
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn situ formation of ultrahigh molecular weight polymers in highly concentrated electrolytes and their physicochemical properties†
Yuji
Kamiyama
a,
Takeshi
Ueki
 ab and
Ryota
Tamate
*ac
ab and
Ryota
Tamate
*ac
aResearch Center for Macromolecules & Biomaterials, National Institute for Materials Science, 1-2-1 Sengen, Tsukuba 305-0047, Japan. E-mail: TAMATE.Ryota@nims.go.jp
bGraduate School of Life Science, Hokkaido University, Kita 10, Nishi 8, Kita-ku, Sapporo, Hokkaido 060-0810, Japan
cPRESTO, JST, 7 Gobancho, Chiyoda-ku, Tokyo 102-0076, Japan
First published on 10th January 2025
Abstract
We developed a facile one-pot method for fabricating physical gels consisting of ultrahigh molecular weight (UHMW) polymers and highly concentrated lithium salt electrolytes. We previously reported physical gels formed from the entanglement of UHMW polymers by radical polymerisation in aprotic ionic liquids. In this study, we found that the molecular weight of methacrylate polymers formed by radical polymerisation increased with the concentration of lithium salts in the organic solvents. Consequently, the synthesis of UHMW polymers with a high monomer conversion was achieved at very low initiator concentrations, leading to the formation of physical gels in highly concentrated electrolytes by the chain entanglement of UHMW polymers. The viscoelastic and mechanical properties of the UHMW gel electrolytes and their self-healing properties were investigated in detail.
Introduction
Gel electrolytes are soft materials that consist of a three-dimensional polymer network swollen with an electrolyte solution. They have been applied in various electrochemical devices, such as lithium secondary batteries, capacitors, sensors, actuators, and transistors.1–6 In recent years, applications in wearable devices and high-energy-density secondary batteries have required gel electrolytes that can withstand repetitive mechanical inputs and intense electrode interface phenomena. To meet this demand, numerous gel electrolytes with excellent mechanical and electrochemical properties have been reported.7–11 Wu et al. developed a gel electrolyte with a double network structure and showed that it can improve the performance of a lithium metal anode.12 Fujii et al. synthesised a gel electrolyte with a uniform network structure via an A–B-type coupling reaction of tetra-branched poly(ethylene glycol) in the electrolyte.13 Recently, we developed a highly tough and stretchable hydrogen-bonded gel electrolyte and applied it as artificial interphase for a lithium metal anode.14 Furthermore, there have been many reports on the preparation of self-healing gel electrolytes via reversible crosslinking such as hydrogen bonding, ionic bonding, and physical entanglement.15–22 For example, Jaumaux et al. created self-healing gel electrolytes via hydrogen bonding and applied them to lithium metal batteries.23We previously reported ultrahigh molecular weight (UHMW) gels formed by chain entanglements of UHMW polymers (>106 Da) formed in situ by radical polymerisation in aprotic ionic liquids (ILs).24 In addition to excellent mechanical strength and recyclability, UHMW gels have an outstanding self-healing ability. In a previous study, we found that UHMW gels can be formed by the one-pot radical polymerisation of vinyl monomers in aprotic ILs at extremely low initiator concentrations (∼0.02 mol% vs. monomer in the feed). This results in the formation of UHMW polymers with nearly 100% monomer conversion. In contrast, in the case of radical polymerisation in toluene, which is a conventional organic solvent, the monomer conversion was low (∼40%) at a low initiator concentration (∼0.02 mol%), and the molecular weight did not reach the UHMW range. It has also been reported that the highly ionic atmosphere in the precursor solution plays a key role in the formation of UHMW polymers.25
In the present study, we fabricated UHMW gel electrolytes using highly concentrated electrolytes (HCEs), which have recently attracted attention for use in lithium secondary batteries owing to their excellent electrochemical properties (Fig. 1). The molecular weight of the methacrylate polymer synthesised via free-radical polymerisation in electrolytes increased with the lithium salt concentration; thus, UHMW polymers could be formed in situ in the HCEs at an extremely low initiator concentration. Physical gels were successfully synthesised in a one-pot process using the abundant chain entanglements of the UHMW polymers in the HCEs. Herein, we discuss the synthetic characteristics as well as the viscoelastic and mechanical properties of UHMW gels in detail.
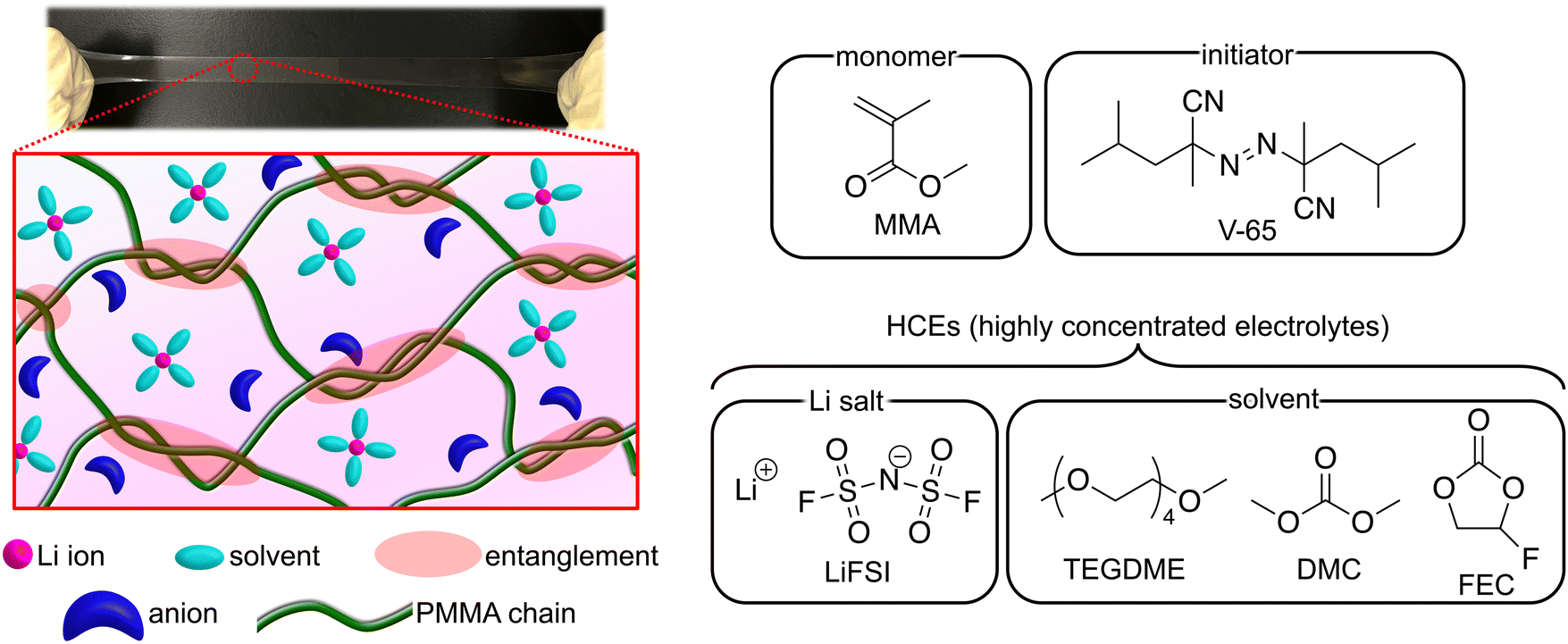 | ||
| Fig. 1 Photograph and schematic of UHMW gel electrolytes containing HCEs, along with the chemical structures of the monomer, initiator, and HCEs. | ||
Experimental
Materials
