge redistribution upon heterojunction formation. This redistribution likely induces a built-in electric field at the NPO/CINS interface, facilitating enhanced charge transfer across the heterojunction.
The oxygen evolution reaction (OER) performance of the NPO/CINS electrocatalyst was evaluated using cyclic voltammetry (CV) in 1 M KOH at a scan rate of 5 mV s−1. Before evaluating the OER activity, the electrocatalysts were activated via CV cycling (20 cycles) in the range of 0.8 to 1.8 V vs. RHE at a scan rate of 100 mV s−1 (Fig. S3a†). This activation step is for the reconstruction of a catalytically active surface by removing adsorbed contaminants or passivating layers that could hinder the catalytic performance of electrocatalyst. As shown in Fig. 3a, the iR-corrected CV curves for NPO, CINS, and the NPO/CINS heterostructure reveal that the NPO/CINS electrocatalyst demonstrates superior OER activity, characterized by the lowest onset potential and highest current density compared to its counterparts. To avoid interference from the Ni2+/Ni3+ oxidation during the positive-going scan (1.3–1.5 V vs. RHE range), the polarization curves from the reverse scan (high to low potential) were analyzed to estimate the overpotentials and assess the OER activity. NPO exhibited notable overpotentials of 315 mV and 410 mV at current densities of 10 mA cm−2 and 100 mA cm−2, respectively. CINS showed slightly better overpotentials of 283 mV and 380 mV at the same current densities. However, the formation of the NPO/CINS heterostructure led to a dramatic decrease in overpotentials, reaching 245 mV and 320 mV at 10 mA cm−2 and 100 mA cm−2, respectively. This remarkable enhancement is attributed to the synergistic interaction within the heterostructure, where interfacial bonding promotes efficient electron transfer between components, significantly enhancing the catalytic activity for OER. Moreover, due to electron transfer from CINS to NPO, the increased electron density on Ni sites reduces the adsorption energy of key intermediates such as OH, OOH, and O, further boosting the OER catalytic efficiency.
 | ||
| Fig. 3 OER performance of the NH4NiPO4·H2O/CdIn2S4/Ni3S2 heterostructure in 1 M KOH. (a) LSV curves of NH4NiPO4·H2O, CdIn2S4/Ni3S2 and NH4NiPO4·H2O/CdIn2S4/Ni3S2 at scan rate of 5 mV s−1. (b) Tafel plots illustrating the kinetics of the OER, (c) EIS spectra at 0.5 V, highlighting the charge transfer resistance, (d) CP curve of NH4NiPO4·H2O/CdIn2S4/Ni3S2 recorded at a constant current density of 10 mA cm−2. | ||
Further kinetic analysis, including the determination of Tafel slopes, highlighted the superior performance of the NPO/CINS electrocatalyst (Fig. 3b). The Tafel slope for the NPO/CINS heterostructure, at just 43.65 mV dec−1, is significantly lower than those of NPO (68.64 mV dec−1) and CINS (68.31 mV dec−1), underscoring its notably faster OER kinetics compared to its components. This low Tafel slope, alongside the reduced overpotential, confirms the rapid kinetic advantages of the NPO/CINS heterostructure, affirming its exceptional potential for efficient water electrolysis. Additionally, electrochemical impedance spectroscopy (EIS) measurements were conducted to further probe the charge transfer dynamics during the OER process (Fig. 3c). The NPO/CINS electrocatalyst exhibited consistently lower charge transfer resistance compared to its counterparts, revealing a significantly accelerated OER rate on its surface. The turnover frequency (TOF) values were derived using the eqn (S1) provided in the ESI and are presented in Fig. S4.† As illustrated in Fig. S4,† the TOF values for NPO, CINS, and NPO-CINS were calculated to be 0.02, 0.07, and 0.276 × 10−3 s−1 per Ni site, respectively. Notably, the TOF value for the NPO/CINS heterostructure is significantly higher than that of the individual parent phases, thereby underscoring the substantial enhancement in intrinsic catalytic activity facilitated by forming the heterostructure between NPO and CINS. The long-term stability of the NPO/CINS electrocatalyst was also evaluated through time-dependent potential measurements (Fig. 3d). Impressively, the electrocatalyst showed only a 2% variation in overpotential during 65 hours of continuous galvanostatic electrolysis at a current density of 10 mA cm−2, demonstrating its remarkable stability and durability for prolonged OER operation.
