
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Physical science of the didodecyldimethylammonium bromide–water system: 1. Equilibrium phase behaviour†‡
Louisa
Reissig§
*ab,
Wim
Pyckhout-Hintzen
 c,
Simon
Dalgleish
a,
Andrew R.
Mount
a,
Michael E.
Cates¶
b,
David J.
Fairhurst
*b and
Stefan E.
Egelhaaf
d
c,
Simon
Dalgleish
a,
Andrew R.
Mount
a,
Michael E.
Cates¶
b,
David J.
Fairhurst
*b and
Stefan E.
Egelhaaf
d
aSchool of Chemistry, The University of Edinburgh, Edinburgh EH9 3JJ, UK. E-mail: Louisa_Reissig@gmx.de
bSUPA, School of Physics and Astronomy, The University of Edinburgh, Edinburgh EH9 3JZ, UK. E-mail: david.fairhurst@ed.ac.uk
cJülich Centre for Neutron Science (JCNS-1), Forschungszentrum Jülich, 52428 Jülich, Germany
dCondensed Matter Physics Laboratory, Heinrich-Heine-University, 40225 Düsseldorf, Germany
First published on 28th January 2025
Abstract
Surfactant molecules in solvents self-assemble into a large variety of structures depending on their chemical composition, concentration and temperature, summarized in the system's equilibrium phase diagram. However, the occurrence of long-lived metastable states can lead to incomplete or partly incorrect phase diagrams. By applying a set of complementary techniques and recording changes on different length scales, we determine an improved aqueous equilibrium phase diagram of the widely used double-chain surfactant didodecyldimethylammonium bromide (DDAB) over a broad concentration range (ϕDDAB = 3–100 wt%). We reveal that DDAB molecules exist as zero-hydrates in the room temperature solid state and decompose above 90 °C: the upper temperature of the phase diagram. Differential scanning calorimetry was used to characterise the transition's heat energy, kinetics and temperature, while the structure of the phases was characterized by small angle X-ray scattering and microscopy. Raman spectroscopy combined with computational techniques provided information regarding the conformational properties of the surfactant molecules. Our results were in good agreement with the literature phase diagram for moderate temperatures and surfactant concentrations. At 16 °C, a transition from a frozen lamellar phase (Lβ) to a fluid lamellar phase Lα has previously been suggested across all concentrations (Dubois et al. 1998), with Tm coinciding with the Krafft temperature (Tc) determined in dilute systems. Here, we characterize for the first time the low temperature equilibrium phase for ϕDDAB > 3 wt% as a crystalline dispersion, and determine the position and shape of the Krafft eutectic. The equilibrium phase below 14.1 °C is now assigned to a coexistence region of surfactant hydrate crystals and water XWn + W. At intermediate temperatures, the crystal hydrates XWn melt gradually into the previously reported Lα phase, leaving a narrow coexistence region in the phase diagram XWn + Lα. In conclusion, an amended broad equilibrium phase diagram is presented, combining our new results with those previously reported in the literature.
Introduction
Dialkyldimethylammonium halides were the first entirely synthetic double-chain amphiphiles found to form bilayer structures similar to those of phospholipids constituting biological membranes.1 They are commonly abbreviated as CmCnDA+X−, where Cm and Cn represent the two long hydrocarbon chains, D (or DM) stands for the two methyl groups, A stands for the quaternary ammonium, and X stands for the counterion, most commonly either chloride or bromide. For symmetric chains, the convention is to abbreviate the number of carbons in each chain, e.g. with D standing for di-dodecyl or di-decyl. Because of their ability to form long-lived vesicles at very low surfactant concentrations, they are commonly used as stable models for membranes.2 Furthermore, they became some of the most commonly used double-chain amphiphiles in applications, such as protein electrochemistry3 and bio-sensors4 due to their excellent ability to simulate the natural environment for biomolecules, their high electrocatalytic activity and great structural stability. Since cationic lipids can also combine electrostatically with a wide variety of oppositely charged biomolecules, cells or other biological structures,5 they were found to be efficient coatings for fast ion exchange chromatography,6 capillary electrophoresis,7 and can be used in gene delivery through complex formation with negatively charged DNA.8 On the other hand, their ability to destroy existing cell membranes containing negatively charged phospholipids make them applicable as immunosuppressant agents9,10 or bactericides.10,11 This cytotoxicity is significantly reduced when coated nanoparticles instead of pure vesicles are used5,12 or neutral helper lipids are added.13 In the chemical industry, cationic surfactants are highly applicable as softeners or hair conditioners, due to their low solubility and ability to neutralize charged surfaces.9,14 They found further application as disinfections agents,9 herbicides,15 or as wetting10,16 and antistatic agents.10One of the most important representatives of this group is the cationic double-chain surfactant didodecyldimethylammonium bromide ((C12H25)2N+(CH3)2Br−, abbreviated as DDAB, also known as dilauryldimethylammonium bromide) (see Fig. 1 top), first synthesized in 1977.1 Its molecular properties and aqueous phases have been studied extensively.17–27 However, the most recently published phase diagram covering a broad ϕDDAB range of the DDAB–water system17 differs in the low temperature regime from those of the other known dialkyldimethylammonium halide water systems28–31 through the occurrence of a stable lamellar gel phase Lβ at low temperatures in contrast to a crystalline dispersion. For dilute systems, where vesicles are observed, it has been reported that both a Lβ to Lα phase transition happens at the same temperature as the melting of DDAB crystals23–26 (determining the systems’ Krafft temperature Tc), and that the long-lived Lβ phase can be induced when excessive overcooling is avoided.24,26
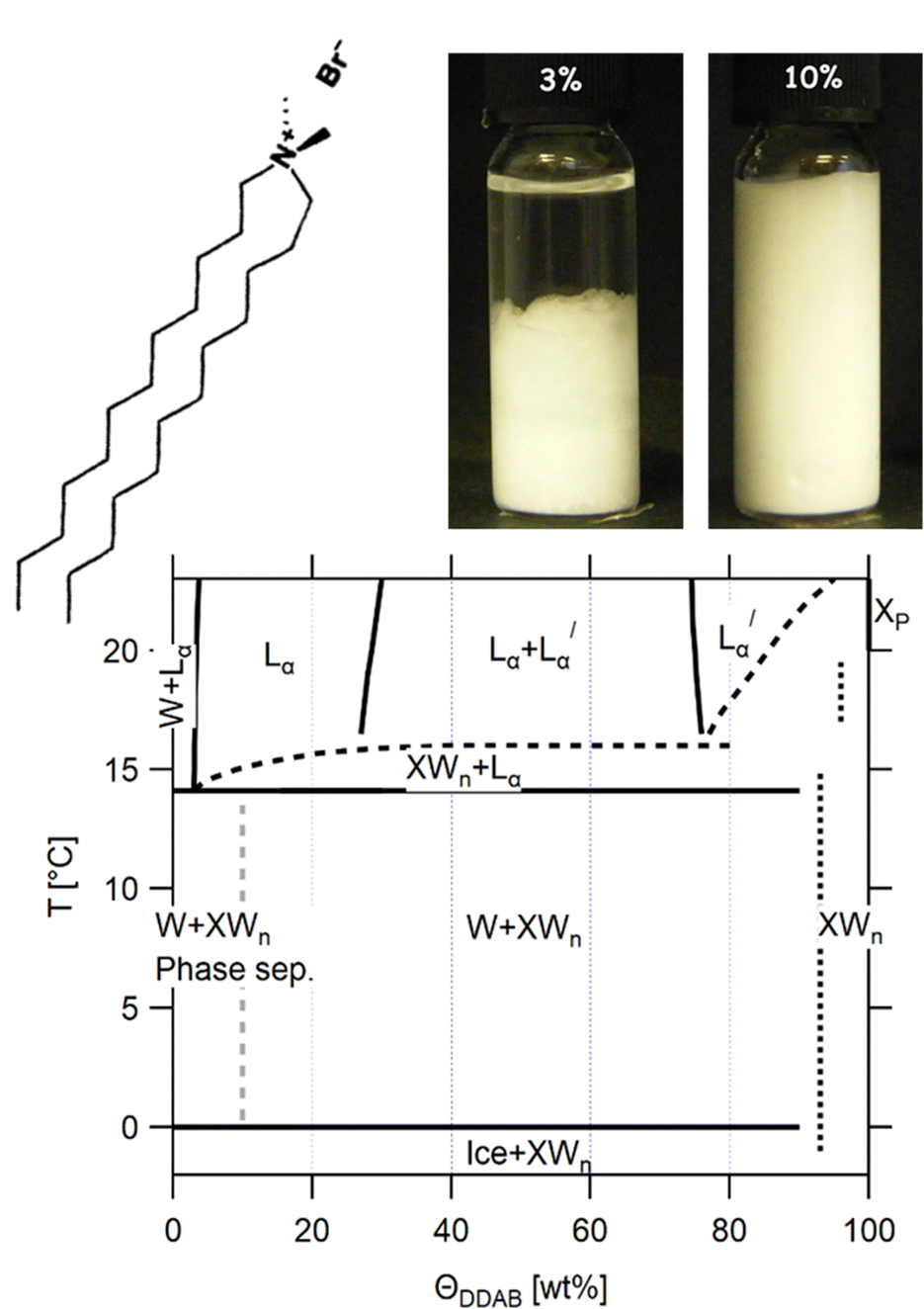 | ||
Fig. 1 (top) Sketch of the DDAB molecule, and photographs of the newly characterised 'white' phase; (bottom) proposed phase diagram of the binary DDAB/water system, where W corresponds to a dilute monomer solution (essentially pure water), Lα and 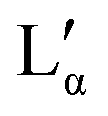 to the swollen and collapsed lamellar phase, respectively, XP to the powder crystal structure, and XWn to a non-zero hydrate low temperature crystal structure. The position of the dotted lines in the diagram corresponding to XWn as well as other non-zero hydrates are only estimated, as the exact number of water molecules are not known. The dashed grey line in the phase diagram limits the dilute region in which the low temperature dispersion of non-zero hydrates XWn in W show a macroscopic phase separation (see left photograph at the top). to the swollen and collapsed lamellar phase, respectively, XP to the powder crystal structure, and XWn to a non-zero hydrate low temperature crystal structure. The position of the dotted lines in the diagram corresponding to XWn as well as other non-zero hydrates are only estimated, as the exact number of water molecules are not known. The dashed grey line in the phase diagram limits the dilute region in which the low temperature dispersion of non-zero hydrates XWn in W show a macroscopic phase separation (see left photograph at the top). | ||
In this paper, we present a revision to the existing broad phase diagram (see Fig. 1 bottom), which replaces the Lβ phase with a region in which DDAB hydrate crystals coexist either with water XWn + W below 14.1 °C or with a lamellar phase XWn + Lα (at intermediate temperatures), drawing it closer to its longer-chain analogues. A set of complementary experimental techniques was used to characterize the obtained phases over many length scales. Furthermore, the stability of the pure DDAB powder sample, which was identified as a zero-hydrate at room temperature (in contrast to previous studies), could only be confirmed up to 90 °C. At higher temperatures, the molecule thermally degrades, setting a new upper limit to any DDAB phase diagram. We contextualise our results with the current literature, which is reviewed in Section 3, and provide reasons for the amendments to the phase diagram.
Experimental
Materials
DDAB was purchased from Aldrich (purity 99%) and Fluka (purity 99%) for comparison, and stored in a desiccator (RH < 40%) at room temperature (T ≈ 20 °C). Around 5 g of DDAB was recrystallised from ethyl acetate following established procedures,17,32 to ensure the results were not affected by impurities. In short, into a conical flask containing DDAB, ethyl acetate was incrementally added by Pasteur pipette (0.5–1 mL), with swirling, until the DDAB just dissolved. Diethyl ether was then added dropwise also with swirling, slowly reducing the polarity of the solvent system, until a precipitate remained upon swirling. The vessel was sealed to limit evaporation, and the solution cooled in a freezer. After 4 h, the flask was removed and the precipitate filtered. The nature of the precipitate was such that a large amount of product remained in the vessel. This was recrystallized from the same solvent system with extensive precipitation induced by an excess of diethyl ether (ca. 2–3 vol. eq.) rather than cooling. The precipitate was filtered to dryness in air and this process repeated twice. The pure product was dried with phosphorous pentoxide under a nitrogen atmosphere. Because of the high capacity of the DDAB powder to absorb water, the sample was dried under vacuum for 48 h, and maintained under pure nitrogen atmosphere prior to measurements.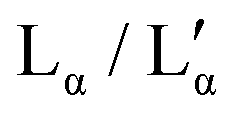 ) were turbid, indicating phase coexistence, while samples above 75 wt% and below 25 wt% were optically clear. The samples were equilibrated for between one week and several months at the required temperature prior to measurement.