Tetraethylene glycol dimethyl ether (TEGDME), dimethyl carbonate (DMC), and fluoroethylene carbonate (FEC) were purchased from Sigma-Aldrich (USA). Lithium bis(fluorosulfonyl)imide (LiFSI), lithium bis(trifluoromethanesulfonyl)imide (LiTFSI), and lithium tetrafluoroborate (LiBF4) were purchased from Kishida Chemicals (Japan). Lithium nitrate (LiNO3) was purchased from the FUJIFILM Wako Pure Chemical Corporation (Japan). All electrolyte solutions were prepared in an Ar-filled glovebox by mixing the lithium salts and solvents, followed by stirring at room temperature overnight. Methyl methacrylate (MMA) was purchased from TCI (Japan) and purified by passing it through a short column loaded with activated alumina prior to use. 2,2′-Azobis(2,4-dimethylvaleronitrile) (V-65) was purchased from FUJIFILM Wako Pure Chemical Corporation (Japan).Synthesis
A typical synthetic procedure for the PMMA-LiFSI-TEGDME system, with the highest molecular weight, was described below. First, HCE (equimolar mixture of LiFSI and TEGDME, 3.5 g), MMA (1.5 g, 15.0 mmol), and V-65 (0.37 mg, 1.50 μmol) were mixed and stirred in a glass vial to obtain a homogeneous transparent solution. The vial was sealed with a rubber septum, bubbled with Ar for 15 min, and then transferred to a glovebox. The precursor solution was injected into a mould consisting of two PET films and one silicone rubber spacer, and then polymerisation was conducted by heating to 70 °C for 24 h. In all the systems, the volume fraction of the MMA monomer was set to 38 vol%.Characterisation
Proton nuclear magnetic resonance (1H NMR) measurements were performed to determine the monomer conversion using an ECS-400 spectrometer (JEOL, Japan), where the sample solutions were prepared by directly dissolving the polymerised gels in acetone-d6. To characterise the molecular weight and molecular-weight distribution of the synthesised polymer, the sample was purified via reprecipitation using acetone and methanol as good and poor solvents, respectively. The purified polymer was analysed via gel permeation chromatography (GPC) using a 10 mM LiBr/N,N-dimethylformamide solution as the eluent. The GPC columns were calibrated using PMMA standards.Differential scanning calorimetry (DSC) was performed using a Discovery DSC 250 (TA Instruments, USA). The samples placed in a semi-sealed Al pan were cooled to −120 °C and then heated to 100 °C. This process was repeated for two cycles. The cooling and heating rate was 10 °C min−1. The glass transition temperature (Tg), which is the midpoint of the heat-capacity change, was determined from the DSC thermogram of the heating process during the second cycle.
Rheological measurements were performed using an MCR 102 rheometer (Anton Paar, Austria). A parallel-plate geometry with a diameter of 8 mm was used, and the sample thickness was approximately 1 mm. Temperature sweep measurements were performed from 10 to 100 °C. The heating rate, frequency, and strain amplitude were set to 1 °C min−1, 1 rad s−1, and 1%, respectively. Time–temperature superposition (tTS) master curves of the storage (G′) and loss (G′′) moduli were constructed using frequency sweep measurements performed over a frequency range of 0.1–100 rad s−1 with a strain amplitude of 1% at different temperatures.
Tensile tests were performed using an AGS-X tester (Shimadzu, Japan) at room temperature. For the tensile tests, the gels were cut into a dumbbell shape (dimensions of the rectangular portion: 2.0 × 12.0 × 1.0 mm3), stretched at a speed of 10 cm min−1, and monitored using a 100-N load cell.
Results and discussion
Fig. 2a shows the relationship between the monomer and thermal initiator (V-65) molar ratio ([M]/[I]) and the number-averaged molecular weight (Mn) for the radical polymerisation of MMA in the organic solvent TEGDME and in the electrolyte with LiFSI dissolved in TEGDME. The detailed characterisation of the polymerisation via GPC and 1H NMR is summarised in Table S1 (ESI†). The GPC traces and 1H NMR spectra of representative results are shown in Fig. S1 and S2 (ESI†). In all the systems, the Mn of PMMA increased as [M]/[I] increased. Notably, the increase in the molecular weight with an increase in the [M]/[I] ratio was larger at a higher LiFSI concentration. cLiFSI = 3.2 M corresponds to an equimolar mixture of LiFSI and TEGDME, which are known as solvate ionic liquids (SILs).26–28 SILs are a special class of HCEs in which almost all the solvent molecules (TEGDME) are present as complex cations coordinated to Li cations, resulting in unique physicochemical properties similar to those of conventional aprotic ILs. The in situ formation of UHMW polymers with molecular weights exceeding 1 million Da was observed at concentrations of cLiFSI = 3.2 M in the high-[M]/[I] regime. Thus, physically crosslinked polymer gel electrolytes formed by the entanglement of UHMW polymers, which are called UHMW gels, can be obtained in this regime. In addition, the monomer conversion was calculated from 1H NMR measurements. It was found that the monomer conversion of MMA in 3.2 M LiFSI-TEGDME reached a very high value over 99% even at the highest [M]/[I] value of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, whereas in pure TEGDME solvent, the monomer conversion was only approximately 80% for [M]/[I] = 10000 (Fig. S1 and Table S1, ESI†). Furthermore, Mn increases with cLiFSI for [M]/[I] = 10000 (Fig. S3, ESI†). These results suggest that the free-radical polymerisation reaction is promoted as the lithium salt concentration increases and that UHMW polymers are formed with a high monomer conversion under high-[M]/[I] conditions.
000, whereas in pure TEGDME solvent, the monomer conversion was only approximately 80% for [M]/[I] = 10000 (Fig. S1 and Table S1, ESI†). Furthermore, Mn increases with cLiFSI for [M]/[I] = 10000 (Fig. S3, ESI†). These results suggest that the free-radical polymerisation reaction is promoted as the lithium salt concentration increases and that UHMW polymers are formed with a high monomer conversion under high-[M]/[I] conditions.
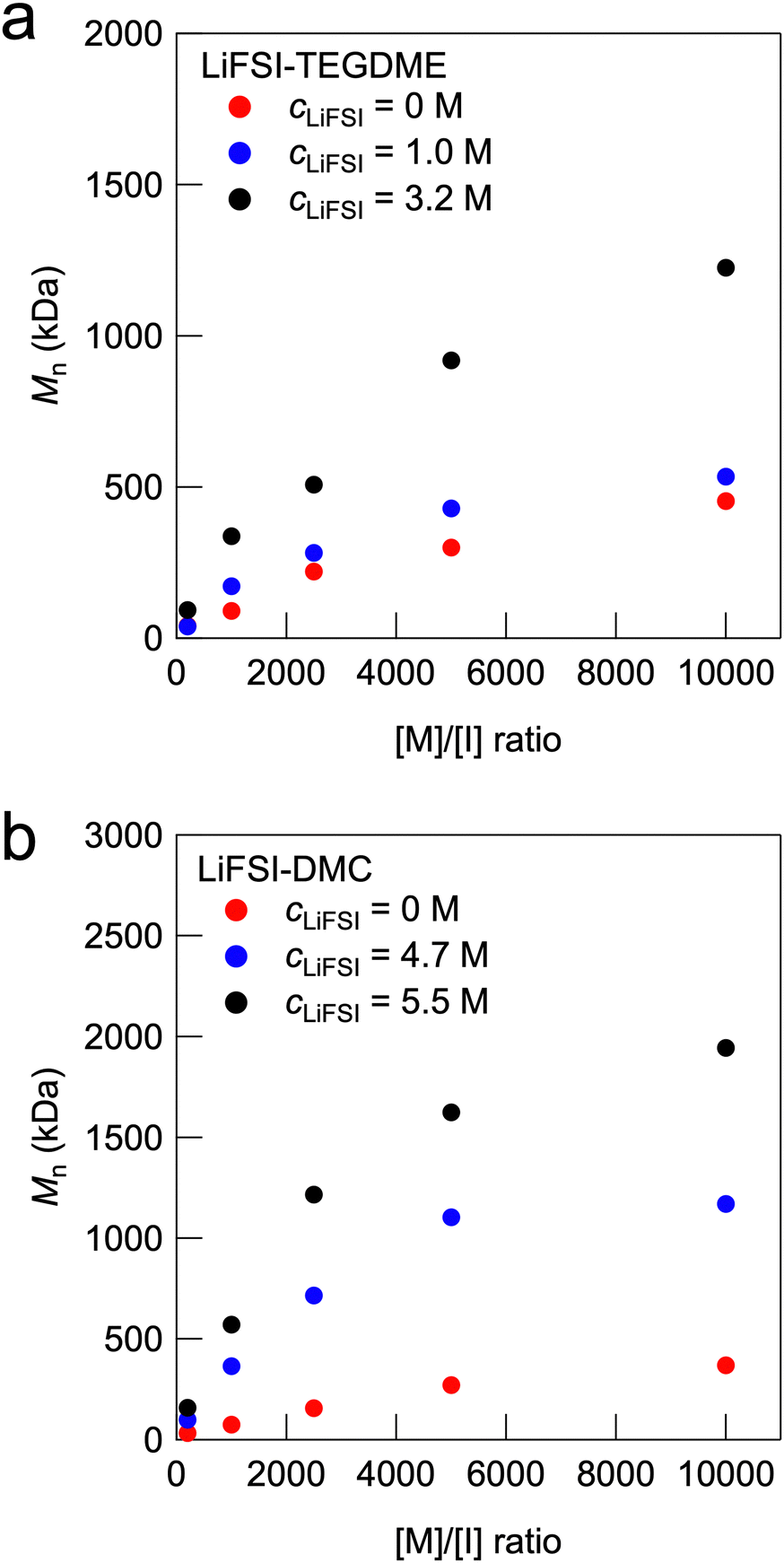 | ||
| Fig. 2 Dependence of Mn on the [M]/[I] ratio with various cLiFSI values for (a) LiFSI-TEGDME and (b) LiFSI-DMC systems. | ||
We previously reported self-healable polymer gels formed by physical entanglement of UHMW polymers formed in situ in aprotic ILs, which are called UHMW ion gels.24 The results of the present study suggest that UHMW gels can be produced not only in aprotic ILs but also in SILs using the same method. Furthermore, UHMW gels can be fabricated using other well-known HCEs. Fig. 2b shows the relationship between the [M]/[I] ratio and the Mn of the synthesised PMMA for the LiFSI-DMC systems. cLiFSI = 4.7 M and 5.5 M correspond to [DMC]:[LiFSI] = 1.5:1 and 1.1:1, respectively, which have been reported to have excellent performance as liquid electrolytes for lithium secondary batteries.29–31 Compared with pure DMC, these LiFSI-DMC HCEs produced UHMW PMMA polymers in the high-[M]/[I] regime with a high monomer conversion (Table S1, ESI†). In addition, UHMW polymers were obtained using the well-known fluorine-donating HCE LiFSI-FEC system (Fig. S4 and Table S1, ESI†).32 These results indicate that the proposed strategy for fabricating UHMW gels can be applied to versatile HCE systems with LiFSI salts. The effect of the anion species was investigated for equimolar TEGDME-lithium salt systems. Table S1 (ESI†) summarises the results of the free-radical polymerisation of MMA in TEGDME-lithium salt equimolar mixtures with [M]/[I] = 10000. It was shown that in all equimolar TEGDME-lithium salt systems, a larger Mn and a higher monomer conversion were realised compared with the pure TEGDME system. However, in the case of NO3 anions, the obtained Mn was lower than those for BF4, TFSI, and FSI anions (Fig. S5, ESI†). The equimolar mixture of TEGDME and NO3 anions differs from other anion systems in that it has a very high percentage of free TEGDME molecules that do not form complex cations owing to the strong Li–NO3 interaction.33 In the case of BF4 anion, polymerisation-induced phase separation occurs, which may also contribute to the observed increase in molecular weight.34,35 Although the detailed mechanism has not been clarified, it is implied that the high ionic moieties of free anions might enhance the rate constant of propagation for free-radical polymerisation, similar to aprotic IL systems.36–38 The increase in molecular weight in aprotic ILs and HCEs can also be attributed to the suppression of termination reactions due to their high viscosity. To investigate the effect of solvent viscosity on molecular weight, we compared the molecular weights in HCE systems and viscosity-tuned non-ionic systems. The viscosity-tuned non-ionic system was prepared by pre-doping a commercial polymer (120 kDa PMMA) with TEGDME at various concentrations (Fig. S6, ESI†). The molecular weight of the synthesised PMMA in the viscosity-tuned TEGDME solutions increased slightly with increasing viscosity but did not reach the UHMW range observed in the aprotic IL and HCE systems. These results suggest that the suppression of termination reaction due to high solvent viscosity has a minor effect, while the enhancement of propagation reaction is the dominant factor in the synthesis of UHMW polymers in HCEs.
This concept is applicable to various HCE systems, making it promising for the development of polymer gel electrolytes formed by physical entanglements of UHMW polymers for electrochemical applications including lithium secondary batteries. It should be noted that, as alternative methods for preparing UHMW gels, pre-synthesised UHMW PMMA could be directly dissolved in HCEs or composited using the cosolvent method.39 In the cosolvent method, a volatile cosolvent is used to dissolve both the UHMW polymers and HCEs, followed by casting and removing only the cosolvent to prepare the UHMW gel. However, due to the extremely slow relaxation kinetics of UHMW PMMA, it is challenging to obtain a homogeneous gel using either method (Fig. S7, ESI†). Therefore, the one-pot fabrication strategy presented here is the superior approach for preparing homogeneous physical gels formed by the chain entanglements of UHMW polymers in HCEs.
Owing to the high degree of chain entanglement, the fabricated UHMW gel electrolytes had a solid-like appearance and were classified as physical gels.40–42 The viscoelastic properties of the UHMW gels based on UHMW PMMA and HCEs were evaluated. The viscoelastic properties of the LiFSI-DMC system could not be evaluated, because of the significant effect of DMC evaporation during the long measurement time required (data not shown). Therefore, we focused on the UHMW gels formed in LiFSI-TEGDME (cLiFSI = 3.2 M). The LiFSI-TEGDME equimolar complex had good thermal stability, which allowed us to evaluate the rheological properties of the LiFSI-TEGDME UHMW gel. Fig. 3 shows the temperature sweep measurements from 10 to 100 °C for PMMA-LiFSI-TEGDME systems with different molecular weights adjusted by changing the initiator concentration. Composites consisting of UHMW PMMA and HCEs, which were UHMW gels, maintained solid-like properties at high temperatures, that is, higher storage moduli (G′) than loss moduli (G′′) over all the measurement temperature ranges. In contrast, a crossover between G′ and G′′ was observed in the high-temperature region for the composites containing moderate molecular weight PMMA and HCEs. This suggests that the relaxation time of physical crosslinking becomes very long owing to the chain entanglements of the UHMW polymers, maintaining solid-like integrity over a wide temperature range.
 | ||
| Fig. 3 Temperature dependence of the storage (G′) and loss (G′′) moduli for PMMA-LiFSI-TEGDME systems with different molecular weights. | ||
The tTS master curves for the G′ and G′′ values, which were constructed using frequency sweep measurements at different temperatures, are presented in Fig. 4a. Although the master curves for G′ were obtained according to the tTS principle, deviations were observed on the high-frequency side of each temperature for G′′. This was related to the temperature dependence of the viscosity of the solvent, which is often observed in gels with ILs as solvents, including UHMW gels.25,41,43,44 In the tTS master curves for LiFSI-TEGDME systems, the composite composed of moderate-molecular-weight PMMA exhibits terminal relaxation behaviour (G′ ∝ ω2, G′′ ∝ ω) in the low frequency region, while the UHMW gels exhibit a wide rubbery plateau region of G′ without crossover between G′ and G′′ in the constructed frequency range. This also indicates that UHMW gels behave as solids over a very long timescale. However, it should be noted that the UHMW gel is classified as a physical gel with a finite relaxation time, which is different from a chemical gel having an infinite relaxation time. Fig. S8a (ESI†) shows the viscoelastic master curve for the PMMA-LiFSI-TEGDME UHMW gel alongside that of the PMMA-[C2mIm][TFSI] UHMW gel, which uses an aprotic IL (C2mIm: 1-ethyl-3-methylimidazolium).24 Both UHMW gels exhibit a wide rubbery plateau region due to the abundant chain entanglements of the UHMW polymers. Compared to the PMMA-[C2mIm][TFSI] UHMW gel, the PMMA-LiFSI-TEGDME UHMW gel displays a significant upturn in the storage (G′) and loss (G′′) moduli in the high-frequency region. This behaviour can be attributed to the higher Tg of the PMMA-LiFSI-TEGDME UHMW gel compared to that of the PMMA-[C2mIm][TFSI] UHMW gel. DSC measurements confirmed that the glass transition temperature of the PMMA-HCE composites was independent of the molecular weight of PMMA (Fig. S9, ESI†). This suggests that the polymer molecular weight affects the dynamics on a long timescale rather than on a short timescale such as segmental motion. The master curves of PMMA-LiFSI-TEGDME with different molecular weights can be constructed with the same shift factor, suggesting that the activation processes of the polymer chain dynamics are similar (Fig. 4b).
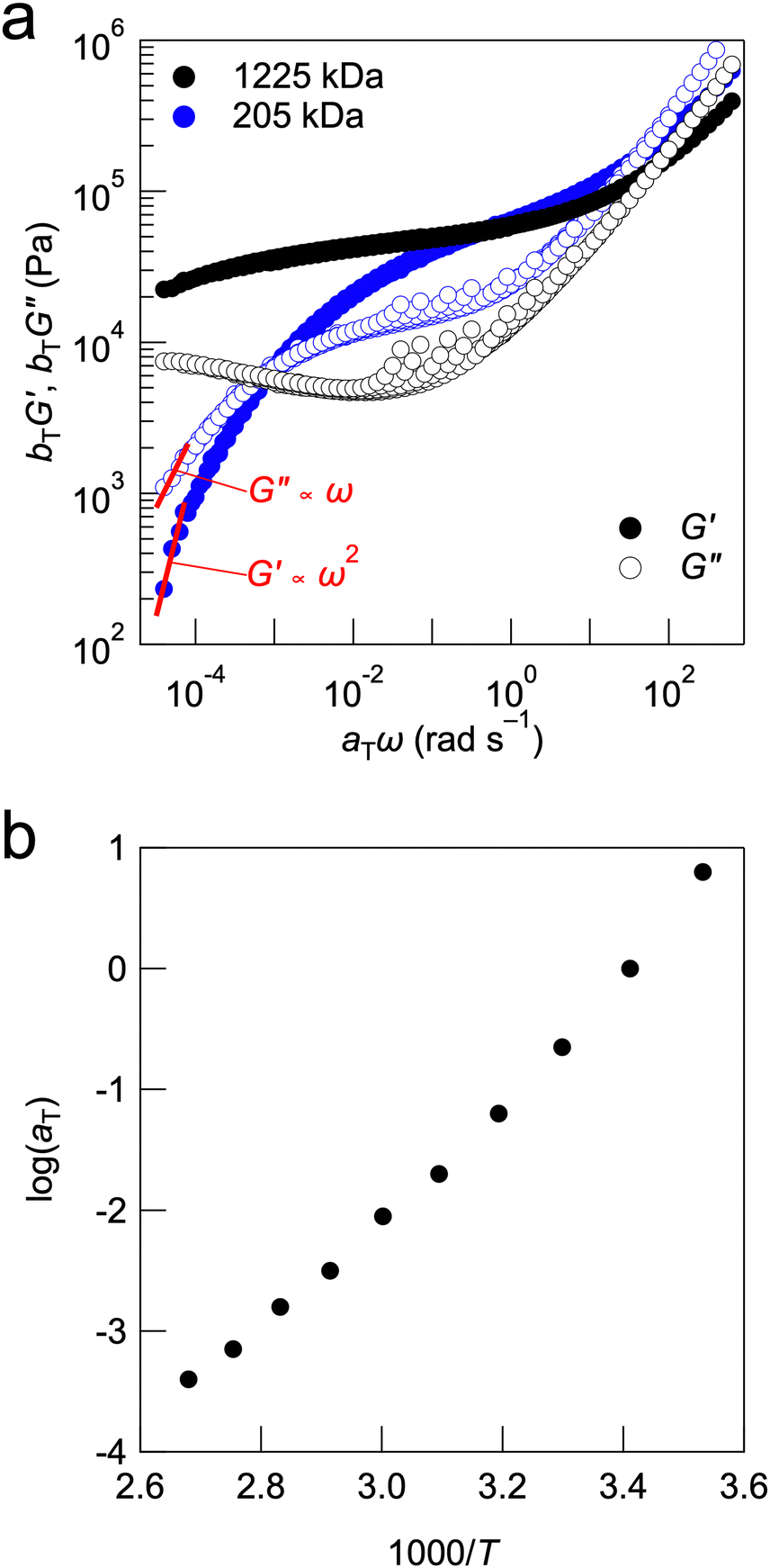 | ||
| Fig. 4 (a) tTS master curves for PMMA-LiFSI-TEGDME composites with moderate molecular weight and UHMW PMMA. The reference temperature is 20 °C. (b) Arrhenius plot of the shift factor (aT) for both PMMA-LiFSI-TEGDME systems. | ||
Uniaxial tensile tests were performed to investigate the mechanical properties of the UHMW gels. Fig. 5a shows the stress–strain curves of PMMA-LiFSI-TEGDME physical gels with moderate molecular weight and UHMW PMMA, as well as a conventional chemically crosslinked gel using a bifunctional ethylene glycol dimethacrylate (EGDMA) crosslinker with the same MMA monomer concentration. The chemically crosslinked gels exhibited brittle properties owing to the spatial heterogeneity of the covalent crosslinking points.45 The PMMA-HCE composites with moderate molecular weights exhibited low maximum stress and very large elongation, which was attributed to the fact that the low-molecular-weight entangled polymers were easily pulled out. In contrast, the UHMW gel exhibited excellent mechanical properties, with a fracture stress of 376 kPa and a fracture elongation of 1069%. Fig. S8b (ESI†) shows the comparison of stress–strain curves for the PMMA-LiFSI-TEGDME UHMW gel and the PMMA-[C2mIm][TFSI] UHMW gel.24 The results indicate that the PMMA-LiFSI-TEGDME UHMW gel exhibits superior fracture stress and strain to the PMMA-[C2mIm][TFSI] UHMW gel. As shown in the viscoelastic master curves in Fig. S8a (ESI†), the PMMA-LiFSI-TEGDME UHMW gel displays higher G′ and G′′ values in the high-frequency region. This might suggest that the improvement in mechanical properties can be attributed to energy dissipation due to segmental motion during the stretching deformation.46 It should also be noted that the compatibility between the UHMW polymer and the HCE influences the mechanical properties of UHMW gels. As mentioned above, polymerisation-induced phase separation is observed during the synthesis of UHMW PMMA in the LiBF4-TEGDME equimolar complex. This phase-separated structure significantly enhances mechanical properties, including high G′ and G′′ values, as well as mechanical strength (Fig. S10a and b, ESI†).
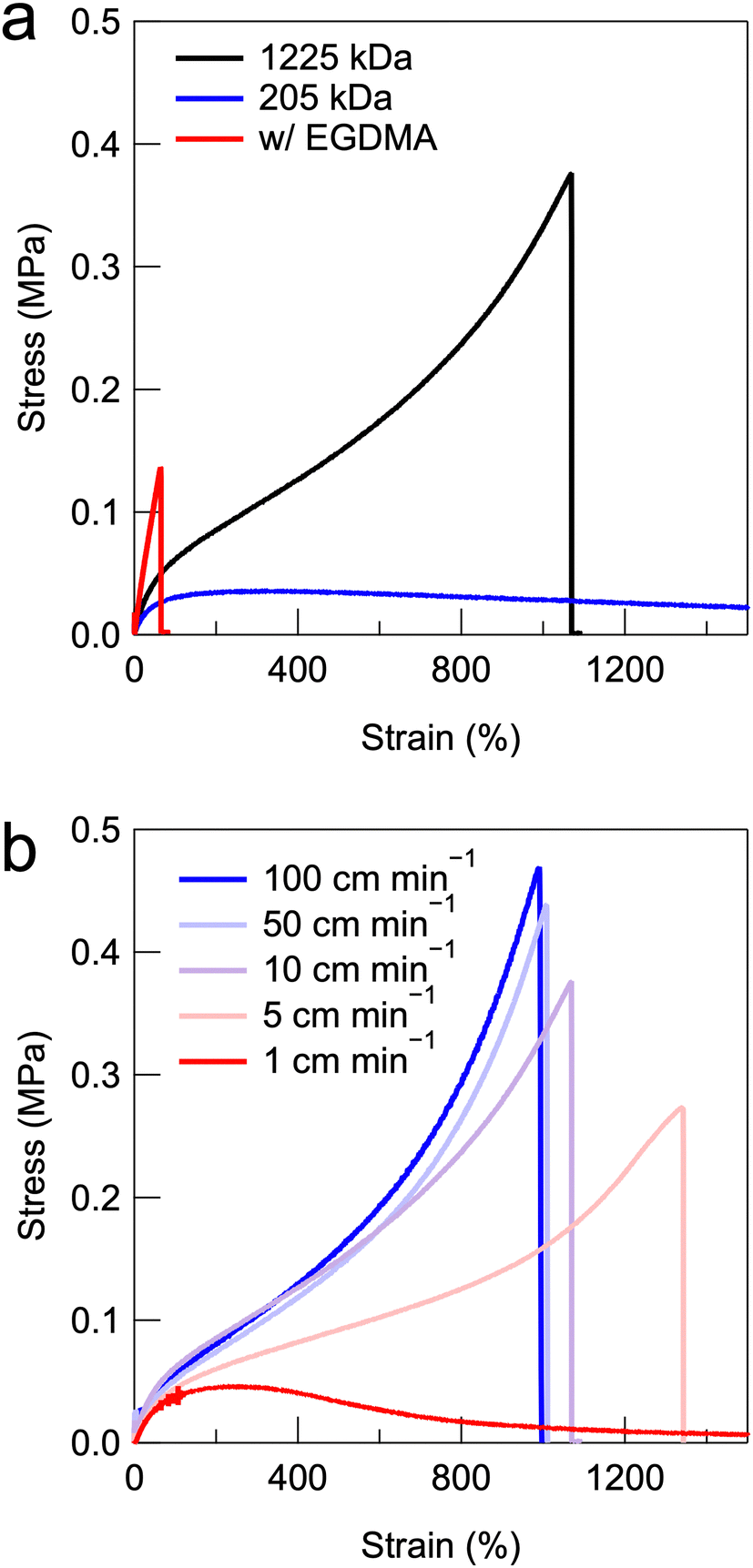 | ||
| Fig. 5 (a) Stress–strain curves of chemically crosslinked gel with EGDMA, and non-chemically crosslinked PMMA-LiFSI-TEGDME composites with moderate molecular weight PMMA and UHMW PMMA. The strain rate is 10 cm min−1. (b) Stress–strain curves of the UHMW PMMA-LiFSI-TEGDME gel electrolyte at different strain rates. | ||
Importantly, the stress–strain curves of the UHMW gels are strain-rate-dependent, owing to the nature of physical crosslinking with a finite relaxation time. A higher strain rate corresponds to a larger stress increase and a lower breaking strain (Fig. 5b). This phenomenon is also observed in other non-covalently bonded polymer gels and elastomers.18,46 In UHMW gels, a lower tensile rate corresponds to a lower fracture stress and higher elongation at break, because the relaxation of the chain entanglements follows the bulk deformation.
UHMW gel electrolytes are expected to have self-healing functions because of the reversible nature of the physical crosslinking point of polymer chain entanglement.47,48 Therefore, the self-healing efficiency based on the recovery of the stress–strain curve was evaluated by cutting the dumbbell-shaped UHMW gel at the centre, immediately re-contacting the cut surfaces, and then leaving it at room temperature for 24 h, followed by tensile testing. Fig. 6 shows the stress–strain curves of the pristine and healed UHMW gels for the PMMA-LiFSI-TEGDME system. The healed UHMW gel exhibited a Young's modulus comparable to that of the pristine sample and could be stretched to more than half the fracture strain of the pristine sample. This self-healing capability is thought to be driven by the reformation of chain entanglements and is promoted by the non-equilibrium nature of the cut surface.24 A trade-off between mechanical strength and healing efficiency has also been reported for UHMW gels with aprotic ILs.25 Therefore, the relatively insufficient self-healing ability of the present PMMA-LiFSI-TEGDME UHMW gel, compared to the previous PMMA-[C2mIm][TFSI] UHMW gel, can be attributed in part to a trade-off with its higher mechanical strength.
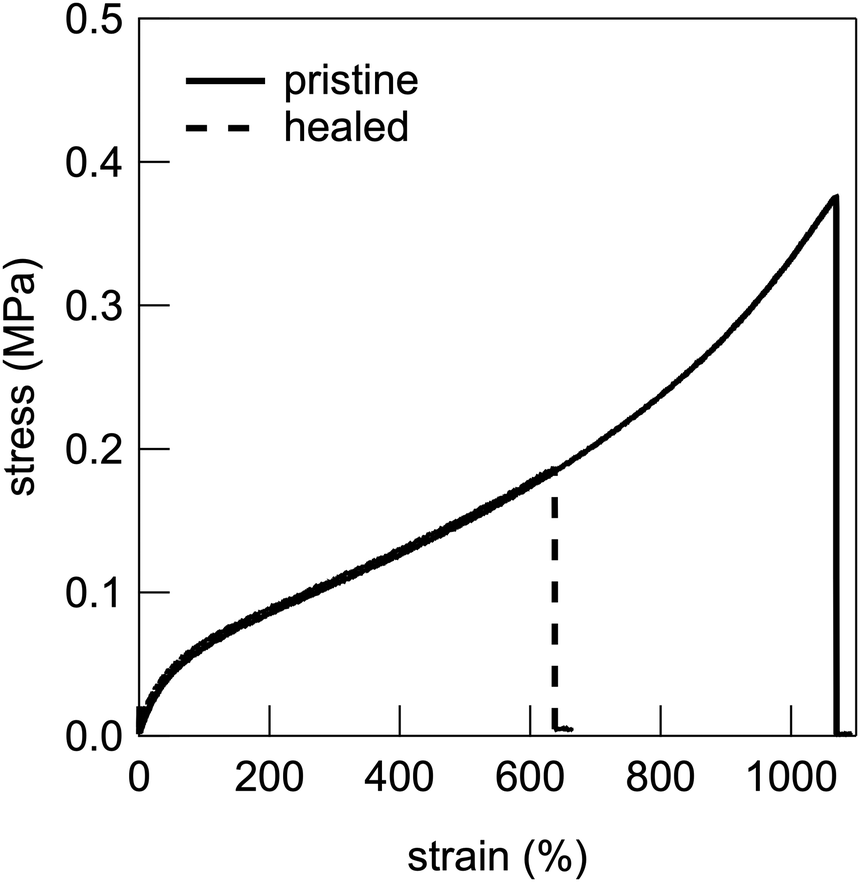 | ||
| Fig. 6 Stress–strain curves of pristine and healed PMMA-LiFSI-PEGDME UHMW gel samples. | ||
The self-healing ability of UHMW gels is a promising function of electrolytes in wearable and flexible devices that require durability against mechanical inputs. Furthermore, their high stretchability and self-healing abilities are expected to be useful for applications in next-generation lithium secondary batteries, such as the protection of lithium metal anodes that undergo severe electrode changes during charging and discharging.49–51 However, the current PMMA-LiFSI-TEGDME UHMW gel, partly due to its relatively high PMMA volume fraction of 38%, exhibits insufficient electrochemical properties, such as low ion conductivity (Fig. S11 and Table S2, ESI†). Therefore, achieving a balance between higher mechanical strength and improved electrochemical properties is required. Additionally, in order to fully utilise the excellent mechanical properties, such as self-healing capability, it is considered that the gel could be applied as a protective coating material for electrodes, such as lithium metal anodes,14 and this is currently under investigation.
Conclusion
We demonstrated that radical polymerisation at extremely low initiator concentrations can generate physical gels in situ, through the entanglement of UHMW polymers in highly concentrated lithium salt electrolytes. UHMW gel electrolytes maintain solid-like properties and high mechanical strength over a long timescale because of the abundant entanglement between the polymer chains. Furthermore, they have self-healing properties driven by the reformation of entanglement at the cut interface. This facile method is useful for the gelation of HCEs, which have attracted considerable attention in recent years as promising electrolytes for next-generation lithium secondary batteries. However, although the UHMW gel can be obtained with an extremely high monomer conversion rate (>99%), even a small amount of residual monomer could potentially affect electrode reactions in battery applications, warranting further detailed investigation. Additionally, for application in next-generation high-energy-density secondary batteries, it is crucial to address challenges, such as improving the gel's ionic conductivity and exploring its use as a protective coating film for electrodes.The present study demonstrates that the concept of UHMW gels is applicable to a wide range of HCEs. However, the mechanism whereby UHMW polymers are formed in HCEs is not yet understood. Therefore, more detailed studies, including simulations such as DFT calculations,52 are needed. The performance of gel electrolytes in practical applications such as rechargeable batteries and other electrochemical devices is also under investigation.
Author contributions
Investigation: Y. K. and R. T. supervision: R. T. writing – original draft: Y. K. and R. T. writing – review and editing: Y. K., R. T., and T. U.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by JSPS KAKENHI (23K26409), JST PRESTO program (JPMJPR2196), COI-NEXT (JPMJPF2016), and Green Technologies of Excellence (GteX) Program (JPMJGX23S3).References
- C. Ma, W. Cui, X. Liu, Y. Ding and Y. Wang, InfoMat, 2022, 4, e12232 CrossRef CAS.
- S. Kundu and Y. Ein-Eli, J. Power Sources, 2023, 553, 232267 CrossRef CAS.
- A. Manuel Stephan, Eur. Polym. J., 2006, 42, 21–42 CrossRef.
- X. Cheng, J. Pan, Y. Zhao, M. Liao and H. Peng, Adv. Energy Mater., 2018, 8, 1702184 CrossRef.
- H. Wang, Z. Wang, J. Yang, C. Xu, Q. Zhang and Z. Peng, Macromol. Rapid Commun., 2018, 39, 1800246 CrossRef PubMed.
- A. J. D’Angelo and M. J. Panzer, Chem. Mater., 2019, 31, 2913–2922 CrossRef.
- C. Chen, J. Zhang, B. Hu, Q. Liang and X. Xiong, Nat. Commun., 2023, 14, 4018 CrossRef CAS PubMed.
- L. Porcarelli, C. Gerbaldi, F. Bella and J. R. Nair, Sci. Rep., 2016, 6, 19892 CrossRef CAS PubMed.
- S. Choudhury, R. Mangal, A. Agrawal and L. A. Archer, Nat. Commun., 2015, 6, 10101 CrossRef CAS PubMed.
- D. G. Mackanic, X. Yan, Q. Zhang, N. Matsuhisa, Z. Yu, Y. Jiang, T. Manika, J. Lopez, H. Yan, K. Liu, X. Chen, Y. Cui and Z. Bao, Nat. Commun., 2019, 10, 5384 CrossRef PubMed.
- Y. Wang, C. J. Zanelotti, X. Wang, R. Kerr, L. Jin, W. H. Kan, T. J. Dingemans, M. Forsyth and L. A. Madsen, Nat. Mater., 2021, 20, 1255–1263 CrossRef CAS PubMed.
- H. Wu, Y. Cao, H. Su and C. Wang, Angew. Chem., Int. Ed., 2018, 57, 1361–1365 CrossRef CAS PubMed.
- S. Matsuura, M. Shibata, J. Han and K. Fujii, ACS Appl. Polym. Mater., 2020, 2, 1276–1282 CrossRef CAS.
- R. Tamate, Y. Peng, Y. Kamiyama and K. Nishikawa, Adv. Mater., 2023, 35, 2211679 CrossRef CAS PubMed.
- R. Tamate, K. Hashimoto, T. Horii, M. Hirasawa, X. Li, M. Shibayama and M. Watanabe, Adv. Mater., 2018, 30, 1802792 CrossRef PubMed.
- L. Xu, Z. Huang, Z. Deng, Z. Du, T. L. Sun, Z.-H. Guo and K. Yue, Adv. Mater., 2021, 33, 2105306 CrossRef CAS PubMed.
- Y. M. Kim, J. H. Kwon, S. Kim, U. H. Choi and H. C. Moon, Nat. Commun., 2022, 13, 3769 CrossRef CAS PubMed.
- W. Li, L. Li, S. Zheng, Z. Liu, X. Zou, Z. Sun, J. Guo and F. Yan, Adv. Mater., 2022, 34, 2203049 CrossRef CAS PubMed.
- Y. Shi, J. Zhang, L. Pan, Y. Shi and G. Yu, Nano Today, 2016, 11, 738–762 CrossRef CAS.
- R. Tamate and M. Watanabe, Sci. Technol. Adv. Mater., 2020, 21, 388–401 CrossRef CAS PubMed.
- R. Tamate and T. Ueki, Chem. Rec., 2023, 23, e202300043 CrossRef CAS PubMed.
- P. Guo, A. Su, Y. Wei, X. Liu, Y. Li, F. Guo, J. Li, Z. Hu and J. Sun, ACS Appl. Mater. Interfaces, 2019, 11, 19413–19420 CrossRef CAS PubMed.
- P. Jaumaux, Q. Liu, D. Zhou, X. Xu, T. Wang, Y. Wang, F. Kang, B. Li and G. Wang, Angew. Chem., Int. Ed., 2020, 59, 9134–9142 CrossRef CAS PubMed.
- Y. Kamiyama, R. Tamate, T. Hiroi, S. Samitsu, K. Fujii and T. Ueki, Sci. Adv., 2022, 8, eadd0226 CrossRef CAS PubMed.
- Y. Kamiyama, R. Tamate, K. Fujii and T. Ueki, Soft Matter, 2022, 18, 8582–8590 RSC.
- K. Yoshida, M. Nakamura, Y. Kazue, N. Tachikawa, S. Tsuzuki, S. Seki, K. Dokko and M. Watanabe, J. Am. Chem. Soc., 2011, 133, 13121–13129 CrossRef CAS PubMed.
- T. Mandai, K. Yoshida, K. Ueno, K. Dokko and M. Watanabe, Phys. Chem. Chem. Phys., 2014, 16, 8761–8772 RSC.
- M. Watanabe, K. Dokko, K. Ueno and M. L. Thomas, Bull. Chem. Soc. Jpn., 2018, 91, 1660–1682 CrossRef CAS.
- J. Wang, Y. Yamada, K. Sodeyama, C. H. Chiang, Y. Tateyama and A. Yamada, Nat. Commun., 2016, 7, 12032 CrossRef CAS PubMed.
- S. Chen, J. Zheng, D. Mei, K. S. Han, M. H. Engelhard, W. Zhao, W. Xu, J. Liu and J. G. Zhang, Adv. Mater., 2018, 30, 1706102 CrossRef PubMed.
- N. Piao, X. Ji, H. Xu, X. Fan, L. Chen, S. Liu, M. N. Garaga, S. G. Greenbaum, L. Wang, C. Wang and X. He, Adv. Energy Mater., 2020, 10, 1903568 CrossRef CAS.
- L. Suo, W. Xue, M. Gobet, S. G. Greenbaum, C. Wang, Y. Chen, W. Yang, Y. Li and J. Li, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 1156–1161 CrossRef CAS PubMed.
- K. Ueno, R. Tatara, S. Tsuzuki, S. Saito, H. Doi, K. Yoshida, T. Mandai, M. Matsugami, Y. Umebayashi, K. Dokko and M. Watanabe, Phys. Chem. Chem. Phys., 2015, 17, 8248–8257 RSC.
- Y. Suzuki, D. S. Cousins, Y. Shinagawa, R. T. Bell, A. Matsumoto and A. P. Stebner, Polym. J., 2019, 51, 423–431 CrossRef CAS.
- Y. Suzuki, Y. Doi, R. Mishima, K. Mayumi and A. Matsumoto, Macromolecules, 2023, 56, 3731–3738 CrossRef CAS.
- S. Harrisson, S. R. Mackenzie and D. M. Haddleton, Chem. Commun., 2002, 2850–2851 RSC.
- K. Hong, H. Zhang, J. W. Mays, A. E. Visser, C. S. Brazel, J. D. Holbrey, W. M. Reichert and R. D. Rogers, Chem. Commun., 2002, 1368–1369 RSC.
- P. Kubisa, Prog. Polym. Sci., 2004, 29, 3–12 CrossRef CAS.
- R. Tamate, R. Usui, K. Hashimoto, Y. Kitazawa, H. Kokubo and M. Watanabe, Soft Matter, 2018, 14, 9088–9095 RSC.
- F. Tanaka and S. F. Edwards, J. Non-Newton. Fluid Mech., 1992, 43, 247–271 CrossRef CAS.
- A. Noro, Y. Matsushita and T. P. Lodge, Macromolecules, 2008, 41, 5839–5844 CrossRef CAS.
- T. L. Sun, T. Kurokawa, S. Kuroda, A. B. Ihsan, T. Akasaki, K. Sato, M. A. Haque, T. Nakajima and J. P. Gong, Nat. Mater., 2013, 12, 932–937 CrossRef CAS PubMed.
- Y. He, P. G. Boswell, P. Bühlmann and T. P. Lodge, J. Phys. Chem. B, 2007, 111, 4645–4652 CrossRef CAS PubMed.
- X. Ma, R. Usui, Y. Kitazawa, R. Tamate, H. Kokubo and M. Watanabe, Macromolecules, 2017, 50, 6788–6795 CrossRef CAS.
- M. Shibayama, Soft Matter, 2012, 8, 8030–8038 RSC.
- L. Chen, T. L. Sun, K. Cui, D. R. King, T. Kurokawa, Y. Saruwatari and J. P. Gong, J. Mater. Chem. A, 2019, 7, 17334–17344 RSC.
- M. Yamaguchi, R. Maeda, R. Kobayashi, T. Wada, S. Ono and S. Nobukawa, Polym. Int., 2012, 61, 9–16 CrossRef CAS.
- R. P. Wool, Soft Matter, 2008, 4, 400–418 RSC.
- K. Liu, A. Pei, H. R. Lee, B. Kong, N. Liu, D. Lin, Y. Liu, C. Liu, P.-C. Hsu, Z. Bao and Y. Cui, J. Am. Chem. Soc., 2017, 139, 4815–4820 CrossRef CAS PubMed.
- Q. Liu, D. Zhou, D. Shanmukaraj, P. Li, F. Kang, B. Li, M. Armand and G. Wang, ACS Energy Lett., 2020, 1456–1464 CrossRef CAS.
- R. Xu, X.-B. Cheng, C. Yan, X.-Q. Zhang, Y. Xiao, C.-Z. Zhao, J.-Q. Huang and Q. Zhang, Matter, 2019, 1, 317–344 CrossRef.
- K. Low, L. Wylie, D. L. A. Scarborough and E. I. Izgorodina, Chem. Commun., 2018, 54, 11226–11243 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sm01248h |
| This journal is © The Royal Society of Chemistry 2025 |