Due to the significantly lower oxidation potential of urea compared to OH−, substituting the oxygen evolution reaction (OER) with the urea oxidation reaction (UOR) at the anode can substantially reduce the cell voltage. Accordingly, the urea oxidation reaction (UOR) efficiency of the NPO/CINS electrocatalyst was assessed via linear sweep voltammetry (LSV) in a 1 M KOH + 0.3 M urea solution, conducted at a scan rate of 5 mV s−1. Like the OER assessment, the electrocatalysts were activated for UOR by CV cycling (20 cycles) within the potential range of 0.8 to 1.8 V vs. RHE at a scan rate of 100 mV s−1, as shown in Fig. S3b.† As depicted in Fig. 4a, the iR-corrected LSV polarization curves for NPO, CINS, and NPO/CINS, reveal the latter's remarkable UOR activity. The NPO/CINS heterostructure demonstrates outstanding performance, requiring impressively low potentials of just 1.26 V and 1.35 V to achieve current densities of 10 mA cm−2 and 100 mA cm−2, respectively. These values are significantly lower than those of NPO (1.33 V and 1.37 V) and CINS (1.30 V and 1.36 V) electrocatalysts. The NPO/CINS heterostructure exhibits an impressively low Tafel slope of just 53 mV dec−1, markedly lower than that of NPO (65.47 mV dec−1) and CINS (61.31 mV dec−1), highlighting its significantly enhanced UOR kinetics compared to the individual components (Fig. 4b). Electrochemical impedance spectroscopy (EIS) measurements (Fig. 4c) were conducted to further investigate charge transfer dynamics during the UOR process. The NPO/CINS electrocatalyst demonstrated consistently lower charge transfer resistance than its counterparts, indicating a notably faster UOR rate on its surface. This confirms the superior UOR kinetics and efficient electron transfer capabilities of the heterostructure. Additionally, durability tests of the NPO/CINS heterostructure, recorded at a current density of 10 mA cm−2 (Fig. 4d), revealed only minimal potential degradation (∼2%) after 20 hours, underscoring its outstanding durability for long-term operation.
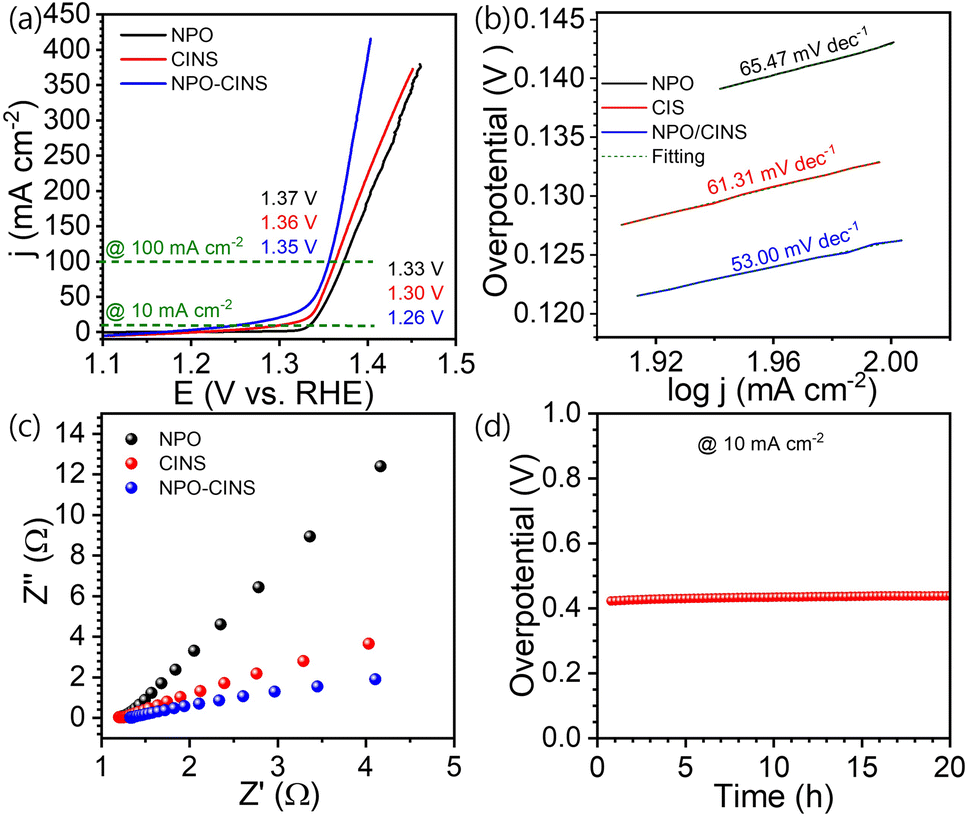 | ||
| Fig. 4 UOR performance of the NH4NiPO4·H2O/CdIn2S4/Ni3S2 heterostructure in 1 M KOH. (a) LSV curves of NH4NiPO4·H2O, CdIn2S4/Ni3S2 and NH4NiPO4·H2O/CdIn2S4/Ni3S2 at scan rate of 5 mV s−1. (b) Tafel plots illustrating the kinetics of the OER, (c) EIS spectra at 0.5 V, highlighting the charge transfer resistance, (d) CP curve of NH4NiPO4·H2O/CdIn2S4/Ni3S2 recorded at a constant j of 10 mA cm−2. | ||
Fig. 5a presents a comparative analysis of the CV curves of NPO/CINS heterostructure electrocatalyst for its OER and UOR activities at a scan rate of 5 mV s−1. Notably, the UOR process on the NPO/CINS electrocatalyst achieves current densities of 100 and 200 mA cm−2 at remarkably low potentials of 1.37 V and 1.39 V, respectively which is 180 mV and 200 mV below the OER potentials of 1.55 V and 1.59 V. This notable decrease in overpotential highlights the superior kinetics and efficiency of the UOR relative to the OER. The UOR performance of NPO/CINS heterostructure not only demonstrates exceptional efficiency but also exceeds that of previously reported oxide and sulfide based heterostructure electrocatalysts, as depicted in Fig. 5b.29–37 To elucidate the enhanced OER and UOR performance of the NPO/CINS electrocatalyst, post-reaction samples were characterized through FESEM and XPS analyses. High-potential electrocatalytic oxidation typically induces surface oxidation, which can alter both the composition and morphology of the catalysts. As shown in the FESEM images (Fig. S5†), the NPO and CINS maintain their structural integrity post-OER and UOR, with minimal morphological changes. However, XPS spectra reveal substantial surface oxidation of NPO/CINS following these reactions. In the Ni 2p spectrum (Fig. 5c), a pronounced increase in the Ni3+ valence state intensity, along with a negative shift in the Ni 2p3/2 and Ni 2p1/2 peaks relative to the untreated sample, signifies extensive redox transformations of Ni ions under OER and UOR conditions. Similarly, the O 1s spectrum (Fig. 5d) shows a significant increase in the M–O peak intensity at 530.8 eV after the reactions, while the M–OH and H2O-bound oxygen peaks decrease, indicating the formation of OOH-like species. A negative shift in O 1s peaks post-reaction suggests nuanced structural reorganization in the oxygen environment, enhancing reaction kinetics. The emergence of a new peak near 500 cm−1 in the Raman spectra (Fig. S6†), attributed to the metal–O band of NiOOH, further supports the partial transformation of the NPO-CINS surface to metal (oxy)hydroxide. Transmission Electron Microscopy (TEM) images provide compelling evidence of the surface transformation of the NPO-CINS heterostructure into a metal (oxy)hydroxide phase (Fig. 6).
