
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Large modulation of the bottlebrush diblock copolymer morphology and structural color through solvent selectivity†
Sanghyun
Jeon
 a,
Yash
Kamble
b,
Zhuang
Xu
c,
Azzaya
Khasbaatar
b,
Changhyun
Hwang
b,
Jong-Hoon
Lee
d,
Jiachun
Shi
b,
Simon A.
Rogers
be,
Damien
Guironnet
bce and
Ying
Diao
*be
a,
Yash
Kamble
b,
Zhuang
Xu
c,
Azzaya
Khasbaatar
b,
Changhyun
Hwang
b,
Jong-Hoon
Lee
d,
Jiachun
Shi
b,
Simon A.
Rogers
be,
Damien
Guironnet
bce and
Ying
Diao
*be
aDepartment Materials Science and Engineering, University of Illinois at Urbana-Champaign, 1304 W Green St, Urbana, IL 61801, USA
bDepartment of Chemical and Biomolecular Engineering, University of Illinois at Urbana-Champaign, 600 South Mathews Avenue, Urbana, Illinois 61801, USA
cDepartment of Chemistry, University of Illinois at Urbana-Champaign, 505 S Mathews Ave, Urbana, IL 61801, USA
dDepartment of Advanced Materials Engineering, Kyonggi University, Suwon 16227, Republic of Korea
eBeckman Institute for Advanced Science and Technology, University of Illinois at Urbana-Champaign, 405 N. Mathews Ave. M/C 251, Urbana, Illinois 61801, USA
First published on 11th February 2025
Abstract
Bottlebrush block copolymers (BBCPs), characterized by densely grafted side chains along their backbone, have emerged as promising materials for structural color applications. Their unique architecture prevents entanglement and facilitates rapid assembly kinetics, enabling the formation of various photonic crystals with high tunability of structural color from the visible to the infrared range. However, accessing non-1D structures has been largely limited to synthetic approaches. In this paper, we report large modulation of a microphase separated morphology of the polystyrene-b-polylactide (PS-b-PLA) BBCP using a single material by exploiting selective solubility of the two blocks in a series of structurally similar solvents. Combining optical spectroscopy, electron microscopy, photoinduced force microscopy and X-ray scattering, we unveil the microstructural evolution as dependent on solvent selectivity. Furthermore, we uncovered the mechanisms underlying the differences in assembly, where highly selective solvents induce sidechain aggregation, altering the volume fraction and facilitating the formation of non-1D structures.
Introduction
Structural color is a phenomenon where colors are produced through the interaction of light with nanostructured materials, resulting in the reflection and interference of light waves, distinct from dyes and pigments which induce coloration through absorption. Structural color is advantageous over dyes and pigments given their environmental stability, biocompatibility,1,2 brilliance and adaptability.3 This coloration strategy is widespread in nature, arising from a diverse range of structures and producing a wide spectrum of colors determined by the scattering mechanism.4–11 One of the most well-known examples of structural color is found in the panther chameleon (Furcifer pardalis),4 whose skin contains multiple layers of well-ordered structures, including face-centered cubic and brick-and-rod configurations. These structures generate dynamic structural colors for communication and reflect infrared light to aid in thermal regulation. Beyond chameleons, numerous species employ diverse well-ordered nanostructures, producing structural color, primarily for camouflage and communication: the cylindrical melanin rods in the peacock's barbules (Pavo muticus),9 the gyroid structures in the green wing of Callophrys rubi,10 and the lamellar structures in the blue wings of Morpho butterflies.11 In addition to well-ordered structures, the random close-packed nanostructures, producing non-iridescence colors, are widespread in nature, appearing in over 250 bird species (approximately 2.5% of all avian species),12 as well as primates (Mandrillus sphinx and Cercopithecus aethiops),13 dragonflies (Odonata),14 and longhorn beetle (Anoplophora graafi).15These diverse structures and their interesting optical properties in nature have inspired numerous biomimicry efforts aimed at replicating these remarkable natural designs. A promising approach for mimicking various nanostructures in nature is the lithographic technique such as two photon polymerization lithography.16–19 However, despite the advantages of lithographic approaches such as high precision, their limitations for photonic coloration are apparent: material selection is limited to cross-linkable materials, resolution below 300 nm still remains challenging, and scaling up production is restricted due to low throughput, hindering mass production efforts. An alternative method to attain structural color is by employing self-assembling materials such as nanocrystals,20,21 cholesteric liquid crystals,22–25 and block copolymers26–28 which assemble into nanostructures with feature size ranging from around 100 to 300 nm, which corresponds to visible range wavelength reflection. Among these, block copolymers have proven to be versatile exhibiting nanostructures ranging from lamellar, cylindrical, gyroid, and spherical morphology based on the volume fraction of the blocks,29–31 resembling those found in nature. However, traditional linear block copolymers faced challenges in achieving sufficiently large domain spacing for visible light reflection due to entanglement.32,33 A breakthrough came with the introduction of bottlebrush block copolymers (BBCPs), which comprise densely grafted sidechains along the backbone. This unique architecture causes the molecules to adopt an extended worm-like cylinder conformation in solution,34–36 which prevents entanglement37,38 even in the ultrahigh molecular weight regime and contributes to the rapid assembly kinetics.38–40 This property enables BBCPs to form photonic structures with large domain spacing, which reflects not only visible but also infrared (IR) wavelengths of up to 1500 nm40,41 usually attained using 1D lamellar structures.
Recently, researchers from Vignolini, Parker, and Song groups have expanded the scope of structural color studies beyond the 1D photonic lamellar structure to include 3D photonic glass structures,42–44 having short-range order nanostructures, to achieve angle-independent and non-iridescent structural colors. On the other hand, existing approaches for controlling the BBCP morphology have largely followed two routes: (1) modifying the length of the backbone or sidechains through chemical synthesis to adjust the volume fraction45,46 or architecture;47–50 (2) designing amphiphilic bottlebrush block copolymers which assemble through micellization.43,51,52 These synthetic approaches, while elegant and versatile, can be labor-intensive and susceptible to reproducibility issues arising from batch-to-batch deviations.
In this study, we present a simple yet surprisingly effective processing method for tuning the nanoscale morphology of the PS-b-PLA BBCP leveraging distinct solubilities of the two blocks (PLA and PS) in a series of structurally similar organic solvents, namely toluene, o-xylene, m-xylene and mesitylene. Despite the structural similarity, the differential solubility of the PLA vs. PS arms is modulated by almost 4 orders of magnitude, thereby varying the nanoscale morphology from lamellar to spherical when increasing solvent selectivity/solubility difference. Correspondingly, the peak reflected wavelength can be modulated from 693 to sub-350 nm, which corresponds to orange to deep-blue color. This is unveiled through extensive solid-state structural analysis using techniques spanning scanning electron microscopy (SEM), photo-induced force microscopy (PiFM), and film small angle X-ray scattering (SAXS). Furthermore, we uncover the assembly pathways of BBCPs and the mechanisms underlying these pathways influenced by solvent selectivity. The differential solubility of each block leads to substantial differences in the volume fraction of BBCPs, leading to various phase separations (cylindrical and spherical) beyond the lamellar structure. This insight is gleaned from examining BBCP solution structures using solution SAXS, dynamic light scattering (DLS), and cryo-transmission electron microscopy (TEM).
