
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hybrid phytoglycogen-dopamine nanoparticles as biodegradable underwater adhesives†
Jiayi
Liu
a,
Dmitrii
Sychev
a,
Nadiia
Davydiuk
a,
Mahmoud
Al-Hussein
ab,
Andreas
Fery
 a and
Quinn A.
Besford
*a
a and
Quinn A.
Besford
*a
aLeibniz-Insitut für Polymerforschung Dresden e. V., Hohe Str. 6, 01069 Dresden, Germany. E-mail: besford@ipfdd.de
bPhysics Department, The University of Jordan, Amman 19942, Jordan
First published on 17th February 2025
Abstract
Developing adhesive materials that can selectively degrade into non-toxic by-products is a key challenge in materials science, particularly for short-term implantable devices and tissue regeneration treatments. Herein, we leverage biodegradable phytoglycogen (PG) nanoparticles (highly branched glucose polysaccharide nanoparticles) as scaffolds for coupling adhesive dopamine motifs to be used as biodegradable underwater adhesives. Phytoglycogen-dopamine (PG-dopa) hybrid nanoparticles could be synthesised in an aqueous solvent, to which the products retained a similar size and particle morphology to the initial PG nanoparticles. The PG-dopa nanoparticles could readily be assembled into dense monolayers on silica substrates through a simple dip-coating procedure. Colloidal probe atomic force microscopy was used to characterise underwater adhesiveness, where it was found that the films produced strain energy release rates approaching 8 mJ m−2 between hard silica materials. Importantly, the PG-dopa films retained the original biodegradability towards glucosidase enzymes, which can degrade the adhesives in fluids containing these enzymes over time (e.g., 45 U mL−1 of α-amylase solution degraded the majority of the adhesive films in 30 min). Given the inherent biocompatibility of glycogen materials, we anticipate these adhesives having application in short-term implantable devices.
Introduction
Mussel feet secrete adhesive proteins that help them firmly stick to rocks in marine environments, where the adhesive proteins form plaques on substrates. A linear protein (mefp-5) with an average molecular weight of about 10 kDa plays a significant role in forming robust mussel plaques, especially in the strong interface connection.1,2 The adhesion ability of mefp-5 is mainly due to L-dopa, which contains a functional catechol group. Hydrogen bonds, metal-coordination and hydrophobic interactions can form between these catechol groups and substrates resulting in strong interface adhesions.3,4These unique underwater adhesive properties of L-dopa have inspired the development of synthetic underwater or wet adhesives, which might find application in the biomedical field. For example, synthetic polymers that mimic mussel adhesive proteins have shown high underwater adhesion strengths.5–7 Cui et al. developed a starch-based tissue adhesive by using dopamine-conjugated starch. This dopamine modified starch can form a hydrogel when hydrogen peroxide with horseradish peroxidase is used as a cross-linker. Catechol groups served as interfacial adhesion and crosslinking segments.8 Du et al. reported a mechanical and chemical robust coating using catechol-modified polyallylamine. The catechol group and amine group contribute to the strong interface adhesion on several substrates and cohesive interaction by forming covalent bonds. The abundant amine groups on the coating provide grafting sites for secondary modification.9 Whilst these materials have demonstrated unique adhesive properties, there is interest in moving towards minimally synthetic, bio-sourced materials as adhesives. Furthermore, for applications involving bio-contact (e.g., implants and tissue adhesives), biocompatibility is necessary. Glycogen is an interesting material in this regard, which has been gaining momentum in various applications,10 particularly as it has been shown to induce minimal inflammation, coagulation, and interactions with the immune system in human blood.11
Glycogen is a randomly hyperbranched polysaccharide nanoparticle that can be obtained from various sources (animals, tissues, and plants). Its structure consists of repeating D-glucose units connected by linear α-(1,4) glycosidic linkages, with branching via α-(1,6) glycosidic linkages. The nanoparticle size, molecular weight, degree of branching and content of proteins are different depending on the source. For instance, phytoglycogen (PG) derived from sweet corn has a diameter of ∼80 nm in water and a high molecular weight of 20 MDa12 (Fig. 1(a)). The hydroxyl groups on glycogen can be modified to produce other functional groups, such as carboxylic acid groups,13 quaternary ammonium groups,14 and allyl groups,15 as examples. The glucose residues can be oxidized to produce an aldehyde group, which can further react with primary amines to conjugate functional groups.16 Previous works have investigated thiolated glycogens as mucosal adhesives and as gold-adhering nanoparticles,17 which have demonstrated the robust properties of adhesive glycogen. Coupling the unique properties of glycogen with underwater adhesive catechol motifs has not been pursued, to our best knowledge.
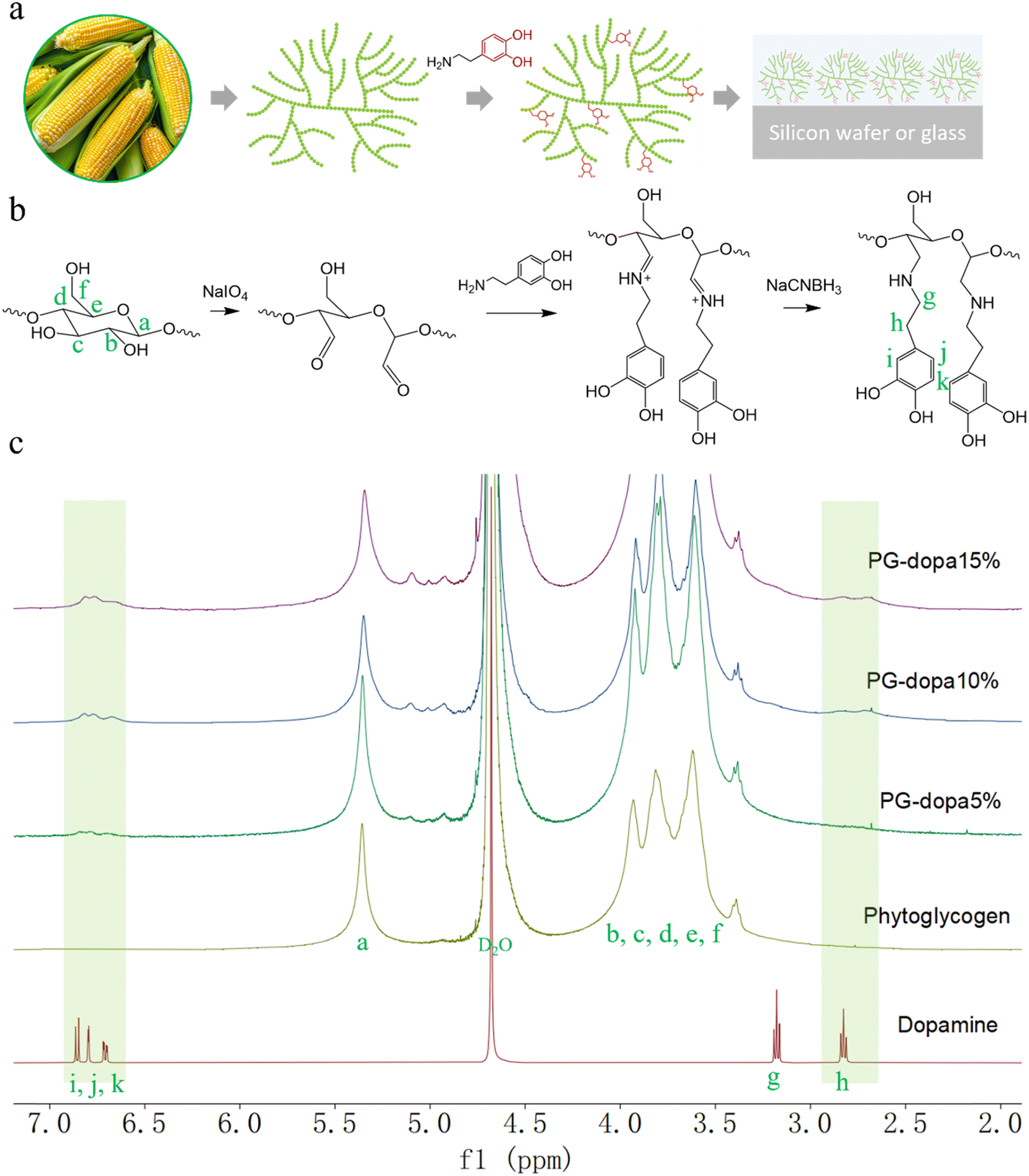 | ||
| Fig. 1 (a) Phytoglycogen (PG) extracted from sweet corn and monolayer dopamine-conjugated phytoglycogen (PG-dopa) on substrates. (b) The synthesis scheme of PG-dopa. (c) 1H NMR (600 MHz, D2O) spectra of dopamine, PG, PG-dopa5%, PG-dopa10% and PG-dopa15%. | ||
