
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemically pre-lithiated/sodiated reduced graphene oxide–antimony oxide composites for high-rate capability and long-term cycling stability in lithium and sodium-ion batteries†
Minseop
Lee‡
 ,
Gi-Chan
Kim‡
and
Seung-Min
Paek
*
,
Gi-Chan
Kim‡
and
Seung-Min
Paek
*
Department of Chemistry, Kyungpook National University, Daegu 41566, Republic of Korea. E-mail: smpaek@knu.ac.kr
First published on 20th February 2025
Abstract
Utilizing ultrasonication and microwave irradiation processes, we present a straightforward synthetic route to microwave-irradiated reduced graphene oxide (MrGO)–antimony oxide (Sb2O3) composites used as anode materials for lithium-ion batteries (LIBs) and sodium-ion batteries (SIBs). Furthermore, after a chemical pre-lithiation (PL) and pre-sodiation (PS) process, PL-MrGO/LixSb2O3 and PS-MrGO/NaxSb2O3 composites incorporating an inorganic solid electrolyte interface (SEI) layer and amorphous LixSb2O3/NaxSb2O3 were prepared by drying in an ambient environment. The inorganic SEI, including Li(Na)OH already formed at the defect site where irreversible Li/Na-ion trapping occurs, inhibits the initial irreversible reaction and provides ∼100% initial coulombic efficiency. In addition, the amorphous LixSb2O3 and NaxSb2O3 formed before the 1st discharge process promote improved cycling stability. For LIBs, the reversible capacity of the PL-MrGO/LixSb2O3 anode is 877.7 mA h g−1 at 100 mA g−1 after 150 cycles and 315.3 mA h g−1 after 3000 cycles at 5000 mA g−1. Also, for SIBs, PS-MrGO/NaxSb2O3 exhibits a reversible capacity of 313.1 mA h g−1 at 1200 mA g−1 after 3000 cycles. This rational structural design, which considers the irreversible reactions that occur during cycling, can be extended to the development of other high-performance anode materials.
1 Introduction
To meet the growing global demand for energy storage solutions while adhering to the principles of sustainability, researchers have increasingly focused on the development of electrochemical energy storage materials and processes.1–6 In particular, the use of chemically pre-lithiated and pre-sodiated anode materials in lithium- and sodium-ion battery (LIB and SIB) systems has emerged as a promising approach to improve the performance and lifespan of batteries. These materials address structural stability and capacity retention issues, enhancing the batteries' overall durability and providing performance benefits throughout their lifecycle.7–13The chemical pre-lithiation/sodiation process involves the incorporation of lithium or sodium ions into the anode material during synthesis. This preconditioning stabilizes the electrode structure and mitigates irreversible capacity losses that typically occur during the initial cycles of battery operation, thus substantially improving operational efficiency. Furthermore, these techniques align closely with the goals of a circular economy by reducing material waste and improving resource efficiency.
Additionally, one of the biggest challenges in current LIB and SIB research is developing anode materials that offer high energy density and long-term cycling stability.14–19 Graphite-based anodes are widely used in LIBs due to their excellent stability and cost-effectiveness. However, the theoretical capacity of graphite is limited to 372 mA h g−1, which falls short of meeting the increasing demand for high-energy-density batteries required for applications such as electric vehicles and large-scale energy storage systems.20–23 Additionally, graphite-based anode materials in SIBs suffer from low capacity, inefficient cycling performance, and poor structural stability due to the large ionic radius of sodium ions.24–27 In contrast, Sb-based anodes, particularly Sb2O3, have garnered significant attention as lithium and sodium storage materials due to their abundant resources and high theoretical capacity of 1102 mA h g−1. However, like most conversion/alloy type-transition metal oxide-based electrode materials, Sb2O3 suffers from low electrical conductivity and significant volume changes during charge/discharge cycles, leading to physical instability and degraded cycling performance.28–35 To address these challenges, graphene-based composites have been extensively studied. Existing literature primarily focuses on uniformly dispersing antimony oxide in a carbon matrix to mitigate volume changes and resolve conductivity issues.36–39 Nevertheless, to address problems related to high-rate performance and long-term cycling stability, new approaches that consider the irreversible reactions occurring during the cycling process must be explored. Specifically, for the practical application of high-energy-density LIBs and SIBs, it is essential to suppress the loss of active lithium/sodium ions in the electrolyte and enhance the initial coulombic efficiency (ICE) of the anodes up to ∼100%.40–45
This study developed a simple synthesis route for chemically pre-lithiated/sodiated MrGO/Li(Na)xSb2O3 composites to suppress irreversible reactions occurring during cycling.
The combined approach of introducing Li(Na)OH·Li(Na)2CO3 to induce an inorganic-rich solid-electrolyte interphase (SEI) and chemically pre-lithiating/sodiating Sb2O3 to enhance electrochemical reversibility in graphene–antimony oxide composites represents a significant advancement over conventional strategies. While traditional carbon coating or alloying techniques primarily focus on modifying the electrode surface, the approach proposed in this study effectively mitigates initial irreversible capacity loss while simultaneously improving long-term cycling performance. The formation of an inorganic-rich SEI layer stabilizes the electrode–electrolyte interface and suppresses continuous electrolyte decomposition, a common issue in conversion-type anodes. Furthermore, the pre-lithiation/sodiation process of Sb2O3 promotes the formation of amorphous LixSb2O3, enhancing the structural stability of the electrode and ensuring excellent cycling longevity. This synergistic effect contributes to improved coulombic efficiency, reduced interfacial resistance, and increased lithium/sodium ion flux uniformity, ultimately leading to superior high-rate performance and long-term cycling stability.46
The PL(S)-MrGO/Li(Na)xSb2O3 composite was synthesized by ultrasonic and microwave treatments, followed by chemical pre-lithiation/sodiation and drying under ambient conditions. The Li(Na)OH·Li(Na)2CO3-based inorganic SEI introduced into the carbon domain of PL(S)-MrGO/Li(Na)xSb2O3 was characterized using (HR)TEM and XPS analysis. Electrochemical analyses confirmed the improved structural stability and suppression of irreversible lithium/sodium ion trapping. Additionally, it was found that the amorphous Li(Na)xSb2O3 formed from crystalline Sb2O3 minimizes irreversible reactions following the initial conversion reaction and can stably participate in lithium/sodium ion storage reactions over long-term cycling.47–49 Notably, PL-MrGO/LixSb2O3 demonstrates exceptional performance as an anode material for LIBs. The PL-MrGO/LixSb2O3 anode exhibits a discharge capacity of 866.7 mA h g−1 at 100 mA g−1, which remains at 576.9 mA h g−1 when the current density increases to 1000 mA g−1. This value represents ∼99.4% of the theoretical capacity of MrGO/Sb2O3 (872 mA h g−1). In addition, the PL-MrGO/LixSb2O3 anode retains its capacity of 877.7 mA h g−1 after 150 cycles at 100 mA g−1. The long-term cycling stability of the PL-MrGO/LixSb2O3 anode is remarkable, maintaining a capacity of 315.3 mA h g−1 even after 3000 cycles at a high current density of 5000 mA g−1. This excellent rate performance and long-term cycling stability are also observed in SIBs. The PS-MrGO/NaxSb2O3 anode retains a capacity of over 78.0 mA h g−1 as the current density increases from 100 mA g−1 to 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mA g−1. Additionally, it retains a discharge capacity of 386.2 mA h g−1 after 300 cycles when the current density returns to 100 mA g−1. Furthermore, in a constant current test conducted at 1200 mA g−1, the PS-MrGO/NaxSb2O3 anode achieves a reversible capacity of 313.5 mA h g−1 after 3000 cycles. Consequently, this simple and rational synthesis strategy of graphene and metal oxide composites, which includes chemical pre-lithiation/sodiation, can effectively enhance long-term cycling stability and energy density.
000 mA g−1. Additionally, it retains a discharge capacity of 386.2 mA h g−1 after 300 cycles when the current density returns to 100 mA g−1. Furthermore, in a constant current test conducted at 1200 mA g−1, the PS-MrGO/NaxSb2O3 anode achieves a reversible capacity of 313.5 mA h g−1 after 3000 cycles. Consequently, this simple and rational synthesis strategy of graphene and metal oxide composites, which includes chemical pre-lithiation/sodiation, can effectively enhance long-term cycling stability and energy density.
2 Experimental
2.1. Materials
Sodium nitrate (NaNO3, ≥99%), sulfuric acid (H2SO4, 95.0–98.0%), graphite (<20 μm), tetrahydrofuran (THF, 99%) and antimony(III) acetate (SbAc, 99.99%) were purchased from Sigma-Aldrich Corporation (USA). Potassium permanganate (KMnO4, ≥99.3%), ethyl alcohol (≥94.0%) and ethylene glycol (EG, ≥99.5%) were acquired from Duksan Pure Chemical Co., Ltd (Korea). Hydrogen peroxide (H2O2, 34.5%) was purchased from Samchun Pure Chemical Co., Ltd (Korea). Antimony(III) oxide (Sb2O3, 99%) was purchased from Alpha Aesar. All reagents were used without further purification.2.2. Synthesis of graphene oxide
Graphene oxide (GO) was synthesized using a modified Hummer's method, involving the chemical exfoliation of graphite with a strong oxidizing agent in an acidic medium.50 Initially, graphite powder (1.0 g), sodium nitrate (NaNO3, 0.75 g), and sulfuric acid (H2SO4, 75 mL) were mixed and stirred in an ice bath to control the exothermic reaction. Potassium permanganate (KMnO4, 4.5 g) was gradually added to the mixture over the course of 1 hour, followed by stirring for two more hours in the ice bath and subsequently at room temperature for five days. The resulting GO suspension was purified by washing with H2SO4, hydrogen peroxide (H2O2), and distilled water to remove residual manganese salts and halt the oxidation process. Finally, the GO was repeatedly washed with distilled water and ethanol until a neutral pH was achieved. Then, it was collected through centrifugation at 15000 rpm for 5 min.
2.3. Synthesis of rGO/Sb(OH)3 and MrGO/Sb2O3
To synthesize rGO by reducing GO, antimony acetate (Sb(Ac)3) and GO are reacted using ultrasonication and microwave treatments. Initially, GO and Sb(Ac)3 are dissolved separately in ethylene glycol (EG) to prepare 1 g per L GO/EG and 4 g per L Sb(Ac)3/EG solutions, respectively. These two solutions are then thoroughly mixed at a volume ratio of GO/EG:Sb(Ac)3/EG = 25:65 and subjected to ultrasonic treatment for 1 hour, resulting in the formation of an rGO/Sb(OH)3/EG solution. This solution is centrifuged and dried to obtain a slurry form of rGO/Sb(OH)3. Finally, the obtained rGO/Sb(OH)3 is subjected to microwave treatment in a microwave oven for 10 min, yielding the final product, MrGO/Sb2O3.
2.4. Chemical pre-lithiation and pre-sodiation
In a glovebox filled with ultrapure Ar, 50 mg of MRGO/Sb2O3 powder is immersed in a 3 mL solution of 1 M lithium-biphenyl (Li-Bp) and sodium-biphenyl (Na-Bp) in THF for 1 minute and 1.2 min, respectively, followed by washing with THF. The resulting powder is vacuum-dried. The powders obtained by vacuum drying are then dried in an oven at 60 °C under an ambient atmosphere for 6 hours. The final products are named PL-MrGO/LixSb2O3 and PS-MrGO/NaxSb2O3. To evaluate the effect of drying conditions on the initial coulombic efficiency (ICE) of PL-MrGO/LixSb2O3 electrodes, samples were dried at 60 °C for different durations (6 and 24 hours) before electrode slurry preparation and initial charge–discharge testing. The results showed that the ICE was ∼99% after 6 hours of drying and remained ∼97% even after 24 hours (Fig. S1†). This indicates that the 60 °C, 6-hour drying conditions were chosen for experimental convenience, and the material maintains stable electrochemical performance even after extended drying.2.5. Calculation of theoretical capacity
The theoretical capacities of MrGO/Sb2O3 as an anode material were calculated based on the weight fractions of its components and their respective theoretical capacities. The weight fraction of Sb2O3 in MrGO/Sb2O3 is 35.68 wt%, and the remaining 64.32 wt% corresponds to MrGO. The theoretical capacity of Sb2O3 is 1102 mA h g−1, while MrGO, assumed to have the same theoretical capacity as graphene, contributes 744 mA h g−1. The total theoretical capacity was calculated as a weighted sum of the contributions from Sb2O3 and MrGO, using the following formula:| CMrGO/Sb2O3 = (wtSb2O3 × CSb2O3) + (wtMrGO × CMrGO) |
| CMrGO/Sb2O3 = (0.3568 × 1102 mA h g−1) + (0.6432 × 744 mA h g−1) = 872 mA h g−1 |
Therefore, the theoretical capacity of the MrGO/Sb2O3 anode is calculated to be 872 mA h g−1.
