
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective dissolution and re-precipitation by pH cycling enables recovery of manganese from surface nodules†
Pravalika
Butreddy
 a,
Sebastian T.
Mergelsberg
a,
Jennifer N.
Jocz
a,
Dongsheng
Li
a,
Venkateshkumar
Prabhakaran
a,
Andrew J.
Ritchhart
a,
Chinmayee V.
Subban
ab,
Jon
Kellar
c,
Scott R.
Beeler
d,
Sarah W.
Keenan
*e and
Elias
Nakouzi
*a
a,
Sebastian T.
Mergelsberg
a,
Jennifer N.
Jocz
a,
Dongsheng
Li
a,
Venkateshkumar
Prabhakaran
a,
Andrew J.
Ritchhart
a,
Chinmayee V.
Subban
ab,
Jon
Kellar
c,
Scott R.
Beeler
d,
Sarah W.
Keenan
*e and
Elias
Nakouzi
*a
aPacific Northwest National Laboratory, Richland, Washington 99354, USA. E-mail: elias.nakouzi@pnnl.gov
bDepartment of Materials Science and Engineering, University of Washington, Seattle, Washington 98195, USA
cDepartment of Materials and Metallurgical Engineering, South Dakota School of Mines and Technology, Rapid City, South Dakota 57701, USA
dEngineering and Mining Experiment Station, South Dakota School of Mines and Technology, Rapid City, South Dakota 57701, USA
eDepartment of Geology and Geological Engineering, South Dakota School of Mines and Technology, Rapid City, South Dakota 57701, USA. E-mail: sarah.keenan@sdsmt.edu
First published on 6th January 2025
Abstract
Meeting global sustainable development and climate goals requires a rapid transition to renewable energy technologies. However, these emerging technologies rely on critical elements whose sourcing presents geopolitical and environmental challenges. In this study, we explore ferromanganese nodules from the Oacoma site in South Dakota as a viable feedstock for sourcing manganese, a critical element used in the production of battery cathodes, consumer electronics, and steel. The nodules are readily accessible from the surface site and primarily consist of rhombohedral metal carbonates, including manganese at 3.5–5.4 at% (9.2–14.1 wt%) relative to all the elements present in the nodules. Based on titration experiments and an equilibrium speciation model, we developed a strategy for extracting the manganese by selectively dissolving carbonate phases in acidic conditions, followed by selectively re-precipitating manganese oxide in alkaline conditions. Specifically, exposing the samples to pH 1.5–2 dissolved almost all the calcium and manganese ions, while retaining a significant portion of the iron and magnesium in the residual nodule powders. Subsequently, increasing the pH of the leachate to 5.7 resulted in the selective re-precipitation of predominantly iron hydroxide. Further increasing the pH of the leachate solution to 10.9 finally produced a relatively pure manganese oxide product. Our pH cycling approach recovered 65.7–74.2% of the manganese in the nodules at 70.3–85.4 at% (81.5–91.0 wt%) purity relative to the other metals, without the need for specialty chemicals, membranes, ligands, or resins, and without generating highly acidic wastes. We further performed a preliminary assessment of the scalability and industrial relevance of this process to explore these nodules as a feedstock for sustainable sourcing of manganese.
Sustainability spotlightThe growth in the need for rechargeable batteries and renewable energy is expected to drastically increase the demand for critical elements, including manganese. Here, we examine manganese extraction from a US domestic resource, namely ancient marine ferromanganese nodules that are accessible from a surface site. Our pH cycling approach extracts 65.7–74.2% of the manganese at 70.3–85.4 at% purity relative to the other metals through a bulk process that uses simple commodity chemicals and avoids highly acidic waste products. This work helps to address UN sustainability goals 6 and 7 on access to affordable green energy. Specifically, our methodology for manganese extraction reduces environmental disruption, particularly to marine resources, and helps support the supply chain of materials needed for clean energy technologies. |
Introduction
A key challenge towards meeting sustainability goals is ensuring the reliable supply and environmentally-friendly sourcing of critical materials essential for green technologies.1–3 One such critical material is manganese (Mn), which is widely used in batteries,4 ferroalloys,5 aluminum alloys, and consumer electronics.6 For example, lithium manganese oxides are used in commercial rechargeable batteries due to their relatively fast charge/discharge cycling.7,8 More recently, the combination of nickel–manganese–cobalt-based cathodes has been shown to enhance battery performance and longevity, further emphasizing the growing importance of manganese for energy storage and sustainability.9,10 The addition of manganese to cathode materials also decreases the required quantities of cobalt; another critical material that is even more fraught with ethical and environmental mining challenges. Manganese also remains an important component with “no known alternatives” in traditional commodities such as fertilizers and steel.11–14Accordingly, the global demand for Mn is anticipated to increase dramatically, particularly with the surge in need for batteries in both stationary and mobile applications. The U.S. Geological Survey (USGS) estimates that U.S. manganese ore imports in January 2024 increased by 65% compared to the previous year.15 The report also highlights the increasing import reliance on manganese and potential supply disruptions due to the limited domestic sources of the ore.13 Based on this assessment, the USGS has designated Mn as a “critical element,” signaling the urgent need to utilize new domestic manganese sources and their safe, scalable, and cost-effective processing.16 Likewise, manganese has been identified as a strategic raw material mineral in the European Critical Raw Materials Act and is on the United Kingdom's Critical Mineral Expert Committee's watchlist.17,18 To meet these growing needs, sourcing Mn must occur in a manner that mitigates environmental damage, meets sustainability goals, and ensures ethical mining practices. The development of chemical separation technologies could bridge this gap by recovering critical elements from feedstocks such as recycled batteries,19 seawater,20 or unconventional mining sources (e.g., water from abandoned mines).
Perhaps the most promising sources of manganese are seabed deposits in the form of polymetallic or ferromanganese nodules, which have been estimated at 3 trillion tons; vastly larger than the approximately 17 billion metric tons of Mn present in conventional terrestrial deposits.13 These marine nodules contain moderate to high manganese content, approximated as 16–24 wt% relative to all other elements.21 The nodules often also include other energy-critical elements such as nickel, cobalt, lithium, copper, molybdenum, and rare earths.22 They are mostly poorly crystalline and have a complex, heterogeneous composition, typically forming concentric layers of iron and Mn oxyhydroxides around a core. Nodule sizes vary from < 1 cm to 20 cm or greater, have ellipsoidal, spherical, tubular, or irregular shapes, and generally have a core or nucleus and one or several concentric layers. They mainly contain phyllomanganates, such as vernadite, birnessite, buserite, and some todorokite, and Fe-bearing minerals, such as goethite, lepidocrocite, ferroxyhite, and hematite, were also reported to be present.23 Other nodules are carbonaceous and contain rhodochrosite, calcite, and siderite.24,25
The two main growth mechanisms for forming manganese nodules are hydrogenetic and diagenetic, with a combination of the two mechanisms most frequently observed. Hydrogenetic precipitation results from the oxidation of dissolved Mn2+ and Fe2+ in oxygen-rich seawater and the subsequent growth of Mn4+ and Fe3+ oxide colloids around a core or nucleus. Diagenetic growth occurs in low-oxygen content pore water of deep-sea sediments.21,22 Hydrogenetic nodules are typified by lower Mn/Fe ratios (typically less than 2.5) compared to diagenetic nodules (2.5 to >10).26,27 Taken together, the mineralogy and the inferred growth mechanisms indicate that ferromanganese nodules can form under a diverse set of pH, redox (Eh), and oxygen conditions.
While ferromanganese nodules are present in most modern oceans, particularly the Pacific Ocean, at about 4000–6000 m depth, they can also be observed at all depths.26,28 In addition to modern nodules actively precipitating in ocean settings globally, there are several deposits of ancient nodules that formed in oceans in the geologic past.24,29,30 At present, these ancient nodules are poorly studied but are known to contain variable and potentially high Mn content, approximated as 5–25 wt% relative to all other elements.25 One such source is the Oacoma Site located near Chamberlain, South Dakota, which represents an extensive area along the Missouri River in central South Dakota where ferromanganese nodules are exposed at the surface.29–31 Nodules were originally deposited in the Western Interior Seaway during the late Cretaceous and are preserved within discrete layers in the Pierre Shale Formation. The deposit has previously been estimated to contain 5 billion metric tons of manganese, however the lateral extent of nodule outcrops and their stratigraphic position within the Pierre Shale Formation is not well understood.25 These ancient nodules were previously considered too low-grade to be utilized economically,29 but developing novel methods to selectively extract Mn from this readily available unconventional source would be beneficial for the current Mn supply scenario. Recent work has investigated bioleaching as a potential method for the sustainable extraction of Mn from this ore, however leaching efficiency was relatively low (∼26.3% of total Mn leached) highlighting the need for continued development of alternative processing methods.32
In the current study, we explored the prospect of sourcing Mn from carbonaceous ferromanganese nodules through selective dissolution and precipitation methods under varying pH conditions. We hypothesized that by targeting the carbonate fraction of the nodules,33 Mn could readily be released and re-precipitated into more concentrated forms. Rather than performing an optimization, engineering, or technoeconomic study, this manuscript aims to (1) characterize these highly interesting, potentially valuable, and drastically understudied surface nodules and (2) demonstrate proof-of-concept for manganese extraction from this real feedstock using simple commodity chemicals, informed by speciation modelling.
Materials and methods
Materials
The ferromanganese nodule samples were acquired from the Oacoma Site in South Dakota (Fig. 1A and B).29 Nodules were collected from the surface in June 2021 and stored at room temperature until subsequent analyses (Fig. 1C). The duration of nodule exposure to modern weathering processes prior to collection is unknown. The samples were crushed using a rolling mill/ball milling, and the resulting powder collected through a sieve of <200 mesh (75 microns) (Fig. 1D–F). | ||
| Fig. 1 Manganese nodules from the field to the microscale. (A and B) Manganese nodules at the Oacoma site along the Missouri River near Chamberlain, South Dakota. (C) Example of the manganese nodule rock samples acquired from the Oacoma site and (D) crushed into a powder. (E) Optical and (F) SEM images of the nodule powder. | ||
The chemical reagents for sample preparation, dissolution, and precipitation were hydrochloric acid (HCl, 37%, Sigma-Aldrich), sodium hydroxide (NaOH, ≥97%, Sigma-Aldrich), and nitric acid (HNO3, fuming 100%, Fluka Analytical), which were used as received. All the solutions were prepared using ultrapure water filtered from a Milli-Q IQ 7000 water purification system (resistivity, 18.2 MΩ cm).
Methods
We prepared different pH solutions ranging from 1.5 to 6.5 at approximately 0.5 pH unit increments by adding the required volumes of 0.1 M HCl stock to 10 mL of de-ionized (DI) water in separate vials while the pH was continuously monitored. Subsequently, 5 mg of the nodules powder was added to each pH solution and left undisturbed for 2 hours, 24 hours, or three days. The residual powder sample was removed from the leachate after centrifugation for 5 min at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm. For the re-precipitation experiments, 0.1 M NaOH stock solution was added to the leachates to increase the pH to the desired values. The solid products were also retrieved by centrifugation. All recovered solids were washed with DI water to remove the surface-attached ions and dried under ambient conditions for instrument characterization.
000 rpm. For the re-precipitation experiments, 0.1 M NaOH stock solution was added to the leachates to increase the pH to the desired values. The solid products were also retrieved by centrifugation. All recovered solids were washed with DI water to remove the surface-attached ions and dried under ambient conditions for instrument characterization.
Characterization
Powder X-ray diffraction (XRD) analysis was performed using Cu Kα radiation (λ = 1.54 Å) on a Rigaku MiniFlexII DESKTOP X-ray diffractometer at a step width of 0.02° with the start and stop angles being 5° and 90° respectively. The diffraction pattern obtained was qualitatively analyzed using the PDXL2 software for peak-matching. The surface chemistry and oxidation states of the nodule powders and final precipitates were investigated using the Quantera Hybrid X-ray photoelectron spectrometer (XPS). The elemental narrow scans have been corrected (binding energy scale corrected) for sample charging and neutralization by referencing the most intense C 1s line to 285.0 eV. Deconvolution of the peaks and spectral line fitting were carried out using the CasaXPS software.Fourier transform infrared (FTIR) spectra of the powder samples were acquired using a Bruker Alpha II spectrometer operating in attenuated total reflection mode at room temperature. The FTIR spectra were recorded for a wavenumber range of 4000–500 cm−1 with a spectral resolution of 4 cm−1. The powder samples were imaged using a Keyence VHX 7000 Optical Microscope. Titrations were carried out with a 40 mg sample and using 0.1 M HCl and NaOH solutions. The pH was measured with a Mettler-Toledo NMR pH electrode S7. Titrants were loaded into 904 Titrando modules (Metrohm) with 20 mL burettes and added using an automated pH titration setup with maximum dosing increments, pH signal drift acceptance of 1 mV min−1, and a maximum waiting time of 40 seconds. Titration curves were produced using the tiamo software (Metrohm, v. 2.5). All other pH measurements were performed using a SevenCompact pH meter (Mettler Toledo, S220).
The samples were also imaged using a ThermoFisher Scientific Apreo 2 scanning electron microscope (SEM) equipped with an Oxford Instruments energy dispersive spectroscopy (EDS) detector, which was used for the elemental analysis. The SEM and EDS mapping on the pristine nodule powder was performed after dispersing the powder on carbon tape. The residual nodule powders and the precipitates were re-dispersed in DI water after washing and drop-cast on silicon wafers. SEM imaging of these samples was performed at low accelerating voltages and beam currents to reduce the charging effects using the Everhart–Thornley detector (ETD) in the secondary electron mode. For the elemental mapping, the instrument was operated at an accelerating voltage of 20 kV at an analytical working distance of 10 mm. We performed an analysis on the EDS maps to evaluate the correlation between the various elements by extracting element-specific intensity maps and using the corr2 function in Matlab.
Transmission electron microscopy (TEM) experiments were carried out with a ThermoFisher Titan TEM (300 KV), equipped with an energy dispersive spectroscopy (EDS) detector (Bruker), to determine the nanoscale sample morphology, elemental chemistry, and crystallography. For this purpose, the sample powder was first dispersed in an ethanol solution and then sonicated for 5 min. A drop of the liquid was cast on a regular TEM copper grid with a carbon film. Upon completing the separation experiments, the mass of the dried precipitates was measured using an analytical balance (Sartorius, BCE224i-1S).
The major and trace elemental composition of the nodule powders and dissolution products were analyzed using a PerkinElmer Optima 8300 Dual View Inductively Coupled Plasma-Optical Emission Spectrometer (ICP-OES) with a PerkinElmer S-10 auto-sampler interface. For these analyses, 1 mg of the nodule and precipitate samples were digested in a hydrochloric and nitric acid (3:1) mixture (aqua regia), as described above. Further, this acid matrix was diluted with ultrapure water by a factor of 20 to reach the final acid concentration of about 2%. Each leachate solution acquired from the different pH solutions was acidified to reach a 2% HCl final concentration and diluted with ultrapure water by a factor of 10 to make up a total volume of 10 mL, containing about 0.54 mL HCl (37%). All the samples were filtered through a 0.22 μm filter (VWR syringe filter, Nylon, 25 mm) in preparation for the ICP-OES analysis. The instrument was calibrated using the High-Purity Standards Corporation standards to generate the calibration curves. The range of calibration curves was 50 ppb to 50 ppm. This calibration was verified with an Initial Calibration Verification (ICV) and during sample analysis with a Continuing Calibration Verification (CCV) run every ten samples, at a minimum, as per Hanford Analytical Quality Assurance Requirements Document (HASQARD) requirements. Calibration blanks were analyzed following each calibration verification solution to ensure background signals and potential carryover effects were not a factor. All calibration verification values were within ±10% of the target concentrations to comply with the quality control requirements as defined in the HASQARD. A 1 ppm Internal Standard (ISTD) solution was added in-line to all samples, standards, and blanks to demonstrate the stability of the instrument and sample introduction system. Samples were run at several dilutions to bring their various elemental concentrations within the optimal analytical ranges of the instrument and to provide another level of result confirmation.
Solution chemistry modeling
Thermodynamic modeling of the titration data and development of the chemical model was carried out using PHREEQC.34,35 Most of the thermodynamic data used in the models presented here are taken from the Lawrence Livermore National Laboratory database, based on EQ3/6,36 except for the siderite solubility data.37 Initial concentrations of titrants were determined by titration of a 0.05 M Na2CO3 solution using HCl to pH 1 and a back-titration to pH 10 using NaOH. The determined concentrations were 0.098 M for HCl and 0.096 M for NaOH, which were used throughout the model. The initial nodule solid was assumed to be primarily a carbonate with a single solid solution phase defined by the Mn/Ca and Fe/Ca measured by EDS. Additionally, amorphous SiO2 was assumed to be present, important for surface protonation equilibria and the titration with NaOH to high pH. Oxidation of the sample by the atmosphere was modeled by assuming 20% oxygen above the sample and 50 moles of air at standard atmospheric pressure. The solution was assumed to not be in equilibrium with atmospheric CO2. Including atmospheric CO2 in the models did not match the data, which is consistent with other small-volume carbonate titrations published previously.38Results and discussion
Crystallographic analysis of manganese nodules
We performed a series of characterization experiments to determine the chemical and crystallographic nature of the nodules (Fig. 1A–D) and inform the strategy for extracting critical elements. The powdered nodules consisted of a heterogeneous mixture of orange, black, brown, reddish-brown, and a few pink/white particles, as observed through the optical microscope (Fig. 1E). Most of the particles were <100 μm in size (Fig. 1F). Powder X-ray diffraction (XRD) analysis showed the presence of rhombohedral metal carbonates of the calcite family (Fig. 2A). The diffraction peaks were slightly shifted compared to pure calcite and in some cases showed double peaks, suggesting the presence of various isomorphs that include magnesium, manganese, and iron. For example, the largest intensity [104] reflection typically occurs at 29.4°, 30.7°, 31.6°, 32.1°, and 32.5° for calcite (CaCO3), ankerite (Ca(Fe,Mg,Mn)(CO3)2), rhodochrosite (MnCO3), kutnohorite (CaMn(CO3)2), siderite (FeCO3), and magnesite (MgCO3), respectively. The nodules samples showed two relatively broad [104] peaks at 31.2° and 31.9°. This result indicates that most of the crystals were likely mixed carbonates with direct substitutions of the constituent metals within the rhombohedral lattice, which produces lattice strain and slight peak shifts. However, it is possible that various carbonates were also present as relatively pure grains within the nodule matrix. The diffractogram also showed a peak at 26.7° corresponding to crystalline quartz (SiO2). Note that the XRD data did not preclude the possibility of amorphous phases or minor amounts of other phases such oxides, hydroxides, and silicates. The TGA analysis (Fig. S2†) further confirms the presence of carbonates in these nodules. | ||
| Fig. 2 Mineralogical and structural characterization of manganese nodule powders. (A) X-ray diffraction, (B) FTIR data indicate the presence of rhombohedral metal carbonates, quartz, and possibly hydroxy(oxide) phases (green triangle marker), (C and D) ex situ TEM images and diffraction pattern showing lattice spacing corresponding to the MnOOH phase. | ||
We further performed bulk characterization on the nodules using FTIR, which showed at least five distinct absorption peaks corresponding to the various phases (Fig. 2B). The strong peak at 1403 cm−1 is attributed to the asymmetric CO32− stretching vibration of the carbonate minerals.39 Two other sharp peaks at 863 cm−1 and 721 cm−1 correspond to the carbonate out-of-plane and in-plane bending modes, respectively.40 The peak at 1032 cm−1 is attributed to the Si–O asymmetrical stretching vibration in quartz crystals but has also been reported for other silicate and clay minerals.41–43 Finally, the absorption peak at 602 cm−1 is plausibly related to Mn–O vibrations from small amounts of manganese oxides, consistent with previous reports on characteristic bands for Mn–O vibrations in the 500–600 cm−1 range.41 The existence of such minor phases in our samples was indeed confirmed by high-resolution TEM imaging along with SAED characterization, which resolved diffraction patterns corresponding to groutite nanocrystals (MnOOH) (Fig. 2C and D), in addition to the more dominant carbonate phases such as calcite and siderite (Fig. S1†). Accordingly, the ferromanganese nodules in the present study differ markedly from other ocean nodules that have been studied more extensively, which consist of polymetallic oxides and hydroxides – rather than carbonates – as the major components.44–46
Elemental composition of manganese nodules
In the next step, we determined the elemental composition of the nodules using a combination of XPS, EDS, and ICP-OES characterization (Table 1 and S1–S3†). XPS analysis was the most conducive to evaluating the lighter elements, specifically 15.6 at% carbon, 58.9 at% oxygen, 9.7 at% silicon, and 0.4 at% phosphorous. These values correspond to a total non-metal content of 84.6 at% or 66.8 wt%; slightly lower than some deep-sea nodules, whose total non-metal composition is 76.1 wt%.21 The manganese content relative to all the elements in the nodules was measured as 3.5 at% (9.2 wt%) using XPS, 4.7 at% (14.1 wt%) using ICP-OES, and 5.4 at% (13.3 wt%) using EDS. Relative to the other metals, rather than the total elemental composition, the manganese content was measured as 23.0–34.1 at% (27.6–41.7 wt%) using the various techniques (Table 1). The remaining metal composition included 15.8–34.8 at% iron, 6.4–20.2 at% calcium, 6.9–19.0 at% magnesium, 2.0–12.7 at% sodium, 4.6–9.5 at% aluminum, and 0–0.2 at% chromium. For a given technique, these values were largely consistent for multiple samples of the nodule powders, indicating that the original heterogeneous rock matrix was sufficiently homogenized by the sample preparation method. Our results validated these nodules as a rich source of manganese, comparable to deep-sea polymetallic sea nodules that are also reported to contain Mn as the largest component and a slightly lower Fe content.47,48 Notice that we did not observe appreciable amounts of copper, nickel, or cobalt, which are often found in deep-sea nodules.| Element | ICP-OES (ppm) | ICP-OES (at%) | XPS (at%) | EDS (at%) | ICP-OES (wt%) | XPS (wt%) | EDS (wt%) |
|---|---|---|---|---|---|---|---|
| Mn | 214 | 35.4 | 23.0 | 34.1 | 43.6 | 27.6 | 39.3 |
| Fe | 115.8 | 18.9 | 34.9 | 31.3 | 23.6 | 42.6 | 36.4 |
| Mg | 20.4 | 22.7 | 10.6 | 7.0 | 4.2 | 5.6 | 3.5 |
| Ca | 100 | 7.6 | 19.2 | 20.2 | 20.4 | 16.9 | 16.7 |
| Na | 38.6 | 15.2 | 2.0 | 1.7 | 7.9 | 1.0 | 0.7 |
| Cr | 1.44 | 0.2 | — | — | 0.3 | — | — |
| Al | — | — | 9.5 | 4.6 | — | 5.6 | 2.5 |
| K | — | — | 0.8 | 1.1 | — | 0.7 | 0.8 |
In addition to the bulk characterization, we also used EDS to visualize the spatial distribution of the various elements in the nodules. The distribution maps of Mn, Fe, Ca, Mg, and Si are presented in Fig. 3A. As expected, multiple metals co-existed in many of the crystals, indicating inclusions and direct substitutions into mixed phases. However, the elemental distributions showed additional subtle features that could be resolved by a spatial correlation analysis (Fig. S3†). Specifically, calcium was mostly localized in spherical grains of approximately 0.53 (±0.21) μm in radius. These particles showed only scarce inclusions of the other elements, indicating the presence of relatively pure calcite crystals. Similarly, the silicon distribution was only slightly correlated with Mn, Fe, Ca, and Mg, consistent with the presence of quartz rather than mixed metal silicates as the primary silicon-rich phase (Fig. 3B). The Mn distribution was highly correlated with both Mg and Fe, confirming that these elements occurred mainly in mixed carbonate phases. However, a significant number of grains – while including multiple metals – were distinctly Mn-rich, as demonstrated by a plot of the corresponding intensities in the EDS maps (Fig. 3C). These heterogeneous distributions have important implications on dissolution-driven separations; particles with high inclusion rates and defect densities tend to dissolve faster at a given undersaturation condition.
 | ||
| Fig. 3 Elemental mapping of manganese nodule powders and correlation analysis. (A) Spatial distribution of various elements in the nodules as measured by EDS mapping, (B) spatial correlation between the elements, (C) Mn versus Fe correlation. | ||
Speciation model and hypothesis for manganese extraction
The existence of predominantly carbonate phases, rather than oxides or hydroxides, suggests that pH cycling can be used to separate the metal components. The underlying rationale is that acidic leaching selectively dissolves certain metal carbonates based on their equilibrium solubilities as a function of pH, thus enriching the leachate relative to the original nodule samples. Subsequently, increasing the pH of the leachate selectively re-precipitates certain metal components according to the relative solubilities of their corresponding (oxy)hydroxides. This cycle of dissolving carbonates and re-precipitating (oxy)hydroxides presents a pathway for extracting critical elements, particularly manganese, from the polymetallic nodules.To evaluate this prospect, we performed calculations of the solution speciation at various pH conditions using PHREEQC. As input parameters, we used the chemical and crystallographic composition of the nodules determined in the previous section as well as the literature-reported solubilities of the relevant phases. The metal ion concentrations in the leachate were modelled as a function of pH (Fig. 4A). The results show that calcite dissolves at a higher pH compared to Fe/Mn/Mg carbonates, releasing most of the calcium ions into the leachate at pH 3–4. As the pH is further decreased, the Mn carbonates dissolve at slightly milder acidic conditions compared to Fe. In principle, this discrepancy can be leveraged to control the leachate composition at various pH values and achieve separations. Note that while this model serves as an excellent starting point for predicting the outcomes of acidic leaching, it does not account for other aspects of the process, including dissolution kinetics, polydisperse grain sizes, or defects in the crystals due to substitutions and inclusions which enhance the rates of dissolution and modify phase solubility.
 | ||
| Fig. 4 Modeled and experimental changes to select elements with changing pH conditions. (A) Equilibrium speciation model showing expected ion concentrations in leachate as the nodules are exposed to acidic solutions, (B) experimental and PHREEQC titration curve of the nodule powder as the pH is decreased, and (C) the leachate solution and PHREEQC model as the pH is increased. The first numerical derivative of the data, dpH/dV, is shown in red underneath the titration curves. | ||
To validate the speciation model, we performed titration experiments of nodule dissolution by 0.1 M HCl and subsequent oxyhydroxide precipitation by 0.1 M NaOH (Fig. 4B and C). Results show very quick dissolution of the powder after minimal addition of HCl (Fig. 4B). This precipitous drop in pH is due to the oxidation of Fe2+ to Fe3+ by oxygen in the air and the subsequent precipitation of ferrihydrite [–Fe(OH)3]. The model does not capture the exact mechanism and varies according to pH and [Fe2+].49,50 The overall reaction, however, is well-fit by the model and suggests each mole of Fe2+ generates approximately 2 moles of H+, quickly dissolving the carbonate solids. This increased reactivity is so rapid that identification of an equivalence point was not possible. The coupled Fe2+ oxidation/precipitation also prevents any oxidation of Mn2+ to Mn4+ at high pH and the formation of any Mn(OH)2. Other Mn phases, such as tobermorite or birnessite, were included in the model but would cause even steeper pH drops not observed in the data. The plateau at low pH is likely due to the dissolution of some ferrihydrite and an overlapping signal from the pHPZC of silica in the sample.
The reverse titration to high pH reveals two major steps from pH 4–8 and another step around pH 10. After the precipitation of Fe3+ at pH < 3 as ferrihydrite, the observed steps correspond to Mn2+ oxidation to MnO4− at pH 7.8 and the formation of MnO42− above pH 9.0. For these calculations, only the Fe3+ precipitates and Mn is not allowed to precipitate. This choice was made because Mn precipitates often have very large surface areas and small particle sizes that (1) keep the solution in good contact with Mn, and (2) modifies the Ksp of any precipitate to the point that bulk phase values cannot be used for modeling precipitation. This assumption results in a reasonable comparison of the thermodynamic model to the experimental titration data (Fig. 4C). In the experiment, the MnO4− and MnO42− species would cause the formation of MnOOH and MnO2 solids, given their limited stability/solubility. This is supported by the first steep increase in pH, which occurs in multiple steps in the experiment and at lower volumes of NaOH, compared to one large increase in the model. This discrepancy likely stems from Mn precipitation and condensation reactions in the experiment that are not captured by the model. Thus, our model supports selective precipitation of Fe at pH < 3–4 and Mn at pH > 4. The solubility of Mn can be further reduced by continuously increasing the pH to eliminate the strongly oxidizing MnO4− species. While this initial assessment was based purely on equilibrium speciation and did not consider kinetic effects, it served as a starting point for pH-driven manganese extraction from the nodules.
Selective dissolution and re-precipitation by pH cycling
Based on the speciation model and characterization results, we attempted to extract manganese by leaching the nodules with hydrochloric acid, followed by re-precipitating the leachate with sodium hydroxide. To test the dissolution step, we performed a series of experiments by placing samples of the nodule powder in HCl solutions at pH 6.5–1.5. After three days, the elemental composition of the residual powder was analyzed using EDS measurements. Within the pH range of 6.5 to 3, the residual powder showed similar composition of Mn, Mg, Fe, and Ca – as well as the other minor constituents – compared to the pristine nodules, indicating that dissolution of the carbonates in this regime did not noticeably alter the ratio of the constituent elements (Fig. 5A). In contrast, at pH 1.5, the relative atomic percent of Fe and Mg increased from 30.6 at% and 7.17 at% to 78.9 at% and 15.7 at%, respectively. By comparison, the atomic percent of Mn was reduced drastically to <6%, while Ca was not detected, confirming that it had been completely leached from the nodules. | ||
| Fig. 5 Elemental composition as a function of pH of (A) the residual powder as determined by EDS analysis after 3 days of acid leaching and (B) the leachate solution as determined by ICP-OES analysis. | ||
In addition to characterizing the residual powder samples after dissolution, we determined the elemental composition of the leachate solutions using ICP-OES measurements (Fig. 5B). As expected, the leachate contained negligible concentrations of Fe and Mg upon leaching at pH 2.5 and 2, which increased to 0.54 mM and 0.22 mM, respectively, at pH 1.5. By comparison, Mn and Ca were already extracted at pH 2.5 with concentrations of 0.83 mM and 0.26 mM, which further increased to 1.65 mM and 0.55 mM, respectively at pH 2. No appreciable amounts of the minor metal components such as chromium or aluminum were observed in the leachate solutions. We concluded that acidic leaching of the nodules at pH ∼ 2 is partially selective, concentrating Mn and Ca relative to the pristine nodules, while leaving a significant portion of the constituent Fe and Mg in the residual powder.
We also examined the role of dissolution kinetics by leaving the nodule powders in the acidic solutions for different periods of time. The optimal duration for extraction was 24 hours since an increased exposure time of three days marginally improved the extraction efficiency, while a reduced exposure time of 2 hours did not sufficiently extract the manganese ions (Fig. S8†). Specifically, EDS analysis showed that the residual powder still contained a high portion of Mn at 25.3 at% after 2 hours of leaching at pH 1.5, compared to 7.6 at% after 24 hours. In contrast, almost all of the Ca was already leached within the first 2 hours.
In the next set of experiments, we recovered the leachates at pH 1.5, filtered out any remaining powders, and added NaOH to increase the pH and precipitate the various metals sequentially. The duration of each precipitation step was duration of 24 hours; we performed additional experiments which showed that 3 days could extract larger quantities of purer products at the expense of longer processing times (Fig. S9†). Multiple precipitation events were observed as a function of pH. Firstly, the leachate changed to an orange-brown color at approximately pH 3, which can be ascribed to the oxidation of Fe2+ to Fe3+ and the formation of colloidal iron hydroxide phases. These particles grow into Fe-rich precipitates between pH 4 and 6, which consist of 78.9 at% iron and 20.1 at% manganese relatively (Fig. 6A and B). We performed XPS measurements to confirm the oxidation state of iron in the products. Indeed, the Fe 2p elemental scan showed two major peaks attributed to the Fe 2p3/2 and Fe 2p1/2 at 711.3 eV and 724.8 eV, respectively (Fig. S5†). Additionally, their corresponding two satellite peaks at 718.9 eV and 733.3 eV indicate the Fe3+ oxidation state.51,52
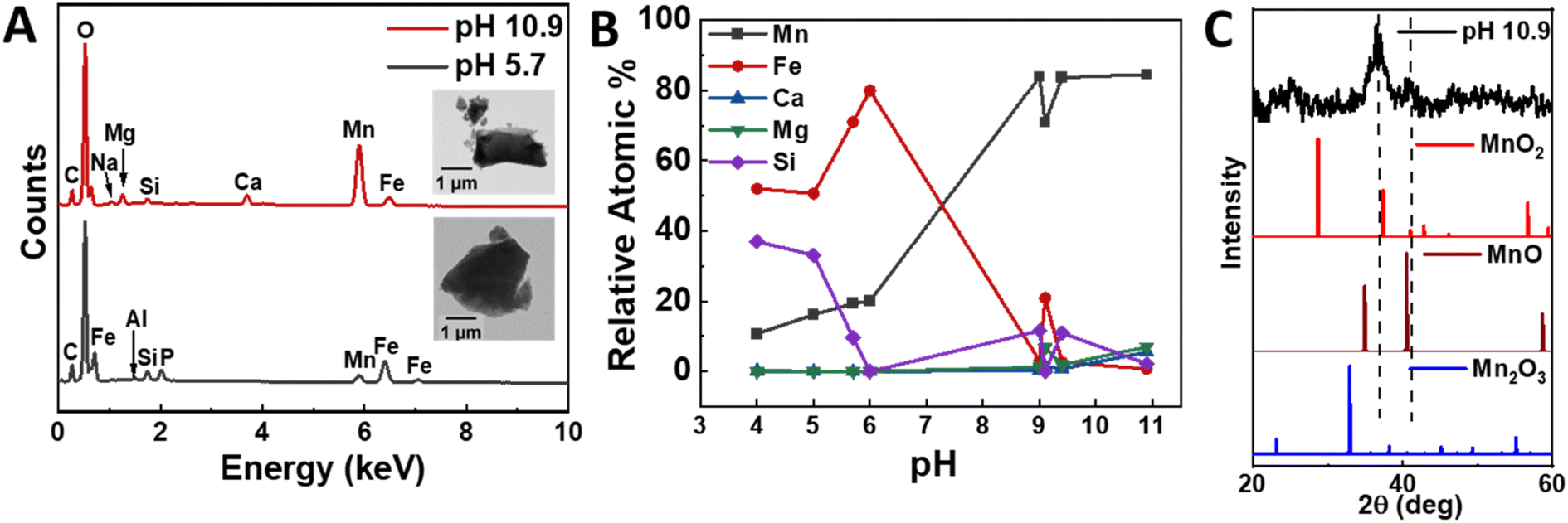 | ||
| Fig. 6 Elemental and chemical characterization of experimentally derived precipitates under select pH conditions. (A) EDS spectra recovered at pH 5.7 and pH 10.9 (inset: TEM images of the precipitates), (B) EDS compositional analysis on precipitates recovered at various pH, and (C) X-ray diffraction pattern of the pH 10.9 precipitate. | ||
After removing the Fe-rich solid products, we increased the pH to 9–11, which resulted in the formation of a denser, brownish-black precipitate. This solid product was extracted for analysis and changed into a darker color upon exposure to air for a duration of a few minutes. XRD measurements showed peaks corresponding to MnO2 (Fig. 6C), while TEM/SAED analysis suggested the trace occurrence of other oxide phases, namely Mn2O3 and MnO (Fig. S7†). We also performed an XPS surface elemental analysis to confirm the manganese oxidation state, which validated MnO2 as the main product. Specifically, the narrow scan of Mn 2p showed the spin-orbit doublet 2p3/2 and 2p1/2 levels centered at 654.56 eV and 642.85 eV with an 11.71 eV peak separation (Fig. 7B), in agreement with the reported data for MnO2.53–56 Moreover, the elemental scan of O 1s showed two major peaks after deconvolution (Fig. 7C). The peak at 530.36 eV indicates the presence of metal oxides (Mn–O)53 and had a full width at half maximum (FWHM) of 1.2.57 The peak at 531.53 eV corresponds to the sp2 C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond, possibly due to small amounts of CaCO3 impurities.58 These results suggest that the initial Mn-rich precipitate was rapidly oxidized to the Mn4+ oxidation state.
O bond, possibly due to small amounts of CaCO3 impurities.58 These results suggest that the initial Mn-rich precipitate was rapidly oxidized to the Mn4+ oxidation state.
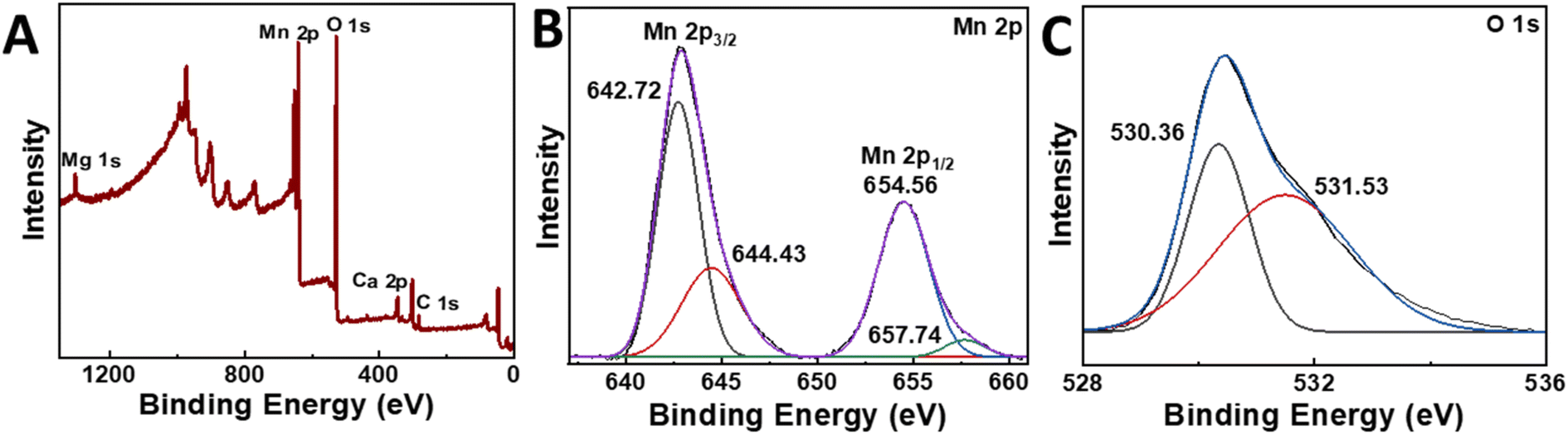 | ||
| Fig. 7 XPS survey spectrum and high-resolution XPS scan spectra. (A) XPS survey spectra, (B) Mn 2p, (C) O 1s narrow scans of the pH 10.9 precipitate. | ||
Furthermore, we reproduced this approach for 30 mg powder samples and evaluated the elemental composition of the product in a preliminary attempt to determine if the current approach can be applied to larger scales. We recovered ∼5.6 mg, whose manganese content was measured by XPS as 26.8 at% (54.0 wt%) relative to all other elements and 80.0 at% (87.2 wt%) relative to the other metals, which included minor amounts of magnesium, calcium, sodium, and iron, as well as silicon (Tables S2 and S3†). As such, 3.0 mg manganese was recovered from an upper estimate of 4.23 mg initially present in the nodules, corresponding to a recovery rate of at least 70.9%. To further validate these results, we performed ICP-OES and EDS analyses of the product. The manganese content was 70.3–85.4 at% (81.5–91.0 wt%) relative to the other metals. Although the lighter elements, namely oxygen and carbon, were difficult to resolve using these techniques, we were able to estimate the manganese recovery rate using some assumptions (Fig. 8, Tables S4 and S5†). Specifically, we considered the manganese as primarily MnO2, as supported by the XPS analysis, and the other elements as hydroxide precipitates. We determined that the total manganese recovery rate was 65.7–74.2%, comparable to the value obtained from the XPS measurements.
 | ||
| Fig. 8 Relative elemental compositions of pristine nodule powder and precipitates using ICP-OES. | ||
Scalability and industrial relevance
Based on the pH-cycling dissolution and selective precipitation method explored in this work, we envision a three-stage process for scaling up the extraction of Fe- and Mn-rich solids from the US domestic supply of carbonaceous ferromanganese nodules (Fig. 9). We then compare this process to common commercial processes for HCl acid-leaching of oceanic ferromanganese nodules (Fig. 10).22,23,59–61 Similar to most other industrial processes for ferromanganese feedstocks, the first stage of the process is a pretreatment that consists of crushing and grinding the nodules into a fine powder to improve the acid-leaching efficiency. In the second stage, the nodule powder is dissolved in the HCl solution, and the leachate is separated from the residual solids. In the third stage, NaOH solution is added stepwise, and the pH-dependent Fe- and Mn-rich precipitates are separated. | ||
| Fig. 9 Proposed pH-cycling process model for critical mineral separation and recovery from carbonaceous ferromanganese (Mn) nodules studied in this work. | ||
 | ||
| Fig. 10 Example of a typical hydrometallurgical acid-leaching process for element separation and recovery from oceanic (or deep-sea) ferromanganese nodules. Additional process steps and reagents are indicated in orange (in comparison to the process in Fig. 9). | ||
The three-stage process for terrestrial carbonaceous ferromanganese nodules contains fewer steps and fewer reagent inputs compared to typical oceanic nodules and should, therefore, be lower in overall processing cost compared to other commercial hydrometallurgical processes for oceanic ferromanganese nodules (Fig. 10).22,23,59–61 Both processes overlap in (1) their use of corrosive HCl solutions to fully dissolve minerals and (2) their crushing, grinding, leaching, precipitation, and solid–liquid separation steps. However, oceanic Mn nodules necessitate additional processing steps and costly reduction and precipitation reagents due to the physical, chemical, and mineralogical differences when compared with the terrestrial carbonaceous Mn nodules. Specifically, oceanic Mn nodules are polymetallic with valuable Co, Ni, and Cu impurities and contain bedrock (gangue) of little to no value. The value-less gangue must be removed after crushing and grinding as part of the beneficiation and enrichment pretreatment processing stage through magnetic, flotation, or gravity separation or a combination thereof, with the goal of improving the mineral grade prior to dissolution and separation. Additionally, the manganese and other valuable impurities in deep-sea nodules commonly exist as stable higher oxides and, as such, are difficult to dissolve in acid without first undergoing an energy-intensive reduction roasting treatment to destroy the oxide structure. Lastly, the presence of Co, Ni, and Cu in the leaching solution increases the complexity of the separation, and various solvents and reagents, such as H2S, are employed to selectively recover each element from the solution. From analyzing these industrial processes, the advantageous process simplicity and, thus, the lower anticipated processing cost of the pH-cycling method examined in this work is undoubtedly a feature of the feedstock's chemical composition and carbonaceous mineral structure, which is more easily dissolved and simpler to separate when compared to other feedstocks including oceanic manganese nodules that consist of highly stable polymetallic oxides mixed with gangue.
From a sustainability perspective, the Oacoma site, which is located on the surface, presents an easier to access resource compared to oceanic nodules. Recovery of oceanic nodules presents significant impacts on environments and ecosystems and often requires the use of diesel-powered machinery to access marine locations.62 This work demonstrates the potential for even low-grade ores to provide a potential source of Mn that would not require extensive mining and would minimize ecosystem disturbance. Here, the use of readily accessible chemicals with low hazard and minimal waste products without the need for specialty reagents, membranes, ligands, or resins provides an easy-to-reproduce and comparatively safer method of Mn extraction and concentration.
Conclusions
In this study, we demonstrated proof-of-concept for Mn recovery from ferromanganese nodules obtained from the Oacoma site in South Dakota. Using simple mechanical milling followed by dissolution and precipitation to recover Mn hydroxides, we were able to recover approximately 65.7–74.2% of the manganese in the form of MnO2 with 70.3–85.4 at% (81.5–91.0 wt%) relative to the other metals. This approach used simple commodity chemicals, namely hydrochloric acid and sodium hydroxide, without the need for advanced materials such as membranes, resins, ligands, or specialty chemicals. The pH cycling approach also did not produce acidic waste products, since the initial leachates were titrated to higher pH in order to recover the manganese. With subsequent calcination, the Mn hydroxides could be turned into commodity Mn oxides suitable for use as precursors in Li-ion battery cathode production. Two key features of the ferromanganese nodules at the Oacoma site show promise for a potentially economically viable source of manganese. Firstly, the nodules are predominantly carbonates rather than oxides and hydroxides, which enabled a relatively facile extraction by pH cycling. Secondly, the site is readily accessible at the surface; a sharp departure from other marine nodules whose sourcing involves disrupting deep-sea environments.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by a collaborative effort funded by the Laboratory Directed Research and Development (LDRD) program at PNNL, under the Non-Equilibrium Transport Driven Separations Initiative (NETS). PNNL is a multi-program national laboratory operated for the U.S. Department of Energy (DOE) by Battelle Memorial Institute under Contract No. DE-AC05-76RLO 1830. Additional funding for sample acquisition and shipping was provided by the Department of Geology and Geological Engineering (SWK).References
- N. R. Council, Minerals, Critical Minerals, and the U.S. Economy, The National Academies Press, Washington, DC, 2008 Search PubMed.
- S. Kalantzakos, Int. Spectator, 2020, 55, 1–16 CrossRef.
- K. Hund, D. La Porta, T. P. Fabregas, T. Laing and J. Drexhage, Minerals for Climate Action: The Mineral Intensity of the Clean Energy Transition, 2023 Search PubMed.
- H. X. Li, W. Zhang, K. N. Sun, J. Guo, K. Yuan, J. J. Fu, T. Zhang, X. K. Zhang, H. W. Long, Z. A. Zhang, Y. Q. Lai and H. Y. Sun, Adv. Energy Mater., 2021, 11, 2100867 CrossRef CAS.
- M. Tangstad, in Handbook of Ferroalloys, ed. M. Gasik, Butterworth-Heinemann, Oxford, 2013, pp. 221–266 Search PubMed.
- W. Shen, A. Hu, S. Liu and H. Hu, J. Alloys Metallurg. Syst., 2023, 2, 100008 CrossRef.
- M. M. Wang, X. H. Zheng, X. Zhang, D. L. Chao, S. Z. Qiao, H. N. Alshareef, Y. Cui and W. Chen, Adv. Energy Mater., 2021, 11, 2002904 CrossRef CAS.
- Y. Tang, S. Zheng, Y. Xu, X. Xiao, H. Xue and H. Pang, Energy Storage Mater., 2018, 12, 284–309 CrossRef.
- M. Malik, K. H. Chan and G. Azimi, Mater. Today Energy, 2022, 28, 101066 CrossRef CAS.
- L. J. Chang, W. Yang, K. D. Cai, X. L. Bi, A. L. Wei, R. F. Yang and J. N. Liu, Mater. Horiz., 2023, 10, 4776–4826 RSC.
- K. Hagelstein, J. Environ. Manage., 2009, 90, 3736–3740 CrossRef CAS PubMed.
- C. Clarke and S. Upson, Neurotoxicology, 2017, 58, 173–179 CrossRef CAS PubMed.
- W. F. Cannon, B. E. Kimball and L. A. Corathers, Manganese, Report 1802L, Reston, VA, 2017 Search PubMed.
- R. Jacob, S. Raman Sankaranarayanan and S. P. Kumaresh Babu, Mater. Today: Proc., 2020, 27, 2852–2858 CAS.
- J. E. K. Justin and Y. Kang, Manganese in March 2024, U.S. Geological Survey, National Minerals Information Center, 2024 Search PubMed.
- J. D. Applegate, 2022 Final List of Critical Minerals, Geological Survey, Federal Register, 2022 Search PubMed.
- Proposal for a Regulation of the European Parliament and of the Council Establishing a Framework for Ensuring a Secure and Sustainable Supply of Critical Raw Materials and Amending Regulations (EU) 168/2013, (EU) 2018/858, 2018/1724 and (EU) 2019/1020, Report 52023PC0160, European Commission, 2023, https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=celex:52023PC0160 Search PubMed.
- Resilience for the Future: the United Kingdom’s Critical Minerals Strategy, Department for Business and Trade; and Department for Business, Energy & Industrial Strategy; HM Government, 2023, https://www.gov.uk/government/publications/uk-critical-mineral-strategy/4acf2ca4-70cf-4834-a081-cf16b7c66959 Search PubMed.
- Q. Wang and E. Nakouzi, Environ. Sci. Technol. Lett., 2023, 10, 1188–1194 CrossRef CAS.
- Q. Wang, E. Nakouzi, E. A. Ryan and C. V. Subban, Environ. Sci. Technol. Lett., 2022, 9, 645–649 CrossRef CAS.
- R. Sharma, in Deep-Sea Mining: Resource Potential, Technical and Environmental Considerations, ed. R. Sharma, Springer International Publishing, Cham, 2017, pp. 229–256 Search PubMed.
- J. Ju, Y. Feng, H. Li, Z. Xue, R. Ma and Y. Li, Sep. Purif. Technol., 2023, 315, 123626 CrossRef CAS.
- G. Senanayake, Miner. Eng., 2011, 24, 1379–1396 CrossRef CAS.
- F. J. González, L. Somoza, R. León, T. Medialdea, T. de Torres, J. E. Ortiz, R. Lunar, J. Martínez-Frías and R. Merinero, Chem. Geol., 2012, 310–311, 56–78 CrossRef.
- E. R. Force and W. F. Cannon, Econ. Geol., 1988, 83, 93–117 CrossRef CAS.
- P. Halbach, U. Hebisch and C. Scherhag, Chem. Geol., 1981, 34, 3–17 CrossRef CAS.
- D. Li, Y. Fu, X. Sun and Z. Wei, Ore Geol. Rev., 2020, 118, 103371 CrossRef.
- A. Usui, H. Hino, D. Suzushima, N. Tomioka, Y. Suzuki, M. Sunamura, S. Kato, T. Kashiwabara, S. Kikuchi, G. I. Uramoto, K. Suzuki and K. Yamaoka, Sci. Rep., 2020, 10, 3558 CrossRef CAS PubMed.
- J. P. Gries and E. P. Rothrock, Manganese Deposits of the Lower Missouri Valley in South Dakota, University of South Dakota, South Dakota Geological Survey, 1941 Search PubMed.
- E. P. Rothrock, Missouri Valley Manganese Deposits between Lower Brule and , University of South Dakota, South Dakota Geological Survey, 1943 Search PubMed.
- F. D. De Vaney, F. D. Lamb and S. M. Shelton, Manganese Investigations: Metallurgical Division. No. 3., Ore-Dressing Studies of Manganese Ores: Concentration of Manganese Nodules from Chamberlain, S. Dak, U.S. Dept. of the Interior, Bureau of Mines, Washington, D.C., 1942 Search PubMed.
- S. R. Beeler, E. A. Rehwinkel and B. N. Carlson, Journal of Sustainable Metallurgy, 2024, 10, 2376–2383 CrossRef.
- V. Singh, T. Chakraborty and S. K. Tripathy, Miner. Process. Extr. Metall. Rev., 2020, 41, 417–438 CrossRef CAS.
- C. A. J. Appelo and D. Postma, Geochemistry, Groundwater and Pollution, CRC Press, 2nd edn, 2005 Search PubMed.
- D. L. Parkhurst and C. A. J. Appelo, Description of Input and Examples for PHREEQC Version 3: a Computer Program for Speciation, Batch-reaction, One-dimensional Transport, and Inverse Geochemical Calculations, Report 6-A43, Reston, VA, 2013 Search PubMed.
- W. F. McKenzie, T. J. Wolery, J. M. Delany, R. J. Silva, K. J. Jackson, W. L. Bourcier and D. O. Emerson, Geochemical Modeling (EQ3/6) Plan: Office of Civilian Radioactive Waste Management Program, 1986 Search PubMed.
- P. Bénézeth, J. L. Dandurand and J. C. Harrichoury, Chem. Geol., 2009, 265, 3–12 CrossRef.
- S. T. Mergelsberg, J. J. De Yoreo, Q. R. S. Miller, F. Marc Michel, R. N. Ulrich and P. M. Dove, Geochim. Cosmochim. Acta, 2020, 289, 196–206 CrossRef CAS.
- R. L. Frost, J. Cejka, G. A. Ayoko and M. J. Dickfos, J. Raman Spectrosc., 2008, 39, 374–379 CrossRef CAS.
- X. R. Bi, N. Yao, X. Meng, M. X. Gou and P. Q. Zhao, Catal. Lett., 2021, 151, 454–462 CrossRef CAS.
- H. Elderfield and G. P. Glasby, Chem. Geol., 1973, 11, 117–122 CrossRef CAS.
- A. P. L. Nunes, A. E. C. Peres, A. P. Chaues and W. R. Ferreira, J. Mater. Res. Technol., 2019, 8, 3623–3634 CrossRef CAS.
- B. J. Saikia, J. Mater. Phys. Chem., 2014, 2, 28–33 CAS.
- K. M. Parida, S. Mallick, B. K. Mohapatra and V. N. Misra, J. Colloid Interface Sci., 2004, 277, 48–54 CrossRef CAS PubMed.
- J. R. Hein, K. Mizell, A. Koschinsky and T. A. Conrad, Ore Geol. Rev., 2013, 51, 1–14 CrossRef.
- A. Koschinsky and P. Halbach, Geochim. Cosmochim. Acta, 1995, 59, 5113–5132 CrossRef CAS.
- X. Wang, U. Schlossmacher, M. Wiens, H. C. Schroder and W. E. Muller, Mar. Biotechnol., 2009, 11, 99–108 CrossRef CAS PubMed.
- G. P. Glasby, N. Z. J. Geol. Geophys., 1976, 19, 707–736 CrossRef CAS.
- W. S. James and J. Morgan, Aquatic Chemistry: Chemical Equilibria and Rates in Natural Waters, John Wiley & Sons, Inc., New York, 3rd edn, 1995 Search PubMed.
- B. Morgan and O. Lahav, Chemosphere, 2007, 68, 2080–2084 CrossRef CAS PubMed.
- H. Chen, Y. Fan, Z. Fan, H. Xu, D. Cui, C. Xue and W. Zhang, Ceram. Int., 2021, 47, 35057–35066 CrossRef CAS.
- T. Yamashita and P. Hayes, Appl. Surf. Sci., 2008, 254, 2441–2449 CrossRef CAS.
- T. Le, Y. Yang, L. Yu, Z.-h. Huang and F. Kang, Sci. Rep., 2016, 6, 37368 CrossRef CAS PubMed.
- S. W. Lee, J. Kim, S. Chen, P. T. Hammond and Y. Shao-Horn, ACS Nano, 2010, 4, 3889–3896 CrossRef CAS PubMed.
- Q. Li, J. Liu, J. Zou, A. Chunder, Y. Chen and L. Zhai, J. Power Sources, 2011, 196, 565–572 CrossRef CAS.
- F. M. John, F. S. William, E. S. Peter and D. B. Kenneth, Handbook of X-Ray Photoelectron Spectroscopy, Perkin-Elmer Corporation, 1992 Search PubMed.
- M. C. Militello and S. W. Gaarenstroom, Surf. Sci. Spectra, 2002, 8, 200–206 CrossRef.
- D. R. Baer and J. F. Moulder, Surf. Sci. Spectra, 1993, 2, 1–7 CrossRef CAS.
- A. A. Baba, L. Ibrahim, F. A. Adekola, R. B. Bale, M. K. Ghosh, A. R. Sheik, S. R. Pradhan, O. S. Ayanda and I. O. Folorunsho, J. Miner. Mater. Charact. Eng., 2014, 02, 230–247 CAS.
- N. S. Randhawa, J. Hait and R. K. Jana, Hydrometallurgy, 2016, 165, 166–181 CrossRef CAS.
- M. K. Sinha and W. Purcell, Hydrometallurgy, 2019, 187, 168–186 CrossRef CAS.
- P. P. E. Weaver and D. Billett, in Environmental Issues of Deep-Sea Mining: Impacts, Consequences and Policy Perspectives, ed. R. Sharma, Springer International Publishing, Cham, 2019, pp. 27–62 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4su00444b |
| This journal is © The Royal Society of Chemistry 2025 |