Herein, we report a rapid, straightforward method to form underwater PG adhesive coatings using PG–dopamine (PG-dopa) hybrids, inspired by the mussel foot protein (Fig. 1(a)). PG nanoparticles, which are sourced from plants (sweet corn kernels), are hydrophilic and have no ability to adhere to underwater substrates.17 However, we found that PG-dopa nanoparticles show remarkable affinity to silicon dioxide surfaces. A homogenous coating consisting of single layer PG-dopa nanoparticles can be formed by simply dip coating into PG-dopa dispersions. The PG-dopa coating showed great adhesiveness to underwater silicon dioxide materials. The maximum average adhesion energy reached 7.20 mJ m−2. Previously, values as high as 2.64 mJ m−2 were reported for dopa-containing mussel-inspired proteins.18 The modification of dopamine did not affect the biodegradability of PG-dopa nanoparticles or coatings. We anticipate these adhesives to have use in short-term implantable devices.
Methods
Materials
Phytoglycogen (PG) sourced from the kernels of sweet corn was purchased from Mirexus (Guelph, Canada). Sodium periodate (≥99.8%), dopamine hydrochloride (≥98%), sodium cyanoborohydride (≥95%), 4-methycatechol (≥95%), α-amylase (powder, ∼30 U mg−1), phenol (≥99%) and deuterium oxide (≥99.9%) were purchased from Sigma-Aldrich. Sulfuric acid (≥95%) was purchased from Fisher Chemical. Absolute ethanol was purchased from VWR chemicals. Methyl sulfoxide (99.7 + %) was purchased from Thermo Scientific. The dialysis tube (14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Daltons cut-off) was purchased from BioDesign Inc. of New York. High-purity (Milli-Q) water with a resistivity of 18.2 MΩ cm was obtained from a Direct-Q® 3UV water purification system (Merck Chemicals GmbH, German).
000 Daltons cut-off) was purchased from BioDesign Inc. of New York. High-purity (Milli-Q) water with a resistivity of 18.2 MΩ cm was obtained from a Direct-Q® 3UV water purification system (Merck Chemicals GmbH, German).
Synthesis of PG-dopa
PG-dopa was synthesized by the Schiff base reaction between dopamine and oxidized PG and then reduced by sodium cyanoborohydride. We synthesized some different conjugates (percentage modifications of PG). Here, we describe the 10% modification (10% of glucose units of PG modified). PG (0.2433 g, 1.50 mmol) was dispersed in 10 mL of Milli-Q water by ultrasonication. Then, sodium periodate (0.0321 g, 0.15 mmol) was added and the dispersion was stirred for 2 h at room temperature in the dark. The dispersion was dialyzed (14 kDa cutoff) against Milli-Q water one night to remove the oxidizing agent. The dispersion was transferred to a round bottom flask. The flask was sealed by a rubber septum and degassed with argon for 20 minutes. Dopamine hydrochloride (0.0569 g, 0.30 mmol) was dissolved in 5 mL of phosphate buffer (0.1 M, pH 5) and was added to the oxidized PG dispersion. After 4 h, sodium cyanoborohydride (0.0471 g, 0.75 mmol) was dissolved in 5 mL of phosphate buffer (0.1 M, pH 5) and was added to the reaction dispersion for one night. Subsequently, the dispersion was precipitated in 40 mL of ethanol twice. The precipitate dispersed in 20 mL of Milli-Q water again and dialyzed (14 kDa cut-off) against Milli-Q water for 3 days. Finally, the dispersion was freeze-dried to obtain the PG-dopa10%. The degree of conjugation was determined by the 1H NMR spectrum and catechol group assay.Nuclear magnetic resonance spectroscopy (NMR)
1H spectra were collected using an Avance III HD 500 MHz spectrometer (Bruker, Germany). Deuterium oxide (D2O) was used as the solvent.Catechol group assay
The UV-vis absorbance spectra were recorded on a SPECORD 40 spectrophotometer (Analytik Jena, Germany) and analyzed using the software WinASPECT. Spectra were acquired with a scan speed of 20 nm s−1. A constant of 0.005 mg mL−1 iron(III) chloride solution, the 4-methylcatechol solution concentration range from 0.01 mg mL−1 to 0.08 mg mL−1 and the total volume of 2 mL of carbonate buffer solution (0.1 M, pH 8.3) samples were prepared for the standard absorbance curve. In detail, a fresh 0.05 mg mL−1 iron(III) chloride and 1 mg mL−1 4-methylcatechol water solution were prepared as stock solutions. To prepare the sample with a 4-methylcatechol concentration of 0.01 mg mL−1, 0.2 mL of 0.05 mg mL−1 iron(III) chloride solution, 0.02 mL of 1 mg mL−1 4-methylcatechol solution and 1.78 mL of Milli-Q water were added to the PMMA cuvette. The sample with a higher 4-methylcatechol concentration was prepared by increasing the volume of 1 mg mL−1 4-methylcatechol solution and decreasing the volume of Milli-Q water. The volume of 0.05 mg mL−1 iron(III) chloride solution and the total volume were always 0.2 mL and 2 mL. The cuvettes were placed in the chamber for the measurement after the solution became homogeneous. A wavelength range from 300 to 800 nm was used.The PG-dopa5%, PG-dopa10% and PG-dopa15% nanoparticles were dispersed in carbonate buffer solution (0.1 M, pH 8.3) with the help of ultrasonication for 30 minutes. All the concentrations were 0.8 mg mL−1. To prepare the PG-dopa samples for the UV-vis spectrometer, 0.2 mL of 0.05 mg mL−1 iron(III) chloride solution and 1.8 mL of 0.8 mg mL−1 PG-dopa5% or PG-dopa10% or PG-dopa15% solution was added to the cuvette. The cuvettes were placed in the chamber for the measurement after the solution became homogeneous. A wavelength range from 300 to 800 nm was used.