2.6. Electrode preparation and Li(Na) half cells assembly
Active electrodes were fabricated by casting a slurry consisting of 70 wt% active material, 20 wt% carbon black, and 10 wt% polyacrylic acid (PAA) in N-methyl-2-pyrrolidone (NMP) onto copper foil. The coated electrodes were dried in a vacuum oven at 110 °C for 12 hours. These electrodes were used for evaluating the electrochemical performance of LIB and SIB anodes. CR2032 coin cells were assembled in an argon-filled glovebox (H2O and O2 levels < 0.1 ppm). Lithium metal was cast onto an aluminum spacer to serve as the counter electrode. A Celgard 3501 separator was used to separate the anode and counter electrode. The electrolyte consisted of a 1 M LiPF6 solution in a 1:1 (v/v) mixture of ethylene carbonate (EC) and diethyl carbonate (DEC). Sodium metal on aluminum foil served as the counter electrode, and a Celgard 3501 separator was employed. The electrolyte comprised 1 M NaPF6 in a propylene carbonate/fluoroethylene carbonate (98:2 w/w) solution.
Galvanostatic charge–discharge tests were performed within a voltage range of 0.01–3.0 V (vs. Li/Li+) and 0.01–2.5 V (vs. Na/Na+) using a battery tester (Maccor K4300). Cyclic voltammetry (CV) was conducted at scan rates of 0.1, 0.3, 0.5, and 0.8 mV s−1 using a potentiostat (WonATech WMPG1000). EIS measurements were performed at open-circuit potential over a frequency range of 10−2 to 105 Hz using a potentiostat (Oneartec ZIVE SP2). For DRT analysis, the MATLAB-based software ‘DRTtools’, available at Ciucci's GitHub repository (https://github.com/ciuccislab/), was utilized.51
2.7. Characterization
The crystalline phases of the samples were analyzed using powder X-ray diffraction (PXRD, Bruker D2 Phaser, USA) with CuKα radiation (λ = 1.54056 Å). FTIR spectra were recorded using a Nicolet iS5 FTIR spectrometer (Thermo Fisher Scientific, USA) with the KBr pellet method. Surface elemental composition and oxidation states were determined through X-ray photoelectron spectroscopy (XPS, K-Alpha, Thermo Fisher Scientific, USA). Thermogravimetric analysis (TGA) was conducted using a Discovery SDT 650 instrument (TA Instruments, USA) over a temperature range of 35–900 °C in an ambient atmosphere at a heating rate of 5 °C min−1. The surface morphology of the samples was characterized via scanning electron microscopy (SEM, Hitachi SU8220, Japan). Transmission electron microscopy (TEM) and selected area electron diffraction (SAED) were performed using a Titan G2 ChemiSTEM Cs Probe (FEI Company, Netherlands).3 Results and discussion
3.1. Synthesis and chemical pre-lithiation and pre-sodiation mechanism
Fig. 1a schematically illustrates the synthesis process of PL(S)-MrGO/Li(Na)xSb2O3, while Fig. 1b shows the analysis of the crystallinity of the products using X-ray diffraction (XRD). In ethylene glycol (EG) solvent, GO nanosheets containing oxygen functional groups exhibit negative surface charges, and the positively charged [Sb(EG)3]3+ complexes are adsorbed through electrostatic interactions. Upon ultrasonic treatment, the temperature of the solution rises significantly due to an exothermic reaction. At elevated temperatures, the oxygen-containing species on the unstable GO nanosheet surfaces react with –OH and are removed, forming reduced graphene oxide (rGO) nanosheets. The XRD pattern of rGO/Sb(OH)3 formed after ultrasonic treatment does not exhibit the peak around 8° associated with GO, nor does it show distinct crystallization peaks attributed to Sb2O3. However, the XRD pattern of microwave-treated MrGO/Sb2O3 reveals clear crystallization peaks due to the formation of Sb2O3 from Sb(OH)3. Consequently, a simple process involving ultrasonic and microwave irradiation successfully fabricated composites containing Sb2O3 nanoparticles within a graphene matrix.52 The chemical reactions occurring during the synthesis of MrGO/Sb2O3 are detailed in Fig. 1c. During the synthesis of MrGO/Sb2O3, the interaction between acetate anions and ethylene glycol in the reaction solution serves as a source of OH−. These generated OH− remove oxygen-containing functional groups from GO, thereby promoting the formation of rGO. | ||
| Fig. 1 (a) Schematic representation of the synthesis route for PL(S)-MrGO/Li(Na)xSb2O3 composites. (b) X-ray diffraction (XRD) patterns of GO, MrGO/Sb(OH)3, MrGO/Sb2O3, PL-MrGO/LixSb2O3, and PS-MrGO/NaxSb2O3. (c) Chemical reactions during the synthesis of MrGO/Sb2O3. (d) Chemical reactions during the pre-lithiation/sodiation process. | ||
Fig. S2† presents the XRD spectra of samples where GO was dispersed in EG and then subjected to ultrasonic treatment (EG–GO) and microwave treatment (EG–MGO) in the absence of antimony acetate. These spectra confirm that no reduction of GO occurs in EG without antimony acetate, demonstrating that antimony acetate is essential for the reduction of GO during the synthesis of MrGO/Sb2O3. Under ambient conditions, the XRD pattern of pre-lithiated/sodiated and dried PL(S)-MrGO/Li(Na)xSb2O3 shows a decrease in peaks attributed to crystalline Sb2O3, indicating the formation of amorphous Li(Na)xSb2O3. The peaks observed at around 41° and 46° in the XRD spectrum of PL(S)-MrGO/Li(Na)xSb2O3 can be indexed to inorganic crystalline phases. The peaks observed around 41° and 46° can be indexed to inorganic crystalline phases. According to the PDF card (database) in Fig. S3,† the peaks near 41° and 46° are likely associated with the crystal planes of Li(Na)OH or Li(Na)2CO3. This suggests that during the pre-lithiation (pre-sodiation) process, reactions between Li(Na)-organic radical reagents, Sb2O3, and MrGO may lead to the presence of residual Li(Na)-ions, which subsequently react with moisture and oxygen in the ambient environment, resulting in the formation of Li(Na)OH and Li(Na)2CO3.
The chemical reactions involved in the pre-lithiation/sodiation process of MrGO/Sb2O3 are illustrated in Fig. 1d. When MrGO/Sb2O3 powder is immersed in a pre-lithiation/sodiation solution containing Li(Na)-Bp under an inert atmosphere, both MrGO and Sb2O3 within MrGO/Sb2O3 undergo a series of chemical reduction reactions. MrGO undergoes chemical reduction via electron transfer from biphenyl anions, with Li(Na)-ions being introduced into the disordered regions of MrGO, forming a lithiated/sodiated host. Additionally, crystalline Sb2O3, which has a higher redox potential than Li-Bp (0.33 V vs. Li+/Li) and Na-Bp (0.12 V vs. Na+/Na), transforms into amorphous Li(Na)xSb2O3.53–61 Therefore, this straightforward pre-activation method effectively prevents the irreversible loss of lithium/sodium and mitigates the initial capacity decrease due to the amorphization of Sb2O3 during the early cycling process. Following the pre-lithiation/sodiation process in an inert atmosphere, excess Li(Na)-ions were removed by washing with THF. Subsequently, during the drying process under ambient conditions, the Li(Na)-ions introduced into the MrGO nanosheets reacted with moisture in the air to form Li(Na)OH phases. This process resulted in the integration of an inorganic SEI domain with enhanced mechanical strength and ionic conductivity into PL(S)-MrGO/Li(Na)xSb2O3. Furthermore, the crystalline LiOH phase incorporated into the carbon domain occupied voids where irreversible Li(Na) ion trapping could occur, thereby contributing to a high ICE.
Fig. S4† shows the Fourier Transform Infrared (FTIR) spectra of the materials utilized during the synthesis process. In the spectrum of GO, peaks corresponding to C–O, C–O–C, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, and CO (carboxylic acid) confirm the presence of oxygen-containing functional groups. However, following ultrasonication treatment of antimony acetate and GO, the peaks associated with oxygen-containing groups almost disappeared or significantly diminished. After microwave irradiation, most of the oxygen-containing functional groups were removed, and only the aromatic CC stretching mode at approximately ∼1550 cm−1, attributed to the MrGO nanosheets, was prominently observed.62–67
C, and CO (carboxylic acid) confirm the presence of oxygen-containing functional groups. However, following ultrasonication treatment of antimony acetate and GO, the peaks associated with oxygen-containing groups almost disappeared or significantly diminished. After microwave irradiation, most of the oxygen-containing functional groups were removed, and only the aromatic CC stretching mode at approximately ∼1550 cm−1, attributed to the MrGO nanosheets, was prominently observed.62–67
3.2. Thermal stability and composition analysis
To quantify the Sb2O3 content in MrGO/Sb2O3, thermogravimetric analysis (TGA) was conducted in an ambient atmosphere (Fig. S5†). The mass loss observed below 600 °C corresponds to the decomposition and combustion of carbonaceous material, with the residual mass of approximately 39.50 wt% attributed to the formation of Sb2O4. After the TGA scan, the XRD pattern of the residue confirmed the conversion of Sb2O3 to Sb2O4 (Fig. S6†). Based on the molar mass of Sb2O4, the estimated Sb2O3 content in MrGO/Sb2O3 was determined to be 37.44 wt%.67–69 Further analysis using inductively coupled plasma (ICP) was performed to assess the Sb content in MrGO/Sb2O3. The Sb2O3 content, calculated from the molar mass of Sb obtained via ICP analysis, was 35.68 wt%. Additionally, X-ray photoelectron spectroscopy (XPS) elemental analysis was conducted to verify the surface composition of MrGO/Sb2O3, revealing an Sb2O3 content of 38.66 wt%. These values are consistent with the results obtained from TGA and ICP analyses, indicating that most Sb2O3 nanoparticles are uniformly adsorbed on the surface of the graphene nanosheets in MrGO/Sb2O3. A summary of the TGA, ICP, XPS, and TEM-EDS elemental analysis results for MrGO/Sb2O3 can be found in Table S1.†3.3. Morphological and structural analysis
The surface morphology of the samples was characterized using scanning electron microscopy (SEM). Fig. 2a illustrates the expanded open structure of MrGO, with its increased interlayer spacing. In contrast, EG–MGO retains oxygen functional groups on the edges and basal planes of the graphene, resulting in a thicker, multilayered structure similar to GO (Fig. 2b and S7†). The SEM images of MrGO/Sb2O3 are distinctly different from those of EG–MGO. Fig. 2c shows that the reduced graphene nanosheets form an open structure, with a rough surface indicating the presence of Sb2O3 nanoparticles on the graphene nanosheets. This structure is expected to exhibit a synergistic effect, where the graphene nanosheets act as conductive channels while the Sb2O3 nanoparticles prevent excessive restacking of the graphene. Fig. 2d and e demonstrate the volumetric expansion due to the formation of lithiated/sodiated graphene and Li(Na)xSb2O3 following the pre-lithiation/sodiation treatment of MrGO/Sb2O3. Therefore, utilizing the already amorphized and expanded samples as active materials through pre-lithiation/sodiation treatments is expected to enhance the physical stability of the electrodes during cycling. | ||
| Fig. 2 (a) Field emission scanning electron microscopy (FE-SEM) images of (a) MrGO, (b) EG–MGO, (c) MrGO/Sb2O3, (d) PL-MrGO/LixSb2O3, and (e) PS-MrGO/NaxSb2O3. | ||
The porosity of the synthesized samples was investigated using nitrogen adsorption–desorption isotherms (Fig. S8 and S9†). The specific surface area calculated by the Brunauer–Emmett–Teller (BET) method and the pore size distribution calculated by the Barrett–Joyner–Halenda (BJH) method are presented in Table S2†. The adsorption isotherms of GO and EG–MGO exhibited very low BET-specific surface areas of 26 m2 g−1 and 21 m2 g−1, respectively. In contrast, MrGO, reduced by microwave treatment under hydrated conditions, displayed a significantly higher BET-specific surface area of 240 m2 g−1 and increased pore volume. This suggests the presence of numerous electrochemically active sites capable of accommodating lithium/sodium ions. However, this also indicates an increase in the active surface area where irreversible reactions at the electrode/electrolyte interface may occur. Therefore, the Sb2O3 nanoparticles adsorbed on the surface of the graphene nanosheets and embedded within the carbon structure can help inhibit irreversible reactions on the electrode surface and within the SEI layer, thereby achieving a high ICE. The BET-specific surface areas of PL-MrGO/LixSb2O3 and PS-MrGO/NaxSb2O3, which underwent pre-lithiation/sodiation treatment, were found to be 11 m2 g−1 and 27 m2 g−1, respectively. The reduction in pore volume and specific surface area can be attributed to the formation of an inorganic SEI layer containing Li(Na)OH within the pores of the carbon structure and the lithiation/sodiation of Sb2O3 nanoparticles.