Results and discussion
The Influence of solvent selectivity on the optical properties of BBCPs
In this section, we explore the impact of solvent selectivity, defined as differential solubilities for the two blocks (PS and PLA), on the structural color of the PS-b-PLA BBCP film. A series of PS-b-PLA BBCPs (Nbb = 300, 400, and 500) were synthesized by sequential graft-through polymerization of macromonomers27,53,54 as shown in Fig. 1a. PS-b-PLA BBCPs with varying backbone lengths were targeted as described in Table 1. Gel permeation chromatography (GPC) analysis revealed that the synthesized materials comprise three main components: residual non-norbornene-functionalized PS, homopolymer PS building blocks, and the target diblock copolymer PS-b-PLA (BBCP) (Fig. S1–S4, ESI†). All synthetic details of BBCPs and homobrush block copolymers are provided in the Materials and methods section, as well as in the ESI† (Material synthesis section, Fig. S1–S4, and Table S1, ESI†).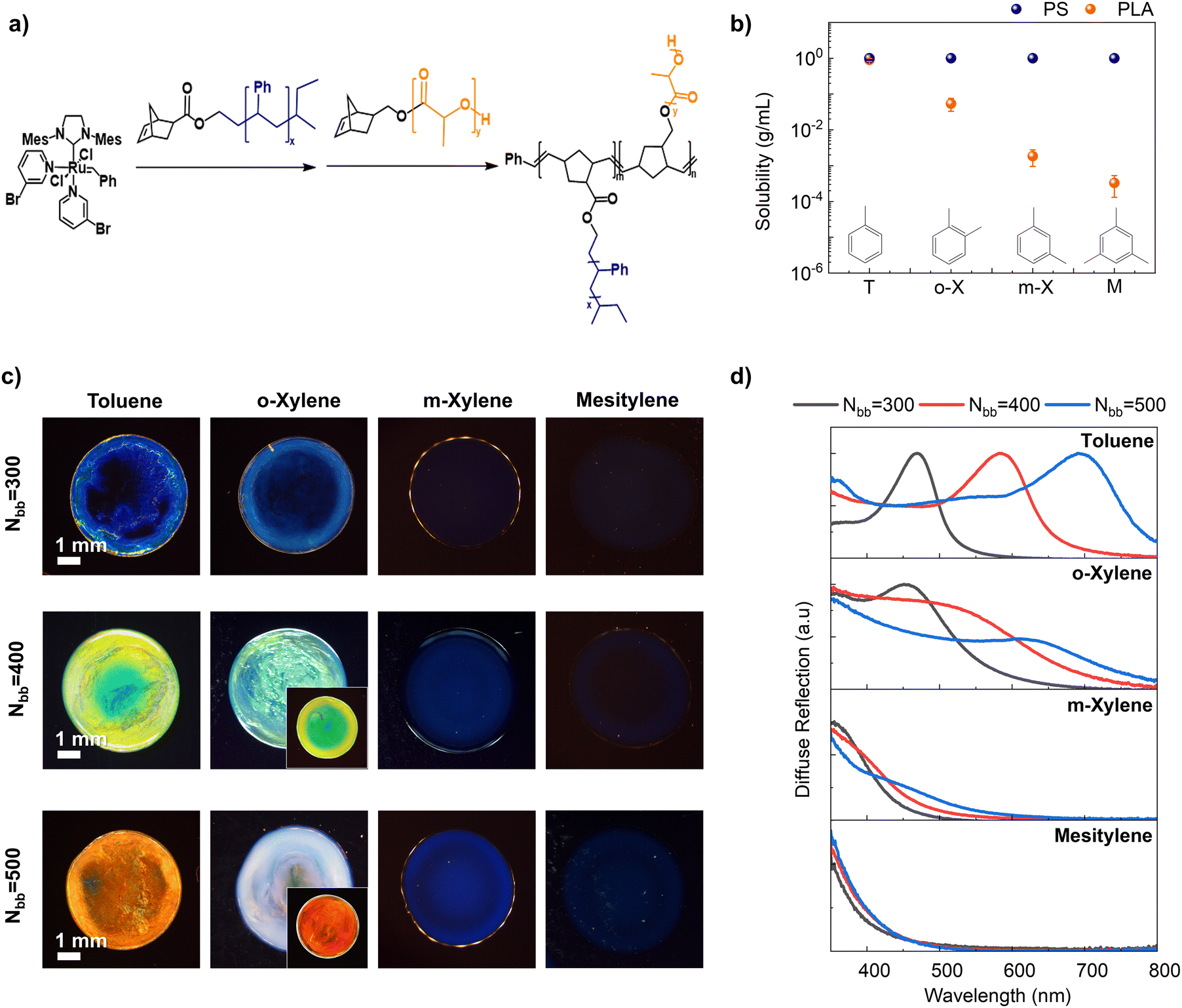 | ||
| Fig. 1 (a) Synthesis of PS-b-PLA BBCPs via sequential ring-opening metathesis polymerization of PS and PLA macromonomers. (b) Solubility measurements employing PS (Nbb = 200, Nsc = 45) and PLA (Nbb = 200, Nsc = 60) homobrush polymers in various solvents (T: toluene, o-X: o-xylene, m-X: m-xylene, and M: mesitylene). The solubility of PLA decreased from toluene to mesitylene, whereas that of PS remained constant. (c) Optical microscope camera images depicting distinct structural colors of drop-cast BBCP films dependent on backbone lengths (Nbb = 300, 400, and 500) and solvent. Inset photos show the structural color when blending the polymer with a broadband absorber (PC71BM). (d) UV-Vis diffuse reflection spectra dependent on backbone length (black: 300, red: 400, and blue: 500) and solvent. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) PS
PSSubsequently, four solvents (toluene, o-xylene, m-xylene, and mesitylene) were selected based on their varying solubility with respect to PS and PLA blocks. Gravimetric solubility measurements using PS (Nbb = 200 and Nsc = 45), and PLA (Nbb = 200 and Nsc = 60) homobrush polymers revealed that all four solvents exhibited good solubility (>1 g mL−1) for PS, while these solvents exhibited markedly different solubility levels for PLA (Fig. 1b). Specifically, the solubility of PLA dramatically decreased with an increase in the number of methyl groups in the benzene ring and variations in their positions (toluene: 0.91 ± 0.09, o-xylene: 0.05 ± 0.02, m-xylene: (1.9 ± 0.9) × 10−4, and mesitylene: (3.3 ± 2.0) × 10−4 g mL−1).
We then investigated the effect of solvent selectivity on the structural color of PS-b-PLA BBCP film samples (Nbb = 300, 400, and 500) drop-cast from the solvent series (see the Materials and methods section). The optical microscopy images of these samples displayed a large modulation of structural colors from red to blue by varying the backbone length and solvent selectivity (Fig. 1c). Specifically, when cast from the non-selective solvent toluene, the structural colors ranged from red (Nbb = 500) to blue (Nbb = 300) upon increasing the BBCP backbone length as expected. It was previously established that longer backbones lead to a red shift in color due to larger domain sizes.32,40,53 Interestingly, regardless of the BBCP length, higher solvent selectivity consistently resulted in a blue shift in structural color. For instance, with Nbb = 400, a non-selective toluene solution produced a green-yellow colored photonic crystal, while a slightly more selective o-xylene solution yielded a photonic crystal with green-blue hues. Highly selective solvents such as m-xylene and mesitylene produced even bluer and faint-blue colors, respectively. Some samples (toluene and o-xylene) exhibited a color gradient from the center to the edge. After the solvent-vapor annealing process, the gradient disappeared, and the color became more uniform, although the overall color remained similar. The details of this phenomenon are discussed in Fig. S5 (ESI†).
To quantitatively assess the structural colors, diffuse reflection spectra of each photonic crystal were obtained using UV-Vis spectroscopy with an integrating sphere (Fig. 1d). We observed that varying solvents led to significant changes in the shape of reflection spectra. Specifically, in reflection spectra of toluene-based films, pronounced reflection peaks appear centered at 470, 585, and 693 nm, respectively, for Nbb = 300, 400, and 500. In o-xylene, these peaks broadened and reduced in intensity exhibiting a blue shift to 456 nm, 529 nm, and 633 nm for Nbb = 300, 400, and 500, respectively. At the same time, background reflection at lower wavelengths significantly rises compared to the toluene case. From o-xylene to m-xylene, the peaks continue to weaken in intensity and blue shift to 460, 395, and 361 nm, respectively, for Nbb = 300, 400, and 500. Lastly, in mesitylene, no discernible peaks were observed in the visible range (350–800 nm) leaving only background reflection in the low wavelength range independent of backbone length. The origin of different reflection spectra and raised reflection at low wavelengths will be explained in later discussion through structural characterization and scattering theory. Generally, the length of the BBCPs affects the peak wavelength of the reflection spectra but does not alter the overall shape of the spectra. In contrast, solvents influence both the shape and peak position of the spectra. As we will discuss later, this optical behavior, influenced by solvent selectivity, arises from distinct nanostructures.
Expanding on this discovery, fine modulations of structural color can be achieved by blending solvents (Fig. S6, ESI†). For instance, blending toluene with m-xylene or mesitylene resulted in intermediate hues, demonstrating a gradual shift in the optical properties of the film from those of toluene-based films towards those of m-xylene or mesitylene films. Also, it was demonstrated that the color palette can be further broadened, and color purity can be enhanced by mitigating broadband reflection in samples exhibiting high reflectance in the low-wavelength region. For example, o-xylene exhibits peak reflectance spanning 456 to 633 nm, corresponding to blue to red structural color, but is obscured by strong blue reflection in the background. We showed that the background reflection can be mitigated by incorporating a broadband absorber, PC71BM, which is a small molecule that can absorb blue light efficiently (Fig. S7a, ESI†). After adopting the broadband absorber, which suppresses the effect of raised reflection at low wavelength, the structural color of samples showed clear red and green colors (Fig. 1c insets, Fig. S7b, ESI†). The structural colors obtained were quantified in the CIExy color space (Fig. S7c, ESI†).
