
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCold plasma activated CO2 desorption from calcium carbonate for carbon capture†
Hongtao
Zhong‡
 *a,
Daniel
Piriaei
a,
Gennaro
Liccardo
b,
Jieun
Kang
a,
Benjamin
Wang
a,
Matteo
Cargnello
b and
Mark A.
Cappelli
a
*a,
Daniel
Piriaei
a,
Gennaro
Liccardo
b,
Jieun
Kang
a,
Benjamin
Wang
a,
Matteo
Cargnello
b and
Mark A.
Cappelli
a
aStanford Plasma Physics Laboratory, Department of Mechanical Engineering, Stanford University, Stanford, CA 94305, USA. E-mail: hongtaoz@stanford.edu
bDepartment of Chemical Engineering, Stanford University, Stanford, CA 94305, USA
First published on 15th January 2025
Abstract
This work investigates the non-equilibrium regeneration of one scalable sorbent material for carbon capture, calcium oxide, in a customized flow reactor coupled to a low-temperature atmospheric-pressure plasma source. The results show that such a plasma is capable of desorbing CO2 from CaCO3, with an operating temperature far below the thermal decomposition temperature of carbonate. The desorbed CO2 is further converted to CO in situ. The energy cost is 1.90 × 103 kWh per tCO2, as the same order of magnitude as the state-of-the-art high temperature regeneration technology. A non-equilibrium kinetic mechanism is proposed in which CO2 desorption is coupled into air plasma chemistry. Electron-impact reactions in air lead to the generation of vibrationally excited nitrogen and ozone. Subsequent quenching of atomic oxygen on the carbonate surface can regenerate CaO, while NOx will pollute the surface. Compared with the previous methods used in sorbent regeneration, plasma-based technologies offer an electrified, sustainable, and low-temperature solution based on the non-equilibrium plasma chemistry. Possible scaling strategies include fluidization, flow pulsation, and plasma catalysis. This work demonstrates the feasibility of non-equilibrium plasma processing of the sorbent material for cyclic capture and regeneration in atmospheric air using thermally low-intensity processes.
Sustainability spotlightA sustainable energy future demands negative CO2 emissions. Current methods require significantly more energy for CO2 desorption and sorbent regeneration than the thermodynamic limit, making carbon capture costly and ineffective. With more renewable electricity available and the need for cost-effective, distributed solutions, non-equilibrium plasma generated by electric discharges is gaining interest for carbon capture and utilization (CCU). We demonstrate that atmospheric air plasma can desorb CO2 from CaCO3 at temperatures far below traditional thermal decomposition. Plasma-based technologies provide an electrified, sustainable, and low-temperature alternative for carbon capture, aligning with the UN SDGs: Affordable and Clean Energy (7), Industry, Innovation, and Infrastructure (9), and Climate Action (13). |
1 Introduction
Carbon capture from the point source or air using sorbent materials1–3 is critical to negative carbon emissions. The state-of-the-art carbon capture technology based on either high-temperature carbonate chemistry or low-temperature amine chemistry typically requires a centralized infrastructure with a high energy input (∼1800–2600 kWh per tCO2).4 The most energy-consuming process in carbon capture is the regeneration of the sorbent materials, consuming over 60% of the energy in one carbon capture cycle.With the increasing availability of renewable electricity and the imperative to decrease capital costs while delivering distributed capabilities, there has been a great interest in using non-equilibrium plasma generated by electric discharges for carbon capture and utilization (CCU) applications. Compared with other electrified techniques including electrochemical and Joule heating,5 plasma-based methods are more adaptable and consistent with the intermittency of renewable electricity, such as solar and wind power. Specifically, plasma reactors can be rapidly powered up and down within the second scale, allowing them to adapt quickly to fluctuations in the energy supply. Plasma reactors can be operated effectively at various power levels and flexible operating conditions, enabling the use of surplus renewable energy during peak production periods. In addition, direct use of electricity to generate plasma reduces the need for intermediate energy conversion steps, minimizing energy losses, and maximizing the effective use of intermittent renewable energy. Recent developments in homogeneous dielectric barrier discharges6 provide more opportunities for scalable plasma applications. In summary, the great adaptability ensures consistent and efficient operation, aligning well with the variable output of renewable energy sources.
However, to date, most studies have focused on applying plasmas for carbon utilization, i.e., converting CO2 into value-added chemicals.5,7–9 Limited studies10–13 discuss the possibility of plasma-assisted regeneration of sorbent materials. Compared with other electrified regeneration techniques of sorbent materials, for example, electrical swing adsorption (ESA), plasma-based regeneration could activate specific chemical bonds at low temperatures, exhibiting mostly kinetic effects instead of simply Joule heating or charging, thus exhibiting minimal thermal inertia and preventing the fast sintering of materials. Li et al. reported that the argon plasma generated from a dielectric barrier discharge (DBD) is capable of desorbing CO2 from a low-temperature sorbent material hydrotalcite. The simultaneous conversion from CO2 to CO was also observed during plasma treatment. In another study by Gorky et al., gentle pulses of helium plasmas were shown to induce a greater amount of desorption of CO2 compared to simple heating for the sorbent material MOF-177. Nevertheless, economically more feasible options, such as plasma discharges generated in ambient air, or the use of plasmas on sorbent materials produced at the gigaton scale,14,15e.g., metal oxide/carbonates, have not yet been discussed. Furthermore, the energy conversion efficiency reported for low temperature plasma-based approaches is several orders of magnitude lower than the traditional high temperature approach,10 and significant improvements are needed to make it a viable technology. Such advances are limited by our incomplete understanding of the interaction of the gas-phase species generated by the plasma while interacting with the active surfaces of the sorbent, which in turn stems from the dearth of advanced plasma reactor development and sensitive diagnostics that can be applied to those reactors.
In the present study, we have customized a surface dielectric barrier discharge (sDBD) reactor for plasma-based regeneration of the sorbent material and probed the composition of the gas and solid phase products. A kinetic mechanism is developed based on the experimental measurements, which provides insights for the chemical interaction between the sorbent surface and the plasma-generated species. The choice of CaO as a model sorbent for CO2 while CaCO3 for CO2 desorption is inspired by its relevance to carbon capture. The wide natural abundance and low cost make it suitable for scaling up. In the calcium loop cycle, the carbonation of CaO captures CO2. This is followed by the calcination of CaCO3 in order to regenerate calcium oxide and collect concentrated CO2 for sequestration or further chemical conversion. The practical use of CaCO3 in the classical adsorptive carbon capture, however, is limited by its lower surface area, slower kinetics, and higher energy requirements needed for high-temperature thermal decomposition. Plasma-based electrified technologies are expected to accelerate the kinetics, reduce the energy barrier, and ultimately improve the scalability of this sorbent material for carbon capture.
2 Experimental
2.1 Plasma setup
The experimental schematic is shown in Fig. 1. The sDBD source consists of a flat high-voltage electrode, a buried electrode, and a dielectric layer sandwiched between the two electrodes. The dielectric layer is an alumina ceramic sheet. The size of the high-voltage electrode is 10 cm × 15 cm, and the corresponding discharge area is 150 cm2. The dielectric thickness is 1.5 mm. The chamber is a 33 cm × 21 cm × 10 cm cube made of aluminum. A thermocouple and a pressure gauge are placed inside to measure the internal thermodynamic conditions. When connected to the supply, a DBD plasma “sheet” was generated at atmospheric pressure and moderate temperatures (below 50 °C) by a modulating alternating-current (AC) power supply that peaked at ±5 kV and a frequency of 20 kHz. | ||
| Fig. 1 Schematic of (a) the customized surface DBD plasma source, UV-vis absorption spectroscopy, (b) sampling of the gas-phase species from the plasma reactor, and the residual gas analyzer (RGA). | ||
The wall-plug power is adjusted by a voltage regulator (VEVOR) and monitored by a powermeter (Poniie PN1500). The energy deposited in the plasma is measured using the Lissajous method detailed elsewhere.16,17 In this work, the deposited energy can be varied from 5 to 50 W. A typical SDBD discharge current waveform consists of a sinusoidal displacement current component and superimposed sharp spikes with a pulse width of 10–50 ns corresponding to isolated discharge events that appear randomly on the surface of the plasma source.
We have used various process gases or combinations thereof (air, O2, Ar, N2), with flow controlled by calibrated mass flow controllers (MKS Instruments) operating in the range of 10 to 100 sccm. Calcium carbonate powders (lab grade, Innovating Science, approxiamte particle size 44 μm) are placed 2–10 mm beneath the plasma layer. For each experiment, an aluminum tray is used to load 10 grams of powder that spanned a surface area of 80 cm2.
2.2 In situ UV-vis absorption spectroscopy (UVAS) and optical emission spectroscopy (OES)
The optical access of the reactor allows UVAS as well as OES. Absorption and emission spectra were recorded over the wavelength (λ) range of 200–800 nm using the combination of a deuterium lamp (Oriel) and a spectrometer (Ocean Optics). The absorption path length was 33 cm. An integration time of 100 ms was used for each collected spectrum, and the time interval between spectra was varied from 300 ms to 2 min. This time resolution of the measurements was sufficient to record the chemical dynamics of ozone and nitrogen oxides (NO, NO2, NO3, N2O4, N2O5), which are long-lived chemical species (lifetime τ ≫ 1 s).It should be noted that other species such as H2O, CO2, N2O, and O2 were also possible species, but they were excluded from the calculation because of their small cross-sections (H2O of 10−25–26, CO2 of 10−25, N2O of 10−24, and O2 of 10−26 [cm2 per molecule]) at our target wavelengths. The absorbance was determined by the Beer–Lambert law as
 | (1) |
2.3 Residual gas analysis (RGA)
The reactor is connected to a residual gas analyzer (RGA, Stanford Research Systems) using a capillary tube located in the downstream wall of the reactor, opposite the wall where the reactant gas is introduced. The RGA was continuously operated to record time-dependent signals associated with CO2, CO, O2, and NO. The ion gauge (IG) signal at different atomic mass units (amu) is calibrated and converted to molar fractions. The peak CO2 RGA signals are in the range of 6 × 10−8–1.0 × 10−7, which correspond to the molar fraction of 2–4%. More details of the reactor system, species calibrations, flow conditions, optical diagnostics, measurement uncertainty, and the material preparation method are described in the ESI.†2.4 X-ray photoelectron spectroscopy (XPS)
The XPS measurements were performed in Stanford Nano Shared Facilities. The penetration depth of the XPS measurement is in the order of nanometers. Wide scans from 0 to 1400 eV have been performed to identify the elements in the sample. Narrow scans of carbon (278–292 eV), nitrogen (398–410 eV), oxygen (524–537 eV) and calcium (340–360 eV) peaks have been completed for the determination of the elemental composition.3 Zero-dimensional chemical kinetic model
Plasma-assisted CO2 desorption has vast time scale differences, from nanoseconds to hours. Our experimental configuration also has vast spatial scale differences, from the sheath length in micrometers to the reactor length in centimeters. Therefore, replicating the exact experimental results using zero-dimensional kinetic modeling with simplified kinetic schemes is inherently unrealistic. The goal of the chemical kinetic model is to identify the dominant kinetic mechanism and provide insights for the observed chemical dynamics in the air plasmas.Due to the above-mentioned scale disparity of the problem, it is intractable to resolve electron energy evolution and simulate plasma kinetics in a detailed fashion as reported previously.18 Instead, three global parameters (summarized in Table 1) are fitted to characterize the two major electron impact reactions.
| Index | Reaction | Fitting parameter |
|---|---|---|
| E-1 | e + O2 → e + O + O | n O |
| E-2 | e + N2 → e + N2(v) | T 0v, τv |
Three fitting parameters include the atomic oxygen number density (nO), the vibrational temperature at steady state (T0v), and the time scale of the vibrational temperature increase (τv). These fitting parameters are determined to reproduce the measured profiles of ozone and NOx density, from the UVAS measurements. The fitted values are reasonable and consistent with previous studies in similar conditions.19 The long time scale of fitted τv may be caused by the collisional radiation by excitation and de-excitation in the air plasma discharge. Then the number density of vibrationally excited nitrogen and the vibrational temperature are calculated as
 | (2) |
| Tv = Tg + T0v(1 − exp(−t/τv)) | (3) |
3.1 Gas-phase kinetics
For gas-phase kinetics, we developed a simple parameterized model with reactions listed in Table 2. The focus is on the evolution of the number density of key species, such as O3 and NOx in a well-mixed control volume without spatial gradient.The loss caused by diffusion and convection is considered in the model by adding the additional loss term with τdif = 40–200 s. It is obtained by measuring the decay rate of the NOx density after the plasma discharge is turned off.
The equations were solved with an ordinary differential equation solver ZDPlaskin.24Fig. 2(a and b) shows a comparison between the measured profile and the model output without the presence of calcium carbonate powders. The fitted parameters are nO = 1.4 × 1011 cm−3, T0v = 7500 K, and τv = 1.2 × 103 s. These parameters were uniquely determined to reproduce the measured ozone density: nO and T0v decide the slope of the increasing rate and the decay rate of the ozone density. The cross-over time was determined by τv. In this case, the cross-over time, the initial O3 number density, steady-state NO and NO2 number density are reasonably well predicted. An increase in NO and NO2 levels is predicted after O3 levels have decreased to low values, primarily due to simplifications in the kinetic scheme. Larger NOx species, such as N2O3, N2O4, and N2O5, are excluded from this scheme, resulting in the omission of pathways for the consumption of NO and NO2.
 | ||
| Fig. 2 A comparison between experimental measurements (a and c) and kinetic modelling (b and d) for the case with and without CaCO3 powders. Fitting parameters: nO = 1.4 × 1011 cm−3. T0v = 7500 K. τv = 1.2 × 103 s for (b). τv = 5.0 × 103 s for (d). | ||
3.2 Surface kinetics
We further develop a simplified and hypothesized surface kinetic mechanism involving atomic oxygen, surface calcium species ( , CaO, CaCO3), and NOx species to explain the CO2 formation in the presence of CaCO3 powders and air plasmas. Here, surface calcium peroxide
, CaO, CaCO3), and NOx species to explain the CO2 formation in the presence of CaCO3 powders and air plasmas. Here, surface calcium peroxide  is a hypothesized intermediate species.25 The simplified model considers the interaction between reactive oxygen and nitrogen species (RONS) and calcium carbonate powder. One example is shown in Fig. 2(c and d). As discussed in Section 4, this proposed model can reasonably predict the formation CO2 and the cross-over time. However, the NOx formation differs from the experimental measurements by almost an order of magnitude. Future detailed modeling is still needed (Table 3).
is a hypothesized intermediate species.25 The simplified model considers the interaction between reactive oxygen and nitrogen species (RONS) and calcium carbonate powder. One example is shown in Fig. 2(c and d). As discussed in Section 4, this proposed model can reasonably predict the formation CO2 and the cross-over time. However, the NOx formation differs from the experimental measurements by almost an order of magnitude. Future detailed modeling is still needed (Table 3).


4 Results and discussions
RGA measurements were recorded while the DBD reactor was operated in air over calcium carbonate (10.0 g) or α-Al2O3 powder (10.0 g), the latter serving as the control. Fig. 3 shows the RGA signals of the desorbed CO2 and other key species (O2, NO, N2/CO – note that N2 cannot be distinguished from CO). The CO2 concentration was at low levels (below 500 ppm) for all cases when the plasma was off. Treatment of alumina powders with air plasmas resulted in background CO2 signals only while treatment of calcium carbonate powders with air plasmas led to a significant increase in CO2 signals. The CO2 signal manifested within the initial 60 minutes, eventually reaching a saturation point. After calibration of the RGA signal, the quantitative concentration of CO2 was determined, increasing from 400 ppm to 4%, indicative of a notable production of CO2. The linear increase of CO2 signals indicates that CO2 desorption may have a pseudo-zero-order kinetic scheme, which may occur by reaction of gas-phase species on a solid surface. | ||
| Fig. 3 Time dynamics of (a) CO2, (b) O2, (c) NO, and (d) N2/CO in the gas phase detected by RGA during the plasma-assisted CO2 desorption. Data were obtained for loading CaCO3 particles or Al2O3 particles (10 g). Reaction conditions: 1 atm, 300 K, total flow rate 10 sccm. | ||
The RGA signal also reveals a strong coupling between CO2 desorption, air plasma chemistry, and treated powders. In the presence of Al2O3 powders, the O2 concentration remains relatively stable throughout the plasma operation, while the NO concentration experiences an abrupt increase after 25 minutes. In contrast, when CaCO3 powders are introduced, there is a pronounced and rapid decrease in O2 signals accompanied by a delayed onset in the rise of NO signals. The presence of calcium carbonates also promotes the RGA signals at amu = 28, attributed to the dissociation of CO2in situ, ultimately forming CO with a concentration of 4–6%.
We also performed experiments with CaCO3 powders in plasmas of inert gas such as argon, or empty loading under the action of an air plasma, resulting in a negligible CO2 yield, similar to the case of powder Al2O3 under air plasma treatment. The results further demonstrate that appropriate plasma species and material surface sites are indispensable for the plasma-assisted regeneration of sorbent materials. Our results are qualitatively consistent with those reported by Giammaria et al.,12 who found that a plasma consisting of inert argon only induced minimal thermal effects on the sorbent material.
It is well-established that the decomposition of calcium carbonates proceeds when heated to a high temperature28 with an activation energy of 100–200 kJ mol−1. However, with a low-temperature air plasma, CO2 desorption proceeds via a non-equilibrium chemical mechanism as the temperature is far below the temperature at which thermal decomposition begins (approximately ∼900 K).
UVAS was performed to probe species in situ to better understand this nonequilibrium decomposition mechanism. A typical absorption spectra is shown in Fig. 4(a). An abrupt transition in the shape of the absorbance spectra during the treatment was recorded. The absorbance in the range of 220–270 nm decreases by two orders of magnitude after firing the plasma for 51–53 minutes, while that in the range of 320–520 nm increases. This reflects a sudden change in the gas phase composition. By fitting the spectra using the known absorption cross-section data for species believed to be active in this region, the temporal evolution of the number density for different species O3, NO, and NO2 is extracted. These are shown in Fig. 4(b).
 | ||
| Fig. 4 (a) Absorption spectra over the O3–NOx transition. Each spectrum is presented with fitted synthetic curves obtained from the absorption cross-section data for O3, NO, NO2, NO3, N2O4, and N2O5. Plotting the absorbance on a logarithmic scale clearly separates different absorption strengths at different wavelengths. (b) The temporal evolution of the number density for O3, NO, and NO2 with and without CaCO3 powders loaded. (c) The corresponding CO2 signals detected by RGA with and without CaCO3 powders loaded. The shaded zone indicated the onset of the NOx poisoning. (d) Optical emission spectroscopy targeting the second positive band of molecular nitrogen (C3Πu → B3Πg) before and after the O3–NOx transition. | ||
In the absence of CaCO3 powders, the relatively high level of O3 concentration is significantly reduced after discharge was activated for 15 minutes. The estimated time for the decay of O3 is found to be much shorter than that associated with a first-order decomposition reaction, suggesting that thermal decomposition of O3 was not the dominant mechanism. In contrast, NO2 and NO were not noticeably produced immediately after plasma ignition, but a rapid increase is seen in their concentrations concomitant with the drop in O3. The NOx concentrations eventually level off at about the 20 minutes mark.
Such chemical mode transitions involving O3 and NOx have been discussed in previous studies.19,29 From the model developed in Section 3, ozone is rapidly formed following electron-impact O2 dissociation reaction (E-1 in Table 1) and the three-body recombination reaction O + O2 + M ↔ O3 + M (G-1 in Table 2). Due to the partial oxidation by O3, the concentration of NO and NO2 is maintained at a low level through subsequent reactions with this O3:
| O3 + NO → O2 + NO2 | (G-4) |
| O3 + NO2 → O2 + NO3 | (G-5) |
| e + N2 → e + N2(v) | (E-2) |
| O + N2(v) → NO + N | (G-3) |
With the introduction of CaCO3 powder, we find that the onset of this transition is delayed by approximately 40 minutes. Coinciding with this period of time leading to this transition is a surge in the CO2 signal detected by the RGA, as shown in Fig. 4(c). The saturation of the CO2 concentration coincides with this delayed O3–NOx mode transition.
In the following, we propose a mechanism to explain this observed chemical dynamics. Following the onset of discharge, the plasma preferentially dissociates O2 and generates O3 and O atoms in the N2–O2 mixture due to different dissociation energy thresholds. These reactive and radical species are then expected to undergo the following reactions:
| O3(g) + M → O2(g) + O(g) + M | (G-6) |
 | (S-1) |
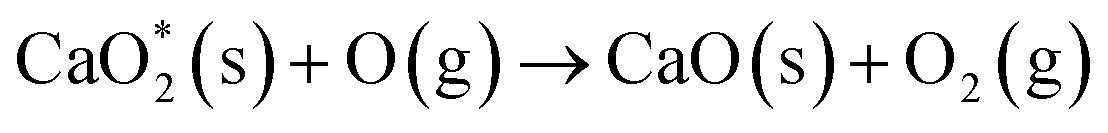 | (S-2) |
 is presumed to be a weakly-physisorbed unstable calcium peroxide complex. From the mechanism, in the presence of calcium carbonates, the oxygen atom is adsorbed by the carbonate surface, which delays the vibrational quenching reaction (G-3) to form NO. NO is a key intermediate for O3 consumption (G-4) and the NOx–O3 mode transition. As evidenced by Fig. 4(b), with the presence of calcium carbonates, NO formation is inhibited and the NOx–O3 mode transition is delayed. Future advanced diagnostics (such as Two-Photon Laser-Induced Fluorescence (TALIF) for gas-phase atomic oxygen measurements) are required to further validate this mechanism.
is presumed to be a weakly-physisorbed unstable calcium peroxide complex. From the mechanism, in the presence of calcium carbonates, the oxygen atom is adsorbed by the carbonate surface, which delays the vibrational quenching reaction (G-3) to form NO. NO is a key intermediate for O3 consumption (G-4) and the NOx–O3 mode transition. As evidenced by Fig. 4(b), with the presence of calcium carbonates, NO formation is inhibited and the NOx–O3 mode transition is delayed. Future advanced diagnostics (such as Two-Photon Laser-Induced Fluorescence (TALIF) for gas-phase atomic oxygen measurements) are required to further validate this mechanism.
Also after discharge onset, there is a gradual accumulation of vibrationally excited nitrogen (N2(v)). As shown in Fig. 4(d), the normalized emission spectra targeting the second positive band of molecular nitrogen (C3Πu → B3Πg) qualitatively show that the intensity of these (v′, v′′) vibrational lines change across this transition. We see that while the intensity of the nonexcited (0, 0) line at (337.1 nm) is largely unchanged, there is an increase in intensity of the (1, 2) line (353.7 nm) and a decrease in intensity of the (0, 1) (357.7 nm) and (0, 2) (380.5 nm) lines after the O3-to-NOx transition, indicating that more vibrationally excited nitrogen states are populated following this chemical transition. Now, NOx becomes important in chemical reactions. The kinetics of NOx species30,31 can be further coupled to reactions with carbonates and water molecules. Possible reaction pathways include:
| H2O(g) + 2NO2(g) → HNO3(g) + HONO(g) | (S-3) |
| 2HNO3(g) + CaCO3(s) → Ca(NO3)2(s) + H2O(g) + CO2(g) | (S-4) |
The adsorption of NOx/O3 onto the carbonate surface with subsequent catalytic chemical reactions at active sites affords a potential path to CO2 release, albeit through the production of relatively stable nitrated surface sites. Once the active sites on the oxide surface are occupied, the non-equilibrium chemistry switches from the gas–solid interface to the gas phase. The O3 to NOx mode transition in this case is delayed for ∼40 minutes. From the above-proposed mechanism, an oxygen plasma (diluted by argon) also has the capability of desorbing CO2 from the carbonate. This is confirmed by our experiments as shown in Fig. 5. However, the CO2 yield in the oxygen plasma case is lower and the corresponding energy cost is higher. A possible strategy to avoid this nitrogen catastrophe is to have an alternating flow with a pulsed discharge, in which air and oxygen plasmas are fired in sequence to eliminate NOx deposition on the carbonate surface.
 | ||
| Fig. 5 The comparison of RGA CO2 signals (amu = 44) under argon, 50% oxygen (balanced by Ar) and air plasmas with the presence of CaCO3 powders. | ||
The above proposed mechanism and discussion are largely focused on the measured gas-phase products. An equally important factor relevant to the interpretation and model development is the structure of the powder surface during treatment. X-ray photoelectron spectroscopy (XPS) was carried out on both untreated and treated samples under exposure to air and oxygen plasmas. The corresponding spectra for C1, N1, and O1 are shown in Fig. 6(a–c). The ratio of the area of those peaks to that of the Ca 2p peaks is provided in the ESI† for the comparison of mixture compositions. Due to limitations in the amount of phase change occurring in the experiment, we were unable to perform X-ray Diffraction (XRD) or Thermogravimetric Analysis (TGA) experiments for this study.
 | ||
| Fig. 6 (a) C 1s, (b) N 1s, (c) O 1s spectra of the powders under three different conditions: non-treated, air–plasma-treated and oxygen–plasma-treated. (d) The comparison between the measurement and the fitting for O 1s spectra treated by the air plasma. | ||
First, when compared with the non-plasma-treated case, a significant reduction of C 1s carbonate peak (Fig. 6(a)) is observed for the plasma-treated samples, confirming the desorption of CO2. Second, under air plasma treatment, the footprint of N 1s is clearly observed in the spectra in Fig. 6(b), confirming nitrogen deposition on the surface. Last but not least, there is a discernible variance in the O 1s peak for three cases, with plasma-processed samples exhibiting a pronounced shift. This shift indicates a compositional change induced by plasma treatment. Further analysis of the spectra using line shape fitting,32 as shown in Fig. 6(d), suggests that the surface includes the presence of both calcium oxide and calcium nitrate following plasma treatment. This further supports the proposed regeneration mechanism, that is, the potentially important role that molecular nitrogen plays in CO2 desorption.
The average power injected into the DBD plasma plate is measured to be 20 watts from the Lissajous plot. Taking into account the overlap area of the plasma discharge and the aluminum tray, the averaged power for CO2 desorption is ∼8 W. Considering the convective heat loss from DBD ceramic, as reported in the previous literature,33 we estimate that most of the energy input is lost in thermal energy and the actual energy in activating chemicals is ∼1 W.
The whole plasma reactor has a volume of 3397 cm3. Given that the maximum CO2 signal (4%) is achieved within 4000 s, while a considerable amount of CO2 (5.7%) is converted into CO in situ, the desorped CO2 mass from the powder is calculated as 0.58 g. Then the energy efficiency is 0.001 kW × 1.11 h/0.58 g CO2 = 1.90 × 103 kWh per tCO2, same order of magnitude compared to current state-of-the-art regeneration technologies.
This nonequilibrium process is diffusion-limited, primarily affecting only the first few monolayers of the calcium carbonate substrate because of the limited penetration depth of reactive species. To enhance scalability, it is crucial to implement active control mechanisms. This involves ensuring a homogeneous distribution and dynamic adjustment of the powder within the plasma, as well as using fluidized bed reactors34 to increase the contact area between the plasma and the powder and improve the mixing of the particles. Efficient catalyst for carbon capture and conversion, for example, dual functional material,35 can further increase the yield. These strategies will make plasma-based CO2 desorption and conversion more efficient, and eventually suitable for industrial-scale applications.
5 Conclusion
In this work, we prototype a flow reactor coupled to a customized surface dielectric barrier discharge (DBD) plasma source, specifically designed for investigating the chemistry of plasma-sorbent interactions. Employing air plasmas, CO2 desorption from CaCO3 powders is achieved at 40 °C, elevating concentrations from 400 ppm to a maximum of 4% within an hour-long time-frame. From residual gas analysis, UV-vis absorption spectroscopy, and X-ray photoelectron spectroscopy, the gas-phase and solid-phase species are strongly coupled. The CO2 desorption from the sorbent material is first facilitated by atomic oxygen generation from the air plasma. Then the mode transition from O3 to NOx interrupts this kinetic process. The NOx species are absorbed by the sorbent material to release CO2. Surface diagnostics further confirm that the solid products include calcium oxide and calcium nitrate. The energy cost is 1.90 × 103 kWh per tCO2, reaching the same order of magnitude compared to the existing state-of-the-art regeneration technologies. The use of low-temperature non-equilibrium plasma allows for CCUS technologies at temperatures significantly lower than those of traditional thermal technologies, robust to the intermittency of renewable energy sources. This can lead to substantial energy savings and make the process more economically viable and sustainable. By reducing the thermal intensity, this approach minimizes the carbon footprint associated with the carbon capture process itself, enables smaller-scale operations that can be distributed closer to emission sources, and reduces the need for extensive transportation and storage infrastructure. Overall, it aligns well with global goals for reducing greenhouse gas emissions and transitioning to cleaner energy systems. Further optimizations include flow pulsation, fluidized bed, and deploying novel catalytic material for carbon capture. By integrating a CO2-selective membrane downstream of the plasma reactor, it is possible to continuously filter and concentrate CO2 for sustainable carbon utilization.Data availability
The data supporting this article have been included as part of ESI.† The code for ZDPlaskin can be found at http://www.zdplaskin.laplace.univ-tlse.fr. The version of the code employed for this study is version 2.0a.Author contributions
HZ: conceptualization, methodology, investigation, writing – original draft, revision. DP: investigation (plasma sources). GL: investigation (XPS diagnostics). JK: investigation (UVAS diagnostics). BW: investigation (plasma sources, reactor design). MC: conceptualization, methodology, supervision. MA: conceptualization, methodology, writing – proofreading, supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Stanford Doerr School of Sustainability (Sustainability Accelerator 269523), and Stanford Woods Institute for the Environment (Grant 198188). G. L. and M. C. acknowledge support from the US Department of Energy, Office of Science, Office of Basic Energy Sciences, under contract no. DE-SC0022197. H. Z. acknowledges helpful discussions with Dr Xiaoyu Song at Columbia University on the line fitting of XPS spectra.References
- X. Shi, H. Xiao, H. Azarabadi, J. Song, X. Wu, X. Chen and K. S. Lackner, Angew. Chem., Int. Ed., 2020, 59, 6984–7006 CrossRef CAS PubMed.
- S. Wang, S. Yan, X. Ma and J. Gong, Energy Environ. Sci., 2011, 4, 3805–3819 RSC.
- S. Choi, J. H. Drese and C. W. Jones, ChemSusChem, 2009, 2, 796–854 CrossRef CAS.
- M. Fasihi, O. Efimova and C. Breyer, J. Clean. Prod., 2019, 224, 957–980 CrossRef CAS.
- R. Snoeckx and A. Bogaerts, Chem. Soc. Rev., 2017, 46, 5805–5863 RSC.
- X. Lu, Z. Fang, D. Dai, T. Shao, F. Liu, C. Zhang, D. Liu, L. Nie and C. Jiang, High Volt., 2023, 8, 1132–1150 CrossRef.
- S. Soldatov, G. Link, L. Silberer, C. M. Schmedt, E. Carbone, F. D'Isa, J. Jelonnek, R. Dittmeyer and A. Navarrete, ACS Energy Lett., 2020, 6, 124–130 CrossRef.
- E. Delikonstantis, M. Scapinello, V. Singh, H. Poelman, C. Montesano, L. M. Martini, P. Tosi, G. B. Marin, K. M. Van Geem and V. V. Galvita, et al. , ACS Energy Lett., 2022, 7, 1896–1902 CrossRef CAS.
- T. Yong, H. Zhong, E. Pannier, C. Laux and M. A. Cappelli, Plasma Sources Sci. Technol., 2023, 32, 115012 CrossRef.
- S. Li and F. Gallucci, Chem. Eng. J., 2022, 430, 132979 CrossRef CAS.
- S. Li, M. Ongis, G. Manzolini and F. Gallucci, Chem. Eng. J., 2021, 410, 128335 CrossRef CAS.
- G. Giammaria, G. Van Rooij and L. Lefferts, Catalysts, 2019, 9, 185 CrossRef.
- F. Gorky, A. Nambo and M. L. Carreon, J. CO2 Util., 2021, 51, 101642 CrossRef CAS.
- J. Blamey, E. Anthony, J. Wang and P. Fennell, Prog. Energy Combust. Sci., 2010, 36, 260–279 CrossRef CAS.
- M. T. Dunstan, F. Donat, A. H. Bork, C. P. Grey and C. R. Müller, Chem. Rev., 2021, 121, 12681–12745 CrossRef CAS.
- I. Biganzoli, R. Barni, A. Gurioli, R. Pertile and C. Riccardi, J. Phys.: Conf. Ser., 2014, 012039 CrossRef.
- R. Wang, Y. Yang, S. Chen, H. Jiang and P. Martin, IEEE Trans. Plasma Sci., 2021, 49, 2210–2216 CAS.
- Y. Sakiyama, D. B. Graves, H.-W. Chang, T. Shimizu and G. E. Morfill, J. Phys. D: Appl. Phys., 2012, 45, 425201 CrossRef.
- S. Park, W. Choe and C. Jo, Chem. Eng. J., 2018, 352, 1014–1021 CrossRef CAS.
- H. Hippler, R. Rahn and J. Troe, J. Chem. Phys., 1990, 93, 6560–6569 CrossRef CAS.
- R. Atkinson, D. Baulch, R. Cox, R. Hampson Jr, J. Kerr, M. Rossi and J. Troe, J. Phys. Chem. Ref. Data, 1997, 26, 1329–1499 CrossRef CAS.
- R. Atkinson, D. Baulch, R. Cox, J. Crowley, R. Hampson, R. Hynes, M. Jenkin, M. Rossi and J. Troe, Atmos. Chem. Phys., 2007, 7, 981–1191 CrossRef CAS.
- M. Capitelli, C. M. Ferreira, B. F. Gordiets and A. I. Osipov, Plasma Kinetics in Atmospheric Gases, Springer Science & Business Media, 2013, vol. 31 Search PubMed.
- S. Pancheshnyi, B. Eismann, G. Hagelaar and L. Pitchford, Bull. Am. Phys. Soc., 2008, 53, MWP1.00060 Search PubMed.
- G. Deffrennes, N. Jakse, C. M. Alvares, I. Nuta, A. Pasturel, A. Khvan and A. Pisch, Calphad, 2020, 69, 101764 CrossRef CAS.
- S. L. Broadley and J. M. Plane, Phys. Chem. Chem. Phys., 2010, 12, 9094–9106 RSC.
- R. Zhu, K.-Y. Lai and M.-C. Lin, J. Phys. Chem., 2012, 116, 4466–4472 CrossRef CAS PubMed.
- B. V. L'vov, Thermochim. Acta, 2002, 386, 1–16 CrossRef.
- T. Shimizu, Y. Sakiyama, D. B. Graves, J. L. Zimmermann and G. E. Morfill, New J. Phys., 2012, 14, 103028 CrossRef.
- H. Li, T. Zhu, D. Zhao, Z. Zhang and Z. Chen, Atmos. Chem. Phys., 2010, 10, 463–474 CrossRef CAS.
- X. Zhao, M. A. Simioni, K. H. Smith, S. E. Kentish, W. Fei and G. W. Stevens, Energy Fuel., 2009, 23, 4768–4773 CrossRef CAS.
- N. Fairley, V. Fernandez, M. Richard-Plouet, C. Guillot-Deudon, J. Walton, E. Smith, D. Flahaut, M. Greiner, M. Biesinger and S. Tougaard, et al. , Appl. Surf. Sci. Adv., 2021, 5, 100112 CrossRef.
- F. Rodrigues, J. Pascoa and M. Trancossi, Exp. Therm. Fluid Sci., 2018, 90, 55–65 CrossRef.
- H. Zhang, Q. Tan, Q. Huang, K. Wang, X. Tu, X. Zhao, C. Wu, J. Yan and X. Li, ACS Sustain. Chem. Eng., 2022, 10, 7712–7725 CrossRef CAS.
- A. C. Faria, R. Trujillano, V. Rives, C. Miguel, A. Rodrigues and L. M. Madeira, J. CO2 Util., 2023, 72, 102476 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4su00491d |
| ‡ Current address: Department of Mechanical Engineering, Michigan State University, East Lansing, MI 48824, US. |
| This journal is © The Royal Society of Chemistry 2025 |