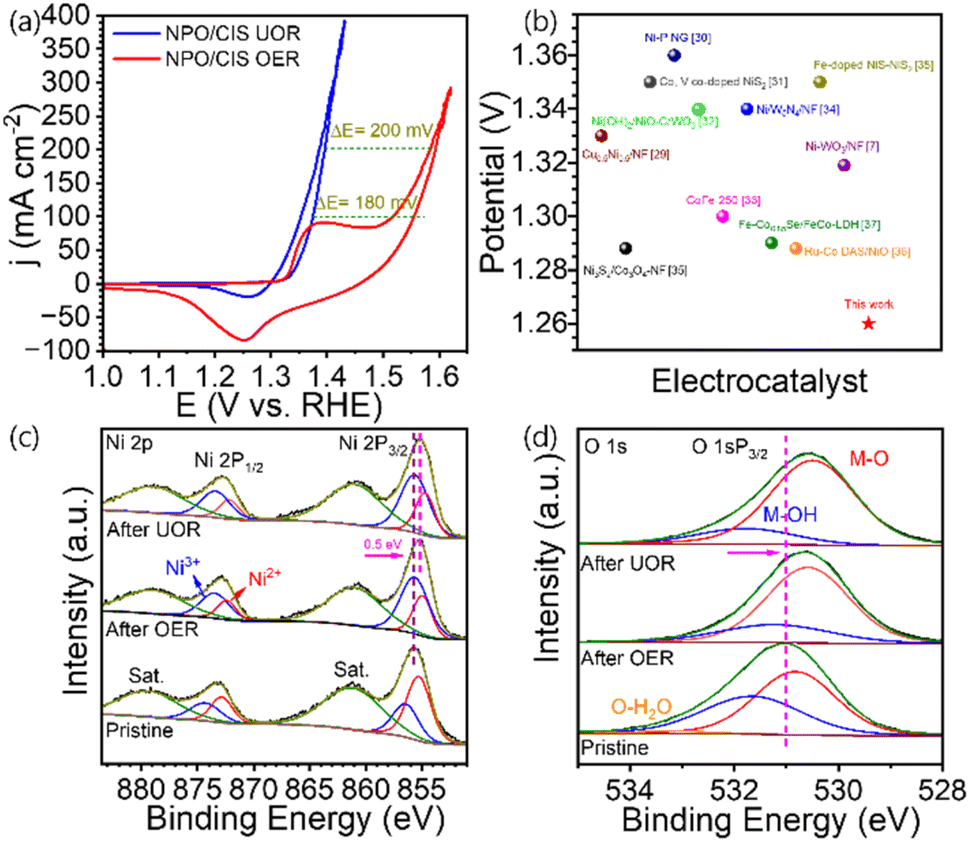 | ||
| Fig. 5 (a) CV curves of NH4NiPO4·H2O/CdIn2S4/Ni3S2 heterostructure for OER and UOR at a scan rate of 5 mV s−1. (b) Comparison of the UOR activity of the NH4NiPO4·H2O/CdIn2S4/Ni3S2 heterostructure with various electrocatalysts. High-resolution XPS spectra of (c) Ni 2p and (d) O 1s before and after OER and UOR activity. | ||
 | ||
| Fig. 6 (a) TEM image of the NH4NiPO4·H2O/CdIn2S4/Ni3S2 heterostructure after the UOR process, highlighting the overall morphology and nanoscale features. (b and c) HRTEM images illustrating the formation of the NiOOH phase via surface reconstruction during UOR. The bottom panel displays TEM elemental mappings, confirming the uniform distribution of elements within the NH4NiPO4·H2O/CdIn2S4/Ni3S2 heterostructure and validating the formation of NiOOH at the reconstructed regions. | ||
The catalytic mechanism for OER and UOR is primarily driven by the in situ formation of Ni3+–OOH species on the catalyst surface, which are recognized as the true active sites for these processes. For OER, the mechanism initiates with the adsorption of hydroxide ions (OH−) from the KOH electrolyte, resulting in OH bond dissociation to form surface-bound *O species. This intermediate *O subsequently binds with another OH− to yield the *OOH species, which then couples with additional hydroxide to release molecular oxygen (*O2) from the catalyst surface.38 XPS and Raman analyses highlight the critical role of the high oxidation state Ni3+ (NiOOH) species in enhancing the electrocatalytic activity of the NPO/CINS electrocatalyst. Ni3+ not only amplifies the intrinsic electronic properties of the catalyst but also optimizes intermediate stability, addressing rate-limiting steps in the reaction pathway and elevating OER performance. The electron transfer from CINS to NPO at the NPO/CINS heterointerface increases electron density on Ni sites, reducing the adsorption energy for intermediates such as OHad, OOHad, and Oad, and thus significantly boosting OER activity. The improved electronic conductivity, coupled with the higher oxidation state of Ni3+, facilitates electron transfer and stabilizes the OER intermediates, thus significantly boosting catalytic activity.