Transmission electron microscopy (TEM) images were analyzed to gain a deeper understanding of the structural characteristics of MrGO/Sb2O3. The high-resolution (HR)TEM images in Fig. S10† clearly illustrate the wide basal spacing of MrGO nanosheets (0.39–0.41 nm) and the integration of small Sb2O3 nanoparticles, measuring a few nanometers, into the graphene. Additionally, Fig. S11† presents HAADF and EDS elemental mapping (C, O, and Sb) images of MrGO/Sb2O3, confirming the uniform distribution of each element. Fig. S12 and S13† show the HRTEM and EDS elemental mapping images of PL-MrGO/LixSb2O3 and PS-MrGO/NaxSb2O3. After pre-lithiation/sodiation, the HRTEM and SAED images reveal the integration of amorphous nanoparticles (LixSb2O3 and NaxSb2O3) into the graphene. The EDS images confirm that uniform chemical pre-lithiation/sodiation was successfully achieved through the lithium/sodium-organic composite solution.70,71
3.4. Chemical bonding and surface chemistry analysis
Fig. 3 and S14a–e† show the X-ray photoelectron spectroscopy (XPS) spectra of MrGO/Sb2O3, PL-MrGO/LixSb2O3, and PS-MrGO/NaxSb2O3 electrodes. The XPS elemental analysis results of PL-MrGO/LixSb2O3 and PS-MrGO/NaxSb2O3 are detailed in Tables S3 and S4.† In Fig. 3a, the C 1s spectra of MrGO/Sb2O3 show a significantly lower relative intensity of oxygen-containing functional groups compared to GO synthesized via the Hummer's method (Fig. S15†). This indicates that the GO nanosheets were effectively reduced during the synthesis of MrGO/Sb2O3. Additionally, the presence of an additional peak at 285.4 eV in the C 1s spectra of MrGO/Sb2O3 can be attributed to the C–O(–Sb) bond, suggesting the formation of strong electronic interactions between Sb2O3 and the MrGO nanosheets.72 In Fig. 3b, the Sb 3d spectra of MrGO/Sb2O3 overlap with the O 1s spectra. The signals at 530.7 eV and 540.1 eV correspond to Sb3+ 3d3/2 and Sb3+ 3d5/2 in c-Sb2O3, respectively.73–76 The O 1s spectral signal at 531.9 eV arises from lattice oxygen and oxygen-containing functional groups on the graphene surface. After pre-lithiation, the C 1s spectra of PL-MrGO/LixSb2O3 reveal an additional peak attributed to Li2CO3, while the Sb 3d and O 1s spectra exhibit new peaks assigned to LiOH (Fig. 3c and d). These findings suggest that Li+-ions react with atmospheric moisture to form LiOH, which subsequently reacts with CO2 in air to form Li2CO3 (Fig. S14d†). Furthermore, the formation of amorphous LixSb2O3 shifts the Sb 3d3/2 and Sb 3d5/2 peaks to lower binding energies (530.4 eV and 539.7 eV, respectively).77–81 In the C 1s spectra of PS-MrGO/NaxSb2O3 presented in Fig. 3e, an additional peak corresponding to Na2CO3 is observed. The Sb 3d, O 1s, and Na 1s spectra confirm the formation of Na2CO3 and NaOH (Fig. 3f and S14e†). Furthermore, the formation of amorphous LixSb2O3 shifts the Sb 3d3/2 and Sb 3d5/2 peaks to lower binding energies (530.4 eV and 539.7 eV, respectively).82–84 The Li(Na)OH·Li(Na)2CO3 formed on the electrode surface after pre-lithiation/sodiation plays a crucial role in guiding inorganic-rich SEI growth during charge/discharge cycles. Typically, Sb2O3-based electrodes undergo significant volume changes during cycling, leading to continuous SEI destruction and reformation. This process often results in the formation of a thick organic-rich SEI layer and accumulation of insoluble byproducts from electrolyte decomposition. Such effects increase interfacial resistance and deteriorate long-term electrochemical stability. However, the inorganic-rich SEI layer exhibits high mechanical strength and effectively mitigates excessive SEI growth. This property helps suppress unnecessary electrolyte consumption, thereby reducing initial irreversible capacity loss and enhancing the long-term cycling stability of the electrode.46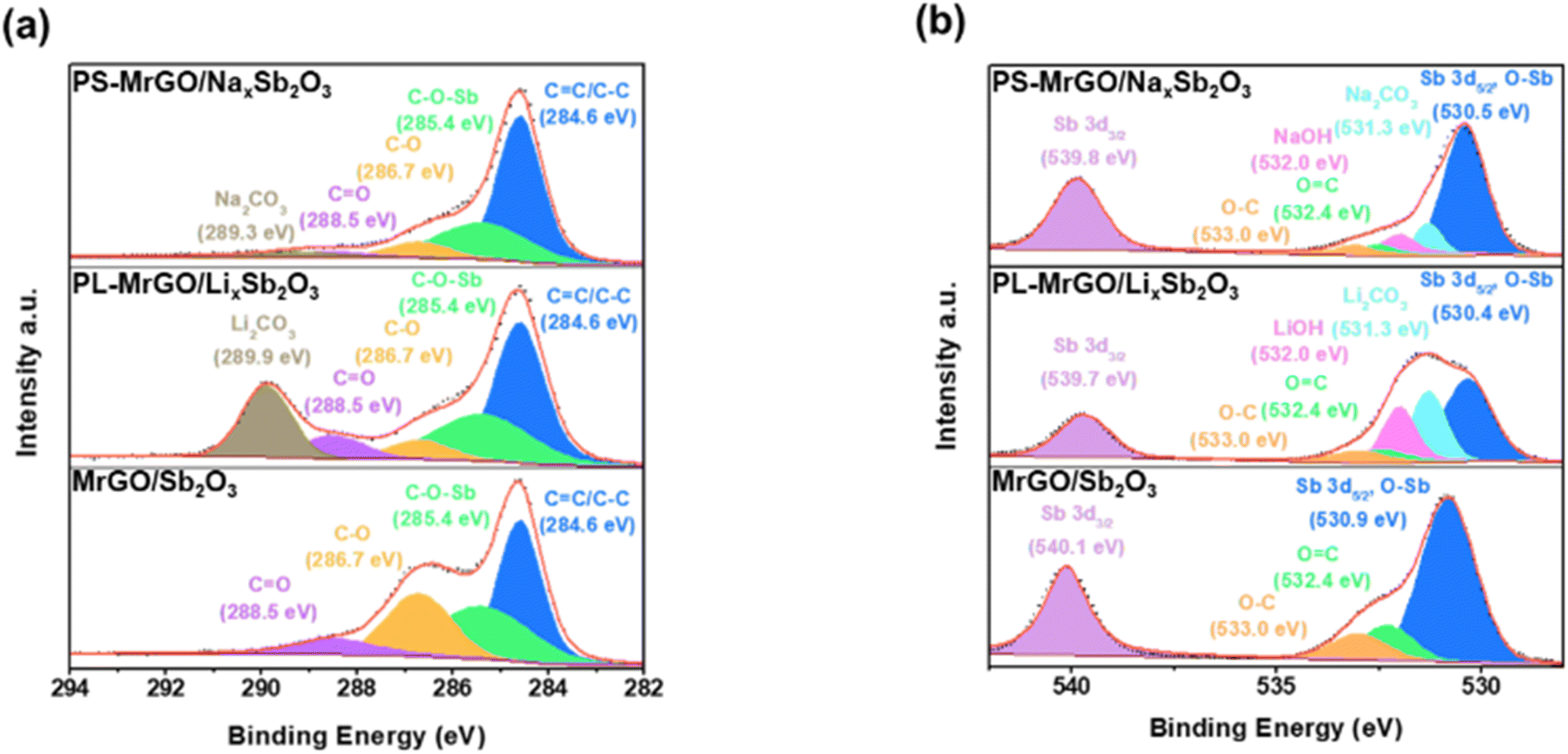 | ||
| Fig. 3 High-resolution XPS spectra of (a) C 1s and (b) Sb 3d & O 1s for MrGO/Sb2O3, PL-MrGO/LixSb2O3, and PS-MrGO/NaxSb2O3. | ||
3.5. Lithium storage performance of PL-MrGO/LixSb2O3
To understand the effects of pre-lithiation and the structural synergy of composites, the electrochemical performance of PL-MrGO/LixSb2O3 was compared with that of MrGO/Sb2O3, MrGO and bare Sb2O3. The rate performance of each electrode was evaluated at current densities ranging from 100 to 1000 mA g−1 (Fig. 4a). The comparison of charge/discharge capacities and capacity retention rates over the initial 35 cycles for PL-MrGO/LixSb2O3, MrGO/Sb2O3, Sb2O3, and MrGO electrodes presented in Fig. 4a is shown in Fig. S16.† The PL-MrGO/LixSb2O3 electrode exhibited reversible capacities of 866.7 mA h g−1 (2nd cycles), 716.3 mA h g−1 (10th cycles), 656.7 mA h g−1 (15th cycles), 611.6 mA h g−1 (20th cycles), 580.8 mA h g−1 (25th cycles), and 573.7 mA h g−1 (30th cycles) at current densities of 100, 300, 500, 700, 900, and 1000 mA g−1, respectively. The capacity retention rates relative to the 2nd cycle were 82.6%, 75.8%, 70.6%, 67.0%, and 66.2%, respectively. The MrGO/Sb2O3 electrode, under the same conditions, showed capacities of 850.8 mA h g−1 (2nd cycles), 678.8 mA h g−1 (10th cycles), 615.8 mA h g−1 (15th cycles), 573.2 mA h g−1 (20th cycles), 541.5 mA h g−1 (25th cycles), and 518.2 mA h g−1 (30th cycles), with retention rates of 79.8%, 72.4%, 67.4%, 63.6%, and 60.9%, respectively.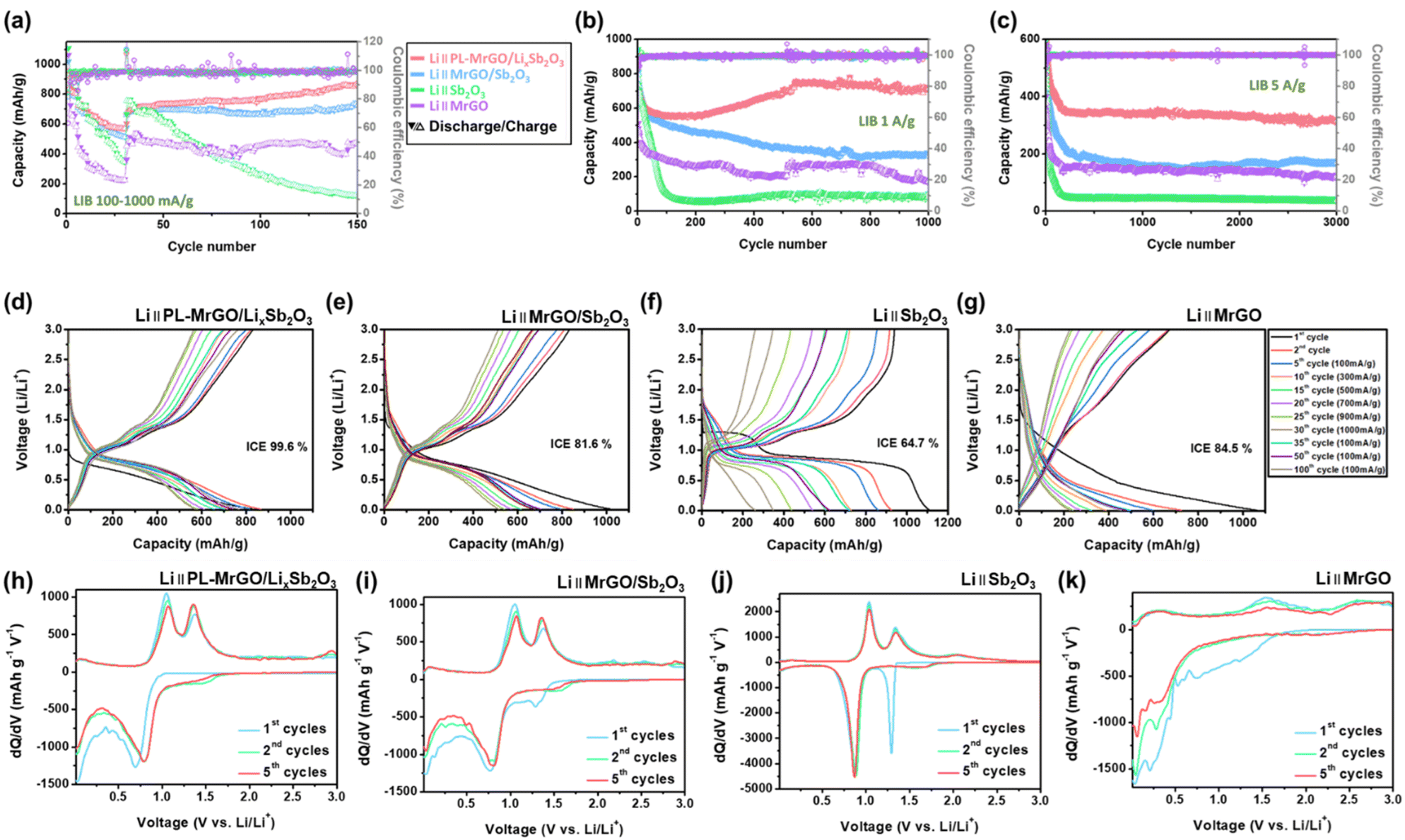 | ||
| Fig. 4 Lithium storage performance: (a) rate performance of the PL-MrGO/LixSb2O3, MrGO/Sb2O3, bare Sb2O3 and MrGO electrodes measured at different current densities (100–1000 mA g−1). Long-term cycling performance at (b) 1000 mA g−1 and (c) 5000 mA g−1. Charge–discharge profiles of (d) PL-MrGO/LixSb2O3, (e) MrGO/Sb2O3, (f) bare Sb2O3 and (g) MrGO electrodes at different current densities (100–1000 mA g−1). dQ/dV curves of (h) PL-MrGO/LixSb2O3, (i) MrGO/Sb2O3, (j) bare Sb2O3 and (k) MrGO electrodes. | ||
At 100 mA g−1, the initial discharge/charge capacities and ICE values for the PL-MrGO/LixSb2O3 and MrGO/Sb2O3 electrodes were 834.0/830.8 mA h g−1 (ICE = 99.6%) and 1020.4/832.2 mA h g−1 (ICE = 81.6%), respectively. The MrGO/Sb2O3 electrode exhibited a lower ICE due to irreversible SEI formation, Li-ion trapping at graphene defect sites, and irreversible reactions between Sb2O3 and lithium ions during the initial discharge. On the other hand, the PL-MrGO/LixSb2O3 electrode suppressed this irreversible reaction by forming amorphous LixSb2O3 and lithiated MrGO after the pre-lithiation process, achieving a high ICE. Furthermore, after reverting the current density to 100 mA g−1, the PL-MrGO/LixSb2O3 and MrGO/Sb2O3 electrodes retained discharge capacities of 877.7 and 736.4 mA h g−1, respectively, at the 150th cycle, with capacity retention rates of 101.3% and 86.6%. These results are especially important compared to previously reported Sb-based anodes, as summarized in Table S5,† which provides a comprehensive comparison of various synthesis strategies and their corresponding electrochemical lithium storage performance. Compared to conventional Sb-based electrodes, PL-MrGO/LixSb2O3 exhibits superior rate capability and long-term cycling stability. These advantages demonstrate that the chemical pre-lithiation strategy and inorganic SEI formation provide a more effective solution to the critical problems of Sb-based anodes.