To compare the absorbance intensity of PG-dopa samples with the 4-methylcatechol standard absorbance curve, the absorbance intensity at a wavelength of 525 nm was used.
Preparation of the PG-dopa10% coating on a silicon wafer
The PG-dopa10% coating on a silicon wafer was obtained by dip coating in a 1 mg mL−1 PG-dopa10% Milli-Q water dispersion without waiting time. The lift speed was 0.48 mm s−1. The coated wafer was rinsed with Milli-Q water to remove the unbounded PG-dopa10% nanoparticles.Atomic force microscopy (AFM)
AFM topography measurements on nanoparticles and coatings were performed in peak-force mode on a Dimension Fastscan AFM (Bruker, Billerica, MA, USA) with a peak-force set point of 30 mV. A Tap300 Al-G cantilever (40 N m−1, 300 kHz, coated by 30 nm Aluminium, BudgetSensors) was employed. All measurements were performed consecutively with the same type of cantilever. The obtained data were processed using Gwyddion software. To obtain the PG and PG-dopa10% nanoparticle AFM topography images, 0.005 mg mL−1 nanoparticle water dispersion was prepared. 0.1 mL of water dispersion was applied to the silicon wafer and dried by slow nitrogen flow. For the thickness of the PG-dopa coating on the silicon wafer, several scratches were prepared using a small syringe needle.Dynamic light scattering (DLS)
Hydrodynamic diameters and zeta potential were measured using a Zetasizer Nano-ZS instrument (Malvern Instrument. Malvern, UK). The concentration of both PG and PG-dopa water dispersion was 1 mg mL−1.Scanning electron microscopy (SEM)
SEM images were recorded using a NEON40 SEM (Carl Zeiss Microscopy Deutschland GmbH, Oberkochen, Germany) operated at an acceleration voltage of 1 kV. No additional conductive coating was necessary to prevent charging of the specimen in the electron beam.Cryo-transmission electron microscopy (cryo-TEM)
Cryo-TEM images were recorded using a Libra 120 microscope (Carl Zeiss Microscopy Deutschland GmbH, Oberkochen, Germany). 4 μL of the specimen was placed onto a holey carbon TEM grid (Quantifoil R3.5/1, 300 mesh), blotted with filter paper and vitrified in liquid ethane at −178 °C using a Grid Plunger (Leica Microsystems GmbH, Wetzlar, Germany). Frozen grids were transferred into a Gatan 626 (Gatan GmbH, München, Germany) cryo-TEM holder. Images were recorded at an accelerating voltage of 120 kV while keeping the specimen at −170 °C.X-ray reflectivity
X-ray reflectivity measurements were performed using a XRD 3300 T-T (Seifert) diffractometer with a copper target (λ = 0.154 nm). The samples were mounted horizontally at the center of a two-circle goniometer and investigated under specular reflection conditions from 0 to 6°. At specular reflectivity, the scattering vector, q = (4π/λ)sinθ, was perpendicular to the film (z-direction), where 2θ is the scattering angle. Hence, the reflected intensity was sensitive to the electron density profile averaged over the footprint of the incident X-ray beam.
PG-dopa coating underwater adhesion measurements by colloidal probe AFM
Adhesion measurements were carried out using an atomic force microscope (MFP-3D, Asylum Research, Oxford Instruments, California, USA). The colloidal probe atomic force microscopy (AFM) technique was utilized to measure the adhesion force and quantify adhesion interactions.19,20 This technique allows quantitative measurements of the adhesion interaction between the adhesive PG-dopa coating and a 20 μm diameter silica spherical particle, the colloidal probe. The advantage of using the micrometer size sphere as an indenter is the defined geometry of the contact, unattainable in the case of the hard tip.21 The borosilicate glass slides with no. 1 thickness (VWR, Radnor, USA) were coated with the adhesive PG-dopa film and used as a sample for the AFM measurements. To fabricate the colloidal probe equipped with a cantilever, the tipless cantilever (CSC 36, Mikromasch Europe, Wetzlar, Germany) was calibrated using the thermal noise technique22 prior to the attachment of the colloidal probe using a micromanipulator, resulting in a spring constant of 2.51 N m−1. Adhesion characterization was carried out by recording the force–distance curves with different maximal loads on the cantilever (50 nN, 100 nN, 215 nN), contact times (0.5–50 s), and retraction rates (100–5000 nm s−1). It is worth noting that the retraction rate was 2000 nm s−1 unless stated otherwise. The measurement was carried out by recording a 4 × 4 point force map on an arbitrarily chosen position on the sample. For every parameter set, two such force maps were recorded (one for the PG-dopa15% coating). Due to the comparable adhesion of the PG-dopa film between the borosilicate glass substrate and the silica colloidal probe, as the functional groups are the same, the material transfer interfered with the measurements, resulting in the measurement of the cohesion and adhesion interaction mixture. To overcome this issue, the parameters of the measurement were varied in different directions for each of the force maps (i.e. for the first set of force maps the load was consecutively increased and for the second set it was consecutively decreased). Within this set, the measured adhesion force (the minimum on the retraction part of the force curve) decreased after several force curves had been recorded. To measure only the adhesive interactions, the values after such drop were omitted, and the cantilever was treated with O2 plasma to achieve a greater contribution of the adhesion interactions to the measured force.Adhesion was quantified by integrating the part of the force curve below zero on the retraction part of the force curve and normalising it to the calculated area of contact from maximal deformation within the framework of Johnson–Kendall–Roberts theory.23 The measurements were conducted at room temperature in Milli-Q water.
Degradation of PG and PG-dopa nanoparticles by α-amylase solution
The enzymatic degradation of PG and PG-dopa10% was assessed using a phenol–sulfuric acid assay.11,24 Dispersions of both nanoparticles in Milli-Q water with a concentration of 1 mg mL−1 were prepared. α-Amylase was dissolved in PBS buffer (0.01 M, pH 7.4) and the concentration was 1 U mL−1. To begin the assay, 70 μL of PG or PG-dopa dispersion was added to 200 μL of enzyme solution. The resulting solution was incubated at 23 °C with gentle agitation for 3 h. Then, the undigested PG and enzyme were separated from the free glucose using spin columns with a pore size of 10 kDa. The filtrate was collected and divided into sample aliquots of 50 μL, to which concentrated sulfuric acid (150 μL) was added, followed by addition of 5 wt% phenol water solution. The samples were subsequently incubated at 90 °C with agitation for 20 minutes. The UV-vis absorbance of the digested solution was then measured at 490 nm with an infinite M200 microplate reader (Tecan, Switzerland) using a 96-well plate (Greiner CELLSTAR, Sigma-Aldrich).To determine the amount of glucose on each particle type, the original PG on each particle type and the original PG solutions (70 μL) were each treated with 0.2 M TFA (200 μL) for 3 h at 80 °C with agitation. The mixtures were then treated and analyzed as described above for the enzymatic experiments. The extent of degradability was determined as the ratio of UV-vis absorbance reading intensity at 490 nm for the enzymatically degraded particles to the trifluoroacetic acid degraded particles.