In parallel, the remarkable UOR activity of the NPO/CINS catalyst is largely attributed to the dynamic formation of the NiOOH phase, which actively oxidizes urea molecules, leading to the efficient evolution of nitrogen (N2) and carbon dioxide (CO2). This highly active NiOOH phase significantly boosts electrochemical performance by amplifying the density of catalytic sites, enhancing conductivity, and optimizing the overall reaction environment for UOR. Moreover, the strategic modulation at the NPO/CINS heterointerface facilitates substantial electron transfer from CINS to NPO, creating high-valence Ni sites with superior intrinsic activity, which further accelerates the reaction.
The HRTEM image of the NPO/CINS electrocatalyst after undergoing the UOR reaction provides compelling evidence of a surface reconstruction phenomenon. As illustrated in Fig. 6a, nanoscale morphological transformations are clearly discernible, showcasing distinct reconstructed NiOOH regions surrounding the NPO and CINS phases. These regions exhibit an amorphous or partially crystalline structure, signifying the dynamic evolution of the material during the reaction process (Fig. 6b and c). Specifically, the HRTEM images in Fig. 6b and c capture the reconstructed NiOOH species around the NPO and CINS phases, respectively. The observed interplanar spacing of 0.14 nm is attributed to the (110) plane of NiOOH, in agreement with the reference pattern (JCPDS no. 00-006-0075). The image unequivocally reveals that the NPO and CINS phases establish interfaces mediated by the reconstructed NiOOH species. Complementary insights from EDX mapping, shown in the bottom panel and Fig. S7,† confirm the uniform elemental distribution of Ni, N, P, and O in the NPO phase, as well as Cd, In, Ni, S, and O in the CINS phase. Notably, Ni and O dominate the surfaces of both phases, further substantiating the formation of NiOOH layers as a result of surface reconstruction around the NPO and CINS structures. This heterointerface is critical for enhanced electron transfer and the formation of high-density active sites, which are crucial for the catalytic activity during UOR.
At the molecular level, the electron-withdrawing C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group in urea exhibits a strong affinity for electron-rich (negatively charged) CINS sites on the catalyst surface, establishing a stable adsorption configuration. Concurrently, the electron-donating amino (–NH2) group is drawn toward electron-deficient (positively charged) regions associated with the NPO component.7 This complementary adsorption mechanism, engaging distinct active sites, optimally aligns the urea molecule and facilitates effective bond dissociation. Together, these synergistic interactions expedite urea decomposition and drive enhanced reaction kinetics, positioning the NPO/CINS catalyst as an exceptional system for efficient urea oxidation.
O group in urea exhibits a strong affinity for electron-rich (negatively charged) CINS sites on the catalyst surface, establishing a stable adsorption configuration. Concurrently, the electron-donating amino (–NH2) group is drawn toward electron-deficient (positively charged) regions associated with the NPO component.7 This complementary adsorption mechanism, engaging distinct active sites, optimally aligns the urea molecule and facilitates effective bond dissociation. Together, these synergistic interactions expedite urea decomposition and drive enhanced reaction kinetics, positioning the NPO/CINS catalyst as an exceptional system for efficient urea oxidation.
4. Conclusion
This study presents a high-performance electrocatalyst based on NH4NiPO4·H4O, integrated with CdIn2S4 and in situ formed Ni3S2 on nickel foam, which is engineered to achieve exceptional OER and UOR efficiencies. The heterostructure's layered architecture and phosphate–sulfide interfacial interactions enhance electron transport, achieving low overpotentials of 245 mV for OER and 1.26 V for UOR at 10 mA cm−2. The exceptional UOR efficiency exceeds that of previously reported oxide and sulfide-based heterostructure electrocatalysts. Tafel slope and EIS data underscore the rapid reaction kinetics and low charge transfer resistance, while stability tests reveal minimal degradation over prolonged operation. The study confirms that synergistic interfacial bonding and optimized electron transfer significantly boost catalytic activity, setting a new standard for durable, scalable catalysts in advanced energy applications.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
D. Patil: conceptualization, writing – original draft, S. Patil: visualization, H. Chavan, A. Lee: methodology. K. Lee: funding, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Korea Institute of Energy Technology Evaluation and Planning (KETEP) and granted financial resources by the Ministry of Trade, Industry & Energy, Republic of Korea (No. 20213030040590) and Korea Basic Science Institute (National Research Facilities and Equipment Center) grant funded by the Ministry of Education (2021R1A6C101A404).References
- I. Roger, M. A. Shipman and M. D. Symes, Nat. Rev. Chem, 2017, 1, 0003 CrossRef CAS.
- E. Fabbri, A. Habereder, K. Waltar, R. Kotz and T. J. Schmidt, Catal. Sci. Technol., 2014, 4, 3800–3821 RSC.
- H. S. Oh, H. N. Nong, T. Reier, A. Bergmann, M. Gliech, J. Ferreira de Araujo, E. Willinger, R. Schlogl, D. Teschner and P. Strasser, J. Am. Chem. Soc., 2016, 138, 12552–12563 CrossRef CAS PubMed.
- S. J. Trasatti, J. Electroanal. Chem. Interfacial Electrochem., 1980, 111, 125–131 CrossRef CAS.
- Y. Zhu, Q. Lin, Y. Zhong, H. Tahini, Z. Shao and H. Wang, Energy Environ. Sci., 2020, 13, 3361–3392 RSC.
- X. Xiao, L. Yang, W. Sun, Y. Chen, H. Yu, K. Li, B. Jia, L. Zhang and T. Ma, Small, 2022, 18, 2105830 CrossRef CAS PubMed.
- H. Shen, T. Wei, Q. Liu, S. Zhang, J. Luo and X. Liu, J. Colloid Interface Sci., 2023, 634, 730 CrossRef CAS PubMed.
- T. Wei, G. Meng, Y. Zhou, Z. Wang, Q. L. J. Luo and X. Liu, Chem. Commun., 2023, 59, 9992 RSC.
- J. Masa, C. Andronescu, H. Antoni, I. Sinev, S. Seisel, K. Elumeeva, S. Barwe, S. Marti-Sanchez, J. Arbiol and B. R. Cuenya, ChemElectroChem, 2019, 6, 235–240 CrossRef CAS.
- H. S. Chavan, J. Yoo, D. R. Patil, J. Kim, Y. Choi and K. Lee, J. Alloys Compd., 2027, 999, 174911 CrossRef.
- M. S. Shah, G. Y. Jang, K. Zhang and J. H. Park, EcoEnergy, 2023, 1, 344–374 CrossRef.
- S. Anantharaj, H. Sugime and S. Noda, ACS Appl. Mater. Interfaces, 2020, 12, 27327–27338 CrossRef CAS PubMed.
- S. Rezaee and S. Shahrokhian, Appl. Catal., B, 2019, 244, 802–813 CrossRef CAS.
- S. Gupta, M. K. Patel, A. Miotello and N. Pateil, Adv. Funct. Mater., 2020, 30, 1906481 CrossRef CAS.
- A. Vazhayil, L. Vazhayal, J. Thomas, S. C. Ashok and N. A. Thomas, Appl. Surf. Sci. Adv., 2021, 6, 100184 CrossRef.
- F. Song, L. Bai, A. Moysiadou, S. Lee, C. Hu, L. Liardet and X. Hu, J. Am. Chem. Soc., 2018, 140, 7748–7759 CrossRef CAS PubMed.
- H. S. Chavan, J. E. Yoo, D. R. Patil, J. Kim, Y. Choi and K. Lee, J. Alloys Compd., 2024, 999, 174911 CrossRef CAS.
- P. Rekha, S. Yadav and L. Singh, Ceram. Int., 2021, 12, 16385–16401 CrossRef.
- X. Guo, N. Li, Y. Cheng, G. Wang, Y. Zhang and H. Pang, Chem. Eng. J., 2021, 411, 128544 CrossRef CAS.