In long-term cycling tests conducted at 1000 mA g−1, the PL-MrGO/LixSb2O3 and MrGO/Sb2O3 electrodes exhibited capacities of 715.7 and 333.5 mA h g−1, respectively, after 1000 cycles (Fig. 4b). Additionally, in cycling tests performed at 5000 mA g−1, the PL-MrGO/LixSb2O3 and MrGO/Sb2O3 electrodes retained capacities of 315.3 and 169.4 mA h g−1, respectively, after 3000 cycles (Fig. 4c). These results emphasize the superior cycling performance of the PL-MrGO/LixSb2O3 electrode compared to MrGO, bare Sb2O3, and MrGO/Sb2O3 electrodes.
The superior long-term cycling performance of the PL-MrGO/LixSb2O3 electrode can be attributed to the formation of an initial SEI layer during the chemical pre-lithiation/sodiation process, which acts as a stable, protective barrier at the electrode–electrolyte interface. Typically, Sb2O3-based electrodes undergo significant volume changes during charge/discharge cycles, leading to continuous SEI cracking and reformation. This process results in the formation of an irregular SEI structure, continuous electrolyte consumption, and an increase in interfacial electronic and ionic resistance, ultimately compromising long-term electrochemical stability. In contrast, the PL-MrGO/LixSb2O3 electrode synthesized in this study forms an inorganic-rich SEI layer composed of Li(Na)OH·Li(Na)2CO3, which provides high mechanical strength and effectively suppresses excessive SEI growth and degradation. As a result, the stability of the SEI layer and the reduction in interfacial resistance ensure that lithium-ion diffusion within the electrode remains efficient throughout extended cycling. Consequently, the PL-MrGO/LixSb2O3 electrode retains a high reversible capacity even after prolonged charge/discharge cycles, demonstrating its excellent long-term cycling stability.
Fig. 4d–g present the charge–discharge profiles of PL-MrGO/LixSb2O3, MrGO/Sb2O3, MrGO, and bare Sb2O3 electrodes, corresponding to Fig. 4a. The bare Sb2O3 electrode exhibits a distinct plateau at ∼0.9 V, whereas the PL-MrGO/LixSb2O3 and MrGO/Sb2O3 electrodes show more linear profiles, indicating more pseudocapacitive behaviour over the test voltage range of 0.01–3.0 V. Furthermore, the average charge potentials for each electrode are shown in Fig. S17.† The PL-MrGO/LixSb2O3 and MrGO/Sb2O3 electrodes, which incorporate a graphene matrix, exhibit a minimal polarization increase even as the current density increases from 100 to 1000 mA g−1, demonstrating the formation of highly conductive graphene–Sb2O3 composites. In contrast, the bare Sb2O3 electrode shows larger polarization and a significant increase in operating potential (∼0.2 V) with increasing current density along with poor capacity retention, highlighting its inferior electrical conductivity properties (Fig. S17†). Fig. 4h–k present the dQ/dV curves derived from the charge–discharge profiles. The MrGO/Sb2O3 and Sb2O3 electrodes exhibit a prominent irreversible lithiation peak at ∼1.3 V during the 1st cycle, attributed to the irreversible conversion reaction of Sb2O3. However, the PL-MrGO/LixSb2O3 electrode, having undergone pre-lithiation, already forms an amorphous LixSb2O3 phase, thereby avoiding such irreversible reactions in the 1st cycle.
The Ragone plots in Fig. S18† illustrate the energy density (W h kg−1) and power density (W kg−1) calculated based on the mass of active material for PL-MrGO/LixSb2O3 and MrGO/Sb2O3 electrodes in LIBs. The PL-MrGO/LixSb2O3 electrode consistently delivers a high energy density across various power density conditions. This observation suggests that the pre-lithiation process significantly enhances the electrode's reversible energy storage capacity and efficiency.
Several factors contribute to the superior performance of the PL-MrGO/LixSb2O3 electrode. Firstly, the pre-lithiation process improves the structural stability of the electrode material, thereby reducing structural damage during repeated charge/discharge cycles. Secondly, the combination of MrGO and Sb2O3 optimizes electron transport pathways and enhances conductivity, thereby maximizing the electrochemical performance of the electrode. Specifically, amorphous LixSb2O3 exhibits high energy density, while graphene nanosheets offer excellent conductivity and a large surface area, resulting in a synergistic effect between these two materials.
3.6. Lithium storage kinetics and reaction mechanism
Fig. 5a–d show the cyclic voltammetry (CV) curves for PL-MrGO/LixSb2O3, MrGO/Sb2O3, bare Sb2O3, and MrGO electrodes over the initial 15 cycles measured at a scan rate of 0.1 mV s−1 within the voltage range of 0.01–3.0 V. In the first cycle, the CV curve for the MrGO/Sb2O3 electrode exhibits two reduction peaks. The peak at 1.15 V corresponds to the partially reversible conversion reaction of Sb2O3 to Sb and the formation of Li2O, while the peak at 0.7 V is attributed to the alloying reaction of Sb and the formation of the SEI.85–87 However, for the PL-MrGO/LixSb2O3 electrode, the reduction peak at 0.7 V associated with the alloying reaction is observed, while the peak corresponding to the partially reversible conversion reaction of Sb2O3 is absent. This absence is due to the pre-lithiation process, which results in the formation of amorphous LixSb2O3. Additionally, the sharp peak around 0.01 V is attributed to the insertion of lithium ions into graphene. The oxidation peaks at 1.18 V for both MrGO/Sb2O3 and PL-MrGO/LixSb2O3 electrodes result from the alloying and de-alloying reactions between Sb and Li, while the peak at 1.48 V is due to the re-oxidation of Sb to Sb2O3. The redox reactions during the first cycle for the MrGO/Sb2O3 electrode can be summarized as follows:47| Sb2O3 + 6Li+ + 6e− → 2Sb + 3Li2O (partially reversible due to the formation of Li2O) |
| Sb + xLi+ + xe− → LixSb (x ≤ 3, reversible) |
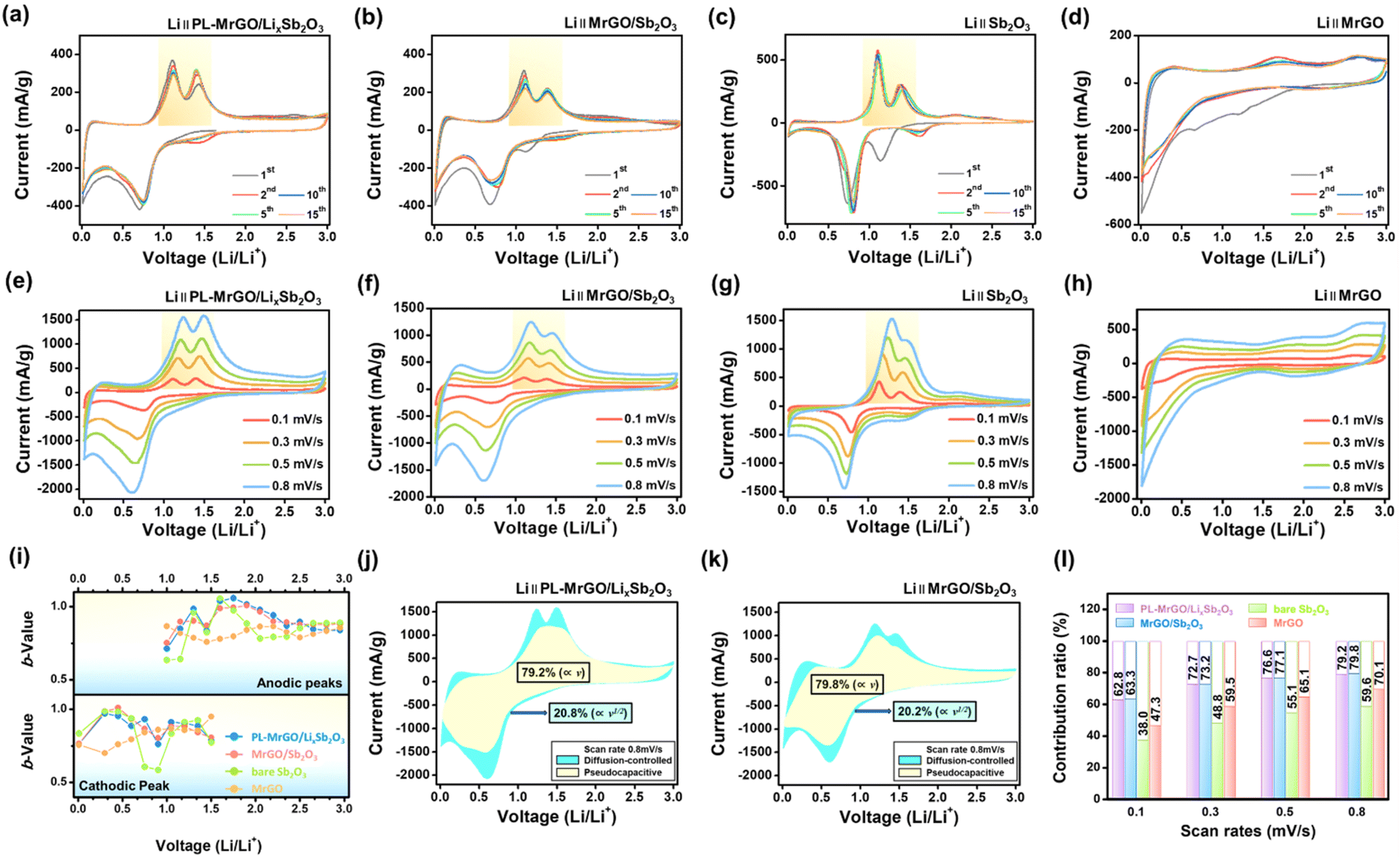 | ||
| Fig. 5 Lithium storage kinetics analysis: cyclic voltammetry (CV) curves of (a) PL-MrGO/LixSb2O3, (b) MrGO/Sb2O3, (c) bare Sb2O3 and (d) MrGO electrodes at a scan rate of 0.1 mV s−1. CV curves of (e) PL-MrGO/LixSb2O3, (f) MrGO/Sb2O3, (g) bare Sb2O3 and (h) MrGO electrodes at various scan rates (0.1–0.8 mV s−1). (i) Comparison of b-values at different voltages (0.01–3.0 V). Separation of the pseudocapacitive and diffusion-controlled charge storage processes at the (j) PL-MrGO/LixSb2O3 and (k) MrGO/Sb2O3 electrodes. (l) Comparison of the pseudocapacitive contributions. | ||
In subsequent cycles, the redox peak pairs corresponding to the conversion and alloying reactions are consistently observed at 1.53/1.48 V and 0.73/1.18 V, indicating the reversible nature of these processes in Sb2O3 nanoparticles. Notably, the oxidation peak at 1.48 V for the PL-MrGO/LixSb2O3 electrode is stronger than those for the MrGO/Sb2O3 and bare Sb2O3 electrodes, indicating a higher degree of reversibility in the conversion/alloying reaction. Furthermore, the CV curves for subsequent cycles overlap well after the 1st cycle, demonstrating the reversible lithium-ion storage behaviour of the electrode. Fig. 5e–h present the CV curves of PL-MrGO/LixSb2O3, MrGO/Sb2O3, MrGO, and bare Sb2O3 electrodes measured at various scan rates ranging from 0.1 to 0.8 mV s−1. The relationship between the peak current (i) and the scan rate (ν, mV s−1) is used to calculate the b-value (eqn (1) and (2)).