Fluorescence
Fluorescence of the PG-dopa coatings was analysed with a JASCO FP-8500 spectrofluorometer instrument. A quartz surface was cut into a 10 mm strip, and coated with PG-dopa10% nanoparticles. This was then placed inside a quartz cuvette with 4 mL of solution, on a 45° angle to the excitation source, and the detection channel (situated perpendicular to each other). The emission wavelength of the excitation spectrum was 340 nm. The excitation wavelength of the emission spectrum was 280 nm. The excitation wavelength and the emission wavelength for the fluorescence intensity of PG-dopa10% coated quartz over time in the solution of α-amylase are 280 nm and 310 nm.Results and discussion
Synthesis of PG-dopa
The PG was first oxidized by sodium periodate to produce an aldehyde group.25 Then, the dispersion was purified by dialysis to remove the oxidizing agent. The aldehyde group in oxidized PG was reacted with the amine of dopamine hydrochloride in phosphate buffer (pH 5.0). Finally, the imine was reduced to a stable secondary amine by reductive amination (Fig. 1(b)).The successful synthesis and degree of conjugation was confirmed by 1H NMR spectroscopy. The i, j and k peaks at δ ∼ 6.8 ppm were assigned to the protons from the aromatic ring of dopamine, which indicated the presence of the catechol group on the modified nanoparticles (Fig. 1(c)). The methylene protons (g and h) from dopamine hydrochloride can also be found at δ 3.1 and 2.8 ppm after modification. The degree of conjugation (DC) of dopamine of the PG-dopa was calculated by the equation DC = A6.8/(3 × A5.3), where A6.8 and A5.3 are the integrations of the peak area at δ = 6.8 (aromatic protons) and δ = 5.3 (the proton at the C1 of glucose repeat unit, a peak), respectively. The DC of different PG-dopa is summarized in Table 1. The number in the term PG-dopa10% is the ratio (mol:mol) of sodium periodate and glucose repeat unit. The degree of conjugation of PG-dopa10% was found to be 4.67%. However, the broadness of the NMR peaks makes absolute quantification difficult for such low conjugation degrees.
| Samples | Degree of conjugation (%) | |
|---|---|---|
| 1H NMR | Catechol assay | |
| PG-dopa5% | 2.67 | 3.35 |
| PG-dopa10% | 4.67 | 5.22 |
| PG-dopa15% | 6.00 | 7.31 |
Given the broadness of the peaks, the degree of conjugation was additionally confirmed using a catechol assay. 4-Methylcatechol was used to obtain the standard curve over the concentration range of 0.01–0.08 mg mL−1 (Fig. S1b, ESI†). The UV-vis absorbance intensity at a wavelength of 525 nm was used to plot the fitting curve (Fig. S1d, ESI†). For the purpose of the assay, we assume that the molecular weight of the glucose repeat unit does not change after the conjugation of dopamine. Then, the DC can be calculated by the following equation:
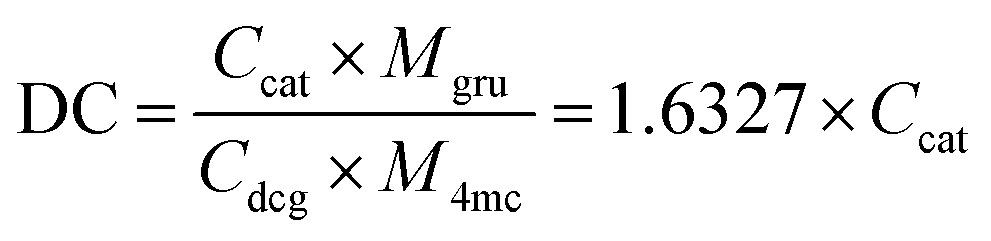
Characterization of PG and PG-dopa nanoparticles