) were turbid, indicating phase coexistence, while samples above 75 wt% and below 25 wt% were optically clear. The samples were equilibrated for between one week and several months at the required temperature prior to measurement.
Analytical techniques
Thin-layer chromatography (TLC), electronspray ionization mass spectroscopy (ESI-MS) and nuclear magnetic resonance spectroscopy (NMR) were used to confirm the purity of the DDAB samples, as well as study their degradation products. The technical details are given in the ESI.‡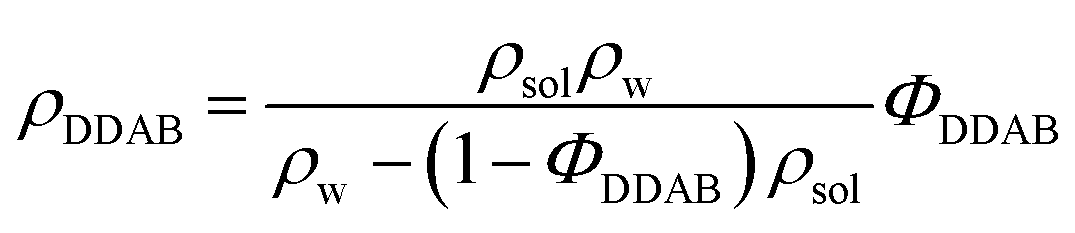 | (1) |
The data were compared to the measured density dependence of pure water in the same range and analysed using Origin8.0.
Raman techniques and band assignment
Raman spectroscopy was used to investigate the conformational properties based on the vibrational states of the DDAB molecules in the aqueous solutions and solid samples, and their dependence on temperature and water concentration.Small-angle X-ray scattering (SAXS)
SAXS was used to obtain information about the dependence of the repeat distance in the lamellar phases on surfactant concentration and temperature. Additionally, the strong forward scattering at low scattering vectors that was observed for the ‘white’ phase samples in the low temperature range was evaluated using the form factor of a particulate (such as a crystal) and the Porod scattering of its smooth interface in water. SAXS measurements were performed on a Nanostar U (Bruker, Karlsruhe), equipped with a rotating Turbo-Xray Source (TXS), operated at 40 kV/40 mA. The CuKα radiation with λ = 1.5418 Å was focussed at the sample position by 2 crossed Göbel mirrors and 3 pinholes and scattering intensities detected on a 2D Hi-Star detector with 1048 × 1048 pixel resolution, positioned at a distance of 1.06 m and allowing a scattering vector range 0.008 < q < 0.2 Å−1 with q = 4π/λ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) sin(θ/2). The beam size was 0.5 mm. Absolute intensities [cm−1] were obtained from comparison with a secondary standard, a thermoplastic polymer FEP1400 (Dupont Inc.), previously calibrated at the European Synchroton Radiation Facility (ESRF), Grenoble, France. The data were obtained from random-oriented samples in glass capillaries (Hilgenberg, nominal thickness 1.5 mm), using an empty glass capillary for background correction. The samples were temperature-controlled in a custom-built cell (ΔT = 0.5 °C). The detected ‘rings’ were radially integrated using Bruker software. The data was background- and transmission-corrected using a Fortran program, and further analysed using IGOR Pro. Uncertainties in the intensity scale are of the order of 5–10% in view of possible thickness variations in the capillaries.
sin(θ/2). The beam size was 0.5 mm. Absolute intensities [cm−1] were obtained from comparison with a secondary standard, a thermoplastic polymer FEP1400 (Dupont Inc.), previously calibrated at the European Synchroton Radiation Facility (ESRF), Grenoble, France. The data were obtained from random-oriented samples in glass capillaries (Hilgenberg, nominal thickness 1.5 mm), using an empty glass capillary for background correction. The samples were temperature-controlled in a custom-built cell (ΔT = 0.5 °C). The detected ‘rings’ were radially integrated using Bruker software. The data was background- and transmission-corrected using a Fortran program, and further analysed using IGOR Pro. Uncertainties in the intensity scale are of the order of 5–10% in view of possible thickness variations in the capillaries.
Microscopy
A Nikkon Eclipse 80i microscope with a 10× phase contrast objective (image size 2.2 mm × 1.5 mm) and an RMA 4376 (Media Cybernetics) camera were used to investigate structures in the micrometer length scale, such as multilamellar vesicles or crystals, in liquid samples depending on surfactant concentration and temperature. To increase contrast, DDAB crystals in solution were recorded under crossed polarizers. If needed the sample was held in a temperature-controlled stage (linkam LTS350/TMS93/LNNP).Differential scanning calorimetry (DSC)
DSC was used to identify the temperature and properties of the thermal phase transitions. DSC measurements on solution samples were carried out on the Mettler DSC30 in disposable crucibles made of aluminium under a nitrogen flow of 20 mL min−1. Before the measurements, the equipment was calibrated using indium, gallium, lead and bismuth with scanning rates of 2, 5 and 10 °C min−1. For samples larger than 10 mg, an aluminium oxide filled reference crucible was used. In order to confirm that the observed melting peak around 15 °C is associated with the transition of the ‘white’ phase and is not present in overcooled clear samples, respective reference samples were loaded into the crucible in a cold room (5 °C), and immediately put into the precooled device. The optimised scanning rate of 1 or 2 °C min−1 was chosen, after studying the dependence of the onset temperature and peak width on scanning rate was measured for rates of 10, 5, 2, 1 and 0.1 °C min−1. Samples were in general scanned between 2 °C and 20 °C with the upper limit expanded for samples with high DDAB concentration. For selected samples, the influence of water freezing was also studied by extending the lower limit of the scan range down to −20 °C. No influence of water freezing on the position or shape of the DDAB melting transition was observed. The onset temperature and peak width were within the experimental error of 0.3 °C for the relative temperature for scanning rates of 1 °C. However, the calibration of the absolute temperature (error about 2 °C) was of comparably low precision. Furthermore, no dependence of peak width on sample mass or relative contact area between crucible and sample was found, indicating that the observed effects were not caused by a temperature gradient across the sample. In addition, control measurements on pure water were performed to ensure the observed effects were not artefacts of the equipment. For comparison of samples with different mass and DDAB concentration, the measured heat flow was normalized by dividing by the sample mass and DDAB volume fraction. The transition enthalpies were estimated by integrating the peak areas over the transition time. The changes in enthalpy observed with concentration lie within the error in the measured enthalpy value of δΔHDDAB = 5 kJ mol−1, which is mostly caused by variations due to scan rate, DDAB freezing temperature or sample history. In the low temperature water freezing runs, the enthalpy value of the DDAB melting transition shifted to the upper range of the measured values, but still lay within the measurement error of non-frozen values.Review of phase studies of the DDAB–water system
In order to put the obtained experimental results into context, we provide below (and as a summary in Table 1) a thorough review of the reported studies of the molecular properties of DDAB and the phases formed in the binary aqueous system, as well as comparisons with other dialkyldimethylammonium halide systems.| Molecular mass m | 461 g mol−1 |
| Molecular volume Vm | 784 Å3 (20 °C), 792 Å3 (40 °C) |
| Density ρ | 0.98 kg cm−3 (20 °C)19 |
| Area per headgroup | 63 Å2 (Lα)–68 Å2 (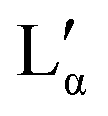 ) (depending on temperature and salt concentration)17,19 ) (depending on temperature and salt concentration)17,19 |
| Chain length lc | 16.68 Å (fully stretched)10,18 |
| Chain volume Vc | 703.7 Å3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 10,18 10,18 |
| Distance between bilayers in fully swollen lamellar phase | 80 nm (decreases with increasing salt concentration)19 |
| Bilayer thickness ab | 24 Å (varies with temperature)19 |
| Ion layer thickness aBr | 7 Å19 |
| Critical vesicle concentration CVC | 0.05 mM (2.3 × 10−3 wt%)24 |
| Critical multilamellar phase concentration CVC2 | 0.7 mM (31 × 10−3 wt%) [0.21 mM (9.5 × 10−3 wt%) as multilamellar vesicles]24 |
The aqueous phase behaviour of DDAB (as well as of other representatives of this group28–31) has been extensively studied in the past.17–24 At temperatures above 16 °C (assigned as the chain melting temperature Tm19,24 or Krafft temperature Tc23,25,26) two optically identical lamellar phases have been reported: the swollen lamellar phase Lα (3–28 wt%17,20 or 3–30 wt%9,36), and the collapsed lamellar phase 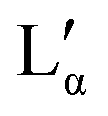 (83–91 wt%,9 >75 wt%,17,20 75–85 wt%36). In contrast to the Lα phase, some counterions in the
(83–91 wt%,9 >75 wt%,17,20 75–85 wt%36). In contrast to the Lα phase, some counterions in the 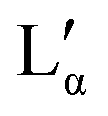 phase are expected to be bound to the bilayers, resulting in a ‘neutral’ bilayer, between which the hydration force and not the electrostatic force can be considered the dominant repulsive force.9,17,37 For intermediate concentrations, both phases coexist, stabilized by the interplay of the two repulsive forces which differ in decay length9,17,37 and compete both with a strong attraction. The existence of such a miscibility region, also known for other systems,37 has only been reported for dialkyldimethylammonium halides with bromide as the counter ion.17,28,30,31 One explanation is that chloride ions are more strongly hydrated compared to bromide, which decreases their binding affinity to the surface of the aggregates.10,17,22,38 An alternative explanation is the strong electrostatic coupling observed for bromide ions when in close proximity to bilayers:39 for such small distances the counterions are assumed to be only influenced by the surface charge of the bilayer, and not by the electrostatic field of the other counterions; for long distances (the Poisson Boltzmann regime) their position is, to a large degree, influenced by the other counterions. For systems showing strong coupling, a separation between two phases with different distances between the “charged plates” can be predicted.39
phase are expected to be bound to the bilayers, resulting in a ‘neutral’ bilayer, between which the hydration force and not the electrostatic force can be considered the dominant repulsive force.9,17,37 For intermediate concentrations, both phases coexist, stabilized by the interplay of the two repulsive forces which differ in decay length9,17,37 and compete both with a strong attraction. The existence of such a miscibility region, also known for other systems,37 has only been reported for dialkyldimethylammonium halides with bromide as the counter ion.17,28,30,31 One explanation is that chloride ions are more strongly hydrated compared to bromide, which decreases their binding affinity to the surface of the aggregates.10,17,22,38 An alternative explanation is the strong electrostatic coupling observed for bromide ions when in close proximity to bilayers:39 for such small distances the counterions are assumed to be only influenced by the surface charge of the bilayer, and not by the electrostatic field of the other counterions; for long distances (the Poisson Boltzmann regime) their position is, to a large degree, influenced by the other counterions. For systems showing strong coupling, a separation between two phases with different distances between the “charged plates” can be predicted.39
When going from the Lα phase to the 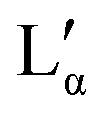 phase a sudden increase in head group area and concomitant decrease in bilayer thickness was observed, which both relax with increasing surfactant concentration.17 The critical point at which the two lamellar phases merge is around 75 °C (72 °C21) at 62.2 wt%.10,32
phase a sudden increase in head group area and concomitant decrease in bilayer thickness was observed, which both relax with increasing surfactant concentration.17 The critical point at which the two lamellar phases merge is around 75 °C (72 °C21) at 62.2 wt%.10,32
At lower surfactant concentration (<3 wt%) (analogous to other dialkyldimethylammonium halide systems) a fully swollen lamellar phase coexists with a dilute liquid phase, often described as a vesicle phase, showing unilamellar vesicles at low concentration,24,40 and multilamellar vesicles with increasing surfactant concentration.10,24,41 However, there is ongoing discussion regarding the equilibrium status of vesicles,42,43 even long-lived ones, due to the bending energy of the surfactant monolayers: thermodynamically bilayers should either be flat (for positive bending rigidity) or extremely curved to make very small vesicles (here rigidity is negative and higher order in the curvature expansion becomes necessary to restore stability). Below the CVC (critical vesicle concentration) of 0.002 wt% a monomer solution has been identified,24 while in related systems small spherical or cylindrical micelles have been reported.30,31 For temperatures above 22 °C, a flow birefringent long-lived metastable sponge phase L3 has also been observed instead of the equilibrium coexistence region.19
Commonly for dialkyldimethylammonium systems, the lamellar phase is limited at low temperature by the Krafft temperature Tc, below which the dilute liquid phase coexists with crystals or crystal hydrates.23,28,29,44,45 A large hysteresis is observed for the freezing of the lamellar phases due to the kinetic hindrance of the crystallization of the surfactant.30,31,46 This leads to the occurrence of metastable phases, which can be of liquid crystalline or crystalline character. In the case of vesicles, the occurrence of gel phases such as the Lβ phase – a lamellar phase, in which the fluid character of the bilayers is highly decreased43,47 – have been frequently reported at low temperatures, which can require long equilibration times for the molecules to rearrange.8,27,48 The melting temperatures Tm of these gel phases often correspond to the Krafft temperature. For the DDAB vesicle system, this was confirmed and it was shown that a long-lived Lβ phase can be formed by avoiding excessive overcooling (>5 °C).24,26 In contrast to other dialkyl dimethylammonium halide systems, in DDAB an Lβ phase has not only been reported for the vesicular solutions, but has been suggested for the bulk lamellar phases17,19,20 in the equilibrium phase diagram. The chain melting temperature Tm in the swollen lamellar phase19 coincides with that observed in vesicle solutions,8,10,24,26,27 while a slightly higher transition temperature of 24 °C has been reported in the case of the collapsed lamellar phase.32 The Lβ lamellar phases were described as clear solutions, with spacings similar to Lα phases, obtained by SAXS.17,19
The behaviour of the solid DDAB phase has been studied by DSC and X-ray diffraction.49 The sample has been reported to degrade at 160 °C (the upper boundary of a DDAB phase diagram) below which it shows three phase transitions: (1) at 30 °C the monohydrate (or di-hydrate50) undergoes polytectic melting to the pure crystal, (2) at 58.6 °C there is a transition into a liquid crystalline phase, and (3) at 76.2 °C occurs a polymorphic transition within the liquid crystal phase.
Results and discussion
When we cooled aqueous solutions of DDAB in the range of ϕDDAB = 3–90% below Tm (≤ 10 °C), the samples turned white (see Fig. 1 top right). These ‘white’ phases were stable up to around 15 °C, i.e. close to their reported chain melting (Tm) or Krafft (Tc) temperatures. The occurrence of a white phase is in contradiction with the description of the “optically clear” Lβ phase,17 which has been suggested in this region in the latest reported broad equilibrium phase diagram.17 In order to assess the equilibrium nature and the properties of the ‘white’ phase, a range of experiments were conducted. As it is known that the low monomer solubility, and thus, low monomer exchange rates between aggregates reported for dialkyldimethyl-ammonium halide systems will lead – especially for crystalline phases – to long equilibration times, the historical treatment of the samples is highly significant.10 Thus, the influence of sample history on the (kinetics of the) phase transitions was investigated in the second part of this study.51DDAB powder (T ≥ 20 °C)
The phase behaviour, especially of the pure DDAB sample, can be strongly influenced by impurities.29,30,52 Three impurities were expected to be found in the DDAB sample: (i) water, caused by the existence of crystal hydrates and/or by the high water affinity of the sample; (ii) DDAB analogues of slightly different chain lengths, which could not be removed by recrystallization, but could be confirmed absent by ESI-MS and NMR analysis of the samples (see the ESI,‡ Fig. S6 and S7); and (iii) degradation products caused by thermal decomposition of DDAB at higher temperatures.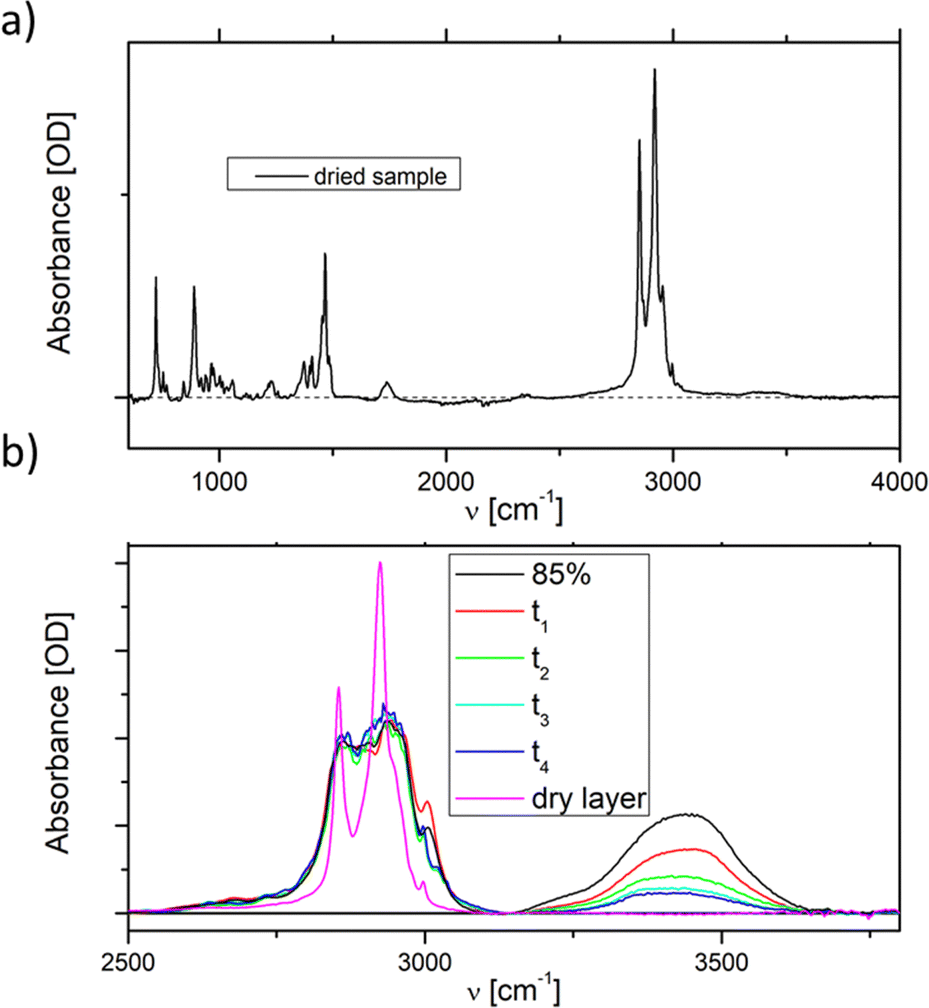 | ||
| Fig. 2 Room temperature ATR FTIR spectra of (a) recrystallised dry DDAB samples and (b) during the evaporation of a condensed DDAB solution, showing a smooth decrease in water peak, until all water is evaporated. | ||
To investigate the appearance of additional stable crystal hydrates at room temperature, an 85 wt% solution ( phase) – containing less than five water molecules per DDAB molecule – was observed during evaporation of its water content at room temperature (Fig. 2b). The water peaks could be fitted with two main broad Gaussians around 3370 cm−1 and 3475 cm−1 corresponding to the symmetrical and asymmetrical stretching modes of water, respectively. During evaporation, a smooth decrease in peak intensity was observed, suggesting limited structural changes in the water. Thus, the results suggest that, in contrast to DODAB45 and DODMAC,29 which show distinct sharp peaks in the vibrational water spectrum, a zero-hydrate X is the stable and only crystalline form of DDAB at room temperature. Thus, the high affinity of water under atmospheric conditions would lead to the dissolution of the surface of some DDAB crystals, resulting in a coexistence region of X with the concentrated
phase) – containing less than five water molecules per DDAB molecule – was observed during evaporation of its water content at room temperature (Fig. 2b). The water peaks could be fitted with two main broad Gaussians around 3370 cm−1 and 3475 cm−1 corresponding to the symmetrical and asymmetrical stretching modes of water, respectively. During evaporation, a smooth decrease in peak intensity was observed, suggesting limited structural changes in the water. Thus, the results suggest that, in contrast to DODAB45 and DODMAC,29 which show distinct sharp peaks in the vibrational water spectrum, a zero-hydrate X is the stable and only crystalline form of DDAB at room temperature. Thus, the high affinity of water under atmospheric conditions would lead to the dissolution of the surface of some DDAB crystals, resulting in a coexistence region of X with the concentrated  phase reported to start in the concentration range of 85–91 wt% (see above).
phase reported to start in the concentration range of 85–91 wt% (see above).