| i = avb | (1) |
| logi = blogv + loga | (2) |
Therefore, the b-values calculated for the reduction and oxidation processes of PL-MrGO/LixSb2O3 and MrGO/Sb2O3 electrodes show that both electrodes exhibit surface charge storage behaviour (Fig. 5i). Integrating the MrGO framework with small Sb2O3 nanoparticles provides rapid electron transport and efficient lithium-ion pathways, enhancing the diffusion-independent pseudocapacitive behaviour. Therefore, the capacitive-controlled and diffusion-controlled currents were calculated using eqn (3) and (4) to analyze the capacitive contribution quantitatively.
| i(V) = k1v + k2v½ | (3) |
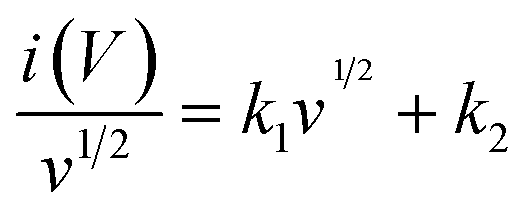 | (4) |
The terms k1v and k2v½ represent the capacitive and diffusion-controlled currents, respectively, allowing the quantification of the capacitive current from the CV curve at specific potentials (Fig. 5j and k).88–91Fig. 5l compares the capacitive contributions of PL-MrGO/LixSb2O3, MrGO/Sb2O3, MrGO, and bare Sb2O3 electrodes at various scan rates. The PL-MrGO/LixSb2O3 and MrGO/Sb2O3 electrodes exhibit the highest capacitive-controlled ratios, reaching 79.2% and 79.8% at 0.8 mV s−1, respectively. Therefore, the excellent cycling stability and rate performance of the PL-MrGO/LixSb2O3 and MrGO/Sb2O3 electrodes can be attributed to the diffusion-independent pseudocapacitive behaviour. However, since the capacitive contributions of the PL-MrGO/LixSb2O3 and MrGO/Sb2O3 electrodes show minimal differences, further EIS analysis was conducted to explain the performance enhancement before and after pre-lithiation.
Fig. 6a–c and S19† show the Nyquist plots of PL-MrGO/LixSb2O3, MrGO/Sb2O3, MrGO, and bare Sb2O3 electrodes measured at various charge/discharge states. All electrodes exhibit a decreasing trend in charge transfer resistance up to the 100th cycle. Fig. 6d–f present the distribution of relaxation times (DRT) plots for a more detailed comparison of the internal resistance changes for each electrode. The MrGO electrode showed the highest increase in resistance due to SEI formation in the high-frequency region after the 1st cycle.92–95 In contrast, the resistance increase due to SEI formation in the PL-MrGO/LixSb2O3 electrode was negligible. Furthermore, the PL-MrGO/LixSb2O3 electrode exhibited lower internal resistance over prolonged cycling compared to MrGO/Sb2O3, indicating its superior electrical conductivity.
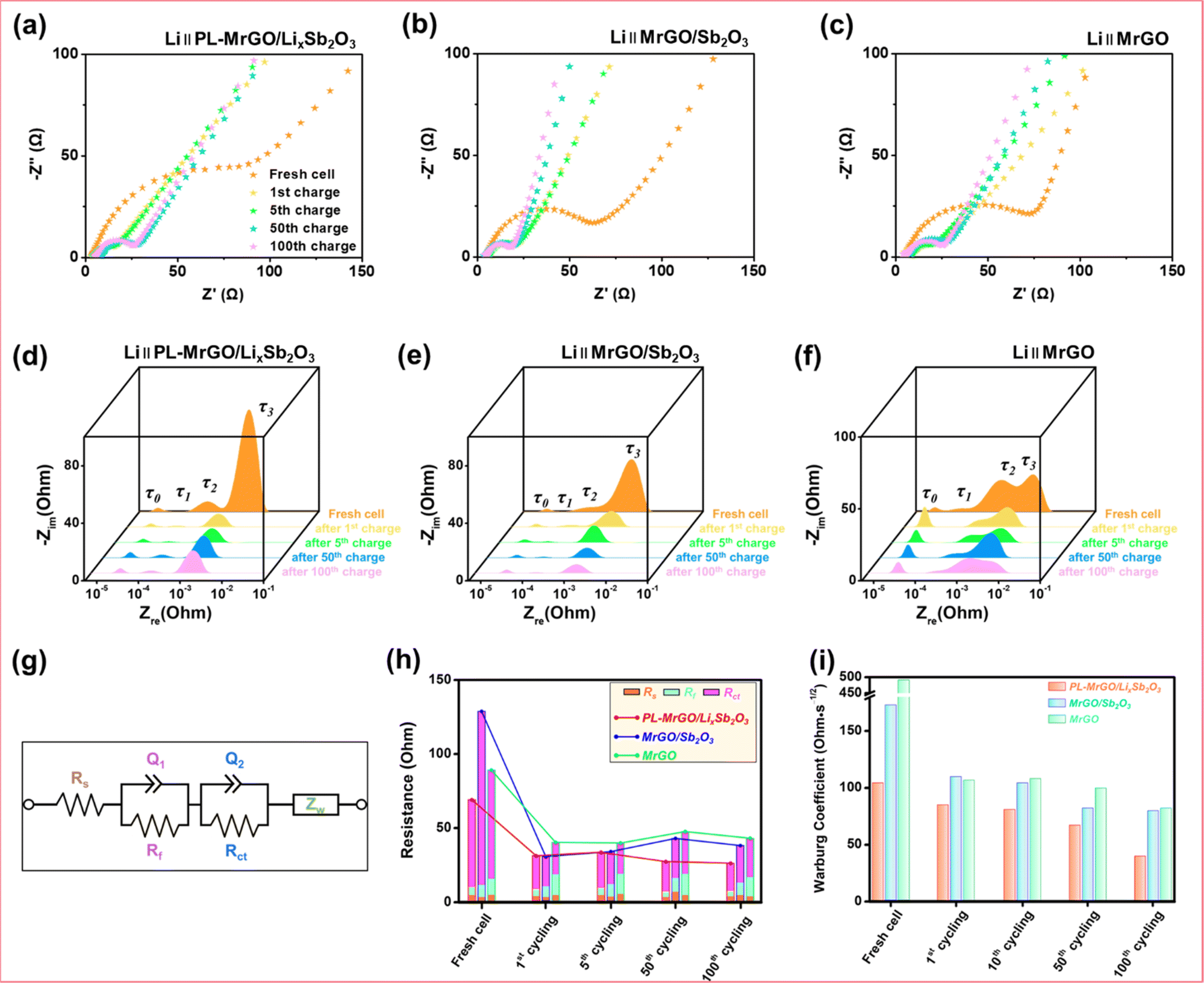 | ||
| Fig. 6 Electrochemical impedance spectroscopy analysis in LIBs: Nyquist plot of the (a) PL-MrGO/LixSb2O3, (b) MrGO/Sb2O3 and (c) MrGO electrodes measured up to the 100th cycle. Distribution of relaxation times (DRT) plot of the (d) PL-MrGO/LixSb2O3, (e) MrGO/Sb2O3 and (f) MrGO electrodes. (g) Equivalent circuit model (ECM) for fitting EIS data (the impedance parameters are provided in Table S6†). (h) Comparison of the parameters from the ECM fits and (i) Warburg coefficients. | ||
Using the Equivalent Circuit Model (ECM) presented in Fig. 6g, the impedance components of each electrode were separated and analyzed (Table S6†). The ECM fitting results for the PL-MrGO/LixSb2O3 electrode exhibit low charge-transfer resistance and SEI resistance, which are associated with the stable interfacial structure formed through chemical pre-lithiation (Fig. 6h). In contrast, the fitting results for the MrGO/Sb2O3 electrode indicate higher resistance values, reflecting the instability of the electrode–electrolyte interface. Additionally, the comparison of Warburg coefficients in Fig. 6i reveals that the PL-MrGO/LixSb2O3 electrode retains a low Warburg coefficient even after extended cycling, indicating efficient lithium-ion diffusion within the MrGO matrix.96–99 Conversely, the bare Sb2O3 electrode shows a sharp increase in the Warburg coefficient, signifying performance degradation related to ion diffusion (Fig. S19†). As a result, the PL-MrGO/LixSb2O3 electrode demonstrates excellent electrochemical performance and long-term stability, attributed to the stabilization of the SEI layer and improved conductivity.
3.7. Sodium storage performance of PS-MrGO/NaxSb2O3
Fig. 7a–c illustrate the performance of PS-MrGO/NaxSb2O3, MrGO/Sb2O3, bare Sb2O3, and MrGO electrodes in SIBs. The PS-MrGO/NaxSb2O3 electrode demonstrates superior rate capability and long-term cycling stability compared to the MrGO/Sb2O3 electrode. As shown in Fig. 7a, when the current density increases from 100 mA g−1 to 20000 mA g−1, the MrGO/Sb2O3, bare Sb2O3, and MrGO electrodes exhibit a sharp decline in capacity performance. In contrast, the PS-MrGO/NaxSb2O3 electrode retains a discharge capacity of over 78.0 mA h g−1, even under high current density conditions of 20000 mA g−1, indicating stable capacity performance at elevated current densities. Furthermore, after the current density is recovered to 100 mA g−1 at the 31st cycle, the PS-MrGO/NaxSb2O3 electrode sustains a discharge capacity of 386.2 mA h g−1 at the 300th cycle. In comparison, the bare Sb2O3 electrode loses most of its capacity performance. The comparison of charge/discharge capacities and capacity retention rates over the initial 60 cycles for PS-MrGO/NaxSb2O3, MrGO/Sb2O3, Sb2O3, and MrGO electrodes presented in Fig. 7a is shown in Fig. S20.†
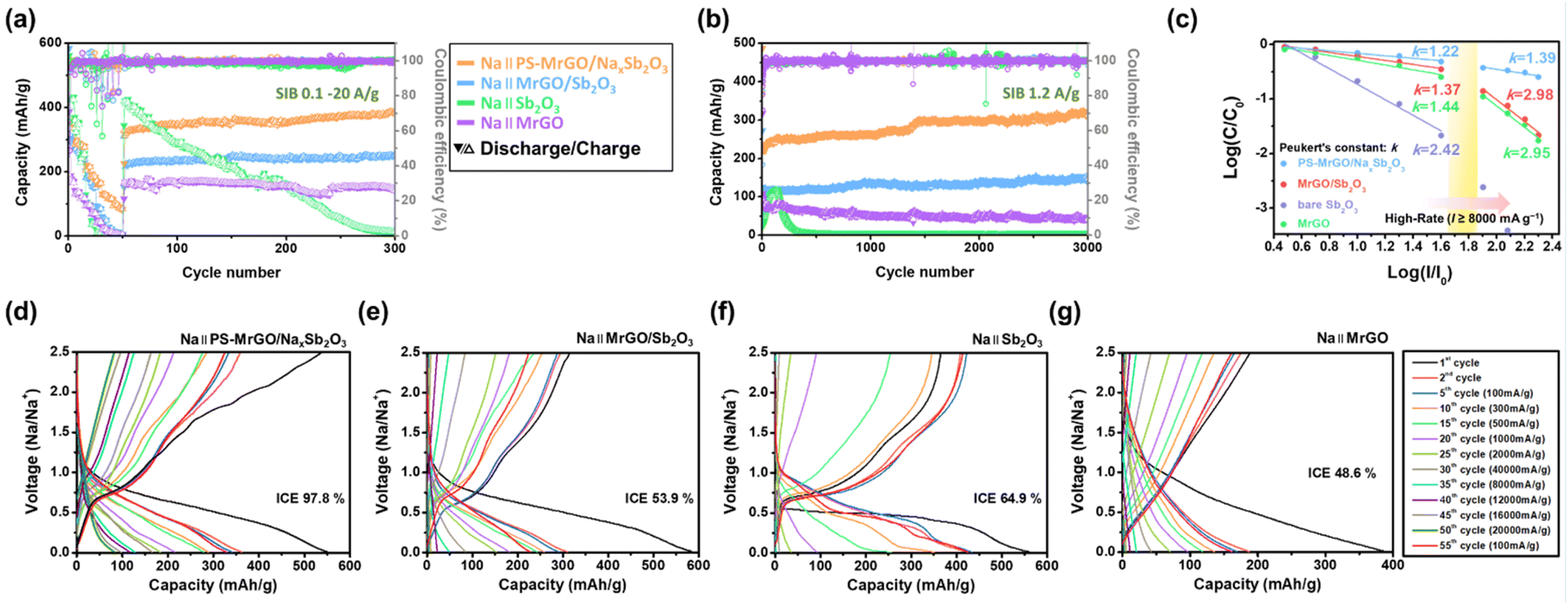 | ||
| Fig. 7 Sodium storage performance: (a) rate performance of PS-MrGO/NaxSb2O3, MrGO/Sb2O3, bare Sb2O3 and MrGO electrodes at different current densities (100–20000 mA g−1). (b) Long-term cycling performance at 1200 mA g−1. (c) Peukert's constant for PS-MrGO/NaxSb2O3, MrGO/Sb2O3, bare Sb2O3 and MrGO electrodes. Charge–discharge profiles of (d) PS-MrGO/NaxSb2O3, (e) MrGO/Sb2O3, (f) bare Sb2O3 and (g) MrGO electrodes at different current densities (100–20000 mA g−1). | ||
The Ragone plot in Fig. S21† illustrates the energy density (W h kg−1) and power density (W kg−1) of PS-MrGO/NaxSb2O3 and MrGO/Sb2O3 electrodes in SIBs. The PS-MrGO/NaxSb2O3 electrode exhibited superior energy density compared to the MrGO/Sb2O3 electrode under various power density conditions.
Fig. 7b illustrates the long-term cycling performance of the electrodes at a high current density of 1200 mA g−1. Due to the formation of an inorganic SEI layer and the amorphization of Sb2O3 through pre-sodiation, the PS-MrGO/NaxSb2O3 electrode exhibits superior long-term cycling stability compared to the MrGO/Sb2O3, bare Sb2O3, and MrGO electrodes. After 3000 cycles, the PS-MrGO/NaxSb2O3 electrode retains a reversible capacity of 313.5 mA h g−1, demonstrating excellent durability. Peukert's constant in Fig. 7c, an indicator used to assess rate performance, also suggests that the PS-MrGO/NaxSb2O3 electrode has a superior rate capability compared to the other electrodes. Additionally, as shown in Fig. 7d–g, the PS-MrGO/NaxSb2O3 electrode achieves an ICE of 97.8%. In contrast, the MrGO electrode displays a lower ICE due to the irreversible trapping of Na-ions.
The performance of PS-MrGO/NaxSb2O3 and MrGO/Sb2O3 electrodes has been compared with the performance metrics of recently reported Sb-based electrodes, as presented in Table S7.†
3.8. Sodium storage kinetics and reaction mechanism
Fig. 8a and b show the CV curves of the PS-MrGO/NaxSb2O3 and MrGO/Sb2O3 electrodes. Additionally, the CV curves of the MrGO and bare Sb2O3 electrodes are presented in Fig. S22.† The CV curves of the MrGO/Sb2O3 electrode exhibit reduction peaks at approximately 0.3 V and 0.6 V and oxidation peaks at 0.8 V and 1.8 V. These peaks correspond to the conversion reactions between antimony oxide and sodium ions.100–102 In contrast, the PS-MrGO/NaxSb2O3 electrode does not show distinct reduction peaks during the first cycle and displays broader peaks compared to the MrGO/Sb2O3 electrode. Furthermore, the CV curve exhibits a nearly rectangular shape, which suggests an increased pseudocapacitive behaviour in the electrode. | ||
| Fig. 8 Sodium storage kinetics analysis: CV profiles of (a) PS-MrGO/NaxSb2O3, and (b) MrGO/Sb2O3 at a scan rate of 0.1 mV s−1. CV curves of (c) PS-MrGO/NaxSb2O3 and (d) MrGO/Sb2O3 electrodes at various scan rates (0.1–0.8 mV s−1); (e) comparison of b-values at different voltage (0.01–2.5 V); separation of the pseudocapacitive and diffusion-controlled charge storage processes at the (f) PS-MrGO/NaxSb2O3 and (g) MrGO/Sb2O3 electrodes; (h) comparison of the pseudocapacitive contributions. | ||
To further analyze the ion storage behaviour in detail, CV curves were measured at various scan rates (0.1–0.8 mV s−1) (Fig. 8c and d). PS-MrGO/NaxSb2O3 and MrGO/Sb2O3 electrodes exhibited a gradual shift in redox peak positions with increasing scan rates, along with a linear increase in current density. The peak position shifts were notably more pronounced in the MrGO/Sb2O3 electrode than in the PS-MrGO/NaxSb2O3 electrode. This observation suggests that the charge storage mechanism in the MrGO/Sb2O3 electrode relies more heavily on diffusion-controlled processes. In contrast, the PS-MrGO/NaxSb2O3 electrode displayed relatively stable peak shifts, indicating that surface-based pseudocapacitive processes more significantly influence the charge storage mechanism. Additionally, the PS-MrGO/NaxSb2O3 electrode demonstrated a b-value of approximately 0.94–0.77, confirming that its charge storage mechanism is predominantly governed by pseudocapacitive reactions (Fig. 8e). These findings clearly indicate that the PS-MrGO/NaxSb2O3 electrode possesses superior reaction kinetics and enhanced ion diffusion rates on the electrode surface compared to the MrGO/Sb2O3 electrode.
In Fig. 8f, the MrGO/Sb2O3 electrode exhibits a pseudocapacitive contribution of approximately 75.8% at a scan rate of 0.8 mV s−1. In contrast, the pseudocapacitive contribution of the PS-MrGO/NaxSb2O3 electrode remains 77.5%, as shown in Fig. 8g. Furthermore, Fig. 8h illustrates that the PS-MrGO/NaxSb2O3 electrode consistently demonstrates higher pseudocapacitive contributions across all scan rates (0.1–0.8 mV s−1) compared to the MrGO/Sb2O3 electrode. These findings highlight the enhanced electrochemical reactivity at the electrode surface, attributed to the robust composite structure formed by integrating PS-MrGO and NaxSb2O3. Additionally, the stabilization of the SEI plays a role in maintaining the high pseudocapacitive behaviour of the PS-MrGO/NaxSb2O3 electrode.
Fig. 9a–c show the Nyquist plots of the PS-MrGO/NaxSb2O3, MrGO/Sb2O3, and MrGO electrodes. The EIS analysis results for the bare Sb2O3 electrode are presented in Fig. S23.† The PS-MrGO/NaxSb2O3 and MrGO/Sb2O3 electrodes exhibit lower high-frequency resistance (film resistance) and mid-frequency charge transfer resistance compared to the MrGO and bare Sb2O3 electrodes during both the initial charge/discharge cycle and the 100th cycle.