Influence of solvent selectivity on the BBCP morphology
In this section, we delve into the nanoscale morphology of assembled BBCP films (Nbb = 400) to unravel the underpinning structure that gives rise to the optical properties observed. We first image the solvent-dependent nanostructure of BBCP films using cross-sectional SEM complemented by transmission SAXS. To discern the chemical composition of the structural features, we resort to photoinduced force microscopy (PiFM) which imparts chemical contrast to AFM through IR excitation. We further rationalize the morphology–optical property relationship through scattering theories.Cross-sectional SEM images revealed the strongly solvent dependent nanoscale morphology of BBCP films (Fig. 2a). Toluene samples showed well-defined lamellar structures with a d-spacing of 187.8 ± 10.7 nm, while o-xylene samples exhibited randomly oriented short cylinders, and the center-to-center distance of nearest neighbors (dNN) is 172.6 ± 22.9 nm. The m-xylene and mesitylene samples both displayed spherical structures with sphere radii of 52.4 ± 8.7, and 30.5 ± 7.1 nm, and dNN values of 165.0 ± 10.2 and 128.0 ± 9.5 nm, respectively. It is evident that as the morphology transitions from lamella to spherical, the characteristic domain spacing decreases progressively consistent with the structural color evolution. The morphological differences depending on the solvent persist regardless of the length of the polymer backbone (Nbb = 300 and 500) (Fig. S8a, ESI†). Furthermore, samples prepared with solvent mixtures (toluene and m-xylene; toluene and mesitylene) displayed a gradual transition from lamellar to spherical structures depending on the ratio, supporting the effects of a mixed solvent on the structural color (Fig. S8b and c, ESI†).
 | ||
Fig. 2 Structural characterization of BBCP films (Nbb = 400) drop-cast from the solvent series. (a) Cross-sectional SEM images displaying variations in nanostructures based on solvents (toluene: lamellar; o-xylene: cylindrical; m-xylene: spherical; mesitylene: spherical). PiFM images showing: (b) 1D profile of transmission SAXS for four drop-cast samples from the solvent series. (c) PiFM signal recorded at an excitation laser wavelength of 1492 cm−1 (corresponding to the aromatic stretching mode of PS), and (d) PiFM signal captured at 1750 cm−1 (corresponding to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching mode of the ester group of PLA). Each absorption band corresponding to the vibrational modes was confirmed by FT-IR analysis using PS and PLA homobrush polymers. The scale remains consistent across all images in the same figure. O stretching mode of the ester group of PLA). Each absorption band corresponding to the vibrational modes was confirmed by FT-IR analysis using PS and PLA homobrush polymers. The scale remains consistent across all images in the same figure. | ||
The film SAXS supported the SEM observation (Fig. 2b). In toluene-based films, a discernible long-range lamellar stacking is evident with a series of regularly spaced peaks in q-space, showcasing a calculated d-spacing of 203.0 ± 0.4 nm. Contrarily, for selective solvents like xylenes and mesitylene, two broad peaks emerge, consistent with the FCC peak inferences, supporting observations from imaging analysis. However, due to the significantly disordered structure, it is impossible to determine the exact packing structure. Assuming a close-packing FCC structure, we estimated dNN based on the peak position of the lowest q: (4.47 ± 0.01) × 10−3, (4.64 ± 0.01) × 10−3, and (5.95 ± 0.02) × 10−3 Å−1 for o-xylene, m-xylene, and mesitylene, respectively. These values correspond to dNN values of 172.2 ± 0.4, 165.8 ± 0.5, and 129.3 ± 0.4 nm for o-xylene, m-xylene, and mesitylene, respectively, in good agreement with our SEM image analysis.
To determine how the blocks distributed across the nanostructures observed, we applied photoinduced force microscopy (PiFM) which is a scanning probe technique that combines atomic force microscopy (AFM) with a tunable infrared laser to impart chemical contrast (Fig. 2c and d). During PiFM measurements, the IR laser is focused on a specific region of the sample where it interacts with the AFM probe. When the sample molecules absorb the light, an electric dipole is induced in the sample, which in turn creates a mirror dipole on the AFM tip. The attractive force between these dipoles allows us to generate a compositional map of the blend film with high resolution, down to approximately 10 nm.55,56 Distinct vibrational signals from each domain (PLA, Fig. 2c and PS, Fig. 2d) were obtained by tuning the excitation laser to specific absorption bands (mutually exclusive infrared signals) at 1492 cm−1 (aromatic stretching mode) for PS and 1750 cm−1 for PLA (CO stretching mode), as identified through Fourier-transform infrared spectroscopy (Fig. S9, ESI†). PiFM unveiled discrete cylindrical and spherical domains exhibited strong PLA signals in samples deposited from o- and m-xylenes and mesitylene, whereas the continuous domains showed strong PS peaks. This clearly indicates that PLA brushes assembled into cylinders or spheres in selective solvents, consistent with the fact that PLA solubility decreases by orders of magnitude with an increase in solvent selectivity.
Based on the structural analysis above, we conclude that with increasing solvent selectivity whereby the solubility towards the PS block is maintained while that towards the PLA block reduces, the BBCP morphology transitions from lamella to cylindrical to spherical structures. At the same time, the PLA domain size and thus center-to-center distance of adjacent domains (dNN), progressively reduces due to a reduction in PLA volume fraction in poorer solvents. These observations are aligned with the expected phase behavior of block copolymers following the volume fraction changes.29,31 Furthermore, we can establish a link between the optical properties of each film by considering the microstructures. Specifically, for lamellar structures (toluene) preferentially aligned normal to the incident light,28,46 a strong reflection peak is usually observed following Bragg's and Snell's laws.32,57 Additionally, there may be a plateau of reflection at lower wavelengths, attributed to misoriented lamellae.54 On the other hand, in disordered (randomly oriented) cylindrical or spherical phases (xylenes, mesitylene), optical behavior can be elucidated through the scattering properties of photonic glasses,58,59 where the color of these morphologies results from both single particles scattering (form factor) and the interference of scattered waves from particle assemblies (structure factor).21 In photonic glass, these scattering phenomena are defined by the form factor, which describes particle scattering according to Mie theory, and the structure factor, accounting for the constructive interference of waves scattered by a particle lattice according to the Percus–Yevick equation.21,60,61 Previous theoretical calculations and experiments of scattering behavior in the photonic glass structure, where the particle refractive index is larger than that of the matrix,60 have indicated a dominant scattering peak along with a rising trend in the lower wavelength region, which is consistent with our UV-vis data (Fig. 1d).
Solvent-dependent concentration-driven assembly pathway of the BBCP
In this section, we elucidate the solvent-dependent assembly pathway of BBCPs to unravel the underlying mechanism of morphology transition dictated by solvent selectivity. Based on our previous reports,26,27,53 we hypothesize that the BBCP assembly during drop casting is driven by a concentration increase from solvent evaporation. When the evaporation rate is relatively low, the assembly essentially traverses equilibrium states at various concentrations. We characterize the equilibrium solution structure in the four solvents using solution SAXS spanning BBCP concentrations from 0.1 mg mL−1 to 300 mg mL−1. Solution scattering is complemented by cryo-TEM to directly image the solution state assemblies as dependent on solvents. Based on these results we deduce solvent-dependent conformation of the BBCP to validate the hypothesis that the observed structural differences arise from distinct assembly pathways influenced by solvent selectivity.Solution SAXS of BBCPs from 0.1 to 300 mg mL−1 across the four solvents clearly shows the evolution of solution-state conformation and assembly, ultimately giving rise to structure factor peaks at a low q value (<0.02 Å−1) corresponding to ordered nanostructures formed in concentrated solutions (Fig. 3a–d). These findings strongly suggest that BBCP molecules assemble in their solution states, which we categorize into three regimes: diluted, disorder-to-order transition, and ordered, based on our prior work on lamella-forming solution structures.53