Conformational properties of DDAB
A stable Raman spectrum of the solid DDAB sample has been obtained (see Fig. 3 black lines and Table 2), in which the majority of peaks could be assigned, in agreement with the literature.33 The spectrum indicates an all-trans extended chain structure: (i) the dominance of the symmetric C–H stretching vibrations (2880 cm−1) over the asymmetric C–H stretching vibration (2850 cm−1) indicates a high order in the hydrocarbon chains, and (ii) the 1060 cm−1 and 1130 cm−1 bands are characteristic of C–C stretching vibrations for the all-trans conformation of the chain.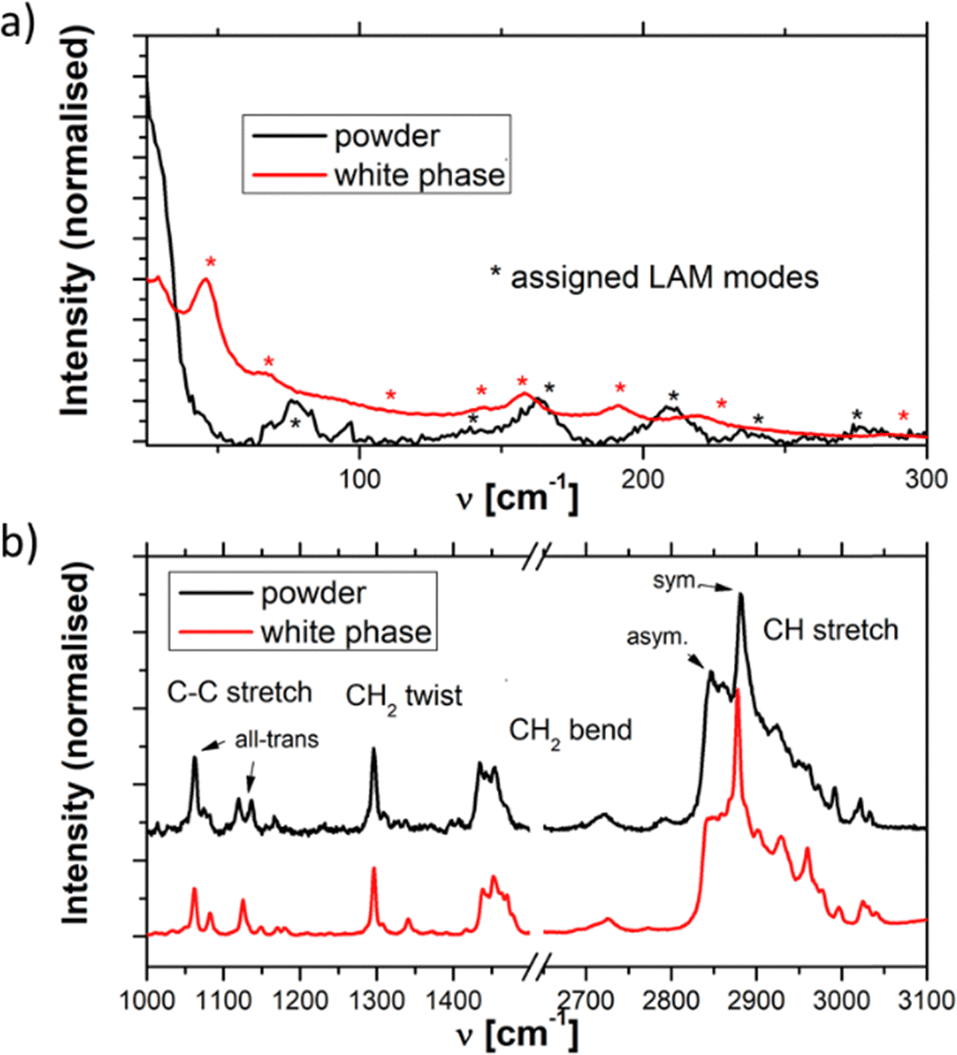 | ||
| Fig. 3 Raman spectra of the DDAB powder (black line) and 'white' phase (red line) in the (a) low and (b) medium-high frequency range. | ||
| ν [cm−1] | Δν [cm−1] | Rel. height | Area (%) | Assignment |
|---|---|---|---|---|
| 77 | 11 | 0.163 | 2 | LAM mode |
| 142 | 18 | 0.047 | 1 | LAM mode |
| 163 | 18 | 0.123 | 2 | LAM mode33 |
| 209 | 12 | 0.122 | 2 | LAM mode |
| 237 | 15 | 0.032 | 0 | LAM mode33 |
| 278 | 12 | 0.054 | 1 | LAM mode33 |
| 417 | 19 | 0.073 | 1 | LAM mode33 |
| 440 | 7 | 0.043 | 0 | Unassigned33 |
| 465 | 4 | 0.073 | 0 | Unassigned33 |
| 479 | 9 | 0.029 | 0 | |
| 521 | 11 | 0.019 | 0 | Unassigned33 |
| 564 | 3 | 0.019 | 0 | Unassigned33 |
| 725 | 10 | 0.062 | 1 | CH3 rock from N+(CH3)2 group33 |
| 761 | 14 | 0.148 | 2 | CH3 rock from N+(CH3)2 group33 |
| 879 | 10 | 0.092 | 1 | |
| 891 | 9 | 0.102 | 1 | CH3 rock & CN+ stretch33 |
| 918 | 3 | 0.021 | 0 | CN+ stretch33 |
| 943 | 10 | 0.020 | 0 | Unassigned33 |
| 979 | 14 | 0.021 | 0 | Unassigned33 |
| 1062 | 8 | 0.376 | 3 | C–C sym stretch & CH2 wag.33 |
| 1120 | 8 | 0.139 | 1 | Unassigned33 |
| 1136 | 6 | 0.140 | 1 | C–C asym stretch & CH2 (gauche) wag.33 |
| 1167 | 5 | 0.055 | 0 | CH2 rock33 |
| 1232 | 1 | 0.033 | 0 | CH2 wag., crystal. & CH2 twist33 |
| 1296 | 8 | 0.403 | 3 | CH2 twist33 |
| 1311 | 7 | 0.066 | 0 | |
| 1332 | 17 | 0.028 | 0 | CH2 wag33 |
| 1403 | 14 | 0.026 | 0 | |
| 1434 | 10 | 0.303 | 3 | CH2 bend33 |
| 1443 | 7 | 0.144 | 1 | CH sym bend N+(CH3)2 group33 |
| 1454 | 15 | 0.291 | 4 | CH2 bend & CH asym bend from N+(CH3)2 group33 |
| 1469 | 10 | 0.081 | 1 | |
| 2719 | 24 | 0.065 | 1 | Unassigned33 |
| 2791 | 23 | 0.053 | 1 | Unassigned33 |
| 2845 | 17 | 0.067 | 8 | CH asym stretch of CH233 |
| 2862 | 19 | 0.572 | 10 | CH asym stretch of CH233 |
| 2882 | 16 | 1 | 16 | CH sym stretch of CH333 |
| 2900 | 23 | 0.417 | 11 | |
| 2924 | 27 | 0.408 | 10 | CH sym stretch of N+(CH3)233 |
| 2950 | 18 | 0.265 | 4 | |
| 2962 | 9 | 0.232 | 2 | CH asym stretch of N+(CH3)233 |
| 2974 | 9 | 0.193 | 1 | |
| 2992 | 6 | 0.144 | 1 | Unassigned33 |
| 3021 | 12 | 0.132 | 2 | |
Thermal stability of DDAB
At temperatures above T ≈ 90 °C the white DDAB powder changes its colour to yellowish-brown (within the period of one week). This colour change is more apparent at higher temperatures and is frequently accompanied by a melting of the DDAB powder. The colour change is likely to be related to the degradation of the quaternary ammonium ion into the thermally stable amine,46,52 and subsequent N-oxidation of the amine (see the ESI,‡ Fig. S8). The degradation pathway of DDAB, expected to be caused by nucleophilic attack by the bromide ion,46 was confirmed using TLC analysis of samples subjected to different temperatures and exposure times, as well as 1H-NMR analysis of the degraded sample.The melting of the coloured DDAB sample, rapid at temperatures of 150 °C (close to the previously assigned degradation temperature49), can be assigned to the decomposition into the amine, which is in a liquid state at this temperature, making the use of melting point analysis for determining the purity of DDAB meaningless, similar to the case of related systems.30,31
In summary, the DDAB sample used in this study could be shown to be of sufficient purity to allow the study of the phase behaviour of the binary DDAB water system at temperatures below the thermal degradation temperature of DDAB (Tdeg ≈ 90 °C). Furthermore, a zero-hydrate is the stable crystalline form of the DDAB powder at room temperature, in which the chains of the DDAB molecule are in an all-trans conformation.
Thermal phase transitions of aqueous DDAB solutions
The thermal properties of aqueous DDAB solutions, which were anticipated to be in either the swollen or collapsed lamellar phase at room temperature, were investigated by DSC in the temperature range between T = −20 °C and +20 °C. Two freezing and melting transitions were identified around 0 °C and 15 °C (Fig. 4a). The large hysteresis between the exothermic cooling and endothermic heating peaks indicates slow kinetics and the existence of stable supercooled phases over a broad temperature range. The reversible transition centred near 0 °C is assigned to the freezing and melting of the water in the samples based on the known temperature and enthalpy values for pure water, and its dependence on sample concentration. The second phase transition around 15 °C has been previously assigned to the Lβ → Lα phase transition,19 with the transition temperature corresponding to the chain melting temperature Tm. Due to the mismatch in optical properties (the Lβ phase has been described as a clear phase, while the low temperature phase observed in this study is white (see Fig. 1)), and the detailed characterization of the white low temperature phase (see the Section “The low temperature phase”), this transition is here assigned to the melting of crystal hydrates in water XWn + W → Lα. It should be noted that this is consistent with the measurements of the Krafft temperature25 for dilute DDAB solutions performed by conductivity measurements. As is commonly observed for systems in which the monomer exchange between aggregates is slow,10 the degree of supercooling of the Lα phase before the start of the Lα → XWn + W transition as well as the transition kinetics depend strongly on the sample history (see the ESI,‡ Fig. S8). This behaviour is common for a nucleation and growth process, where the amount of surviving nucleation centres often determines the kinetics (see also part II of this study51), and supports that a crystallization process occurs during this phase transition. Furthermore, the heat required for the XWn + W → Lα phase transition is proportional to the surfactant concentration, supporting its assignment to the DDAB molecules and not the water phase (see Fig. 4b). Over the whole concentration range, transition enthalpy values ΔHDDAB = (38 ± 0.5) kJ mol−1 could be extracted. The kinetics of the melting transition have a non-cooperative character (characteristic for a non-isothermal phase transition) (see also part II of this study51).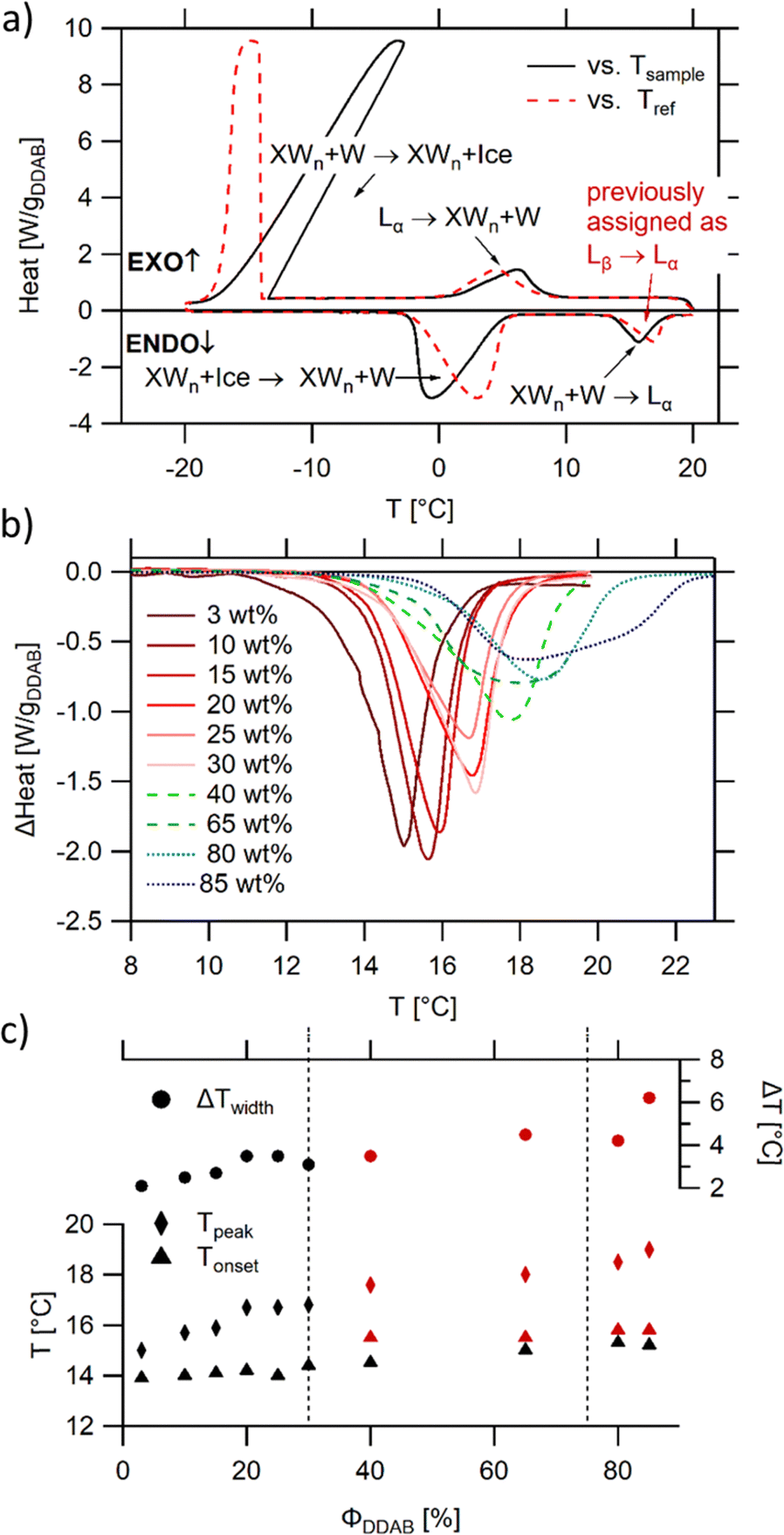 | ||
| Fig. 4 DSC analysis of aqueous DDAB solution: (a) representative scan in the −20 °C to 20 °C temperature range showing two melting and two freezing transitions (ΦDDAB = 25 wt%) with the black and red lines displaying the data against the sample or reference temperature, respectively; (b) dependence of the peak assigned to the melting of hydrated DDAB crystals on DDAB concentration; (c) dependence on transition onset (determined using a line fit), peak temperature and peak width on DDAB concentration. For concentrations above 30 wt%, a broadening of the peak occurs (red markers), and the second onset displays the beginning of the underlying peak. The dashed lines indicate the phase boundaries between the swollen and collapsed lamellar phase at high temperature, as well as their coexistence region. | ||
The broadening of the peaks which is observed for concentrations above 30 wt% (samples in the 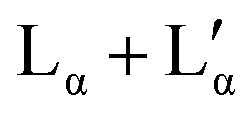 coexistence region or pure
coexistence region or pure 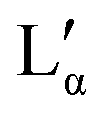 phase) can be either a result of several underlying transitions or the change in the melting pathway as proposed by the new phase diagram (which is a direct consequence of the rules of phase science for a eutectic phase transition – see below). For ΦDDAB < 30 wt% (but above 3 wt%) the melted crystals would form the Lα phase. In contrast, for ΦDDAB > 75% the melting would first result in the formation of a small fraction of Lα phase. The remaining crystals would then melt into the
phase) can be either a result of several underlying transitions or the change in the melting pathway as proposed by the new phase diagram (which is a direct consequence of the rules of phase science for a eutectic phase transition – see below). For ΦDDAB < 30 wt% (but above 3 wt%) the melted crystals would form the Lα phase. In contrast, for ΦDDAB > 75% the melting would first result in the formation of a small fraction of Lα phase. The remaining crystals would then melt into the 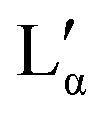 phase. For samples in the intermediate concentration range, crystals would first melt with increasing temperature into the Lα phase, and then above a critical temperature transform into the Lα or
phase. For samples in the intermediate concentration range, crystals would first melt with increasing temperature into the Lα phase, and then above a critical temperature transform into the Lα or  phase at a ratio given by the phase boundaries. The later transformation has an iso-thermic characteristic, but would still appear broadened in DSC scans. The non-isothermic characteristic of the phase transition is further studied in part II of this study.51
phase at a ratio given by the phase boundaries. The later transformation has an iso-thermic characteristic, but would still appear broadened in DSC scans. The non-isothermic characteristic of the phase transition is further studied in part II of this study.51
Structural properties of the lamellar phases
As already mentioned, the lamellar phases have already been extensively studied in the past.19,20,53 For the sake of comparison, some of the reported measurements were repeated in order to directly assess the changes in structure and shape of the aggregates in the low temperature phase. The existence of the lamellar phase could be confirmed through its birefringent character when observed through crossed polarizers, except at very low concentration, when the birefringence was comparably weak, giving the sample the appearance of an isotropic solution. The concentration range of the swollen lamellar phase was therefore investigated using SAXS in the range of 5–25 wt%. This range was limited by the detector limit (Bragg peaks for concentrations below 5 wt% were covered by the beam stop), hampering the comparison of the onset of the lamellar phase with literature values. As expected, the observed Bragg peaks, and therefore the repeat distance in the lamellar phase D* = aw + δ, where aw is the water layer thickness and δ the bilayer thickness, depend on sample concentration (Fig. 5a). This dependence can be approximated using a simple geometrical dependence | (2) |
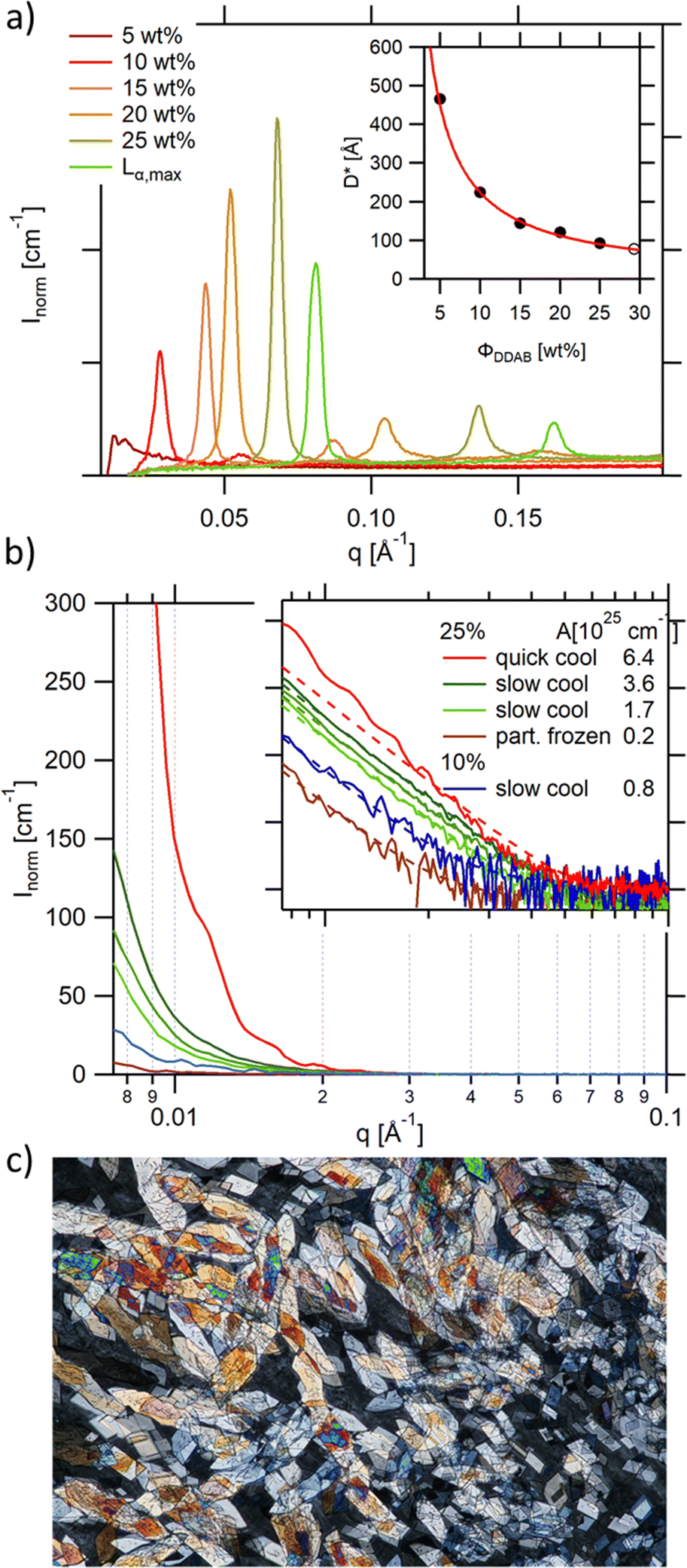 | ||
| Fig. 5 (a) Bragg peaks seen in the SAXS spectra of aqueous DDAB solutions being in the Lα phase showing a decrease in repeating distance with increasing DDAB concentration (inset: the line shows the least squares fit to the data giving a bilayer thickness of δ = (22.4 ± 0.4) Å); (b) scattering intensity at small q values for a number of samples containing the ‘white’ phase. The q−4 behaviour can be explained by scattering of single crystals in the phase. The Guinier regime can only be resolved for comparably small crystals obtained with fast cooling rates (red trace), while in slower cooled samples only the Porod scattering can be observed. The scattering intensity changes with cooling rate, and decreases with decreasing DDAB concentration, as well as for partially frozen samples. The inset shows the log–log presentation of the data. The prefactor (given in the figure legend) of the q−4 dependence was obtained by fitting the data with eqn (4) (dashed lines); (c) DDAB crystals of the 'white' phase observed under crossed polarizers (image size 2.2 × 1.5 mm). | ||
The high concentration limit of the swollen lamellar phase Lα was estimated by phase separating a sample in the 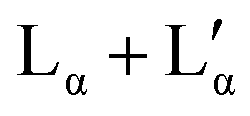 coexistence region by centrifugation (3000 rpm, 8 h, 20 °C), and measuring the repeating distance D* of the dilute phase. The obtained value corresponded to a surfactant concentration of approximately ΦDDAB = (29.5 ± 0.5) wt%, which is in good agreement with the literature value of 28%17,20 or 30%.9,36 Finally, it should be mentioned that the Bragg peak signal did not change within measurement error in the measured temperature range, even when the Lα phase was supercooled. It should be noted that the SAXS characterisation of the supercooled Lα phase, as well as its optically clear appearance, agree with the characterisation of the previously assigned equilibrium Lβ phase,17 with the exception that no phase transition can been measured between the two phases by DSC, suggesting that during their study, the structural characterisation experiments were conducted on the non-equilibrium long-live supercooled Lα phase.
coexistence region by centrifugation (3000 rpm, 8 h, 20 °C), and measuring the repeating distance D* of the dilute phase. The obtained value corresponded to a surfactant concentration of approximately ΦDDAB = (29.5 ± 0.5) wt%, which is in good agreement with the literature value of 28%17,20 or 30%.9,36 Finally, it should be mentioned that the Bragg peak signal did not change within measurement error in the measured temperature range, even when the Lα phase was supercooled. It should be noted that the SAXS characterisation of the supercooled Lα phase, as well as its optically clear appearance, agree with the characterisation of the previously assigned equilibrium Lβ phase,17 with the exception that no phase transition can been measured between the two phases by DSC, suggesting that during their study, the structural characterisation experiments were conducted on the non-equilibrium long-live supercooled Lα phase.
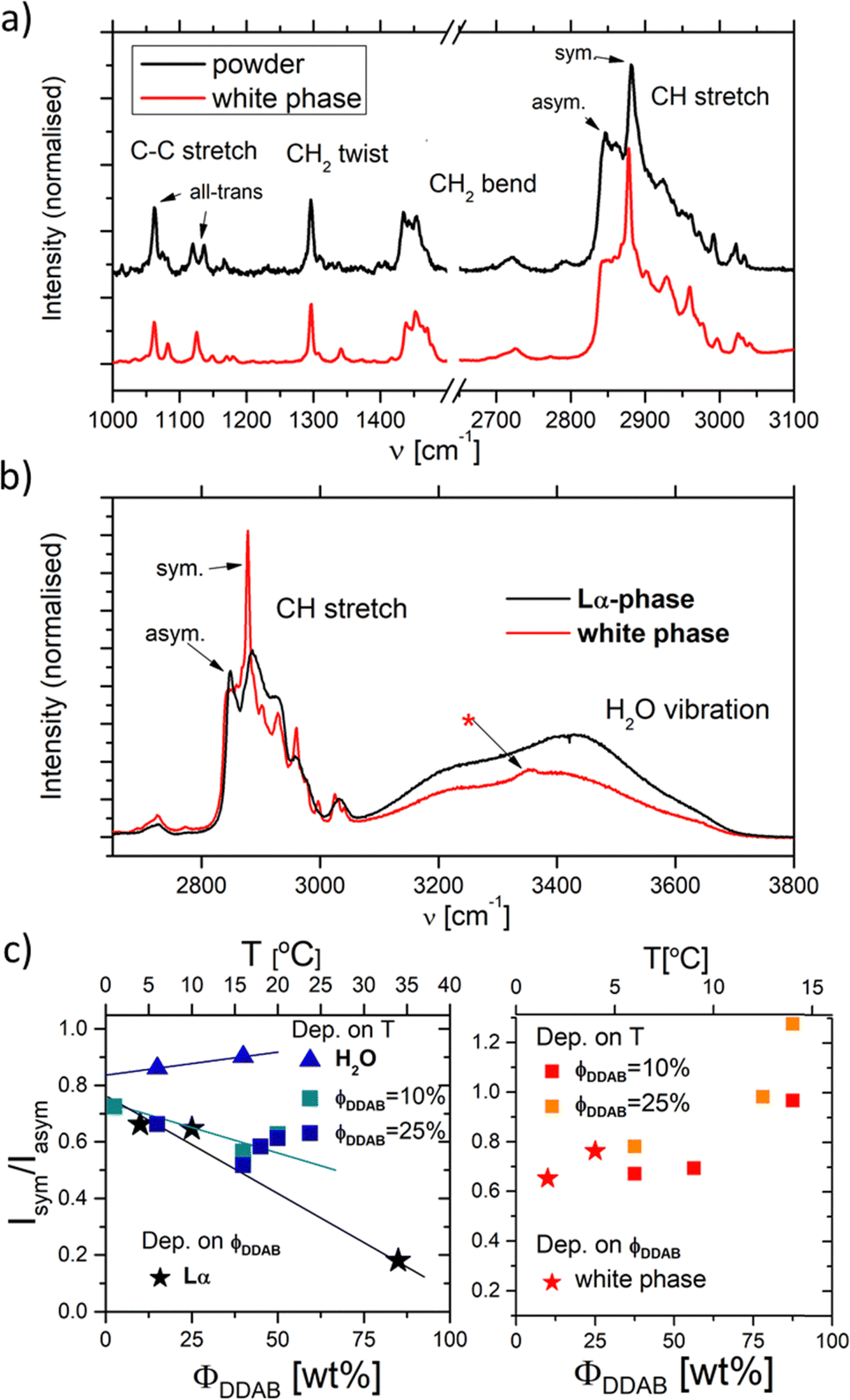 | ||
| Fig. 6 (a) and (b) Raman spectra of aqueous DDAB solution in the lamellar phase (black) compared to the 'white' phase (red). (c) Ratio of the Isym/Iasym water peaks depending on DDAB concentration, phase and temperature (see also the ESI,‡ Table S1). | ||
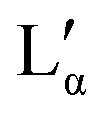 phase at room temperature
phase at room temperature
| ν [cm−1] | Δν [cm−1] | Rel. Height | Area (%) | Assignment |
|---|---|---|---|---|
| 772 | 34 | 0.040 | 2 | |
| 840 | 19 | 0.024 | 2 | CH2 rock in CH2–N+(CH3)2 group33 |
| 848 | 12 | 0.018 | 3 | |
| 872 | 16 | 0.035 | 4 | |
| 890 | 15 | 0.064 | 4 | CH3 rock & CN+ stretch33 |
| 910 | 27 | 0.034 | 4 | CN+ stretch33 |
| 928 | 16 | 0.027 | 4 | |
| 1066 | 32 | 0.136 | 3 | C–C sym stretch & CH2 wag.33 |
| 1084 | 14 | 0.096 | 8 | C–C stretch (gauche conformation)33 |
| 1127 | 19 | 0.039 | 8 | C–C asym stretch & CH2 (gauche) wag.33 |
| 1301 | 18 | 0.207 | 5 | C–C twist33 |
| 1321 | 52 | 0.076 | 8 | |
| 1437 | 14 | 0.287 | 6 | CH2 bend33 |
| 1455 | 32 | 0.439 | 5 | CH2 bend & CH asym bend from N+(CH3)2 group33 |
| 2849 | 17 | 0.897 | 6 | CH asym stretch of CH233 |
| 2880 | 45 | 1 | 6 | CH sym stretch of CH333 |
| 2910 | 52 | 0.656 | 7 | |
| 2931 | 22 | 0.417 | 7 | |
| 2964 | 45 | 0.469 | 3 | CH asym stretch of N+(CH3)233 |
| 3033 | 29 | 0.090 | 4 | |
| 3219 | 210 | 0.590 | — | H2O sym stretch34 |
| 3434 | 243 | 1 | — | H2O asym stretch34 |
| 3620 | 101 | 0.120 | — | H2O |
Furthermore, apart from predictable changes in the relative intensity of the DDAB to water peaks, no dependence on DDAB concentration or type of lamellar phase could be observed (see the ESI,‡ Fig. S10). The homogeneity of the spectra throughout the sample was confirmed using Raman microscopy. As expected, an increased gauche content in the hydrocarbon chains and thus an increased fluidity in the bilayers of the lamellar phase, compared to the powder sample, was observed as the appearance of a strong band at 1088 cm−1 in the C–C bending region of the spectrum,33 as well as an increase in the ratio I2850/I2880 of the asymmetric to the symmetric C–H stretching vibration. The more fluid character of the sample is also supported by the observed broadening of the vibrational bands and by the disappearance of the LAM frequencies in the spectrum. Interestingly, the Raman signal also showed no effect of the degree of supercooling of the lamellar phase in the peaks corresponding to the DDAB molecules, supporting the assignment as a supercooled Lα phase.
In contrast to the DDAB molecule, the structure of the water molecules is affected by the DDAB concentration and sample temperature, as evident in the changes in the shape of the water peak above 3000 cm−1 (see Fig. 6c left and the ESI,‡ Table S1). With increasing surfactant concentration, the vibration assigned to the asymmetric stretching mode of water molecules also increased, which is commonly associated with an increase in order of the water molecules.34 When comparing to pure water solution even at low temperature, the relative intensity of the symmetric stretching mode compared to the asymmetric stretching mode is already low in dilute samples, but further dramatically reduces upon increasing DDAB concentration. Interestingly, the supercooled lamellar phase shows opposing behaviour, suggesting that with the degree of supercooling, water in the lamellar phase exists with a more fluidlike characteristic.
Structural properties of the low temperature phase ('white' phase)
As already mentioned, if the aqueous DDAB solution is cooled sufficiently, a dramatic increase in turbidity is observed resulting in a whitish appearance (Fig. 1 top right). This whitening takes a long time (up to several months at temperatures close to the phase transition temperature, while only minutes at T < 5 °C). The white colour suggests microscopic phase coexistence due to scattering from the multiple phase boundaries. For thin samples (below 100 μm), transmission of light was sufficiently increased that crystals could be resolved under a microscope (Fig. 5c). Once a white sample is heated above the phase transition temperature, it returns to its original optically clear appearance.For the white low temperature samples with surfactant concentration ΦDDAB ≤ 10 wt% macroscopic phase separation was observed (see the left photograph in Fig. 1) by sedimentation of the denser white phase, containing dispersed DDAB, from a clear liquid phase (indicated by a grey broken line in the proposed phase diagram in Fig. 1). This phase separation could be enhanced by centrifugation. The ratio of clear phase to white phase decreased with ΦDDAB until, for higher surfactant concentrations, no phase separation was observed, even with prolonged centrifugation. No DDAB molecules could be identified in the clear liquid using analytical methods. Therefore, this phase was denoted as W. The exact position of the boundary in the phase diagram below which macroscopic phase separation could be observed was determined by removing the clear liquid phase from the sedimented white phase in phase separated samples. The samples were heated to room temperature to transform into their corresponding lamellar phase and their repeating distance was probed by SAXS. The measured repeating distance corresponded to a lamellar phase of ΦDDAB = 10 wt%.
The density of samples in the ‘white’ phase was measured, and the density corresponding to the DDAB molecules ρDDAB,white = (1.1 ± 0.2) kg dm−3 extracted, a value about 15% higher than that of the lamellar phase of ρDDAB,Lα = (0.97 ± 0.01) kg dm−3, which is in good agreement with the reported literature value19 (Table 1). Therefore, the comparably high density of DDAB in the ‘white’ phase, compared to water, can explain the sedimentation behaviour discussed above.
The SAXS spectrum of the ‘white’ phase itself (Fig. 5b) does not show any series of Bragg peaks, which is incompatible with the existence of an Lβ phase in this temperature range.19 In contrast, high forward scattering intensities at small q values (q < 0.05 A−1) and a decay following a q−4 envelope, characteristic for the hard-soft interface scattering of small single crystals in the sample are observed. In samples in which the freezing was performed comparably rapidly (resulting in comparably small crystallites) the signature of an incompletely resolved Guinier regime (with missing plateau) at around q ∼ 0.007 A−1i.e.
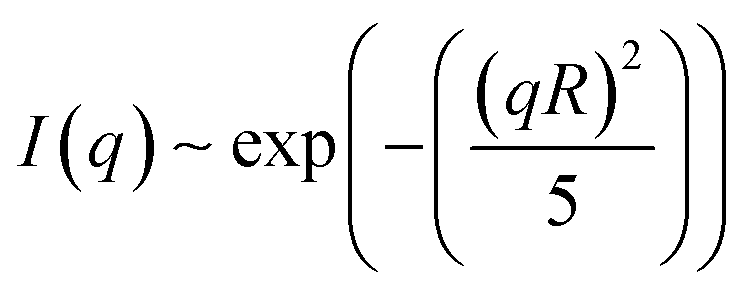 | (3) |
The q−4 Porod scattering can also be seen for the other samples displayed in Fig. 5b (visible in the log–log presentation in the inset) in which the large crystal size prohibits a clear observation of the Guinier regime and can be used to estimate the surface-to-volume S/V independently using
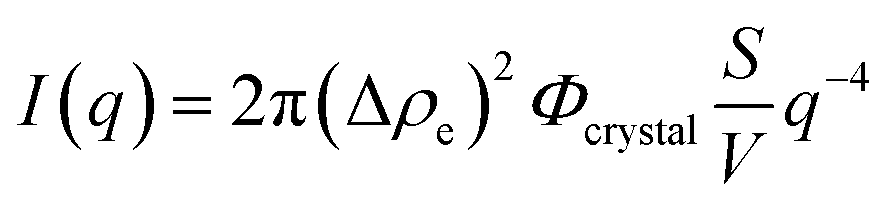 | (4) |
| ν [cm−1] | Δν [cm−1] | Rel. height | Area (%) | Assignment |
|---|---|---|---|---|
| 46 | 8 | 0.263 | 4 | LAM mode |
| 68 | 7 | 0.067 | 2 | LAM mode |
| 111 | 7 | 0.032 | 2 | LAM mode |
| 141 | 5 | 0.036 | 2 | LAM mode |
| 158 | 16 | 0.109 | 2 | LAM mode33 |
| 190 | 11 | 0.079 | 2 | LAM mode |
| 241 | 14 | 0.019 | 2 | LAM mode |
| 288 | 18 | 0.009 | 2 | LAM mode |
| 372 | 13 | 0.030 | 2 | LAM mode |
| 391 | 10 | 0.021 | 1 | LAM mode |
| 415 | 15 | 0.024 | 2 | LAM mode33 |
| 446 | 19 | 0.010 | 1 | Unassigned33 |
| 469 | 12 | 0.021 | 2 | Unassigned33 |
| 509 | 11 | 0.034 | 2 | |
| 523 | 4 | 0.008 | 2 | Unassigned33 |
| 770 | 7 | 0.033 | 2 | CH3 rock from N+(CH3)2 group33 |
| 845 | 8 | 0.054 | 0 | CH3 rock from CH2–NCH3 group33 |
| 885 | 6 | 0.048 | 2 | |
| 891 | 8 | 0.088 | 1 | CH3 rock & CN+ stretch33 |
| 1034 | 8 | 0.022 | 2 | |
| 1062 | 6 | 0.297 | 1 | C–C sym stretch & CH2 wag.33 |
| 1083 | 7 | 0.140 | 0 | C–C stretch (gauche conformation)33 |
| 1126 | 8 | 0.216 | 1 | C–C asym stretch & CH2 (gauche) wag.33 |
| 1149 | 7 | 0.052 | 1 | |
| 1170 | 7 | 0.046 | 1 | |
| 1181 | 9 | 0.045 | 1 | |
| 1297 | 6 | 0.42 | 2 | CH2 twist33 |
| 1308 | 8 | 0.056 | 1 | |
| 1341 | 8 | 0.089 | 1 | |
| 1438 | 8 | 0.204 | 2 | CH2 bend33 |
| 1445 | 13 | 0.174 | 2 | CH sym bend from N+(CH3)2 group33 |
| 1453 | 8 | 0.263 | 2 | CH2 bend & CH asym bend from N+(CH3)2 group33 |
| 1461 | 9 | 0.199 | 2 | |
| 1470 | 9 | 0.195 | 2 | |
| 2842 | 13 | 0.551 | 2 | CH asym stretch of CH233 |
| 2853 | 15 | 0.463 | 2 | CH asym stretch of CH233 |
| 2863 | 14 | 0.474 | 2 | CH asym stretch of CH233 |
| 2871 | 8 | 0.399 | 2 | |
| 2877 | 6 | 1 | 3 | CH sym stretch of CH333 |
| 2886 | 18 | 0.646 | 2 | CH sym stretch of CH333 |
| 2903 | 17 | 0.413 | 2 | |
| 2917 | 19 | 0.252 | 2 | |
| 2930 | 17 | 0.354 | 2 | |
| 2943 | 31 | 0.213 | 2 | |
| 2960 | 5 | 0.119 | 2 | |
| 2961 | 16 | 0.277 | 2 | CH asym stretch of N+(CH3)233 |
| 2976 | 15 | 0.181 | 1 | |
| 2997 | 9 | 0.105 | 2 | Unassigned33 |
| 3026 | 12 | 0.146 | 2 | |
| 3041 | 14 | 0.063 | 2 | |
| 3250 | 260 | 1.004 | — | H2O sym stretch34 |
| 3356 | 71 | 0.268 | — | H2O |
| 3445 | 188 | 1 | — | H2O asym stretch34 |
| 3596 | 122 | 0.258 | — | H2O |
Preliminary powder X-ray diffraction measurements performed on the powder and the low temperature phase confirm the existence of at least two stable crystalline forms in the DDAB samples in the investigated temperature range (see the ESI,‡ Fig. S11). We were not able to grow suitable crystals for single-crystal XRD analysis. The distinct diffraction patterns suggest the existence of one or several stable crystal hydrates at low temperature and/or conformational changes of the DDAB molecule compared to the zero-hydrate crystal.
In order to correlate the changes in the Raman spectra with conformational changes in the DDAB molecule, single molecule calculations have been carried out. The appearance of the band at around 1082 cm−1 in the low temperature crystalline phase, suggests a distinct gauche conformation in the DDAB chain,33 however less pronounced than in the lamellar phase (Fig. 6, red line), which is also supported from the reduced ratio of the I2850/I2880 modes. Raman frequencies were calculated for several DDA+ conformations, which differ in the gauche content of the chains. The Raman spectrum of the most stable DDA+ configuration featuring all-trans chains (upper left corner in Fig. 7) differs remarkably, especially in the region of the C–H stretching modes, from the experimentally obtained spectra of both the powder and the ‘white’ phase. This is reasonable, because calculations were performed on isolated molecules in the gas phase, which neglect the effects of packing in solid and lamellar structures. However, the following trends can still be deduced from these calculations: (i) the introduction of gauche kinks in the chain influences the ratio of the peaks IChsym/ICHasym; (ii) gauche kinks at positions closer to the head group lead to a peak shape similar to the peaks in the powder or the 'white' phase, whilst kinks towards the end seem to be rather characteristic of the Lα phase; (iii) the structure of the three bands between 1000 cm−1 and 1150 cm−1 support such an assignment, with the middle peak likely corresponding to the experimental peak at 1080 cm−1 which does not vanish for the all-trans conformation of the alkyl chain, but decreases with positioning of the gauche kinks closer to the head group (Fig. 7). It should also be noted, that the conformation of DDAB molecules with a kink close to the head group is similar to the crystal structure obtained for its slightly longer analogues DODAB45 and DODMAC.43
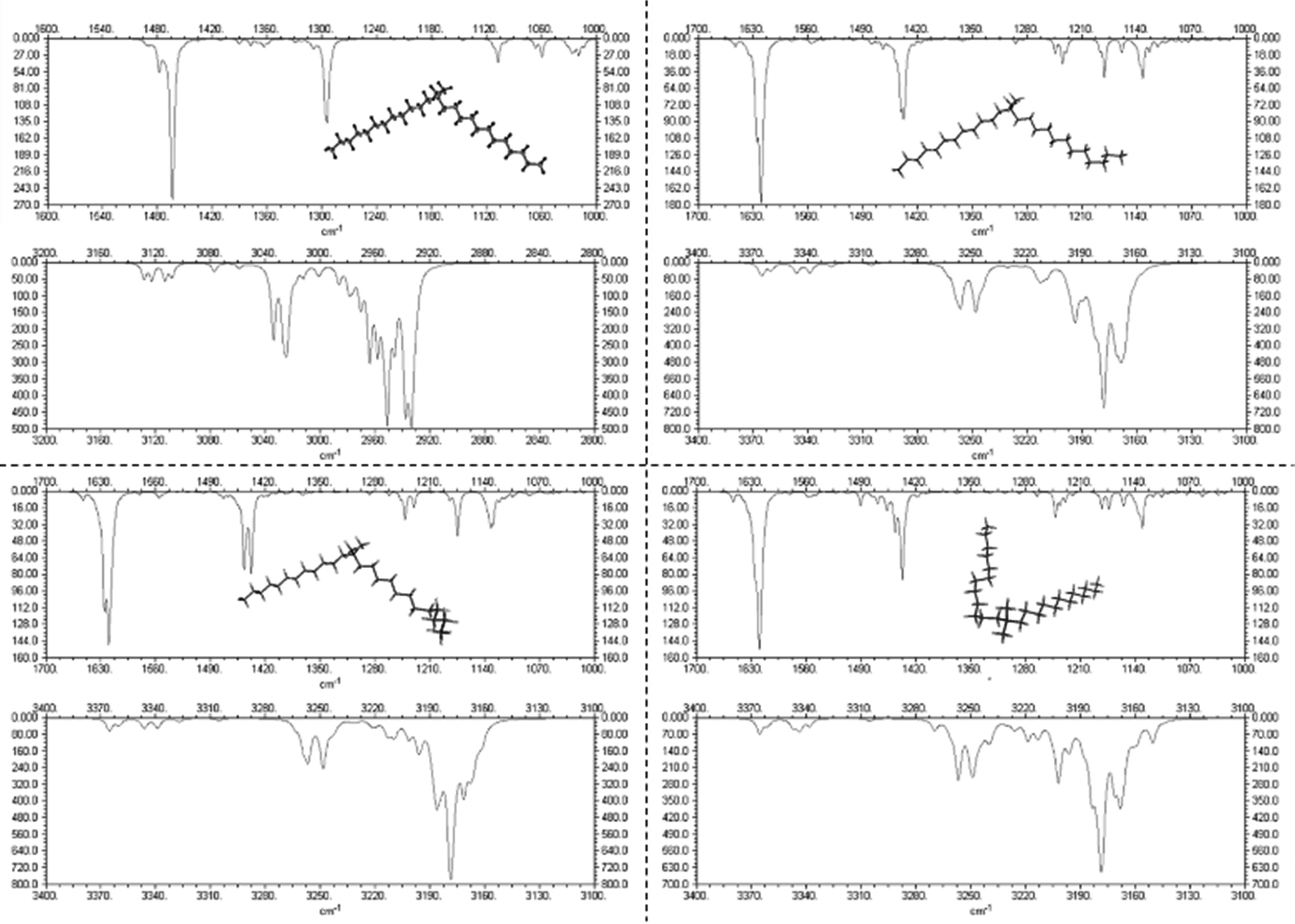 | ||
| Fig. 7 Calculated Raman spectra of DDA+ conformations differing in the position of gauche kinks in the hydrocarbon chain. The most stable conformation (all-trans) is shown in the top left corner. | ||
It should also be noted, that the water peak in the ‘white’ phase is distinct from that in the Lα phase (see the red line in Fig. 6b). An additional sharp peak around 3350 cm−1 could be identified which increases with decreasing temperatures. This peak might suggest the existence of a crystal hydrate, as sharp water bands are characteristic for bound water molecules. Furthermore, in contrast to the lamellar phase, an increase in the surfactant concentration or temperature leads to an increase of the ratio Isym/Iasym water stretch (Fig. 6c right). This suggests that the disorder in the water phase increases with increasing temperature (as expected for pure water), but also with increasing surfactant concentration. This also offers support for the observation that dense phases cannot be separated by sedimentation or centrifugation.
In summary, all the performed characterizations of the low temperature phase suggest that the equilibrium phase is a coexistence phase of a crystal hydrate with a very dilute liquid phase XWn + W. Further study of the temperature dependence of the structure at low water concentration, ensuring sufficiently long equilibration times, is necessary to obtain greater insight into the hydration state of the low-temperature crystals and its transformation temperature into the zero-hydrate form. In summary, all the performed characterizations of the low temperature phase suggest that the equilibrium phase is a coexistence phase of a crystal hydrate with a very dilute liquid phase XWn + W. A further study of the temperature dependence of the structure at low water concentration, ensuring sufficiently long equilibration times, is necessary to obtain greater insight into the hydration state of the low-temperature crystals and its transformation temperature into the zero-hydrate form.
The proposed new phase diagram
Combining the results obtained in this study with information in the literature, a modified broad phase diagram for the low temperature region of the aqueous DDAB system is proposed (see Fig. 1). The previously suggested Lβ phase is replaced by a coexistence region of a crystalline phase XWn and a very dilute (near pure water) monomer solution W, which we draw at ΦDDAB = 0%. This is consistent with the phase transition for W to Ice seen at 0 °C, and the invariance in XWn composition in the low temperature range. The hydration state of the crystal hydrate phase at low temperatures could not be resolved, which would fix the position of the vertical line separating the W + XWn and XWn regions at high DDAB concentration. However, the crystalline form differs in structure from the zero-hydrate, depicted as XP, found in the DDAB powder at room temperature. Furthermore, the occurrence of a distinct and sharp water peak in the Raman spectrum suggests the formation of a hydrate rather than a polymorph of the zero-hydrate. The accurate transition temperature between the two crystalline phases as well as the number and character of intermediate crystal structures still has to be determined, and, therefore, this part of the phase diagram is represented with broken lines.At higher temperatures, and compositions below 85 wt%, the phase diagram is consistent with the phase diagram reported in the literature. Two lamellar phases can be found, the swollen Lα phase and the collapsed 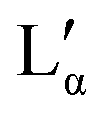 phase, which coexist over a broad temperature and concentration range. Based on the rules of phase science, both the Lα phase and the
phase, which coexist over a broad temperature and concentration range. Based on the rules of phase science, both the Lα phase and the 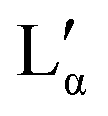 phase, need to have at their lower temperature limits a eutectic discontinuity.43,52,54 The eutectic discontinuity of the
phase, need to have at their lower temperature limits a eutectic discontinuity.43,52,54 The eutectic discontinuity of the 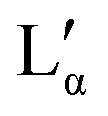 phase could, in general, lay above or below the eutectic of the Lα phase. Both versions have been reported for other systems.55 The observed results, however, suggest that there is a region in the phase diagram in which the Lα phase coexists with the crystalline phase (see also part II of this study51). Therefore, it can be assumed that the eutectic limiting the Lα phase corresponds to the Krafft eutectic, whilst the lower limit of the
phase could, in general, lay above or below the eutectic of the Lα phase. Both versions have been reported for other systems.55 The observed results, however, suggest that there is a region in the phase diagram in which the Lα phase coexists with the crystalline phase (see also part II of this study51). Therefore, it can be assumed that the eutectic limiting the Lα phase corresponds to the Krafft eutectic, whilst the lower limit of the 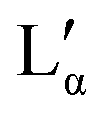 phase can be found at higher temperatures, which is also in agreement with our DSC data as well as the previously reported transition temperature of 24 °C for the
phase can be found at higher temperatures, which is also in agreement with our DSC data as well as the previously reported transition temperature of 24 °C for the 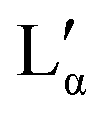 phase.36 The exact position of the discontinuity limiting the collapsed
phase.36 The exact position of the discontinuity limiting the collapsed 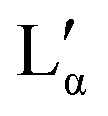 phase can, however, not be determined from the performed experiments. Furthermore, the thermal stability limit of DDAB is close to 90 °C, which is therefore the upper limit of the phase diagram.
phase can, however, not be determined from the performed experiments. Furthermore, the thermal stability limit of DDAB is close to 90 °C, which is therefore the upper limit of the phase diagram.
Conclusions
A detailed study of the equilibrium phase behaviour of the binary DDAB/water system has been presented. The samples used in this study were shown to be of sufficient purity such that the reported properties did not change with additional purification. The obtained results, based on a range of experimental techniques, characterizing the samples at different length scales, could not be explained within the frame of the latest reported broad equilibrium phase diagram.17 The low temperature phase, which was previously suggested to be an Lβ phase, was replaced by a coexistence region of a crystal hydrate phase XWn and a dilute monomer solution W. This phase differs not only in optical properties (being white in contrast to the clear Lβ phase), but also its crystalline character could be confirmed by Raman spectroscopy, small and wide angle X-ray scattering and microscopy. Our results suggest that the previously proposed Lβ phase for concentrations above 3 wt% is likely to correspond to the non-equilibrium supercooled Lα phase. Our results do not disagree with the characterization and report of the Lβ phase in the dilute vesicle system, which has already been reported as a long-lived non-equilibrium phase only obtained when the formation of crystals is avoided by limited overcooling below Tc.24,26 A detailed study of the thermal phase transition involving the crystal/water coexistence region and the Lα phase is presented in part II of this study,51 elucidating the exact position of the boundary between the Lα phase and its coexistence with the crystals. In future, a detailed study of the high concentration region is needed in order to obtain the exact position of the lower temperature limit of the collapsed phase as well as the hydration state and thermal stability limit of the crystalline phase found at low temperatures.
phase as well as the hydration state and thermal stability limit of the crystalline phase found at low temperatures.
Data availability
The data supporting this article have been included as part of the ESI.‡Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors want to thank the Mass Spec (Mr Alan T. Taylor) and NMR (Dr Maria DeCremoux) services at Edinburgh University for their help in characterization. Thanks to the group of Arlene Sloan (Glasgow University) for the use of their ATR FTIR equipment, to the group of Eleonar Campbell (Edinburgh University) for the use of their ATR microscope, and to the group of Alan Cooper (Glasgow University) for their assistance with the density measurements. We are grateful for the sharing of expertise in the interpretation and conduction of Raman experiments to Hugh Vass (Edinburgh University) and of DSC experiments to Peter Roloff (Heinrich Heine University). Thanks to Andy Turner (Edinburgh University) for his help in the single molecule calculations. Thanks to CSEC for the availability of the X-ray diffraction equipment. Many thanks to the late expert on surfactant phase behaviour Robert G. Laughlin PhD, deceased 15th February 2015, who greatly contributed to the interpretation of the obtained results, to the experimental design of the analytical methods and to the establishment of the new phase diagram. We also thank P. Clegg and A. Schofield for helpful discussions. The School of Chemistry is part of the EaStCHEM joint Chemistry Research School. We acknowledge the financial support of the Scottish Funding Council. This work was funded in part by EPSRC EP/E030173. L. R. acknowledges financial support from the Marie Curie Early Stage Training Network on “Biomimetic Systems” (Contract MEST-CT-2004-504465).References
- T. Kunitake, et al. , Chem. Lett., 1977, 387 CrossRef CAS.
- F. Lindstroem, et al. , Phys. Chem. Chem. Phys., 2002, 4, 5524 Search PubMed; M. I. Viseu, T. I. Carvallo and S. M. B. Costa, Biophys. J., 2004, 86, 2392 CrossRef CAS PubMed.
- X. Chen, et al. , J. Electroanal. Chem., 2000, 480, 26 CrossRef CAS; Y. Shao, et al. , Biosens. Bioelectron., 2005, 20, 1373 CrossRef PubMed; J. F. Rusling and A.-E. F. Nassar, J. Am. Chem. Soc., 1993, 115, 11891 CrossRef; M. Zhao, et al. , Microchim. Acta, 2009, 165, 45 CrossRef.
- A. Ignaszak, et al. , J. Pharm. Biomed. Anal., 2009, 49, 498 CrossRef CAS PubMed; V. Shumyantseva, et al. , Electroanalysis, 2009, 21, 530 CrossRef; Y. Peng, et al. , Sens. Actuators, B, 2009, 137, 656 CrossRef.
- N. Lincopan, et al. , BMC Biotechnol., 2009, 9 Search PubMed.
- D. Connolly and B. Paull, J. Chromatography, 2002, 953, 299 CrossRef CAS PubMed; P. Hatsis and C. A. Lucy, Anal. Chem., 2003, 75, 995 CrossRef PubMed; D. Connolly, D. Victor and B. Paull, J. Sep. Sci., 2004, 27, 912 CrossRef PubMed; S. Pelletier and C. A. Lucy, J. Chromatogr., 2006, 1125, 189 CrossRef PubMed.
- J. E. Melanson, N. E. Baryla and C. A. Lucy, Anal. Chem., 2000, 72, 4110 CrossRef CAS PubMed; N. E. Baryla, et al. , Anal. Chem., 2001, 73, 4558 CrossRef PubMed; A. G. Diress and C. A. Lucy, J. Chromatogr. A, 2004, 1027, 185 CrossRef PubMed; M. M. Yassine and C. A. Lucy, Anal. Chem., 2004, 76, 2983 CrossRef PubMed; S. Mohabbati, S. Hjerten and D. Westerlung, Anal. Bioanal. Chem., 2008, 390, 667 Search PubMed; Q. Liu, Y. Yang and Sh. Yao, J. Chromatogr. A, 2008, 1187, 260 Search PubMed.
- J. P. M. Silva, P. J. G. Coutinho and M. E. C. D. R. Oliveira, J. Fluoresc., 2009, 18, 555 CrossRef CAS PubMed; E. Feitosa, et al. , Langmuir, 2006, 22, 3579 CrossRef PubMed.
- G. Montalvo and A. Khan, Langmuir, 2002, 18, 8330 CrossRef CAS.
- E. F. Marques, et al. , Adv. Colloid Interface Sci., 2003, 102, 83 CrossRef.
- N. Muthukumar, S. Maruthamuthu and N. Palaniswamy, Colloids Surf., B, 2007, 57, 152 CrossRef CAS PubMed; E. M. A. Pereira, et al. , J. Phys. Chem. B, 2008, 112, 9301 CrossRef PubMed; H. Rosa, D. F. S. Petri and A. M. Carmona-Ribeiro, J. Phys. Chem. B, 2008, 112, 16422 CrossRef PubMed.
- Y. Li, Q. Liu and S. Yao, Talanta, 2008, 75, 677 CrossRef CAS PubMed; D. Li, et al. , Biomaterials, 2009, 30, 1382 CrossRef PubMed.
- P. Callow, et al. , Langmuir, 2009, 25, 4184 CrossRef CAS PubMed; C. N. C. Sobral, M. A. Soto and A. M. Carmona-Riberio, Chem. Phys. Lipids, 2008, 152, 38 CrossRef PubMed.
- C. Groth, J. Bender and M. Nyden, Colloids Surf., A, 2003, 228, 64 CrossRef CAS.
- T. Undabeytia, S. Nir and M. J. Gomara, Langmuir, 2004, 20, 6605 CrossRef CAS PubMed.
- T. F. Svitova, R. M. Hill and C. J. Radke, Langmuir, 2001, 17, 335 CrossRef CAS.
- M. Dubois, et al. , J. Chem. Phys., 1998, 108, 7855 CrossRef CAS.
- G. G. Warr, et al. , J. Phys. Chem., 1988, 92, 774 CrossRef CAS.
- M. Dubois and T. Zemb, Langmuir, 1991, 7, 1352 CrossRef CAS.
- M. Dubois, et al. , J. Chem. Phys., 1992, 96, 2278 CrossRef CAS.
- F. Caboi and M. Monduzzi, Langmuir, 1996, 12, 3548 CrossRef CAS.
- H. N. Patrick and G. G. Warr, J. Phys. Chem., 1996, 100, 16268 CrossRef CAS.
- M. Goto, et al. , Langmuir, 2011, 27, 1592 CrossRef CAS PubMed.
- E. Feitosa, J. Colloids Interface Sci., 2010, 344, 70 CrossRef CAS PubMed; E. Feitosa, R. D. Adati and F. R. Alves, Colloids Surf., A, 2015, 480, 253 CrossRef.
- S. Segota, S. Heimer and D. Tezak, Colloids Surf., A, 2006, 274, 91 CrossRef CAS.
- M. J. Blandamer, et al. , J. Chem. Soc., Faraday Trans., 1997, 93, 453 RSC.
- E. F. Marques, A. Khan and B. Lindman, Thermochim. Acta, 2002, 394, 31 CrossRef CAS; E. Feitosa, J. Jansson and B. Lindman, Chem. Phys. Lipids, 2006, 142, 128 CrossRef PubMed.
- P. C. Schulz, et al. , J. Therm. Anal., 1951, 49 Search PubMed.
- R. G. Laughlin, et al. , J. Phys. Chem., 1990, 94, 2546 CrossRef CAS.
- S. Haas, PhD thesis, University of Bayreuth, 1998.
- S. Haas, et al. , Colloid Polym. Sci., 1999, 277, 856 Search PubMed.
- M. Aratono, et al. , J. Phys. Chem. B, 2007, 111, 107 CrossRef CAS PubMed; Y. Ono, et al. , J. Colloid Interface Sci., 2005, 287, 685 Search PubMed; A. Bumajdad, et al. , Langmuir, 2004, 20, 11223 CrossRef PubMed.
- R. Foucault, R. L. Birke and J. R. Lombardi, Langmuir, 2003, 19, 8818 CrossRef CAS.
- P. F. Bernath, The spectroscopy of water vapour: Experiment, theory and applications, Phys. Chem. Chem. Phys., 2002, 4, 1501–1509 RSC.
- Gaussian03, https://www.ch.cam.ac.uk/computing/software/gaussian-03.
- Th. Zemb, et al. , Europhys. Lett., 1993, 21, 759 CrossRef CAS.
- J. Ockelford, et al. , J. Phys. Chem., 1993, 97, 6767 Search PubMed.
- D. Horinek, S. I. Mamatkulov and R. Netz, J. Chem. Phys., 2009, 130, 124507 CrossRef PubMed.
- H. Boroudjerdi, et al. , Phys. Rep., 2005, 416, 129 CrossRef CAS.
- M. I. Viseu, et al. , Langmuir, 2000, 16, 2105 CrossRef CAS; T. Matsumoto, Colloid Polym. Sci., 1992, 270, 492 CrossRef.
- A. Caria and A. Khan, Langmuir, 1996, 12, 6282 CrossRef CAS; M. Youssry, et al. , J. Colloid Interface Sci., 2008, 321, 459 CrossRef PubMed; A. Stenstam, et al. , J. Phys. Chem. B, 2003, 107, 12331 CrossRef.
- R. Laughlin, Colloids Surf., A, 1997, 128, 27 CrossRef CAS; M. Bergmeier, H. Hoffmann and C. Thunig, J. Phys. Chem. B, 1997, 101 Search PubMed.
- R. Zana, Dynamics of surfactant self-assemblies: micelles, microemulsions, vesicles and lyotropic phases, Surfactant Science Series, Taylor & Francis Group, Boca Raton, 2005, vol. 125 Search PubMed.
- K. Okuyama, et al. , Chem. Lett., 1984, 2117 CrossRef CAS.
- K. Okuyama, et al. , Bull. Chem. Soc. Jpn., 1988, 61, 1485 CrossRef CAS.
- R. G. Laughlin, The aqueous phase behaviour of surfactants, Academic Press, London, 1994 Search PubMed.
- B. R. Lentz, Y. Barenholz and T. E. Thompson, Biochemistry, 1976, 15, 4521 CrossRef CAS PubMed; A. Blume, Thermochim. Acta, 1991, 193, 299 CrossRef; B. L. Silver, The physical chemistry of membranes, Allen & Unwin and The Solomon Press, 1986 Search PubMed.
- E. Feitosa, P. C. A. Barreleiro and G. Olofsson, Chem. Phys. Lipids, 2000, 105, 201 CrossRef CAS PubMed.
- M. Godlewska, et al. , Mol. Cryst. Liq. Cryst. Sci. Technol., Sect. A, 1997, 300, 113 CrossRef CAS; P. Dynarowicz, M. Godlewska and W. Witko, Mol. Cryst. Liq. Cryst. Sci. Technol., Sect. C, 1997, 8, 307 Search PubMed; M. Godlewska, et al. , Mol. Mater., 1998, 9, 217 Search PubMed.
- P. C. Schulz and J. E. Puig, Colloids Surf., A, 1993, 71, 83 CrossRef.
- Manuscript in preparation, L. Reissig.
- D. N. Rubingh and P. M. Holland, Cationic surfactants: Physical chemistry, Surfactant Science Series, Marcel Dekker Inc., New York, 1991, vol. 37 Search PubMed.
- K. K. Karukstis, C. A. Zieleniuk and M. J. Fox, Langmuir, 2003, 19, 10054 CrossRef CAS.
- D. F. Evans and H. Wennerstrom, The colloidal domain: where physics, chemistry, biology and technology meet, Wiley VCH, New York, 1994 Search PubMed; R. DeHoff, Thermodynamics in materials science, Taylor & Francis Group, Boca Raton, 2006 Search PubMed.
- F. N. Rhinne, Phase diagrams in metallurgy, McGraW-Hill Book Company Inc., New York, 1956 Search PubMed; R. G. Laughlin, Private communication, 2009.
Footnotes |
| † This work is dedicated to the memory of our colleague and coauthor, Stefan Egelhaaf, who contributed scientific rigour, infectious enthusiasm and supportive nature to the project. |
| ‡ Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sm01320d |
| § Present address: Freie University Berlin, Germany. |
| ¶ Present address: University of Cambridge, UK. |
| This journal is © The Royal Society of Chemistry 2025 |