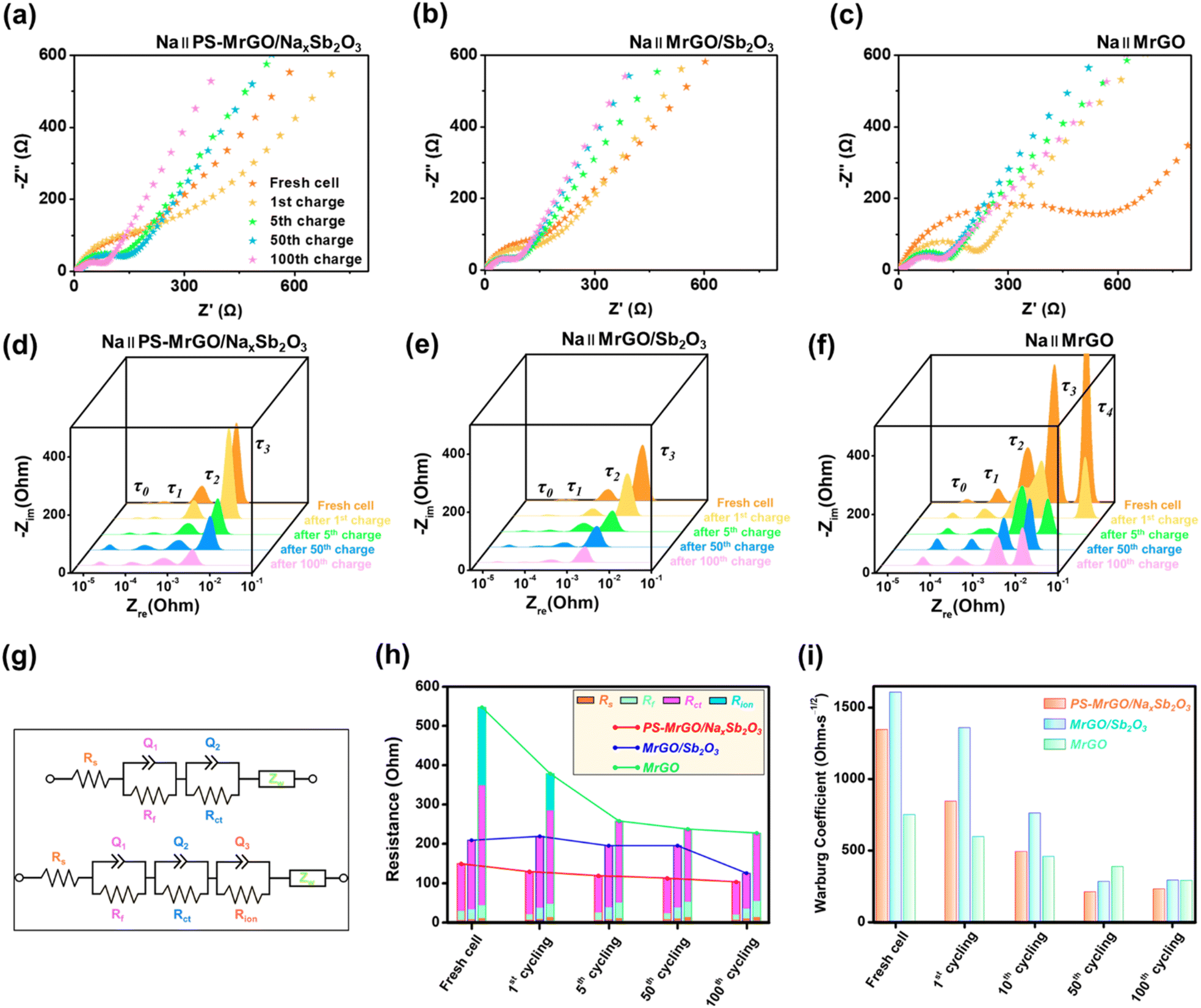 | ||
| Fig. 9 Electrochemical impedance spectroscopy analysis in SIBs: Nyquist plot of the (a) PS-MrGO/NaxSb2O3, (b) MrGO/Sb2O3 and (c) MrGO electrodes measured up to the 100th cycle. DRT plot of the (d) PS-MrGO/NaxSb2O3, (e) MrGO/Sb2O3 and (f) MrGO electrodes. (g) ECM for fitting EIS data (the impedance parameters are provided in Table S8†). (h) Comparison of the parameters from the ECM fits and (i) Warburg coefficients. | ||
According to the DRT analysis, the PS-MrGO/NaxSb2O3 electrode exhibits relatively low resistance across all frequency ranges (Fig. 9d–f). In particular, this electrode shows a minimal resistance increase due to the formation of the SEI layer. These results indicate that the PS-MrGO/NaxSb2O3 electrode possesses high electrical conductivity and stable interfacial properties, with low kinetic barriers for electrode reactions. In contrast, the MrGO and bare Sb2O3 electrodes exhibit significant resistance increases attributed to SEI layer formation, as revealed by the DRT analysis (Fig. S23†). This suggests that these electrodes have non-uniform electrochemical active sites and suffer from substantial degradation in reactivity after cycling. Compared to the PS-MrGO/NaxSb2O3 electrode, the increased SEI resistance observed in the bare Sb2O3 electrodes reflects structural deterioration. These factors act as major limitations to the long-term performance stability of these electrodes. As shown in Fig. 9g and h, ECM fitting was performed to quantitatively separate the electrochemical resistance components of each electrode. The impedance parameters obtained from the EIS data fitting are provided in Table S8.† The PS-MrGO/NaxSb2O3 electrode exhibited the lowest charge transfer resistance and SEI resistance, which can be attributed to the formation of a stable and highly conductive interfacial structure facilitated by chemical pre-sodiation. Furthermore, the PS-MrGO/NaxSb2O3 electrode exhibited a low Warburg coefficient even after 100 cycles, indicating a reduction in diffusion resistance during cycling (Fig. 9i).
In conclusion, the PS-MrGO/NaxSb2O3 electrode demonstrated excellent capacity performance and long-term cycling stability in SIBs, achieved through the stabilization of the SEI layer, low charge transfer resistance, and enhanced ionic conductivity.
4 Conclusions
In this study, PL(S)-MrGO/Li(Na)xSb2O3 electrodes, exhibiting excellent performance in LIBs and SIBs, were developed through a simple and efficient synthesis method involving ultrasonication, microwave processing, and chemical pre-lithiation/sodiation. The PL(S)-MrGO/Li(Na)xSb2O3 electrode achieved an ICE close to 100%, with the formation of an inorganic SEI layer and amorphization of Sb2O3 contributing to the suppression of irreversible Li/Na-ion trapping. These features minimized irreversible reactions during charge/discharge cycles, enhancing long-term cycling stability and electrical conductivity. Notably, the PL(S)-MrGO/Li(Na)xSb2O3 electrode demonstrated superior long-term cycling stability and high-rate performance compared to the MrGO/Sb2O3, bare Sb2O3, and MrGO electrodes. This approach presents a promising solution to address the initial capacity loss and low ICE issues frequently observed in conventional reduced graphene oxide and antimony oxide-based electrodes.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Minseop Lee: conceptualization, methodology, synthesis, characterization, writing – original draft. Gi-Chan Kim: investigation, synthesis, characterization, Seung-Min Paek: methodology, supervision, validation, funding acquisition, writing – review & editing.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This study was supported by grants from the Basic Science Research Program (RS-2024-00345599).References
- P. Simon and Y. Gogotsi, Nat. Mater., 2020, 19, 1151–1163 CrossRef CAS.
- T. Cao, C. Zhu, X. Wang, Z. Ji, H. Liang, J. Shi, W. Tian, J. Chen, J. Wu and H. Wang, Sustain. Energy Fuels, 2024, 8, 4790–4798 Search PubMed.
- F. Huang, P. Xu, G. Fang and S. Liang, Adv. Mater., 2024, 36, 2405310 CrossRef CAS.
- P. Xu, F. Huang, Y. Sun, Y. Lei, X. Cao, S. Liang and G. Fang, Adv. Funct. Mater., 2024, 34, 2406080 CrossRef CAS.
- J. Khan, A. Ahmed, M. I. Saleem and A. A. Al-Kahtani, Sustain. Energy Fuels, 2024, 8, 4355–4364 RSC.
- M. Lee, J. Xie, J.-M. Oh and S.-M. Paek, Chem. Eng. J., 2025, 506, 159671 CrossRef CAS.
- P. Kulkarni, H. Jung, D. Ghosh, M. Jalalah, M. Alsaiari, F. A. Harraz and R. G. Balakrishna, J. Energy Chem., 2023, 76, 479–494 CrossRef CAS.
- L. Jin, C. Shen, Q. Wu, A. Shellikeri, J. Zheng, C. Zhang and J. P. Zheng, Adv. Sci., 2021, 8, 2005031 CrossRef CAS PubMed.
- Y. Wang, J. Lu, W. Dai, X. Cheng, J. Zuo, H. Lei, W. Liu and Z. Fu, Adv. Funct. Mater., 2024, 34, 2403841 CrossRef CAS.
- J. S. Ko, B. Tan, M. W. Logan, S. A. Langevin and K. Gerasopoulos, J. Mater. Chem. A, 2024, 12, 14354–14359 RSC.
- T. Panneerselvam, R. Murugan and O. V. Sreejith, Sustain. Energy Fuels, 2024, 8, 1704–1711 RSC.
- S. Lin, H. Zhang, C. Shu, W. Hua, X. Wang, Y. Zhao, J. Luo, Z. Tang, Y. Wu and W. Tang, Adv. Funct. Mater., 2024, 34, 2409628 CrossRef CAS.
- Q. Liu, J. Chen, D. Du, S. Zhang, C. Zhu, Z. Zhang, C. Wang, L. Yin and R. Wang, J. Mater. Chem. A, 2023, 11, 17491–17502 RSC.
- C. Guo, B. Zhang, M. Xiao, M. Hao, L. Zhao, X. Zhang, H. Zhang and R. Wang, Sustain. Energy Fuels, 2024, 8, 837–842 RSC.
- Z. Lin, H. Zhang, C. Yang, Z. Liu, D. Wen, X. Peng, S. Li and X. Wu, Sustain. Energy Fuels, 2024, 8, 934–941 RSC.
- Z. Li, M. Du, X. Guo, D. Zhang, Q. Wang, H. Sun, B. Wang and Y. A. Wu, Chem. Eng. J., 2023, 473, 145294 CrossRef CAS.
- M. Yasoubi, A. Habibi, S. Hoornam, Z. Sanaee and S. Mohajerzadeh, Sustain. Energy Fuels, 2024, 8, 3419–3437 RSC.
- M. Lee, S. Park, B. Bae, Y. K. Jeong, J.-M. Oh, J. K. Park and S.-M. Paek, Chem. Eng. J., 2023, 477, 147072 CrossRef CAS.
- R. Wang, L. Wang, R. Liu, X. Li, Y. Wu and F. Ran, ACS Nano, 2024, 18, 2611–2648 CrossRef CAS PubMed.
- R. Mo, F. Li, X. Tan, P. Xu, R. Tao, G. Shen, X. Lu, F. Liu, L. Shen, B. Xu, Q. Xiao, X. Wang, C. Wang, J. Li, G. Wang and Y. Lu, Nat. Commun., 2019, 10, 1–10 CrossRef CAS PubMed.
- C. S. Bongu, S. Tasleem, M. R. Krishnan and E. H. Alsharaeh, Sustain. Energy Fuels, 2024, 8, 4039–4070 RSC.
- C.-Y. Wang, T. Liu, X.-G. Yang, S. Ge, N. V. Stanley, E. S. Rountree, Y. Leng and B. D. McCarthy, Nature, 2022, 611, 485–490 CrossRef CAS PubMed.
- M. Lee and S.-M. Paek, Nanomaterials, 2022, 12, 1507 CrossRef CAS.
- M. Lee, M.-S. Kim, J.-M. Oh, J. K. Park and S.-M. Paek, ACS Nano, 2023, 17, 3019–3036 CrossRef CAS.
- C. Wang, S. Xue, X. Lei, J. Wen, X. Pan, F. Zhang, C. Zou and Y. Tang, Chem. Eng. J., 2023, 470, 144043 CrossRef CAS.
- D. Zhao, Z. Zhang, J. Ren, Y. Xu, X. Xu, J. Zhou, F. Gao, H. Tang, S. Liu, Z. Wang, D. Wang, Y. Wu, X. Liu and Y. Zhang, Chem. Eng. J., 2023, 451, 138882 CrossRef CAS.
- J. Rehman, J. Gao, T. Yu, A. El-marghany and G. Yang, J. Mater. Chem. A, 2024, 12, 6703–6711 RSC.
- S. Liang, Y.-J. Cheng, J. Zhu, Y. Xia and P. Müller-Buschbaum, Small Methods, 2020, 4, 2000218 CrossRef CAS.
- S. Sarkar and S. C. Peter, J. Mater. Chem. A, 2021, 9, 5164–5196 RSC.
- W. T. Jing, C. C. Yang and Q. Jiang, J. Mater. Chem. A, 2020, 8, 2913–2933 RSC.
- L. Feng, J. Chen, Y. Li, S. Zhou, R. A. Soomro, P. Zhang and B. Xu, Chem. Eng. J., 2024, 489, 151396 CrossRef CAS.
- X. Shi, W. Liu, H. Xue, B. Chen, C. Wang, L. Sun, L. Chang, Y. Cheng and L. Wang, J. Power Sources, 2021, 506, 230074 CrossRef CAS.
- A. Xu, C. Huang, G. Li, K. Zou, H. Sun, L. Fu, J. Ju, Y. Song, S. Wu, Z. Xu and Y. Yan, J. Mater. Chem. A, 2021, 9, 12169–12178 RSC.
- H. Bryngelsson, J. Eskhult, L. Nyholm, M. Herranen, O. Alm and K. Edström, Chem. Mater., 2007, 19, 1170–1180 CrossRef CAS.
- S. Kim, S. Qu, R. Zhang and P. V. Braun, Small, 2019, 15, 1900258 CrossRef.
- J. Zheng, Y. Yang, X. Fan, G. Ji, X. Ji, H. Wang, S. Hou, M. R. Zachariah and C. Wang, Energy Environ. Sci., 2019, 12, 615–623 RSC.
- J. Song, P. Yan, L. Luo, X. Qi, X. Rong, J. Zheng, B. Xiao, S. Feng, C. Wang, Y.-S. Hu, Y. Lin, V. L. Sprenkle and X. Li, Nano Energy, 2017, 40, 504–511 CrossRef CAS.
- Y. Li, Z. Song, T. Sun, Y. Shen, X. Lv, D. Xu and H.-G. Wang, Int. J. Hydrogen Energy, 2021, 46, 26308–26317 CrossRef CAS.
- Z. Yi, Q. Han, P. Zan, Y. Wu, Y. Cheng and L. Wang, J. Power Sources, 2016, 331, 16–21 CrossRef CAS.
- B. Jin, K. Zhang, G. Gao, Q. Zhao, X. Jiang, D. Cui, K. Chen, X. Lin, L. Liu, R. Yan, B. Yang and Y. Yao, Chem. Eng. J., 2024, 493, 152542 CrossRef CAS.
- R. Yi, S. Hu, L. Zheng, Y. Li, W. Luo, H. Zhang, J. Ge, Y. Shen and L. Chen, Chem. Eng. J., 2024, 500, 157201 CrossRef CAS.
- X. Li, X. Sun, X. Hu, F. Fan, S. Cai, C. Zheng and G. D. Stucky, Nano Energy, 2020, 77, 105143 CrossRef CAS.
- D. Liu, Y. Qiu, Y. Du, J. Yang, X. Jing, X. Peng, Q. Song and F. Xu, J. Mater. Chem. A, 2024, 12, 16695–16703 RSC.
- H. He, D. Sun, Y. Tang, H. Wang and M. Shao, Energy Storage Mater., 2019, 23, 233–251 CrossRef.
- L. Sun, Y. Liu, J. Wu, R. Shao, R. Jiang, Z. Tie and Z. Jin, Small, 2022, 18, 2102894 CrossRef CAS.
- M. Liu, F. Wu, Y. Gong, Y. Li, Y. Li, X. Feng, Q. Li, C. Wu and Y. Bai, Adv. Mater., 2023, 35, 2300002 CrossRef CAS PubMed.
- R. Shao, Z. Sun, L. Wang, J. Pan, L. Yi, Y. Zhang, J. Han, Z. Yao, J. Li, Z. Wen, S. Chen, S.-L. Chou, D.-L. Peng and Q. Zhang, Angew. Chem., 2024, 136, 2320183 Search PubMed.
- J. Yang, J. Li, T. Wang, P. H. L. Notten, H. Ma, Z. Liu, C. Wang and G. Wang, Chem. Eng. J., 2021, 407, 127169 CrossRef CAS.
- H. Li, K. Qian, X. Qin, D. Liu, R. Shi, A. Ran, C. Han, Y.-B. He, F. Kang and B. Li, J. Power Sources, 2018, 385, 114–121 CrossRef CAS.
- W. S. Hummers Jr and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef.
- T. H. Wan, M. Saccoccio, C. Chen and F. Ciucci, Electrochim. Acta, 2015, 184, 483–499 CrossRef CAS.
- W. Zhang, H. Xu, F. Xie, X. Ma, B. Niu, M. Chen, H. Zhang, Y. Zhang and D. Long, Nat. Commun., 2022, 13, 1–10 CAS.
- J. Jang, I. Kang, J. Choi, H. Jeong, K.-W. Yi, J. Hong and M. Lee, Angew. Chem., 2020, 59, 14473–14480 CrossRef CAS PubMed.
- G. Wang, F. Li, D. Liu, D. Zheng, Y. Luo, D. Qu, T. Ding and D. Qu, ACS Appl. Mater. Interfaces, 2019, 11, 8699–8703 CrossRef CAS PubMed.
- J. Peng, D. Wu, P. Lu, Z. Wang, Y. Du, Y. Wu, Y. Wu, W. Yan, J. Wang, H. Li, L. Chen and F. Wu, Energy Storage Mater., 2023, 54, 430–439 CrossRef.
- Y. Kim, J. Jung, H. Yu, G.-T. Kim, D. Jeong, D. Bresser, S. J. Kang, Y. Kim and S. Passerini, Adv. Funct. Mater., 2020, 30, 2001249 CrossRef CAS.
- X. Liu, Y. Tan, T. Liu, W. Wang, C. Li, J. Lu and Y. Sun, Adv. Funct. Mater., 2019, 29, 1903795 CrossRef CAS.
- K. Zou, W. Deng, P. Cai, X. Deng, B. Wang, C. Liu, J. Li, H. Hou, G. Zou and X. Ji, Adv. Funct. Mater., 2021, 31, 2005581 CrossRef CAS.
- G. Zheng, Q. Lin, J. Ma, J. Zhang, Y.-B. He, X. Tang, F. Kang, W. Lv and Q.-H. Yang, InfoMat, 2021, 3, 1445–1454 CrossRef CAS.
- M. Liu, J. Zhang, S. Guo, B. Wang, Y. Shen, X. Ai, H. Yang and J. Qian, ACS Appl. Mater. Interfaces, 2020, 12, 17620–17627 CrossRef CAS PubMed.
- M. Liu, Z. Yang, Y. Shen, S. Guo, J. Zhang, X. Ai, H. Yang and J. Qian, J. Mater. Chem. A, 2021, 9, 5639–5647 RSC.
- R. Mei, X. Song, Y. Hu, Y. Yang and J. Zhang, Electrochim. Acta, 2015, 153, 540–545 CrossRef CAS.
- D. Jiao, Z. Xie, Q. Wan and M. Qu, J. Energy Chem., 2019, 37, 73–81 CrossRef.
- S. Wu, G. Fu, W. Lv, J. Wei, W. Chen, H. Yi, M. Gu, X. Bai, L. Zhu, C. Tan, Y. Liang, G. Zhu, J. He, X. Wang, K. H. L. Zhang, J. Xiong and W. He, Small, 2018, 14, 1702667 CrossRef PubMed.
- X. Dong, L. Li, C. Zhao, H.-K. Liu and Z. Guo, J. Mater. Chem. A, 2014, 2, 9844–9850 RSC.
- W.-J. Lee, H.-R. Jang, M.-J. Kim, H.-M. Kim, J.-M. Oh and S.-M. Paek, J. Alloys Compd., 2019, 778, 382–390 CrossRef CAS.
- G. Huang, P. Song, L. Liu, D. Han, C. Ge, R. Li and Q. Guo, Carbon, 2016, 98, 689–701 CrossRef CAS.
- K. P. Lakshmi, R. Deivanayagam and M. M. Shaijumon, J. Alloys Compd., 2021, 857, 158267 CrossRef CAS.
- N. Li, Y. Xia, Z. Mao, L. Wang, Y. Guan and A. Zheng, Polym. Degrad. Stab., 2012, 97, 1737–1744 CrossRef CAS.
- X. Yang, Y. Zhu, D. Wu, M. Li, Y. He, L. Huang and M. Gu, Adv. Funct. Mater., 2022, 32, 2111391 CrossRef CAS.
- L. Zhang, Y. Song, Y. Hu, H. Ruan, J. Bai, S. Li, Y. Liu and S. Guo, J. Alloys Compd., 2022, 890, 161913 CrossRef CAS.
- X. Zhou, Z. Zhang, X. Lu, X. Lv, G. Ma, Q. Wang and Z. Lei, ACS Appl. Mater. Interfaces, 2017, 9, 34927–34936 CrossRef CAS PubMed.
- K. Pfeifer, M. F. Greenstein, D. Aurbach, X. Luo, H. Ehrenberg and S. Dsoke, Chemelectrochem, 2020, 7, 3487–3495 CrossRef CAS.
- O. A. Jaramillo-Quintero, R. V. Barrera-Peralta, A. Baron-Jaimes, R. A. Miranda-Gamboa and M. E. Rincon, RSC Adv., 2021, 11, 31566–31571 RSC.
- Z. Han, J. Zheng, F. Kong, S. Tao and B. Qian, Nano Sel., 2021, 2, 425–432 CrossRef CAS.
- M. B. Costa, F. W. S. Lucas, M. Medina and L. H. Mascaro, ACS Appl. Energy Mater., 2020, 3, 9799–9808 CrossRef CAS.
- F. Zhang, W. Zhang, J. A. Yuwono, D. Wexler, Y. Fan, J. Zou, G. Liang, L. Sun and Z. Guo, Nat. Commun., 2024, 15, 1–11 CAS.
- B. Zhao, J. Li, M. Guillaume, J. Dendooven and C. Detavernier, J. Energy Chem., 2022, 66, 295–305 CrossRef CAS.
- S. Malmgren, K. Ciosek, R. Lindblad, S. Plogmaker, J. Kühn, H. Rensmo, K. Edström and M. Hahlin, Electrochim. Acta, 2013, 105, 83–91 CrossRef CAS.
- J. P. Tonks, M. O. King, E. C. Galloway and J. F. Watts, J. Nucl. Mater., 2017, 484, 228–235 CrossRef CAS.
- N. Hornsveld, B. Put, W. M. M. Kessels, P. M. Vereecken and M. Creatore, RSC Adv., 2017, 7, 41359–41368 RSC.
- M. L. Kalapsazova, E. N. Zhecheva, G. T. Tyuliev, D. D. Nihtianova, L. Mihaylov and R. K. Stoyanova, J. Phys. Chem. C, 2017, 121, 5931–5940 CrossRef CAS.
- C. Hou, Z. Yue, H. Sun, L. Zhai, H. Xie, H. Tian, Y. Qu, X. Wang and J. Hou, J. Mater. Sci.: Mater. Electron., 2024, 35, 956 CrossRef CAS.
- B. Qin, A. Schiele, Z. Jusys, A. Mariani, T. Diemant, X. Liu, T. Brezesinski, R. J. Behm, A. Varzi and S. Passerini, ACS Appl. Mater. Interfaces, 2020, 12, 3697–3708 CrossRef CAS PubMed.
- Z. Wang, Y. Cheng, Q. Li, L. Chang and L. Wang, J. Power Sources, 2018, 389, 214–221 CrossRef CAS.
- Z. Wang, F. Zeng, D. Zhang, X. Wang, W. Yang, Y. Cheng, C. Li and L. Wang, Electrochim. Acta, 2021, 395, 139210 CrossRef CAS.
- O. A. Jaramillo-Quintero, M. Benítez-Cruz, J. L. García-Ocampo, A. Cano and M. E. Rincón, J. Alloys Compd., 2019, 807, 151647 CrossRef CAS.
- V. Augustyn, P. Simon and B. Dunn, Energy Environ. Sci., 2014, 7, 1597 RSC.
- C. Choi, D. S. Ashby, D. M. Butts, R. H. DeBlock, Q. Wei, J. Lau and B. Dunn, Nat. Rev. Mater., 2019, 5, 5–19 CrossRef.
- M. Lee, J.-H. Park and S.-M. Paek, New J. Chem., 2024, 48, 2381–2388 RSC.
- J.-H. Jang, M. Lee, S. Park, J.-M. Oh, J. K. Park and S.-M. Paek, J. Mater. Chem. A, 2023, 11, 13320–13330 RSC.
- Y. Lu, C.-Z. Zhao, J.-Q. Huang and Q. Zhang, Joule, 2022, 6, 1172–1198 CrossRef CAS.
- X. Zhou, Z. Pan, X. Han, L. Lu and M. Ouyang, J. Power Sources, 2019, 417, 188–192 CrossRef CAS.
- Y. Lu, C.-Z. Zhao, R. Zhang, H. Yuan, L.-P. Hou, Z.-H. Fu, X. Chen, J.-Q. Huang and Q. Zhang, Sci. Adv., 2021, 7, eabi5520 CrossRef CAS PubMed.
- J.-H. Jang, M. Lee, J.-H. Koo and S.-M. Paek, Int. J. Mol. Sci., 2022, 23, 11766 CrossRef CAS PubMed.
- B. Manikandan, V. Ramar, C. Yap and P. Balaya, J. Power Sources, 2017, 361, 300–309 CrossRef CAS.
- M.-S. Kim, M. Lee, M.-J. Kim, Y. K. Jeong, J. K. Park and S.-M. Paek, J. Mater. Chem. A, 2020, 8, 17790–17799 RSC.
- M. Lee, M.-S. Kim, J.-M. Oh, J. K. Park and S.-M. Paek, ChemSusChem, 2021, 14, 3244–3256 CrossRef CAS PubMed.
- M. Liang, H. Zhang, B. Chen, X. Meng, J. Zhou, L. Ma, F. He, W. Hu, C. He and N. Zhao, Adv. Mater., 2023, 35, 2307209 CrossRef CAS PubMed.
- M. Hu, Y. Jiang, W. Sun, H. Wang, C. Jin and M. Yan, ACS Appl. Mater. Interfaces, 2014, 6, 19449–19455 CrossRef CAS PubMed.
- B. Selvaraj, C.-C. Wang, Y.-F. Song, H.-S. Sheu, Y.-F. Liao and N.-L. Wu, J. Mater. Chem. A, 2020, 8, 22620–22625 RSC.
- W. Ma, J. Wang, H. Gao, J. Niu, F. Luo, Z. Peng and Z. Zhang, Energy Storage Mater., 2018, 13, 247–256 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5se00172b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |