
Probing nanopores: molecular dynamics insights into the mechanisms of DNA and protein translocation through solid-state and biological nanopores
Yuanshuo
Zhang
a and
Mingming
Ding
 *ab
*ab
aSchool of Chemical Engineering and Light Industry, Guangdong University of Technology, Guangzhou 510006, P. R. China. E-mail: mmding@gdut.edu.cn
bJieyang Branch of Chemistry and Chemical Engineering Guangdong Laboratory, Jieyang 515200, P. R. China
First published on 11th March 2025
Abstract
Nanopore sequencing technology has revolutionized single-molecule analysis through its unique capability to detect and characterize individual biomolecules with unprecedented precision. This perspective provides a comprehensive analysis of molecular dynamics (MD) simulations in nanopore research, with particular emphasis on comparing molecular transport mechanisms between biological and solid-state platforms. We first examine how MD simulations at atomic resolution reveal distinct characteristics: biological nanopores exhibit sophisticated molecular recognition through specific amino acid interactions, while solid-state nanopores demonstrate advantages in structural stability and geometric control. Through detailed analysis of simulation methodologies and their applications, we show how computational approaches have advanced our understanding of critical phenomena such as ion selectivity, conformational dynamics, and surface effects in both nanopore types. Despite computational challenges including limited simulation timescales and force field accuracy constraints, recent advances in high-performance computing and artificial intelligence integration have significantly improved simulation capabilities. By synthesizing perspectives from physics, chemistry, biology, and computational science, this perspective provides both theoretical insights and practical guidelines for developing next-generation nanopore platforms. The integration of computational and experimental approaches discussed here offers promising directions for advancing nanopore technology in applications ranging from DNA/RNA sequencing and protein post-translational modification analysis to disease diagnosis and drug screening.
1 Introduction
Nanopore technology has emerged as a revolutionary single-molecule analytical platform, offering unprecedented capabilities in biomolecular detection and characterization. The fundamental principle involves the electrophoretic translocation of charged molecules through nanoscale pores under an applied voltage, where the passage of molecules generates characteristic ionic current blockades that serve as molecular signatures (Fig. 1(a)).1–3 The foundation of this field was established by the pioneering work of Kasianowicz et al., who first demonstrated the electrophoretic translocation of single-stranded RNA/DNA through α-hemolysin (α-HL) nanopores and proposed the principle of polymer length measurement using current blockade duration.4 Early investigations by Meller et al. systematically characterized DNA translocation properties and voltage-dependent transport dynamics,5–7 while Akeson et al. demonstrated the microsecond-level discrimination capability of RNA homopolymers.8 Over the past three decades, this technology has evolved remarkably, as comprehensively reviewed by Deamer et al.9 This label-free detection method has demonstrated remarkable versatility in various applications, from nucleic acid sequencing to protein analysis. Its exceptional sensitivity enables detection of subtle structural variations at the single-molecule level, discriminating between different nucleotides (Fig. 1(b)) and amino acids (Fig. 1(c)).10–14 | ||
| Fig. 1 Schematic illustration of nanopore sequencing technology and its applications. (a) Basic principle of nanopore sensing showing the translocation of charged molecules through a nanopore under applied voltage, generating characteristic ionic current blockades that serve as molecular signatures. Created in BioRender. Y. Zhang (2024), https://BioRender.com/j42j080. (b) DNA homopolymer discrimination capabilities. Representative ionic current traces showing DNA homopolymer translocation through COF-1.1 nanopores at 300 mV bias voltage, where the black line indicates the threshold for translocation events.15 Copyright 2022 American Chemical Society. (c) Amino acid discrimination capabilities. Representative ionic current traces showing distinct blockade patterns for different amino acids, demonstrating characteristic molecular signatures for each analyte.14 Copyright 2020 Royal Society of Chemistry. (d) Structural comparison of three widely used biological nanopores. Top: Side view of α-HL, MspA, and CsgG. Bottom: Top view showing the distinct pore geometries and constriction sites (marked with red arrows) with their respective diameters. Created in BioRender. Y. Zhang (2024), https://BioRender.com/j42j080. (e) Representative solid-state nanopore materials. Atomic structures of various 2D materials used for nanopore fabrication, including graphene, h-BN, MoS2, WS2, and MXenes, showing their distinct atomic compositions and pore configurations.16 Copyright 2021 American Chemical Society. | ||
Biological nanopores, derived from transmembrane protein channels, constitute a fundamental category in nanopore technology, with their molecular transport properties refined through millions of years of evolutionary optimization. These sophisticated protein-based channels, exemplified by α-HL from Staphylococcus aureus,4,17Mycobacterium smegmatis porin A (MspA),18,19 and curli specific gene G (CsgG),20,21 exhibit precisely orchestrated molecular architectures that facilitate selective transport across cellular membranes (Fig. 1(d)). Their intricate structural organization, characterized by strategically positioned amino acid residues and specifically engineered binding sites, enables exceptional molecular recognition capabilities at the atomic scale. This inherent molecular specificity has proven particularly advantageous in DNA sequencing applications, where biological nanopores demonstrate remarkable precision in discriminating between individual nucleotides through distinct ionic current signatures.4,22,23 However, biological nanopores face several critical experimental challenges that limit their broader application.24,25 First, these protein-based channels exhibit limited stability under non-physiological conditions, often resulting in protein denaturation. Second, structural modifications aimed at optimizing pore functionality frequently compromise protein stability, as evidenced by mutations in the α-HL constriction region.26 Third, the scalability of biological nanopore production remains problematic due to low yields of functional channels and challenges in membrane integration. Additionally, conformational fluctuations during measurements and batch-to-batch variations pose significant obstacles to achieving reproducible and standardized sensing platforms.
Solid-state nanopores, fabricated in synthetic materials such as SiO2, Si3N4, or two-dimensional materials (Fig. 1(e)), have emerged as a promising alternative platform that addresses several inherent limitations of biological nanopores.27,28 These synthetic architectures exhibit superior chemical and mechanical stability, enabling sustained operation under extreme conditions including high temperatures (>100 °C), broad pH ranges (2–12), and diverse organic solvents.22,29 Their fabrication process, primarily based on electron beam lithography and controlled dielectric breakdown, enables precise engineering of pore geometry (1–100 nm in diameter) and surface properties, facilitating customization for specific applications.30 Recent advances in two-dimensional materials, particularly graphene and MoS2, have revolutionized the field by enabling the creation of atomically thin nanopores with enhanced sensitivity for molecular detection.31–35 However, solid-state nanopores face several critical experimental challenges that require careful consideration. The primary limitation lies in their inability to achieve the molecular specificity exhibited by biological counterparts, resulting in rapid and poorly controlled molecular translocation events (typically 10–100 times faster than biological nanopores) that compromise signal resolution.36,37 Surface charge heterogeneity and mechanical deformation during fabrication often lead to asymmetric ion transport and variable sensing performance. Additionally, these synthetic platforms encounter challenges in reproducible nanopore formation, with electron beam-induced damage and uncontrolled pore expansion during operation affecting long-term stability.38 The integration of surface modifications to enhance molecular specificity frequently results in reduced pore diameter and irregular surface chemistry.39
Molecular dynamics (MD) simulation, a sophisticated computational methodology that enables the modeling and prediction of complex molecular systems at atomic resolution and femtosecond timescales, has emerged as an indispensable tool in addressing the experimental challenges encountered in nanopore technology. By numerically solving Newton's equations of motion for interacting particles, MD simulations provide detailed trajectories of atomic movements and molecular interactions, offering unprecedented insights into nanoscale phenomena. In response to the experimental challenges in both biological and solid-state nanopore systems, these computational approaches provide atomic-resolution insights otherwise inaccessible through experimental methods alone. For biological nanopores, MD simulations enable the systematic investigation of protein stability under various environmental conditions,40,41 allowing researchers to predict structural changes and optimize operating parameters without extensive experimental trials. These simulations can also guide the rational design of protein mutations by evaluating their impact on pore stability and function before experimental implementation, significantly reducing the time and resources required for optimization.42,43 In the context of solid-state nanopores, MD simulations offer precise control over pore geometry and surface properties in solid-state nanopore, facilitating the optimization of design parameters to achieve desired molecular specificity and controlled translocation dynamics.44–46 The computational approach provides detailed mechanistic understanding of rapid translocation events (at femtosecond resolution) and surface charge effects, enabling the development of strategies to regulate molecular transport. Furthermore, MD simulations allow for direct visualization and quantitative analysis of ion transport mechanisms, protein-pore interactions, and conformational dynamics during molecular translocation, providing crucial insights for improving both biological and solid-state nanopore designs. The integration of these computational insights with experimental approaches has led to significant advances in addressing the fundamental challenges of nanopore technology, particularly in optimizing molecular recognition, controlling translocation dynamics, and enhancing signal resolution.
This perspective provides a comprehensive analysis of MD simulations in biological and solid-state nanopore systems, following a systematic progression through interconnected sections. Beginning with MD simulation fundamentals, we detail the theoretical frameworks and computational methodologies essential for modern molecular simulations. Through extensive analysis of MD applications, we examine the distinct characteristics and operational mechanisms of both nanopore types, with particular emphasis on their molecular transport properties. Throughout these sections, we highlight how MD simulations have advanced our understanding of critical phenomena such as ion selectivity, conformational dynamics, and surface effects, while demonstrating how computational predictions guide experimental optimization of nanopore design and operation. Recent technological advances, including deep learning approaches for automated signal analysis and hybrid simulation methods combining quantum and classical mechanics, are expanding the capabilities of computational modeling in nanopore research. By examining the complementary advantages and limitations of these two nanopore categories through integrated computational-experimental approaches, we offer insights into their optimization and future development, particularly addressing key challenges such as limited simulation timescales, force field accuracy, and the need for improved molecular recognition in solid-state platforms. This computational perspective proves crucial for advancing nanopore technology, especially in enhancing molecular specificity, controlling translocation dynamics, and improving sequencing accuracy.
