
Response of a polyelectrolyte under oscillatory shear of low frequency†
Hao
Peng
ab,
Chao
Zhou
ab,
Wu
Zhou
ab,
Jingfa
Yang
ab and
Jiang
Zhao
 *ab
*ab
aBeijing National Research Center for Molecular Sciences, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China. E-mail: jzhao@iccas.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
First published on 29th May 2025
Abstract
It is still a challenging task to gain a comprehensive understanding of the physical mechanism of rheological properties of polyelectrolyte solutions. For the purpose of investigating the molecular picture, a study is conducted to examine the counterion distribution of the charged main chain under oscillatory shear of low frequency, using single molecule fluorescence spectroscopy. Adopting sodium polystyrene sulfonate (NaPSS) as the model polyelectrolyte, the local concentration of hydronium ions around the PSS− chain is measured in situ as a function of shear strain and shear frequency, by monitoring the fluorescence intensity of a pH-responsive fluorophore attached to the PSS− chain end, by single molecule confocal fluorescence spectroscopy. The results not only show that the counterions depart from the charged main chain when shear is applied but also that the response increases nonlinearly with shear strain and shear frequency. Systematic investigations are performed with molecular weight, salt concentration and polyelectrolyte concentration. The discoveries indicate that the shear-induced counterion departure depends on the strength of the inter-chain electrostatic interaction, which determines the rigidity or resilience of the “cage-like” structure formed by multiple neighboring charged chains.
Introduction
Charged macromolecules, including synthetic polyelectrolytes and biomacromolecules, are among the most important but the least understood macromolecules. Different to neutral polymers, the main chain of charged macromolecules carries multiple permanent charges or ionizable groups, which can be charged upon dissolution in polar solvents.1–5 Polyelectrolytes are very useful and found in many applications, such as drug delivery,6–8 water processing,9,10 functional hydrogels,11,12 robust coating,13,14 and so forth. Biomacromolecules, such as DNA and proteins, play important roles in rendering biological functions.15 Charged macromolecules exhibit unique properties distinct from their neutral counterparts, due to the long-range electrostatic interaction and the presence of numerous counterions. Unique dynamics and rheological properties of charged macromolecules are essentially important in a variety of industrial and biological processes, including oil drilling, battery performance, food and cosmetics, enzyme activities, biological fluids, and so forth.16–22The dynamics and rheological properties of charged macromolecules have long been attracting research attention23–27 while there are still lots of unsolved puzzles, such as the physical origins of the non-Gaussian diffusive motion of charged main chains,28 the extraordinary–ordinary transition of the dynamical modes,29 the well-known concentration dependence of the viscosity of polyelectrolyte solutions,16 the concentration dependence of the shear-thinning process,30 and so forth. It has long been recognized that it is important to have access to molecular information during the rheological process, such as chain conformation,25,26 effective charges and counterion distribution.31,32
The effective charge of the main chain is believed to be the key property-determining factor.33,34 In contrast to the long-predicted fixed charge density of the main chain,33 recent investigations have demonstrated that the effective charge depends on a number of factors,34 such as salt concentration,35 polymer concentration,36 molecular weight,37,38 as well as molecular topography.41 Related to chain dynamics and rheological properties, it was recently discovered that the counterions of polyelectrolytes depart from the main chains under steady shear, leading to a state of promoted effective charge amount of the main chain. Under this kind of situation, the inter-chain electrostatic repulsion is enhanced, providing a stronger driving force pushing the chains back to the original equilibrium state.31
The results of the previous investigations strongly suggest that the effect of shear is not by acting on the individual charge chains but instead on the transient network formed by multiple chains inside the solution, due to the long-range inter-chain electrostatic repulsion.31,32 The effect by shear is through the distortion of the network and the amplitude of the shear-induced effect depends highly on the salt concentration and molecular weight of polyelectrolytes. It is anticipated that the effect of shear acting on such a dynamic network should rely heavily on shear strain and frequency, and the investigation in this direction should provide rich information on the dynamical response of the system. In the current study, the effect of oscillatory shear on the distribution of counterions of a model polyelectrolyte, sodium polystyrene sulfonate (NaPSS), is investigated. A home-built rheo-microspectrometer is used to access the information of counterion distribution by monitoring the fluorescence emission intensity of a pH-responsive fluorophore as the indicator of the local concentration of hydronium ions, that serve partially as the counterions of the PSS− chain. Systematic investigations into the dependence on molecular weight, salt concentration, and polyelectrolyte concentration have been conducted. The results reveal the nonlinear dependence of the response of the counterion distribution of the charged main chain to external shear, on the shear strain and frequency, providing experimental information helping to understand the molecular mechanism of the rheological properties of charged macromolecules.
Experimental section
Materials
NaPSS was prepared by sulfonation of the parent amino-terminated polystyrene (PS, purchased from Polymer Source). The PS sample had three molecular weights (Mn = 11 × 103 g mol−1, Mw/Mn = 1.11; Mn = 29 × 103 g mol−1, Mw/Mn = 1.08; Mn = 58 × 103 g mol−1, Mw/Mn = 1.08). A well-established protocol was adopted to sulfonate the PS samples and the degree of sulfonation was measured to be almost 100%, without detectable degradation or cross-linking.43 The sulfonated samples were later treated by dialysis and freeze-drying and the final products were denoted as NaPSS21k, NaPSS58k, and NaPSS115k, respectively.The NaPSS samples were further labeled with a pH-responsive fluorescent molecule, Oregon Green 514 (OG 514, Thermo Fisher), through the terminating amino group by reacting with the succinimidyl ester group of the fluorescent molecule. In solution, the OG 514 probe is estimated to be approximately 1 nm away from the chain end.42 The chemical structure of NaPSS labeled with OG 514 is shown in the ESI.† Due to the association–dissociation process, the fluorescence emission intensity of OG514 depends on the pH value and therefore it serves as an effective way of measuring the hydronium ion concentration around it.31,32,41 After fluorescence labeling, the sample was purified by polyacrylamide gel chromatography and ultrafiltration through a filter with a cutoff molecular weight of 2000 Dalton. The purification by ultrafiltration was terminated when no fluorescence signal was detected inside the filtrate at the single molecular level. To prepare the sample solution for measurements, the concentrated sample solution was further diluted by deionized water with a resistivity of 18.2 MΩ cm (Millipore, Milli-Q).
Fluorescence correlation spectroscopy and laser light scattering
The labeling efficiency was determined by fluorescence correlation spectroscopy (FCS) and laser light scattering (LLS). FCS was used to measure the diffusion coefficient and concentration of fluorescence-labeled NaPSS chains. The principles of FCS have been described and reviewed previously.44–46 In brief, the FCS experimental setup is based on a confocal microscope and it measures the dynamics of fluorescent molecules by monitoring the fluorescence fluctuations inside the confocal volume (the excitation-detection volume) due to the dynamics of the systems under investigation. The confocal volume has an ellipsoid shape with a radius of ∼250 nm and a half-length of ∼2.0 μm. The autocorrelation function of the fluorescence fluctuation is measured and later fitted using suitable theoretical models, from which the parameters of the dynamical process are calculated. In the case of translational diffusion, the diffusion coefficient and average concentration of the fluorescent molecule are calculated by fitting with the diffusion model.47The FCS experiments were performed on a commercial setup (LSM 780, Zeiss). A water-immersion objective lens (UPlanApo 40×, number aperture = 1.20, working distance = 0.25 mm) was used. To conduct measurements, the dimension of the excitation-detection volume was firstly calibrated by measuring and fitting the autocorrelation function of a standard sample (Rhodamine 6G) with the known diffusion coefficient and pre-determined concentration. Afterward, FCS measurements were conducted with the fluorescence-labeled NaPSS and the values of diffusion coefficient and concentration were determined.
LLS measurements were conducted to establish a method to measure the concentration of unlabeled NaPSS. The experiments were performed with a particle size analyzer (Zetasizer Pro, Malvern Panalytical). The expression of intensity of scattered light (Is) is Is = (2π2(1 + cos2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) θ)n02/λ4r2)(dn/dc)2(cM/NA)I0, where θ is the scattering angle, n0 is the refractive index of solvent, λ is the wavelength of laser light, r is the distance between the detector and scattering center, n is the refractive index of the solution, c is the concentration, M is the molecular weight, NA is Avogadro's constant, and I0 is the incident light intensity, respectively.48 For the NaPSS sample with a known molecular weight, Is is proportional to c with fixed values of θ, λ, r and I0. By obtaining the Is values of un-labeled NaPSS solution with pre-determined concentrations, the concentration of fluorescence-labeled NaPSS solution can be readily determined by comparing its Is value with the un-labeled NaPSS solution. Combining the results of FCS and LLS, the labeling efficiency was determined. In the current study, the labeling efficiencies of NaPSS21k-OG 514, NaPSS58k-OG 514, and NaPSS115k-OG 514 were 85%, 71%, and 80%, respectively.
θ)n02/λ4r2)(dn/dc)2(cM/NA)I0, where θ is the scattering angle, n0 is the refractive index of solvent, λ is the wavelength of laser light, r is the distance between the detector and scattering center, n is the refractive index of the solution, c is the concentration, M is the molecular weight, NA is Avogadro's constant, and I0 is the incident light intensity, respectively.48 For the NaPSS sample with a known molecular weight, Is is proportional to c with fixed values of θ, λ, r and I0. By obtaining the Is values of un-labeled NaPSS solution with pre-determined concentrations, the concentration of fluorescence-labeled NaPSS solution can be readily determined by comparing its Is value with the un-labeled NaPSS solution. Combining the results of FCS and LLS, the labeling efficiency was determined. In the current study, the labeling efficiencies of NaPSS21k-OG 514, NaPSS58k-OG 514, and NaPSS115k-OG 514 were 85%, 71%, and 80%, respectively.
Rheo-microspectrometer and the application of oscillatory shear
The investigation into counterion redistribution of the NaPSS singe chain under oscillatory shear was performed on a home-built rheo-microspectrometer. A detailed description of this apparatus was provided in a previous publication,31,32 while it is briefly introduced here. The apparatus integrates a shearing device with single molecule fluorescence microscopy and spectroscopy, including fluorescence correlation spectroscopy, photon counts histogram spectroscopy, singe molecule fluorescence imaging, and single molecule fluorescence emission spectroscopy. It is composed of an advanced rheometer (MCR 302 WESP, Anton Paar) and an inverted optical microscope (IX71, Olympus), to which an EMCCD camera (Evolve 512, Photometrics), two single photon counting modules (H7421, Hamamatsu) and a spectrometer (SR303i-A, Andor) with an EMCCD (DU970P-BVF, Andor) were equipped.In the current study, the circularly polarized laser beam of 473 nm served as the excitation light source. A water-immersion objective lens (Plan Apochromat 60×, NA = 1.2) was used to focus excitation light into the sample under shear and to collect fluorescence emissions from the sample at the same time. The fluorescence excitation and collection were done through a window at the bottom of the sample cell of the rheometer, which was precisely positioned above the microscope. The sample solution was held between the cone plate of the rheometer (diameter: 40 mm, cone angle: 2°) and the bottom plate, which was made of a circular cover glass with a thickness of ∼200 μm supported on the rigid frame. The fluorescence signal collected by the objective lens passed the optimized optical path and a pinhole of 50 μm in diameter and then split into two components of nearly identical intensity and finally recorded by the single photon counting modules.
In the experiments, the sample was a mixture of fluorescence labeled NaPSS with a concentration of 1.0 × 10−8 M (10 nM) and unlabeled NaPSS (5 nM) and the pH of the sample solution was 6.5. The pH value of the sample was adjusted by adding the HCl and NaOH solution to minimize the effect brought by salt. First of all, the sample solution (0.54 mL) was applied into the sample cell and afterwards a small amount of decane was added around the outer rim so that a thin layer of decane was formed at the edge to minimize the effect of dissolution of airborne CO2 into the sample solution. Control experiments were performed to check the conductivity of the samples before and after shear measurements and the results showed slight increases in conductivity, indicating the occurrence of minor CO2 dissolution. The laser intensity at the objective lens was ∼20 μW and the excitation-detection volume was located at 30 μm above the bottom of the sample cell.
Oscillatory shear was actuated by the rheometer with a certain shear strain (γ) and frequency (ν) and the fluorescence intensity was detected at the time resolution of 20 ms for 20 s and the averaged value (photon counts per second) was taken as the signal value with the subtraction of the background signal from the pure water.
Results and discussions
When shear is applied to the sample solution with different strain and frequency, it is immediately discovered that fluorescence emission from OG514 attached to the NaPSS chain end increases, compared with the situation of zero shear. As an example, Fig. 1a shows that the fluorescence intensity from NaPSS58k-OG514 increases about 30% at the strain of 50% and frequency of 5 Hz – the temporal profile of the fluorescence intensity agrees well with the time period of shear application, denoted by two dashed lines in the figure. The fluorescence intensity shows an immediate increase when shear is applied and a gradual decrease when shear is terminated. As described above, the OG514 probe is pH-responsive as its fluorescence emission increases at the elevation of the pH value around it.31,32,36–38 Therefore, the increase of fluorescence intensity indicates a decrease in local concentration of hydronium ions at the vicinity of the PSS− chain when shear is applied, showing that the counterions depart from the chain induced by shear.31,32 The results of Fourier transformation analysis of the temporal variation of fluorescence under different shear frequency are displayed in Fig. 1b, in which the characteristic peaks at the doubles of the shear frequency of each case are discovered. This fact provides direct evidence that the effect is merely due to the shear and indicates the effect shows up twice within one shear circle, i.e., it occurs once in each shear direction, meaning that it does not depend on the shear direction.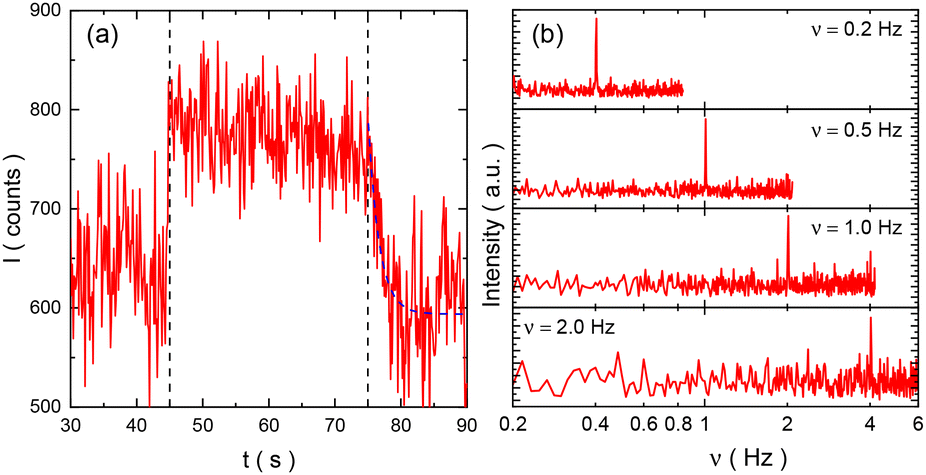 | ||
| Fig. 1 (a) An example of the temporal profile of fluorescence intensity of the solution of NaPSS58k-OG514 with and without oscillatory shear with γ of 50% and ν of 5 Hz. The black dashed line at 45 s denotes the moment of shear application and that at 75 s denotes the shear termination. The blue dashed line denotes the result of fitting by exponential decay. (b) Power spectra of the temporal profile under different shear frequency. The shear strain is 100% and the frequencies are specified in the figure. | ||
The dependence of local concentration of hydronium ions (expressed as −log[H+]local)31,32,35 on shear strain and frequency under different salt concentrations are displayed in Fig. 2. The values of −log[H+]local are determined by comparing the fluorescence intensity of OG514 attached to the NaPSS chain with that of free OG514 under different pH values, as detailed in previous publications.31,32,35–41 Just as demonstrated by previous investigations, the initial value of −log[H+]local increases with salt concentration for each molecular weight sample, as a result of enhanced counterion adsorption, creating a smaller difference between the concentration of hydronium ions around the PSS− chain and that in the bulk solution, which is at the infinite distance to the chain.31,32 Under each salt concentration, the initial −log[H+]local value is lower for the sample of the higher molecular weight, also due to the stronger electrostatic attraction from the longer charged main chain, just as previously demonstrated by the experimental evidence of the higher local hydronium concentration and higher adsorption rate of counterions.31,32,37–40
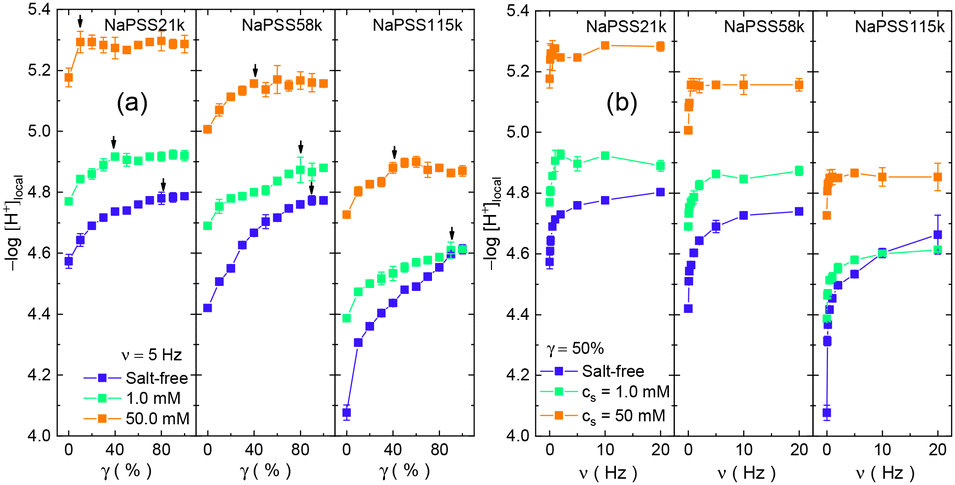 | ||
| Fig. 2 (a) The local concentration of hydronium ions at the vicinity of PSS− chains (expressed as −log[H+]local) as a function of shear strain at the frequency of 5 Hz, under different salt concentrations. The molecular weight and salt concentration are specified in the figure. The black arrows denote the saturation points of the shear effect. (b) The value of −log[H+]local as a function of shear frequency at the strain of 50%, under different salt concentrations. | ||
Data in Fig. 2 show that the −log[H+]local value increases monotonously with shear strain and frequency, and it saturates at high enough shear strain and frequency. These are observed for all conditions, i.e., under different salt concentrations and for all molecular weights, indicating the continuous decreases in hydronium concentration under shear. The amplitude of the shear effect decreases with the elevation of salt level. As an example in Fig. 2, for the case of NaPSS21k, the initial −log[H+]local value at zero strain is 4.57 and it increases to 4.79 at γ of 100% under salt-free conditions, corresponding to a reduction in local hydronium concentration, [H+]local, of 1.7-fold. For 1.0 mM salt concentration, the change in hydronium concentration is 1.4-fold, and it becomes 1.2-fold for a 50.0 mM salt concentration. The changes expressed by the ratio between local hydronium concentration between zero and 100% shear strain for all conditions are listed in Table 1, in which the smaller changes in [H+]local under higher salt concentrations are evidenced. Another important observation is that the changes in [H+]local are bigger with higher molecular weight samples.
| c s | [H+]localatγ=0/[H+]localatγ=100% |
[H+]localatν=0/[H+]localatν=20Hz |
||||
|---|---|---|---|---|---|---|
| NaPSS21k | NaPSS58k | NaPSS115k | NaPSS21k | NaPSS58k | NaPSS115k | |
| Salt-free | 1.7 | 2.2 | 3.3 | 1.6 | 1.7 | 2.2 |
| 1.0 mM | 1.4 | 1.6 | 1.7 | 1.2 | 1.4 | 1.4 |
| 50.0 mM | 1.2 | 1.4 | 1.4 | 1.1 | 1.2 | 1.1 |
Similar behaviors are found in the response of the molecule to shear frequency, as shown in Fig. 2b for the case of γ = 50%. The −log[H+]local value increases with shear frequency and saturates at high enough frequency. It is again observed that the shear effect decreases when the salt concentration is increased. The results are summarized in Table 1, in which the reduced effect at elevated salt level and the enhanced effect at higher molecular weight are clearly demonstrated. (Control experiments using NaPSS labeled with a non-pH-responsive fluorophore (Alexa 488), OG 514 along, and unlabeled NaPSS mixed with OG 514, do not show any change in fluorescence emission upon shear applications.) Although the data here only show the decrease of concentration of hydronium ions near the chain end, it is envisioned that the same process occurs at the other location of the main chain, because a previous study has demonstrated a higher local concentration of counterions at the middle of the chain than the end and that they change simultaneously.41
An interesting feature of the data in Fig. 2 is that the response starts to saturate at high shear strains. The point at which the shear effect saturates, either by strain or frequency, has lower values under higher salt concentrations. As demonstrated in Fig. 2a, for each molecular weight sample, the point of saturation shifts to lower γ values at elevated salt level, and at each salt level, the point shifts to lower γ values for lower molecular weights. In Fig. 2b, the data show a sharp response at the low frequency region, typically below 5 Hz, beyond which the increase of −log[H+]local value with frequency becomes gradual and, in some cases, even becomes constant at higher salt concentrations. This behavior is also discovered to have a molecular weight dependence: the increase in −log[H+]local value with frequency is more gradual for the higher molecular weight sample compared with the lower one.
The dependence of the response of the NaPSS molecule to shear on strain and frequency indicates the alternation of the solution structure formed at the equilibrium condition, i.e., the condition without external shear. As has been well-documented, the long-range inter-chain electrostatic repulsion creates a “cage-like” structure inside the solution, i.e., every charged chain is surrounded by its nearest neighboring chains.28,49–57 Under such an equilibrated solution structure, a stabilized distribution of the electric field is also resulted in, as demonstrated in the left panel in Fig. 3. This kind of distribution is dynamical because the PSS− chains are constantly undergoing diffusive motion. Following the distribution of the electric field, the distribution of the counterions, that are constantly adsorbing on and desorbing off the main chain, is established. This process results in a stabilized local concentration of the counterions surrounding each chain and therefore its equilibrated effective charged state. When external shear is applied to the solution, this “cage-like” structure is perturbed or changed by the mutual displacement of the chains between the neighboring shear planes. (The low Raynolds number of the system excludes the situation of turbulence in the shear process.31) When the mutual positions of neighboring chains are changed by shear, the distribution of the electric field among them is also changed and therefore the distribution of the counterions accordingly (right panel in Fig. 3). From a microscopic point of view, the mutual inter-chain distance is the largest at equilibrium. As the shear is applied, the inter-chain distance must be shortened. When two PSS− chains are brought closer to each other from their equilibrated positions, their electric fields overlap (right panel, Fig. 3), and for a counterion previously attracted “solely” by one PSS− chain, it becomes attracted by two chains simultaneously and therefore has a higher probability of departing from the original “host” chain and exploring more space in the solution.31,32 As a consequence, the local counterion concentration around the PSS− chain decreases, and meanwhile, it causes an increase in the effective charges of the main chain. From the viewpoint of free energy, the shear-induced perturbation of the solution structure results in a lower entropy of the system and the counterions are released to compensate the free energy cost due to this entropy reduction.
 | ||
| Fig. 3 A schematic illustration of the physical mechanism of the shear-induced counterion departure of polyelectrolytes. Each blue circle represents the electric field of a polyelectrolyte molecule (denoted by the curves in red in the centers of the blue circles). Left: The distribution of the polyelectrolyte molecules at the equilibrium state (without shear); right: the distribution of the molecules at the out-of-equilibrium state under shear. The overlap of the electric fields is demonstrated. | ||
This process results in a stronger inter-chain repulsion, which in turn can promote this out-of-equilibrium state to recover back to the original equilibrium. This kind of recovery process is reflected in the gradual decay of fluorescence intensity with a characteristic time constant of ∼2 s after the termination of the shear, as displayed in Fig. 1. This time scale agrees with the frequency (∼10−1 Hz) at which the shear effect starts to saturate.
It is estimated that the recovery of the solution structure takes a much longer time compared with the mere diffusion process. The diffusion coefficient of NaPSS21k, NaPSS58k and NaPSS115k at the concentration of 15 nM under salt-free conditions is 65.0, 34.2 and 12.8 μm2 s−1, respectively. (Measurements were conducted by FCS and the data are detailed in the ESI.†) Considering the experimental condition of 50% shear strain, the shear-induced displacement of the chain is about 300 nm. Therefore, the time for the chain to diffuse back cross such a distance is about 0.35, 0.66 and 1.76 ms, respectively. This time scale is much shorter than that corresponding to the frequency at which the shear effect starts to saturate (∼10−1 Hz). It shows that the re-establishment of the solution structure should involve more processes, such as collective motions of the multiple chains. Previous investigations have shown that under electrostatic repulsion, the diffusion of the polyelectrolyte chain experiences abnormality, depending on a few factors of the strength of the electrostatic interaction as salt concentration and molecular weight. The results shown here imply that more complicity in diffusion of the charged chains can be brought about by shear, although no direct experimental data can be provided at this stage due to the difficulties in measuring diffusion under shear.
The dependence of the change in local hydronium concentration on shear strain and frequency shows how resilient the solution structure is to the external shear. When the shear-induced displacement of the chains brings about an alternation of the “cage-like” structure or the dynamical network inside the NaPSS solution, it is against the recovery process driven by the inter-chain repulsion, together with the randomization process via diffusion. The time for the solution structure to recover is dependent on the extent of its distortion. Under a fixed shear frequency, where the time of reaching a certain displacement is fixed, the effect of shear should depend on the extent of the structural distortion as well as the extent of its recovery. If the strain is not too big for the system to recover on time, the shear effect should depend monotonically on the strain. At high enough shear strains under a fixed frequency, when the alternation of the structure or network goes too large for the structure to recover on time, the shear effect saturates. The point of saturation is highly dependent on the strength of the inter-chain electric interaction. Under the condition of a lower salt level or higher molecular weight, the stronger inter-chain electrostatic repulsion makes the “cage-like” structure or network more rigid and more resilient to external shear. When the salt concentration is raised, the inter-chain repulsion is weakened by electrostatic screening and reduced effective charge of the main chain due to enhanced counterion adsorption, making the “cage-like” structure easier to change and harder to recover. As a result, the shear-induced effect saturates at much smaller shear strains, compared with the salt-free condition. Because of the same reason, this process can also be found in the saturation of shear effect regarding the molecular weight of NaPSS – the saturation points are lower for smaller molecular weight samples.
The same principle applies to the frequency dependence of the shear response of the molecules. As demonstrated by the previous study,32 the recovery of the solution structure after the distortion is faster under the stronger inter-chain repulsion, because of the stronger driving force. In this sense, the dynamic network inside the solution should be more resilient to high shear rate, exhibiting a much larger response to shear frequency and much higher saturation point under the condition of lower salt concentrations and higher molecular weights.
The shear-induced counterion departure also depends on the concentration of polyelectrolytes (cp). The shear effect as a function of shear strain and frequency under different NaPSS concentration is demonstrated in Fig. 4, in which the sharp decreases in the local hydronium concentration are discovered at low strain and frequency, and the response gets more gradual at higher strains and frequencies. The ratio between the local hydronium concentration between zero and 100% shear strain and that between zero shear and 20 Hz shear frequency for three NaPSS concentrations are listed in Table 2. The data show that the shear effect is weakened at elevated NaPSS concentrations. The changes in local hydronium concentration are the biggest for cp of 15 nM and the changes become much smaller when cp increases to 100 and 200 nM. The shear effect is enhanced for higher molecular weight samples, as demonstrated by the larger difference in local hydronium concentration between zero and 100% shear strain as well as 20 Hz shear frequency. It is also noticed that the shear effect is very much magnified for the high molecular sample (NaPSS115k). (It is noted that the sample concentrations are orders of magnitude lower than the calculated overlap concentration.58)
 | ||
| Fig. 4 The value of −log[H+]local of NaPSS as a function of (a) shear strain and (b) shear frequency, under different NaPSS concentrations for three molecular weights. The concentrations and molecular weights are specified in the figure. | ||
| c p (nM) | [H+]localatγ=0/[H+]localatγ=100% |
[H+]localatν=0/[H+]localatν=20Hz |
||||
|---|---|---|---|---|---|---|
| NaPSS21k | NaPSS58k | NaPSS115k | NaPSS21k | NaPSS58k | NaPSS115k | |
| 15.0 | 1.66 | 2.24 | 3.39 | 1.70 | 2.09 | 3.80 |
| 100.0 | 1.55 | 1.90 | 2.14 | 1.55 | 2.00 | 2.14 |
| 200.0 | 1.51 | 2.04 | 1.91 | 1.55 | 2.00 | 1.95 |
The concentration dependence of the shear-induced counterion departure has exposed further evidence of the effect of the strength of inter-chain electrostatic repulsion on the shear-induced response, originated from the alternation of the “cage-like” structure. The data in Fig. 4 show that the initial local concentration of hydronium ions around the PSS− chain is considerably higher for lower NaPSS concentrations, as a result of the lower adsorption of the counterions on the main chain, resulting in the lower neutralization of the original charges on the main chain.36 When the NaPSS concentration is increased, the reduced entropy penalty promotes counterion adsorption and results in a weaker chain-counterion attraction, and therefore a lower local counterion concentration around the chain.39 The response to shear is much more pronounced for a lower NaPSS concentration, especially for high molecular weight samples, for example, NaPSS115k (Fig. 4a and b). This again demonstrates the stronger inter-chain electrostatic repulsion has established a more rigid or a more resilient “cage-like” structure inside the solution. It is noted that although the initial local hydronium ion concentration is higher at a concentration of 15 nM than those at concentrations of 100 and 200 nM, it eventually becomes the lowest at high enough shear strain and frequency. This is because the total amount of counterions presented in the solution is smaller due to the smaller total number of the charges on the NaPSS chains.
A subtle point is whether the shear effect is due to shear-induced chain stretching or orientation. These possibilities are excluded because these processes are much faster than the time scale of the shear adopted in the current. Detailed discussions on the large difference between the characteristic time of the shear and the Zimm time of the polyelectrolyte chains have been provided in previous publications.31,32
Conclusions
When oscillatory shear is applied to polyelectrolyte solutions, the counterions are found to depart from the charged main chain, as a result of the alternation of the mutual position of the charged chains from their equilibrium. The increase of the overlap extent of the electric fields of neighboring chains increases the probability of the delocalization of the counterions. The amplitude of the response of the counterion redistribution relies on the rigidity or resilience of the “cage-like” structures inside the solution, due to the strength of the inter-chain electrostatic repulsion, which has the dependence on the molecular weight, salt concentration and polyelectrolyte concentration. This process of counterion departure increases the effective charges of the main chain, helping the solution to recover back to its original structure under equilibrium. It is noted that the results of the current study are only related to the shear of low frequency while studies under high shear rate should be more relevant to practical processes. Future investigations will be conducted in this direction.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the National Key R&D Program of China (2023YFE0124500) and Beijing National Research Center for Molecular Science are appreciated.Notes and references
- F. Oosawa, Polyelectrolytes, Dekker, 1971 Search PubMed.
- M. Muthukumar, Physics of Charged Macromolecules: Synthetic and Biological Systems, Cambridge University Press, 2023 Search PubMed.
- A. V. Dobrynin and M. Rubinstein, Prog. Polym. Sci., 2005, 30, 1049–1118 CrossRef CAS.
- S. Förster and M. Schmidt, Polyelectrolytes in solution, Physical Properties of Polymers. Advances in Polymer Science, Springer, Berlin, Heidelberg, 1995, pp. 51–133 Search PubMed.
- C. Holm, J. Joanny, K. Kremer, R. Netz, P. Reineker, C. Seidel, T. A. Vilgis and R. Winkler, Polyelectrolytes with defined molecular architecture II, Springer, 2004, pp. 67–111 Search PubMed.
- O. Boussif, F. Lezoualch, M. A. Zanta, M. D. Mergny, D. Scherman, B. Demeneix and J. P. Behr, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 7297–7301 CrossRef CAS.
- D. W. Pack, A. S. Hoffman, S. Pun and P. S. Stayton, Nat. Rev. Drug Discovery, 2005, 4, 581–593 CrossRef CAS PubMed.
- R. Deng, Y. Yue, F. Jin, Y. C. Chen, H. F. Kung, M. C. M. Lin and C. Wu, J. Controlled Release, 2009, 140, 40–46 CrossRef CAS PubMed.
- H. N. Xiao and N. Cezar, J. Colloid Interface Sci., 2005, 283, 406–413 CrossRef CAS PubMed.
- D. A. Palacio, B. F. Urbano and B. L. Rivas, J. Water Process Eng., 2022, 46, 102582 CrossRef.
- T. L. Sun, T. Kurokawa, S. Kuroda, A. Bin Ihsan, T. Akasaki, K. Sato, M. A. Haque, T. Nakajima and J. P. Gong, Nat. Mater., 2013, 12, 932–937 CrossRef CAS PubMed.
- D. Morales, E. Palleau, M. D. Dickey and O. D. Velev, Soft Matter, 2014, 10, 1337–1348 RSC.
- E. Vazquez, D. M. Dewitt, P. T. Hammond and D. M. Lynn, J. Am. Chem. Soc., 2002, 124, 13992–13993 CrossRef CAS.
- D. V. Andreeva, E. V. Skorb and D. G. Shchukin, ACS Appl. Mater. Interfaces, 2010, 2, 1954–1962 CrossRef CAS.
- R. H. Garrett and C. M. Grisham, Biochemistry, Brooks/Cole, 2013 Search PubMed.
- R. M. Fuoss, J. Polym. Sci., 1948, 3, 603–604 CrossRef CAS.
- H. Eisenberg and J. Pouyet, J. Polym. Sci., 1954, 13, 85–91 CrossRef.
- J. Cohen, Z. Priel and Y. Rabin, J. Chem. Phys., 1988, 88, 7111–7116 CrossRef CAS.
- J. Yamanaka, H. Matsuoka, H. Kitano, M. Hasegawa and N. Ise, J. Am. Chem. Soc., 1990, 112, 587–592 CrossRef CAS.
- D. F. Hodgson and E. J. Amis, J. Chem. Phys., 1991, 94, 4581–4586 CrossRef CAS.
- H. Vink, Polymer, 1992, 33, 3711–3716 CrossRef CAS.
- D. C. Boris and R. H. Colby, Macromolecules, 1998, 31, 5746–5755 CrossRef CAS.
- C. G. Lopez, R. H. Colby, P. Graham and J. T. Cabral, Macromolecules, 2017, 50, 332–338 CrossRef CAS.
- C. G. Lopez, A. Matsumoto and A. Q. Shen, Soft Matter, 2024, 20, 2635–2687 RSC.
- M. Villetti, R. Borsali, O. Diat, V. Soldi and K. Fukada, Macromolecules, 2000, 33, 9418–9422 CrossRef CAS.
- E. Mendes, S. Viale, O. Santin, M. Heinrich and S. J. Picken, J. Appl. Crystallogr., 2003, 36, 1000–1005 CrossRef CAS.
- A. J. Han and R. H. Colby, Macromolecules, 2021, 54, 1375–1387 CrossRef CAS.
- K. Chen, K. K. Zheng, G. F. Xu, J. F. Yang and J. Zhao, Macromolecules, 2019, 52, 3925–3934 CrossRef CAS.
- M. Muthukumar, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 12627–12632 CrossRef CAS.
- A. J. Han and R. H. Colby, Macromolecules, 2021, 54, 1375–1387 CrossRef CAS.
- K. K. Zheng, K. Chen, W. Ren, J. F. Yang and J. Zhao, Macromolecules, 2022, 55, 1647–1656 CrossRef CAS.
- C. Zhou, K. Chen, K. K. Zheng, J. F. Yang and J. Zhao, Macromolecules, 2024, 57, 5739–5746 CrossRef CAS.
- G. S. Manning, J. Chem. Phys., 1969, 51, 924–933 CrossRef CAS.
- M. Muthukumar, J. Chem. Phys., 2004, 120, 9343 CrossRef CAS PubMed.
- S. Q. Wang, S. Granick and J. Zhao, J. Chem. Phys., 2008, 129, 241102 CrossRef PubMed.
- K. K. Zheng, K. Chen, W. B. Ren, J. F. Yang and J. Zhao, Macromolecules, 2018, 51, 4444–4450 CrossRef CAS.
- G. F. Xu, S. Luo, Q. B. Yang, J. F. Yang and J. Zhao, J. Chem. Phys., 2016, 145, 144903 CrossRef.
- G. F. Xu, J. F. Yang and J. Zhao, J. Chem. Phys., 2018, 149, 163329 CrossRef PubMed.
- P. X. Jia, Q. B. Yang, Y. K. Gong and J. Zhao, J. Chem. Phys., 2012, 136, 084904 CrossRef PubMed.
- Y. Shi, H. Peng, J. F. Yang and J. Zhao, Macromolecules, 2021, 54, 4926–4933 CrossRef CAS.
- S. J. Luo, X. B. Jiang, L. Zou, F. Wang, J. F. Yang, Y. M. Chen and J. Zhao, Macromolecules, 2013, 46, 3132–3136 CrossRef CAS.
- J. Zhao, J. Chem. Phys., 2020, 153, 170903 CrossRef CAS.
- H. Vink, Die Makromol. Chem., 1981, 182, 279–281 CrossRef CAS.
- D. Magde, E. Elson and W. W. Webb, Phys. Rev. Lett., 1972, 29, 705–708 CrossRef CAS.
- E. L. Elson and D. Magde, Biopolymers, 1974, 13, 1–27 CrossRef CAS.
- R. Rigler, U. Mets, J. Widengren and P. Kask, Eur. Biophys. J. Biophys. Lett., 1993, 22, 169–175 CAS.
- S. Q. Wang and J. Zhao, J. Chem. Phys., 2007, 126, 091104 CrossRef PubMed.
- B. Chu, Laser Light Scattering: Basic Principles and Practice, Academic Press, 1991 Search PubMed.
- A. L. Thorneywork, R. E. Rozas, R. P. Dullens and J. Horbach, Phys. Rev. Lett., 2015, 115, 268301 CrossRef PubMed.
- P. N. Pusey, J. Phys. A: Math. Gen., 1977, 11, 119–135 CrossRef.
- W. Vanmegen, S. M. Underwood and I. Snook, J. Chem. Phys., 1986, 85, 4065–4072 CrossRef CAS.
- F. B. Khorasani, R. Poling-Skutvik, R. Krishnamoorti and J. C. Conrad, Macromolecules, 2014, 47, 5328–5333 CrossRef CAS.
- M. Sedlak, Langmuir, 1999, 15, 4045–4051 CrossRef CAS.
- M. Drifford and J.-P. Dalbiez, J. Phys. Chem., 1984, 88, 5368–5375 CrossRef CAS.
- M. Muthukumar, J. Chem. Phys., 1997, 107, 2619–2635 CrossRef CAS.
- J. F. Li, T. Ngai and C. Wu, Polym. J., 2010, 42, 609–625 CrossRef CAS.
- L. G. Feng, J. F. Yang, J. Zhao, D. P. Wang, K. Koynov and H.-J. Butt, J. Chem. Phys., 2013, 138, 214902 CrossRef PubMed.
- C. Wandrey, Langmuir, 1999, 15, 4069–4075 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sm00254k |
| This journal is © The Royal Society of Chemistry 2025 |