Atomic force microscopy (AFM) was used to measure the dry state morphology and height of PG and PG-dopa10% nanoparticles (Fig. 2(a) and (b), respectively). The height of both PG and PG-dopa10% nanoparticles was ∼12 nm from AFM topography images (Fig. S2, ESI†). It should be noted that the AFM measurements were done in a dry state, which were then compared to cryo-transmission electron microscopy (TEM) and dynamic light scattering (DLS) which were in frozen and liquid water, respectively. Cryo-TEM images of PG and PG-dopa10% nanoparticles (Fig. 2(c) and (d), respectively) revealed the shape of the nanoparticles at high resolution, which are consistent with an irregular sphere. DLS revealed a shift in the intensity peak towards larger hydrodynamic diameters, where the Z-average diameter increased from ∼72 nm to ∼110 nm, and the dispersity (PDI) also increased from 0.084 to 0.239 (Table S1, ESI†). Given the bias of DLS towards larger particles, we computed the average diameters of both particle types from the cryo-TEM images (Fig. 2(f)). We found that the PG nanoparticles had diameters in the range of 40 nm to 45 nm, whereas for PG-dopa10% this increased slightly towards 50 nm to 55 nm. This indicated the average size increased after modification. Furthermore, the column graph shape of PG nanoparticles was sharper than that of PG-dopa10% nanoparticles, indicating that the size distribution also increased after modification. This trend was consistent with the diameter population distribution curve from DLS measurements (Fig. 3(e)). The graph shows the relative intensity of scattering light of the nanoparticles with different sizes. We note that the diameters between cryo-TEM and DLS are different in definition (observable size vs. hydrodynamic size). The PG-dopa10% nanoparticles were found to be negatively charged with a zeta potential of −15.0 mV, possibly due to the deprotonation of the catechol groups. The negative charge can aid the nanoparticle stability in water.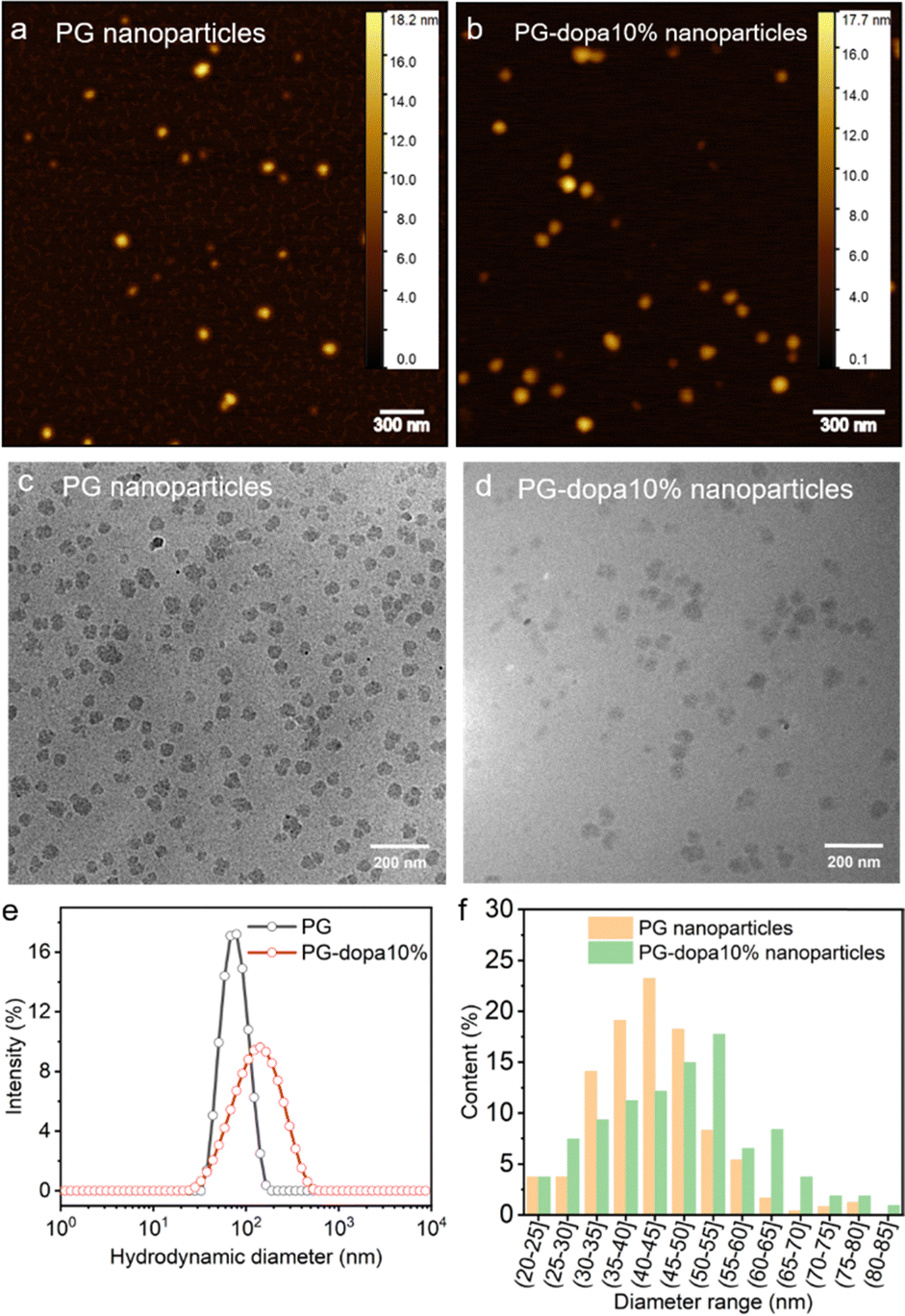 | ||
| Fig. 2 (a) Dry state AFM topography image of PG nanoparticles. (b) Dry state AFM topography image of PG-dopa nanoparticles. (c) TEM image of PG nanoparticles. (d) TEM image of PG-dopa10% nanoparticles. (e) DLS hydrodynamic diameter populations of PG and PG-dopa nanoparticles. (f) Calculated diameter range populations of PG and PG-dopa10% nanoparticles from Fig. 3(c) and (d). | ||
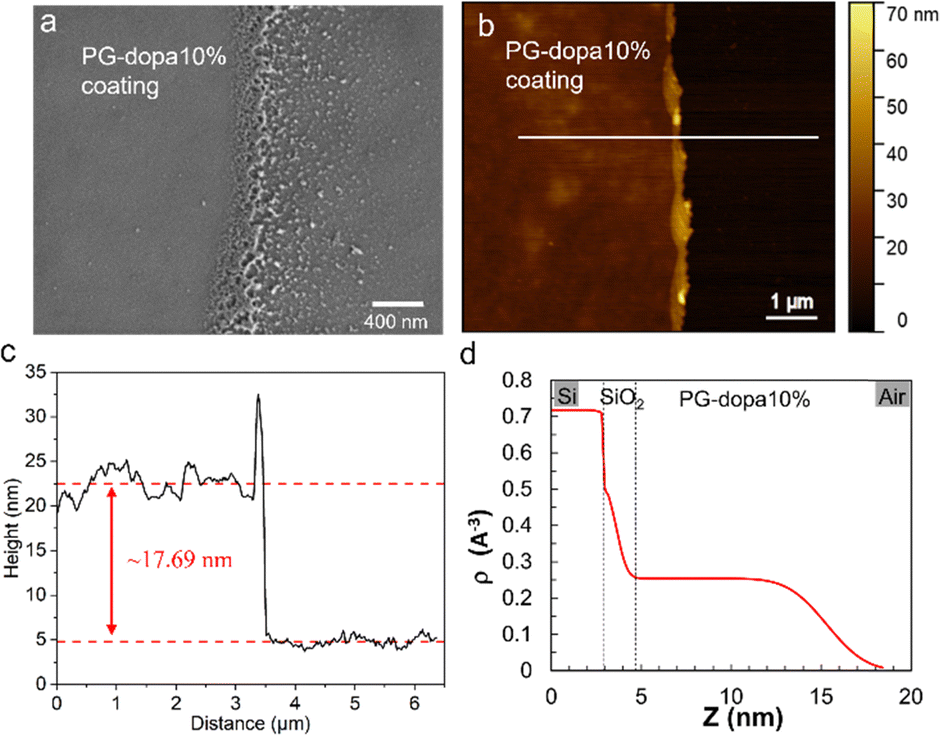 | ||
| Fig. 3 (a) The SEM image of the PG-dopa10% coating on the silicon wafer prepared by dip coating (the left part is the coating and the edge of the coating is in the middle). (b) The AFM topography image of a scratch of the PG-dopa10% coating on the silicon wafer. (c) The height of the white line in the AFM topography image. (d) XRR curve of the PG-dopa coating on the silicon wafer. The inset shows the electron density profile extracted from the best fit of the XRR curve. | ||
Morphology and thickness of the PG-dopa10% coating
Scanning electron microscopy (SEM) was used to observe the PG-dopa10% coating on silicon wafers. The coating was continuous and smooth (the left area of Fig. 3(a)), whereas the edge of the film (limit of the dip-coating) was porous, with some aggregate nanoparticles. This indicated that the PG-dopa10% nanoparticles showed affinity to the silica material and the interactions between nanoparticles are negligible. The AFM topography image revealed that a continuous, dense PG-dopa10% coating of ∼18 nm in thickness formed on the silicon wafer by dip coating into the PG-dopa10% water dispersion (Fig. 3(b) and (c)). The thickness of the PG-dopa10% coating was slightly larger than the height of individual PG-dopa10% nanoparticles (∼12 nm) (Fig. S2, ESI†), from AFM topography images but less than the double height of the nanoparticles. To obtain further insight into the structure of PG-dopa and PG-dopa coating, wide-angle X-ray scattering and X-ray reflectivity measurements were performed using established methods.26 Fig. S3a and b (ESI†) shows the amorphous nature of the PG-dopa10% which is significantly different from crystalline cellulose fibres. The obtained X-ray reflectivity curves together with its corresponding fit data are shown in Fig. S4 (ESI†). The weakly damped Kiessig fringes observed throughout the curve indicated a relatively smooth surface with a uniform coating thickness. A good fit of the experimental curve was obtained using a single-layer model onto a thin native silicon oxide layer with a thickness of 0.74 nm. The electron density profile used to obtain the best fit (Fig. 3(d)) corroborates the uniformity of the coating with a thickness and a RMS roughness of 11.7 nm and 1.7 nm, respectively.The underwater adhesion performance of PG-dopa coatings
The colloidal probe AFM technique was used to characterize the adhesion properties of the underwater PG-dopa10% coating (Fig. 4(a)). A colloidal probe (spherical silica particles with a 20 μm diameter) was brought into contact with PG-dopa coatings on glass substrates, and the force–distance curves were recorded, in water solution. Fig. 4(b) shows a schematic representation of the force distance curve from our experiments. The contact time as well as the maximal load on the cantilever and the retraction rate were varied. As shown previously, the thermodynamic work of adhesion in the case of the interactions of soft polymeric bodies is often very challenging to measure due to the very rapid pull-off event and requires specific experimental setups that are not suitable for every experimental system.27 In order to avoid misunderstanding, we use here the strain energy release rate G instead of the work of adhesion, as we use a pull-off experiment to determine the adhesion properties of the PG-dopa coatings. The strain energy release rate G, the area on the retraction part of the force–distance curve (Fig. S6, ESI†) below zero normalized on the calculated area of contact, was used to characterize the adhesion performance of the PG-dopa coatings (Fig. 4(c) and (d)).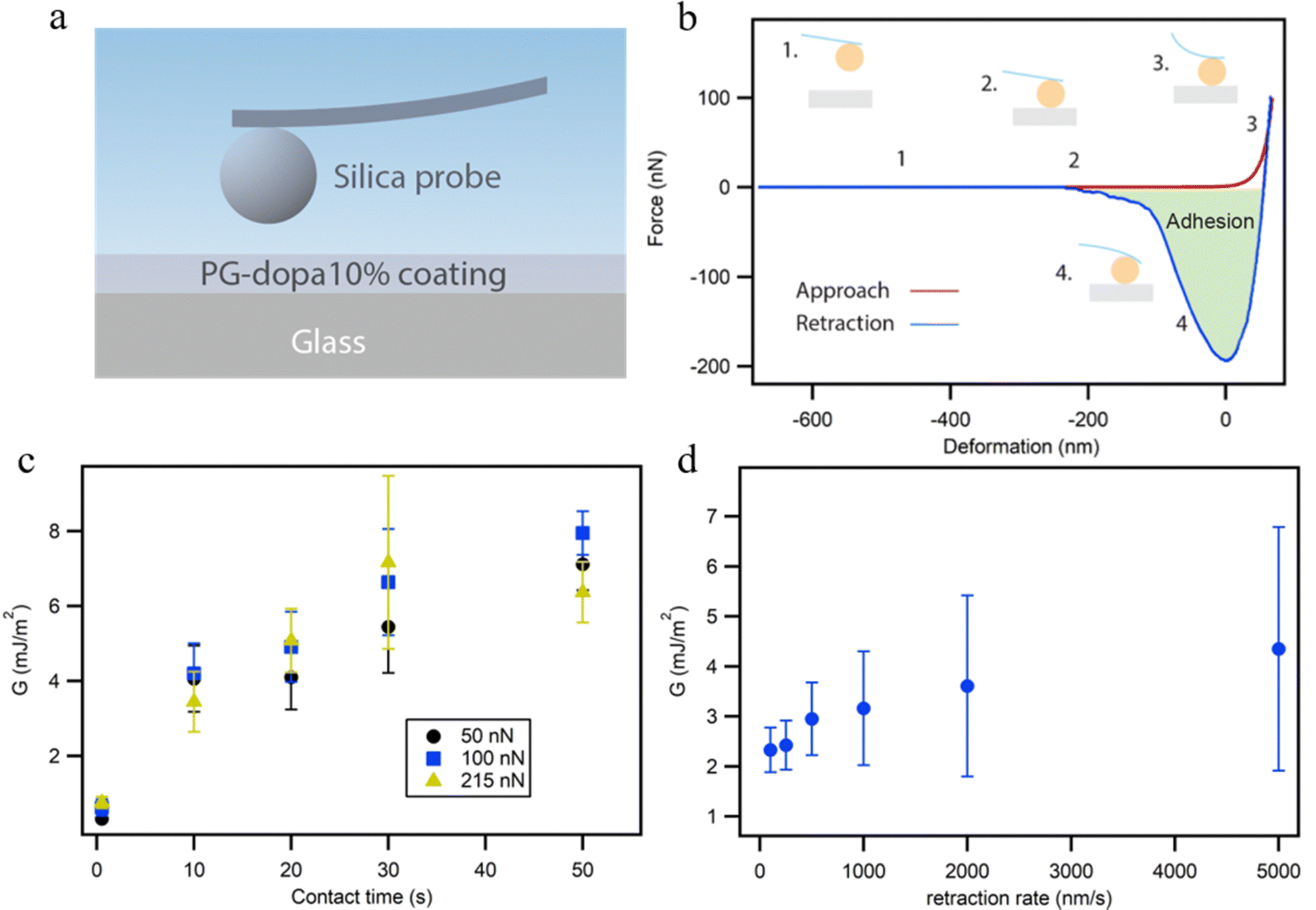 | ||
| Fig. 4 (a) Schematic of the colloidal probe AFM measurement setup. (b) A typical force-deformation approaching and retraction curve from our colloidal probe AFM experiment of PG-dopa10% films. (c) The strain energy release rate G of PG-dopa10% coating dependence on contact time for the maximal load force. (d) Strain energy release rate of the PG-dopa10% coating as a function of retraction rate. | ||
In the case of the interaction of soft bodies, the increase of adhesion on contact time, retraction rate, and maximal load is very common.27–30 A longer contact time allows additional bonds and interactions between the two surfaces to develop. Larger maximal loads increase the deformation of the soft bodies, thereby increasing the contact area and the total number of bonds between the two surfaces. Higher retraction rates are connected with the higher rates for the separation of two surfaces influencing the pull-off force drastically.31,32 In our case, the increase of the contact time results in a drastic increase of G from below 1 mJ m−2 to around 7 mJ m−2 on average for each maximal load used (Fig. 5(c)).
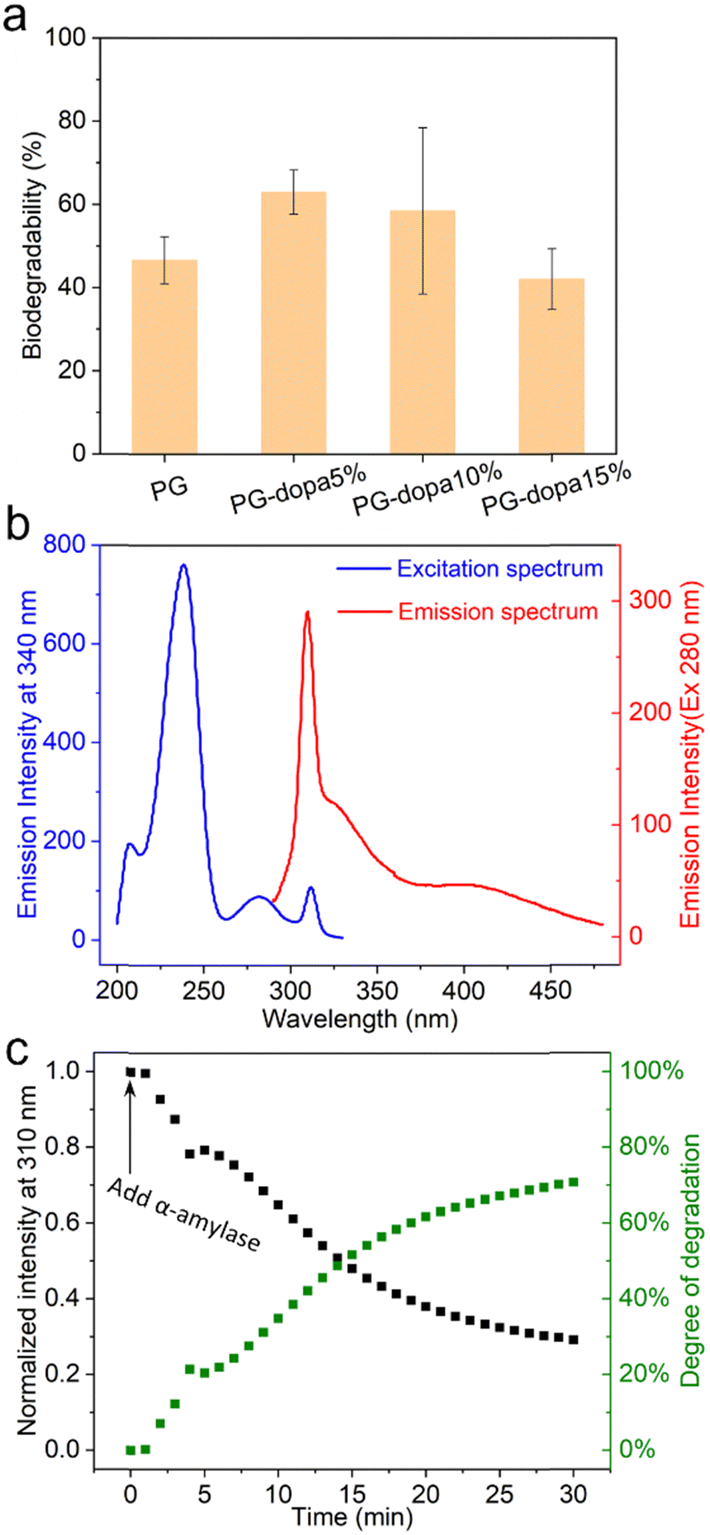 | ||
| Fig. 5 (a) The degradation of PG and PG-dopa nanoparticles in 1 U α-amylase PBS solution after 3 h. (b) Fluorescence excitation and emission spectra of PG-dopa10% films. (c) PG-dopa10% coated quartz incubated in α-amylase (45 U mL−1) PBS solution, with fluorescence intensity monitored as a function of time. The excitation and emission wavelengths are 280 nm and 310 nm. | ||
While the data for the influence of the retraction rate on G have a large standard deviation due to the deposition of the matter on the colloidal probe, which is inevitable in the case of the single particle experiment, the trends can still be seen. The average values of G more than doubled from 100 nm s−1 to 5000 nm s−1.
The influence of the maximal load, however, was insignificant in our case. The reason for this behavior can be understood by comparing the maximum deformation of the PG-dopa10% coating generated at each maximal load (Fig. S5, ESI†). The increase of the maximal load did not result in a substantial increase in the deformation. The load applied to a soft deformable surface causes a certain amount of deformation. The simplest theory describing the relationship between the applied load F and the resulting deformation δ is the Hertz model. According to this model, F ∼ δ3/2. Therefore, an increase in load should cause additional deformation. The fact that the deformation remains the same and is not zero suggests that a very hard substrate (i.e., glass) is covered by a relatively soft layer. This layer is deformed until the colloidal probe reaches the glass substrate. After this, the cantilever is too soft to indent and measure the deformation of the glass and the silica colloidal probe. Therefore, a maximal load of 50 nN was sufficient to reach the glass substrate under the PG-dopa10% coating. The resulting values of the maximum deformation in this case can be used as an indication of the thickness of the PG-dopa10% coating under water. The maximum deformation of the coating was around 75 nm (Fig. S5, ESI†), which is consistent with the nanoparticle size from the DLS measurements.
The adhesion performance of the different PG-dopa coatings was also characterized, namely PG-dopa5% and PG-dopa15% (Fig. S7, ESI†). PG-dopa5% showed very low adhesion and made the interactions of the colloidal probe with the substrate weaker than in the case of the borosilicate glass (data not shown). PG-dopa15% showed comparable performance to PG-dopa10% at all contact times except 50 s, where the adhesion of PG-dopa15% was about 50% greater than that of PG-dopa10%. However, comprehensive investigation of the adhesion performance of PG-dopa15% was limited by the very high colloidal probe contamination, which was significantly greater than that of PG-dopa10%. The thickness of PG-dopa15% was close to that of PG-dopa5% and less than that of PG-dopa10% (Fig. S8 and S9, ESI†). An increased tendency for colloidal probe contamination in the case of PG-dopa15% and lower thickness compared to PG-dopa10% may indicate the lower layer uniformity and stability of PG-dopa15%.
Degradation of PG-dopa nanoparticles and PG-dopa10% coatings
The degradation of PG and PG-dopa nanoparticles in a 1 U mL−1 α-amylase PBS solution (pH 7.4, 0.01 M) over 3 h was assessed using a phenol–sulfuric acid assay (Fig. 5(a)). It was found that the functionalized PG-dopa nanoparticles retained good biodegradability towards α-amylase as the starting material PG nanoparticles. The PG-5% and PG-10% nanoparticles even showed a higher degradation degree than PG nanoparticles, perhaps due to modifications in the branching structure due to the synthetic additions. To glean insight into the degradability of the PG-dopa films, a different assay was needed, due to an inability to ascertain the exact amount of PG-dopa10% in the films. We found that the PG-dopa10% has fluorescence properties, whereby under 280 nm excitation, significant fluorescence was observed, peaking at 310 nm (Fig. 5(b)). This emission maximum at 310 nm was used to assess the degradability of the films in real-time, after soaking in α-amylase (45 U mL−1) solution (Fig. 5(c)). It was found that there was a kinetic decrease in the fluorescence of the film, decreasing from ∼1800 counts and saturating at abut ∼500 counts after 30 minutes. This highlights that the adhesive PG-dopa materials are still biodegradable towards glucosidase enzymes with time.The lyophilised PG-dopa10% was found to be stable under refrigeration for years. The color of the PG-dopa10% water dispersion changed from white foggy to slightly brown (Fig. S10a, ESI†) because of the gradual oxidization of the catechol groups.33 However, no aggregated particles were observed from DLS measurements after storing in the 4 °C fridge for a week or 9 months (Fig. S10b, ESI†). The reason for decreasing PG-dopa10% particle sizes after 9 months might be the degradation of the glycogen nanoparticles over time.
Together, these underwater adhesive nanoparticles can offer a renewable biomaterial alternative to highly synthetic mussel-foot protein mimics, where the degradability yields glucose and a small amount of dopamine. Our study has focused on contact between ideal surfaces (plane and smooth sphere) and how it changes between rough surfaces is not yet known. Furthermore, the adhesiveness towards other materials, such as soft, elastic tissues, is currently the subject of our future investigations.