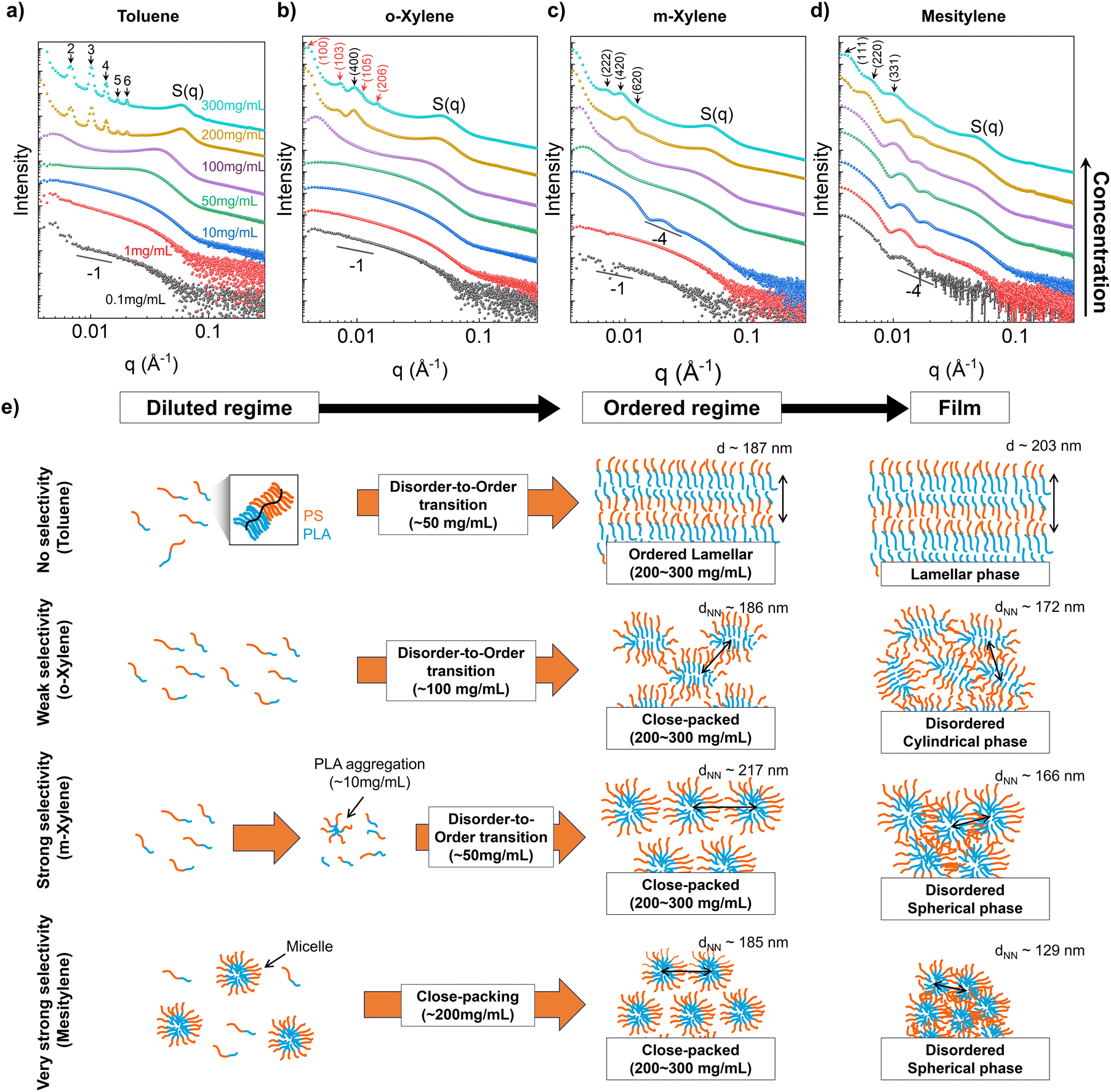 | ||
| Fig. 3 Solution SAXS data for BBCPs (Nbb = 400) in (a) toluene, (b) o-xylene, (c) m-xylene, and (d) mesitylene at various concentrations (black: 0.1, red: 1, blue: 10, green: 50, purple: 100, ocher: 200, and turquoise: 300 mg mL−1). Scattering curves are vertically shifted for clarity. S(q) denotes the structure factor peak from the ordered packing of BBCP molecules along the short axis of the backbone. Unit cell structures (red: HCP and black: FCC) are analyzed and diffraction peaks are indexed for the samples with the highest concentration in each solvent. (e) Schematic depicting the concentration-driven assembly pathways of BBCPs in four solvents. Orange lines denote the PS brush and blue lines denote the PLA brush. The domain spacing and dNN of concentrated solution were derived from the diffraction peaks in the low q range, and those of films were calculated from structural analysis in Fig. 2. | ||
In the dilute regime of a toluene solution (0.1–10 mg mL−1, Fig. 3a), the scattering curve showed the typical form factor of a rod- or cylinder-like object with a slope of −1 in the low q regime. We will further demonstrate this in a later section with curve fitting, and imaging. When the solution concentration reaches 50 mg mL−1, a structural peak (S(q)) emerges around 0.03 Å−1, corresponding to a length scale of 20.9 ± 2.1 nm. This peak is attributed to lateral chain interactions (short axis of the backbone), with the length scale corresponding to the lateral stacking distance of BBCP. At 100 mg mL−1, the S(q) becomes sharper and red shifts, indicating that more BBCP chains are laterally packed tightly due to increased crowding. Additionally, a new structural feature appears at a lower q value of around 0.005 Å−1, corresponding to a length scale of 127.7 ± 0.4 nm, which is attributed to the spacing along the long axis of BBCPs. This transition marks the beginning of the assembly of BBCPs from individual worm-like chains to more defined structures through disorder-to-order transition. As the concentration further increases over 200 mg mL−1, the scattering becomes dominated by the structure factor. Specifically, in the low-q regime, prominent structure factor peaks associated with a well-ordered lamellar structure are observed, and the estimated lamellar spacing at 300 mg mL−1 is increased to 187.2 ± 0.4 nm, while the lateral stacking distance decreases to 10.2 ± 0.8 nm. These changes at higher concentrations are explained by backbone stretching of BBCP, resulting from the increased number of block–block contacts within molecules in solution, which in turn leads to larger lamellar domain sizes and reduced lateral stacking distances.27
The scattering curves varied notably depending on the solvent used, unlike toluene, which exhibits lamellar packing, the o-xylene showed a closed-packed structure with hybrid features of hexagonal close-packed (HCP, red), and face-centered cubic (FCC, black) structures at concentrations above 200 mg mL−1 (Fig. 3b). The dNN was estimated using the peak position at the lowest q, and which increased from 139.1 ± 0.5 to 186.2 ± 0.8 nm as the concentration increased from 100 to 300 mg mL−1. In the case of m-xylene (Fig. 3c), the differences in assembly are more evident. At very low concentration below 1 mg mL−1, the scattering curve exhibited a form factor of the flexible cylinder, however, as the concentration increased beyond 10 mg mL−1, a spherical form factor (q−4) appeared, indicating an abrupt transition to spherical shapes from a cylinder. We propose that the transition occurs due to the aggregation of PLA brushes into small-particle-like aggregates near the solubility limit of PLA brushes in m-xylene (Fig. 1b), a hypothesis we will confirm through imaging techniques later. Additionally, disorder-to-order transition begins at 50 mg mL−1, where the dNN value is 139.4 ± 0.8 nm, and in the ordered regime (>200 mg mL−1), the scattering peaks predominantly correspond to an FCC structure, with a dNN value of 217.2 ± 1.1 nm at 300 mg mL−1.
As for mesitylene, the most selective solvent, the scattering curve exhibited a spherical form factor even at the lowest concentration, with no distinct dilute regime nor clear disorder-to-order transition before showing FCC close-packed structure peaks in the ordered regime (>200 mg mL−1) (Fig. 3d). The spherical form factor originates from micelle formation at low concentrations, consistent with previous findings on the high micelle stability and low critical micelle concentration of BBCP,51 which we later confirm through imaging techniques. This suggests that in a highly selective solvent, the BBCP forms micelles that pack into a close-packed structure as the concentration increases, rather than undergoing a disorder-to-order transition. The calculated dNN value at 300 mg mL−1 is 185.3 ± 0.7 nm. Additional details on peak indexing in the ordered regime (300 mg mL−1) are provided in Fig. S10 (ESI†).
Comparing the calculated domain sizes derived from solution SAXS with those from film SAXS provides insights into the evaporation-driven assembly from the ordered regime in solution to the film state. For toluene samples, the estimated lamellar domain spacing was 187.2 ± 0.4 nm for the concentrated solution (300 mg mL−1) and 203.0 ± 0.4 nm for film samples, indicating lamellar expansion. This expansion mainly arises from backbone stretching as the packing density increases, as previously reported.26,27,53 Conversely, in selective solvents, the domain spacing consistently contracts. For o-xylene, the dNN value reduced from 186.2 ± 0.8 nm for solution samples to 172.2 ± 0.4 nm for film samples; for m-xylene, the dNN values were 217.2 ± 1.1 nm for solution samples and 165.8 ± 0.5 nm for film samples; similarly, for mesitylene, the dNN values were 185.3 ± 0.7 nm for solution samples and 129.3 ± 0.4 nm for film samples. Upon calculating the ratio of domain size changes (Fig. S11, ESI†), it became evident that when the solvent selectivity increases, the domain size tends to decrease more from 300 mg mL−1 solution to film. This phenomenon can be comprehended by considering the interplay between backbone extension due to increased block-to-block contact; leading to an increase in domain size with an increase in solution concentration;27 and deswelling due to solvent removal. As the solvent evaporates, the two factors compete and determine the domain size. In non-selective solvents, our prior research indicates that backbone extension outweighs deswelling, leading to domain expansion.53 In contrast, for selective solvents where PLA is already aggregated in solution, limited room for PLA extension with increasing concentrations results in the dominance of deswelling, causing a reduction in dNN. We also observed that solution SAXS generally exhibits sharper and more prominent peaks compared to film SAXS, which we attributed to greater structural disorder induced by solvent evaporation. This increased disorder arises from spatial constraints during drying, compressing molecular structures and resulting in highly disordered arrangements, as further supported by the UV-vis reflection spectral comparison between the solution and film (Fig. S12, ESI†). The proposed overall assembly pathways from solution to the film are summarized in Fig. 3e.
To validate the proposed solution state aggregation and infer the molecular conformation in the dilute regime, we employ cryo-TEM to directly image down to the molecular level at a concentration of 10 mg mL−1 (Fig. 4a, and Fig. S13, ESI†). In the case of toluene and o-xylene, we observed individual chains without any discernible aggregation as expected. In contrast, for m-xylene, we observed single chains as well as spherical aggregates of 43.2 ± 5.1 nm in diameter consistent with SAXS. In mesitylene, we observed micelles with a core diameter of 66.2 ± 8.1 nm and a shell of 33.3 ± 6.8 nm in thickness coexistent with individual chains. Notably, the aggregate size in the m-xylene solution was smaller than the size of the spherical domain observed in SEM images of dried films. In contrast, the micelle size in the mesitylene solution closely matched that of the dried films (Fig. 2a). This suggests that in m-xylene, small aggregates grow into larger micelles during the disorder-to-order transition, while in mesitylene, the micelles remain stable and simply pack together due to their high stability. These observations further support the assembly pathway proposed in Fig. 3.
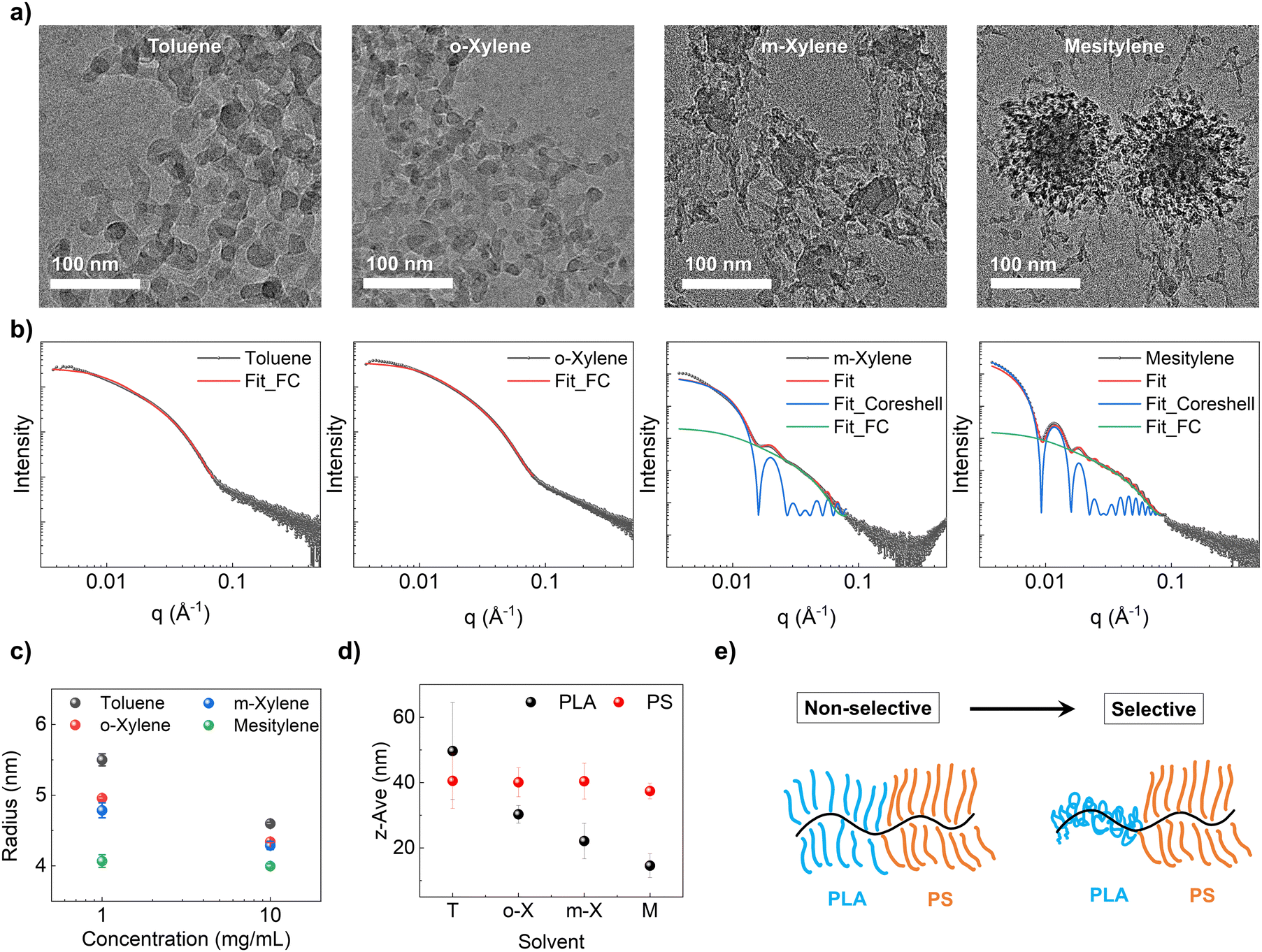 | ||
| Fig. 4 (a) Cryo-TEM images of the BBCP in four different solvents (toluene, o-xylene, m-xylene, and mesitylene) at a concentration of 10 mg mL−1. (b) SAXS fitting curves of 10 mg mL−1 solutions (black dots). For toluene and o-xylene, the flexible cylinder (FC) model was used for fitting (red curve). For m-xylene and mesitylene, a hybrid model comprising the core–shell model (blue curve) and flexible cylinder model (green curve) was employed for fitting. The red curves represent the overall fitting curve. (c) Fitted BBCP radius in different solvents (toluene: black, o-xylene: red, m-xylene: blue, and mesitylene: green) at solution concentrations of 1 and 10 mg mL−1. (d) The measured z-average size of PLA (Nbb = 200 and Nsc = 60, shown in black) and PS (Nbb = 200 and Nsc = 45, shown in red) homo bottlebrush polymers with different solvents in DLS experiments. (e) Schematic depicting the conformation of BBCPs in each solvent. | ||
Due to limitations in image resolution attributed to the susceptibility of BBCP materials to e-beam damage, we did not pursue quantitative analyses of single chain conformation through cryo-TEM. Instead, we resorted to solution SAXS, and selected fitting models based on cryo-TEM observations to analyze polymer conformation (Fig. 4b and Fig. S13, ESI†). Specifically, we chose the flexible cylinder model to fit wormlike chains observed in toluene and o-xylene. For m-xylene and mesitylene, we employed a hybrid model involving both flexible cylinder and core–shell models to capture the co-existence of both wormlike chains and micelles with a PLA core and PS shell. In each fitting for the flexible cylinder model, we fixed the contour length at 2480 nm estimated by multiplying the backbone degree of polymerization by the length of the norbonyl unit (∼0.62 nm).62 As the estimated contour length significantly exceeded the detectable range in SAXS (low-q limit), and the presence of core–shell contributions made it challenging to analyze below the q-range of 0.01 Å−1 (specifically for m-xylene and mesitylene), the precise determination of Kuhn length became challenging. Therefore, we only focused on fitting for the radius associated with the Guinier knee discernible within the 0.01–0.1 Å−1 range. Full details about fitting results are available in the ESI,† Fig. S14.
At 1 mg mL−1, the calculated radius was 5.5 ± 0.9, 5.0 ± 0.2, 4.8 ± 1.1, and 4.1 ± 0.9 nm for toluene, o-xylene, m-xylene, and mesitylene, respectively. At 10 mg mL−1, the radius was calculated as 4.6 ± 0.2, 4.3 ± 0.1, 4.3 ± 0.6, and 4.0 ± 0.3 nm for each solvent (Fig. 4c). Scattering curves at 0.1 mg mL−1 were excluded for fitting due to weak signal to noise. The calculated radius of single molecules showed a decreasing trend as selectivity increased, and it also decreased as the concentration went higher. These fitting results were cross-verified using Guinier–Porod model fitting, yielding similar trends in the radius of gyration (Rg) fitted in the 0.01–0.1 Å−1 range (Fig. S15, ESI†). This is further supported by DLS experiments, where the measured z-average size of the PLA homo bottlebrush polymer decreased from 49.7 ± 14.8 nm (toluene) to 14.6 ± 3.7 nm (mesitylene) as selectivity increased, while the size of the PS homo bottlebrush polymer remained relatively constant at around 40 nm (Fig. 4d). This trend is consistent with our hypothesis that reducing PLA solubility going from toluene to mesitylene leads to the collapse of the PLA block and thus reduced volume fraction of the PLA block and overall smaller radius of the wormlike cylinder (Fig. 4e). The inferred PLA block conformation change dependent on solvent selectivity ultimately leads to different assembly pathways, resulting in distinct film morphologies and structural colors.