2 Molecular dynamics fundamentals
The investigation of molecular transport mechanisms in biological and solid-state nanopores requires computational methods capable of capturing both atomic-level interactions and system-wide dynamics. Among various computational approaches, MD simulation has emerged as a particularly powerful tool for comparing and analyzing these two distinct nanopore categories. The complete workflow of MD simulations, from initial structure preparation through force field parameterization to trajectory analysis and data processing, follows a systematic process (Fig. 2(a)). | ||
| Fig. 2 Fundamental principles and methodologies of MD simulations. (a) Complete workflow of MD simulations illustrating the systematic process from initial structure preparation through force field parameterization to trajectory analysis and data processing. Created in BioRender. Y. Zhang (2024), https://BioRender.com/j42j080. (b) Comparison of different molecular simulation approaches across temporal and spatial scales: (i) AIMD47 Copyright 2023 Royal Society of Chemistry, (ii) QM/MM48 Copyright 2018 American Chemical Society, (iii) all-atom MD49 Copyright 2015 American Chemical Society, and (iv) CG MD50 Copyright 2016 American Chemical Society, highlighting their respective applications and limitations. (c) Schematic representation of force field components showing bonded interactions (bond length, angle, Urey–Bradley, dihedral, and improper terms) and non-bonded interactions (van der Waals and electrostatic forces). Created in BioRender. Y. Zhang (2024), https://BioRender.com/j42j080. | ||
Different simulation methodologies offer distinct advantages in nanopore research, varying in their temporal and spatial scales (Fig. 2(b)). Ab initio molecular dynamics (AIMD), combining quantum mechanical principles with MD simulations, excels in predicting electronic properties and charge distributions during nanopore interactions.51–55 Classical all-atom molecular dynamics (CMD), based on Newton's equations of motion, relies on carefully parameterized force fields, which incorporate various molecular interactions including bonded terms (bond length, angle, Urey–Bradley, dihedral, and improper terms) and non-bonded terms (van der Waals and electrostatic forces), as illustrated in Fig. 2(c). This approach enables the study of larger systems over extended timescales.56,57 The quantum mechanics/molecular mechanics (QM/MM) hybrid approach bridges the gap between quantum and classical descriptions, particularly useful for studying chemical reactions in nanopore systems, while coarse-grained (CG) approaches facilitate the simulation of even larger-scale phenomena.58–61
Beyond these methods, Monte Carlo (MC) and Langevin dynamics (LD) simulations offer complementary approaches to nanopore research. MC methods, which explore configurational space through random sampling, are particularly valuable for studying thermodynamic equilibrium properties in nanopore technology. For example, Chen et al. demonstrated through MC simulations that a rotating electric field could effectively control polynucleotide translocation kinetics, showing that at low frequencies, the translocation time becomes inversely proportional to the field frequency, which could potentially improve nanopore sequencing resolution.62 However, MC methods lack temporal evolution information and are primarily suited for static thermodynamic quantities.
LD incorporates random noise forces and viscous damping terms to approximate solvent effects through mean-field treatment, enabling accelerated simulations. In nanopore translocation studies, LD is particularly useful for investigating large-molecule diffusion processes in coarse-grained models (reaching tens of microseconds timescales). For instance, Luo et al. employed LD simulations to investigate sequence-dependent DNA translocation dynamics, revealing that translocation time exponentially decreases with the volume fraction of bases having weaker pore interactions, while also demonstrating that the waiting time patterns of individual bases could potentially enable efficient sequence detection.63 However, LD's implicit solvent treatment overlooks important effects such as ionic hydration shell screening and local charge distribution perturbations at interfaces, limiting its applicability in modeling electroosmotic flow or specific molecular recognition processes.
Compared to MC and LD, CMD offers unique advantages through its all-atom resolution and explicit integration of Newton's equations of motion, enabling simultaneous capture of ion migration, water hydrogen bond restructuring, and biomolecular conformational changes with femtosecond temporal resolution. While traditional MD faces timescale limitations, enhanced sampling techniques effectively extract critical saddle points and potential well structures from free energy landscapes. These advantages in dual spatial-temporal resolution are particularly crucial for modeling interfacial hydration and electrostatic field gradients, which directly establish structure–function relationships between nanopore material properties and translocation signals.
Among these approaches, CMD has become the predominant method for comparative studies of biological and solid-state nanopores, owing to its ability to efficiently simulate systems containing hundreds of thousands of atoms while maintaining atomic resolution. This approach has proven particularly valuable in elucidating the distinct characteristics of molecular transport in different nanopore systems, from protein conformational changes in biological nanopores to surface–molecule interactions in solid-state platforms. The widespread adoption of CMD is further supported by robust software packages such as GROMACS,64 NAMD,65 GROMOS,66 CHARMM,67 and AMBER,68 which provide comprehensive tools for simulating both types of nanopore systems.
In the following sections, we focus on CMD simulations and their applications in nanopore research, presenting a detailed overview of simulation methodologies and comparing the specific settings required for biological and solid-state nanopore systems. This analysis will highlight how CMD simulations contribute to understanding the fundamental differences in molecular transport mechanisms between these two nanopore categories.
2.1 Force field theory
CMD simulations are fundamentally grounded in Newtonian mechanics, employing numerical integration of Newton's equations of motion to elucidate the temporal evolution of molecular systems. In MD simulations, the interactions between molecules are described by potential energy functions, which, along with their associated parameter sets, constitute what is known as a force field. These force fields determine the nature and strength of interactions between particles in the system, with their parameters typically derived from both empirical data and ab initio calculations.CMD force fields comprise three main components that are essential for accurately simulating nanopore systems. The total potential energy function can be expressed as:
| U = Ubonds + Unon-bonds + Uspecial | (1) |
In nanopore sequencing technology, several CMD force fields have been widely employed, each with distinct advantages for different aspects of nanopore simulation. CHARMM has demonstrated particular strength in simulating protein-based biological nanopores and their interactions with translocating molecules.69–71 AMBER excels in modeling nucleic acid translocation through nanopores, making it valuable for DNA/RNA sequencing applications.72–74 GROMOS, with its united-atom approach, offers computational efficiency particularly beneficial for large-scale simulations of solid-state nanopore systems.75,76 OPLS shows advantages in parameterizing small molecules, which is useful for studying molecular modifications of nanopore surfaces.77,78
This diverse array of force fields provides researchers with powerful tools for investigating the distinct characteristics of biological and solid-state nanopores. The choice of force field significantly influences the accuracy and efficiency of nanopore simulations, particularly in capturing the unique aspects of molecular transport mechanisms in different nanopore systems. Understanding these differences is crucial for selecting the most appropriate force field for specific nanopore applications, whether studying protein channel dynamics in biological nanopores or surface interactions in solid-state systems.
Among these force fields, CHARMM,71 initially developed within the framework of the CHARMM software package,79 has emerged as a particularly versatile tool for nanopore simulations. Its comprehensive parameterization encompasses proteins, nucleic acids, lipids, and small organic molecules, making it especially suitable for studying complex nanopore systems. The force field's ability to accurately represent both structural and dynamic properties has proven invaluable in simulating biomolecule translocation through different types of nanopores. In biological nanopore simulations, CHARMM has demonstrated exceptional performance in modeling the conformational dynamics of protein channels such as α-HL, where it accurately captures the essential protein–substrate interactions during DNA translocation and the critical role of specific amino acid residues in molecular recognition.80–82 For instance, CHARMM simulations have revealed how charged residues in the constriction region of α-HL interact with DNA phosphate groups, influencing translocation dynamics and contributing to sequence discrimination. In solid-state nanopore applications, CHARMM effectively models surface–molecule interactions and the behavior of functionalized pore surfaces, particularly in systems with modified graphene nanopores.83,84 These simulations have provided valuable insights into how surface charge distribution and chemical modifications affect biomolecule transport and orientation during translocation, enabling the optimization of pore surface properties for enhanced molecular detection.
2.2 Simulation methods
The implementation of MD simulations in biological and solid-state nanopore systems follows distinct pathways that reflect their unique structural and functional properties. For biological nanopores, the simulation process begins with the preparation of protein structures, typically obtained from the Protein Data Bank (PDB).85 These protein structures are then carefully embedded into lipid membranes and solvated with water molecules and ions. The protein–membrane complex requires meticulous equilibration to ensure structural stability and proper protein–lipid interactions. In contrast, solid-state nanopore simulations commence with the construction of synthetic materials such as Si3N4 or graphene, followed by precise pore geometry optimization and surface modification when required.Despite their differences, both systems share a common framework of essential simulation steps. The process begins with system construction and energy minimization to eliminate unfavorable atomic contacts. This is followed by system equilibration under constant particle number, pressure, and temperature (NPT) conditions to achieve a stable configuration. Once equilibrated, production runs are conducted under applied electric fields to study molecular transport phenomena. The final step involves comprehensive trajectory analysis to extract meaningful physical quantities. Notably, biological nanopore simulations typically demand longer equilibration periods, often exceeding 10 nanoseconds, to stabilize the complex protein–membrane interface. While solid-state systems might achieve stability more rapidly, they require careful attention to surface charge distributions and boundary conditions.
The characterization of nanopore performance relies on several key physical quantities that can be computed from simulation trajectories. Ionic current, a fundamental measure of nanopore conductance, can be calculated either by directly counting ion movements across the pore.86–88 The direct counting method considers the displacement of individual ions along the pore axis over time:
 | (2) |
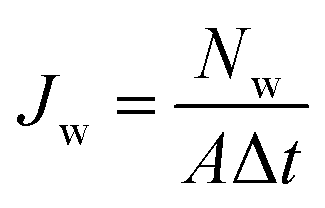 | (3) |
G(z) = −kBT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln〈P(z)〉 ln〈P(z)〉 | (4) |
 | (5) |
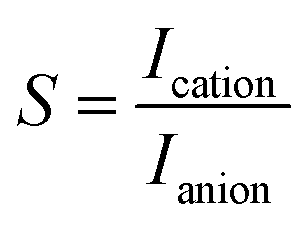 | (6) |
| τ = texit − tentrance | (7) |
The selection of specific analysis methods is often guided by the unique features of each nanopore type. Studies of biological nanopores frequently emphasize protein conformational dynamics and specific binding interactions, necessitating detailed free energy calculations and residence time analysis. For solid-state nanopores, research often focuses on surface charge effects and rapid transport phenomena, requiring careful analysis of ionic current fluctuations and water flux patterns. The combination of these analytical approaches with appropriate visualization techniques provides comprehensive insights into the distinct transport mechanisms characteristic of different nanopore systems, ultimately guiding the optimization of nanopore design for specific applications.
3 Molecular dynamics applications
3.1 Protein channel mechanisms
Biological nanopore technology has revolutionized single-molecule analysis through the strategic utilization of protein channels with distinct structural and functional characteristics (Fig. 1(d)). The most widely studied biological nanopores include α-HL, MspA, and CsgG, each offering unique advantages for specific applications. α-HL, the pioneering nanopore for DNA sequencing,4,17 features a stable β-barrel structure with an inner diameter of 1.4 nm and a well-defined constriction site that enables precise nucleotide discrimination. While its robust structure and specific binding sites make it ideal for DNA analysis, challenges in controlling translocation speed have led to the exploration of alternative protein channels. MspA, characterized by its distinctive funnel-shaped structure and narrower sensing region (1.2 nm),18,98 has demonstrated superior spatial resolution and significantly improved sequencing accuracy.99 The CsgG nanopore has emerged as a versatile platform,20,21 showing particular promise in RNA sequencing and addressing the challenging issue of homopolymer sequence recognition through its innovative combination with CsgF N-terminal residues,100 which has increased single-read accuracy from 25% to 70%.101 Beyond nucleic acid analysis, these biological nanopores have been adapted for an expanding range of applications, including protein sequencing and peptide analysis.102 Despite challenges posed by the diverse physicochemical properties of amino acids, innovative approaches such as peptide–DNA conjugation with controlled translocation through MspA using DNA helicase have achieved remarkable single amino acid resolution.103 The technology's versatility continues to expand, with recent applications in microRNA detection and other small molecule analyses,81,104,105 demonstrating its broad potential in molecular sensing and characterization.MD simulations have emerged as an essential tool for understanding and optimizing biological nanopore applications, providing atomic-resolution insights that complement experimental observations. In α-HL systems, simulations have revealed quantitative details of DNA–protein interactions through PMF calculations, identifying key residues like D127 and K131 that create strong electrostatic interactions (−2.5 kcal mol−1 per nucleotide) with DNA phosphate groups.106 These regions serve as potential targets for pore optimization, where the protein–DNA interactions involve complex combinations of electrostatic and short-range interactions, with water molecules often mediating these interactions. These computational findings have guided experimental modifications of the constriction region, leading to the development of mutant α-HL pores with improved nucleotide discrimination capabilities.107,108
In MspA research, MD simulations have revealed the critical role of the 1.2 nm constriction zone in molecular recognition. Detailed analysis of size exclusion and pore binding effects showed that single amino acid variations in peptides can produce distinct ionic current signatures, enabling variant detection with remarkably low error rates (<10−6).11 MD simulations have provided atomic-level insights into these interactions, revealing specific contacts between DNA phosphate groups and bases with arginine residues in the constriction region (Fig. 3(a)).109 These insights led to the development of variable voltage protocols, where the driving voltage was varied between 100 and 200 mV. This approach improved single-passage base-calling accuracy from 62.7 ± 0.5% to 79.3 ± 0.3%.98
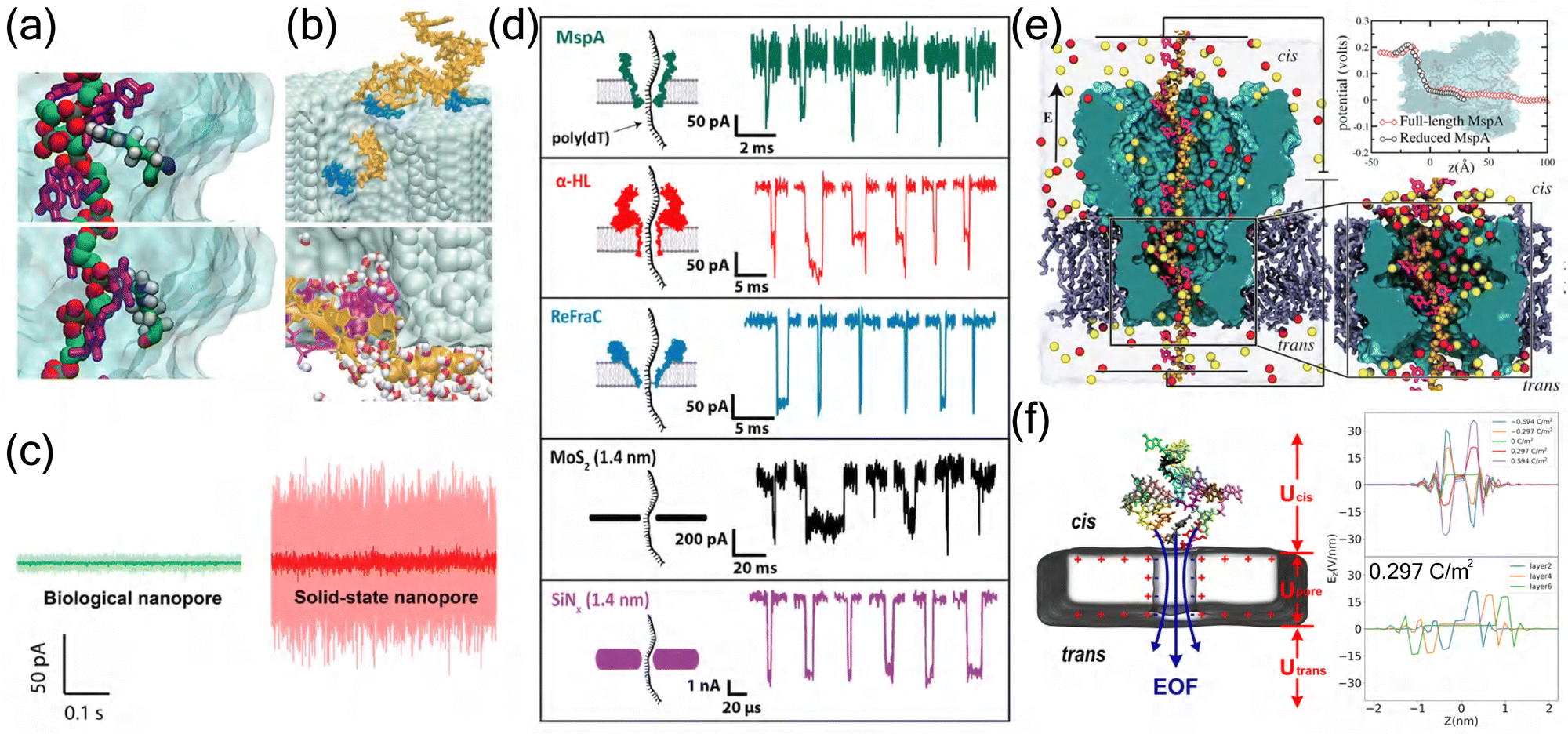 | ||
| Fig. 3 Comparative analysis of biological and solid-state nanopores. (a) MD simulations revealing distinct DNA–protein interactions in biological nanopores. Top: Single phosphate group interaction with arginine residue. Bottom: Cytosine base interaction with arginine residue, demonstrating the specific molecular recognition mechanisms in biological nanopores.109 Copyright 2012 American Chemical Society. (b) DNA–surface interactions in solid-state nanopores. Top: Single-stranded DNA adhesion to Si3N4 surface. Bottom: Spontaneous base-pair splitting of double-stranded DNA within the pore, illustrating the non-specific interactions characteristic of solid-state nanopores.110 Copyright 2004 The Biophysical Society. Published by Elsevier Inc. (c) Comparison of ionic current noise levels between biological (green) and solid-state (red) nanopores under identical experimental conditions (100 mV, 1 M KCl, pH 7), demonstrating the superior noise characteristics of biological nanopores.111 Copyright 2020 American Chemical Society. (d) Representative ionic current traces from different nanopore systems. Comparison of DNA translocation events through biological nanopores (MspA, α-HL, ReFraC) and solid-state nanopores (MoS2, SiN4), showing distinct current signatures and temporal characteristics.111 Copyright 2020 American Chemical Society. (e) Electrostatic potential analysis of MspA nanopore. Left: Cross-sectional view showing ion distribution and DNA conformation. Right: Comparison of electrostatic potential profiles between full-length and reduced MspA systems.109 Copyright 2012 American Chemical Society. (f) Electric field analysis in graphene nanopore systems. Left: ssDNA translocation through a graphene nanopore. Right: Z-axis electric field intensity profiles under varying graphene layer and surface charge densities configurations.84 Copyright 2024 American Chemical Society. | ||
The synergy between computational predictions and experimental validation has proven particularly valuable in nanopore engineering. MD simulations have revealed that pore geometry and amino acid properties at the constriction site play crucial roles in ionic and molecular transport.112 For instance, MspA's conical shape has demonstrated advantages over α-HL's cylindrical structure in detecting small analytes, showing systematically larger event amplitudes (up to 10 pA compared to 1–3 pA).113 Furthermore, simulation studies of protein translocation through α-HL have identified robust translocation intermediates associated with the rearrangement of secondary structural elements,114 while computational analysis of homopeptide transport has shown that pore clogging is significantly influenced by amino acid volume, hydrophobicity, and net charge.115 These computational insights have guided experimental strategies, such as the substitution of key amino acids at pore constrictions to modulate electrostatic potential and ionic current,112 and the optimization of measurement pH (from 6.8 to 8.0) to extend single ion binding events by 46-fold.113 Such simulation-guided approaches have proven valuable for developing high-performance nanopores.112
Recent advances in MD simulations have also facilitated the design of novel biological nanopores for specific applications. Computational studies have revealed that CsgG nanopores exhibit distinct transport properties, characterized by a sudden drop in electrostatic potentials at their constriction site, in contrast to the gradual changes observed in α-HL.112 These insights have guided experimental modifications, such as the integration of CsgG with the N-terminal region of CsgF to create a dual-constriction pore. This innovative design, with constrictions separated by 2.5 nm, has improved single-read accuracy by 25–70% for homopolymer sequences up to 9 nucleotides long.21 Furthermore, the strategic substitution of key amino acids at pore constrictions, particularly with charged residues, has proven effective in modulating pore electrostatic potential and ionic current,112 demonstrating how computational insights can guide nanopore engineering for enhanced sensing capabilities.
3.2 Synthetic pore transport
Solid-state nanopores, fabricated in synthetic materials such as Si3N4, graphene, h-BN, MoS2, and WS2, have emerged as powerful tools for single-molecule analysis.3,116 These engineered nanopores offer unique advantages through their tunable pore geometry, surface chemistry, and mechanical stability. The development of advanced fabrication techniques, particularly transmission electron microscopy (TEM) and controlled dielectric breakdown (CDB), has enabled precise control over pore dimensions and improved production efficiency.117,118 This technological advancement has facilitated diverse applications in biomolecular analysis, from nucleic acid sequencing to protein characterization.119 Recent innovations in detection modalities, including field-effect transistor-based sensing, quantum tunneling, and plasmonic methods, have significantly expanded the analytical capabilities of solid-state nanopores.3 The technology has demonstrated particular promise in detecting various biomolecules, including carbohydrates,120 though challenges remain in achieving specific molecular recognition in complex biological samples.121MD simulations have proven instrumental in addressing experimental challenges and optimizing solid-state nanopore design, providing atomic-resolution insights into molecular transport mechanisms. For Si3N4 nanopores, all-atom simulations have revealed critical details about DNA translocation dynamics, particularly the role of ion–nucleotide interactions in sequence discrimination. Detailed simulations have shown that both single-stranded and double-stranded DNA can interact strongly with the Si3N4 surface, where nucleotide bases can adhere to the pore wall and Watson–Crick base pairs may split spontaneously during translocation (Fig. 3(b)).110 These studies demonstrated that local ion concentrations near the pore surface (typically 2–3 times higher than bulk) create distinct electrostatic environments that significantly influence DNA transport.122 These insights have guided the optimization of pore surface chemistry, leading to enhanced DNA capture rates and improved signal-to-noise ratios in experimental measurements.
In graphene-based systems, MD simulations have revolutionized nanopore design through the development of novel multilayer architectures. Studies of asymmetric double-layer structures revealed how strategically positioned nanopores create distinct energy barriers for precise molecular transport control. The simulations revealed that molecules translocate from the smaller to larger pore, the dwell time increases threefold compared to the reverse direction, demonstrating enhanced control over translocation dynamics.123 Atomistic simulations of DNA translocation through graphene nanopores reveal that hydrophobic interactions suppress nucleotide fluctuations during transport. The resulting ion current signals show nucleotide-specific features, though molecular orientation significantly affects detection accuracy, suggesting the need for optimized surface engineering.83 Furthermore, investigations of graphene surface-step defects revealed directional molecular transport, where DNA molecules exhibited preferential movement along defect edges and downward across steps compared to upward motion, enabling controlled biomolecule delivery to nanopores.124
Particularly noteworthy are the simulation-guided advances in protein analysis using MoS2 nanopores. Microsecond-scale non-equilibrium MD simulations of α-synuclein peptide translocation revealed distinct current blockade patterns (I ranging from 0.08–0.90 nA) corresponding to specific amino acid sequences.125 These computational insights led to experimental optimizations, such as the development of DNA scaffold carriers for controlled protein translocation,126 improving detection efficiency by up to 60% and reducing translocation speed variations by a factor of 4.
Recent computational studies have focused on surface modification strategies for enhanced molecular specificity. Simulations of functionalized nanopores demonstrated that variations in surface charge density can create localized electric field gradients (>6 mV nm−1), increasing capture rates by an order of magnitude while maintaining selectivity.127 The integration of MD simulations with machine learning approaches has further enhanced signal interpretation, enabling automated classification of molecular events with accuracy exceeding 95%.128,129 These computational advances have also facilitated the design of novel sensing modalities, such as plasmonic nanopores, where engineered electromagnetic fields enable enhanced optical detection and precise temperature control.130
The synergy between MD simulations and experimental validation has led to significant improvements in nanopore design. For instance, MD simulations have revealed how atomic-level structural changes affect nanopore conductivity and expansion rates, guiding the development of chemical modification strategies to enhance pore stability.131 Similarly, computational studies of bio-inspired solid-state nanopores with thin constrictions have demonstrated improved spatial resolution comparable to 2D material nanopores while reducing noise levels, providing valuable insights for DNA sequencing applications.132
3.3 Transport mechanism comparison
MD simulations reveal fundamental distinctions between biological and solid-state nanopores in their structural characteristics, molecular recognition mechanisms, and transport properties. Biological nanopores, evolved through natural selection and refined through protein engineering, demonstrate sophisticated molecular recognition through specific amino acid interactions.81,133 MD simulations have revealed that protein structure creates well-defined binding sites with precise electrostatic and hydrophobic interactions, enabling exquisite control over molecular transport (Fig. 2(a)).12 The dynamic nature of protein channels, captured through microsecond-scale simulations, shows coordinated conformational changes that facilitate molecular recognition, with characteristic timescales ranging from nanoseconds (side-chain movements) to microseconds (backbone fluctuations).134In contrast, solid-state nanopores, fabricated from materials ranging from Si3N4 to emerging 2D materials, exhibit distinct transport mechanisms dominated by geometric confinement and surface interactions.135,136 MD simulations demonstrate that their rigid structure creates well-defined electric field distributions,46 with molecular transport primarily governed by electrophoretic forces rather than specific binding interactions.137 The atomic-scale thickness of 2D materials (0.3–1.0 nm) enables unique transport phenomena, with simulations revealing discrete hydration layer structures and ion distributions that significantly influence molecular translocation.
Stability analysis through MD simulations highlights the complementary advantages of these systems. Biological nanopores maintain remarkable functional stability under physiological conditions, with simulations showing stable protein conformations and consistent ion conductance over microsecond timescales.138 This stability is reflected in their superior noise characteristics compared to solid-state counterparts under identical experimental conditions (Fig. 3(c)).111 Their engineered variants demonstrate enhanced stability through strategic mutations, though environmental sensitivity remains a limitation.139 Solid-state nanopores, simulated across broader condition ranges, maintain structural integrity with negligible deformation (<0.1 nm) under various pH and temperature conditions.135 The distinct properties of biological and solid-state nanopores are particularly evident in their DNA translocation characteristics, where different nanopore systems exhibit unique current signatures and temporal patterns (Fig. 3(d)).111
The molecular recognition mechanisms and controllability aspects, elucidated through detailed simulations, reveal distinct approaches in different nanopore systems. Biological nanopores utilize specific amino acid arrangements to create energy barriers and binding sites, enabling precise control over molecular transport,12 while exhibiting sophisticated gating mechanisms controlled by protein conformational changes that modulate pore conductance.134 This is particularly evident in MspA nanopores, where electrostatic potential analysis reveals distinct voltage drops and ion distributions that facilitate molecular recognition (Fig. 3(e)).109 In contrast, solid-state systems achieve selectivity through engineered surface chemistry and geometric constraints, with recent innovations in surface charge density modulation extending DNA base residence times by up to 30-fold in the sensing region.46 These synthetic systems offer direct geometric and surface chemistry control, exemplified by carbon nanotube porins (CNTPs) where external voltage precisely regulates ion flux through well-defined electric field distributions.140 The unique characteristics of 2D material nanopores, such as graphene, are particularly evident in their distinct electrostatic potential distributions (Fig. 3(f)), which contribute to their enhanced sensing capabilities.16
These MD-derived insights guide the optimization of both platforms for specific applications. Biological nanopores excel in applications requiring precise molecular discrimination through evolved recognition mechanisms, while solid-state nanopores dominate in scenarios demanding robust operation and geometric control. The integration of simulation-derived understanding continues to advance both platforms, suggesting potential hybrid approaches that combine their complementary strengths.
3.4 Current challenges
Despite significant advances in MD simulations of nanopore systems, several fundamental challenges persist in accurately modeling molecular transport processes. These challenges span multiple scales, from atomic-level interactions to system-wide phenomena, affecting both biological and solid-state nanopore simulations. A primary limitation lies in the computational demands of high-precision simulations. Current studies must balance between system size and simulation duration, particularly challenging when modeling complete translocation events. For instance, comprehensive modeling of nanopore–peptide interactions requires sophisticated conductivity models (σ′) that account for molecular position, orientation, and conformational changes.141 These simulations typically remain constrained to nanosecond to microsecond timescales, often insufficient for capturing complete biomolecule translocation events, which can span milliseconds in experimental settings.The accuracy of molecular recognition presents another significant challenge, particularly in distinguishing structurally similar molecules. In biological nanopores, the subtle differences in current blockade signatures between amino acids in α-HL systems,142 making reliable protein sequencing predictions extremely challenging. This limitation becomes more pronounced when dealing with post-translational modifications or similar amino acid residues, where current signatures may overlap significantly. The complexity increases further when considering the dynamic nature of protein–pore interactions, where conformational fluctuations can mask or alter characteristic signals.121,143 Similarly, solid-state nanopores face substantial challenges in molecular discrimination, primarily due to rapid translocation speeds. The lack of specific binding sites in solid-state systems, often results in stochastic transport behaviors that complicate sequence identification.16 These limitations necessitate the development of enhanced sampling techniques and more accurate force fields to capture the nuanced dynamics of molecular transport, particularly in regions where traditional force fields may not accurately represent quantum mechanical effects.
Signal processing and analysis pose additional challenges across both nanopore types, with particular emphasis on noise reduction and signal enhancement. The detection of transient current pulses, requires sophisticated high-gain, low-noise amplifiers to distinguish weak signals from background noise.144 Traditional threshold-based algorithms often lack objectivity in feature extraction, leading to potential biases in data interpretation, particularly when dealing with overlapping signals or multiple molecule translocations.145 These challenges are especially acute in solid-state systems, where higher noise levels (typically 2–3 times greater than biological nanopores) and faster translocation speeds (often exceeding 1 base per ns) significantly complicate signal analysis. The situation becomes even more challenging when considering effects such as DNA–surface interactions, ion current rectification, and varying hydration patterns, all of which can modulate the measured signals in complex and often unpredictable ways.
To address these limitations, several innovative approaches have emerged in recent years. The integration of GPU acceleration has achieved order-of-magnitude improvements in simulation speed,146,147 while the development of implicit solvent models has significantly expanded accessible simulation scales.148,149 Enhanced sampling techniques have improved the exploration of conformational space,150,151 enabling more comprehensive investigation of molecular transport mechanisms. Particularly promising is the integration of machine learning approaches with traditional MD simulations. Deep learning models have enhanced signal processing and classification capabilities,152 while CNNs and RNNs have improved feature extraction from complex time-series data.145 These AI-driven approaches have enabled real-time analysis of translocation events,153,154 significantly advancing our ability to interpret nanopore signals.
4 Conclusions and future perspectives
This perspective examines the critical role of MD simulations in advancing nanopore sequencing technology, highlighting their contributions to understanding molecular transport mechanisms and optimizing nanopore design. Biological nanopores, with their sophisticated molecular recognition mechanisms and high selectivity, have demonstrated remarkable success in DNA sequencing applications. The integration of MD simulations has been instrumental in elucidating the atomic-level interactions that govern their exceptional specificity, despite challenges in computational cost and parameter complexity (Fig. 4(a)). Looking forward, engineered biological nanopores show particular promise for direct detection of protein post-translational modifications and enhanced sequence discrimination. The development of specialized force fields, optimized for protein–substrate interactions and incorporating quantum mechanical corrections, may further improve the accuracy of biological nanopore simulations. | ||
| Fig. 4 Future perspectives in nanopore technology development. (a) Comparative advantages (red) and limitations (black) of biological and solid-state nanopores in MD simulations. Left: Biological nanopores featuring high molecular specificity and precise recognition sites while being constrained by limited size, structure flexibility, high computational cost, and complex parameter settings. Right: Solid-state nanopores offering controllable size, high structural stability, low computational cost, and easy parameter settings while suffering from poor molecular specificity and limited chemical modification. Created in BioRender. Y. Zhang (2024), https://BioRender.com/j42j080. (b) Integration of artificial intelligence in nanopore sensing. Workflow of deep learning implementation showing event extraction, model training using SteviNet architecture, and practical applications in molecular classification, illustrating the potential of AI-enhanced nanopore analysis.155 Copyright 2024 American Chemical Society. (c) Bio-inspired nanopore designs. Left: Structure of biological nanopore showing characteristic constriction site and vestibule regions. Right: Bio-inspired solid-state nanopore incorporating key biological features through a trilayer architecture with controlled constriction and vestibule dimensions.132 Copyright 2024 American Chemical Society. (d) Hybrid nanopore-membrane system design. Integration of extended pillararene macrocycle (EPM) nanopore within a biological POPC lipid bilayer, demonstrating the successful combination of synthetic channels with biological membranes. The system shows the EPM channel's key structural features including constriction region (green) and peptide side chains (purple), surrounded by water molecules and ions (K+ in yellow, Cl− in cyan) in a 1 M KCl electrolyte environment. Inset displays the structural details of the EPM channel from top and side perspectives.156 Copyright 2021 American Chemical Society. | ||
Solid-state nanopores, particularly those fabricated from emerging two-dimensional materials like graphene and MoS2, offer complementary advantages in terms of chemical stability, mechanical robustness, and scalability. While currently lagging in molecular recognition accuracy compared to their biological counterparts, their potential for large-scale production and commercialization makes them attractive for various applications. These systems benefit from simpler parameter settings and lower computational costs in MD simulations, though they face challenges in achieving molecular specificity (Fig. 4(a)). MD simulations have proven crucial in optimizing their design and understanding transport mechanisms, though challenges remain in achieving single-base resolution for electronic DNA sequencing.
The future advancement of nanopore technology will likely be driven by several key developments in computational methodology. The refinement of force fields, balancing computational efficiency with accuracy, remains crucial for reliable predictions of molecular transport phenomena. Enhanced sampling techniques and more efficient algorithms will be essential for extending simulation timescales beyond the current nanosecond to microsecond limitations. The integration of artificial intelligence methods with MD simulations represents a particularly promising direction, as demonstrated by recent developments in deep learning frameworks for nanopore signal analysis (Fig. 4(b)), potentially enabling direct analysis of complex biological samples and automated interpretation of sequencing data. These computational advances have already begun to bridge the gap between biological and solid-state nanopore systems, suggesting new possibilities for hybrid approaches.
The convergence of high-performance computing architectures and interdisciplinary methodologies is positioning nanopore technology research at the frontier of revolutionary breakthroughs. The next generation of hybrid computing platforms, integrating CPU, GPU, and quantum computing units, coupled with enhanced sampling algorithms, shows promise in overcoming the spatiotemporal limitations of CMD simulations. This advancement is expected to enable precise simulation of complex biomolecular translocation processes at millisecond timescales within the next decade.
We envision four key pathways toward this goal. First, AI-driven multiscale modeling frameworks will integrate machine learning force fields, such as Deep Potential, with specialized nanopore material databases to train dedicated neural networks. This integration will specifically address the precision bottlenecks in heterogeneous interface charge distribution and molecular recognition site prediction. Second, the evolution of quantum-classical hybrid computing paradigms will open new dimensions in free energy landscape calculations, enabling more accurate predictions of molecular transport mechanisms. Third, inter-institutional collaborative feedback systems, incorporating spiking neural networks (SNN) for real-time signal processing, will establish closed-loop optimization networks connecting electric field parameters, molecular trajectories, and pore current signals. Fourth, high-throughput screening strategies combining generative adversarial networks (GANs) with MD will advance intelligent interface design for single-molecule recognition, exemplified by programmable optimization of MXene pore edge functionalization.
While current challenges persist in hardware performance limitations and multimodal data integration complexity, the cross-pollination of artificial intelligence, quantum computing, and microfluidic technologies promises unprecedented insights into single-molecule transport physics. These advances will establish theoretical foundations for the rational design of personalized diagnostic nanopore devices and efficient membrane separation materials.
Indeed, hybrid systems combining the advantages of both biological and solid-state nanopores have emerged as an exciting frontier in nanopore research. These innovative platforms aim to leverage the precise molecular recognition of biological pores while maintaining the robustness and scalability of solid-state platforms. Bio-inspired design strategies have successfully incorporated key biological features, such as precise constriction sites and vestibule regions, into solid-state architectures (Fig. 4(c)). Furthermore, the integration of synthetic nanopores with biological membranes has shown promising results, as demonstrated by the successful embedding of extended pillararene macrocycle (EPM) nanopores within lipid bilayers (Fig. 4(d)). The optimization of such hybrid systems requires sophisticated multiscale simulation approaches, integrating atomic-level details with larger-scale phenomena. This integration of different length and time scales represents a crucial step toward more comprehensive and accurate modeling of complex nanopore systems.
To support these ambitious developments, the continued advancement of specialized computing hardware and simulation methodologies remains essential. As computational capabilities expand, we anticipate significant improvements in our ability to model and optimize nanopore-based technologies across multiple scales. These advances are expected to catalyze applications beyond traditional sequencing, extending to environmental monitoring, biomedical research, and personalized medicine. The growing synergy between experimental and computational approaches, particularly in the development of hybrid systems, will be crucial in realizing the full potential of nanopore technology and addressing current challenges in molecular analysis.
Looking to the future, we identify two critical research directions that merit intensive investigation through the synergistic integration of theoretical and experimental approaches. The first direction focuses on hybrid nanopore system design, where MD simulations can systematically optimize bio-solid interfaces, particularly emphasizing the exploration of novel two-dimensional materials (such as MXenes and MOFs) in conjunction with biological nanopores. Through iterative interplay between computational modeling and experimental validation, this approach aims to achieve sub-angstrom resolution in molecular detection by understanding atomic–scale interactions at interfaces and optimizing chemical functionalization for enhanced selectivity. The second direction involves developing advanced signal processing methodologies through deep learning approaches, where modern architectures such as transformers and graph neural networks can establish universal molecular feature prediction models. These sophisticated models, trained and validated using diverse experimental sequencing data, aim to achieve recognition accuracies exceeding 95% through effective combination of multiple data streams and real-time feedback between measurements and predictions.
Data availability
All data mentioned in this perspective are from reported studies and are correctly cited.Conflicts of interest
The authors declare no interest conflict.Acknowledgements
This work is financially supported by the National Natural Science Foundation of China (no. 22373025 and 22227804).References
- J. D. Prajapati, S. Pangeni, M. A. Aksoyoglu, M. Winterhalter and U. Kleinekathöfer, ACS Nano, 2022, 16, 7701–7712 CrossRef CAS PubMed.
- M. Xiong, M. Graf, N. Athreya, A. Radenović and J. Leburton, ACS Nano, 2020, 14, 16131–16139 CrossRef CAS PubMed.
- L. Xue, H. Yamazaki, R. Ren, M. Wanunu, A. Ivanov and J. Edel, Nat. Rev. Mater., 2020, 5, 931–951 CrossRef CAS.
- J. J. Kasianowicz, E. Brandin, D. Branton and D. W. Deamer, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 13770–13773 CrossRef CAS PubMed.
- A. Meller, L. Nivon, E. Brandin, J. Golovchenko and D. Branton, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 1079–1084 CrossRef CAS PubMed.
- A. Meller, L. Nivon and D. Branton, Phys. Rev. Lett., 2001, 86, 3435–3438 CrossRef CAS PubMed.
- A. Meller and D. Branton, Electrophoresis, 2002, 23, 2583–2591 CrossRef CAS PubMed.
- M. Akeson, D. Branton, J. J. Kasianowicz, E. Brandin and D. W. Deamer, Biophys. J., 1999, 77, 3227–3233 CrossRef CAS PubMed.
- D. Deamer, M. Akeson and D. Branton, Nat. Biotechnol., 2016, 34, 518–524 CrossRef CAS PubMed.
- X. Wei, T. Penkauskas, J. Reiner, C. Kennard, M. Uline, Q. Wang, S. Li, A. Aksimentiev, J. Robertson and C. Liu, ACS Nano, 2023, 17, 16369–16395 CrossRef CAS PubMed.
- H. Brinkerhoff, A. S. W. Kang, J. Liu, A. Aksimentiev and C. Dekker, Science, 2021, 374, 1509–1513 CrossRef CAS PubMed.
- L. Yu, X. Kang, F. Li, B. Mehrafrooz, A. Makhamreh, A. Fallahi, J. C. Foster, A. Aksimentiev, M. Chen and M. Wanunu, Nat. Biotechnol., 2021, 41, 1130–1139 CrossRef PubMed.
- Y. Wang, S. Zhang, W. Jia, P. Fan, L. Wang, X. Li, J. Chen, Z. Cao, X. Du and Y. Liu, et al. , Nat. Nanotechnol., 2022, 17, 976–983 CrossRef CAS PubMed.
- B. Yuan, S. Li, Y.-L. Ying and Y.-T. Long, Analyst, 2020, 145, 1179–1183 RSC.
- X.-L. Xing, Z.-C. He, S. A. Ahmed, Q. Liao, L.-R. Guo, S. Ren, K. Xi, L.-N. Ji, K. Wang and X.-H. Xia, Anal. Chem., 2022, 94, 9851–9855 CrossRef CAS PubMed.
- H. Qiu, W. Zhou and W. Guo, ACS Nano, 2021, 15, 18848–18864 CrossRef CAS PubMed.
- L. Song, M. R. Hobaugh, C. Shustak, S. Cheley, H. Bayley and J. E. Gouaux, Science, 1996, 274, 1859–1865 CrossRef CAS PubMed.
- M. Faller, M. Niederweis and G. E. Schulz, Science, 2004, 303, 1189–1192 CrossRef CAS PubMed.
- T. Z. Butler, M. Pavlenok, I. M. Derrington, M. Niederweis and J. H. Gundlach, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 20647–20652 CrossRef CAS PubMed.
- B. Cao, Y. Zhao, Y. Kou, D. Ni, X. C. Zhang and Y. Huang, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E5439–E5444 CrossRef CAS PubMed.
- S. E. V. D. Verren, N. Gerven, W. Jonckheere, R. Hambley, P. Singh, J. Kilgour, M. Jordan, J. Wallace, L. Jayasinghe and H. Remaut, Nat. Biotechnol., 2020, 38, 1415–1420 CrossRef PubMed.
- W. Shi, A. K. Friedman and L. A. Baker, Anal. Chem., 2017, 89, 157–188 CrossRef CAS PubMed.
- A. Asandei, A. E. Rossini, M. Chinappi, Y. Park and T. Luchian, Langmuir, 2017, 33, 14451–14459 CrossRef CAS PubMed.
- D. Branton, D. W. Deamer, A. Marziali, H. Bayley, S. A. Benner, T. Butler, M. Di Ventra, S. Garaj, A. Hibbs, X. Huang, S. B. Jovanovich, P. S. Krstic, S. Lindsay, X. S. Ling, C. H. Mastrangelo, A. Meller, J. S. Oliver, Y. V. Pershin, J. M. Ramsey, R. Riehn, G. V. Soni, V. Tabard-Cossa, M. Wanunu, M. Wiggin and J. A. Schloss, Nat. Biotechnol., 2008, 26, 1146–1153 CrossRef CAS PubMed.
- S. Majd, E. C. Yusko, Y. N. Billeh, M. X. Macrae, J. Yang and M. Mayer, Curr. Opin. Biotechnol, 2010, 21, 439–476 CrossRef CAS PubMed.
- L. S. R. Adamson, N. Tasneem, M. P. Andreas, W. Close, E. N. Jenner, T. N. Szyszka, R. Young, L. C. Cheah, A. Norman, H. I. MacDermott-Opeskin, M. L. OMara, F. Sainsbury, T. W. Giessen and Y. H. Lau, Sci. Adv., 2022, 8, eabl7346 CrossRef CAS PubMed.
- J. Li, D. Stein, C. McMullan, D. Branton, M. J. Aziz and J. A. Golovchenko, Nature, 2001, 412, 166–169 CrossRef CAS PubMed.
- J. Li, M. Gershow, D. Stein, E. Brandin and J. A. Golovchenko, Nat. Mater., 2003, 2, 611–615 CrossRef CAS PubMed.
- R. Hu, X. Tong and Q. Zhao, Adv. Healthcare Mater., 2020, 9, 2000933 CrossRef CAS PubMed.
- J. P. Fried, J. Swett, B. P. Nadappuram, J. A. Mol, J. Edel, A. Ivanov and J. R. Yates, Chem. Soc. Rev., 2021, 50, 4974–4992 RSC.
- K. Liu, J. Feng, A. Kis and A. Radenovic, ACS Nano, 2014, 8, 2504–2511 CrossRef CAS PubMed.
- J. Feng, K. Liu, R. D. Bulushev, S. Khlybov, D. Dumcenco, A. Kis and A. Radenovic, Nat. Nanotechnol., 2015, 10, 1070–1076 CrossRef CAS PubMed.
- S. Garaj, W. Hubbard, A. Reina, J. Kong, D. Branton and J. A. Golovchenko, Nature, 2010, 467, 190–193 CrossRef CAS PubMed.
- G. F. Schneider, S. W. Kowalczyk, V. E. Calado, G. Pandraud, H. W. Zandbergen, L. M. K. Vandersypen and C. Dekker, Nano Lett., 2010, 10, 3163–3167 CrossRef CAS PubMed.
- M. D. Fischbein and M. Drndić, Appl. Phys. Lett., 2008, 93, 113107 CrossRef.
- C. Plesa, N. van Loo, P. Ketterer, H. Dietz and C. Dekker, Nano Lett., 2014, 15, 732–737 Search PubMed.
- C. Plesa, S. W. Kowalczyk, R. Zinsmeester, A. Y. Grosberg, Y. Rabin and C. Dekker, Nano Lett., 2013, 13, 658–663 CrossRef CAS PubMed.
- M. Puster, J. A. Rodrguez-Manzo, A. Balan and M. Drndic, ACS Nano, 2013, 7, 11283–11289 CrossRef CAS PubMed.
- T. Deng, M. Li, Y. Wang and Z. Liu, Sci. Bull., 2015, 60, 304–319 CrossRef CAS.
- A. Aksimentiev and K. Schulten, Biophys. J., 2005, 88, 3745–3761 CrossRef CAS PubMed.
- J. Mathé, A. Aksimentiev, D. R. Nelson, K. Schulten and A. Meller, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 12377–12382 CrossRef PubMed.
- K. Willems, D. Ruić, F. L. R. Lucas, U. Barman, N. Verellen, J. Hofkens, G. Maglia and P. Van Dorpe, Nanoscale, 2020, 12, 16775–16795 Search PubMed.
- C. Cao, M.-Y. Li, N. Cirauqui, Y.-Q. Wang, M. Dal Peraro, H. Tian and Y.-T. Long, Nat. Commun., 2018, 9, 2823 CrossRef PubMed.
- M. Wanunu, J. Sutin, B. McNally, A. Chow and A. Meller, Biophys. J., 2008, 95, 4716–4725 CrossRef CAS PubMed.
- S. W. Kowalczyk, A. Y. Grosberg, Y. Rabin and C. Dekker, Nanotechnology, 2011, 22, 315101 CrossRef PubMed.
- Y.-S. Zhang, Z.-Y. Qi, M.-M. Ding, M.-L. Li and T.-F. Shi, Chin. J. Polym. Sci., 2024, 42, 2048–2058 CrossRef CAS.
- A. Kiakojouri, I. Frank and E. Nadimi, Phys. Chem. Chem. Phys., 2023, 25, 13452–13464 RSC.
- G. T. Feliciano, C. Sanz-Navarro, M. D. Coutinho-Neto, P. Ordejon, R. H. Scheicher and A. R. Rocha, J. Phys. Chem. B, 2018, 122, 485–492 CrossRef CAS PubMed.
- M. Belkin, S.-H. Chao, M. P. Jonsson, C. Dekker and A. Aksimentiev, ACS Nano, 2015, 9, 10598–10611 CrossRef CAS PubMed.
- S. Pud, S.-H. Chao, M. Belkin, D. Verschueren, T. Huijben, C. Van Engelenburg, C. Dekker and A. Aksimentiev, Nano Lett., 2016, 16, 8021–8028 CrossRef CAS PubMed.
- H. McFarland, T. Ahmed, J.-X. Zhu, A. Balatsky and J. T. Haraldsen, J. Phys. Chem. Lett., 2015, 6, 2616–2621 CrossRef CAS PubMed.
- A. Kiakojouri, I. Frank and E. Nadimi, Phys. Chem. Chem. Phys., 2021, 23, 25126–25135 RSC.
- M. Abasifard, V. Ahmadi, B. Fotouhi and R. Roohi, J. Phys. Chem. C, 2019, 123, 25309–25319 CrossRef CAS.
- A. Pérez, R. G. Amorim, C. Villegas, A. Rocha and A. Rocha, Phys. Chem. Chem. Phys., 2020, 22, 27053–27059 RSC.
- A. Smolyanitsky and B. Luan, Phys. Rev. Lett., 2020, 127, 138103 CrossRef PubMed.
- A. Singharoy, C. Maffeo, K. H. Delgado-Magnero, D. J. Swainsbury, M. Sener, U. Kleinekathöfer, J. W. Vant, J. Nguyen, A. Hitchcock, B. Isralewitz, I. Teo, D. E. Chandler, J. E. Stone, J. C. Phillips, T. V. Pogorelov, M. I. Mallus, C. Chipot, Z. Luthey-Schulten, D. P. Tieleman, C. N. Hunter, E. Tajkhorshid, A. Aksimentiev and K. Schulten, Cell, 2019, 179, 1098–1111 CrossRef CAS PubMed.
- K. Göpfrich, C.-Y. Li, M. Ricci, S. P. Bhamidimarri, J. Yoo, B. Gyenes, A. Ohmann, M. Winterhalter, A. Aksimentiev and U. F. Keyser, ACS Nano, 2016, 10, 8207–8214 CrossRef PubMed.
- S. J. Marrink, H. J. Risselada, S. Yefimov, D. P. Tieleman and A. H. De Vries, J. Phys. Chem. B, 2007, 111, 7812–7824 CrossRef CAS PubMed.
- M. Fyta, S. Melchionna, S. Succi and E. Kaxiras, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2008, 78, 036704 CrossRef PubMed.
- N. Basdevant, D. Dessaux and R. Ramirez, Sci. Rep., 2019, 9, 15740 CrossRef PubMed.
- D. Dessaux, J. Mathé, R. Ramirez and N. Basdevant, J. Phys. Chem. B, 2022, 126, 4189–4199 CrossRef CAS PubMed.
- C.-M. Chen and E.-H. Peng, Appl. Phys. Lett., 2003, 82, 1308–1310 Search PubMed.
- K. Luo, T. Ala-Nissila, S.-C. Ying and A. Bhattacharya, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2008, 78, 061911 CrossRef PubMed.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1, 19–25 Search PubMed.
- J. C. Phillips, D. J. Hardy, J. D. C. Maia, J. E. Stone, J. V. Ribeiro, R. C. Bernardi, R. Buch, G. Fiorin, J. Hénin, W. Jiang, R. McGreevy, M. C. R. Melo, B. K. Radak, R. D. Skeel, A. Singharoy, Y. Wang, B. Roux, A. Aksimentiev, Z. Luthey-Schulten, L. V. Kalé, K. Schulten, C. Chipot and E. Tajkhorshid, J. Chem. Phys., 2020, 153, 044130 CrossRef CAS PubMed.
- W. R. Scott, P. H. Hünenberger, I. G. Tironi, A. E. Mark, S. R. Billeter, J. Fennen, A. E. Torda, T. Huber, P. Krüger and W. F. Van Gunsteren, J. Phys. Chem. A, 1999, 103, 3596–3607 CrossRef CAS.
- B. R. Brooks, C. L. Brooks III, A. D. Mackerell Jr, L. Nilsson, R. J. Petrella, B. Roux, Y. Won, G. Archontis, C. Bartels and S. Boresch, et al. , J. Comput. Chem., 2009, 30, 1545–1614 Search PubMed.
- D. A. Case, H. M. Aktulga, K. Belfon, I. Ben-Shalom, S. R. Brozell, D. S. Cerutti, T. E. Cheatham III, V. W. D. Cruzeiro, T. A. Darden and R. E. Duke, et al., Amber 2021, University of California, San Francisco, 2021 Search PubMed.
- W. Lu, X. Zhao, M. Li, Y. Li, C. Zhang, Y. Xiong, J. Li, H. Zhou, X. Ye and X. Li, et al. , ACS Nano, 2024, 18, 12412–12426 CrossRef CAS PubMed.
- K. Shimizu, B. Mijiddorj, M. Usami, I. Mizoguchi, S. Yoshida, S. Akayama, Y. Hamada, A. Ohyama, K. Usui and I. Kawamura, et al. , Nat. Nanotechnol., 2022, 17, 67–75 Search PubMed.
- K. Vanommeslaeghe, E. Hatcher, C. Acharya, S. Kundu, S. Zhong, J. Shim, E. Darian, O. Guvench, P. Lopes and I. Vorobyov, et al. , J. Comput. Chem., 2010, 31, 671–690 CrossRef CAS PubMed.
- W. Stephenson, R. Razaghi, S. Busan, K. M. Weeks, W. Timp and P. Smibert, Cell Genomics, 2022, 2, 100097 Search PubMed.
- T. J. Oweida, H. S. Kim, J. M. Donald, A. Singh and Y. G. Yingling, J. Chem. Theory Comput., 2021, 17, 1208–1217 Search PubMed.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- J. Wong-Ekkabut and M. Karttunen, J. Chem. Theory Comput., 2012, 8, 2905–2911 CrossRef CAS PubMed.
- C. Oostenbrink, A. Villa, A. E. Mark and W. F. Van Gunsteren, J. Comput. Chem., 2004, 25, 1656–1676 Search PubMed.
- D. Conde, P. F. Garrido, M. Calvelo, Á. Piñeiro and R. García-Fandiño, Int. J. Mol. Sci., 2022, 23, 3158 CrossRef CAS PubMed.
- W. L. Jorgensen, D. S. Maxwell and J. Tirado-Rives, J. Am. Chem. Soc., 1996, 118, 11225–11236 CrossRef CAS.
- A.-P. Hynninen and M. F. Crowley, J. Comput. Chem., 2014, 35, 406–413 CrossRef CAS PubMed.
- S. Wang, Z. Zhao, F. Haque and P. Guo, Curr. Opin. Biotechnol, 2018, 51, 80–89 CrossRef CAS PubMed.
- C. Cao and Y. Long, Acc. Chem. Res., 2018, 51, 331–341 CrossRef CAS PubMed.
- C. Lynch, S. Rao and M. Sansom, Chem. Rev., 2020, 120, 10298–10335 CrossRef CAS PubMed.
- D. B. Wells, M. Belkin, J. Comer and A. Aksimentiev, Nano Lett., 2012, 12, 4117–4123 CrossRef CAS PubMed.
- Y. Zhang, M. Ding, M. Li and T. Shi, J. Phys. Chem. Lett., 2025, 16, 357–364 CrossRef CAS PubMed.
- S. K. Burley, H. M. Berman, G. J. Kleywegt, J. L. Markley, H. Nakamura and S. Velankar, Methods Mol. Biol., 2017, 1607, 627–641 CrossRef CAS PubMed.
- P. S. Crozier, D. Henderson, R. L. Rowley and D. D. Busath, Biophys. J., 2001, 81, 3077–3089 Search PubMed.
- A. Aksimentiev, J. B. Heng, G. Timp and K. Schulten, Biophys. J., 2004, 87, 2086–2097 CrossRef CAS PubMed.
- A. Aksimentiev and K. Schulten, Biophys. J., 2005, 88, 3745–3761 CrossRef CAS PubMed.
- J. Shen, J. Chen Li, F. Liu, L. Zhang, L. Liang, H. Wang and J. Wu, J. Membr. Sci., 2020, 595, 117611 CrossRef CAS.
- Y. Deng and B. Roux, J. Phys. Chem. B, 2009, 113, 2234–2246 CrossRef CAS PubMed.
- G. Hu, A. Ma and J. Wang, J. Chem. Inf. Model., 2017, 57, 918–928 CrossRef CAS PubMed.
- J. P. Thiruraman, K. Fujisawa, G. Danda, P. M. Das, T. Zhang, A. Bolotsky, N. Peréa-López, A. Nicolaï, P. Senet, M. Terrones and M. Drndić, Nano Lett., 2018, 18, 1651–1659 CrossRef CAS PubMed.
- K. Göpfrich, C.-Y. Li, M. Ricci, S. P. Bhamidimarri, J. Yoo, B. Gyenes, A. Ohmann, M. Winterhalter, A. Aksimentiev and U. Keyser, ACS Nano, 2016, 10, 8207–8214 CrossRef PubMed.
- Z. He, J. Zhou, X. Lu and B. Corry, ACS Nano, 2013, 7, 10148–10157 CrossRef CAS PubMed.
- Y. Fu, S. Su, N. Zhang, Y. Wang, X. Guo and J. Xue, ACS Appl. Mater. Interfaces, 2020, 12, 24281–24288 CrossRef CAS PubMed.
- S. W. Kowalczyk, D. Wells, A. Aksimentiev and C. Dekker, Nano Lett., 2012, 12, 1038–1044 CrossRef CAS PubMed.
- M. Belkin and A. Aksimentiev, ACS Appl. Mater. Interfaces, 2016, 8, 12599–12608 Search PubMed.
- M. Noakes, H. Brinkerhoff, A. Laszlo, I. Derrington, K. W. Langford, J. W. Mount, J. L. Bowman, K. S. Baker, K. Doering, B. I. Tickman and J. Gundlach, Nat. Biotechnol., 2019, 37, 651–656 CrossRef CAS PubMed.
- Y. Wang, Y. Zhao, A. Bollas, Y. Wang and K. F. Au, Nat. Biotechnol., 2021, 39, 1348–1365 CrossRef CAS PubMed.
- T. Thi Nhu Thao, F. Labroussaa, N. Ebert, P. Vkovski, H. Stalder, J. Portmann, J. Kelly, S. Steiner, M. Holwerda and A. Kratzel, et al. , Nature, 2020, 582, 561–565 CrossRef CAS PubMed.
- S. E. Van der Verren, N. Van Gerven, W. Jonckheere, R. Hambley, P. Singh, J. Kilgour, M. Jordan, E. J. Wallace, L. Jayasinghe and H. Remaut, Nat. Biotechnol., 2020, 38, 1415–1420 CrossRef CAS PubMed.
- Z.-L. Hu, M.-Z. Huo, Y.-L. Ying and Y.-T. Long, Angew. Chem., 2021, 133, 14862–14873 CrossRef.
- Z. Chen, Z. Wang, Y. Xu, X. Zhang, B. Tian and J. Bai, Chem. Sci., 2021, 12, 15750–15756 RSC.
- S. Cai, J. Y. Y. Sze, A. Ivanov and J. Edel, Nat. Commun., 2019, 10, 1797 CrossRef PubMed.
- Y.-L. Ying, Z.-L. Hu, S. Zhang, Y. Qing, A. Fragasso, G. Maglia, A. Meller, H. Bayley, C. Dekker and Y.-T. Long, Nat. Nanotechnol., 2022, 17, 1136–1146 CrossRef CAS PubMed.
- R. M. Manara, A. T. Guy, E. J. Wallace and S. Khalid, J. Chem. Theory Comput., 2015, 11, 810–816 CrossRef CAS PubMed.
- D. Xi, J. Shang, E. Fan, J. You, S. Zhang and H. Wang, Anal. Chem., 2016, 88, 10540–10546 CrossRef CAS PubMed.
- D. Stoddart, A. Heron, J. W. Klingelhoefer, E. Mikhailova, G. Maglia and H. Bayley, Nano Lett., 2010, 10, 3633–3637 CrossRef CAS PubMed.
- S. Bhattacharya, I. M. Derrington, M. Pavlenok, M. Niederweis, J. H. Gundlach and A. Aksimentiev, ACS Nano, 2012, 6, 6960–6968 CrossRef CAS PubMed.
- A. Aksimentiev, J. B. Heng, G. Timp and K. Schulten, Biophys. J., 2004, 87, 2086–2097 CrossRef CAS PubMed.
- A. Fragasso, S. Schmid and C. Dekker, ACS Nano, 2019, 14, 1338–1349 CrossRef PubMed.
- W. Zhou, H. Qiu, Y. Guo and W. Guo, J. Phys. Chem. B, 2020, 124, 1611–1618 CAS.
- S. Wang, J. Cao, W. Jia, W. Guo, S. Yan, Y. Wang, P. Zhang, H. Chen and S. Huang, Chem. Sci., 2019, 11, 879–887 RSC.
- E. L. Bonome, F. Cecconi and M. Chinappi, Nanoscale, 2019, 11, 9920–9930 RSC.
- G. D. Muccio, A. E. Rossini, D. D. Marino, G. Zollo and M. Chinappi, Sci. Rep., 2019, 9, 6440 CrossRef PubMed.
- B. Shen, P. Piskunen, S. Nummelin, Q. Liu, M. A. Kostiainen and V. Linko, ACS Appl. Bio Mater., 2020, 3, 5606–5619 CrossRef CAS PubMed.
- H. Yang, M. Saqib and R. Hao, Front. Chem., 2021, 9, 664820 CrossRef CAS PubMed.
- M. Waugh, K. Briggs, D. Gunn, M. Gibeault, S. King, Q. Ingram, A. M. Jimenez, S. Berryman, D. Lomovtsev, L. Andrzejewski and V. Tabard-Cossa, Nat. Protoc., 2019, 15, 122–143 CrossRef PubMed.
- A. Singh, Nat. Methods, 2023, 20, 1870 CrossRef CAS PubMed.
- G. Yao, W. Ke, B. Xia and Z. Gao, Chem. Sci., 2024, 15, 6229 RSC.
- M. Ahmad, J.-H. Ha, L. A. Mayse, M. F. Presti, A. J. Wolfe, K. J. Moody, S. N. Loh and L. Movileanu, Nat. Commun., 2023, 14, 1374 Search PubMed.
- M. Farshad and J. C. Rasaiah, ACS Appl. Nano Mater., 2020, 3, 1438–1447 CrossRef CAS.
- Y. Zhou and H. Wang, ACS Omega, 2022, 7, 16422–16429 Search PubMed.
- M. Shankla and A. Aksimentiev, Nat. Nanotechnol., 2019, 14, 858–865 CrossRef CAS PubMed.
- A. Nicola, A. Rath, P. Delarue and P. Senet, Nanoscale, 2020, 12, 22743–22753 RSC.
- J. Kong, N. A. W. Bell and U. Keyser, Nano Lett., 2016, 16, 3557–3562 Search PubMed.
- Y. He, M. Tsutsui, C. Fan, M. Taniguchi and T. Kawai, ACS Nano, 2011, 5, 8391–8397 CrossRef CAS PubMed.
- F. Mouvet, J. Villard, V. Bolnykh and U. Rothlisberger, Acc. Chem. Res., 2022, 55, 221–230 CrossRef CAS PubMed.
- S. Chmiela, H. E. Sauceda, K. Müller and A. Tkatchenko, Nat. Commun., 2018, 9, 3887 CrossRef PubMed.
- D. Garoli, H. Yamazaki, N. Maccaferri and M. Wanunu, Nano Lett., 2019, 19, 7553–7562 Search PubMed.
- H. Yan, G. Hu, Z. Wu, T. Chen, L. Wu, Z. Lu and J. Tu, Chem. Eng. J., 2024, 499, 156560 CrossRef CAS.
- W. Zhou, Y. Guo, W. Guo and H. Qiu, J. Phys. Chem. Lett., 2024, 15, 5556–5563 CrossRef CAS PubMed.
- Y. Ying and Y. Long, J. Am. Chem. Soc., 2019, 141, 15720–15729 CrossRef CAS PubMed.
- R. P. Thomsen, M. Malle, A. Okholm, S. Krishnan, S. Bohr, R. S. Sørensen, O. Ries, S. Vogel, F. Simmel, N. Hatzakis and J. Kjems, Nat. Commun., 2019, 10, 5655 CrossRef CAS PubMed.
- Y. He, M. Tsutsui, Y. Zhou and X.-S. Miao, NPG Asia Mater., 2021, 13, 48 CrossRef.
- Z. Wang, T. yi Lv, Z. Shi, S.-S. Yang and Z. Gu, Dalton Trans., 2021, 50, 13608–13619 RSC.
- M. Shankla and A. Aksimentiev, ACS Appl. Mater. Interfaces, 2020, 12, 26624–26634 Search PubMed.
- D. F. Cairns-Gibson and S. L. Cockroft, Chem. Sci., 2021, 13, 1869–1882 RSC.
- C. Cao, D.-F. Liao, J. Yu, H. Tian and Y. Long, Nat. Protoc., 2017, 12, 1901–1911 CrossRef CAS PubMed.
- Y.-C. Yao, Z. Li, A. J. Gillen, S. Yosinski, M. A. Reed and A. Noy, J. Chem. Phys., 2021, 154, 204704 CrossRef CAS PubMed.
- M.-Z. Huo, M.-Y. Li, Y.-L. Ying and Y.-T. Long, Anal. Chem., 2021, 93, 11364–11369 Search PubMed.
- G. Di Muccio, A. E. Rossini, D. Di Marino, G. Zollo and M. Chinappi, Sci. Rep., 2019, 9, 6440 CrossRef PubMed.
- A. K. Thakur and L. Movileanu, Nat. Biotechnol., 2019, 37, 96–101 CrossRef CAS PubMed.
- Y. Wang, J. Yuan, H. Deng, Z. Zhang, Q. D. Ma, L. Wu and L. Weng, Biosensors, 2022, 12, 1152 CrossRef CAS PubMed.
- D. Dematties, C. Wen, M. D. Pérez, D. Zhou and S.-L. Zhang, ACS Nano, 2021, 15, 14419–14429 CrossRef CAS PubMed.
- R. L. Hayes, J. Buckner and C. L. Brooks III, J. Chem. Theory Comput., 2021, 17, 6799–6807 CrossRef CAS PubMed.
- S. Páll, A. Zhmurov, P. Bauer, M. Abraham, M. Lundborg, A. Gray, B. Hess and E. Lindahl, J. Chem. Phys., 2020, 153, 134110 Search PubMed.
- J. Kleinjung and F. Fraternali, Curr. Opin. Struct. Biol., 2014, 25, 126–134 CrossRef CAS PubMed.
- S. Izadi, R. Anandakrishnan and A. Onufriev, J. Chem. Theory Comput., 2016, 12, 5946–5959 CrossRef CAS PubMed.
- A. S. Kamenik, S. M. Linker and S. Riniker, Phys. Chem. Chem. Phys., 2021, 24, 1225–1236 RSC.
- V. Rizzi, S. Aureli, N. Ansari and F. Gervasio, J. Chem. Theory Comput., 2023, 19, 5731–5742 Search PubMed.
- C. Wen, D. Dematties and S.-L. Zhang, ACS Sens., 2021, 6, 3536–3555 CrossRef CAS PubMed.
- M. Tsutsui, T. Takaai, K. Yokota, T. Kawai and T. Washio, Small Methods, 2021, 5, 2100191 CrossRef CAS PubMed.
- M. Taniguchi, S. Minami, C. Ono, R. Hamajima, A. Morimura, S. Hamaguchi, Y. Akeda, Y. Kanai, T. Kobayashi and W. Kamitani, et al. , Nat. Commun., 2021, 12, 3726 CrossRef CAS PubMed.
- M. Li, J. Wang, C. Zhang, X. Zhao, Y. Xiong, Y. Cao, D. Wang, X. Li, X. Liang and G. Qing, ACS Nano, 2024, 18, 25155–25169 CrossRef CAS PubMed.
- D. Qiao, H. Joshi, H. Zhu, F. Wang, Y. Xu, J. Gao, F. Huang, A. Aksimentiev and J. Feng, J. Am. Chem. Soc., 2021, 143, 15975–15983 Search PubMed.
| This journal is © The Royal Society of Chemistry 2025 |