Conclusions
We have reported on the synthesis of phytoglycogen-dopamine hybrid materials. The catechol group of dopamine endowed the phytoglycogen nanoparticles with an ability to adhere to underwater silicon dioxide materials. It was shown that the phytoglycogen-dopamine nanoparticles adsorbed readily and densely onto silicon dioxide substrates, forming continuous monolayers from a water dispersion. The thickness of the coating was found to be ∼18 nm in the dry state and ∼78 nm when solvated. The adhesive properties of the coating were striking, with a maximum strain energy release rate of 7.2 mJ m−2 when contacting surfaces of underwater silicon dioxide. Moreover, the modification did not affect the biodegradability of phytoglycogen. This sticky phytoglycogen coating might find applications in short-term implantable devices or as “micro” adhesives for the assembly of nanoparticles.Data availability
The authors confirm that the data supporting the findings of this study are available within the article.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We sincerely thank Dr. Petr Formanek and Michael Göbel for assistance with TEM and SEM measurements, respectively, and Dr. Hartmut Komber for NMR measurements. The authors additionally would like to thank Dr. Alexander Münch, Qiong Li and Dr. Ilka Hermes for their help with UV-vis spectroscopy and AFM measurements, and Thomas Witzmann and Dr. Günter Auernhammer for discussions. Prof. Mahmoud Al-Hussein thanks the University of Jordan and Leibniz-Institut for Polymer Research Dresden (IPF Dresden) for financial support. This work received funding from the European Union's Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement 872233 (“PEPSA-MATE”). J. L. gratefully acknowledges the Chinese Scholarship Council (CSC) for funding.References
- E. W. Danner, Y. Kan, M. U. Hammer, J. N. Israelachvili and J. H. Waite, Biochemistry., 2012, 51, 6511–6518 CrossRef CAS PubMed.
- J. H. Waite and X. Qin, Biochemistry., 2001, 40, 2887–2893 CrossRef CAS PubMed.
- H. Lee, N. F. Scherer and P. B. Messersmith, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 12999–13003 CrossRef CAS PubMed.
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Science, 2007, 318, 426–430 CrossRef CAS PubMed.
- M. A. North, C. A. Del Grosso and J. J. Wilker, ACS Appl. Mater. Interfaces, 2017, 9, 7866–7872 CrossRef CAS PubMed.
- J. Yu, B. Cheng and H. Ejima, J. Mater. Chem. B, 2020, 8, 6798–6801 RSC.
- B. Cheng, J. Yu, T. Arisawa, K. Hayashi, J. J. Richardson, Y. Shibuta and H. Ejima, Nat. Commun., 2022, 13, 1892 CrossRef CAS PubMed.
- R. Cui, F. Chen, Y. Zhao, W. Huang and C. Liu, J. Mater. Chem. B, 2020, 8, 8282–8293 RSC.
- Z. Du, F. Qiao, L. Tong, W. Zhang, X. Mou, X. Zhao, M. F. Maitz, H. Wang, N. Huang and Z. Yang, The Innovation, 2024, 5, 100671 CrossRef CAS PubMed.
- Q. A. Besford, Soft Matter, 2024, 20, 3577–3584 RSC.
- N. Davydiuk, V. Londhe, M. F. Maitz, C. Werner, A. Fery and Q. A. Besford, Nanoscale, 2025, 17, 252–260 RSC.
- Q. A. Besford, F. Cavalieri and F. Caruso, Adv. Mater., 2020, 32, 1904625 CrossRef CAS PubMed.
- T. Isogai, T. Saito and A. Isogai, Biomacromolecules, 2010, 11, 1593–1599 CrossRef CAS PubMed.
- S. A. Engelberth, N. Hempel and M. Bergkvist, Bioconjugate Chem., 2015, 26, 1766–1774 CrossRef CAS PubMed.
- M. Rabyk, M. Hruby, M. Vetrik, J. Kucka, V. Proks, M. Parizek, R. Konefal, P. Krist, D. Chvatil, L. Bacakova, M. Slouf and P. Stepanek, Carbohydr. Polym., 2016, 152, 271–279 CrossRef CAS PubMed.
- M. Perrone, A. Lopalco, A. Lopedota, A. Cutrignelli, V. Laquintana, J. Douglas, M. Franco, E. Liberati, V. Russo, S. Tongiani, N. Denora and A. Bernkop-Schnürch, Eur. J. Pharm. Biopharm., 2017, 119, 161–169 CrossRef CAS PubMed.
- P. Pacchin Tomanin, J. Zhou, A. Amodio, R. Cimino, A. Glab, F. Cavalieri and F. Caruso, J. Mater. Chem. B, 2020, 8, 4851–4858 RSC.
- S. Arias, S. Amini, J. Horsch, M. Pretzler, A. Rompel, I. Melnyk, D. Sychev, A. Fery and H. G. Börner, Angew. Chem., Int. Ed., 2020, 59, 18495–18499 CrossRef CAS PubMed.
- H.-J. Butt, Biophys. J., 1991, 60, 1438–1444 CrossRef CAS PubMed.
- W. A. Ducker, T. J. Senden and R. M. Pashley, Nature, 1991, 353, 239–241 CrossRef CAS.
- Y. Jiang and K. T. Turner, Extreme Mech. Lett., 2016, 9, 119–126 CrossRef.
- J. L. Hutter and J. Bechhoefer, Rev. Sci. Instrum., 1993, 64, 1868–1873 CrossRef CAS.
- K. L. Johnson, K. Kendall, A. D. Roberts and D. Tabor, Proc. R. Soc. London, Ser. A, 1971, 324, 301–313 CAS.
- Q. A. Besford, A. C. G. Weiss, J. Schubert, T. M. Ryan, M. F. Maitz, P. P. Tomanin, M. Savioli, C. Werner, A. Fery, F. Caruso and F. Cavalieri, ACS Appl. Mater. Interfaces, 2020, 12, 38976–38988 CrossRef CAS PubMed.
- M. Bertoldo, G. Zampano, L. Suffner, E. Liberati and F. Ciardelli, Polym. Chem., 2013, 4, 653–661 RSC.
- M. Al-Hussein, Y. Séréro, O. Konovalov, A. Mourran, M. Möller and W. H. de Jeu, Macromolecules, 2005, 38, 9610–9616 CrossRef CAS.
- D. Sychev, S. Schubotz, Q. A. Besford, A. Fery and G. K. Auernhammer, J. Colloid Interface Sci., 2023, 642, 216–226 CrossRef CAS PubMed.
- I. U. Vakarelski, A. Toritani, M. Nakayama and K. Higashitani, Langmuir, 2001, 17, 4739–4745 CrossRef CAS.
- R. Buzio, A. Bosca, S. Krol, D. Marchetto, S. Valeri and U. Valbusa, Langmuir, 2007, 23, 9293–9302 CrossRef CAS PubMed.
- D. Pussak, D. Ponader, S. Mosca, T. Pompe, L. Hartmann and S. Schmidt, Langmuir, 2014, 30, 6142–6150 CrossRef CAS PubMed.
- D. Maugis and M. Barquins, J. Phys. D: Appl. Phys., 1978, 11, 1989 CrossRef.
- A. N. Gent and J. Schultz, J. Adhes., 1972, 3, 281–294 CrossRef CAS.
- J. Ju, S. Jin, S. Kim, J. H. Choi, H. A. Lee, D. Son, H. Lee and M. Shin, ACS Appl. Mater. Interfaces, 2022, 14, 25115–25125 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sm01454e |
| This journal is © The Royal Society of Chemistry 2025 |