
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLanthanide-based metal–organic frameworks (Ln-MOFs): synthesis, properties and applications
Kankan
Patra
 *ab and
Haridas
Pal
*bc
*ab and
Haridas
Pal
*bc
aNuclear Recycle Board, Bhabha Atomic Research Centre, Tarapur 401504, India. E-mail: kankan.patra2010@gmail.com
bHomi Bhabha National Institute, Anushaktinagar, Mumbai 400 094, India. E-mail: hpal59@hbni.ac.in
cChemistry Group, Bhabha Atomic Research Centre, Mumbai 400 085, India. E-mail: hpal@barc.gov.in
First published on 3rd January 2025
Abstract
Micro- and meso-porous solid materials based on metal–organic frameworks (MOFs) have been gaining significant attention for the last three decades as they offer diverse applications in a large number of areas. An advantage of these materials is that they can be rationally designed with desired characteristics using several metal ions belonging either to the s-, p-, d-, or f-block elements of the periodic table, in combination with suitable polytopic organic linkers (multidentate ligands), resulting in various structural and application aspects. Among the MOFs, those composed of lanthanide ions {Ln(III)}, commonly referred to as Ln-MOF systems, have attracted enormous attention because they display favorable characteristics, like large structural diversity, tailorable structural designs, tunable porosity, large surface area, high thermal stability, and immense chemical stability. All these characteristics are very useful for their widespread applications in diverse areas. Since Ln(III) ions possess higher coordination numbers compared to transition metal (TM) ions, Ln-MOF materials are generally more porous, offering better applications. Further, hybrid MOF systems consisting of both Ln(III) and TM ions (Ln–TM-MOF systems) can introduce additional features to these mixed metal porous materials for their much wider applications. Luminescence and magnetic properties of Ln(III) ions make these materials ideal for various display and sensing applications, in addition to their porosity-related applications. In this review article, our aim is to discuss the basic aspects, preparation methodologies, important properties, and utilizations of MOF materials with a special emphasis on Ln(III)-based MOF systems. Initially, a short introduction is provided on MOF systems, which is followed by other aspects of these materials as mentioned above. Subsequently, we sequentially highlight the interesting characteristics of these materials, including their structural aspects, porosity, magnetic properties, and luminescence behavior. Finally, some of the potential uses of these systems have been presented with special emphasis on their gas storage, catalysis and luminescence-based chemical sensing applications.
Sustainability spotlightIn recent times, porous Ln-MOF (lanthanide-based metal–organic framework) materials have attracted immense academic and applied interests owing to their many advantages, including their unique framework structures, extensive porosity, large surface area, ample opportunities for derivatization with functional groups, and excellent chemical stability, in comparison to other conventional porous materials. We have presented potential features of Ln-MOFs along with their synthesis procedures, important structural characteristics and relevant categorization of these solid porous network systems. The important properties that have been discussed are the porous characteristics of these materials, their magnetic properties, and their luminescence characteristics. Furthermore, we have explored the potential applications of Ln-MOF materials, including gas separation and storage, catalysis, and luminescence-based chemical sensing. |
1. General introduction to metal–organic frameworks (MOFs)
The unique crystalline and highly porous inorganic–organic hybrid materials constituted through regular arrays of metal nodes (metal ions or their clusters) interconnected via coordination bonds created by the involvement of organic linker molecules are designated as metal–organic framework (MOF) materials, or simply MOFs.1 In these materials, metal nodes are tightly held together in three dimension with regular arrangements, where organic linkers provide the anchoring arms and act as the spacers for the metal nodes to maintain their three-dimensional highly ordered arrangements, providing these materials with very unique network structures that possess numerous rigid inbuilt pores, useful for diverse applications. As mentioned before, the metal nodes in these materials can be either individual metal ions, clusters of metal ions, or even some type of metal compounds that can provide required coordination sites to hold multiple organic linkers, leading to the formation of the desired MOF network. Since the last three decades, MOF materials have been realized as the most intriguing and promising inorganic–organic hybrid solid-state materials owing to their unique characteristics, like high porosities, controllable size and shapes of their inbuilt pores, tailormade network structures, high crystallinity, high thermal and chemical stability, and ready functionalization of their pore walls.2,3It is possible to prepare MOF materials using cations of several metallic elements belonging either to the s, p, d, or f block elements, which can display varying coordination numbers, diverse coordination geometries, and different oxidation states. These metal ions can form suitable metal nodes or clusters, which can subsequently undergo interconnections among themselves involving suitable polytopic organic linker molecules, resulting in the formation of a very well-defined, highly regular and extremely ordered network. Consequently, this family of porous MOF materials can display many interesting properties for wide applications.4 The most common metal ions employed in the synthesis of conventional MOF materials include Ti(III), Co(II), Fe(III), Zn(II), Zr(IV), Cu(II), Cd(II), Al(III), Mg(II), and Ca(II) ions. MOF materials can also be obtained using different trivalent lanthanide [Ln(III)] ions, and this special category of MOF systems is designated as the lanthanide-based MOFs, or simply Ln-MOFs. Metal ions or their suitable clusters can offer different structural geometries for coordination, such as tetrahedral, pyramidal, octahedral, trigonal bipyramidal, and square pyramidal, which are essential for the construction of the MOF networks through the formation of coordination bonds with multitopic organic linkers, resulting in the formation of the desired hybrid solid materials possessing inbuilt pores having various sizes and shapes.5,6 Metal ions such as Ag(I), Co(II), and Cd(II) are also known to satisfy the above-mentioned criteria to produce MOF networks; however, the use of these ions is not encouraged in the literature, mainly because of their substantial toxicity.7 In the synthesis of various MOF materials, multitopic (or multidentate) organic linker molecules having amine, sulfonate, carboxylate, or phosphate units as their anchoring terminals are considered as the useful ligand systems for various metal ions and/or metal nodes.8–10 The most common method for the synthesis of conventional MOF and Ln-MOF systems is the bottom-up approach, where organic compounds and metal ions are allowed to interact directly in the reaction medium. This maintains a dynamic equilibrium between the reactant and product systems, thus ensuring the slow growth of the desired MOF materials with a well-ordered structure and crystallinity. With the given synthesis condition, and because the MOF materials are formed through the formation of coordination bonds between the organic linker molecules and metal ions or metal nodes, the formation of these porous network materials occurs in a reversible manner, resulting in the construction of MOF materials with highly-ordered frameworks via the bottom-up approach.11,12
In recent times, the potential application of porous MOF materials has been demonstrated in diverse areas such as analyte sorption, gas storage, catalysis, ion exchange, luminescence sensing, nonlinear optical devices, drug delivery, environmental pollution control, and many others.13–23 It has been found that many MOF materials have outstanding stability under critical conditions such as high acidity, high radiation field, and presence of high concentration of interfering ions, which is very favorable for the application of these materials under harsh conditions.24 In the case of large scale applications, it is essential to develop protocols for the synthesis of the required MOF materials involving very facile and economically viable methods. In this context, it is important to note that most MOF materials offer quite simple synthesis processes with low-cost methodologies.25 Generally, porous MOF materials possess a very large Brunauer–Emmett–Teller (BET) surface area (1000 to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 m2 g−1), which is much larger than that of the traditional adsorbent materials, making the MOF systems very advantageous for various applications in diverse areas.4
000 m2 g−1), which is much larger than that of the traditional adsorbent materials, making the MOF systems very advantageous for various applications in diverse areas.4
MOF materials having a variety of network structures and possessing numerous types of permanent pores having regular patterns, definite sizes and well-defined shapes are considered very promising materials for adsorption-based applications compared to other common porous materials such as activated carbons, activated alumina, silica gels, and zeolites. This is because the pores in the latter materials are very irregular in regard to their size and shape. The other advantages of MOF materials compared to the common porous materials include their tunable physiochemical properties, tailormade orderliness of their porous structures, and high BET surface area. Owing to these advantageous characteristics of MOF materials, they display greater performances as adsorbents, with exceptionally high adsorption capacity for various analytes, including small molecules, toxic organic and inorganic species, and hazardous metal ions. In the last few decades, several studies have been reported in the literature demonstrating various applications of MOF-based materials in diverse areas of chemical sciences.13–21,26–33
From a general point of view, transition metal (TM) and lanthanide (Ln) ions are suitable to form the requisite metal nodes for the construction of various MOF structures. This is because TM and Ln ions are capable of not only displaying multiple coordination numbers but also creating different coordination geometries with the binding of their organic linker molecules, giving rise to the formation of MOF systems with diverse network structures. Compared to TM ions, Ln ions are generally capable of offering more advantages in the formation of MOF systems, given that these ions have distinct electronic, magnetic and optical properties due to the presence of their 4f electrons. Further, Ln ions can also form a substantially higher number of coordination bonds than TM ions, offering more diverge coordination geometries for binding with the organic linkers in the construction of the MOF networks.34 It should be mentioned that although the common coordination numbers for transition-metal ions are typically 4 to 6, in the case of lanthanide ions, this number is characteristically in the range of 6 to 13. Due to these high coordination numbers, MOF systems based on lanthanide ions (Ln-MOFs) have been found to display much better stability than the corresponding MOF materials formed using other metal ions. Evidently, Ln-MOF systems can offer much greater opportunities for the development of smart multifunctional porous MOF materials, which can be efficiently utilized in the areas such as chemical sensing, heterogeneous catalysis, biomedical applications, environmental pollution control, and many others.26–28
In the literature, a numerous review articles have appeared covering the fundamental aspects of the MOF systems.35–40 To date, several reviews and monographs have summarized the fundamental theory of MOFs and their synthesis and important physicochemical properties.41–43 The adsorption of hazardous organic pollutants such as toxic chemicals from industry, poisonous volatile organic compounds, and harmful gases onto MOF materials and their adsorption mechanisms have also been summarized in some of these reviews.44 However, limited review articles have been reported in the literature on the removal or separation of toxic/radioactive metal ions using such porous MOF materials.29,33,42,45 Recently, a review article on the advances of Ln-MOF systems for application in ratiometric fluorescent sensing was reported.46 Mahmoud et al. reported a review on Ln-MOF systems for the luminescence-based sensing of toxic metal ions.47 Quite recently, a review article on the progress in Ln-MOFs and their derivatives was reported, focusing on their catalytic applications.48 Also, recently, Ren et al. published a review paper on proton conductive Ln-MOFs, describing their synthesis strategies, structural features, and recent progress.49 In general, most of the review articles published thus far on transition metal-based MOFs or Ln-MOFs have dealt only with some specific applications of these materials. In contrast, a limited number of review articles have summarized the fundamental aspects of Ln-MOF materials systematically, covering their synthesis, important properties and diverse applications within a single platform. Hence, we felt that it is necessary to present an inclusive review on Ln-MOF-based porous materials, discussing their synthesis, properties and diverse applications comprehensively. Thus, in the present review, we aim to systematically discuss the different characteristics of MOF materials, with special emphasis on lanthanide ion-based Ln-MOF systems. As can be seen, a general introduction to MOF materials has been presented in this section. In the forthcoming section, we present the major procedures for the synthesis of MOF materials, followed by a discussion on the major structural aspects and relevant classifications of the solid porous MOF materials. In the subsequent section, we highlight and discuss the important properties of MOF materials, covering both conventional MOF and Ln-MOF systems, highlighting the crucial roles of their properties towards their various applications. The important properties that will be discussed in this context are the porous characteristics of these materials, their magnetic properties, and their luminescence characteristics. Following this, the next section presents some important applications of MOF materials, emphasizing the applications of Ln-MOF systems, including gas separation and storage, catalysis, and also luminescence-based chemical sensing. Finally, we provide our concluding remarks with future perspectives on the R&D activities in the domain of MOF materials. An overview of the various aspects discussed in the present article is schematically presented in Fig. 1 for quick comprehension. We expect that the presentation and discussion made in this impactful review covering from synthesis to applications of both conventional MOF and Ln-MOF systems will produce significant academic and applied impetus, paving the way for a better understanding and enough attention towards Ln-MOF systems, especially. The new R&D activities in this subject domain are expected to inspire many advanced structural design strategies for these porous materials for the development of high-performance porous solid adsorbent systems that can meet all the major needs for their superior utilization in diverse applied areas.
 | ||
| Fig. 1 Overview of various aspects of Ln-MOF systems presented and discussed in this article. | ||
2. Synthesis methods for the conventional MOF and Ln-MOF materials
Various synthesis methodologies have been adopted in the literature for the preparation of conventional MOF and Ln-MOF materials. All these methods enable the facile formation of porous MOF materials having very complex but highly ordered structural networks, maintaining the unique composition of their constitutional components, and ascertaining the high reproducibility of the formation of these materials with desired crystallinity. The general strategy adopted in most of these cases is the bottom-up approach, where the metal nodes and organic linkers are allowed to react under suitable reaction conditions, as conceptually represented in Fig. 2A. Further, the various strategies adopted in the literature to enhance the performance of MOF materials for advanced applications in diverse areas are schematically represented in Fig. 2B for quick visualization of these concepts. In the following part of this section, we present various aspects of the reported synthesis methodologies for different porous MOF systems, highlighting the advantages and difficulties associated with different approaches. | ||
| Fig. 2 (A) General scheme for the synthesis of MOF materials via the bottom-up approach involving the direct reaction of metal nodes and organic linkers under suitable reaction conditions. (B) Different strategies reported for boosting the performance of MOF materials by their suitable functionalizations. | ||
2.1. Solvothermal or hydrothermal synthesis method
Solvothermal or hydrothermal reactions are considered the standard procedures among the synthetic methods applied for the preparation of both conventional MOF and Ln-MOF materials. This synthetic method involves heating the appropriate metal salts together with organic ligands under pressure for hours or even days, as required, inside a confined container.50 The primary variable that can be controlled to build porous MOF networks is the reaction temperature. Typically, two conditions of the reaction temperature, namely the non-solvothermal temperature and solvothermal temperature, may be applied. The former situation describes the reactions that occur in a closed vessel under a pressure that ensures that the temperature reaches above the boiling point of the solvent used, whereas the latter situation describes the reactions that occur below or at the boiling point of the solvent under normal pressure.51 In the preparation of MOF systems, the solvothermal or hydrothermal reactions are deliberately tuned to proceed very slowly for a long period, satisfying the condition that the dynamics of coordination bond formations between the metal ions and multidentate organic ligands would occur very slowly, proceeding through the formation, breaking, and reformation of these bonds in a reversible manner. The reversible nature of formation of the coordination bonds effectively ensures the rectification of the erroneous bonds that might have been created in the initial stages of the reaction, leading to the formation of the desired MOF materials in a highly crystalline condition, along with exceedingly ordered network and controlled size and shape of their inbuilt pores. In the solvothermal/hydrothermal method, to satisfy the above-mentioned reversibility condition, the reactions are always carried out using substantially dilute solutions of the reactants, allowing the reactions to take place for a very long period of time, often hours to days, to obtain the intended MOF materials as the high-quality crystalline products.8 Because reversibility is the major concern in the solvothermal synthesis of MOF materials, solvothermal/hydrothermal synthesis is generally very sensitive to small variations in the reaction conditions such as temperature, pH, nature of the solvent, reaction time, and concentration of the reagents. Consequently, these external conditions can also be used as a very useful handle to control the reactions for the preparation of desired high-quality MOF materials with specific topological frameworks, high crystallinity, desired size and shape of the pores, and also with crystalline structures with phase purity. To obtain conventional MOF and Ln-MOF materials with the required morphology, topology, and particle sizes, it is vital to regulate the fundamental components that affect their formation, most prominently the reaction temperature and pressure. In solvothermal or hydrothermal synthesis, the nucleation process of MOF materials is affected most significantly by the applied reaction temperature.In solvothermal synthesis, it is of utmost importance that the precursors (metal salts and multitopic organic linkers) are adequately solubilized in the solvent used, especially at the elevated temperatures applied for the synthesis. Further, in solvothermal synthesis, clear information about the associated balanced reactions of the precursors is also very important to know, given that these balanced reactions can lead to noteworthy time-dependent changes in the reaction mixture during the progress of the MOF formation in some cases, affecting the growth of the desired MOF materials. For example, when a metal chloride (MCln) is used as the precursor in combination with a multitopic carboxylic acid-based linker, a stoichiometric amount of HCl is liberated during the reaction of the metal precursor with the linker. Given that HCl is a very strong acid, this liberated HCl can easily cause partial dissolution of the MOF crystals already formed in the solution, disturbing the growth of the MOF crystals, and thereby causing the formation of the desired MOF materials to be exceedingly slow. Alternatively, if in the place of a metal chloride a metal acetylacetonate [M(ACAC)n] is used as the precursor, the byproduct formed would be acetylacetone, which is a mild acid (pKa ≈ 9), and accordingly this biproduct will hardly cause any interference in the formation and crystalline growth of the MOF materials. Thus, it is apparent that the selection of the precursors is a very important criterion to obtain the desired MOF materials with high crystallinity.
In some situations during the solvothermal synthesis of MOF materials, especially when the coordination bond between the metal ion and the organic linker is very strong, simple mixing of the precursors does not ensure the dynamic reversibility of the MOF formation, because the reactions occur at an unusually fast rate. This fast reaction does not allow sufficient time for the formed MOF materials to undergo crystallization, leading to their easy precipitation. In these situations, it is often necessary to use a suitable chemical modulator as an additional competitive binder, usually in the form of simple monotopic ligands, which would undergo quicker bond formation with the metal ion but forming a thermodynamically weaker bond than that of the multidentate ligand of the interest. Thus, the added modulator will slow down the formation of the desired MOF system having greater thermodynamic stability, and thus ensures sufficient time for the perfect crystallization of the MOF material.51–54 It should be mentioned in the present context that in some cases when a modulator is needed to control the growth of the MOF material, the as-obtained final product often contains some defect sites in its structure. Understandably, this is due to the presence of some metal coordination sites devoid of any coordination with the structural linkers but occupied by simple ligands such as hydroxyl, water, and chloride.54 These defects are not often considered undesirable, given that they can favorably influence the crystal growth, topology of the framework, effective BET surface area, pore characteristics, catalytic activity, etc., improving the usefulness of the as-formed porous MOF materials.51
Yaghi and colleagues were the pioneers for the solvothermal synthesis of Ln-MOF materials. These authors developed an extended Tb-BDC framework material with a microporous structure by dissolving Tb(III) nitrate pentahydrate and 1,4-benzenedicarboxylic acid (H2BDC) in methanol/DMF and mildly heating the mixture in a closed vial.50 Thermogravimetric (TG) analysis was performed to examine the thermal stability of the prepared Tb-BDC-based MOF crystals. The weight loss of the sample (46.87 mg) from 120 °C to 223 °C was consistent with the loss of 1.97 DMF molecules per formula unit of the MOF material, Tb2(BDC)3. The stability of the synthesized Ln-MOF system was established by the fact that there was no additional weight loss observed up to 320 °C. These authors also employed the solvothermal process to prepare a Tb-BDC-based MOF system by heating terbium nitrate and H2BDC in the presence of triethylamine at 140 °C for 12 h. The initially formed hydrated Tb2(BDC)3·(H2O)4 system was converted to a microporous MOF system, Tb2(BDC)3, following heat treatment and the material was stable up to 450 °C, showing good thermal stability.
Other organic ligands with carboxyl binding groups together with Ln(III) ions are also anticipated to generate Ln-MOF materials with more advantages in terms of thermal stability. Thus, Liu et al. utilized a tricarboxylic ligand, 1,3,5-benzenetricarboxylic acid (H3BTC), in combination with Eu(III) nitrate hydrate in a water–DMF solvent mixture for the solvothermal synthesis of the hierarchical topologies of Eu-BTC-based MOF systems, Eu(BTC)(H2O)DMF.55 By varying the molar ratio of the H2O/DMF mixed solvent, size-tunable behavior was also observed for the synthesized Eu-BTC-based MOF materials. The normal Eu-BTC MOF particles, which had an average length of 35 ± 6 mm, transformed into a micro crystal having an average size of 10 ± 5 mm when the H2O/DMF volumetric ratio was varied from 0.2 to 5. The observed results were rationalized by considering that H2O acts both as a molecular coordinator for the metal ions, and also as an adjustor for the solubility of the metal salt in the presence of DMF co-solvent. The as-formed Eu-MOF crystals demonstrated good thermal stability up to about 600 °C. However, large weight loss was observed above 600 °C due to the breakdown of the organic linkers in the Eu-MOF system. To realize the synthesis of Ln-MOF systems with the desired morphology and stability, surfactants and polymer materials have also been employed to act as the modifying agents. Using surfactants as modifiers, different porphyrin-based Ln-MOF compounds were synthesized. To control the growth of Ln-MOF nanocrystals, Xia et al. utilized polyvinylpyrrolidone (PVP) as a modifier in a one-pot solvothermal synthesis process, and consequently a square-like Ln-MOF structure was produced.56
2.2. Microwave-assisted synthesis
In microwave-assisted synthesis, the precursors are dissolved in a suitable solvent and placed in a sealed container, typically in a Teflon vessel, and the mixture is placed in the oscillating electric field of a microwave heater.57 As the medium is irradiated with microwaves, coupling of the microwave frequency with the permanent dipoles of the molecules occurs, mainly that of the solvent molecules, causing rapid vibrational and rotational motions of these polar molecules in the solvent medium. Accordingly, this leads to the rapid heating of the reaction medium, and thus triggers the reactions amongst the precursors present in the medium, enabling the formation of the MOF systems.In microwave-based synthesis, the favorable conditions that have been realized to help the overall synthesis process are as follows: (1) the heating process acts directly on the materials; (2) the heating is introduced remotely in the reaction container; (3) the heating process requires a shorter reaction time; and (4) it easy to reach a temperature that is higher than the boiling point of the conventional solvents. Due to these favorable conditions, microwave-assisted synthesis processes can often employ reaction temperatures much higher than the boiling point of the solvents used. This directly helps in significantly reducing the reaction time compared to that required in the conventional solvothermal synthesis method.58,59 Extremely uniform size and morphologies of Ln-MOF materials were produced by Klinowski et al. using the microwave-assisted solvothermal method, thereby reducing the reaction time significantly, and also improving the energy economy effectively.60 Following microwave-assisted synthesis, Bag et al. produced a series of isostructural microporous Ln-MOF materials, a total of about forty different Ln-TTTPC systems (Ln = La, Ce, Pr, Nd, Eu, Tb, Dy, Ho, Yb; H3TTTPC = 1,1',1''-tris(2,4,6-trimethylbenzene-1,3,5-triyl)-tris(methylene)-tris(pyridine-4 carboxylic acid)).61 It is important to note that for the above-mentioned synthesis, the microwave-assisted method took only about 5 min to produce the microcrystalline solids, whereas the traditional solvothermal method required about 2 days for heating the reaction mixture and an additional 5 days for evaporation of the solvents to obtain the material with the same conformation and comparable yield. It has been demonstrated in general that the microwave-assisted approach can largely help in scaling up the synthesis of the Ln-MOF systems, which is often quite difficult using the traditional solvothermal method.
Microwave-assisted synthesis has not only been found to be useful in improving the product yield but also in improving the purity of the synthesized materials.62–64 Thus, Savyasachi and colleagues reported the assembly of hepta-dentated complexes of Eu(III) and Tb(III), formed through the use of cyclen and 1,3,5-benzene-trisethynylbenzoate as the ligands.64 The development of these Ln-MOF systems was possible due to the high capacity of the above-mentioned ligands for binding the Ln(III) ions, and also due to their adaptable structural features. In the microwave-assisted synthesis of some Ln-MOF materials, amino acids have often been used as the modulating agents to slow down the reaction of the desired multidentate ligands with the metal ions. Proline was used to modify the coordination sphere of Tb(III) ions, because this amino acid only has one carboxylic group, and hence it acts as a weaker ligand but with a faster kinetics for binding with the metal ions, ensuring the slow formation of the thermodynamically stable Ln-MOF system involving the multidentate ligands. It was observed in this case that with an increase in the proline concentration, it was possible to change the shape of the produced Tb-MOF nanomaterials from pillar-like rods to rod-like nanocrystalline structures. In many cases, it was also realized that the formation of the crystal structures of the Ln-MOF systems was significantly influenced by the pH and concentration of the modifying/capping agents used in the synthesis process.65
2.3. Precipitation synthesis method
One of the simplest strategy for the synthesis of Ln-MOF systems is the precipitation method. The benefits of the precipitation method compared to the solvothermal method include a high product yield, no energy requirements, and a quick synthesis process.66 In this method, the precipitation of the Ln-MOF is induced by the use of a poor solvent, and the following two simple approaches are typically used. One approach involves dissolving the precursors (metal ions and organic linkers) separately in different solvents, and subsequently mixing them to induce the formation of the Ln-MOF system and its precipitation. The other approach involves the dissolution of the precursors in one solvent, and then transferring the solution containing the soluble MOF formed to a different solvent that induces the precipitation process.67 The Ln-MOFs prepared through the precipitation method can be obtained in crystalline form by suitably modifying the precursor concentration, solution pH, and an adequate choice of the precipitating solvent. The primary objective of adjusting these parameters is to vary the solubility of the precursors and that of the final MOF particles.68 This method has been used to synthesize a variety of conventional MOF and Ln-MOF systems.69,70 It should be mentioned that the first nanoscale Ln-MOF system was in fact produced in 2008 using this method, whereby the Tb(III) ion and c,c,t-(diamminedichlorodisuccinato)Pt(IV) (DSCP) linker were used to prepare a nanoscale Ln-MOF-based coordination polymer.71 The PXRD data of the prepared material suggested that the Tb-MOF material was formed in the amorphous state.The Tb(III)-based Ln-MOF material Tb(1,3,5-BTC)(H2O)·3H2O was synthesized by Zhang and co-workers72via the mixing and vigorous shaking of an ethanol–water solution of 1,3,5-H3BTC and aqueous solution of Tb(NO3)3 at normal temperature. The resultant Tb-MOF material was formed as a one-dimensional nanostructure with a sheaf-like construction, which was assumed to be formed due to the splitting growth mechanism. The authors also used this material as a sensor for the detection of metal ions and acetone molecules.73 Liu et al. synthesized nano- and micro-sized coordination polymers, Ln(1,3,5-BTC)(H2O)6, where Ln = Eu3+, La3+, and Ce3+, through the vigorous shaking of an aqueous solution of Ln(NO3)3 with a water–ethanol solution of 1,3,5-H3BTC at room temperature.74 Novel 3D flower-like superstructures were obtained by varying the synthesis conditions, such as precursor concentration, molar ratio of the reactants, use of surfactants as modifiers, and solvent. It was also possible to control the photoluminescence color-tunability of the synthesized co-doped Tb(III) and Eu(III)-based Ln-MOF materials, displaying emission from red, orange, yellow and green-yellow to green, by adjusting the composition of the two Ln(III) ions.
2.4. Mechanochemical synthesis method
Mechanical force imparted by milling or grinding of solid precursors can also lead to chemical reactions, resulting in the synthesis of various conventional MOF and Ln-MOF systems.75 Upon the application of mechanical forces, the boundaries of the reacting components get activated, causing phenomena such as flattening, cold-welding, fracturing, and rewelding effects on the solid materials, leading to their structural deformation. Thus, the crystallographic linkages of the solid materials are broken, promoting the creation of new surfaces and interfaces, causing the constituent components in the material to change their structures in a variety of ways, and leading to the formation of new materials.75 Some of the benefits of the mechanochemical method for the synthesis of MOF materials are as follows: (1) a solvent-free reaction, which can minimize the need for hazardous organic solvents, (2) a reduction in the reaction time quite substantially, (3) metal oxides can be used directly, avoiding the use of metal salts and the consequent formation of side products, and (4) high-energy mechanical milling can ensure the reproducibility of the synthesis process.76 The synthesis of MOF systems via mechanochemical grinding was first reported by Pichon et al.77 in 2006, confirming the utilization of this method for the preparation of higher-dimensional, porous coordination networks. There are three categories of mechanochemical synthesis procedures adopted. The first is the dry grinding, which refers to the grinding of solid precursors without using a solvent. However, this straightforward approach was found to be quite ineffective in many cases. The second is the liquid-assisted grinding (LAG) method. This is a highly efficient method, which can facilitate the incorporation of suitable guest molecules within the MOF network structure. The third approach is the ion and liquid-assisted grinding (ILAG), which was found to be useful in the catalytic production of MOF systems through the introduction of some adjuvant ions and liquids.78Yuan and co-workers effectively synthesized several homometallic and mixed-metallic Ln-MOF systems involving trivalent rare earth ions using the simple LAG method.79 Several rare earth carbonates and H3BTC linker systems were considered in the synthesis of different homometallic Ln-MOF systems, using DMF as the liquid to assist the grinding process. The synthesized Ln-MOF materials showed comparable PXRD characteristics to that of the materials prepared through the solvothermal method. Heterometallic Ln-MOF systems were synthesized using the LAG method for the mixing of gadolinium carbonate with Sm, Eu, Tb, or Dy carbonates in the presence of H3BTC linker. PXRD patterns for these heterometallic materials were quite consistent with those of the homometallic Ln-MOF systems prepared. The mechanochemical synthesis of these Ln-MOF systems was accomplished more easily by using rare earth acetates than their oxides. Three luminous Ln-MOFs based on an MIL-78 network were developed by Alammar et al. in 2018 using a solvent-free mechanochemical process.80 The as-prepared materials based on Eu(III), Tb(III), and Dy(III) ions showed excellent colour purity for red, green, and yellow emission, as demonstrated by their CIE diagrams, respectively. The ball milling method was also used by Balema and co-workers to prepare heterometallic MIL-78 network-based Ln-MOF systems that prominently showed magnetocaloric effects.81
2.5. Electrochemical synthesis method
Electrochemical synthesis is an important method for the preparation of various conventional MOF and LN-MOF systems. Moderate reaction conditions and shorter reaction times are the two important benefits of electrochemical synthesis,76 which can be carried out either by anodic dissolution or cathodic deposition methods.82 In anodic dissolution, the electrode directly acts as the source of metal ions produced through the oxidation of a metal under an applied anodic voltage. Subsequently, the metal ions combine with the organic ligands present in the solution to generate a thin MOF film that encircles the electrode. In cathodic deposition, under an appropriate cathodic potential, the pro-base used in the solution undergoes reduction to generate a base (OH−), which increases the pH locally at the electrode surface, causing the deprotonation of the organic ligands for their coordination with the metal ions in the solution, which results in the formation of an MOF film on the cathode surface.83 Generally, the electrochemical method for the preparation of MOF materials is very efficient. Most importantly, this method helps to avoid the use of metal salts, which are the source of corrosive anions that often alter the MOF formation kinetics.76,82–85 Additionally, the electrochemical method works at a much lower temperature compared to other conventional methods. The MOF materials prepared through the electrochemical method are generally produced in reasonably high yields.76,82–85 Further, these materials are also produced with highly crystalline nature and display relatively smaller crystal sizes compared to that prepared through other synthesis methods.51,76,82–85The synthesis of MOF systems using the electrochemical method was first reported by Mueller et al.86 in 2006. Bulk copper plates were used as the anode in this study, which was immersed in a 1,3,5-benzanetricarboxylic acid containing methanolic solution. The as-produced Cu-BTC MOF acted as a more potent adsorbent than the material prepared using the traditional method.86 Subsequently, this simple and affordable synthesis protocol was also employed to prepare luminous Ln-MOF materials. For example, Li and colleagues87 described electrochemical-assisted microwave deposition technology for the preparation of luminescent films of Ln-MOF materials, where electrochemical deposition was first used to synthesize a lanthanide hydroxide layer on the surface of conductive glass, namely the fluorine-doped tin oxide (FTO) substrate, and then the hydroxide layer was converted into an MOF film following the microwave-induced synthesis. As reported by these authors, Ln-MOF films of various colours could be obtained by adjusting the Tb/Eu ratio during the synthesis. These luminescent films were predicted to find applications as structural probes, luminescence sensors, anti-counterfeiting barcodes, etc.
2.6. Sonochemical synthesis method
Sonochemical synthesis using high-intensity ultrasonic irradiation provides exceptional advantages over conventional solvothermal synthesis, namely shorter reaction time, environmentally friendly method, ease of operation, and excellent efficiency.88 This method relies on the interaction of ultrasound with the reactive liquid because the wavelength of the ultrasonic wave is larger than the size of the molecules in the medium.89 As the high-energy ultrasonic waves propagate through the solution, the interaction of ultrasound with the solvent molecules results in the development of alternating high pressure (compression) and low pressure (rarefaction) regions in the medium. Often the pressure in the low-pressure region drops below the vapor pressure of the solvent, leading to the formation of small bubbles in the solution. These bubbles sequentially grow with time as more solvent vapor diffuses into them under the influence of the alternating pressure created in the liquid by the ultrasound irradiation. In this process, the ultrasonic energy gets largely accumulated into these growing bubbles, and eventually they reach to their critical size, where they become unstable and suffer a sudden collapse as an explosion. These processes of formation, growth, and collapse of the liquid bubbles under the influence of ultrasound irradiation as a whole are known as the cavitation process, through which a huge amount of the accumulated energy is released suddenly in the solution, resulting in a large increase in the temperature and pressure in a localized region. Thus, the cavitation process effectively creates “hot spots” in the solution, leading to the creation of activate chemical species, e.g., free radicals, which finally promote the desired synthesis process. Given that the temperature and pressure conditions with the “hot spots” are very favorable for a very short period, the precursors can quickly react inside these hot spots, resulting in the formation of very high-quality MOF materials with a relatively small crystal size and significantly high yield of the product.51,57 Evidently, microwave-assisted synthesis often occurs at a much quicker rate, typically about 10 to 100 times faster than the conventionally used solvothermal method,90,91 making this method very useful for the synthesis of many conventional MOF and Ln-MOF systems.The initial use of the sonochemical method was reported by Son et al.92 for the synthesis of the MOF-5 system. Although the use of high boiling point inorganic solvents is the desired choice in the sonochemical synthesis method, high boiling point organic solvents such as 1-methyl-2-pyrrolidone (NMP) can also be successfully used, as demonstrated by Son et al.92 A HKUST-1-based MOF system was also synthesized by Li et al.93 using the ultrasonic approach and this material exhibited superior porosity than the MOF system produced through the solvothermal method, as revealed by the BET studies. Comprehensive details on the important aspects of the sonochemical synthesis method have been compiled and reported in the literature by Argirusis and co-workers.94
The synthesis of the isostructural porous Ln-MOF systems [Ln(BTC)(H2O)4·3H2O], where Ln = Ce, Tb, and Y, and BTC = 1,3,5-benzenetricarboxylate was reported by Khan et al.95 using sonochemical synthesis. The produced Ln-MOF nanomaterials had a comparatively smaller particle size and larger surface area than the corresponding materials produced through the solvothermal method. The sonochemically synthesized Ln-MOF system, [Tb(1,3,5-BTC)(H2O)6]n, having a nanowire morphology and enhanced luminous characteristics was reported by Hu et al.,96 which was achieved by adjusting the reaction time. The as-prepared material demonstrated very selective sensing for organic amines. Subsequently, Xiao et al.97 also reported the synthesis of another luminous Tb-MOF system using the same sonochemical method, and the as-obtained material showed high selectivity for sensing picric acid. The different synthesis methods discussed thus far for the preparation of Ln-MOF materials are schematically presented in Fig. 3 for their quick visualization.
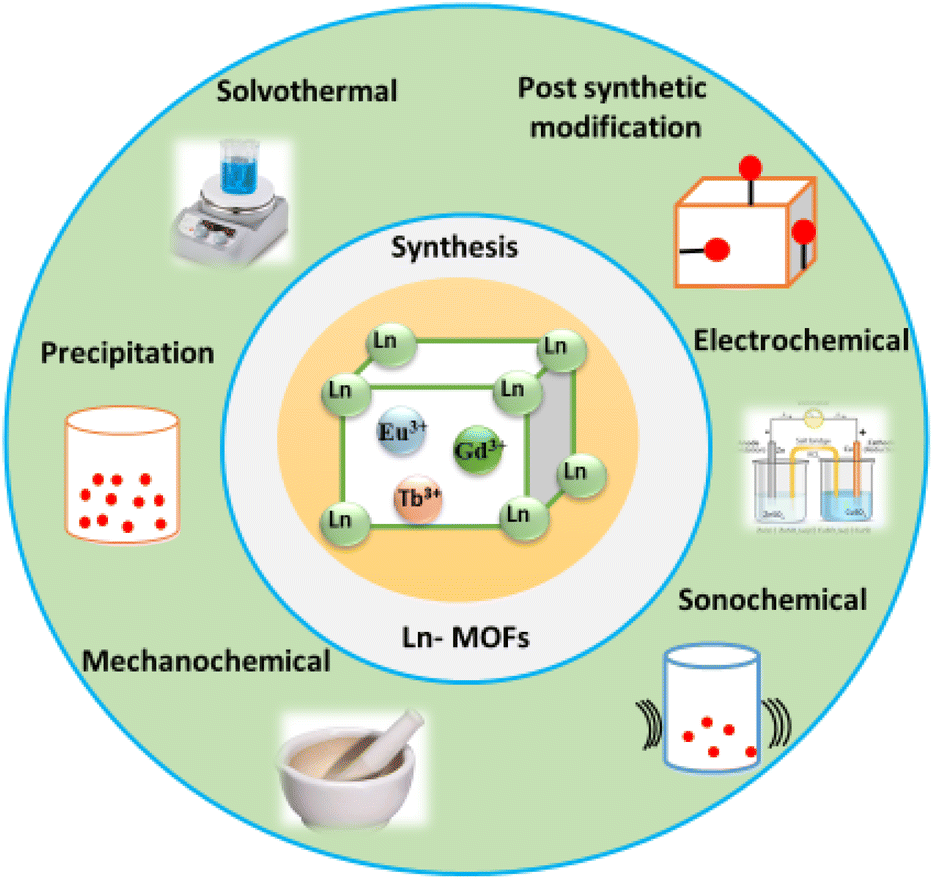 | ||
| Fig. 3 Schematic of the different methods used for the synthesis of Ln-MOF materials. | ||
3. Post-synthetic modification methods for the MOF materials
In general, direct synthetic approaches are found to be less effective in meeting the growing needs of various MOF and Ln-MOF systems that can display advanced characteristics and superior performances for diverse applications. In this respect, post-synthetic modification (PSM) methods have opened up enormous opportunities to develop numerous porous network materials with advanced characteristics. Different PSM methods including covalent-, dative-, encapsulation-, and tandem-based PSMs are often applied either for the introduction of new functional groups or to carry out post-processing of MOF systems to achieve their target-specific utilization. Generally, a PSM relies on the introduction of new functional groups or the incorporation of additional functional materials such as organic molecules and specific metal ions, with the aim to obtain modified porous materials with better performances towards practical applications. This is achieved through their selective bonding, preferred coordinate bond formation, and/or improvement in encapsulation for the identification and sequestration of the target analytes.67 Chemical substitutions within known MOF networks can be achieved either through the incorporation of suitable functional modifications in the linker molecules in advance, before their utilization in the synthesis of MOF systems, or the already synthesized MOF materials can be modified through the incorporation of the desired additional functional groups into the pores of these MOF materials by adopting some suitable post-synthesis treatments. Due to these functional modifications and because of many other advantageous and promising properties, such conventional MOF and Ln-MOF systems have been identified as the important porous solid materials for their utilization as superior sorbent materials compared to their traditional porous material counterparts.Lanthanide-based MOF systems show imense potentials for the fabrication of light-emitting products.98 Using the PSM, Abdelhameed and co-workers fabricated an IRMOF-3-based Ln-MOF system displaying both infrared and visible light emitting characteristics.99 Due to the presence of high porosity and large number of free amino groups on the benzene dicarboxylate linker, which are active towards chemical modification, the isoreticular IRMOF-3 system was selected as the primary backbone material for the PSM of the system through the introduction of diethyl-(ethoxy methylene)-malonate, methyl vinyl ketone, 2-chloroacetic acid, and glyoxylic acid groups in the frameworks. Subsequently, these newly introduced anchoring groups were used to coordinatively bind Eu3+ and Nd3+ ions to develop the desired Ln-MOF systems.99 In a different report, transparent lanthanide-functionalized luminescent MOF thin films were prepared by Ma and Yan,100 following PSM of a UiO (University of Oslo)-based MOF system, namely UiO-67, aiming to apply the obtained materials in sensing. In this case, the non-destructive PSM strategy was utilized to modify the UiO-67 system through the introduction of lanthanide ions together with 2-thenoyltrifluoroacetone (TTA) and 1,1,1-trifluoropentane-2,4-dione (TAA) linkers into the framework of the backbone material to obtain the lanthanide-functionalized UMOF systems, Ln(TTA/TAA)@UMOF, where Ln = Eu, Tb, Er and Nd ions. Subsequently, the synthesized Ln-MOF materials were assembled on organo-silane linker-modified Al2O3 substrates using lanthanide ions as the bridge to obtain Ln-MOF-based luminous thin films. The as-prepared thin films were seen to be very useful for luminescence-based sensor applications. One of these films was seen to provide a superior performance towards ammonia sensing, demonstrating advantageous features such as quick response, outstanding selectivity, and high sensitivity (LOD = 9 ppm).
4. Distinctive characteristics of lanthanide-based MOF systems
Given that the structures of MOF systems are significantly influenced by the coordination environments of their metal systems and the coordination environments of the lanthanide ions are different from that of the conventional metal ions, Ln-MOF systems in general exhibit quite distinctive characteristics in many respects compared to conventional MOF systems. In the following section, initially we discuss some of the important characteristics pertaining to the lanthanide ions in Ln-MOF systems, and then the important structural characteristics of different MOF systems.4.1. Basic structural characteristics pertaining to Ln-MOF systems
4.2. Characteristics pertaining to Ln-MOF systems
Ln-MOF systems are distinguished by their unique properties originating from both the nature of their lanthanide ions and organic ligands. The important properties of Ln-MOF systems that are valuable in various scientific and industrial applications are discussed here.5. Structural characteristics of metal–organic framework (MOF) materials
In materials chemistry, porous materials play a very important role. The repertoire of porous materials represents a vast range of substances, which can be classified as natural, synthetic, organic, inorganic, crystalline, and amorphous materials, and they can be of either neutral or ionic character. Up to the mid-nineties, only two types of porous materials, one belonging to inorganic substances and the other belonging to carbon-based materials, are known to be investigated reasonably substantially considering their promising industrial applications. The most significant inorganic porous materials showing important industrial applications are the porous aluminosilicates, commonly known as zeolites. These materials possess complex crystallographic structures and substantial porosity.31 Among the carbon-based porous materials, various activated carbons have found extensive applications in chemical laboratories and different industries.101,102Zeolites are crystalline solids with very complex crystallographic structures, whereas activated carbon are amorphous materials. However, interestingly, activated carbons have significantly higher porosity, and hence they display much higher BET surface area compared to zeolite materials.101–103 Presently, along with zeolites and activated carbon materials, various other porous solid materials have also been extensively explored for their applications in different industrial processes. The important new types of porous solid materials are the porous organic polymers (POPs), which are formed through strong covalent linkages between different organic building blocks, resulting porous 3D network structures, and the metal–organic framework (MOF) materials, which are formed through the linking of metal ions or metal nodes by multidentate linker molecules, creating porous 3D network structures. All these new generation of porous materials are indicated to play an important role in industry in the future and will certainly influence the global economy significantly.
MOF systems can be broadly considered as a class of organic–inorganic hybrid materials; however, in reality, they are different in regard to their constitution. This is because the constituent inorganic and organic components of MOF systems are arranged in very regular, ordered and well-defined 3D structures, resulting in the formation of porous solid framework materials. The most important feature of MOF materials is that they possess exceedingly large porosity, a special characteristic that promotes their uses as high-performance porous sorbent systems. Owing to their structural rigidity, regularity and orderliness, different varieties of 3D network structures can be formed by these materials. Accordingly, MOF-based materials are emerging as the superior sorbent materials for many exotic applications in industry, chemical laboratory, and also in common life.104 Unlike conventional porous materials such as zeolites, MOF-based materials are quite easy to be modified through suitable derivatization or functionalization,105,106 improving their performance in many specific applications. In fact, through such functionalization, many new porous MOF materials, especially those belonging to Ln-MOF systems have been developed with superior porosity and specific anchoring groups, improving their sorption capacity, luminescence behavior, magnetic characteristics, and many other properties.105–111 It is important to mention in the present context that MOF-based materials having very regular pore structures are normally seen to display very high performances towards the selective adsorption of various metal ions, especially those having environmental and/or strategic importance.105–113
Considering the diversity of metal ions or metal nodes interconnecting through different multitopic organic linkers, a large variety of MOF materials has been reported in the literature. In this respect, the coordination of metal ions with neutral multidentate linkers possessing nitrogen donor groups such as 4,4′-bipyridine (BPy) have produced a variety of cationic MOF materials. However, the 3D network structures formed in this type of MOF systems are not very stable and their network structures often collapse during the attempted removal or exchange of their entrapped guests, limiting the widespread applications of these systems.114,115
Generally, MOF materials with stable network structures are achieved through the use of multidentate carboxylate-based linkers. These linkers have the ability to form M–O–C types of metal clusters or metal nodes, acting as the unique secondary building units (SBU) for further interlinking with the multidentate linkers, resulting in the construction of exceptional structural features in the MOF materials formed.53,114,115 Stable MOF structures have also been reported to be formed by the involvement of other polytopic ligands such as imidazolate-based linker molecules.115–118Fig. 4 summarizes the topologies of some of the typical MOF systems together with their associated metal nodes (SBUs) and the polydentate ligands involved in their construction.116
 | ||
| Fig. 4 List of some of the MOF systems, along with their associated metal node and the organic linker constituents. Color codes: Cl = pink, Zr = green, N = light blue, Cr = light purple, Zn = dark red, Mg = blue, Cu = royal blue, C = grey, O = red, and Fe = yellow. The figure is redrawn from ref. 116 with permission from the American Chemical Society. | ||
As indicated above, polycarboxylate-based ligands are very useful for the construction of different stable MOF systems (both conventional and Ln-MOF systems) with varying network structures.116–119 In the synthesis of MOF systems, the molecular geometry, molecular length, presence of other functional groups, and relative ratio of different anchoring groups present in the carboxylate-based linker molecules play a very significant role in determining the structures of the M–O–C metal nodes (SBUs), and accordingly the construction of the network structures formed for the MOF systems. Accordingly, many conventional MOF and Ln-MOF materials with a large variety of shapes and sizes of the inbuilt pores have been prepared by using different combinations of metal ions together with various polycarboxylate based ligands. A list of the important polycarboxylate-based ligands used in the preparation of various conventional and lanthanide-based MOF systems is shown in Fig. 5 for easy visualization. Regarding the structures of the MOF materials, although in many cases the structural characteristics of Ln-MOF systems drastically differ compared to that of the conventional MOF systems, there are also many cases where conventional MOF and Ln-MOF systems display very common structural features. Such common structural features are also important for the lanthanide-based MOF systems that contain both transition metal (TM) ions and lanthanide (Ln) ions to create the relevant SBUs for the construction of their network structures. With these perspectives, we felt that a discussion on the structural aspects of the conventional MOF and Ln-MOF systems separately would be better to appreciate the similarity and differences between these two types of MOF systems. Accordingly, in the following section, we present a discussion on the structural characteristics of the conventional MOF systems first, and then in the next subsection, we discuss the structural characteristics of Ln-MOF systems.
 | ||
| Fig. 5 List of some of the polycarboxylate linkers used in the construction of various MOF systems through their coordination with suitable metal ions or metal nodes. Reproduced from ref. 118 with permission from the Royal Society of Chemistry. | ||
5.1. Important structural characteristics of conventional MOF systems
In the ongoing research on MOF-based materials, a major breakthrough occurred with the initial synthesis of two organic–inorganic porous materials, one was the MOF-5 system,5 which was later characterized as the IRMOF-1 material belonging to the isoreticular metal–organic framework (IRMOF) series,113 and other one was the HKUST-1 material,12 where HKUST represents “Hong Kong University of Science and Technology”, with the material belonging to the HKUST-n series of MOF systems.109 Both of these porous materials displayed high crystallinity and demonstrated much higher BET surface area compared to that of the conventional porous solids. The crystal structure analysis of the IRMOF-n and HKUST-n series of materials revealed the presence of regular arrangements of some metal nodes interconnected with multitopic linker molecules. Although in some of the MOF systems the individual metal ions can also act as the metal nodes, in most of the systems, in both the conventional and Ln-MOF categories, the formation of some ligand-assisted M–O–C unit-based metal ion clusters invariably occurs. These clusters act as the metal nodes in the formation of the regular frameworks, serving as the secondary building units (SBUs) in the construction of the 3D network structures of MOF systems through their interconnection with the multidentate linker molecules.In the IRMOF-n series of porous materials, the formation of the M–O–C-based metal nodes (SBUs) was achieved by using various types of dicarboxylate-based ligands, namely acetate-, benzoate- and pivalate-based molecules.1,120–126 The first-generation of IRMOF-n materials, namely the IRMOF-1 or MOF-5 system, was synthesized and characterized by Yaghi and co-workers.1 This material has the stoichiometric composition of Zn4(O)(BDC)3, where BDC stands for 1,4-benzenedicarboxylate ligand. In its network structure, the tetra-nuclear super-tetrahedral Zn2+ clusters were identified as the metal nodes (SBUs), which were interconnected through the adequate coordination with BDC ligands constituting the 3D network structure, as schematically shown in Fig. 6a.123 In this network structure, six Zn4O(CO2)6 clusters, each one having an octahedral (Oh) geometry, are inter-connected through BDC linkers, and thereby square-shaped pores are developed in the material with pore openings of about 9 Å.124,125 The MOF materials in the IRMOF-n series display substantially high thermal stability and possess quite open skeletal frameworks with controlled pore dimensions and large BET surface area.1,120–123 In the case of the IRMOF-n series, the inner walls of the pores can be modified easily by using differently functionalized dicarboxylate ligands having different types of substituents.12,117 In fact, the systematic modulation of the pore size from about 2.8 to 28.8 Å was achieved for the IRMOF-n materials by changing the fused aromatic moieties, like biphenyl, tetrahydropyran, pyrene, and terphenyl units, in their dicarboxylate ligands, as schematically shown in Fig. 6b.117 The IRMOF-n series of materials have found applications in the areas such as catalysis,5 gas storage,106 analyte separation,109 electrochemistry,85 and many others. These materials have also shown prospective uses in biology and medicine, especially in drug carrier applications.123
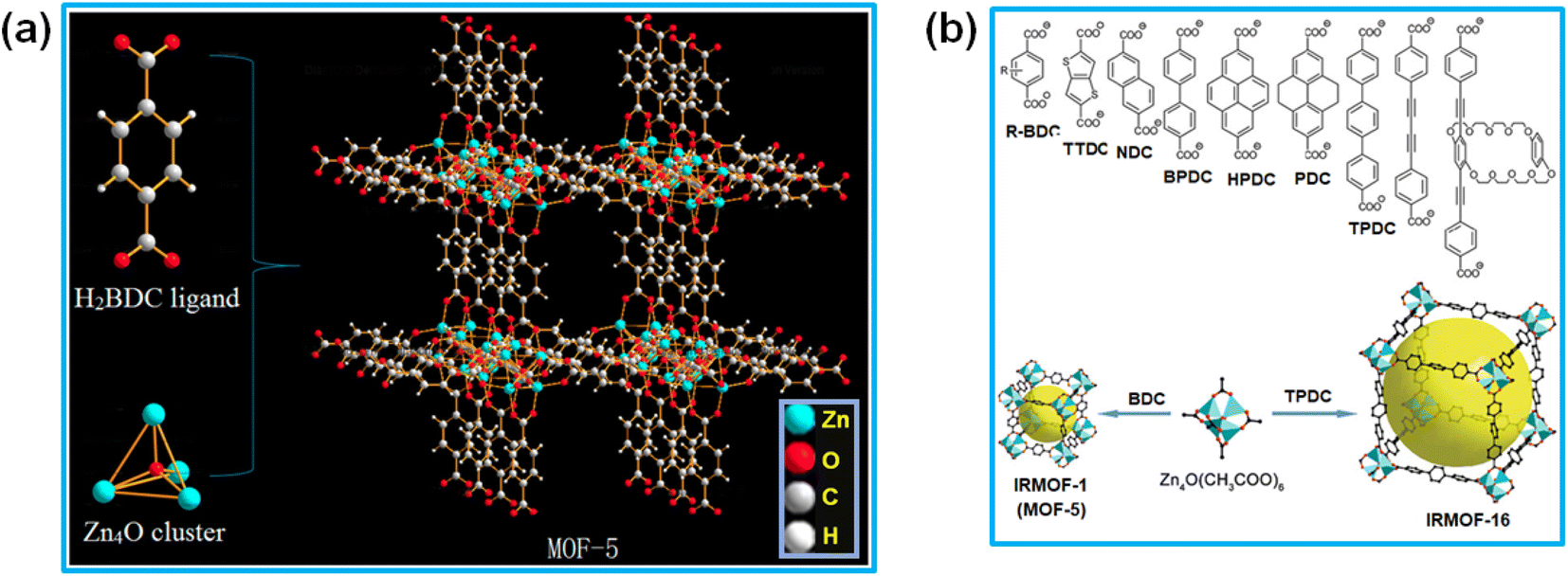 | ||
| Fig. 6 (a) Schematic of the network structures in the MOF-5 system. The figure is reproduced from ref. 123 with permission from Multidisciplinary Digital Publishing Institute (MDPI), Basel, Switzerland. (b) List of different fused groups in the dicarboxylate ligands used for the preparation of IRMOF-1 to IRMOF-16 networks. Color codes: polyhedral Zn cluster (greenish-blue); O (red); C (black). Square-shaped pores are indicated by large yellow balls. The figure is redrawn from ref. 117 with permission from the Royal Society of Chemistry. | ||
Compared to the IRMOF-n systems, the HKUST-n systems possess different 3D network architectures, which are formed by the Td-octahedron-structured metal nodes constituted by grouping of four square planar di-copper-based paddle-wheel structured SUB units, and connecting these metal nodes involving the tri-topic linker like 1,3,5-benzenetricarboxylate (BTC) ligand.12,117 Using various tricarboxylate linkers, a variety of HKUST-n materials was synthesized, particularly to mention are those obtained by using 4,4′,4′′-s-triazine-2,4,6-triyl-tribenzoate (TATB), 4,4′,4′′-(benzene-1,3,5-triyl-tris(benzene-4,1-diyl))tribenzoate (BBC), 4,4′,4′′-s-triazine-1,3,5-triyltri-p-aminobenzoate (TATAB), and 4,4′,4′′-(1,3,4,6,7,9,9-heptaazaphenalene-2,5,8-triyl)tribenzoate (HTB) as linkers, as are shown schematically in Fig. 7.12,117 All these materials have similar topologies but significantly different pore sizes, showing diverse applications.
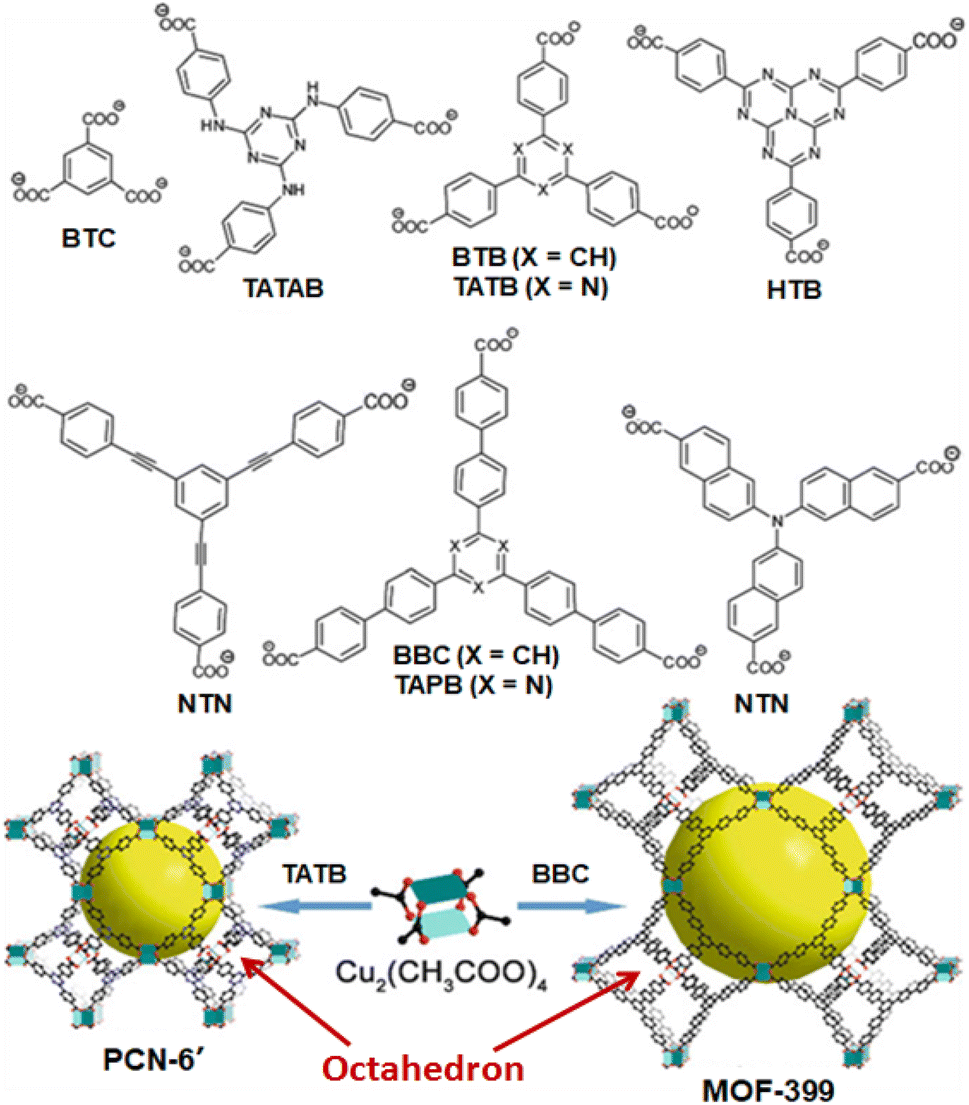 | ||
| Fig. 7 Examples of MOF systems constructed from tri-topic carboxylate linkers and di-copper paddle-wheel SBUs. Color scheme: Cu (turquoise); O (red); C (black). Large yellow balls indicate the pores formed in these materials. The figure is redrawn from ref. 117 with permission from the Royal Society of Chemistry. | ||
The MIL-n series of MOF materials, where MIL stands for “Materials Institute Lavoisier”, are also realized as the unique MOF materials, where the metal nodes are formed as a type of super-octahedron involving three metal octahedra (formed by coordinating with di- or tri-carboxylate ligands), acting as the SBUs, and the 3D networks are developed by the interconnection of these metal nodes involving tricarboxylate ligands.127–129 Depending on the multi-topic ligands used, the MIL-n series of materials was reported to be formed as microporous to mesoporous systems with pore sizes in the range of 5 to 30 Å. In these materials, the 3D network structures lead to the formation of two different types of cages. While one has a significantly smaller size with pentagonal pore openings, the other one is significantly larger in size with hexagonal pore openings.127–129 The network structures of the MIL-100 and MIL-101 systems formed using BTC and BDC as the linkers are schematically shown in Fig. 8 for easy comparison and visualization.
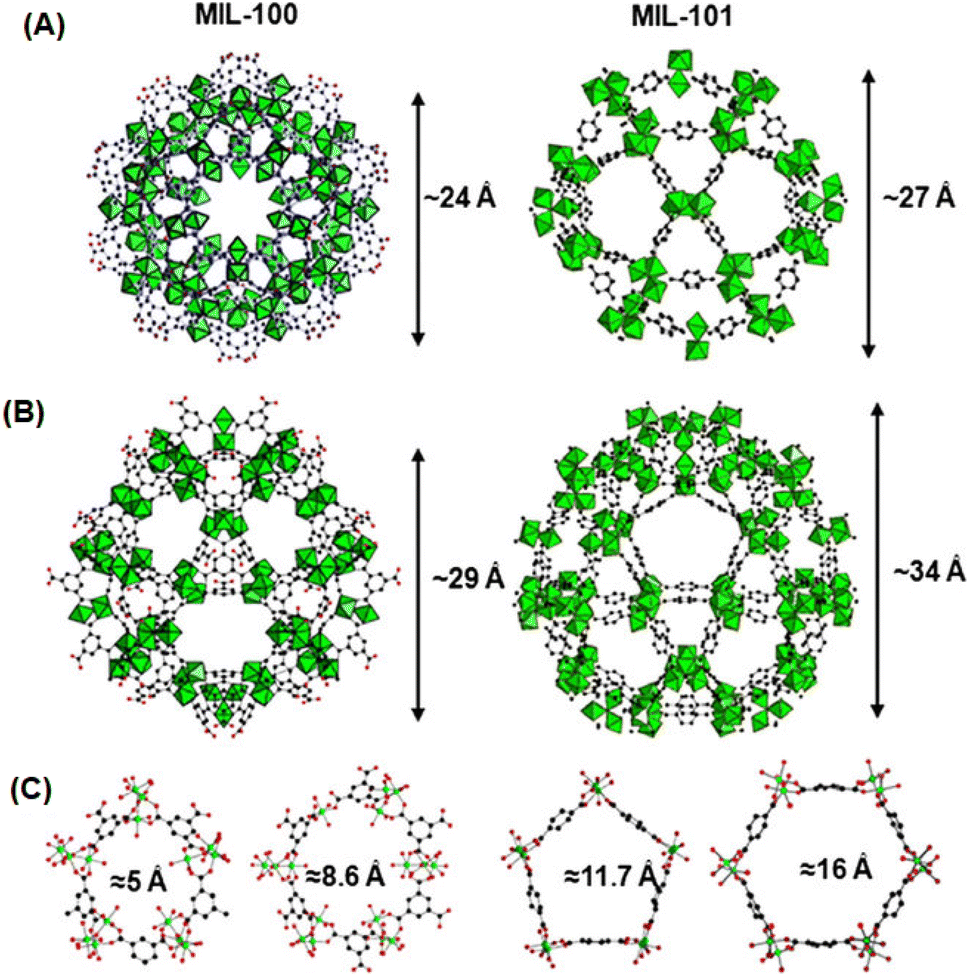 | ||
| Fig. 8 Schematic of the network structures of MIL-100(Cr) and MIL-101(Cr). (A) and (B) Represent the views of the small and large cages formed in these two materials. (C) Represents the views of the pentagonal and hexagonal windows for the pores created in these materials. Color codes: Cr octahedra = green and C = black. The figure is redrawn from ref. 129 with permission from Elsevier. | ||
5.2. Important structural characteristics of Ln-MOF systems
While discussing the general structural characteristics of Ln-MOF systems, it important to note that they can involve either homometallic clusters or heterometallic clusters of lanthanide ions as the metal nodes in the construction of their topological structures. Accordingly, it was felt that for the convenience of presentation, the structural characteristics of the Ln-MOF systems formed based on homometallic and heterometallic clusters are better to be discussed seperately. Thus, in the following two subsections, we seperately present the general structural characteristics of the Ln-MOF systems formed involving homometallic and heterometallic clusters as the metal nodes, respectively.In Ln-MOF systems, different types of homometallic clusters of Ln(III) ions are realized to participate as the metal nodes, coordinating with suitable multitopic linkers. Many of these Ln-MOF systems are often considered as simple Ln(III) ion-based coordination polymers, given that these materials are formed by interlinking the Ln(III) ions directly through the sharing of multitopic linkers. Several of these coordination polymers were reported in the literature involving different Ln(III) ions (Ln = La to Tm) together with the di-topic linker BDC and having the general formula of [Ln2(BDC)3(H2O)4]n.130 In the construction of the [Ln2(BDC)3(H2O)4]n structural motifs, the Ln(III) ions formed octa-coordinated metal centers, with six of them arising through the binding of six oxygen atoms provided by six BDC ligands and the remaining two arising through the binding of oxygen atoms from two water molecules, as schematically shown in Fig. 9. In these Ln-MOF systems, each BDC linker interconnects four Ln(III) ions simultaneously, establishing the formation of a 3D network in the desired Ln-MOF system. In these as-formed Ln-MOF materials, when heavier Ln(III) ions such as Tb(III) to Tm(III) are involved, the easy dehydration/removal of their coordinating water molecules can occur, triggering the easy phase transformation of these materials. This happens because the water-free Ln(III) ions in these materials participate in the formation of new types of coordination structures involving the μ-carboxylate bridges. However, interestingly, no such phase transformation is observed for Ln-MOF materials composed of lighter Ln(III) ions, such as La(III) to Eu(III). In this case, the concerned Ln-MOF materials maintain their original crystal structures even when subjected to dehydration by removing their coordinating water molecules.130
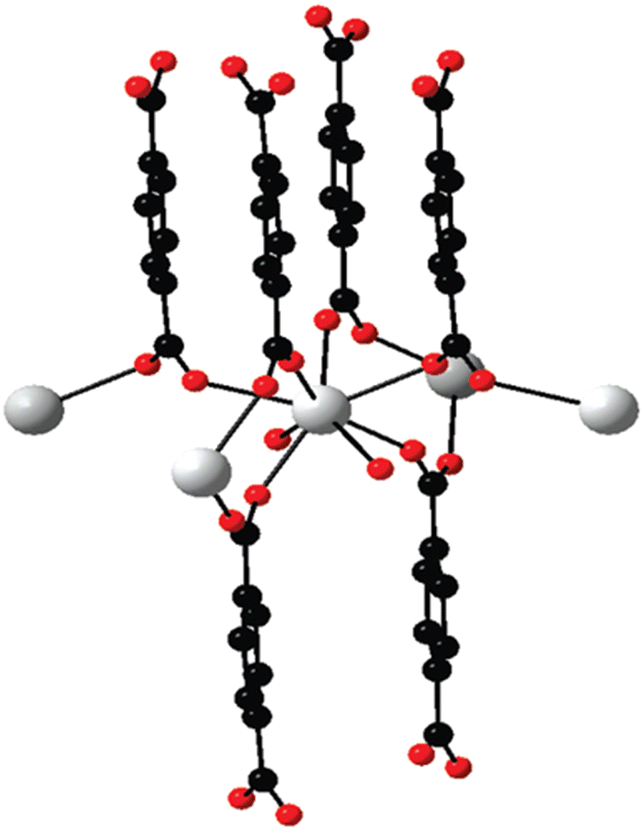 | ||
| Fig. 9 Projection of an extended asymmetric unit of [Tb2(BDC)3(H2O)4]n. Color codes: Ln (white); O (red); and C (black). Reproduced from ref. 130 with permission from the American Chemical Society. | ||
Ln-MOF materials with interpenetrated metal nodes in their network structures were reported by Zhou and co-workers.134 A typical example of these Ln-MOF materials is the PCN-17(Ln) system, where PCN stands for porous coordination network and Ln stands for Dy, Er, Y, and Yb ions. As revealed by these authors, in the PCN-17(Ln) materials, their SBUs are constituted by using square-planar [Ln4(μ4-H2O)] clusters as the basic structural motifs or metal nodes, where all four Ln atoms are on the same plane. In the [Ln4(μ4-H2O)] motifs, all the Ln atoms are hepta-coordinated, two due to two O atoms of a bridging sulfate group, one due to the O atom of the μ4-H2O molecule, which interconnects all four Ln atoms, and four due to the four O atoms arising from four different TATB linkers. In the case of each of these TATB linkers, two of their carboxylate groups are interconnected with two Ln atoms of the same [Ln4(μ4-H2O)] motif, while their third carboxylate group is utilized to connect an Ln atom of a nearby Ln4(μ4-H2O) motif.134 With this coordination pattern, each of the [Ln4(μ4-H2O)] motifs are interconnected to eight different TATB linkers together with four different bridging sulfate groups, leading to the formation of an assembly of six [Ln4(μ4-H2O)] motifs in a truncated octahedral-shaped cage structure, acting as the secondary building unit (SBU) of the PCN-17(Ln) systems. In the above-mentioned SBUs, each of the [Ln4(μ4-H2O)] motifs are placed at the corners (or vertices) of the octahedron and eight TATB linker are placed on the eight faces of the octahedron. Each of these octahedrons participate in the formation of the extended network by sharing their six corners with six other adjacent octahedrons involving the bridging sulfate groups. The as-formed simple MOF network creates the inscribed cuboctahedron cages. However, two of these simplified networks can undergo interpenetration with each other, stabilized and supported by the involvement of the π–π interactions of the TATB ligands, giving rise to the formation of a doubly interpenetrated MOF network structure, referred as the PCN-9 system. Subsequently, these doubly interpenetrated networks can also undergo more complex interconnections among their octahedron units involving the sulfate-bridged coordinative linkages, resulting in the formation of a directly bridged interpenetrated 3D framework, which is referred to as the system. A schematic presentation of the aforementioned non-interpenetrated and interpenetrated Ln-MOF networks is shown in Fig. 10 for easy visualization.
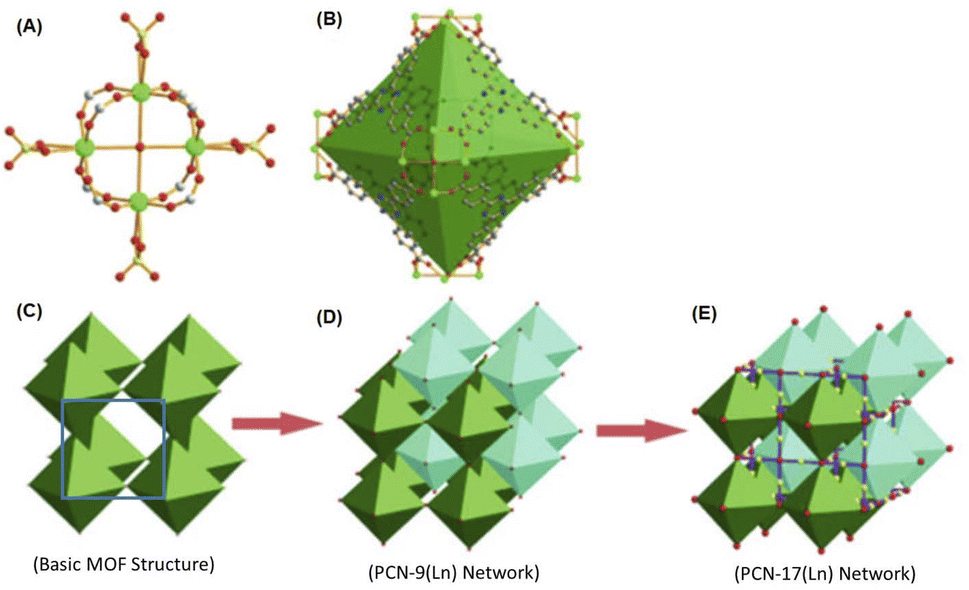 | ||
| Fig. 10 (A) Schematic of the structure of [Ln4(μ4-H2O)] motif. (B) Truncated octahedral cage inscribed by the SBUs formed by six units of [Ln4(μ4-H2O)] motifs, as indicated by the green octahedron. (C) Non-interpenetrated basic Ln-MOF network with cuboctahedron cage (blue square) formed by the interconnection of six corners of the octahedrons. (D) Doubly interpenetrated PCN-9(Ln) structure with reasonably complex pore structures. (E) Bridged interpenetrated PCN-17(Ln) network with very complex arrangements for the SBUs and the pore structures. Color codes: Ln = green, C = gray, N = blue, O = red, and S = yellow. The figure is redrawn from ref. 134 with permission from Wiley Inter Science. | ||
As indicated from the above discussion, the PCN-n(Ln) series of Ln-MOF systems can form with two types of inbuilt cages, where the larger ones are the truncated octahedral cages, and the smaller ones are the cuboctahedron cages, enabling different types of applications for these Ln-MOF materials. The unique interpenetrated frameworks produced in the PCN-17(Ln) systems, which are supported by coordinatively linked sulfate-bridges, demonstrated exceptionally high thermal stability (up to ∼480 °C). These material also possessed a substantially reduced pore size (∼3.5 Å) compared to most other MOF materials. Accordingly, the PCN-17(Ln) systems were realized to display substantially selective adsorption properties, namely for O2 gas over N2; H2 gas over N2; and H2 gas over CO. These selective adsorption properties suggested the potentials of the PCN-n(Ln) series of Ln-MOF systems for selective gas adsorption and separation applications. For more details on the structural aspects of various other Ln-MOF materials possessing homometallic Ln clusters, readers are referred to the monograph that was published a few years ago by Li and Chen.135
Generally, Ln-TM-MOF systems possessing Ln-linker-TM-based heterometallic clusters are produced using multidentate ligands having both N-donor- and O-donor-based arms. This is required because the O-donor ligands display a relatively stronger affinity for Ln ions, while the N-donor ligands exhibit comparatively stronger binding with TM ions due to the hard–soft Lewis acid–base interactions.107,131 The typical multidentate ligands possessing both N- and O-donor sites are pyridine-2,5-dicarboxylate (PyDC), pyrazine-2,4-dicarboxylate (PZDC) and several amino acid molecules. In some cases, isolated N-donors such as bipyridine (BPy) and their derivatives and isolated O-donor ligands such as isophthalate (IP, also known as 1,3-bezenedicarboxylate (BDC)) were also used to obtain heterometallic cluster-based Ln-TM-MOF systems.107,136 In this regard, three types of Ln-Cu-MOF systems containing heterometallic clusters of Ln(III) (Ln = Er, Y and Eu) and Cu(II) ions were developed by Zheng and co-workers,136 using BPy and IP as the independent N- and O-donors, respectively. In one category of these Ln-Cu-MOF systems, rod-shaped octanuclear heterometallic chains were formed, involving six Ln(III)-based nodes, which were linked through IP ligands, and both ends of this Ln(III) cluster were terminated by two Cu(BPy) moieties, giving the SBU compositional formula of [Ln6Cu2(BPy)2(O2C)11]. In the second category of these Ln-Cu-MOF systems, rod-shaped pentanuclear chains were formed involving a central Cu(II)-based node, which was connected from both sides by two Ln(III)-based nodes involving IP ligands and both the ends of this cluster were terminated by two Cu(BPy) moieties, giving the effective SBU with formula of [Ln2Cu3(BPy)2(O2C)12]. In the above-mentioned two categories of the Ln-Cu-MOF systems, each SBU was interconnect with six other SBUs via IP bridges, producing a very complex 3D network structure in the Ln-Cu-MOF materials. In the third category of Ln-Cu-MOF systems, 2D hexagonal networks were formed by the arrangement of the heterometallic clusters, where six Ln(III)-based nodes were placed at the corners and six Cu(II)-based nodes were placed at the edges of the hexagons. Stacking of these 2D sheets in the ABC fashion, where the IP linkers acted as the pillars to separate the 2D sheets, resulted in the formation of the final 3D framework. A schematic of the above-mentioned three types of Ln-Cu-MOF systems is shown in Fig. 11. Various other Ln-TM-MOF systems with complex structural features have been reported in the literature and more details on these structural characteristics can be obtained from the previously published review articles.107,137,138
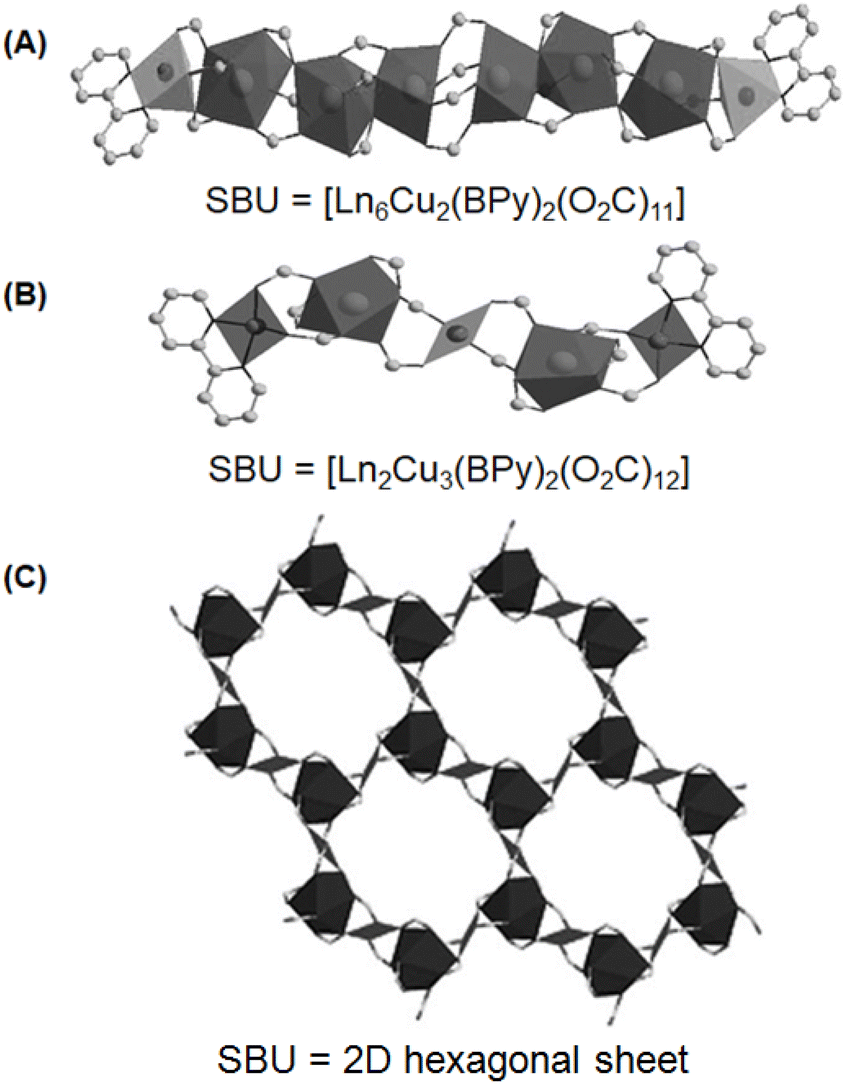 | ||
| Fig. 11 Two different rod-shaped SBUs with constructions as (A) [Ln6Cu2(BPy)2(O2C)11], and (B) [Ln2Cu3(BPy)2(O2C)12], and (C) the 2D hexagonal sheet, as obtained by using IP and BPy as the O-donor and N-donor ligands, respectively, for the synthesis of Ln-TM-MOF systems involving Ln(III) (Ln = Er, Y and Eu) and Cu(II) ions. The figure is redrawn from ref. 136 with permission from Wiley Inter Science. | ||
6. General properties of MOF-based porous materials
MOF systems with tunable network structures and adaptable properties have recently gained significant attention owing to their unique and favorable features towards many advanced applications.139 In MOF systems, the incorporation of inorganic metal nodes and suitable functional moieties in the organic linkers can result in many advanced characteristics, leading to superior applications. In this respect, Ln-MOF materials offer more advantages than the conventional TM-MOF systems, mainly owing to the presence of 4f electrons and the higher and varying coordination numbers of the Ln ions. Although properties such as high porosity and efficient sorption are common features for both conventional and Ln-MOF materials, the latter systems offer additional attractive features such as magnetism and luminescence, making them more suitable for broader applications than conventional MOF systems. In the following section, we discuss some of the general properties of various MOF-based porous materials related to their applications.6.1. Porosity of MOF-based porous materials
High porosity in the network structures of MOF-based materials is an important property, and consequently these materials can adsorb/hold large quantities of analytes/guests in their inbuilt pores, making them super adsorbents. Furthermore, they display a high BET surface area, which increases the interaction probability of the analytes/guests with the active sites embedded inside their pores, enhancing their catalytic activity. Their pore sizes and the characteristics of their active sites can be modulated suitably by introducing adequate substituents/functional groups in the organic linker molecules, which can improve the performance of these materials in the adsorption and catalytic activities.114 Given that MOF-based porous materials can be obtained with large variations in their pore sizes, they can be classified broadly into two categories, namely microporous MOF materials, having pore sizes less than about 2 nm, and mesoporous MOF materials, having pore sizes typically in the range of 2–50 nm.140Han et al. reported the synthesis of different categories of isostructural microporous Ln-MOF systems with complex network structures using linkers such as 1,4-dibenzoate (BDC) and 4,4′-azodibenzoate (ADB) and different Ln ions such as La(III), Ce(III) and Nd(III).141 In the category of these materials having the compositional formula of [Ln6(BDC)9-(DMF)6(H2O)3·3DMF], different types of Ln(III) ions were present in regard to their coordination numbers, three Ln(III) ions with nine-coordination, two with eight-coordination and one with seven-coordination. Although the seven-coordinated Ln(III) ion (marked as Ln6) had a unique coordination pattern, each of the three nine-coordinated (marked as Ln1, Ln2 and Ln3) and two eight-coordinated Ln(III) ions (marked as Ln4 and Ln6) had significantly different coordination patterns, as schematically shown in Fig. 12A.141,142 Combinations of these Ln(III) nodes resulted in the formation of two different types of 1D infinite chains acting as the SUBs and both had the general formula of [La4(CO2)12]n (Fig. 12B). The interconnection of these SUBs resulted in the formation of 3D networks, where the ditopic ligands acted as the spacers for the 1D infinite chains. The Ln-MOF materials displayed enormous microporous channels having rhomboidal openings with a size of about 4 Å × 7 Å (Fig. 12C and D).141
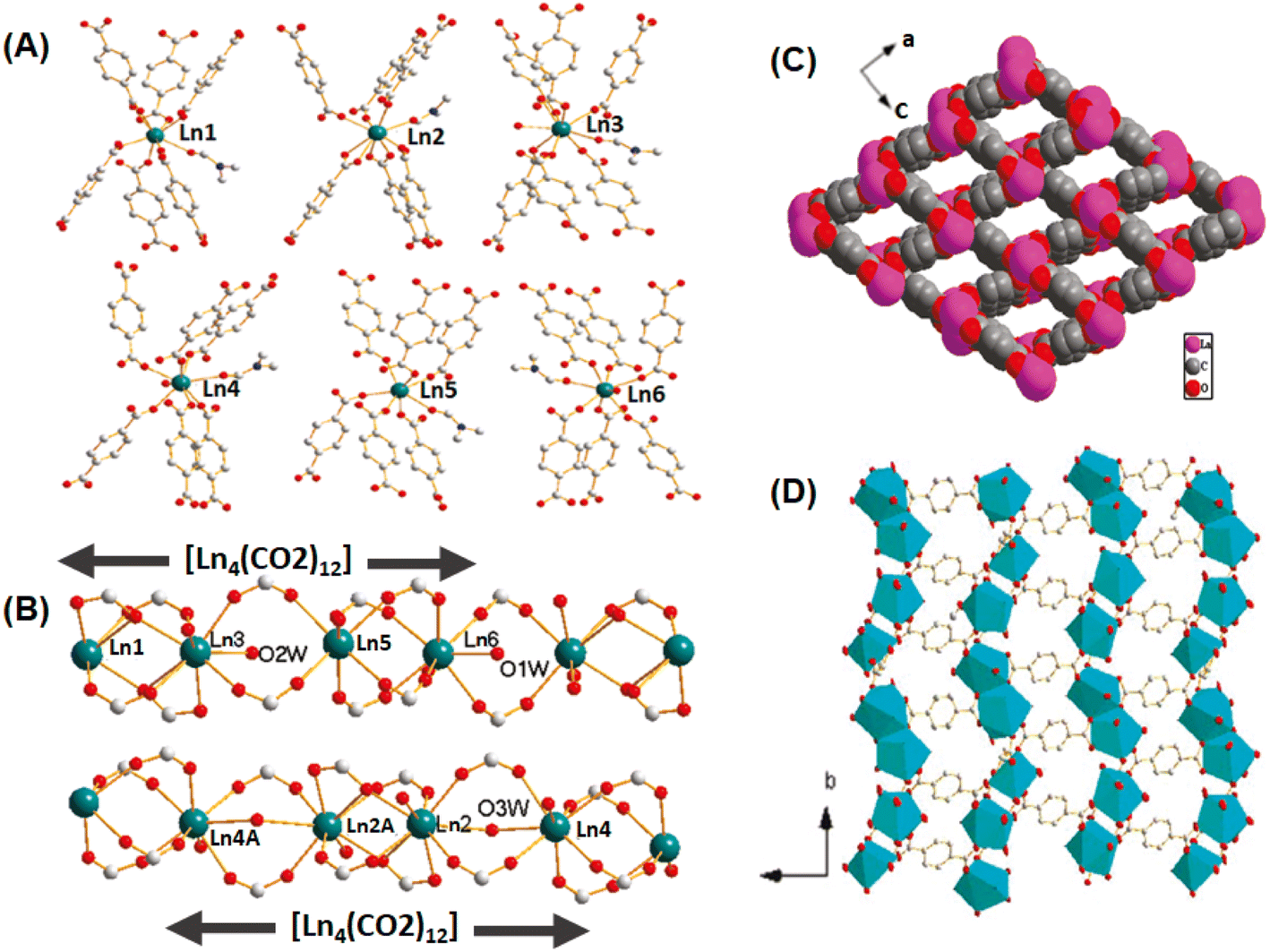 | ||
| Fig. 12 (A) Different types of coordinations of the La(III) ions with the BDC linkers in the Ln-MOF networks. (B) 1D rod-shaped La(III) clusters acting as the SBUs; color codes: C = gray, O = red, and Ln = green. (C) Open channel representation of the Ln-MOF network in the [0 1 1] direction. (D) Polyhedral representation of the Ln-MOF framework in the [0 0 1] direction. The figure is redrawn from ref. 141 with permission from the American Chemical Society. | ||
Another important category of microporous Ln-MOF materials was reported by Zhou and co-workers, designated as the PCN-n(Ln) systems.134,143 Part of the discussion on these systems was already presented in Section 5.2.1 with reference to Fig. 10. In these systems, different Ln(III) ions (Ln = Dy, Er, Y, Yb) were coordinated with tri-topic TATB linkers, resulting in the formation of microporous interpenetrated frameworks, namely, PCN-9(Ln) and PCN-17(Ln) systems, as already discussed in Section 5.2.1 (Fig. 10). In these materials, their pore opening was either blocked or quite tiny due to the interpenetration of the networks. For example, in the pristine PCN-17(Ln) material, effectively no open pores existed along its (1 0 0) and (1 1 0) crystal directions, and only tiny pores with an opening of ∼2.0 Å were observed along its (1 1 1) crystal direction. However, upon activation (removing the μ4-H2O molecules), some pore developed in this material with openings of ∼3.5 Å, which are suitable for the sorption of small gas molecules. The space-filling models of the unactivated and activated PCN-17(Ln) materials, as viewed from the (1 0 0) crystal direction, are shown in Fig. 13A and B, respectively.144
 | ||
| Fig. 13 (A) Space-filling model of an unactivated PCN-17(Ln) material as viewed from the (1 0 0) direction. (B) Space-filling model of an activated PCN-17(Ln) material showing tiny open pores as viewed from the (1 0 0) direction. Color code: Ln = Pink, C = gray, O = red, S = yellow. The figure is redrawn from ref. 143 with permission from the American Chemical Society. | ||
To date, only a few robust mesoporous Ln-MOFs materials have been reported in the literature. This is mainly due to the diverse coordination of the Ln(III) ions, which allows them to retain their coordinated solvent molecules in many systems, thereby blocking their pores substantially. The first examples of mesoporous Ln-MOF systems were reported by Park et al.,145 having the compositional formula of [Tb16(TATB)16-(DMA)24]·(DMA)91(H2O)108. The structural characterization revealed that these materials were formed through the interconnections of the equilateral triangle-shaped [Tb4] clusters, where one Tb atom was at the center and three Tb atoms occupied the corners of the triangle. Four [Tb4] clusters were arranged to form the truncated super-tetrahedron (ST)-shaped SBUs, which were interconnected by TTAB linkers to give the diamond-like interpenetrated network structures, as schematically shown in Fig. 14. In these materials, there were two types of mesoscopic cages, some were relatively smaller (S cages), having an internal diameter of ∼39.1 Å, and the others were relatively larger (L cages), having an internal diameter of ∼47.1 Å. The S cages were surrounded by 20 ST units, creating 12 pentagonal windows (diameter of ∼13.0 Å) and the L cages were surrounded by 28 ST units, forming a total of 12 pentagonal (diameter of ∼13.0 Å) and 4 hexagonal (diameter of ∼17.0 Å) windows.
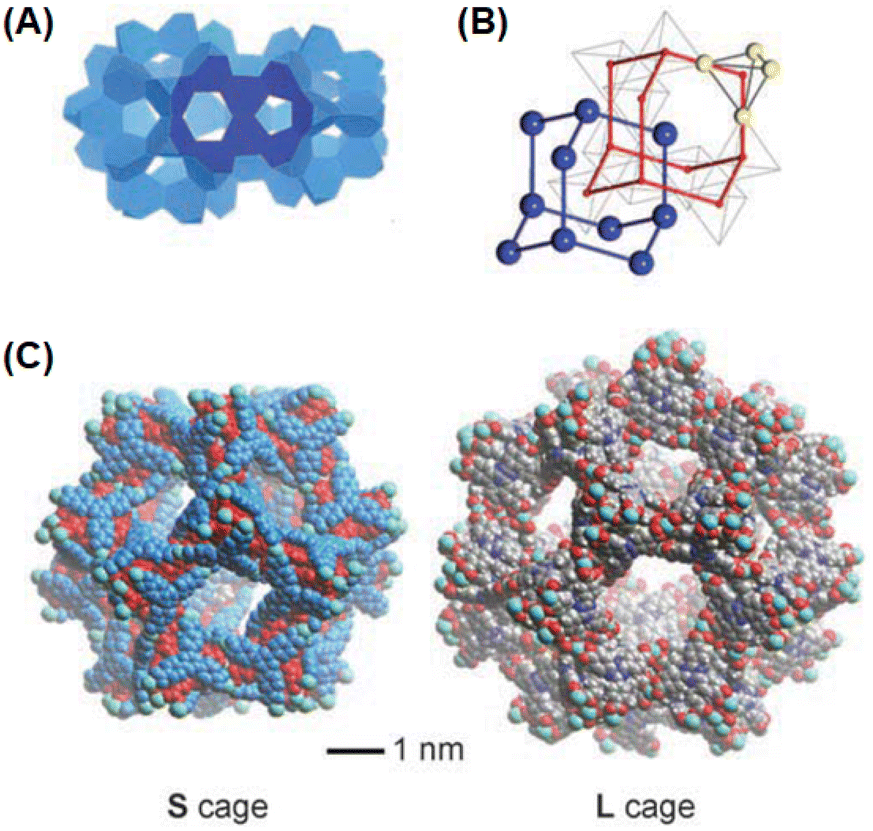 | ||
| Fig. 14 (A) Fused small (S) and large (L) size mesoscopic cages formed by the arrangements of STs as the subunits. (B) Diamond-like doubly interpenetrating nets; blue one is formed directly by L cages and red one is formed by the S cages (connecting the centers of tetrahedra of the four Ln4 clusters, shown in yellow). (C) Schematic of the S and L mesoscopic cages formed in the concerned Ln-MOF systems. Color codes: C = gray, H = white, N = blue, O = red, and Ln = light blue. The figure is redrawn from ref. 145 with permission from Wiley Inter Science. | ||
Another series of mesoporous Ln-MOF based materials, designated as the UTSA-61 systems, where UTSA stands for the University of Texas-San Antonio, was reported by He and co-workers,4,146 which was prepared using a hexa-topic dendritic ligand such as 1,3,5-tris(3,5-di(4-carboxyphenyl) phenyl) benzene (DHC) and Ln(III) ions. In these materials, octahedral-shaped cages (diameter ∼24 Å) were inscribed with the support of the dendritic DHC linkers. Also, there was no interpenetration of the SBUs, and the materials always maintained their mesoporous nature permanently, displaying very good sorption capacity for gases such as N2, H2, CO2 and CH4. The construction of the SBUs and the creation of the octahedral cages through the interconnection of the SBUs, resulting the final 3D network of the UTSA-61 systems, are schematically shown in Fig. 15.
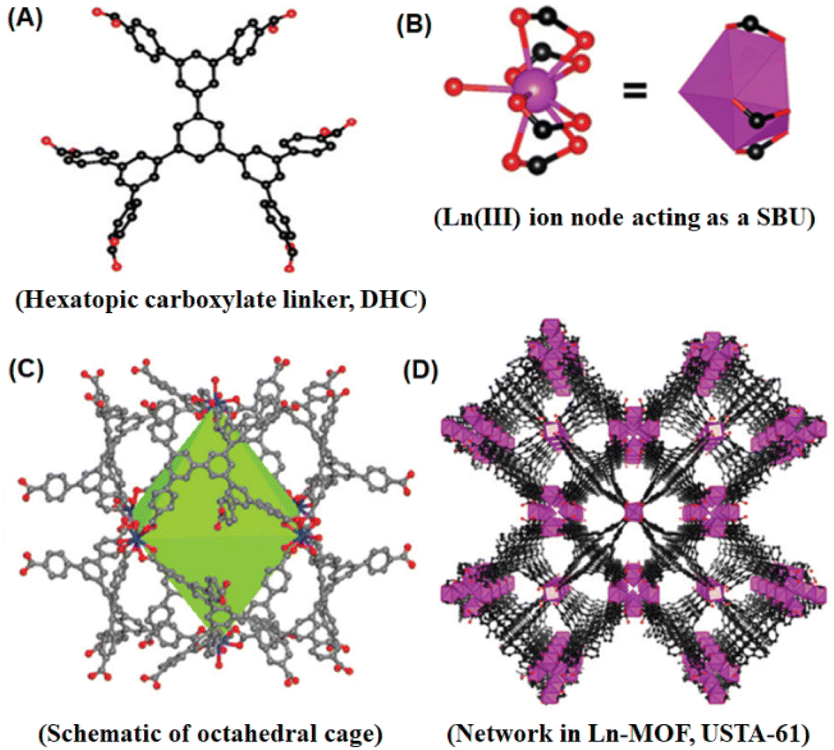 | ||
| Fig. 15 Schematics of (A) hexatopic carboxylate linker DHC, (B) Ln(III) ion node as SBU involving DHC ligands, (C) octahedral cage involving six Ln(III) ion nodes and eight organic linkers, and (D) construction of the overall mesoporous network for the USTA-61 materials. The figure is redrawn from ref. 4 & 146 with permission from the Royal Society of Chemistry. | ||
6.2. Magnetic properties of MOF-based materials
A major challenge in the field of magnetism is the design of materials that can act as molecular ferromagnets.147 A step toward this is developing molecular systems that possess high spin multiplicity in their ground state. In this quest, the majority of the studies involving MOF-based materials have been devoted to TM-based MOF systems, with attention to their unpaired d-electrons. Alternatively, in the case of Ln-MOF materials, there are only limited studies reported with respect to their magnetic properties. Magnetic properties in Ln-TM-MOF materials are very interesting because (i) many Ln ions have large J values owing to their unquenched orbital contributions, (ii) Ln ions can display a substantial extent of easy-axis magnetic anisotropy, and (iii) Ln ions can undergo interesting magnetic interactions with nearby TM ions.135 Among the Ln-MOF- and Ln-TM-MOF-based materials, magnetic properties have been investigated mostly with systems that contain Gd(III) as the lanthanide ion because Gd(III) has relatively less magnetic anisotropy compared to the other Ln(III) ions.107,144 A detailed discussion on the magnetic properties of Ln-based MOF systems is beyond the scope of the present article. However, considering the prospects of the magnetism-based applications of Ln-MOF- and Ln-TM-MOF-based materials, we present a brief discussion on the magnetic aspects of these materials.In the literature, attempts were also made to obtain advanced Ln-TM-MOF materials to achieve coupling between the 3d and 4f metal ions present in these systems. Some representative examples are the extended Ln-TM-MOF systems such as [Cu(BPy)Ln3(IP)5(HIP)(H2O)], [Cu3(BPy)2Ln2(IP)6]·5H2O and [Cu3Ln2(IP)6], where Ln represents the Er(III), Yb(III) and Eu(III) ions for the first system, Yb(III) Gd(III) and Tb(III) ions for the second system, and Eu(III) and Gd(III) ions for the third system, while BPy and IP represent the 2,2′-bipyridine and isophthalate ligands, respectively.136 All these systems displayed very weak antiferromagnetic interaction and the results were rationalized based on the following factors: (i) large separation between the Ln(III) ions, (i) well-shielding of the 4f orbitals of the Ln(III) ions by their 5s and 5p orbitals, and (iii) relatively less covalent characters of the Ln-ligand bonds. All these factors minimized the Ln–Ln interaction, causing the Ln(III)–Cu(II) interaction to be displayed by the studied Ln-TM-MOF systems.
To date, most of the Ln-TM-MOFs investigated in regard to their magnetic behaviors are those obtained by using the pyridine-based carboxylic acid ligands possessing both N- and O-donor groups. A typical example is the [Ln14(μ6-O)(μ3-OH)20(INA)22Cu6Cl4(H2O)8]·6H2O system, where Ln represents Y(III), Gd(III) and Dy(III) ions and INA is the isonicotinic acid ligand.152 In this material, the Ln14(μ6-O)(μ3-OH)20 clusters act as the SBUs, which are linked by the INA ligands to form 3D network structures. The resulting materials showed very weak antiferromagnetic interaction between the Ln(III) ions, which was rationalized based on the above-mentioned factors listed for the previous systems.
A few other heterometallic Ln-TM-MOF systems have also been reported in the literature, e.g., {[Co(H2O)6]·[Ln2(ODA)6Co2]·6H2O}n, {[Ln2(ODA)6Cd3(H2O)6]·mH2O}n and {[Cd(H2O)6]·[Ln2(ODA)6Cd2]·mH2O}n systems, where Ln = Gd, Dy, and Er ions for the first system, Pr, Nd, Sm, Eu, and Dy ions for the second system with m = 9, 6, or 3 in different cases, and Dy, Ho, Er, Tm, and Lu ions for the third system with m = 6 or 12 in different cases, while ODA is the oxy-diacetate ligand.153 All these materials possessed isoreticular Ln–Cd or Ln–Co moieties in their network structures and displayed reasonable ferromagnetic interaction between the Ln and TM centers, albeit only at a very low temperature (1.8 K). Based on various studies, it was inferred that in the Ln-TM-MOF materials, some extent of ferromagnetic coupling between the Ln and TM is generally observed. However, in most of the Ln-TM-MOF systems, the antiferromagnetic interaction between the Ln and TM ions is negligible due to the large physical separation between these ions.
6.3. Luminescence characteristics of Ln-MOF based materials
Traditional luminescent materials such as organic dyes and inorganic phosphor materials have been used extensively in different applications including displays, chemical sensing, optical devices, and many others. The rare earth ions, which include lanthanides together with yttrium, display unique luminescence behavior with vary narrow emission bands and high color purity.154 The electronic transitions that contribute in the luminescent properties of the rare earth ions include their inner shell 4f–4f transitions, the 4f–5d charge transfer (CT) processes, and other CT phenomena such as ligand-to-metal charge transfer (LMCT) and metal-to-ligand charge transfer (MLCT) processes.155 Although La(III) and Lu(III) ions are known to be non-emissive in nature, the other Ln(III) ions are well known for their characteristic emissions. Thus, Eu(III) emits red light, Tb(III) gives green light, Sm(III) emits in orange, Tm(III) emits in blue, and Yb(III), Nd(III), and Er(III) show emission in the near-infrared region.154 The different emissive components present in Ln-MOF systems, namely, coordinated Ln(III) ions, bound organic linkers possessing chromophoric moieties, entrapped chromophoric guests, and physically entrapped Ln(III) ions, can suitably contribute to the observed luminescence behavior of these materials. Given that the emission from each of these components can be tuned suitably either by incorporating a structural modification of the components or by changing/replacing the components as a whole, Ln-MOF systems can offer enormous opportunities for the development of many new and advanced luminescent materials for useful applications in chemical sensing, light-emitting devices, and many others.156 In fact, an abundance of Ln-MOF systems with diverse luminescence characteristics has been reported in the literature, and an in-depth discussion on all these materials is beyond the scope of the present article. However, here, we discuss the luminescent characteristics of a few representative Ln-MOF systems, highlighting some of the important features of these materials in regard to their application potential.A series of 4,4′-(hexafluoroisopropylidene)-bis(benzoate) (FIPBB) ligand-based Ln-MOF systems having the compositional formula of Ln2(FIPBB)3, where Ln represents the trivalent Y, La, Ce, Pr, Nd, Sm, Eu, Gd, Tb, Dy, Ho, Er and Yb ions was investigated in the literature for their luminescence characteristics.157 Most of these materials displayed bluish-white emission upon UV excitation, excluding those containing Eu(III), Tb(III), and Gd(III) ions, which showed emission in the other spectral regions. In the case of the bluish-white emitting materials, it was realized that their emission originated from their ligand excited states. In the latter cases involving Eu(III), Tb(III), and Gd(III) ions, the observed emissions were characteristically found to be red, green and white, respectively, upon UV excitation. These emissions were revealed to arise either from the direct excitation of the Ln ions or due to the energy transfer from the excited ligands to the Ln ions.157 In these cases, the emission intensities were found to be substantially stronger, which was attributed to the negligible luminescence quenching due to the considerable separation between the Ln ions in the chain-like SBUs present in these materials.
Given that the 4f orbitals of the Ln(III) ions are largely shielded by their outer shell 5s and 5p orbitals, these 4f orbitals are largely devoid of interactions with the surrounding matrix. Accordingly, the emissions arising from the 4f–4f transitions in Ln(III) ions are generally very sharp. However, the direct excitation of Ln(III) ions is often very inefficient because the absorption coefficients for the 4f–4f transitions are very low. Accordingly, many luminescence studies involving Ln-MOF systems were attempted by introducing the “antenna effect”, where the excitation of the chromophoric units in the ligands was used to sensitize the excitation of the Ln(III) ions through a non-radiative energy transfer process.158 In this context, the Ba2(H2O)4[LnL3(H2O)2](H2O)nCl series of Ln-TM-MOF systems is of worth mentioning, where Ln represents the trivalent Sm, Eu, Gd, Tb and Dy ions, and ligand L stands for 4,4′-disulfo-2,2′-bipyridine-N′N′′-dioxide. In these materials, the excited ligand can successfully sensitize all the Ln(III) ions present, excluding Gd(III) ions.159 Taking a clue from these systems, it is quite apparent that with the incorporation of suitable Ln(III) ions together with the appropriate organic ligands for effective photosensitization, it is possible to obtain a variety of luminescent Ln-TM-MOF materials that can provide interesting emission characteristics for many useful applications. For further details on luminescent Ln-MOF and Ln-TM-MOF materials, readers are suggested to read the important review article published by Song et al.160
7. Applications of the MOF-based porous solid materials
Owing to their advantages of adequate pore sizes and high BET surface area, MOF-based porous solid materials have prospective usage in different applied areas such as gas adsorption and storage, separation of various analytes, catalysis, construction of magnetic property-based devices, luminescence-based devices, luminescence sensors, and many others.161 Additionally, given that the structures and chemical properties of Ln-MOF materials can also be easily modulated through the selection of appropriate Ln(III) ions and/or using adequate linker molecules, these materials offer advanced applications compared to many conventional porous solids. In the following sections, we discuss some of the important applications of MOF-based porous materials as highlighted in the literature.7.1. Applications of lanthanide-based MOFs in gas storage
Extensive studies have shown that the conventional TM-based MOF materials can exhibit huge gas storage capacities owing to their high porosity and exceptionally large BET surface area. The network structures and pore sizes of these materials can also be easily modulated to enhance their gas storage capacities and specificities for a targeted gas. However, the conventional TM-based MOF materials often suffer from limited thermal and chemical stability, especially when they contain Zn, Cu and Co ions. In terms of thermal and chemical stability, Ln-MOF and Ln-TM-MOF systems are superior to TM-MOF systems, and thus are more suitable for various gas absorption/storage applications. Coordination of the Ln(III) ions, especially with carboxylate linkers, results in the formation of very strong metal–oxygen bonds, owing to the strong affinity of Ln(III) ions for O-donor ligands, enhancing the stability of the obtained MOF materials. Different advanced properties can also be introduced in the Ln-MOF frameworks by incorporating suitable chemical functionalization within their pores, making these materials advantageous for gas storage applications. Further, by introducing coordinatively unsaturated metal centers (UMCs) in Ln-MOF systems, it is also possible to enhance their sorption capacities for the absorption/storage of specific gases to a very large extent. In the following, we present some important aspects related to the gas storage applications of Ln-MOF systems, restricting our discussion to the storage of CO2, H2, CO and N2 gases.As reported in the literature, the interpenetrated PCN-17(Ln) systems, whose structural characteristics have been discussed before (Fig. 10), have shown exceptionally high gas adsorption capacities.134 The inbuilt pores in the PCN-17(Ln) systems are relatively small in size, typically ∼3.5 Å, and their BET surface area is significantly large, in the range of ∼820 m2 g−1. The adsorption characteristics of these materials for O2, H2, CO and N2 gases were investigated extensively, and among them, it was found that they show the greatest selectivity towards O2.134 This higher O2 selectivity was explained in terms of the size selectivity of their pores (∼3.5 Å opening), which are very suitable for O2 molecules compared to the other gas molecules.
Another Ln-MOF system, NJU-Bai11 (NJU-Bai stands for Bai's group at Nanjing University), having the compositional formula of [Y2(TPBTM)(H2O)2]·xG, where Y is the Y(III) ion, G is a guest molecule, and TPBTM is the acylamide-functionalized linker N,N′,N′′-tris(isophthalyl)-1,3,5,-benzenetricarboxylate, was also reported to show a high gas sorption capacity, displaying reasonable selectivity for CO2.162,163 The enhanced efficiency of this material for CO2 uptake resulted from the amide groups incorporated in the walls of its pores, given that amide groups display strong CO2–philic interaction mediated through the polarization of CO2 molecules. The NJU-Bai11 system showed a large BET surface area of ∼1152 m2 g−1 (calculated from N2 isotherm at 77 K), and its CO2 sorption capacity was ∼130 cm3 g−1 at 273 K and 1 bar. However, this material showed a low uptake for N2, only ∼4.9 cm3 g−1 at 273 K and 1 bar, a feature which was very advantageous to achieve high CO2 selectivity over N2.
Ln-MOF systems having the compositional formula of Ln(BTC)(H2O)·(DMF)1.1, where Ln = Y(III), Tb(III), Dy(III), Er(III) and Yb(III) ions and BTC = 1,3,5-benzenetricarboxylate ligand, were also investigated for their gas sorption capabilities.164,165 These materials contained a substantial degree of coordinatively unsaturated metal centers (UMCs) exposed within their porous networks and these UMCs were useful to exhibit selectivity for some specific gases. These isostructural Ln-MOF materials displayed quite high thermal stability with moderate BET surface area, ranging from ∼774 cm3 g−1 with Er(III) to ∼1080 cm3 g−1 with Y(III), as calculated using Ar isotherms at 87 K. These Ln-MOF materials possessing different Ln(III) ions were reported to show moderate H2 uptake, ranging from ∼1.4 wt% to ∼1.8 wt%.
Various types of Ln-MOF materials, with the compositional formula of either [Ln2(2,5-PyDC)2(2,5-PipDC)(H2O)2]n, where Ln = Ce (MOF 1), Pr (MOF 2) and Eu (MOF3), 2,5-PyDC = 2,5-pyridinedicarboxylate ligand and 2,5-PipDC = 2,5-piperazinedicarboxylate ligand, or (H2pip)n[Ln2(2,6-PyDC)4(H2O)2]n, where Ln = Ce (MOF 4), Pr (MOF 5), Eu (MOF 6) and Sm (MOF 7), 2,6-PyDC = 2,6-pyridinedicarboxylate ligand and H2Pip = protonated piperazine ligand, have been reported recently in regard to their gas storage properties.166 X-ray crystallographic studies revealed that the [Ln2(2,5-PyDC)2(2,5-PipDC)(H2O)2]n system possessed 3-D coordination network structures, while the (H2Pip)n[Ln2(2,6-PyDC)4(H2O)2]n system possessed 1-D coordination network structures. Among these materials, the MOF 7 system showed the highest storage capacity for H2 and CO2 gases.166 Another Ln-MOF material, namely the MOF-76(Tb) system, has also been reported recently to show promises for hydrogen storage and humidity sensing applications.163 This material contains M–O–C based metal cores consisting of four Tb(III) ions, which are interconnected by benzene-1,3,5-tricarboxylate linkers. Each of the four Tb(III) ions are also coordinated with an aqua ligand. The framework structure of the MOF-76(Tb) material produces 1D channels with the typical size of 6.6 Å × 6.6 Å, propagating along its c crystallographic axis. The hydrogen storage capacity for the MOF-76(Tb) system was found to be ∼0.6 wt% at 77 K and 20 bar pressure.163Scheme 1 shows a timeline of the development of some important MOF-based porous solid systems, which are highlighted in regard to their uses in the separation of some representative gases.163,167,168
 | ||
| Scheme 1 Timeline of some important MOF and Ln-MOF systems developed and highlighted in regard to their uses in the separation of some representative gases. The scheme is redrawn in accordance with ref. 168 with permission from Elsevier. | ||
7.2. Applications of lanthanide-based MOFs in catalysis
Similar to TM-MOF systems, various Ln-MOF materials have also been recognized to display substantial catalytic effects to support various chemical reactions. A few of these reactions are as follows: (i) hydrolysis reactions, where esters or other substrates can be hydrolyzed catalytically by the Ln-MOF systems due to the Lewis acid properties of the lanthanide ions present in these systems, (ii) oxidation reactions, where substrates such as alcohols can be oxidized catalytically by the Ln-MOF systems, assisted by the redox properties of lanthanide ions, and (iii) photocatalytic reactions, where energy conversion as well as pollutant degradation can be achieved by the catalytic effect of Ln-MOF systems utilizing their unique photophysical properties. Scheme 2 shows a schematic representation of the different catalytic applications of Ln-MOF systems. | ||
| Scheme 2 Schematic of different catalytic applications of Ln-MOF systems. | ||
Different mechanisms can be involved in the catalytic actions of Ln-MOF systems. These mechanistic aspects of the catalytic activities of Ln-MOF systems can be broadly realized based on the following important features associated with these materials.
In regard to their catalytic activity, the most important Ln-MOF systems reported in the literature are those possessing SBUs with coordinatively unsaturated metal centers (UMCs), arising mainly due to the variable coordination numbers of the Ln(III) ions. Together with UMCs, the presence of organic linkers that display Lewis acid characteristics, and thus are adaptable for easy functionalization using Lewis bases or similar other functional groups, are also important for Ln-MOF systems to display catalytic activity.169,170 Given that the coordination geometry and frameworks of Ln-MOF systems are very tenable and strong, and because a large number of Ln(III) ions can be involved in the formation of these materials, Ln-MOF systems are very capable of displaying promising and superior catalytic activity.171
Different Ln-MOF systems have been investigated in the literature for their catalytic properties, exhibiting high thermal and chemical stability owing to their framework structures.169,171,172 An important Ln-MOF series in this regard is the [Ln2(DPA)3(H2O)]·2H2O system, where Ln represents the trivalent La, Ce, Pr, Nd, Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, and Yb ions, while DPA represents the 1,4-phenylene diacetate linker. These materials were stable up to about 450 °C and displayed substantial chemical stability towards repeated hydration and dehydration cycles. Among them, the material having a Tb2(DPA)3 motif in its structure showed outstanding catalytic activity, especially for the conversion of benzaldehyde to 1,1-dimethoxytoluene in the presence of methanol with ∼78% yield in just about 10 h. The other Ln-MOFs in the series showed comparatively lower efficiency in the same reaction.170 It was evident from these studies that an efficient Ln-MOF in an isostructural series for catalyzing a desired chemical reaction can be identified by screening the materials involving different Ln(III) ions.
In the context of the role of variations in the catalytic sites and/or the changes in the microenvironments around these sites, the succinate (SC) and sulfate (SO4−)-based mixed ligand containing RPF-16 systems (RPF stands for rare earth polymeric frameworks) having the compositional formula of [Ln2(SC)2(SO4)(H2O)2], where Ln = La(III), Pr(III), Nd(III) and Sm(III) ions, have attracted reasonable interest in the literature.173 These Ln-MOF materials possess both redox and acid centers as the active sites within their pores, and both the sites can adequately participate in the catalytic activity for appropriate chemical reactions. Thus, the RPF-16 systems can catalytically carry out highly selective hydrogenation of nitro groups in aromatic molecules. It was revealed that the catalytic action of the RPF-16 materials is initiated at their acid centers and selective hydrogenation occurs at their redox centers in a subsequent step. The catalytic acid centers basically promote the heterolytic rupture of the H2 molecules, forming Ln-hydrides as intermediates, which then interact with the nitro groups, resulting in their selective hydrogenation, with a conversion efficiency of close to 100%. The RPF-16 materials were also investigated for the catalytic oxidation of sulfide groups, but this attempt was not successful. It was revealed that in the RPF-16 materials, the coordination number for the Ln(III) ions is at the highest permissible limit of 9, and accordingly the system did not allow any additional sulfide coordination required to catalyze the oxidation of the sulfide groups.173
Three important RPF categories of materials, namely, RPF-21, RPF-22, and RPF-23 systems, were also reported by D'Vries et al.174 for their catalytic activities. The compositional formulae of these systems are [Ln(DSB)(H2O)5], [Ln(DSB)(H2O)3] and [Ln3(DSB)2(OH)3(H2O)3], respectively, where DSB = 3,5-sulfobenzoate linker, Ln = La(III), Pr(III), and Nd(III) ions for the RPF-21 system, Ln = La(III), Pr(III), Nd(III), Sm(III), and Eu(III) ions for the RPF-22 system, and Ln = Pr(III), Nd(III), and Eu(III) ions for the RPF-23 system. All these materials displayed outstanding thermal stability up to about 460–540 °C and demonstrated excellent catalytic activity towards the solvent-free cyanosilylation reaction (CSR) of aldehyde groups. The small differences in the catalytic activity in the three categories of the RPF systems were understood to be due to the differences in their Ln(III) coordination numbers, which were 9 for the RPF-21 series, 8 for the RPF-22 series and both 8 and 9 for the RPF-23 series. It was inferred that the CSR reaction apparently proceeded through the displacement of the labile H2O molecules from the coordination spheres (UMC sites) by the aldehyde groups of the substrates, which effectively initiated the activation of the aldehyde groups for their cyanosilylation by the presence of these Ln-MOF catalysts. It was also further realized that the catalytic activity of the studied series of MOF materials is determined largely by their network structures, with only a minute influence arising from the nature of the Ln(III) ions present in these systems.174
Similar to the RPF systems, another set of Ln-MOF materials having the compositional formula of [Ln2(MELL)(H2O)6], where MELL = mellitic acid, a hexacarboxylate linker, and Ln = La(III), Eu(III), Tb(III) and Gd(III) ions, was reported by Batista et al.175 for their catalytic activity towards CSR reactions of the aldehyde groups. The results showed the substantial catalytic activity of these materials for the addition of a trimethylsilyl-cyanide group to the aldehydes, and the reactions occurred with exceptionally short reaction times together with excellent yields, especially with the Ln-MOF systems possessing Eu(III) ions.175
Quite recently, two Ln-MOF-589 series of isostructural materials with the general formula of [La(BIPA-TC)]·H2O, where Ln = La3+, Ce3+ ions and BIPA-TC is the tetratopic linker, benzoimidephenanthroline tetracarboxylate ligand, were reported by Tran et al.176 in regard to their catalytic effects on the oxidation of olefins. The structural characterization revealed that these materials possess [Ln2(–COO)6(–COOH)2(H2O)6] functional units, which act as a Lewis acid, while the naphthalene diimide functional cores of the BIPA-TC linker act as the electron-attracting units and oxidation centers. The combined effect of these two functional groups resulted in the promising catalytic activity of these Ln-MOF materials for the oxidation reaction of olefins. Between the two isostructural Ln-MOF materials studied, the Ce-MOF-589 material exhibited an outstanding performance for styrene and cyclohexene oxidation (efficiency of about 94% and 90%, respectively) together with very good selectivity towards the formation of styrene oxide (∼85%) and 2-cyclohexen-1-one (∼95%) in the respective cases.
Ln-MOF based porous materials can also display excellent photocatalytic activity, provided their active sites can absorb a sufficient extent of the used light. However, in the case of most MOF systems, given that their active sites are deep inside bulk materials, the used light can hardly reach these active sites. Thus, to realize an efficient photocatalytic effect, the MOF system has to be designed to be easily dispersed in a solvent to assist substantial light absorption by its active sites. In this respect, a dispersible MOP system structurally resembling MOF systems was developed and investigated for its photocatalytic activity, which was abbreviated as MOP-Ru-Co, by Shi and co-workers.177 The developed MOP system contained the [Ru(bpy)3]2+ group as the photosensitive units, where bpy stands for 2,2′-bipyridine ligand. This MOP-Ru system can combine with Co2+ ions easily to form the MOP-Ru-Co system as the resultant photocatalytic material. The MOP-Ru-Co system could be dispersed into discrete cages in methanol solvent, greatly enhancing the light absorption ability of its active sites. This dispersed MOP-Ru-Co system exhibited better catalytic activity for the photoreduction of CO2 than analogous MOF systems or the undispersed MOP-Ru-Co system. This study revealed that CO2 photoreduction by the dispersed MOP-Ru-Co system occurred through the absorption of light by the [Ru(bpy)3]2+ group and the subsequent transfer of the photogenerated electrons to the Co catalytic site of the MOP-Ru-Co system, causing the photoreduction of CO2 to occur very efficiently.177
7.3. Applications of lanthanide-based MOF systems in chemical sensing
Owing to their excellent 3D frameworks, tunability of their porous networks, and their intriguing luminescence properties, Ln-MOF materials in general exhibit many advantages compared to other conventional chemosensory materials. Luminescence is one of the most important properties of Ln-MOF systems, which has attracted immense attentions towards the use of these materials as luminescent-based sensors for many chemical species. The Ln(III) ions present in the metal nodes, the entrapped Ln(III) ions, the chromophoric moieties attached to the linkers, and also the emissive molecules entrapped as the guests can jointly contribute, leading to the different situations of the luminescence characteristics of MOF systems, as schematically shown in Fig. 16.46 | ||
| Fig. 16 Schematic of (A) four possible emissive components and (B–D) three typical scenarios for the construction of dual-emitting ratiometric fluorescent sensors with Ln-MOF systems. The figure is redrawn from ref. 46 with permission from Frontiers Media S. A., Switzerland. | ||
Given that small organic molecules, metal cations, inorganic anions, and other chemical species can be adsorbed into and released from the Ln-MOF pores easily in a reversible manner, they can influence the luminescence characteristics of the host materials, forming the basis for luminescence-based chemical sensing. Owing to the suitable dimensions of their pores and the strong host–guest bonding rendered by their functional sites, Ln-MOF systems show promise to be very effective and quite selective for luminescence-based sensing applications.46,154
Ln-MOF systems can detect gases such as CO2 and NO2, and volatile organic compounds, through changes in their luminescence behavior. Ln-MOFs can also sense various pollutants or toxic substances in the environment, which can easily alter their luminescence properties, providing the desired sensing and detection. Ln-MOFs can also be functionalized suitably for the specific detection of biological analytes such as proteins or DNA through luminescence-based sensing. In all these cases, the sensing process can be interpreted and visualized in terms of the following important reasons.
With respect to metal ion sensing, an Eu(III)-based Ln-MOF system having the compositional formula of [Eu(PDC)1.5(DMF)]·(DMF)0.5(H2O)0.5, where PDC = pyridine-3,5-dicarboxylate ligand and DMF = N,N-dimethylformamide solvent, was reported to be very useful.178 The pyridyl moiety present in this system acts as a Lewis base and coordinates with the analyte metal ion, forming the corresponding Lewis adduct, and thereby quenching the luminescence of the Ln-MOF system. Given that alkali and alkaline earth metal ions did not coordinate with the pyridyl moieties, they did not cause any significant luminescence quenching in this Ln-MOF material. In contrast, transition metal ions including Cu2+, Mn2+ and Co2+ induced significant luminescence quenching in the Ln-MOF system, enabling the very effective sensing of these transition metal ions.
The Ln-MOF-76 system having the compositional formula of [Tb(BTC)(H2O)1.5]·(DMF), where BTC represents the 1,3,5-benzenetricarboxylate (BTC) ligand, was also found to be quite promising for luminescence-based chemical sensing.179,180 This material possessed rod-like infinite length SBUs consisting of Tb(III) ions having 7-coordinations, six involving the carbonyl groups of the BTC ligands and the other one involving one water molecule. The as-formed Ln-MOF-76 system contains square-shaped 1D channels, having dimensions of about 6.6 Å × 6.6 Å. These materials were investigated for their prospects in luminescence-based anion sensing applications.180 It was found that the luminescence intensity of the Ln-MOF material was enhanced quite substantially by F− ions, demonstrating its potential in the sensing of this anion in particular together with different other anions in general.
The Eu(III) based Ln-MOF system [Eu(BTC)(H2O)]·1.5H2O was found to display quite appreciable luminescence-based responses towards the sensing of different small organic analytes.181 Thus, different conventional solvents including 2-propanol, chloroform, ethanol, 1-propanol, methanol, DMF, acetone, methanol, tetrahydrofuran, and acetonitrile were seen to induce significant modulations in the photoluminescence intensity of this material, indicating its possible application in sensing these chemical species. It was realized that the existence of the open Ln(III) sites were mainly responsible for the observed luminescence behavior of this material. As the analytes entered the pores of this system, the luminescence characteristics of the host material were altered, leading to the sensing of the particular analytes. Although the mechanistic details of the concerned processes were not understood very clearly, the results undoubtedly indicated that this material has enormous potential for uses in many luminescent-based sensor applications.
The isostructural Ln-MOF systems CUST-623, CUST-624, CUST-625, CUST-626, and CUST-627, having the compositional formula of [Ln(BDPO)(H2O)4], where BDPO is the N,N′ bis(3,5-dicarboxyphenyl)-oxalamide ligand and Ln stands for Eu(III), Tb(III), Gd(III), Dy(III) and Sm(III) ions, respectively, for the five respective systems, were reported by Wang et al.182 in regard to their luminescence-based sensing applications. All these materials showed excellent temperature-dependent changes in their luminescence intensity, demonstrating their high-performance temperature sensing application with sensitivity of about 3.7% K−1, in the temperature range of 303–423 K. Further, the CUST-623 and CUST-624 materials showed prospects for uses as multi-response luminescent sensors for the detection of Fe3+, Cr2O72−, CrO42− and TNP as analytes.183 Very recently, a series of Ln-TM-MOF materials, designated as MIL-125(Ti–Ln) systems, where Ln = Eu(III) or Tb(III) ions and the linker molecules were the typical bidentate carboxylate ligands, was also reported to exhibit highly selective fluorescence quenching in response to Cu(II) and Fe(III) ions.183
Regarding the use of MOF materials in sensing applications, the challenge is the accommodation of the target analytes within the pores of these materials. In this case, a novel molecular imprinting strategy was adopted by Shi and co-workers184 to introduce target-specific recognition sites into a tailormade multicomponent Ln-MOF system, NKU-66-EuGdTb-P, having the compositional formula of {[Eu0.0067Gd0.992Tb0.0013(FDA)0.5(BETC)0.5(H2O)2]}n, where FDA and BETC represent the 2,5-furandicarboxylate and 1,2,4,5-benzenetetracarboxylate linkers, respectively. The initially obtained NKU-66-EuGdTb systems contained entrapped DMF molecules in their pores, which on removal by methanol exchange, resulted in the NKU-66-EuGdTb-P system, possessing a template for bioanalytes such as methylglyoxal (MGO), a reactive carbonyl molecule having implication in metabolic activity. This material displayed a strong interaction with MGO, making it useful for the sensing and detection of this bio-analyte. It was revealed that at lower MGO concentrations of below ∼15 μM L−1, the entrapped MGO caused dynamic quenching of the Ln-MOF luminescence, showing a systematic reduction in its emission lifetime, which is useful for the selective trace-level detection of MGO. Alternatively, at a higher concentration of MGO, the large amount of entrapped MGO caused a significant extent of static quenching in the luminescence of the Ln-MOF, enabling the detection of a higher concentration of the MGO analyte. This work established a new molecular imprinting strategy for obtaining advanced Ln-MOF materials useful for the trace-level sensing and detection of bio-analytes.
In luminescence-based sensing applications, Ln-MOF systems possessing multiple emission centers are considered to be emerging materials that can enable the ratiometric sensing of analytes with high sensitivity and high selectivity. In these materials, the energy transfer between the existing light-absorbing groups and multiple emission centers can be utilized judicially to design very effective ratiometric sensors for analyte detection. The functionalization of suitable MOFs with multiple lanthanide centers, organic chromophores, carbon dots, or other suitable emissive groups can enable the development of multiple emission center-based Ln-MOF systems. According to this perspective, the various synthesis strategies and ratiometric sensing-based applications of different Ln-MOF systems were summarized and discussed by Cheng and co-workers185 in their recent review article. A few of the important Ln-MOF systems that should be mentioned in the present contexts are the {[Eu2(L)3(H2O)2(DMF)2]·16H2O}n system used for humidity sensing, {[Cd1.5(EDDA)]·(H3O)·3(H2O)}n system used for pH sensing, Eu0.47Tb0.53-CTP-COOH system used for Fe3+ and volatile organic compound (VOC) sensing, Eu2(5-bop)3(H2O)2 system for F− sensing, and MIL-125(Ti)-NH2-Eu system for PO4− sensing. In general, luminescent Ln-MOF and Ln-TM-MOF systems have been found to be very important porous solid materials for various types of emission-based sensing applications.186
Quite recently, five different structurally similar Ln-MOF systems having the compositional formulae of {[Pr10L6(OH)3Cl(H2O)6]·4C2H8N}n, {[Nd10L6(OH)4(H2O)9]·4C2H8N}n, {[Gd10L6(OH)4(H2O)3]·4C2H8N}n,{[Ho10L6(OH)4(H2O)3]·4C2H8N}n and {[Er10L6(OH)4(H2O)6]·4C2H8N}n were reported by Chai et al.187 in regard to their sensing applications, though in these cases their responses were based on their electrochemical sensing. All five Ln-MOF systems were crystallized with the trigonal R3 space group, as established from their single-crystal X-ray analysis. These materials possessed 3D mesoporous structures, featuring the coexistence of binuclear and tetranuclear species as the inorganic building units. The studied materials were found to very useful for the electrochemical detection of trinitrophenol, a strongly explosive chemical, with excellent sensitivity.187
In the literature, various kinds of applications of lanthanide-based Ln-MOF systems have been reported. However, a full account of all these applications is beyond the scope of the present article. Thus, we restricted our discussion to a limited number of important applications of Ln-MOF systems, focusing on the type of sensing mechanisms involved in their different applications. For a quick reference, some more examples on the applications of Ln-MOF materials are listed in Table 1 together with the properties of these Ln-MOF systems that are directly involved in their sensing responses.
| Area of application | Details of the application | Properties of the Ln-MOFs utilized | Specific examples | Ref. |
|---|---|---|---|---|
| Gas storage and separation | Storage and selective separation of gases | High porosity, tunable pore sizes, and selective adsorption | Capture/storage/separation of hydrogen, CO2, and methane | 140 |
| Catalysis | Used as catalysts | Redox properties of lanthanide ions and high surface area and stability of Ln-MOFs | Oxidation reactions, polymerizations, and hydrolysis reactions | 48 |
| Environmental monitoring | Remediation of pollutants and heavy metals | Luminescence and magnetic properties of Ln-MOFs | Sensing, detection and removal of toxic metals and pollutants | 188 |
| Biomedical imaging | Imaging and tracking of biological processes | Luminescence properties and biocompatibility of Ln-MOFs | Used as MRI contrast agents and fluorescence imaging of biosystems | 189 |
| Biomolecular sensing | Detection of biomolecules and biological systems | Luminescence properties and specificity for biomolecules | Protein detection and DNA/RNA sensing | 190 |
| Drug delivery | Targeted therapeutic uses | Stability, luminescence, and biocompatibility | Controlled release at targeted sites in the body for therapeutic effects | 191 |
| Cancer diagnosis and therapy | Targeting; NIR triggered TPA-PDT; FL-imaging | Tunable size, high porosity, and large specific surface area | MB@THA-NMOF-76@RCD | 192 |
| Spectroscopy | Spectroscopic analysis of materials | Photoluminescence and sharp spectral lines | Sensing, imaging, analyte detections, environmental monitoring, and time-resolved emission-based spectroscopy | 193 |
| Luminescent probes | Sensitively and selectively respond to cations/anions or small molecules | Exhibiting effective luminescent response | [Eu(BTPCA)(H2O)]·2DMF·3H2O targeted Zn2+ ions | 194 |
| Ratiometric fluorescent sensing | Eu3+/Tb3+-based MOFs have been widely employed for the construction of dual-emission ratiometric fluorescent sensors | Highly designable multifunctional luminescent materials as lanthanide ions, organic ligands, and guest metal ions | [Zn4(pta)3(H2O)1.5] (ZJU-108) MOF for detection of tryptophan in aqueous solution | 195 |
| Magnetic devices | Development of Ln-MOF-based magnetic devices | Magnetic (paramagnetic, ferromagnetic, or antiferromagnetic) properties of Ln-MOF systems | Magnetic resonance imaging (MRI), and data storage | 196 |
| Information storage and processing | Spintronics and molecular electronics | On-off switching of single-molecule magnet | Molecular spintronics using single-molecule magnets | 197 and 198 |
| Energy-related applications | Stable and conductive MOFs | Strong coordination between Ln(III) ions and rigid carboxylic linkers supported by hydrogen bonds enhance the robustness, stability and electron transport process | Ln4-MOFs, i.e. [{Ln4(μ4-O) (μ3OH)3 (INA)3(GA)3} (CF3SO3)(H2O)6]n; Ln = Gd, Tm, and Lu, INA = isonicotinic acid, GA = glycolic acid | 199 |
8. Summary and future perspectives
In this review article we attempted to provide a brief overview of the basic perceptions, preparation procedures, properties, and applications of lanthanide-based MOF systems (Ln-MOFs). Although the design and control of conventional MOF systems involving transition metal (TM) ions are relatively easy and well-documented in the literature, the corresponding design and control of lanthanide-based metal–organic frameworks are quite challenging, mainly due to the quite unspecific coordination, larger coordination number, and multidimensional coordination properties of the lanthanide ions. Although Ln-MOF systems have great potential in diverse applications due to their intriguing reticular frameworks, large stability of their network structures, high porosity, promising luminescence characteristics, intriguing magnetic properties, etc., these systems, need further exploration to reveal their advanced applications in different areas. The studies carried out thus far have demonstrated that Ln-MOF materials can function as ideal templates for gas capture and storage, sorption and separation of analytes, catalyzing various chemical reactions, luminescence-based sensing of analytes, inventing magnetic property-based devices, and various other applications. The introduction of appropriate functional groups in Ln-MOF systems, and thus modifying the chemical nature of the inner walls of their pores, and also their pore sizes through either pre- or post-synthetic strategies, makes it possible to increase their selectivity and sorption capacity for sensing various analytes such as different inorganic ions, gaseous species, and different organic molecules. Although significant progress has been made in the application of Ln-MOF materials for gas capture/storage and analyte sorption/separation, there are still many unsolved issues and challenges in exploring the versatility of their practical uses. A few challenges are as follows: (i) Ln-MOF systems perform suitably in a narrow range of pH in aqueous solution; however, they generally do not display the desired stability either in acidic or in alkaline pH conditions, which is based on the characteristics of the materials used. Considering this, extensive research is still warranted for the development of suitable Ln-MOF materials that are stable at least under moderate acidic conditions, particularly if the MOF materials are intended to use for the treatment of nuclear waste. (ii) The sorption mechanisms and the relationships between the structures of Ln-MOF systems and their performances towards different sorption-related applications are yet to be understood clearly. Therefore, sufficient attention is needed in understanding these mechanistic details to explore their wider applications, especially to develop Ln-MOF-based methodologies for harsh conditions, such as the those relevant to the separation of radioactive metal ions. (iii) The cost-effective, large-scale and eco-friendly preparation of Ln-MOF materials is still to be realized satisfactorily, and thus many more studies are necessary to achieve the goal in the aforementioned aspects. This is especially important for the successful deployment of these materials in commercial and industrial applications. (iv) The adequate management of MOF materials after their useful life is also an important area of research, which is apparently missing in the current research trends. Overall, it is quite understandable that dedicated efforts are needed to design ecofriendly and commercially viable methods for the synthesis, utilization, recycling and disposal of various MOF materials to write the success story for these fascinating porous 3D network materials. Evidently, intense basic and applied research is necessary to fully utilize the potential of these materials, and thus efforts should continue to build on their existing applications in the literature.Data availability
Data will be available on request.Conflicts of interest
The authors declare that there is no known competing interest to influence the work reported in this review article.Abbreviations
| ACAC | Acetylacetonate |
| ADB | 4,4′-Azodibenzoate |
| BDC | 1,4-Benzenedicarboxylate |
| BTC | 1,3,5-Benzenetricarboxylate |
| BBC | 4,4′,4′′-(Benzene-1,3,5-triyl-tris(benzene-4,1-diyl))tribenzoate |
| BPDC | 4,4′-Biphenyldicarboxylate |
| BET | Brunauer–Emmett–Teller |
| CSR | Cyanosilylation reaction |
| BDPO | N,N′-Bis(3,5-dicarboxyphenyl)-oxalamide |
| DEF | N,N-Diethylformamide |
| DMA | Dimethylacetamide |
| DMF | N,N-Dimethylformamide |
| DPA | 1,4-Phenylenediacetate |
| DSB | 3,5-Disulfobenzoate |
| BIPA-TC | Benzoimidephenanthroline tetracarboxylate |
| HFIPBB | 4,4′-(Hexafluoroisopropylidene)bis(benzoate) |
| H2Pip | Protonated piperazine or piperazinium |
| HTB | 4,4′,4′′-(1,3,4,6,7,9,9-Heptaazaphenalene-2,5,8-triyl)tribenzoate |
| HKUST | Hong Kong University of Science and Technology |
| INA | Isonicotinic acid |
| IP | Isophthalate or 1,3-benzenedicarboxylate |
| IRMOF | Isoreticular metal–organic framework |
| Ln | Lanthanide |
| Ln-MOF | Lanthanide MOF |
| Ln-TM-MOF | Lanthanide-transition metal MOF |
| LMCT | Ligand-to-metal charge transfer |
| MOF | Metal–organic framework |
| MIL | Materials of Institute Lavoisier |
| MELL | Mellitic acid |
| MLCT | Metal–ligand charge transfer |
| MDIP | Methylenediisophthalate |
| NJU | Nanjing University |
| ODA | Oxydiacetate |
| PAM | Pamoic acid [or, 4,4′-methylenebis(3-hydroxy-2-naphthalenecarboxylate)] |
| PCN | Porous coordination network |
| PDC | Pyridine-3,5-dicarboxylate |
| PyDC | Pyridine-2,5-dicarboxylate |
| 2,5-PyDC | 2,5-Pyridinedicarboxylate |
| 2,6-PyDC | 2,6-Pyridinedicarboxylate |
| 2,5-PipDC | 2,5-Piperazinedicarboxylate |
| PZDC | Pyrazine-2,4-dicarboxylate |
| RPF | Rare earth polymeric frames |
| SBU | Secondary building unit |
| TATB | 4,4′,4′′-(s-Triazine-2,4,6-triyl)-tribenzoate |
| TATAB | 4,4′,4′′-(s-Triazine-1,3,5-triyl)-tri-p-aminobenzoate |
| TM | Transition metal |
| UMC | Unsaturated metal center |
Acknowledgements
Authors are grateful to the host institute, Bhabha Atomic Research Centre, for all the generous financial and logistic supports during the course of the present work. Authors are also thankful to Shri U. Dani, C.E., NRB, BARC, Dr Arijit Sengupta, RCD, BARC, Dr P. K. Mohapatra, RCD, BARC for their constant encouragement and support. HP is especially obliged to the Department of Atomic Energy, for the reward of the Raja Ramanna Fellowship, and is extremely thankful to Homi Bhabha National Institute, for proving him all the logistic supports.References
- H. Li, M. Eddaoudi, M. O'Keeffe and O. M. Yaghi, Nature, 1999, 402, 276–279 CrossRef CAS.
- S. Kitagawa, R. Kitaura and S. I. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS PubMed.
- M. Zhang, Y.-P. Chen, M. Bosch, T. Gentle III, K. Wang, D. Feng, Z. U. Wang and H.-C. Zhou, Angew. Chem., Int. Ed., 2014, 53, 815–818 CrossRef CAS PubMed.
- F. Saraci, V. Quezada-Novoa, P. R. Donnarumma and A. J. Howarth, Chem. Soc. Rev., 2020, 49, 7949–7977 RSC.
- J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. B. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 Search PubMed.
- L. J. Murray, M. Dinca and J. R. Long, Chem. Soc. Rev., 2009, 38, 1294–1314 RSC.
- X.-J. Kong and J.-R. Li, Engineering, 2021, 7, 1115–1139 CrossRef CAS.
- Y.-R. Lee, J. Kim and W.-S. Ahn, Korean J. Chem. Eng., 2013, 30, 1667–1680 CrossRef CAS.
- T. A. Makal, D. Yuan, D. Zhao and H.-C. Zhou, in Metal–Organic Frameworks,World Scientific, Singapore, 2011, pp. 37–64 Search PubMed.
- E. Biemmi, S. Christian, N. Stock and T. Bein, Microporous Mesoporous Mater., 2009, 117, 111–117 Search PubMed.
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469–472 CrossRef CAS PubMed.
- S. S.-Y. Chui, S. M.-F. Lo, J. P. H. Charmant, A. G. Orpen and I. D. Williams, Science, 2002, 283, 1148–1150 CrossRef PubMed.
- P. Kumar, A. Deep and K. Kim, TrAC, Trends Anal. Chem., 2015, 73, 39–53 CrossRef CAS.
- A. Douvali, A. C. Tsipis, S. V. Eliseeva, S. Petoud, G. S. Papaefstathiou, C. D. Malliakas, I. Papadas, G. S. Armatas, I. Margiolaki and M. G. Kanatzidis, Angew. Chem., Int. Ed., 2015, 127, 1671–1676 CrossRef.
- S. Horike, S. Shimomura and S. Kitagawa, Nat. Chem., 2009, 1, 695–704 CrossRef CAS PubMed.
- D. Bradshaw, J. Claridge, E. Cussen, T. Prior and M. Rosseinsky, Acc. Chem. Res., 2005, 38, 273–282 CrossRef CAS PubMed.
- O. K. Farha and J. T. Hupp, Acc. Chem. Res., 2010, 43, 1166–1175 CrossRef CAS PubMed.
- S. Zheng, T. Wu, J. Zhang, M. Chow, R. A. Nieto, P. Feng and X. Bu, Angew. Chem., Int. Ed., 2010, 49, 5362–5366 CrossRef CAS PubMed.
- J. M. Taylor, T. Komatsu, S. Dekura, K. Otsubo, M. Takata and H. Kitagawa, J. Am. Chem. Soc., 2015, 137, 11498–11506 CrossRef CAS PubMed.
- R. Kaur, K. Kim, A. Paul and A. Deep, J. Mater. Chem. A, 2016, 4, 3991–4002 RSC.
- K. Patra, S. A. Ansari and P. K. Mohapatra, J. Chromatogr., 2021, 1655, 462491 Search PubMed.
- X. Li, X. Yang, H. Xue, H. Pang and Q. Xu, EnergyChem, 2020, 2, 100027 CrossRef.
- C. Duan, K. Liang, Z. Zhang, J. Li, T. Chen, D. Lv, L. Li, L. Kang, K. Wang, H. Hu and H. Xi, Nano Mater. Sci., 2022, 4, 351–365 Search PubMed.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef PubMed.
- S. Rapti, A. Pournara, D. Sarma, I. T. Papadas, G. S. Armatas, Y. S. Hassan, M. H. Alkordi, M. G. Kanatzidis and M. J. Manos, Inorg. Chem. Front., 2016, 3, 635–644 RSC.
- A. J. Howarth, M. J. Katz, T. C. Wang, A. E. Platero-Prats, K. W. Chapman, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2015, 137, 7488–7494 CrossRef CAS PubMed.
- K. Yee, N. Reimer, J. Liu, S. Cheng, S. Yiu, J. Weber, N. Stock and Z. Xu, J. Am. Chem. Soc., 2013, 35, 7795–7798 CrossRef PubMed.
- M. Jian, H. Wang, R. Liu, J. Qu, H. Wang and X. Zhang, Environ. Sci.: Nano, 2016, 3, 1186–1194 RSC.
- W. Shi, W. Li, W. Nguyen, W. Chen, J. Wang and M. Chen, Mater. Today Adv., 2022, 15, 100273 CrossRef CAS.
- X. Liu, H. Yang, Y. Diao, Q. He, C. Lu, A. Singh, A. Kumar, J. Liu and Q. Lan, Chemosphere, 2022, 307, 135729 CrossRef CAS PubMed.
- B. Jie, H. Lin, Y. Zhai, J. Ye, D. Zhang, Y. Xie, X. Zhang and Y. Yang, Chem. Eng. J., 2023, 454, 139931 CrossRef CAS.
- W. Sun, X. Zhao, E. Webb, G. Xu, W. Zhang and Y. Wang, J. Mater. Chem. A, 2023, 11, 2092–2127 RSC.
- S. Kamal, M. Khalid, M. S. Khan and M. Shahid, Coord. Chem. Rev., 2023, 474, 214859 CrossRef CAS.
- E. Echenique-Errandonea, R. F. Mendes, F. Figueira, D. Choquesillo-Lazarte, G. Beobide, J. Cepeda, D. Ananias, A. Rodríguez-Diéguez, F. A. Almeida Paz and J. M. Seco, Inorg. Chem., 2022, 61, 12977–12990 CrossRef CAS PubMed.
- M. Rubio-Martinez, C. Avci-Camur, A. W. Thornton, I. Imaz, D. Maspoch and M. R. Hill, Chem. Soc. Rev., 2017, 46, 3453–3480 RSC.
- T. Kitao, Y. Zhang, S. Kitagawa, B. Wang and T. Uemura, Chem. Soc. Rev., 2017, 46, 3108–3133 RSC.
- X. Liu, Y. Zhou, J. Zhang, L. Tang, L. Luo and G. Zeng, ACS Appl. Mater. Interfaces, 2017, 9, 20255–20275 CrossRef CAS PubMed.
- L. Jiang, Y. Dong, Y. Yuan, X. Zhou, Y. Liu and X. Meng, Chem. Eng. J., 2022, 430, 132823 CrossRef CAS.
- X. Huang, Z. Gong and Y. Lv, TrAC, Trends Anal. Chem., 2022, 153, 116644 CrossRef CAS.
- N.-C. Chiu, K. T. Smith and K. C. Stylianou, Coord. Chem. Rev., 2022, 459, 214441 CrossRef CAS.
- M. Rubio-Martinez, C. Avci-Camur, A. W. Thornton, I. Imaz, D. Maspoch and M. R. Hill, Chem. Soc. Rev., 2017, 46, 3453–3480 RSC.
- T. Kitao, Y. Zhang, S. Kitagawa, B. Wang and T. Uemura, Chem. Soc. Rev., 2017, 46, 3108–3133 RSC.
- X. Liu, Y. Zhou, J. Zhang, L. Tang, L. Luo and G. Zeng, ACS Appl. Mater. Interfaces, 2017, 9, 20255–20275 CrossRef CAS PubMed.
- N. S. Bobbitt, M. L. Mendonca, A. J. Howarth, T. Islamoglu, J. T. Hupp, O. K. Farha and R. Q. Snurr, Chem. Soc. Rev., 2017, 46, 3357–3385 RSC.
- K. Patra, A. Sengupta, V. K. Mittal and T. P. Valsala, Mater. Today Sustain., 2023, 24, 100489 Search PubMed.
- T. Sun, Y. Gao, Y. Du, L. Zhou and X. Chen, Front. Chem., 2021, 8, 624592 CrossRef PubMed.
- M. E. Mahmoud, Z. Moussa, T. Prakasam, L. Li, M. G. Abiad, D. Patra and M. Hmadeh, J. Solid State Chem., 2020, 281, 121031 CrossRef CAS.
- Y. Zhang, S. Liu, Z.-S. Zhao, Z. Wang, R. Zhang, L. Liu and Z.-B. Han, Inorg. Chem. Front., 2021, 8, 590–619 RSC.
- H.-M. Ren, H.-W. Wang, Y.-F. Jiang, Z.-X. Tao, C.-Y. Mu and G. Li, Top. Curr. Chem., 2022, 380(9), 1–58 Search PubMed.
- T. M. Reineke, M. Eddaoudi, M. O’keeffe and O. M. Yaghi, Angew. Chem., Int. Ed., 1999, 38, 2590–2594 CrossRef CAS PubMed.
- N. Stock and S. Biswas, Chem. Rev., 2012, 112, 933–969 CrossRef CAS PubMed.
- A. Schaate, P. Roy, A. Godt, J. Lippke, F. Waltz, M. Wiebcke and P. Behrens, Chem.–Eur. J., 2011, 17, 6643–6651 CrossRef CAS PubMed.
- F. Vermoortele, B. Bueken, G. L. Bars, B. V. de Voorde, M. Vandichel, K. Houthoofd, A. Vimont, M. Daturi, M. Waroquier, V. V. Speybroeck, C. Kirschhock and D. E. De Vos, J. Am. Chem. Soc., 2013, 135, 11465–11468 CrossRef CAS PubMed.
- A. J. Howarth, A. W. Peters, N. A. Vermeulen, T. C. Wang, J. T. Hupp and O. K. Farha, Chem. Mater., 2017, 29, 26–39 CrossRef CAS.
- B. Xu, H. Guo, S. Wang, Y. Li, H. Zhang and C. Liu, CrystEngComm, 2012, 14, 2914–2919 RSC.
- J. Xia, Y. Xue, B. Lei, L. Xu, M. Sun, N. Li, H. Zhao, M. Wang, M. Luo, C. Zhang, B. Huang, Y. Du and C.-H. Yan, Natl. Sci. Rev., 2021, 8(221), 1–10 Search PubMed.
- N. A. Khan and S. H. Jhung, Coord. Chem. Rev., 2015, 285, 11–23 CrossRef CAS.
- B. L. Foster and M. E. Cournoyer, ACS Chem. Health Saf., 2005, 12, 27–32 CrossRef CAS.
- Y. Qiao and E. J. Schelter, Acc. Chem. Res., 2018, 51, 2926–2936 CrossRef CAS PubMed.
- J. Klinowski, F. A. A. Paz, P. Silva and J. Rocha, Dalton Trans., 2011, 40, 321–330 RSC.
- P. P. Bag, X.-S. Wang and R. Cao, Dalton Trans., 2015, 44, 11954–11962 RSC.
- W. Liang and D. M. D'Alessandro, Chem. Commun., 2013, 49, 3706–3708 RSC.
- P. Amo-Ochoa, G. Givaja, P. J. S. Miguel, O. Castillo and F. Zamora, Inorg. Chem. Commun., 2007, 10, 921–924 CrossRef CAS.
- A. J. Savyasachi, D. F. Caffrey, K. Byrne, G. Tobin, B. D'Agostino, W. Schmitt and T. Gunnlaugsson, Front. Chem. Sci. Eng., 2019, 13, 171–184 CrossRef CAS.
- W. Yang, J. Feng, S. Song and H. Zhang, ChemPhysChem, 2012, 13, 2734–2738 CrossRef CAS PubMed.
- S. A. Younis, N. Bhardwaj, S. K. Bhardwaj, K.-H. Kim and A. Deep, Coord. Chem. Rev., 2021, 429, 213620 CrossRef CAS.
- D. Hu, Y. Song and L. Wang, J. Nanopart. Res., 2015, 17, 1–21 CrossRef CAS.
- S. Wang, C. M. McGuirk, A. d'Aquino, J. A. Mason and C. A. Mirkin, Adv. Mater., 2018, 30, 1800202 CrossRef PubMed.
- L. Huang, H. Wang, J. Chen, Z. Wang, J. Sun, D. Zhao and Y. Yan, Microporous Mesoporous Mater., 2003, 58, 105–114 CrossRef CAS.
- D. J. Tranchemontagne, J. R. Hunt and O. M. Yaghi, Tetrahedron, 2008, 64, 8553–8557 CrossRef CAS.
- W. J. Rieter, K. M. Pott, K. M. Taylor and W. Lin, J. Am. Chem. Soc., 2008, 130, 11584–11585 CrossRef CAS PubMed.
- K. Liu, H. You, Y. Zheng, G. Jia, Y. Song, Y. Huang, M. Yang, J. Jia, N. Guo and H. Zhang, J. Mater. Chem., 2010, 20, 3272–3279 RSC.
- W. Yang, J. Feng and H. Zhang, J. Mater. Chem., 2012, 22, 6819–6823 RSC.
- K. Liu, Y. Zheng, G. Jia, M. Yang, Y. Song, N. Guo and H. You, J. Solid State Chem., 2010, 183, 2309–2316 CrossRef CAS.
- B. Szczęśniak, S. Borysiuk, J. Choma and M. Jaroniec, Mater. Horiz., 2020, 7, 1457–1473 RSC.
- M. Rubio-Martinez, C. Avci-Camur, A. W. Thornton, I. Imaz, D. Maspoch and M. R. Hill, Chem. Soc. Rev., 2017, 46, 3453–3480 RSC.
- A. Pichon, A. Lazuen-Garay and S. L. James, CrystEngComm, 2006, 8, 211–214 RSC.
- T. Friščić, J. Mater. Chem., 2010, 20, 7599–7605 RSC.
- W. Yuan, J. O'Connor and S. L. James, CrystEngComm, 2010, 12, 3515–3517 RSC.
- T. Alammar, I. Z. Hlova, S. Gupta, V. Balema, V. K. Pecharsky and A. V. Mudring, Dalton Trans., 2018, 47, 7594–7601 RSC.
- N. Singh, S. Gupta, V. Pecharsky and V. Balema, J. Alloys Compd., 2017, 696, 118–122 CrossRef CAS.
- W. Li, J. Feng, Z. Lin, Y. Yang, Y. Yang, X. Wang, S. Gao and R. Cao, Chem. Commun., 2016, 52, 3951–3954 RSC.
- M. Varsha and G. Nageswaran, J. Electrochem. Soc., 2020, 167, 155527 CrossRef CAS.
- A. M. Joaristi, J. Juan-Alcañiz, P. Serra-Crespo, F. Kapteijn and J. Gascon, Cryst. Growth Des., 2012, 12, 3489–3498 CrossRef.
- H. Al-Kutubi, J. Gascon, E. J. R. Sudhölter and L. Rassaei, ChemElectroChem, 2015, 2, 462–474 CrossRef CAS.
- U. Mueller, M. Schubert, F. Teich, H. Puetter, K. Schierle-Arndt and J. Pastre, J. Mater. Chem., 2006, 16, 626–636 RSC.
- W. Li, J. Feng, Z. Lin, Y. Yang, Y. Yang, X. Wang, S. Gao and R. Cao, Chem. Commun., 2016, 52, 3951–3954 RSC.
- Z. Li, T. Zhuang, J. Dong, L. Wang, J. Xia, H. Wang, X. Cui and Z. Wang, Ultrason. Sonochem., 2021, 71, 105384 CrossRef CAS PubMed.
- S. Li, L. Tan and X. Meng, Adv. Funct. Mater., 2020, 30, 1908924 CrossRef CAS.
- S. H. Jhung, J. H. Lee, J. W. Yoon, C. Serre, G. Férey and J.-S. Chang, Adv. Mater., 2007, 19, 121–124 CrossRef CAS.
- R. Kerner, O. Palchikan and A. Gedanken, Chem. Mater., 2001, 13, 1413–1419 CrossRef CAS.
- W. J. Son, J. Kim, J. Kim and W. S. Ahn, Chem. Commun., 2008, 6336–6338 RSC.
- Z. Q. Li, L. G. Qiu, T. Xu, Y. Wu, W. Wang, Z. Y. Wu and X. Jiang, Mater. Lett., 2009, 63, 78–80 CrossRef CAS.
- C. Vaitsis, G. Sourkouni and C. Argirusis, Ultrason. Sonochem., 2019, 52, 106–119 CrossRef CAS PubMed.
- N. A. Khan, M. M. Haque and S. H. Jhung, Eur. J. Inorg. Chem., 2010, 4975–4981 CrossRef CAS.
- S. M. Hu, H. L. Niu, L. G. Qiu, Y. P. Yuan, X. Jiang, A. J. Xie, Y. H. Shen and J. F. Zhu, Inorg. Chem. Commun., 2012, 17, 147–150 CrossRef CAS.
- J. D. Xiao, L. G. Qiu, F. Ke, Y. P. Yuan, G. S. Xu, Y. M. Wang and X. Jiang, J. Mater. Chem. A, 2013, 1, 8745–8752 RSC.
- K. A. White, D. A. Chengelis, M. Zeller, S. J. Geib, J. Szakos, S. Petoud and N. L. Rosi, Chem. Commun., 2009, 4506–4508 RSC.
- R. M. Abdelhameed, L. D. Carlos, A. M. Silva and J. Rocha, New J. Chem., 2015, 39, 4249–4258 RSC.
- W. P. Ma and B. Yan, Dalton Trans., 2020, 49, 15663–15671 RSC.
- R. Xu, W. Pang, J. Yu, Q. Huo and J. Chen, in Chemistry of Zeolites and Related Porous Materials: Synthesis and Structure, John Wiley and Sons, Singapore, 2007 Search PubMed.
- N. Muttil, S. Jagadeesan, A. Chanda, M. Duke and S. K. Singh, Appl. Sci., 2023, 13(257), 1–15 Search PubMed.
- R. C. Bansal and M. Goyal, in Activated Carbon Adsorption, Taylor & Francis Group, CRC Press, Boca Raton, FL, 2005 Search PubMed.
- A. U. Czaja, N. Trukhan and U. Muller, Chem. Soc. Rev., 2009, 38, 1284–1293 RSC.
- X. Feng, W. Pisula and K. Müllen, Pure Appl. Chem., 2009, 81, 2203–2224 CrossRef CAS.
- X. Lin, N. Champness and M. Schröder, Hydrogen, methane and carbon dioxide adsorption in metal-organic framework materials, in Functional metal-organic frameworks: Gas storage, separation and catalysis, Topics in Current Chemistry, ed. M. Schröder, Springer, Berlin/Heidelberg, 2010, vol. 293, pp. 35–76 Search PubMed.
- Y.-G. Huang, F.-L. Jiang and M.-C. Hong, Coord. Chem. Rev., 2009, 253, 2814–2834 CrossRef CAS.
- M. Kurmoo, Chem. Soc. Rev., 2009, 38, 1353–1379 RSC.
- J.-R. Li, J. Sculley and H.-C. Zhou, Chem. Rev., 2012, 112, 869–932 CrossRef CAS PubMed.
- S. A. Younis, N. Bhardwaj, S. K. Bhardwaj, K.-H. Kim and A. Deep, Coord. Chem. Rev., 2021, 421, 213620 CrossRef.
- W. Shi, W. Li, W. Nguyen, W. Chen, J. Wang and M. Chen, Mater. Today Adv., 2022, 15, 100273–100294 CrossRef CAS.
- G. Cai, P. Yan, L. Zhang, H.-C. Zhou and H.-L. Jiang, Chem. Rev., 2021, 121, 12278–12326 CrossRef CAS PubMed.
- S. Kitagawa and R. Matsuda, Coord. Chem. Rev., 2007, 251, 2490–2509 Search PubMed.
- D. J. Tranchemontagne, J. L. Mendoza-Cortes, M. O'Keeffe and O. M. Yaghi, Chem. Soc. Rev., 2009, 38, 1257–1283 RSC.
- M. Eddaoudi, D. B. Moler, H. Li, B. Chen, T. M. Reineke, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2001, 34, 319–330 CrossRef CAS PubMed.
- Y.-P. He, Y.-X. Tan and J. Zhang, Coord. Chem. Rev., 2020, 420, 213354 Search PubMed.
- W. Lu, Z. Wei, Z.-Y. Gu, T.-F. Liu, J. Park, J. Park, J. Tian, M. Zhang, Q. Zhang, T. Gentle III, M. Bosch and H.-C. Zhou, Chem. Soc. Rev., 2014, 43, 5561–5593 RSC.
- Y. Bai, Y. Dou, L.-H. Xie, W. Rutledge, J.-R. Li and H.-C. Zhou, Chem. Soc. Rev., 2016, 45, 2327–2367 RSC.
- O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef CAS PubMed.
- H. Montes-Andrés, G. Orcajo, C. Mellot-Draznieks, C. Martos, J. A. Botas and G. Calleja, J. Phys. Chem. C, 2018, 122, 28123–28132 Search PubMed.
- V. Stavila, M. E. Foster, J. W. Brown, R. W. Davis, J. Edgington, A. I. Benin, R. A. Zarkesh, R. Parthasarathi, D. W. Hoyt, E. D. Walter, A. Andersen, N. M. Washton, A. S. Lipton and M. D. Allendorf, Chem. Sci., 2019, 10, 9880–9892 RSC.
- H. Montes-Andrés, G. Orcajo, C. Martos, J. A. Botas and G. Calleja, Int. J. Hydrogen Energy, 2019, 44, 18205–18213 Search PubMed.
- G. Chen, J. Luo, M. Cai, L. Qin, Y. Wang, L. Gao, P. Huang, Y. Yu, Y. Ding, X. Dong, X. Yin and J. Ni, Molecules, 2019, 24(3369), 1–18 Search PubMed.
- G. Blăniţă, O. Ardelean, D. Lupu, G. Borodi, M. Miheţ, M. Coroş, M. Vlassa, I. Mişan, I. Coldea and G. Popeneciu, Rev. Roum. Chim., 2011, 56, 583–588 Search PubMed.
- M. de Toni, P. Pullumbi, F.-X. Coudert and A. H. Fuchs, J. Phys. Chem. C, 2010, 114, 21631–21637 Search PubMed.
- H. Deng, S. Grunder, K. E. Cordova, C. Valente, H. Furukawa, M. H. F. Gándara, A. C. Whalley, Z. Liu, S. Asahina, H. Kazumori, M. O'Keeffe, O. Terasaki, J. F. Stoddart and O. M. Yaghi, Science, 2012, 336, 1018–1023 CrossRef CAS PubMed.
- G. Férey, C. Serre, C. Mellot-Draznieks, F. Millange, S. Surblé, J. Dutour and I. Margiolak, Angew. Chem., Int. Ed., 2004, 43, 6296–6301 Search PubMed.
- N. V. Maksimchuk, O. V. Zalomaeva, I. Y. Skobelev, K. A. Kovalenko, V. P. Fedin and O. A. Kholdeeva, Proc. R. Soc. A, 2012, 468, 2017–2034 CrossRef CAS.
- T. K. Trung, N. A. Ramsahye, P. Trens, N. Tanchoux, C. Serre, F. Fajula and G. Férey, Microporous Mesoporous Mater., 2010, 134, 134–140 Search PubMed.
- C. Daiguebonne, N. Kerbellec, O. Guillou, J.-C. Bünzli, F. Gumy, L. Catala, T. Mallah, N. Audebrand, Y. Gérault, K. Bernot and G. Calvez, Inorg. Chem., 2008, 47, 3700–3708 Search PubMed.
- R. G. Pearson, Coord. Chem. Rev., 1990, 100, 403–425 Search PubMed.
- Y. Zhu, M. Zhu and L. Xia, Sci. Rep., 2016, 6, 29728 Search PubMed.
- T. Gorai, W. Schmitt and T. Gunnlaugsson, Dalton Trans., 2021, 50, 770–784 Search PubMed.
- S. Ma, X.-S. Wang, D. Yuan and H.-C. Zhou, Angew. Chem., Int. Ed., 2008, 47, 4130–4133 CrossRef CAS PubMed.
- B. Li and B. Chen, in Structure and Bonding, Springer, Berlin, Heidelberg, 2015, vol. 163, pp. 75–107 Search PubMed.
- F. Luo, S. R. Batten, Y. Che and J.-M. Zheng, Chem.–Eur. J., 2007, 3, 4948–4955 Search PubMed.
- J. C. G. Bünzli, Acc. Chem. Res., 2006, 39, 53–61 CrossRef PubMed.
- C. E. Plecnik, S. Liu and S. G. Shore, Acc. Chem. Res., 2003, 36, 499–508 Search PubMed.
- M. Zhang, M. Bosch, T. Gentle III and H. C. Zhou, CrystEngComm, 2014, 16, 4069–4083 RSC.
- G. Férey, Chem. Soc. Rev., 2008, 37, 191–214 Search PubMed.
- Y. Han, X. Li, L. Li, C. Ma, Z. Shen, Y. Song and X. You, Inorg. Chem., 2010, 49, 10781–10787 Search PubMed.
- A. Wang, Y. Zhang, L. Lu, M. Zhu, C. Yuan and S. Feng, Dalton Trans., 2022, 51, 12324–12333 Search PubMed.
- S. Ma, D. Yuan, X.-S. Wang and H.-C. Zhou, Inorg. Chem., 2009, 48, 2072–2077 CrossRef CAS PubMed.
- R. J. Holmberg, L. T. A. Ho, L. Ungur, I. Korobkov, L. F. Chibotaru and M. Murugesu, Dalton Trans., 2015, 44, 20321–20325 RSC.
- Y. K. Park, S. B. Choi, H. Kim, K. Kim, B.-H. Won, K. Choi, J.-S. Choi, W.-S. Ahn, Na. Won, S. Kim, D. H. Jung, S.-H. Choi, G.-H. Kim, S.-S. Cha, Y. H. Jhon, J. K. Yang and J. Kim, Angew. Chem., Int. Ed., 2007, 46, 8230–8233 CrossRef CAS PubMed.
- Y. He, H. Furukawa, C. Wu, M. O'Keeffe and B. Chen, CrystEngComm, 2013, 15, 9328–9331 Search PubMed.
- Y. Pei, Y. Journaux, O. Kahn, A. Dei and D. Gatteschi, J. Chem. Soc. Chem. Commun., 1986, 16, 1300–1301 Search PubMed.
- S. Su, W. Chen, C. Qin, S. Song, Z. Guo, G. Li, X. Song, M. Zhu, S. Wang, Z. Hao and H. Zhang, Cryst. Growth Des., 2012, 12, 1808–1815 Search PubMed.
- S. Biswas, H. S. Jena, S. Goswami, S. Sanda and S. Konar, Cryst. Growth Des., 2014, 14, 1287–1295 CrossRef CAS.
- J.-M. Zhou, H.-M. Li, N. Xu and P. Cheng, Inorg. Chem. Commun., 2013, 37, 30–33 CrossRef CAS.
- A. Bencini, C. Benelli, A. Caneschi, R. L. Carlin, A. Dei and D. Gatteschi, J. Am. Chem. Soc., 1985, 107, 8128–8136 CrossRef CAS.
- M.-B. Zhang, J. Zhang, S.-T. Zheng and G.-Y. Yang, Angew. Chem., Int. Ed., 2005, 44, 1385–1388 Search PubMed.
- X.-F. Huang, J.-X. Ma and W.-S. Liu, Inorg. Chem., 2014, 53, 5922–5930 CrossRef CAS PubMed.
- Y. Cui, Y. Yue, G. Qian and B. Chen, Chem. Rev., 2011, 112, 1126–1162 CrossRef PubMed.
- J.-C. G. Bünzli, Chem. Rev., 2010, 110, 2729–2755 CrossRef PubMed.
- Y. Chen and S. Ma, Rev. Inorg. Chem., 2012, 32, 81–100 CAS.
- F. Gándara, A. de Andrés, B. Gómez-Lor, E. Gutiérrez-Puebla, M. Iglesias, M. A. Monge, D. M. Proserpio and N. Snejko, Cryst. Growth Des., 2008, 8, 378–380 CrossRef.
- R. E. Whan and G. A. Crosby, J. Mol. Spectrosc., 1962, 8, 315–327 CrossRef CAS.
- B. D. Chandler, D. T. Cramb and G. K. H. Shimizu, J. Am. Chem. Soc., 2006, 128, 10403–10412 CrossRef CAS PubMed.
- X.-Z. Song, S.-Y. Song and H.-J. Zhang, Struct. Bonding, 2015, 163, 1–36 Search PubMed.
- P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Férey, R. E. Morris and C. Serre, Chem. Rev., 2011, 112, 1232–1268 CrossRef PubMed.
- K. Tang, R. Yun, Z. Lu, L. Du, M. Zhang, Q. Wang and H. Liu, Cryst. Growth Des., 2013, 13, 1382–1385 CrossRef CAS.
- A. Garg, M. Almáši and R. Saini, Environ. Sci. Pollut. Res., 2023, 30, 98548–98562 CrossRef CAS PubMed.
- J. Luo, H. Xu, Y. Liu, Y. Zhao, L. L. Daemen, C. Brown, T. V. Timofeeva, S. Ma and H.-C. Zhou, J. Am. Chem. Soc., 2008, 130, 9626–9627 CrossRef CAS.
- S. Fordham, X. Wang, M. Bosch and H.-C. Zhou, Struct. Bonding, 2015, 163, 1–27 CAS.
- B. Ay, E. Yildiz, M. Enomoto, A. Okazawa and N. Kojima, Polyhedron, 2022, 226, 116110 CrossRef CAS.
- W. Fan, S. Yuan, W. Wang, L. Feng, X. Liu, X. Zhang, X. Wang, Z. Kang, F. Dai, D. Yuan, D. Sun and H.-C. Zhou, J. Am. Chem. Soc., 2020, 142, 8728–8737 CrossRef PubMed.
- H. Li, L. Li, R.-B. Lin, W. Zhou, Z. Zhang, S. Xiang and B. Chen, EnergyChem, 2019, 1, 100006 Search PubMed.
- Y. Zhang, S. Liu, Z.-S. Zhao, Z. Wang, R. Zhang, L. Liu and Z.-B. Han, Inorg. Chem. Front., 2021, 8, 590–619 RSC.
- Y.-W. Ren, J.-X. Liang and J.-X. Lu, Eur. J. Inorg. Chem., 2011, 4369–4376 CrossRef CAS.
- M. Shibasaki, K.-I. Yamada, N. Yoshikawa, Lanthanide lewis acids catalysis, in Lewis Acids in Organic Synthesis, ed. H. Yamamoto, Wiley-VCH Verlag GmbH, Weinheim, 2000, pp. 911–944 Search PubMed.
- J. Gascon, A. Corma, F. Kapteijn and F. X. L. i Xamena, ACS Catal., 2013, 4, 361–378 CrossRef.
- R. F. D'Vries, M. Iglesias, N. Snejko, S. Alvarez-Garcia, E. Gutiérrez-Puebla and M. Á. Monge, J. Mater. Chem., 2012, 22, 1191–1198 Search PubMed.
- R. F. D'Vries, V. A. de la Peña-O’Shea, N. Snejko, M. Iglesias, E. Gutiérrez-Puebla and M. Á. Monge, Cryst. Growth Des., 2012, 12, 5535–5545 CrossRef.
- P. K. Batista, D. J. M. Alves, M. O. Rodrigues, G. F. de Sá, S. A. Junior and J. A. Vale, J. Mol. Catal. A: Chem., 2013, 379, 68–71 CrossRef CAS.
- Y. B. N. Tran and P. T. K. Nguyen, New J. Chem., 2021, 45, 2090–2102 RSC.
- J. Li, Y. Liu, K. Ma, C. Li and Z. Shi, Inorg. Chem. Front., 2024, 11, 3626–3632 RSC.
- B. Chen, L. Wang, Y. Xiao, F. R. Fronczek, M. Xue, Y. Cui and G. Qian, Angew. Chem., Int. Ed., 2009, 48, 500–503 CrossRef CAS.
- N. L. Rosi, J. Kim, M. Eddaoudi, B. Chen, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 1504–1518 CrossRef CAS PubMed.
- B. Chen, L. Wang, F. Zapata, G. Qian and E. B. Lobkovsky, J. Am. Chem. Soc., 2008, 130, 6718–6719 CrossRef CAS PubMed.
- B. Chen, Y. Yang, F. Zapata, G. Lin, G. Qian and E. B. Lobkovsky, Adv. Mater., 2007, 19, 1693–1696 CrossRef CAS.
- S. Wang, B. Sun, Z. Su, G. Hong, X. Li, Y. Liu, Q. Pan and J. Sun, Inorg. Chem. Front., 2022, 9, 3259–3266 RSC.
- C.-L. Yang, G.-H. Yu, A.-X. Yu, D.-Y. Du and Z. Su, Cryst. Growth Des., 2022, 22, 6960–6966 CrossRef CAS.
- H. Min, Z. Han, T. Sun, K. Wang, J. Xu, P. Yao, S. Yang, P. Cheng and W. Shi, Sci. China:Chem., 2023, 66, 3511–3517 CrossRef CAS.
- S. Wu, H. Min, W. Shi and P. Cheng, Adv. Mater., 2020, 32, 1805871 CrossRef CAS PubMed.
- Q. Wang, J. Dong, Z. Li, X. Wang, Y. He, B. Chen and D. Zhao, Inorg. Chem., 2023, 62, 14439–14447 CrossRef CAS PubMed.
- H.-M. Chai, J.-L. Yan, G.-Q. Zhang, J.-X. Wang, Y.-X. Ren and L.-J. Gao, Inorg. Chem., 2022, 61, 7286–7295 CrossRef CAS PubMed.
- P. Majee, P. Daga, D. K. Singha, D. Saha, P. Mahata and S. K. Mondal, J. Photochem. Photobiol., A, 2020, 402, 112830 Search PubMed.
- H.-S. Wang, Coord. Chem. Rev., 2017, 349, 139–155 CrossRef CAS.
- N. Wu, C. Bo and S. Guo, ACS Sens., 2024, 9, 4402–4424 Search PubMed.
- W. B. Liang, P. Wied, F. Carraro, C. J. Sumby, B. Nidetzky, C. K. Tsung, P. Falcaro and C. J. Doonan, Chem. Rev., 2021, 121, 1077–1129 CrossRef CAS PubMed.
- J. Jia, Y. Zhang, M. Zheng, C. Shan, H. Yan, W. Wu, X. Gao, B. Cheng, W. Liu and Y. Tang, Inorg. Chem., 2018, 57, 300–310 CrossRef CAS PubMed.
- S. Sahoo, S. Mondal and D. Sarma, Coord. Chem. Rev., 2022, 470, 214707 CrossRef CAS.
- H. Xu, C. Cao, X. Kang and B. Zhao, Dalton Trans., 2016, 45, 18003–18017 Search PubMed.
- J. Zhang, Y. K. Huang, D. Yue, Y. J. Cui, Y. Yang and G. D. Qian, J. Mater. Chem. B, 2018, 6, 5174–5180 RSC.
- L.-L. Li, S.-S. Chen, S. Liu, Z.-H. Yong, D.-K. Zhang, S.-S. Zhang and Y.-C. Xin, J. Mol. Struct., 2023, 1276, 134777 CrossRef CAS.
- L. Bogani and W. Wernsdorfer, Nat. Mater., 2008, 7, 179–186 CrossRef CAS PubMed.
- F. Manna, M. Oggianu, N. Avarvari and M. L. Mercuri, Magnetochem, 2023, 9(190), 1–17 Search PubMed.
- C. Chen, C. Wang, X. Zheng, R. Zhang, Y. Xu, G. Zhuang, L. Long, L. Zheng, X. Kong and Y. Cao, J. Am. Chem. Soc., 2023, 145(31), 16983–16987 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |