
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
High sulfur content composite materials from renewable fatty acid cellulose esters (FACEs) via inverse vulcanization†
Timo
Sehn
a,
Julian
Fanelli
 a,
Lisa
Wahl
a and
Michael A. R.
Meier
*ab
a,
Lisa
Wahl
a and
Michael A. R.
Meier
*ab
aInstitute of Biological and Chemical Systems – Functional Molecular Systems (IBCS-FMS), Karlsruhe Institute of Technology (KIT), Karlsruhe 76131, Germany. E-mail: m.a.r.meier@kit.edu
bInstitute of Organic Chemistry (IOC), Karlsruhe Institute of Technology (KIT), Kaiserstraße 12, Karlsruhe 76131, Germany
First published on 21st October 2024
Abstract
Herein, we introduce an efficient inverse vulcanization of fully renewable cellulose-based monomers. Therefore, biobased fatty acid cellulose esters (FACEs) with different degrees of substitution (0.38 ≤ DS ≤ 0.62) were inversely vulcanized to obtain high sulfur content composite materials (∼95 wt% sulfur). In-depth structural characterization of the crosslinked sections via differential scanning calorimetry (DSC), thermogravimetric analysis (TGA), energy dispersive X-ray (EDX) spectroscopy, and scanning electron microscopy (SEM) revealed an increased amount of covalently incorporated sulfur (5.67 wt% ≤ sulfur wt% ≤ 56.2 wt%) with a higher DS of FACEs. Investigating structure–property relationships further revealed an increase in thermal stability (227 °C ≤ Td,5% ≤ 247 °C) accompanied by a decreased wettability (87° ≤ θ ≤ 99°) with DS. The obtained materials showed application possibilities for water purification, i.e. for mercury extraction (70% ≤ Hgremoval2+ ≤ 95%).
Sustainability spotlightSulfur is an abundant waste material, it is directly used as the main component (95% content) in inverse vulcanized materials based on a fully renewable polymer network derived from fatty acids and cellulose. Material properties and especially the network structure of the composite materials were characterized in detail. The obtained materials showed promising application possibilities in the field of water purification, related to the important sustainable development goal of clean water. The described materials were able to efficiently remove mercury ions. |
Introduction
The development of more sustainable synthetic approaches, targeting polymeric materials with tailor made properties, remains of high importance. The 12 principles of green chemistry, established by Anastas and Warner in 1998,1,2 have to serve as guidelines and should not be contradicted to achieve this goal. Key aspects of these principles include the utilization of renewable feedstocks and catalysts accompanied by an improved energy efficiency along with the avoidance of waste and toxic chemicals.1,2The ongoing extensive depletion of petroleum resources underscores the importance of renewable feedstocks in material synthesis. Thus, cellulose, as the most abundant biopolymer on earth (1.5 × 1012 tons p.a.), is a relevant renewable platform chemical, as it offers unique properties such as biodegradability, biocompatibility and high mechanical and thermal stability.3–5 However, cellulose modification remains challenging due to the limited processability and solubility in common organic solvents.6–8 At a molecular level, extensive intra- and intermolecular hydrogen bonding interactions between the hydroxyl groups of the cellulose backbone (consisting of β-1,4 linked glucose units) induce the limited processability and solubility.9
Several heterogeneous and homogeneous modification approaches for cellulose derivatization are established.3,10,11 On one hand, cellulose modification in a heterogeneous fashion is mainly employed in industry and suffers from drawbacks such as backbone degradation induced by harsh reaction conditions or uneven distribution of the introduced side chains along the polymer backbone.3,11 In contrast, homogeneous modification approaches for cellulose using polar solvent systems such as LiCl/DMAc,12,13 ionic liquids (ILs, e.g. 1-butyl-3-methyl-imidazoliumchloride14 or 1-ethyl-3-methyl-imidazolium 2-pyridinolate),9 or so-called switchable solvent systems overcome the above mentioned challenges, i.e. backbone degradation and uneven distribution of the side chains, and furthermore allow for a straightforward adjustment of the degree of substitution (DS).3,5 The DS is defined as the amount of substituted hydroxyl groups per anhydroglucose unit (AGU) and can vary between 0 and 3 for native and fully modified cellulose, respectively.
Cellulose esters (CE) have emerged as the most important cellulose derivatives, since this material class is already employed as a biobased alternative to fossil-based polymers in membranes, food packages and coatings.3,15,16 Especially in terms of sustainability, fully renewable materials such as fatty acid cellulose esters (FACEs), where cellulose is modified with a second biobased substrate derived from a vegetable oil, have received significant attention within the last few years.7,15 However, to truly classify materials such as FACEs as more sustainable than others, the complete synthetic approach must be considered.
To better quantify the environmental impact of chemical processes, the E-factor (taking reagents, yield, side products and solvent losses into account), which is defined as the mass ratio of the desired product and the produced waste, was introduced.17 Nevertheless, the E-factor does not provide any information about toxicity, which is a key aspect of the 12 principles of green chemistry and is crucial in terms of adhering to safety regulations.1,2
Accordingly, when assessing how sustainable certain FACEs are, especially the recyclability of the solvent system and the fatty acid derivative used in the approach must be considered. In most of the synthetic procedures for the synthesis of FACEs, activated fatty acid derivatives, i.e. fatty acid chlorides,18,19N,N′-carbonyldiimidazole (CDI) activated fatty acids20,21 or fatty acid anhydrides,22 are employed. These activated fatty acid derivatives suffer from drawbacks including highly unsustainable synthesis procedures, i.e. including phosgene derived activating agents such as CDI, or the release of toxic or corrosive by-products during the modification processes, i.e. hydrochloric acid (HCl).15,23 More sustainable reagents for the synthesis of FACEs are vinyl and methyl fatty acid esters, as their production can be achieved in a considerably more sustainable manner and the released compounds can be captured during the modification processes.24,25 Regarding the recyclability of the employed solvent system, especially ILs possess limitations, due to their easy contamination.3,26 However, according to the literature, the derivative approach of the switchable solvent system, where microcrystalline cellulose is converted in the presence of carbon dioxide (CO2) and a guanidine superbase, i.e. 1,8-diazabicyclo[5.4.0]undec-7-en (DBU) or 1,5,7-triazabicyclo[4.4.0]dec-5-en (TBD), into a DMSO soluble carbonate species, results in low E-factors (1.92) for cellulose acetate synthesis. This approach is therefore frequently used as a more sustainable alternative to using ILs.3,27
Depending on the synthetic strategy and employed substrates, FACEs can contain functional groups such as double bonds.7,28,29 It is well established that fatty acids can be efficiently modified by applying a broad variety of synthetic reaction protocols,30e.g. epoxidation,31 thiol–ene32,33 or metathesis.34,35 Another efficient modification strategy fulfilling several aspects of green chemistry is inverse vulcanization.36–38 Herein, homolytic cleavage of the highly abundant and practically non-toxic waste material elemental sulfur (70 Mt excess p.a. as a byproduct in petroleum refining) at elevated temperatures (140 to 185 °C) enables a radical reaction with alkene-bearing comonomers towards high sulfur content materials.37,39
These materials show broad applicability, for instance in energy storage systems such as Li–S batteries, in oil spill remediation, in mercury capture, or as antimicrobial materials.37,40–42 Alkene bearing comonomers from renewable feedstocks, such as terpenes or triglycerides, for inverse vulcanization have already been investigated.40,43–49 The inverse vulcanization of alkene bearing cellulose derivatives is however rarely reported.29,50,51 In 2019, Smith et al. presented the synthesis of cellulose–sulfur composites (80 wt% ≤ sulfur ≤ 99 wt%) by inverse vulcanization of a cellulose ester (CE) containing pendant alkene functionalities.50 From a sustainability point of view, the approach however suffers from the use of halogenated substrates, i.e. 3-bromo-3methylpropene (rated as environmental hazard), and the release of corrosive byproducts, i.e. hydrobromic acid (HBr), during the cellulose modification. Subsequently, in 2020 Smith et al. developed a more sustainable three-step procedure for the synthesis of cellulose–sulfur composites (80 wt% ≤ sulfur ≤ 90 wt%), where the C6-position of native cellulose was first selectively oxidized using sodium hypochlorite, then esterified with a terpenoid, i.e. geraniol, and subsequently inversely vulcanized.51 By applying the previously described synthetic method, a total atom economy of 90% could be achieved over all three steps. However, particularly the reaction time required for the C6 oxidation of cellulose, i.e. 14 days, and the following tedious work up remain critical points in terms of energy efficiency and waste production in the approach.
Thus, we herein take advantage of a more sustainable one step synthesis protocol targeting fully renewable FACEs with adjustable DS, which are prominent comonomers for inverse vulcanisation by using a DMSO/TBD/CO2 switchable solvent system and biobased methyl-10-undecenoate as a transesterification agent.7 High sulfur content (95%) composite materials have been synthesized via inverse vulcanization (E-factor = 0) including FACEs with three different DS (0.38 ≤ DS ≤ 0.62) as comonomers. In-depth structural characterization, i.e. differential scanning calorimetry (DSC), thermogravimetric analysis (TGA), energy dispersive X-ray (EDX) spectroscopy, and scanning electron microscopy (SEM), has been performed especially for the crosslinked sections, supplemented by the investigation of structure–property relationships, i.e. thermal stability (via TGA), hydrophobicity (via WCA) and mercury uptake of the high content sulfur composite materials.
Results and discussion
Using the so-called derivative approach (see Introduction) for cellulose modification in a DMSO/TBD/CO2 switchable solvent system, cellulose (microcrystalline, dried at 100 °C at 10 mbar overnight) was esterified with biobased methyl-10-undecenoate (Scheme 1A). By applying different amounts of methyl-10-undecenoate (i.e. 2.00, 3.50 and 9.00 equivalents), FACE-1, FACE-2 and FACE-3, respectively, were obtained.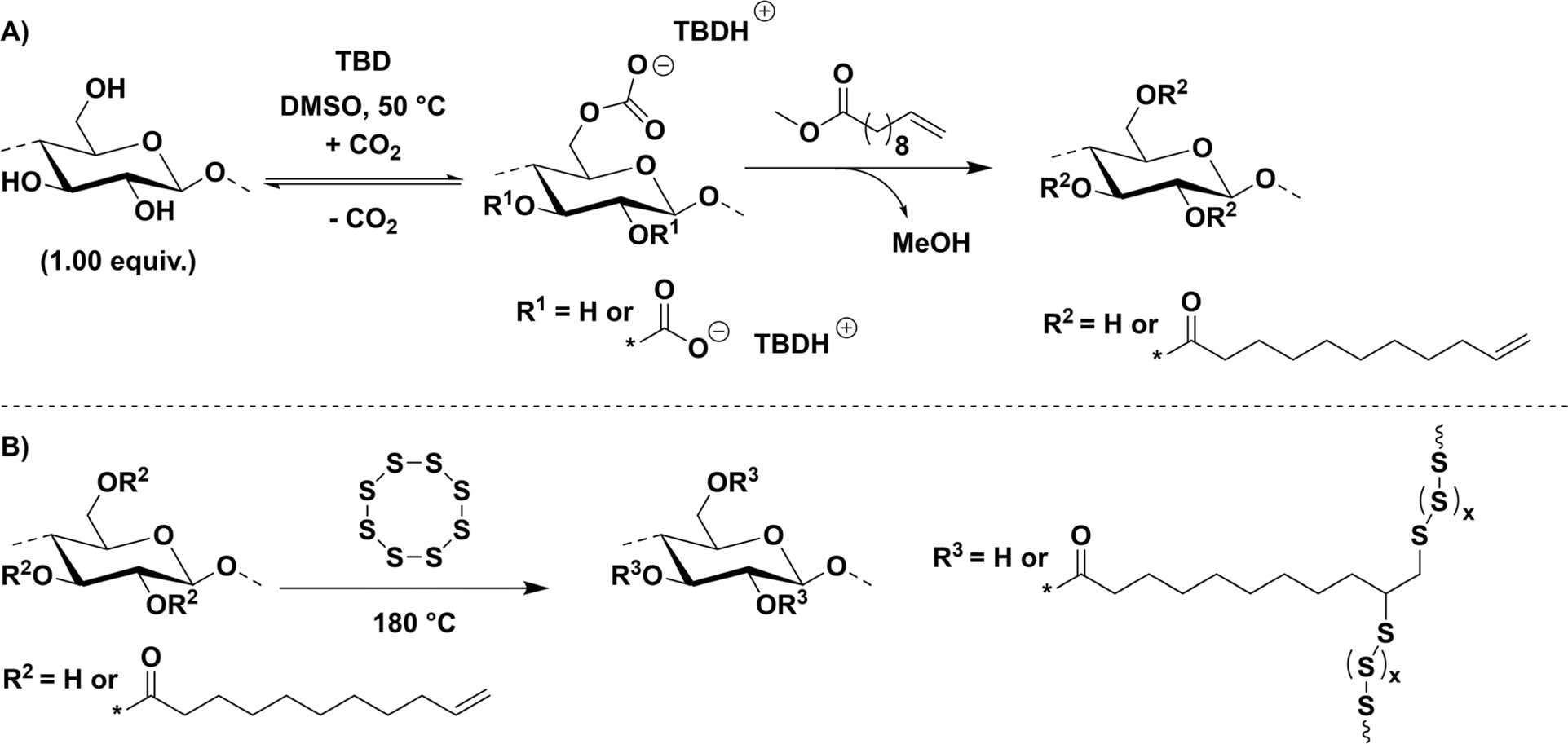 | ||
| Scheme 1 (A) General concept for cellulose dissolution in a DMSO/TBD/CO2 switchable solvent system with subsequent transesterification using methyl-10-undecenoate to fatty acid cellulose esters (FACE). (B) Synthetic approach for the inverse vulcanization of FACEs towards high sulfur content composite materials. | ||
The structural characterization of these FACEs was performed by attenuated total reflection infrared (ATR-IR) spectroscopy supplemented by 1H and 31P nuclear magnetic resonance (NMR) spectroscopy. ATR-IR spectra of all synthesized FACEs (Fig. S1†) revealed characteristic stretching vibration bands at ∼1641 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, Fig.S1†) and ∼1730 cm−1 (CO, Fig. S1†), indicating the successful incorporation of ester and alkene functional groups into cellulose. In 1H NMR (Fig. S2–S4†), magnetic resonances appearing from 2.95 to 5.64 ppm, assigned to the unmodified protons of the (AGU), and magnetic resonances at 4.97 ppm and 5.80 ppm, attributed to the introduced double bond protons, additionally confirmed the success of the synthetic approach. The following DS determination of the synthesized FACE materials was conducted according to an established 31P NMR spectroscopy method (Fig. S5–S7†). Thus, the free hydroxyl groups of the respective FACE were converted into phosphite esters by reaction with the phosphitylation agent 2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane (2-Cl-TMDP). A subsequent quantitative determination of the corresponding phosphite esters via31P NMR spectroscopy using an internal standard (endo-N-hydroxy-5-norbornene-2,3-dicarboximide) allowed the calculation of the average DS as previously reported by Kilpeläinen et al. (eqn (S3), (S4) and Fig. S5–S7†).52 Unsurprisingly, the DS of the synthesized FACEs correlated with the amount of employed transesterification agent, i.e. methyl-10-undecenoate. Thus, FACE-1 appeared as the material with the lowest DS, i.e. 0.38, followed by FACE-2 (DS = 0.42) and FACE-3 (DS = 0.62), respectively. Conventional inverse vulcanization is usually conducted at elevated temperatures above 160 °C. Therefore, to evaluate if the synthesized FACEs can potentially be employed as comonomers for inverse vulcanization, the thermal properties of the respective materials were investigated via TGA (Fig. S8†) and DSC (Fig. S9†). As shown in Fig. S8 of the ESI,† TGA curves revealed a higher thermal stability for FACEs with lower DS. The higher degradation temperatures, which are defined as the temperature of 5% weight loss (Td,5%), can be explained by the inherently existing strong intra- and intermolecular interactions (predominantly hydrogen bonding) between the hydroxyl groups of the cellulose backbones. An increased amount of incorporated bulky alkyl side chains, i.e. a higher DS, affords a partial interruption of the intra- and intermolecular interactions between the cellulose backbones and therefore explains the slightly lower thermal stability of FACE-3 (Td,5% = 221 °C). DSC measurements of all synthesized FACEs did not show any thermal transition. Size exclusion chromatography (SEC) was performed to determine the molecular weight of the FACEs. Unfortunately, FACE-1 was not soluble in the available SEC eluent, i.e. hexafluoro isopropanol (HFIP), and therefore no measurement could be conducted. SEC traces of FACE-2 and FACE-3 (Fig. S10†) revealed high molecular weights (53 kDa ≤ Mn ≤ 64 kDa; 1.85 ≤ Đ ≤ 1.99), a result of the mild reaction conditions during the modification approach using the switchable solvent system,3i.e. less backbone degradation, which is also beneficial for the material properties. The thermal and structural characterization of the FACEs revealed that they are potential fully renewable comonomers for inverse vulcanization, which was an aim of this study (Scheme 1B).
C, Fig.S1†) and ∼1730 cm−1 (CO, Fig. S1†), indicating the successful incorporation of ester and alkene functional groups into cellulose. In 1H NMR (Fig. S2–S4†), magnetic resonances appearing from 2.95 to 5.64 ppm, assigned to the unmodified protons of the (AGU), and magnetic resonances at 4.97 ppm and 5.80 ppm, attributed to the introduced double bond protons, additionally confirmed the success of the synthetic approach. The following DS determination of the synthesized FACE materials was conducted according to an established 31P NMR spectroscopy method (Fig. S5–S7†). Thus, the free hydroxyl groups of the respective FACE were converted into phosphite esters by reaction with the phosphitylation agent 2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane (2-Cl-TMDP). A subsequent quantitative determination of the corresponding phosphite esters via31P NMR spectroscopy using an internal standard (endo-N-hydroxy-5-norbornene-2,3-dicarboximide) allowed the calculation of the average DS as previously reported by Kilpeläinen et al. (eqn (S3), (S4) and Fig. S5–S7†).52 Unsurprisingly, the DS of the synthesized FACEs correlated with the amount of employed transesterification agent, i.e. methyl-10-undecenoate. Thus, FACE-1 appeared as the material with the lowest DS, i.e. 0.38, followed by FACE-2 (DS = 0.42) and FACE-3 (DS = 0.62), respectively. Conventional inverse vulcanization is usually conducted at elevated temperatures above 160 °C. Therefore, to evaluate if the synthesized FACEs can potentially be employed as comonomers for inverse vulcanization, the thermal properties of the respective materials were investigated via TGA (Fig. S8†) and DSC (Fig. S9†). As shown in Fig. S8 of the ESI,† TGA curves revealed a higher thermal stability for FACEs with lower DS. The higher degradation temperatures, which are defined as the temperature of 5% weight loss (Td,5%), can be explained by the inherently existing strong intra- and intermolecular interactions (predominantly hydrogen bonding) between the hydroxyl groups of the cellulose backbones. An increased amount of incorporated bulky alkyl side chains, i.e. a higher DS, affords a partial interruption of the intra- and intermolecular interactions between the cellulose backbones and therefore explains the slightly lower thermal stability of FACE-3 (Td,5% = 221 °C). DSC measurements of all synthesized FACEs did not show any thermal transition. Size exclusion chromatography (SEC) was performed to determine the molecular weight of the FACEs. Unfortunately, FACE-1 was not soluble in the available SEC eluent, i.e. hexafluoro isopropanol (HFIP), and therefore no measurement could be conducted. SEC traces of FACE-2 and FACE-3 (Fig. S10†) revealed high molecular weights (53 kDa ≤ Mn ≤ 64 kDa; 1.85 ≤ Đ ≤ 1.99), a result of the mild reaction conditions during the modification approach using the switchable solvent system,3i.e. less backbone degradation, which is also beneficial for the material properties. The thermal and structural characterization of the FACEs revealed that they are potential fully renewable comonomers for inverse vulcanization, which was an aim of this study (Scheme 1B).
Therefore, the corresponding FACEs (5 wt%) were mixed with elemental sulfur (95 wt%) and reacted for 24 h at 180 °C. The optical appearance of the final composite materials, i.e. FACE-1S, FACE-2S and FACE-3S (Fig. 1) was brown to black. At this point it is worth mentioning that the synthesis of the cellulose-based high sulfur content composite materials was achieved without the employment of any solvent, toxic chemicals, purification steps, and most importantly without the production of any waste, and thus an E-factor of 0 is the result of this synthesis procedure. First, the thermal properties of the obtained inverse vulcanizates were investigated by conducting DSC measurements (Fig. S11†). Herein, the synthesized composite materials revealed a thermal transition, i.e. a significant melting peak, at 120 °C, indicating the presence of unreacted crystalline elemental sulfur.
 | ||
| Fig. 1 Optical appearance of the synthesized high sulfur content composite materials FACE-1S, FACE-2S, and FACE-3S. | ||
Accordingly, DSC measurements did not confirm if the expected reaction between the FACE comonomers and elemental sulfur proceeded successfully. For a more detailed understanding of the network structure, unreacted elemental sulfur was thus removed from the synthesized materials to obtain and characterize the formed crosslinked networks. Hence, a washing procedure implementing carbon disulfide (CS2), which is known to dissolve elemental sulfur at room temperature, was applied to the obtained inverse vulcanizates. Subsequent DSC measurements of the potentially crosslinked residual materials, i.e. FACE-1S-washed, FACE-2S-washed, and FACE-3S-washed, did not show any melting peak at 120 °C (Fig. S12†), indicating that no crystalline elemental sulfur remained after the washing procedure. However, potentially covalent incorporation of sulfur could not be confirmed via DSC. Interestingly, a two-step degradation behaviour was observed by TGA after sulfur extraction (Fig. 2A). It can be assumed that the first degradation step corresponds to the degradation of covalently incorporated S–S moieties, whereas the second one is attributed to the degradation of the FACE backbone. Correspondingly, with increasing DS of the FACEs, the intensity of the first degradation step (corresponding to the decomposition of S–S bonds) also increased from 5.64 wt% in FACE-1S-washed to 56.2 wt% for FACE-3S-washed. In other words, the amount of covalently incorporated sulfur was related to the DS of the employed FACE comonomer and increased with increasing DS. The previously investigated phenomenon might be explained by the more hydrophobic character of the FACE containing higher DS, which enabled an improved solubility in molten sulfur and thus a more efficient reaction, i.e. covalent implementation of sulfur.
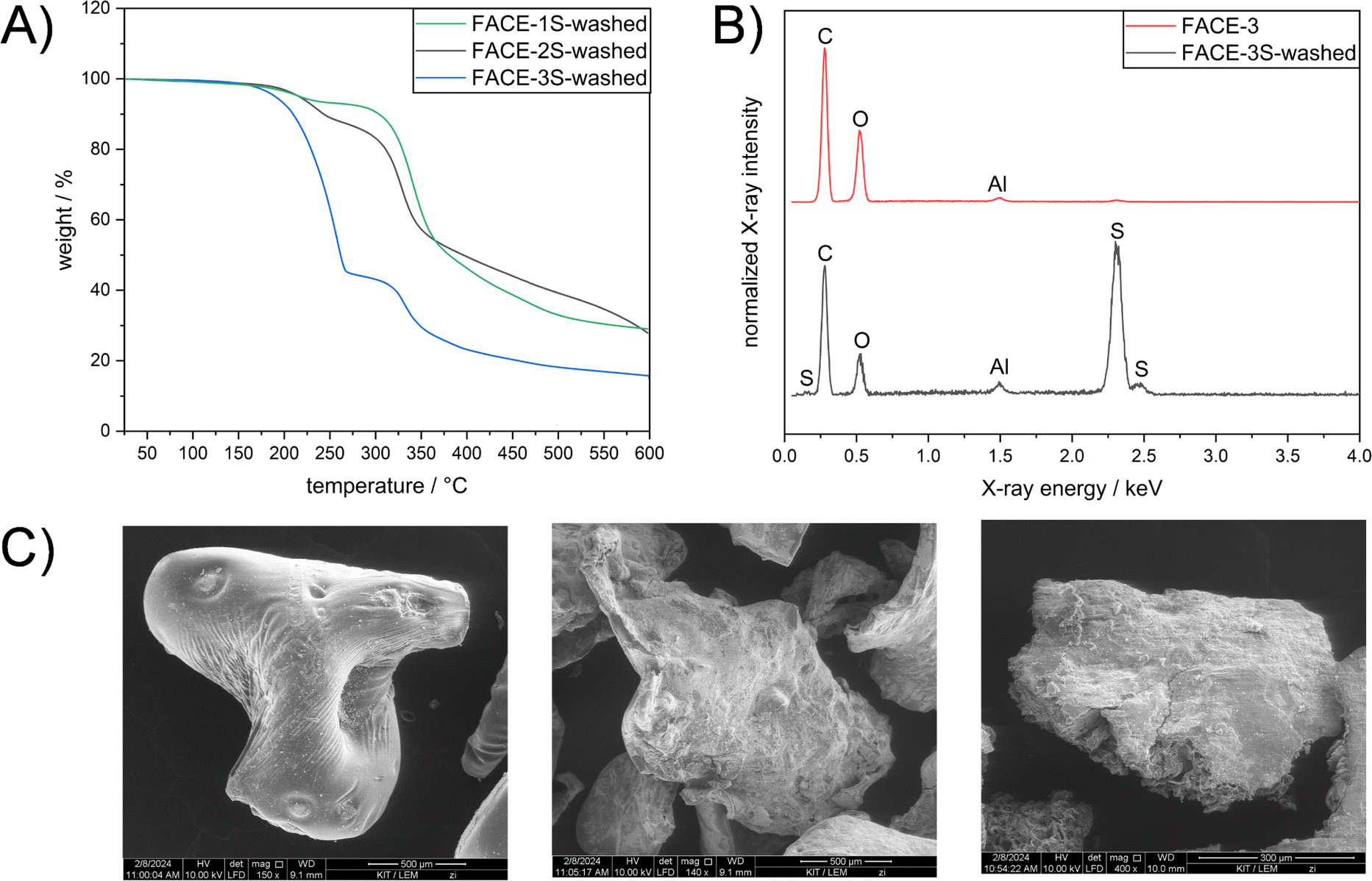 | ||
| Fig. 2 (A) TGA curves of FACE-1S-washed (green line), FACE-2S-washed (black line), and FACE-3S-washed (blue line) from 25 to 600 °C with a heating rate of 10 K min−1 under a nitrogen flow. (B) EDX spectra of FACE-3 (red line) and FACE-3S-washed (black line). (C) SEM pictures of FACE-1S-washed (middle), FACE-2S-washed (right) and FACE-3S-washed (left). | ||
In order to verify the presence of covalently incorporated sulfur in the residue materials, FACE-XS-washed (X = 1, 2 or 3) were investigated by energy-dispersive X-ray (EDX) spectroscopy. EDX spectra of the FACE comonomers before inverse vulcanization revealed characteristic bands for the elements carbon and oxygen (Fig. 2B, S13 and S14†), which was expected according to their chemical structure (depicted in Scheme 1A). Additional bands for FACE-XS-washed particularly appeared between 2.0 and 2.5 keV (Fig. 2B, S13 and S14†), confirming the existence of sulfur in the washed materials. The absence of sulfur crystals in supplemental SEM images of the previously mentioned residue materials combined with smooth surface areas confirmed that the sulfur species, which were detected by EDX, were covalently incorporated (Fig. 2C).
After understanding the formed network structure and confirming that cross-linked composite materials were formed, structure–property relationships were examined. An increased degradation temperature (temperature at which 5% weight loss occurred) was observed by TGA if FACEs with higher DS were incorporated as comonomers (Fig. 3A). More precisely, FACE-1S (DSFACE-1 = 0.38) revealed the lowest degradation temperature (Td,5% = 227 °C) followed by FACE-2S (Td,5% = 233 °C) and FACE-3S (Td,5% = 247 °C), respectively. The improved thermal stability of the composite materials including FACEs with higher DS resulted from the increased amount of covalently bonded sulfur (5.67, 12.2 and 56.2 wt% for FACE-1S, FACE-2S, and FACE-3S, respectively) and thus more extensive crosslinking. Interestingly, the composite materials show a one-step degradation behaviour, resulting from the high sulfur content that prevents the observation of the FACE degradation step. The hydrophobic character of FACE-1S, FACE-2S, and FACE-3S was investigated by conducting static WCA measurements (Fig. 3B). In the current literature,53 it has been frequently described that an increased DS leads to a higher hydrophobicity for CEs. Hence, it was not surprising that the static contact angles increased from 87° for FACE-1S (DSFACE-1 = 0.38) to 99° for FACE-3S (DSFACE-2 = 0.62).
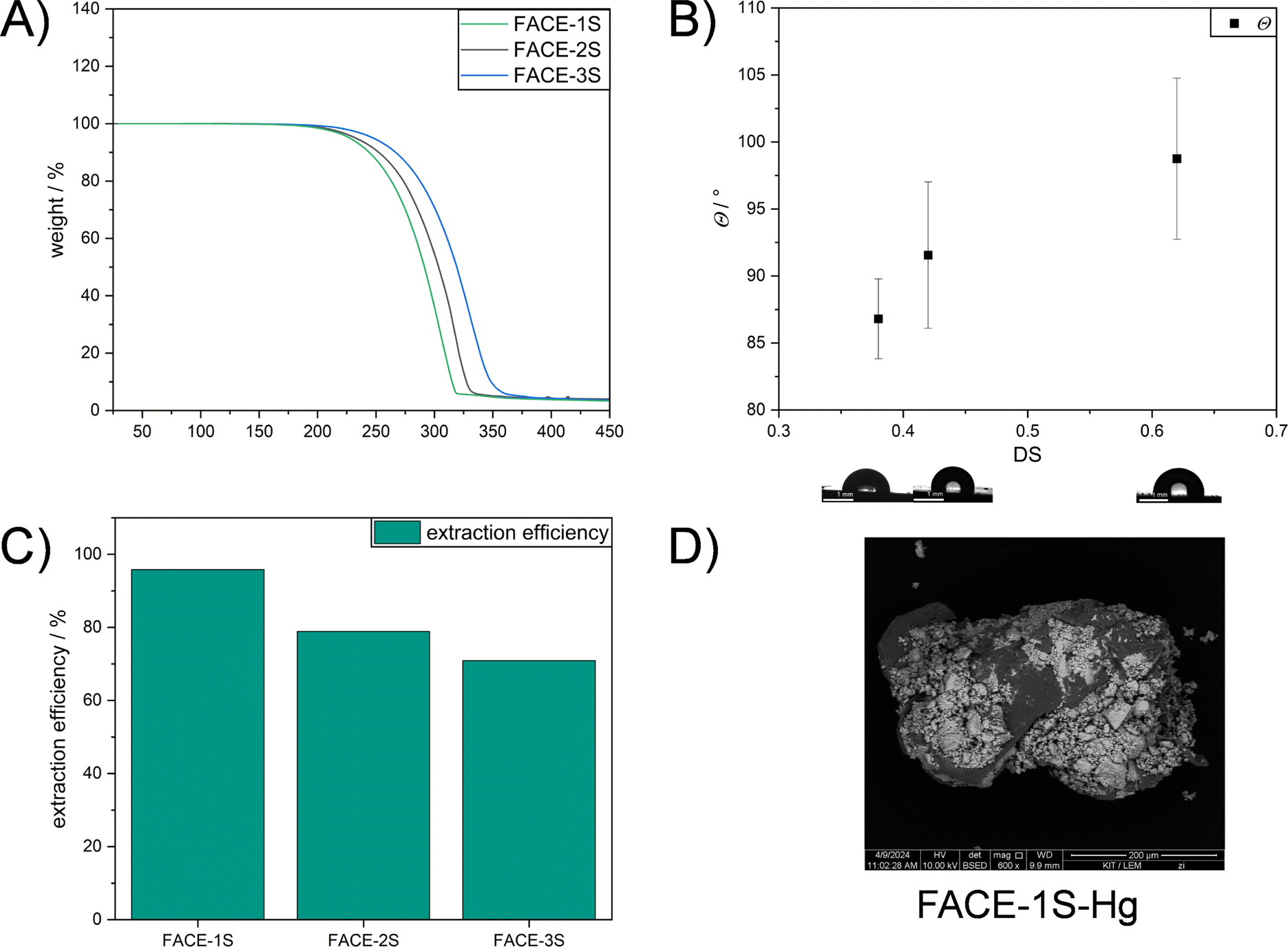 | ||
| Fig. 3 (A) TGA curves of FACE-1S (green line), FACE-2S (black line), and FACE-3S (blue line). (B) Static WCA measurements of FACE-1S, FACE-2S, and FACE-3S. (C) Mercury removal efficiency of FACE-1S, FACE-2S, and FACE-3S from an aqueous HgCl2 solution (1.00 mg L−1) after 24 h at ambient temperature. (D) Backscattered electron image of FACE-1S after mercury uptake via SEM. | ||
Inverse vulcanized materials are known as excellent heavy metal sorbents and can therefore be applied in water purification processes.45–47,54–56 Thus, to emphasize a potential application for the synthesized high sulfur content composite materials, their potential for mercury uptake from an aqueous HgCl2 solution (Ci = 1 mg L−1) was investigated. The chemical structure, i.e. the DS of the incorporated FACE comonomers, revealed a significant impact on the extraction efficiency and capacity of the materials (Table 1). As shown in Fig. 3C, FACE-1S synthesized from FACE-1 occurred as the most efficient mercury sorbent with an extraction efficiency up to 95%, followed by FACE-2S (78%) and FACE-3S (70%), respectively. The equilibrium mercury concentrations were subsequently employed for the calculation of the distribution coefficient (Kd), which assess the affinity of the synthesized sorbents for Hg2+ ions. Kd values can generally be calculated according to eqn (1):
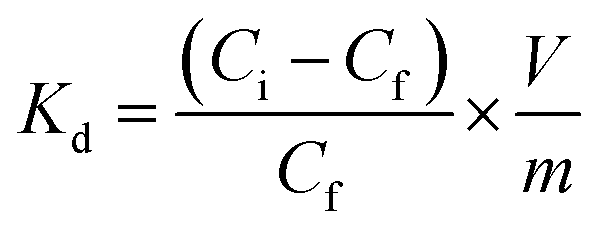 | (1) |
The equilibrium extraction capacities (qe) of all synthesized composite materials can be calculated according to eqn (2):
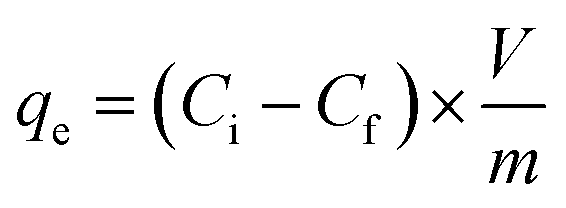 | (2) |
Unsurprisingly, FACE-1S showed the highest qe (0.48 mg g−1) compared to FACE-2S (0.39 mg g−1) and FACE-3S (0.35 mg g−1). As a final proof for the adsorption of mercury at the surfaces of the sorbent materials, i.e. FACE-1S, FACE-2S, and FACE-3S, backscattered electron (BSE) images after mercury uptake were recorded via SEM (Fig. 3D, S16A and S16B†, respectively). Fig. 3D exemplarily reveals bright and dark surface areas, where the dark spots correspond to the initial composite material and the bright spots represent areas with adsorbed mercury.
Conclusions
In the current work, microcrystalline cellulose was solubilized in a DMSO/TBD/CO2 switchable solvent system and subsequently transesterified by using the biobased transesterification agent methyl-10-undecenoate by adopting a literature known procedure.7 Upon the synthesis of renewable FACEs containing three different DS (0.38 ≤ DS ≤ 0.62), in-depth structural characterization, i.e.1H NMR, 31P NMR, ATR-IR, DSC and TGA, was conducted. The subsequent inverse vulcanization of the synthesized FACEs resulted in high sulfur content composite materials (∼95 wt% sulfur). A detailed investigation of the structural composition of the crosslinked sections via DSC, TGA, EDX, XPS and SEM showed an increased amount of covalently implemented sulfur (5.67 wt% ≤ wt% sulfur ≤ 56.2 wt%), depending on the DS of the employed FACE. Additionally, it was also shown that the structural compositions of the FACEs, i.e. their DS, also influence the material properties of the synthesized high sulfur content composite materials. More precisely, a higher DS of the used FACEs afforded a higher thermal stability (227 °C ≤ Td,5% ≤ 247 °C), an elevated surface hydrophobicity (87° ≤ θ ≤ 99°) and a lower mercury extraction efficiency (70% ≤ Hgremoval2+ ≤ 95%).Experimental part
Materials
Microcrystalline cellulose (MCC, Sigma-Aldrich) was dried under reduced pressure at 100 °C for 24 hours prior to use. Dimethyl sulfoxide (DMSO, dried and stored over molecular sieves, Acros Organics, >99.7%), 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TCI, ≥98%), methanol (VWR, ≥99.8%), methyl-10-undecenoate (Sigma-Aldrich, 96%), endo-N-hydroxy-5-norbornene-2,3-dicarboximide (Alfa Aesar, 97%), DMSO-d6 (Eurisotop, 99.8%), and CDCl3 (Eurisotop, 99.8%) were used without further purification. 2-Chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane (2-Cl-TMDP) was synthesized according to a literature known procedure.General procedure for the synthesis of FACEs
Fatty acid cellulose esters (FACEs) were synthesized according to a literature known procedure.7Accordingly, microcrystalline cellulose (MCC, 0.50 g, 1.50 mmol) was suspended in 10 mL anhydrous DMSO in a two neck round bottom flask. Subsequently, TBD (1.28 g, 9.25 mmol) was added, and the reaction mixture was stirred after applying a continuous CO2 flow for 20 minutes at 50 °C. The homogeneous solution was then heated to 95 °C and methyl-10-undecenoate (2.00 equiv., 3.50 equiv. and 9.00 equiv. per AGU for FACE-1, FACE-2, and FACE-3, respectively.) was added in a dropwise manner. The dark brown mixture was stirred for 6 h under air flow, diluted with 10 mL of anhydrous DMSO and precipitated under vigorous stirring in 200 mL of water. The residue was filtered, stirred in methanol, again filtered and subsequently dried under reduced pressure at 80 °C. The corresponding yields were determined depending on the DS31P according to eqn (S1) and (S2).†
FACE-1 (DS = 0.38): yield: 43% ATR-IR: ν (cm−1) = 3644–3035 ν(O–H), 3009–2789 ν(C–H), 1728 ν(CO), 1639 ν(CC), 1016 ν(C–O)AGU. 1H NMR (400 MHz, DMSO-d6) δ (ppm) = 5.79 (br, Hf, 1H), 5.52–2.96 (m, AGU, 7H), 4.96 (br, He, 2H), 2.32 (br, Hd, 2H), 2.01 (br, Hc, 2H), 1.51 (br, Hb, 2H), 1.38–1.17 (br, Ha, 10H).
FACE-2 (DS = 0.42): yield 51.6% ATR-IR: ν (cm−1) = 3653–3029 ν(O–H), 3003–2799 ν(C–H), 1728 ν(CO), 1639 ν(CC), 1016 ν(C–O)AGU. 1H NMR (400 MHz, DMSO-d6) δ (ppm) = 5.79 (br, Hf, 1H), 5.56–2.96 (m, AGU, 7H), 4.96 (br, He, 2H), 2.32 (br, Hd, 2H), 2.01 (br, Hc, 2H), 1.51 (br, Hb, 2H), 1.40–1.15 (br, Ha, 10H).
FACE-3 (DS = 0.62): yield 58.6% ATR-IR: ν (cm−1) = 3651–3105 ν(O–H), 3013–2793 ν(C–H), 1734 ν(CO), 1639 ν(CC), 1016 ν(C–O)AGU. 1H NMR (400 MHz, DMSO-d6) δ (ppm) = 5.78 (br, Hf, 1H), 5.61–2.82 (m, AGU, 7H), 4.94 (br, He, 2H), 2.32 (br, Hd, 2H), 2.00 (br, Hc, 2H), 1.51 (br, Hb, 2H), 1.40–1.15 (br, Ha, 10H).
General procedure for DS determination via31P NMR spectroscopy
An exact amount of the corresponding FACE (20 mg) was dissolved in 1.00 mL of pyridine. Subsequently, 1.00 mL of CDCl3 and 200 μL of 2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane (2-Cl-TMDP, 1.26 mmol) were added and the reaction mixture was stirred until a visibly homogeneous solution was obtained. After adding the internal standard endo-N-hydroxy-5-norbornene-2,3-dicarboximide (125 μL, 105.59 mM in pyridine/CDCl3 = 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, 0.0132 mmol) the solution was again stirred for at least 10 minutes before 0.60 mL was transferred into an NMR tube and 31P NMR was conducted. The DS values were calculated according to Kilpeläinen et al.52 by applying eqn. (S3) and (S4).†
:2, 0.0132 mmol) the solution was again stirred for at least 10 minutes before 0.60 mL was transferred into an NMR tube and 31P NMR was conducted. The DS values were calculated according to Kilpeläinen et al.52 by applying eqn. (S3) and (S4).†
General procedure for the synthesis of FACES95
A crimp vial was charged with elemental sulfur (2.85 g, 95.0 wt%) and the corresponding FACE (150 mg, 5.00 wt%). Subsequently, the reaction mixture was heated to 180 °C and stirred for 24 h until the black reaction medium appeared homogeneous. After cooling to room temperature, the targeted composite materials were obtained.FACE-1S: yield: 100%
FACE-2S: yield: 100%
FACE-3S: yield: 100%
General procedure for mercury sorption studies
200 mg of the respective high sulfur content composite material was crushed into a powder and subsequently added to 100 mL of an aqueous HgCl2 solution (Ci = 1.00 mg L−1). After stirring the solution for 24 h at ambient temperature, the inverse vulcanized material was filtered off and the mercury concentration in the solution was determined via Cold Vapor Atomic Absorption spectroscopy.Instrumentation
000 Å), PSS PFG Micro (25.0 × 0.46 cm, 1000 Å), and PSS PFG Micro (25.0 × 0.46 cm, 100 Å). The system was calibrated with linear poly(methyl methacrylate) standards (Polymer Standard Service, Mp: 102–981 kDa).
Data availability
The data supporting this article have been included within the article and as part of the ESI.†Author contributions
Timo Sehn: conceptualization, formal analysis, investigation, writing – original draft; Julian Fanelli: investigation (synthesis), writing – review & editing; Lisa Wahl: investigation (synthesis); Michael A. R. Meier: project administration, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge Prof. P. Levkin (WCA) and Soft Matter Synthesis Laboratory (SML), i.e. Prof. P. Théato, Prof. J. Lahann and Prof. S. Bräse (TGA), for the access of diverse analytical characterization facilities. Additionally, Volker Zibat and Dr Ute Schwotzer are acknowledged for SEM, EDX and AAS measurements, respectively.References
- P. Anastas and J. Warner, Green Chemistry: Theory and Practice, Oxford University Press, New York, 1998 Search PubMed.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- J. Wolfs and M. A. R. Meier, Green Chem., 2021, 23, 4410–4420 RSC.
- D. Klemm, B. Heublein, H.-P. Fink and A. Bohn, Angew. Chem., Int. Ed., 2005, 44, 3358–3393 CrossRef CAS PubMed.
- T. Sehn and M. A. R. Meier, Biomacromolecules, 2023, 24, 5255–5264 CrossRef CAS PubMed.
- S. Mi, Z. Yao, F. Liu, Y. Li, J. Wang, H. Na and J. Zhu, Green Chem., 2022, 24, 8677–8684 RSC.
- E. Esen, P. Hädinger and M. A. R. Meier, Biomacromolecules, 2021, 22, 586–593 CrossRef CAS PubMed.
- Z. Chen, J. Zhang, P. Xiao, W. Tian and J. Zhang, ACS Sustainable Chem. Eng., 2018, 6, 4931–4939 CrossRef CAS.
- S. B. W. Kusuma, D. Hirose, A. Yoshizawa, L. Szabó, D. Ina, N. Wada and K. Takahashi, ACS Sustainable Chem. Eng., 2021, 9, 8450–8457 CrossRef CAS.
- A. Schenzel, A. Hufendiek, C. Barner-Kowollik and M. A. R. Meier, Green Chem., 2014, 16, 3266–3271 RSC.
- H. Steinmeier, Macromol. Symp., 2004, 208, 49–60 CrossRef CAS.
- C. L. McCormick, P. A. Callais and B. H. Hutchinson Jr, Macromolecules, 1985, 18, 2394–2401 CrossRef CAS.
- O. A. El Seoud, G. A. Marson, G. T. Ciacco and E. Frollini, Macromol. Chem. Phys., 2000, 201, 882–889 CrossRef CAS.
- R. P. Swatloski, S. K. Spear, J. D. Holbrey and R. D. Rogers, J. Am. Chem. Soc., 2002, 124, 4974–4975 CrossRef CAS PubMed.
- P. Willberg-Keyriläinen and J. Ropponen, Heliyon, 2019, 5, e02898 CrossRef PubMed.
- X. Cao, S. Sun, X. Peng, L. Zhong, R. Sun and D. Jiang, J. Agric. Food Chem., 2013, 61, 2489–2495 CrossRef CAS PubMed.
- R. A. Sheldon, Green Chem., 2007, 9, 1273–1283 RSC.
- T. Kulomaa, J. Matikainen, P. Karhunen, M. Heikkilä, J. Fiskari and I. Kilpeläinen, RSC Adv., 2015, 5, 80702–80708 RSC.
- L. Duchatel-Crépy, N. Joly, P. Martin, A. Marin, J.-F. Tahon, J.-M. Lefebvre and V. Gaucher, Carbohydr. Polym., 2020, 234, 115912 CrossRef PubMed.
- M. A. Hussain, T. Liebert and T. Heinze, Macromol. Rapid Commun., 2004, 25, 916–920 CrossRef CAS.
- M. C. Nagel and T. Heinze, Lenzinger Ber., 2012, 90, 85–92 CAS.
- H. Nawaz, R. Casarano and O. A. El Seoud, Cellulose, 2012, 19, 199–207 CrossRef CAS.
- H. A. Staab, Angew Chem. Int. Ed. Engl., 1962, 1, 351–367 CrossRef.
- M. Jebrane, N. Terziev and I. Heinmaa, Biomacromolecules, 2017, 18, 498–504 CrossRef CAS PubMed.
- K. D. Mekonnen and Z. B. Sendekie, ACS Omega, 2021, 6, 24082–24091 CrossRef CAS PubMed.
- M. Gericke, P. Fardim and T. Heinze, Molecules, 2012, 17, 7458–7502 CrossRef PubMed.
- C. Libretti and M. A. R. Meier, Macromolecules, 2023, 56, 7532–7542 CrossRef CAS.
- K. N. Onwukamike, S. Grelier, E. Grau, H. Cramail and M. A. R. Meier, ACS Sustainable Chem. Eng., 2018, 6, 8826–8835 CrossRef CAS.
- H. Kato, F. Nakatsubo, K. Abe and H. Yano, RSC Adv., 2015, 5, 29814–29819 RSC.
- U. Biermann, U. T. Bornscheuer, I. Feussner, M. A. R. Meier and J. O. Metzger, Angew. Chem., Int. Ed., 2021, 60, 20144–20165 CrossRef CAS PubMed.
- J. Chen, M. de Liedekerke Beaufort, L. Gyurik, J. Dorresteijn, M. Otte and R. J. M. Klein Gebbink, Green Chem., 2019, 21, 2436–2447 RSC.
- Y. H. Zhao, S. Hupin, L. Lecamp, D. Vuluga, C. Afonso, F. Burel and C. Loutelier-Bourhis, RSC Adv., 2017, 7, 3343–3352 RSC.
- M. Ge, J.-T. Miao, K. Zhang, Y. Wu, L. Zheng and L. Wu, Polym. Chem., 2021, 12, 564–571 RSC.
- D. Le, C. Samart, K. Tsutsumi, K. Nomura and S. Kongparakul, ACS Omega, 2018, 3, 11041–11049 CrossRef CAS PubMed.
- J. Hobich, B. Huber, P. Theato and H. Mutlu, Macromol. Rapid Commun., 2021, 42, 2100118 CrossRef CAS PubMed.
- F. G. Müller, L. S. Lisboa and J. M. Chalker, Adv. Sustainable Syst., 2023, 7, 2300010 CrossRef.
- W. J. Chung, J. J. Griebel, E. T. Kim, H. Yoon, A. G. Simmonds, H. J. Ji, P. T. Dirlam, R. S. Glass, J. J. Wie, N. A. Nguyen, B. W. Guralnick, J. Park, Á. Somogyi, P. Theato, M. E. Mackay, Y.-E. Sung, K. Char and J. Pyun, Nat. Chem., 2013, 5, 518–524 CrossRef CAS PubMed.
- M. J. H. Worthington, R. L. Kucera and J. M. Chalker, Green Chem., 2017, 19, 2748–2761 RSC.
- T. Lee, P. T. Dirlam, J. T. Njardarson, R. S. Glass and J. Pyun, J. Am. Chem. Soc., 2022, 144, 5–22 CrossRef CAS PubMed.
- A. Hoefling, Y. J. Lee and P. Theato, Macromol. Chem. Phys., 2017, 218, 1600303 CrossRef.
- M. K. Lauer, A. G. Tennyson and R. C. Smith, Mater. Adv., 2021, 2, 2391–2397 RSC.
- A. D. Tikoalu, N. A. Lundquist and J. M. Chalker, Adv. Sustainable Syst., 2020, 4, 1900111 CrossRef CAS.
- A. D. Smith, C. D. McMillen, R. C. Smith and A. G. Tennyson, J. Polym. Sci., 2020, 58, 438–445 CrossRef CAS.
- J. Jia, J. Liu, Z.-Q. Wang, T. Liu, P. Yan, X.-Q. Gong, C. Zhao, L. Chen, C. Miao, W. Zhao, S. Cai, X.-C. Wang, A. I. Cooper, X. Wu, T. Hasell and Z.-J. Quan, Nat. Chem., 2022, 14, 1249–1257 CrossRef CAS PubMed.
- M. P. Crockett, A. M. Evans, M. J. H. Worthington, I. S. Albuquerque, A. D. Slattery, C. T. Gibson, J. A. Campbell, D. A. Lewis, G. J. L. Bernardes and J. M. Chalker, Angew. Chem., Int. Ed., 2016, 55, 1714–1718 CrossRef CAS PubMed.
- D. J. Parker, H. A. Jones, S. Petcher, L. Cervini, J. M. Griffin, R. Akhtar and T. Hasell, J. Mater. Chem. A, 2017, 5, 11682–11692 RSC.
- M. J. H. Worthington, R. L. Kucera, I. S. Albuquerque, C. T. Gibson, A. Sibley, A. D. Slattery, J. A. Campbell, S. F. K. Alboaiji, K. A. Muller, J. Young, N. Adamson, J. R. Gascooke, D. Jampaiah, Y. M. Sabri, S. K. Bhargava, S. J. Ippolito, D. A. Lewis, J. S. Quinton, A. V. Ellis, A. Johs, G. J. L. Bernardes and J. M. Chalker, Chem.–Eur. J., 2017, 23, 16219–16230 CrossRef CAS PubMed.
- M. J. H. Worthington, C. J. Shearer, L. J. Esdaile, J. A. Campbell, C. T. Gibson, S. K. Legg, Y. Yin, N. A. Lundquist, J. R. Gascooke, I. S. Albuquerque, J. G. Shapter, G. G. Andersson, D. A. Lewis, G. J. L. Bernardes and J. M. Chalker, Adv. Sustainable Syst., 2018, 2, 1800024 CrossRef.
- C. V. Lopez, M. S. Karunarathna, M. K. Lauer, C. P. Maladeniya, T. Thiounn, E. D. Ackley and R. C. Smith, J. Polym. Sci., 2020, 58, 2259–2266 CrossRef CAS.
- M. K. Lauer, T. A. Estrada-Mendoza, C. D. McMillen, G. Chumanov, A. G. Tennyson and R. C. Smith, Adv. Sustainable Syst., 2019, 3, 1900062 CrossRef CAS.
- M. K. Lauer, A. G. Tennyson and R. C. Smith, ACS Appl. Polym. Mater., 2020, 2, 3761–3765 CrossRef CAS.
- A. W. T. King, J. Jalomäki, M. Granström, D. S. Argyropoulos, S. Heikkinen and I. Kilpeläinen, Anal. Methods, 2010, 2, 1499–1505 RSC.
- M. Ioelovich, Polymers, 2021, 13, 1241 CrossRef CAS PubMed.
- X. Wu, J. A. Smith, S. Petcher, B. Zhang, D. J. Parker, J. M. Griffin and T. Hasell, Nat. Commun., 2019, 10, 647 CrossRef PubMed.
- M. J. H. Worthington, M. Mann, I. Y. Muhti, A. D. Tikoalu, C. T. Gibson, Z. Jia, A. D. Miller and J. M. Chalker, Phys. Chem. Chem. Phys., 2022, 24, 12363–12373 RSC.
- J. M. Chalker, M. Mann, M. J. H. Worthington and L. J. Esdaile, Org. Mater., 2021, 03, 362–373 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4su00424h |
| This journal is © The Royal Society of Chemistry 2025 |