Conclusion
In summary, this study showcases the ability to significantly modulate the nanoscale morphology and optical properties of PS-PLA bottlebrush block copolymers (BBCPs) by leveraging the differential solubility of their constituent blocks in structurally similar organic solvents. By using a series of solvents, toluene, o-xylene, m-xylene, and mesitylene, with varying selectivities for the PS and PLA blocks, we achieved a diverse range of morphologies from lamellar to spherical, corresponding to a broad spectrum of structural colors from orange to deep blue. Extensive structural analysis in both solution and solid phases revealed that the assembly pathways vary significantly with solvent selectivity. In less-selective solvents such as toluene and o-xylene, BBCP molecules undergo disorder-to-order transition, resulting in lamellar or cylindrical close-packed structures. In contrast, more selective solvents like m-xylene and mesitylene promote the formation of micelles or aggregates in dilute solutions, resulting in spherical close-packed structures. The observed differences in assembly pathways are attributed to the differential volume fraction of BBCPs in solution, driven by sidechain aggregation, which promotes various phase separations beyond the lamellar structure. The resulting structures exhibit distinct structural colors, which can be finely tuned over a wide range by blending different solvents or incorporating broadband absorbers. These findings underscore the critical role of solvent selectivity in shaping block copolymer morphology.Materials and methods
Material synthesis and characterization
All reactions were performed in an argon-filled glovebox (O2 < 0.5 ppm, H2O < 0.5 ppm) at room temperature using oven-dried glassware. THF was dried using a commercial solvent purification system. rac-Lactide (Aldrich), sec-butyllithium solution (sec-BuLi, 1.3 mol L−1 in cyclohexane/hexane (92/8), ACROS Organics), ethylene oxide solution (2.5–3.3 mol L−1 in THF, Aldrich) was used as received. 1,8-Diazabicyclo [5.4.0] undec-7-ene (DBU) (Aldrich) was distilled over CaH2 and storage under argon at −20 °C. Styrene was passed through a basic alumina plug and stored under argon at −20 °C. [(H2IMes)(3-Brpy)2(Cl)2RuCHPh], G3 was synthesized according to literature.63Exo-5-norbornene-2-carboxylic acid, endo-/exo-5-norbornene-2-methanol (M3OH) and exo-5-norbornene-2-carbonyl chloride was synthesized according to literature.64,65 Gel permeation chromatography (GPC) was performed using a Tosoh ECOSEC HLC-8320GPC at 40 °C fitted with a guard column (6.0 mm ID × 4.0 cm) and two analytical columns (TSKgel GMHHR-H, 7.8 mm ID × 30 cm × 5 μm). A flow rate of 1 mL-min−1 was used for both the analytical columns and the reference flow. THF (HPLC grade) was used as the eluent, and polystyrene standards (15 points ranging from 500 MW to 8.42 million MW) were used as the general calibration. UV detector was recorded at 266 nm. The detailed synthesis procedure is provided in the “Material synthesis” section of the ESI.†
Preparation of BBCP drop-cast samples
Samples for optical and structural characteristic analysis were prepared using the drop-casting method. Initially, stock solutions of BBCPs were prepared by direct dissolution in solvents at a concentration of 150 mg mL−1, and stirred for a minimum of 48 hours at 40 °C before being allowed to settle for several hours at room temperature prior to drop-casting. Glass substrates were rinsed in toluene, acetone, and isopropanol before being dried using a nitrogen gun. Using a pipette, a 10-microliter drop of the solution was placed onto the substrate and allowed to air dry at room temperature. Upon complete solvent evaporation, the samples underwent annealing for 4 hours at 60 °C.Gravimetric solubility measurement of homobrush polymer solution
An increasing amount of homobrush polymer powder was added to 1 mL of solvent in a vial until the solution became saturated, indicated by the presence of undissolved powder (solution turning opaque). The solution was then mildly centrifuged at 500 rpm for 5 minutes and allowed to stand at room temperature on the benchtop for 24 hours. Carefully pipette 100 microliters of the clear supernatant from the top of the solution and transfer it into a pre-weighed vial. After complete evaporation of the solvent, measure the mass of the remaining homobrush polymer in the vial to calculate the solubility of the polymer in the solution. This procedure was repeated five times, and the average solubility value was calculated.Characterization of the optical and structural properties of BBCP
Optical microscopy images were captured using a top-mounted optical microscope at low magnification (1.6×) under diffuse (ring) light. UV-Vis diffuse reflection spectra were acquired using a Varian Cary 5G spectrophotometer equipped with an integrating sphere attachment at the Illinois Material Research Laboratory (MRL). DLS was performed using the Malvern Zetasizer at MRL, Illinois, to determine the harmonic intensity-weighted average particle diameter (Z-average size). For the measurements, 1 mg mL−1 solutions of PLA and PS homobrush polymers were prepared. To isolate individual bottlebrush block copolymer chains, a mild centrifugation step (1000 rpm, for 10 min) was applied before the measurements, and the supernatant was used for analysis. SEM imaging was conducted using a Hitachi S4800 instrument at the Illinois MRL. The sample was prepared on cleaned silicon substrates. To explore the sample's vertical orientation, prints on the silicon substrate were precisely cut perpendicular to the by inducing controlled crack propagation (initiated with a diamond glass scriber). Subsequently, the sample was affixed to a 90-degree angled SEM pin stub, and micrographs were acquired using a low accelerating voltage (3–5 keV) with a beam current ranging from approximately 10 to 20 nano-amperes. The acquired images underwent image-processing using the ImageJ software package to calculate the domain spacing. PiFM imaging was performed using a Molecular Vista PiFM-Raman microscope at the Illinois MRL, and the samples for PiFM imaging were prepared on a silicon substrate using the same method employed for optical characterization. PiFM images of PLA and PS rich domains were acquired at 1 line per s over a 1 × 1 μm area with 256 × 256 pixels. Herein, the infrared signal of each bottlebrush molecule was obtained using an FT-IR analyzer (Alpha, Bruker). The cryo-TEM experiment was carried out in MRL Illinois. Samples were frozen using 200 mesh holey carbon copper grids (SPI Supplies, 3620C-MB) using an FEI MarkIV Vitrobot. For each grid, 3 μL of sample was applied to the grid at 22 °C and 90–95% ambient temperature and humidity, respectively. Grids were blotted once for 3 seconds with a blot force of 0–2 and plunged rapidly into liquid ethane for freezing. Once frozen, grids were clipped into autogrids and imaged using a ThermoFisher Glacios CryoTEM at 200 kV using EPU and Velox software.Small-angle X-ray scattering (SAXS)
Films and solutions examined through transmission small-angle X-ray scattering were analyzed at Argonne National Laboratory (Lemont, IL) on beamline 12-ID-B of the Advanced Photon Source, utilizing a beam energy of 13.3 keV and a Pilatus 2M 2D detector. Film samples were prepared by drop-casting the sample, as described previously, with solvent-washed polyimide sheets (Kapton – American Durafilm) used as the substrate. Solution samples were prepared using the same methods described previously.53 The prepared solutions were sequentially loaded and irradiated from the lowest to the highest concentration, starting with the pure solvent. This was achieved using a single 1 mm quartz capillary (Charles Supper Company)/Teflon tubing flow cell for each polymer. In cases where the solution exhibited high viscosity due to a high concentration, the solution was loaded into the capillary by applying gentle centrifugal force using a centrifuge. Curve fitting was carried out using the SasView software package.Author contributions
The manuscript was written through contributions from all authors. All synthetic procedures and chemical characterization were carried out by Y. K. with guidance from D. G. Scattering experiments and analysis were led by S. J. with assistance from A. K. and Z. X. and guidance from Y. D. Optical property measurements were performed by S. J., with assistance from C. H. and J.-H. L. under the guidance of Y. D. Structural property analysis and theoretical studies were performed by S. J. with support from J. S. and S. A. R., under the guidance of Y. D. The initial draft of the manuscript was prepared by S. J. and revised with contributions from all authors. All authors have given approval to the final version of the manuscript.Data availability
All relevant data supporting the findings of this study are included in the manuscript and its ESI.† Raw data are available from the authors upon request.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors would like to thank H. Kang, and C. E. Sing of the University of Illinois at Urbana-Champaign and B. Lee at the Argonne National Lab for intellectual discussions regarding this work. This work was supported by the NSF under DMREF award no. DMR-2119172. Experiments were carried out, in part, in the MRL Central Research Facilities, University of Illinois. This work was also supported by a PPG-MRL Graduate Research Assistantship. This research used resources of the Advanced Photon Source; a U.S. Department of Energy (DOE) Office of Science User Facility under contract no. DE-AC02-04CH11231 operated for the DOE Office of Science by the Argonne National Laboratory. This work benefited from the use of the SasView application.References
- A. Gürses; K. Güneş and E. Şahin, Removal of Dyes and Pigments from Industrial Effluents, Green Chemistry and Water Remediation: Research and Applications, Elsevier, 2020, pp. 135–187 DOI:10.1016/B978-0-12-817742-6.00005-0.
- Y. Zhao, Z. Xie, H. Gu, C. Zhu and Z. Gu, Bio-Inspired Variable Structural Color Materials, Chem. Soc. Rev., 2012, 41(8), 3297–3317, 10.1039/c2cs15267c.
- L. Shang, W. Zhang, K. Xu and Y. Zhao, Bio-Inspired Intelligent Structural Color Materials, Mater. Horiz., 2019, 6(5), 945–958, 10.1039/c9mh00101h.
- J. Teyssier, S. V. Saenko, D. Van Der Marel and M. C. Milinkovitch, Photonic Crystals Cause Active Colour Change in Chameleons, Nat. Commun., 2015, 6, DOI:10.1038/ncomms7368.
- Y. Fu, C. A. Tippets, E. U. Donev and R. Lopez, Structural Colors: From Natural to Artificial Systems, Wiley Interdisciplinary Reviews: Nanomedicine and Nanobiotechnology, Wiley-Blackwell, 2016, pp. 758–775 DOI:10.1002/wnan.1396.
- J. Sun, B. Bhushan and J. Tong, Structural Coloration in Nature, RSC Adv., 2013, 14862–14889, 10.1039/c3ra41096j.
- H. Butt, A. K. Yetisen, D. Mistry, S. A. Khan, M. U. Hassan and S. H. Yun, Morpho Butterfly-Inspired Nanostructures, Adv. Opt. Mater., 2016, 4(4), 497–504, DOI:10.1002/adom.201500658.
- P. Vukusic and J. R. Sambles, Photonic Structures in Biology, Nature, 2003, 424(6950), 852–855, DOI:10.1038/nature01941.
- J. Zi, X. Yu, Y. Li, X. Hu, C. Xu, X. Wang, X. Liu, R. Fu and Y. R. Shen, Coloration Strategies in Peacock Feathers, Proc. Natl. Acad. Sci. U. S. A., 2003, 100(22), 12576–12578 CrossRef CAS PubMed.
- B. Winter, B. Butz, C. Dieker, G. E. Schröder-Turk, K. Mecke and E. Spiecker, Coexistence of Both Gyroid Chiralities in Individual Butterfly Wing Scales of Callophrys rubi, Proc. Natl. Acad. Sci. U. S. A., 2015, 112(42), 12911–12916, DOI:10.1073/pnas.1511354112.