- P. Zhang, L. Gong and Y. Tan, Sustainable Energy Fuels, 2021, 5, 5581–5593 RSC.
- L. Chen, L. Wang, J.-T. Ren, H.-Y. Wang, W.-W. Tian, M.-L. Sun and Z.-Y. Yuan, Small Methods, 2024, 2400108 CrossRef CAS PubMed.
- P. Zhai, Y. Zhang, Y. Wu, J. Gao, B. Zhang, S. Cao, Y. Zhang, Z. Li, L. Sun and J. Hou, Nat. Commun., 2020, 11, 5462 CrossRef CAS PubMed.
- X.-Z. Ren, X.-H. Li, Y.-J. Peng, G.-Z. Wang, J. Yin, X.-C. Zhao, W. Wang and X.-B. Wang, Rare Met., 2022, 41(12), 4127 CrossRef CAS.
- Z. Li, L. Cao, T. Yang, J. He, Z. Wang, J. He, Y. Zhao and Z. Chai, J. Colloid Interface Sci., 2025, 677, 677 CrossRef CAS PubMed.
- W. Xu, W. Tian, L. Meng, F. Cao and L. Li, Adv. Mater. Interfaces, 2020, 7, 1901947 CrossRef CAS.
- J. Wang, T. Zhou, Y. Zhang, L. Li, C. Zhou, J. Bai, J. Li, H. Zhu and B. Zhou, ACS Appl. Mater. Interfaces, 2022, 14, 45392–45402 CrossRef CAS PubMed.
- Q. Che, Q. Ma, J. Wang, Y. Zhu, R. Shi and P. Yang, Catal. Commun., 2020, 140, 106006 CrossRef CAS.
- B. Ma, X. Guo, X. Zhang, Y. Chen, X. Fan, Y. Li, F. Zhang, G. Zhang and W. Peng, Energy Technol., 2019, 7, 1900063 CrossRef.
- K. J. Zhang, S. X. Wang, X. Y. Li, H. H. Li and Y. H. Ni, Small, 2023, 19(28), 2300959 CrossRef CAS PubMed.
- C. Q. Pei, S. Q. Chen, T. C. Zhao, M. Li, Z. T. Cui and B. A. Sun, Adv. Mater., 2022, 34(26), 200850 CrossRef PubMed.
- Z. J. Ji, Y. J. Song, S. H. Zhao, Y. Li, J. Liu and W. P. Hu, ACS Catal., 2022, 12(1), 569–579 CrossRef CAS.
- J. Y. Zhao, Y. Zhang, H. R. Guo, J. K. Ren, H. T. Zhang and Y. H. Wu, Chem. Eng. J., 2022, 433, 134497 CrossRef CAS.
- Q. Zhang, M. S. Sun, J. Zhu, S. D. Yang, L. Chen and X. L. Yang, Chem. Eng. J., 2022, 432, 134275 CrossRef CAS.
- Y. M. Zhou, B. X. Chu, Z. J. Sun, L. H. Dong, F. Wang and B. Li, Appl. Catal., B, 2023, 323, 122168 CrossRef CAS.
- S. S. Huang, Q. Zhang, P. J. Xin, J. Zhang, Q. C. Chen and J. Fu, Small, 2022, 18(14), 2106841 CrossRef CAS PubMed.
- X. B. Zheng, J. R. Yang, P. Li, Z. Jiang, P. Zhu and Q. S. Wang, Angew. Chem., Int. Ed., 2023, 62(22), e202217449 CrossRef CAS PubMed.
- H. Z. Yu, S. Q. Zhu, Y. X. Hao, Y. M. Chang, L. L. Li and J. Ma, Adv. Funct. Mater., 2023, 33(12), 2212811 CrossRef CAS.
- M.-Y. Ma, H.-Z. Yu, L.-M. Deng, L.-Q. Wang, S.-Y. Liu, H. Pan, J.-W. Ren, M. Y. Maximov, F. Hu and S.-J. Peng, Tungsten, 2023, 5, 589 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4se01754d |
| This journal is © The Royal Society of Chemistry 2025 |