- S. Kinoshita, S. Yoshioka and K. Kawagoe, Mechanisms of Structural Colour in the Morpho Butterfly: Cooperation of Regularity and Irregularity in an Iridescent Scale, Proc. R. Soc. B, 2002, 269(1499), 1417–1421, DOI:10.1098/rspb.2002.2019.
- R. O. Prum and R. Torres, Structural Colouration of Avian Skin: Convergent Evolution of Coherently Scattering Dermal Collagen Arrays, J. Exp. Biol., 2003, 206(14), 2409–2429, DOI:10.1242/jeb.00431.
- R. O. Prum and R. H. Torres, Structural Colouration of Mammalian Skin: Convergent Evolution of Coherently Scattering Dermal Collagen Arrays, J. Exp. Biol., 2004, 207(12), 2157–2172, DOI:10.1242/jeb.00989.
- R. O. Prum, J. A. Cole and R. H. Torres, Blue Integumentary Structural Colours in Dragonflies (Odonata) Are Not Produced by Incoherent Tyndall Scattering, J. Exp. Biol., 2004, 207(22), 3999–4009, DOI:10.1242/jeb.01240.
- B. Q. Dong, X. H. Liu, T. R. Zhan, L. P. Jiang, H. W. Yin, F. Liu and J. Zi, Structural coloration and photonic pseudogap in natural random close-packing photonic structures, Optical Express, 2010, 18, 14430–14438 CrossRef CAS PubMed.
- Y. Liu, H. Wang, J. Ho, R. C. Ng, R. J. H. Ng, V. H. Hall-Chen, E. H. H. Koay, Z. Dong, H. Liu, C. W. Qiu, J. R. Greer and J. K. W. Yang, Structural Color Three-Dimensional Printing by Shrinking Photonic Crystals, Nat. Commun., 2019, 10, 4340, DOI:10.1038/s41467-019-12360-w.
- G. Zyla, A. Kovalev, M. Grafen, E. L. Gurevich, C. Esen, A. Ostendorf and S. Gorb, Generation of Bioinspired Structural Colors via Two-Photon Polymerization, Sci. Rep., 2017, 7, 17622, DOI:10.1038/s41598-017-17914-w.
- H. Gu, X. Liu, Z. Mu, Q. Wang, H. Ding, X. Du and Z. Gu, Wide-Gamut Biomimetic Structural Colors from Interference-Assisted Two-Photon Polymerization, ACS Appl. Mater. Interfaces, 2021, 13(50), 60648–60659, DOI:10.1021/acsami.1c18604.
- S. O’Halloran, A. Pandit, A. Heise and A. Kellett, Two-Photon Polymerization: Fundamentals, Materials, and Chemical Modification Strategies, Adv. Sci., 2023, 10, 2204072, DOI:10.1002/advs.202204072.
- Y. Zhang, L. Zhang, C. Zhang, J. Wang, J. Liu, C. Ye, Z. Dong, L. Wu and Y. Song, Continuous Resin Refilling and Hydrogen Bond Synergistically Assisted 3D Structural Color Printing, Nat. Commun., 2022, 13, 7095, DOI:10.1038/s41467-022-34866-6.
- A. F. Demirörs, E. Poloni, M. Chiesa, F. L. Bargardi, M. R. Binelli, W. Woigk, L. D. C. de Castro, N. Kleger, F. B. Coulter, A. Sicher, H. Galinski, F. Scheffold and A. R. Studart, Three-Dimensional Printing of Photonic Colloidal Glasses into Objects with Isotropic Structural Color, Nat. Commun., 2022, 13, 4397, DOI:10.1038/s41467-022-32060-2.
- Z. Zhang, C. Wang, Q. Wang, Y. Zhao and L. Shang, Cholesteric Cellulose Liquid Crystal Ink for Three-Dimensional Structural Coloration, Proc. Natl. Acad. Sci. U. S. A., 2022, 119(23), 2204113119, DOI:10.1073/pnas.2204113119.
- J. A. H. P. Sol, H. Sentjens, L. Yang, N. Grossiord, A. P. H. J. Schenning and M. G. Debije, Anisotropic Iridescence and Polarization Patterns in a Direct Ink Written Chiral Photonic Polymer, Adv. Mater., 2021, 33, 2103309, DOI:10.1002/adma.202103309.
- K. George, M. Esmaeili, J. I. Wang, N. Taheri-Qazvini, A. I. Abbaspourrad and M. Sadati, 3D Printing of Responsive Chiral Photonic Nanostructures, Proc. Natl. Acad. Sci. U. S. A., 2023, 120(12), e2220032120, DOI:10.1073/pnas.2220032120.
- C. L. C. Chan, I. M. Lei, G. T. van de Kerkhof, R. M. Parker, K. D. Richards, R. C. Evans, Y. Y. S. Huang and S. Vignolini, 3D Printing of Liquid Crystalline Hydroxypropyl Cellulose—toward Tunable and Sustainable Volumetric Photonic Structures, Adv. Funct. Mater., 2022, 32, 2108566, DOI:10.1002/adfm.202108566.
- B. B. Patel, D. J. Walsh, D. H. Kim, J. Kwok, B. Lee, D. Guironnet and Y. Diao, Tunable Structural Color of Bottlebrush Block Copolymers through Direct-Write 3D Printing from Solution, Sci. Adv., 2020, 6, 7202–7212, DOI:10.1126/sciadv.aaz7202.
- S. Jeon, Y. L. Kamble, H. Kang, J. Shi, M. A. Wade, B. B. Patel, T. Pan, S. A. Rogers, C. E. Sing, D. Guironnet and Y. Diao, Direct-Ink-Write Cross-Linkable Bottlebrush Block Copolymers for on-the-Fly Control of Structural Color, Proc. Natl. Acad. Sci. U. S. A., 2024, 121(9), e2313617121, DOI:10.1073/pnas.2313617121.
- B. M. Boyle, T. A. French, R. M. Pearson, B. G. McCarthy and G. M. Miyake, Structural Color for Additive Manufacturing: 3D-Printed Photonic Crystals from Block Copolymers, ACS Nano, 2017, 11(3), 3052–3058, DOI:10.1021/acsnano.7b00032.
- Y. Mai and A. Eisenberg, Self-Assembly of Block Copolymers, Chem. Soc. Rev., 2012, 41(18), 5969–5985, 10.1039/c2cs35115c.
- M. Stefik, S. Guldin, S. Vignolini, U. Wiesner and U. Steiner, Block Copolymer Self-Assembly for Nanophotonics, Chem. Soc. Rev., 2015, 44(15), 5076–5091, 10.1039/c4cs00517a.
- F. S. Bates and G. H. Fredrickson, Block Copolymer Thermodynamics: Theory and Experiment, Annu. Rev. Phys. Chem., 1990, 41, 525–557, DOI:10.1146/annurev.pc.41.100190.002521.
- A. L. Liberman-Martin, C. K. Chu and R. H. Grubbs, Application of Bottlebrush Block Copolymers as Photonic Crystals, Macromol. Rapid Commun., 2017, 38, 1700058, DOI:10.1002/marc.201700058.
- D. P. Song, G. Jacucci, F. Dundar, A. Naik, H. F. Fei, S. Vignolini and J. J. Watkins, Photonic Resins: Designing Optical Appearance via Block Copolymer Self-Assembly, Macromolecules, 2018, 51(6), 2395–2400, DOI:10.1021/acs.macromol.7b02288.
- T. Pan, S. Dutta and C. E. Sing, Interaction Potential for Coarse-Grained Models of Bottlebrush Polymers, J. Chem. Phys., 2022, 156, 014903, DOI:10.1063/5.0076507.
- S. Dutta, T. Pan and C. E. Sing, Bridging Simulation Length Scales of Bottlebrush Polymers Using a Wormlike Cylinder Model, Macromolecules, 2019, 52(13), 4858–4874, DOI:10.1021/acs.macromol.9b00363.
- T. Pan, B. B. Patel, D. J. Walsh, S. Dutta, D. Guironnet, Y. Diao and C. E. Sing, Implicit Side-Chain Model and Experimental Characterization of Bottlebrush Block Copolymer Solution Assembly, Macromolecules, 2021, 54(8), 3620–3633, DOI:10.1021/acs.macromol.1c00336.
- M. Hu, Y. Xia, G. B. McKenna, J. A. Kornfield and R. H. Grubbs, Linear Rheological Response of a Series of Densely Branched Brush Polymers, Macromolecules, 2011, 44(17), 6935–6943, DOI:10.1021/ma2009673.
- T. Pan, S. Dutta, Y. Kamble, B. B. Patel, M. A. Wade, S. A. Rogers, Y. Diao, D. Guironnet and C. E. Sing, Materials Design of Highly Branched Bottlebrush Polymers at the Intersection of Modeling, Synthesis, Processing, and Characterization, Chem. Mater., 2022, 34(5), 1990–2024, DOI:10.1021/acs.chemmater.1c04030.
- R. Verduzco, X. Li, S. L. Pesek and G. E. Stein, Structure, Function, Self-Assembly, and Applications of Bottlebrush Copolymers, Chem. Soc. Rev., 2015, 2405–2420, 10.1039/c4cs00329b.
- B. R. Sveinbjörnsson, R. A. Weitekamp, G. M. Miyake, Y. Xia, H. A. Atwater, R. H. Grubbs and T. J. Watson, Rapid Self-Assembly of Brush Block Copolymers to Photonic Crystals, Proc. Natl. Acad. Sci. U. S. A., 2012, 109(36), 14332–14336, DOI:10.1073/pnas.1213055109.
- R. J. Macfarlane, B. Kim, B. Lee, R. A. Weitekamp, C. M. Bates, S. F. Lee, A. B. Chang, K. T. Delaney, G. H. Fredrickson, H. A. Atwater and R. H. Grubbs, Improving Brush Polymer Infrared One-Dimensional Photonic Crystals via Linear Polymer Additives, J. Am. Chem. Soc., 2014, 136(50), 17374–17377, DOI:10.1021/ja5093562.
- D. P. Song, T. H. Zhao, G. Guidetti, S. Vignolini and R. M. Parker, Hierarchical Photonic Pigments via the Confined Self-Assembly of Bottlebrush Block Copolymers, ACS Nano, 2019, 13(2), 1764–1771, DOI:10.1021/acsnano.8b07845.
- T. H. Zhao, G. Jacucci, X. Chen, D. P. Song, S. Vignolini and R. M. Parker, Angular-Independent Photonic Pigments via the Controlled Micellization of Amphiphilic Bottlebrush Block Copolymers, Adv. Mater., 2020, 32, 2002681, DOI:10.1002/adma.202002681.
- Z. Wang, R. Li, Y. Zhang, C. L. C. Chan, J. S. Haataja, K. Yu, R. M. Parker and S. Vignolini, Tuning the Color of Photonic Glass Pigments by Thermal Annealing, Adv. Mater., 2023, 35, 2207923, DOI:10.1002/adma.202207923.
- H. F. Fei, B. M. Yavitt, X. Hu, G. Kopanati, A. Ribbe and J. J. Watkins, Influence of Molecular Architecture and Chain Flexibility on the Phase Map of Polystyrene-Block-Poly(Dimethylsiloxane) Brush Block Copolymers, Macromolecules, 2019, 52(17), 6449–6457, DOI:10.1021/acs.macromol.9b00843.
- Y. Gai, D. P. Song, B. M. Yavitt and J. J. Watkins, Polystyrene-Block-Poly(Ethylene Oxide) Bottlebrush Block Copolymer Morphology Transitions: Influence of Side Chain Length and Volume Fraction, Macromolecules, 2017, 50(4), 1503–1511, DOI:10.1021/acs.macromol.6b01415.
- S. Cui, B. Zhang, L. Shen, F. S. Bates and T. P. Lodge, Core-Shell Gyroid in ABC Bottlebrush Block Terpolymers, J. Am. Chem. Soc., 2022, 144(47), 21719–21727, DOI:10.1021/jacs.2c09674.
- R. J. Sánchez-Leija, J. A. Mysona, J. J. de Pablo and P. F. Nealey, Phase Behavior and Conformational Asymmetry near the Comb-to-Bottlebrush Transition in Linear-Brush Block Copolymers, Macromolecules, 2024, 57(5), 2019–2029, DOI:10.1021/acs.macromol.3c02180.
- K. Kawamoto, M. Zhong, K. R. Gadelrab, L. C. Cheng, C. A. Ross, A. Alexander-Katz and J. A. Johnson, Graft-through Synthesis and Assembly of Janus Bottlebrush Polymers from A-Branch-B Diblock Macromonomers, J. Am. Chem. Soc., 2016, 138(36), 11501–11504, DOI:10.1021/jacs.6b07670.
- K. E. L. Husted, A. Herzog-Arbeitman, D. Kleinschmidt, W. Zhang, Z. Sun, A. J. Fielitz, A. N. Le, M. Zhong and J. A. Johnson, Pendant Group Modifications Provide Graft Copolymer Silicones with Exceptionally Broad Thermomechanical Properties, ACS Cent. Sci., 2023, 9(1), 36–47, DOI:10.1021/acscentsci.2c01246.
- R. Fenyves, M. Schmutz, I. J. Horner, F. V. Bright and J. Rzayev, Aqueous Self-Assembly of Giant Bottlebrush Block Copolymer Surfactants as Shape-Tunable Building Blocks, J. Am. Chem. Soc., 2014, 136(21), 7762–7770, DOI:10.1021/ja503283r.
- Y. Wang, F. Shao, E. R. Sauvé, C. M. Tonge and Z. M. Hudson, Self-Assembly of Giant Bottlebrush Block Copolymer Surfactants from Luminescent Organic Electronic Materials, Soft Matter, 2019, 15(27), 5421–5430, 10.1039/c9sm00931k.
- B. B. Patel, T. Pan, Y. Chang, D. J. Walsh, J. J. Kwok, K. S. Park, K. Patel, D. Guironnet, C. E. Sing and Y. Diao, Concentration-Driven Self-Assembly of PS-b-PLA Bottlebrush Diblock Copolymers in Solution, ACS Polym. Au, 2022, 2(4), 232–244, DOI:10.1021/acspolymersau.1c00057.
- B. B. Patel, D. J. Walsh, K. Patel, D. H. Kim, J. J. Kwok, D. Guironnet and Y. Diao, Rapid, Interface-Driven Domain Orientation in Bottlebrush Diblock Copolymer Films during Thermal Annealing, Soft Matter, 2022, 18(8), 1666–1677, 10.1039/d1sm01634b.
- K. L. Gu, Y. Zhou, W. A. Morrison, K. Park, S. Park and Z. Bao, Nanoscale Domain Imaging of All-Polymer Organic Solar Cells by Photo-Induced Force Microscopy, ACS Nano, 2018, 12(2), 1473–1481, DOI:10.1021/acsnano.7b07865.
- A. Khasbaatar, A. L. Jones, P. S. Fernando, H. Sai, C. Zhu, E. Gann, J. R. Reynolds and Y. Diao, Tuning the Solution Aggregate Structure of a PM7-Based Conjugated Polymer to Enable Green Solvent Processing of Organic Solar Cells, Chem. Mater., 2024, 36(6), 2819–2834, DOI:10.1021/acs.chemmater.3c03055.
- A. C. Edrington, A. M. Urbas, P. Derege, C. X. Chen, T. M. Swager, N. Hadjichristidis, M. Xenidou, L. J. Fetters, J. D. Joannopoulos, Y. Fink and E. L. Thomas, Polymer-Based Photonic Crystals, Adv. Mater., 2001, 13(6), 421–425, DOI:10.1002/1521-4095(200103)13:6<421::AID-ADMA421>3.0.CO;2-%23.
- P. D. García, R. Sapienza and C. López, Photonic Glasses: A Step beyond White Paint, Adv. Mater., 2010, 22(1), 12–19, DOI:10.1002/adma.200900827.
- L. Schertel, L. Siedentop, J. M. Meijer, P. Keim, C. M. Aegerter, G. J. Aubry and G. Maret, The Structural Colors of Photonic Glasses, Adv. Opt. Mater., 2019, 7, 1900442, DOI:10.1002/adom.201900442.
- V. Hwang, A. B. Stephenson, S. Barkely, S. Brandt, M. Xiao, J. Aizenberg and V. N. Manoharan, Designing Angle-Independent Structural Colors Using Monte Carlo Simulations of Multiple Scattering, Proc. Natl. Acad. Sci. U. S. A., 2021, 118(4), e2015551118, DOI:10.1073/pnas.2015551118.
- J. K. Percus and G. J. Yevick, Analysis of Classical Statistical Mechanics by Means of Collective Coordinates, Phys. Rev., 1958, 110(1), 1–13 CrossRef CAS.
- S. J. Dalsin, T. G. Rions-Maehren, M. D. Beam, F. S. Bates, M. A. Hillmyer and M. W. Matsen, Bottlebrush Block Polymers: Quantitative Theory and Experiments, ACS Nano, 2015, 9(12), 12233–12245, DOI:10.1021/acsnano.5b05473.
- J. A. Love, J. P. Morgan, T. M. Trnka and R. H. Grubbs, A Practical and Highly Active Ruthenium-Based Catalyst That Effects the Cross Metathesis of Acrylonitrile, Angew. Chem., Int. Ed., 2002, 41(21), 4035–4037, DOI:10.1002/1521-3773(20021104)41:21<4035::aid-anie4035>3.0.co;2-i.
- S. C. Radzinski, J. C. Foster, R. C. Chapleski, D. Troya and J. B. Matson, Bottlebrush Polymer Synthesis by Ring-Opening Metathesis Polymerization: The Significance of the Anchor Group, J. Am. Chem. Soc., 2016, 138(22), 6998–7004, DOI:10.1021/jacs.5b13317.
- M. A. Wade, D. Walsh, J. C. W. Lee, E. Kelley, K. Weigandt, D. Guironnet and S. A. Rogers, Color, Structure, and Rheology of a Diblock Bottlebrush Copolymer Solution, Soft Matter, 2020, 16(21), 4919–4931, 10.1039/d0sm00397b.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sm01495b |
| This journal is © The Royal Society of Chemistry 2025 |