
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTropospheric methane remediation by enhancing chlorine sinks
Qingchun
Yuan
 *a,
Bo
Xiao
b,
Renaud
de Richter
*c,
Wei
Li
d,
Raul
Quesada-Cabrera
e and
Tingzhen
Ming
f
*a,
Bo
Xiao
b,
Renaud
de Richter
*c,
Wei
Li
d,
Raul
Quesada-Cabrera
e and
Tingzhen
Ming
f
aChemical Engineering and Applied Chemistry, Aston University, Birmingham, B4 7ET, UK. E-mail: q.yuan@aston.ac.uk
bSchool of Chemistry and Chemical Engineering, Queen's University Belfast, Belfast, BT9 5AG, UK. E-mail: b.xiao@qub.ac.uk
cTour-Solaire.fr, 8 Impasse des Papillons, Montpellier, 34090, France. E-mail: renaud.derichter@gmail.com
dInstitute for Materials and Processes, School of Engineering, University of Edinburgh, Edinburgh, EH9 3FB, UK. E-mail: wei.li@ed.ac.uk
eDepartment of Chemistry, Institute of Environmental Studies and Natural Resources (iUNAT), University of Las Palmas de Gran Canaria, Campus de Tafira, Las Palmas, 35017, Spain. E-mail: raul.quesada@ulpgc.es
fSchool of Civil Engineering and Architecture, Wuhan University of Technology, Wuhan, 430070, PR China. E-mail: tzming@whut.edu.cn
First published on 12th February 2025
Abstract
To tackle global warming, the Paris Agreement (2015) strategically proposed achieving net-zero emissions of greenhouse gases (GHGs) by 2050 and limiting the global temperature rise below 2 °C. This requires a substantial reduction of all GHG emissions across all sectors over the next few decades. Methane has come into the spotlight as the second most potent GHG for its contribution to global warming. The Global Methane Pledge announced at COP26 (2021) proposed to reduce 30% of anthropogenic methane emissions by 2030 compared to the 2020 level. However, studies show that methane emissions will continue to increase even with the planned reductions and therefore the atmospheric methane concentration also. Effective methane removal technologies are urgently required for atmospheric methane remediation. This work evaluates the feasibility of atmospheric methane removal by enhancing the chlorine atom sink (i.e. a natural sink of methane in the lower troposphere) at a significant scale, considering that atomic chlorine initiates methane oxidation 16 times faster than the major natural methane sink of hydroxyl radicals in the atmosphere. Atomic chlorine is proposed to be generated by electrolysis of brine for chlorine gas followed by photolysis. This methane removal technology could be integrated with the state-of-the-art industrial chlor-alkali processes. Such integrated technology is evaluated for the potential of negative GHG emissions and their costs, with attention given to cost-efficient measures, i.e., the use of alternative renewable sources. A brief discussion is included on potential risks, side effects, benefits to the atmospheric methane remediation by 2050 and key required developments.
Sustainability spotlightBy removing atmospheric methane at 2 ppm, and also when applied to point sources by reducing new methane emissions, the technology proposed can slow down global warming, both by remediation and mitigation (SDG13), helping to keep alive the 2015 Paris Agreement goal of limiting global warming well below 2.0 °C, in order to permit better life below water (SDG 14), life on land (SDG 15), and good health and well-being (SDG 3) , and reduce inequalities (SDG 10) as it is proved that global warming will hit the poorest the hardest, and climate change risks both increasing existing economic inequalities and causing people to fall into poverty (SDG 1). |
1. Introduction
Restraining the global temperature rise below 2.0 °C, as set out in the Paris Agreement (2015) and reaffirmed in 2021 during the 26th Conference of Parties (COP) in the Glasgow Climate Pact, aims to maintain CO2 concentrations in the atmosphere below 410 ppm and a total CO2-eq concentration below 450 ppm.1 Nevertheless, achieving these targets is already challenging as these thresholds have been reached in recent years (e.g. 420 ppm CO2 in January 2024 and 523 ppm CO2-eq in 2022).2 According to the latest report from the Intergovernmental Panel on Climate Change (IPCC), the global warming in 2010–2019 relative to 1850–1900 (the pre-industrial era) due to CO2 emissions is about 0.75 °C, and about 0.5 °C due to methane emissions (CH4) (Fig. 1a).3,4 The temperature rise caused by the well-mixed GHGs reached 1.5 °C (Fig. 1b).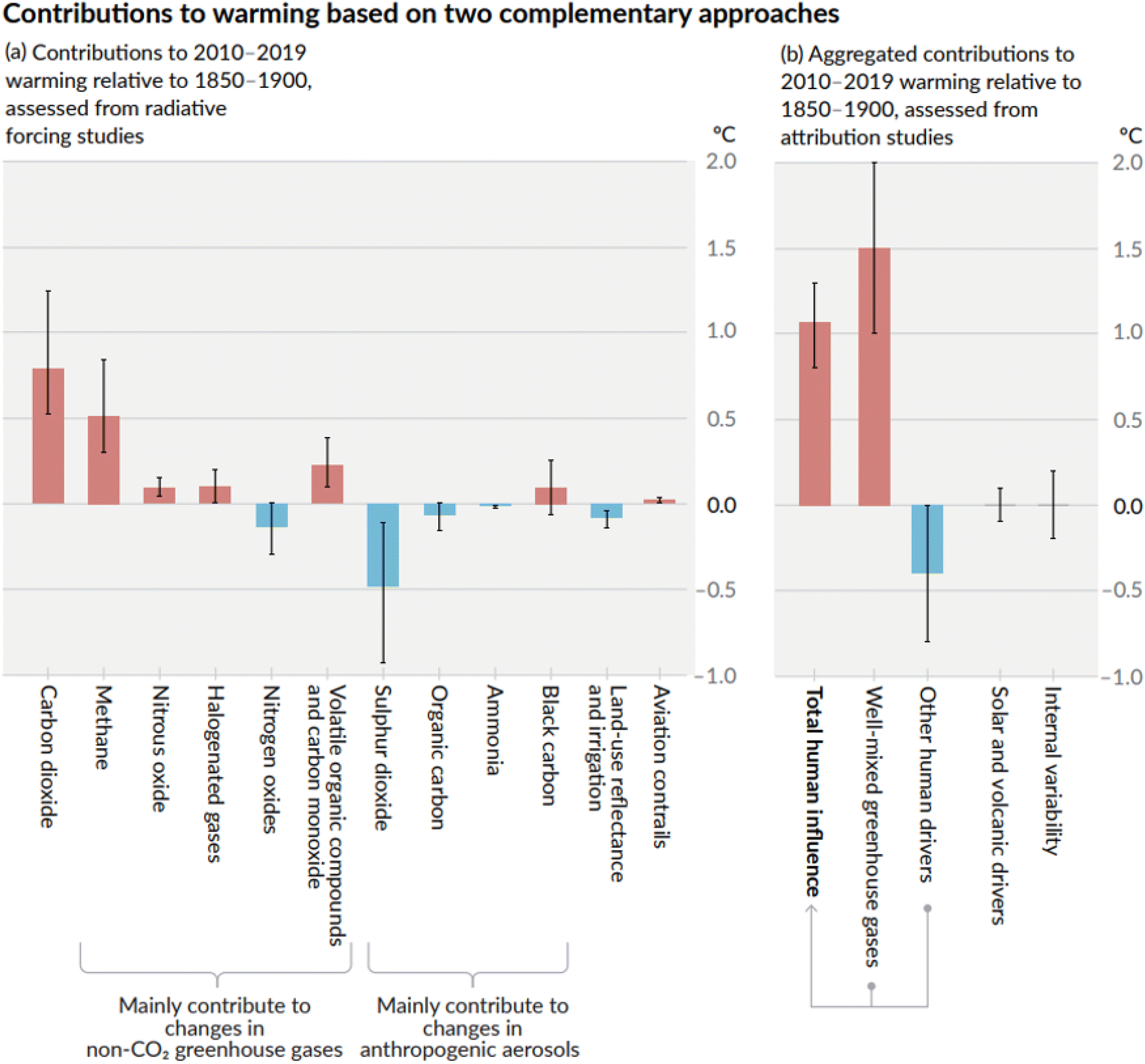 | ||
| Fig. 1 Comparison of the effect of climate forcers on global warming as assessed by the IPCC AR6 report (2021),3,4 showing the significant contributions of carbon dioxide and methane to global warming. | ||
Despite the world-wide progress and further developments in preventing additional anthropogenic GHGs from reaching the atmosphere,4 GHG remediation technologies for removing GHGs already in the atmosphere are urgently required to achieve the net-zero emission goal by 2050. Atmospheric remediation such as direct air capture (DAC) technologies have been under fast development to capture the ∼420 ppm atmospheric CO2 directly, but the process efficiency is still limited, and costs are very high.5
The atmospheric methane concentration was approximately ∼715 ppb in the pre-industrial era,6 and increased to ca. 1900 ppm in 2021.7 This concentration is not high compared with that of CO2 in the atmosphere, but the global warming potency of methane is 84 times that of CO2 over 20 years and 27–35 times over 100 years.8 The relatively short lifespan and high warming potency of atmospheric methane facilitates the development of methane removal technologies for faster warming control compared to CO2 capture and sequestration, especially over a relatively short time of 20 years.9
The natural remediation of atmospheric methane proceeds by its oxidation into CO2 and relies mainly on three types of natural sinks to initiate the oxidation, namely ∼90% by hydroxyl radicals (˙OH), ∼1–4% by chlorine radicals (Cl˙, Cl2˙−) and ∼6–9% by plants, forests, soils, minerals and dust, mainly due to their microorganism content.10 Oxidizing methane into CO2 significantly reduces the overall radiative forcing despite producing a small amount of CO2. Unfortunately, currently these natural sinks cannot deal with all anthropogenic methane emissions. The total annual emission and natural remediation of methane are estimated at ca. 576 and 538 Tg per years, respectively.10 The balance between sources and sinks represents an increase of atmospheric methane at ca. 38 Tg CH4 per years. This explains that the atmospheric methane concentration is seeing a significant net increase year by year. The present stock of methane in the atmosphere is estimated at about 5600 Tg.10
Methane oxidation to CO2 in the atmosphere occurs through a series of radical reactions: CH4 → CH3˙ → CH3OO˙ → H2CO˙ → CO → CO2, of which the first step is the slowest one in the radical propagation reactions that follow.11 Atomic chlorine (i.e. the chlorine radical Cl˙) can initiate methane oxidation reactions 16 times faster than hydroxyl radicals.11 Furthermore, generating atomic Cl needs less energy than generating ˙OH.12 These promising scientific factors encouraged us to develop a new energy-efficient methane removal technology based on promoting the Cl radical sink for large-scale atmospheric methane remediation purposes.13
The current work explores the potential large-scale methane remediation upon the generation of Cl atoms from sea salt (sodium chloride, NaCl) or brine, via a combined electrolysis and photolysis process. It covers the removal of atmospheric methane (at ca. 1900 ppb) as well as relatively high concentration methane at point sources, e.g. coal mines in semi-closed ventilation systems. Deployment scenarios and critical research are discussed, together with technical cost assessments and cost-effective safe operation.
2. Methods proposed for atmospheric methane removal
To the best of our knowledge, four methods have been proposed for the remediation of methane already in the atmosphere so far: thermal- and/or photo-catalytic oxidation,14–17 spraying iron-based salt aerosols in the lower troposphere to generate chlorine radicals,18,19 and also enhancing the population of hydroxyl radicals.122.1 Thermal catalytic oxidation of methane
Methane has a lower flammability limit of 4.4% in air. With a concentration lower than this and higher than 2000 ppm, methane oxidation can be achieved in regenerative thermal oxidisers at a temperature of 800–900 °C.20 The oxidation of low concentration methane in the atmosphere needs even higher temperatures. Solid catalysts prepared from zeolites and metal organic frameworks can lower the oxidation temperature.15 Inexpensive and abundant zeolite that is prepared from clay and doped with Cu catalysed the oxidation of atmospheric and low-level methane at relatively low temperatures of 200–300 °C.15,17Studies show that thermal catalytic removal of methane in the concentration range of 1.9 ppm to 1000 ppm can be excessively energy intensive larger than >100 GJ per tonneCO2eq.21 For atmospheric methane removal, the energy as well as cost penalty to move the air passing through solid catalysts will be rather high.22 To limit the penalty, the following methods can be deployed: (1) in conjunction with large air-flow infrastructures built for other purposes, such as solar chimney power plants for CO2-free renewable electricity generation, or direct air CO2-capture (DAC), which are still under development; (2) with low cost-effective catalysts to operate at a temperature ideally close to ambient temperatures.
2.2 Direct photocatalytic oxidation of methane
The direct photocatalytic oxidation of methane on metal-oxide semiconductor surfaces has been widely reported.23 Metal-oxide semiconductors as photocatalysts can generate hydroxyl radicals under sunlight irradiation, particularly upon absorption of UV photons. The engineering of efficient photocatalysts is still challenging due to their typically poor photon absorption and high charge recombination, which affects their overall efficiency. Nevertheless, photocatalysis of metal-oxide semiconductors is still promising for oxidizing atmospheric methane under ambient conditions. The photocatalytic activity of zinc oxide increases when the particle size is reduced down to the nanoscale, and when further enhanced by nano-silver decoration, showed an impressive quantum yield of 8% at wavelengths <400 nm.24 The test of such a catalyst in an optimally configured continuous reaction system at 25 °C demonstrated the feasibility,25e.g., attaching the photocatalysts to the inner part of the greenhouse glass ceiling of solar chimney power plants as proposed in ref. 14. A highly dispersed CuOx decorated ZnO photocatalyst showed similar catalytic activity.17 More research is still required to develop effective catalysts to improve processing efficiency.2.3 Enhancing the hydroxyl radical sink
Typically, ∼90% of the tropospheric methane is oxidized by hydroxyl radicals naturally generated in the atmosphere. Hydroxyl radicals can be artificially generated in the gas-phase through photolysis of ambient water vapor by ultraviolet (UV) irradiation at 185 nm.12,26 One mole of water vapour can be dissociated into two moles of hydroxyl radicals in the presence of oxygen in the air. With the cost reduction of UV Light-Emitting Diodes (LEDs) in recent years, UV light-based hydroxyl radical generators are commercially available as purifiers for disinfection purposes of air and surfaces.27,28 Recently, a perspective paper proposed several methods to enhance the amounts of hydroxyl radicals directly in the troposphere,12 but no cost estimations were provided by the authors as these methods are at a very early stage of development. Meanwhile, a very recent study suggests that to remove 1 Tg CH4 per year, 8.8 Tg per year of hydroxyl radicals is required with precursors such as ozone and hydrogen peroxide.292.4 Spraying of iron-aerosols in the lower troposphere
Several iron-containing particles have shown photocatalytic ability in converting sodium chloride to atomic Cl under sunlight.30 The photocatalytic cycle between Fe(II) and Fe(III) species is believed to facilitate the redox reaction of NaCl aerosols of sea brine at pH below 3 directly to atomic Cl.30,31 Combined experimental, quantum chemical, and chemical equilibrium model studies demonstrate the photocatalytic chloride-to-chlorine conversion by ionic iron in aqueous aerosols.32In a 3.0 m3 smog chamber, the turnover frequency of the active catalytic site reached ∼78 h−1, (i.e. nearly 78 Cl atoms generated per atom of Fe in an hour).31 Some authors thus proposed spraying iron-salt aerosols in the lower troposphere under the marine boundary layer to sink tropospheric methane,33 and suggested that to remove 1 Tg CH4 per year, 4.8 Tg per year of Cl˙ is required.29 The spray of FeCl3 can be achieved by using ships, balloons, towers or existing infrastructure such as container vessels.18,19 From naturally formed FeCl3 from desert dust, sea salt spray and acidity, a recent study suggests that 3.8 Tg(Cl) per year has been produced over the North Atlantic, which has participated in the oxidation of CH4 in the troposphere.34
It has also been demonstrated that in regions with high NOx pollution, an enhancement of the Cl chemistry occurs, which enhances the atmospheric oxidation capacity and the elevation of O3.35 But in pristine areas with low NOx levels the Cl chemistry destroys O3 and therefore reduces the amount of OH and increases the lifetime of CH4.34 Therefore, when generating Cl atoms by FeCl3, there is a threshold in the amount of iron that must be added to remove methane or below this threshold the CH4 will increase instead.36 Simulation shows that 630, 1250, and 1880 Tg(Cl) per year to remove 20%, 45%, or 70% global methane by 2050 can respectively decrease the surface temperature by 0.2, 0.4, and 0.6 °C.37
3. Promoting chlorine sinks for atmospheric methane removal
As mentioned earlier, the natural sink based on hydroxyl radicals (˙OH) typically contributes to 90% of atmospheric methane removal and the natural chlorine radicals sink only ∼1 to 4%.38 However, the tropospheric Cl concentrations are uncertain by ∼2 orders of magnitude,39 and the apparent kinetic isotope effect of the methane atmospheric sink has shown a magnitude larger in the extratropical Southern Hemisphere than expected if the sink were the hydroxyl radical alone without the inclusion of Cl radicals.40 Modelling studies show that in coastal areas (i.e. marine boundary layer), the chlorine radical sink can account for over 20% of methane removal due to enhanced Cl˙ generation of sea-salt aerosol dichlorination.39,40 This suggests the potential in terms of health and safety to enhance the chlorine sink in the air for removing atmospheric methane at a large scale.3.1 Natural generation of atomic chlorine
Atomic Cl is mainly generated from the photolysis of Cl2 gas and chlorine-containing species including Cl− (natural or anthropogenic), with a concentration of up to 0.04 ppt (106 atoms per cm3).41 There are some 23 Tg of molecular chlorine gas (Cl2) in the atmosphere or about 4 ppt (parts per trillion in the troposphere).42 The lifetime of molecular chlorine in the atmosphere is ca. 7 minutes at noon near the equator.43 If the concentration of Cl2 molecules is sufficiently high, their photolysis can provide an important source of atomic Cl in the lower atmosphere. For a given 0.1 ppb Cl2 concentration in polluted areas where nitrous oxides (NOx) exists, for example, Cl radical formation rates can be as high as 0.016 ppt s−1 (4 × 105 Cl radicals cm−3 s−1). These figures are significant considering the average Cl2 concentrations in coastal areas; for instance, the average concentration in coastal areas in Hong Kong was estimated at ca. 400 ppt, with peaks as high as 1000 ppt.43The release of active chlorine from marine aerosols is widely known and involves dissolved-gas species in aqueous droplets and at the interface between two phases.44 The formation of Cl2 gas across the interface of a bulk aqueous NaCl solution (in clouds or fog for instance) was demonstrated upon the reaction of gas phase ozone as well as hydroxyl radicals (eqn (1) and (2)).45 The process includes acid displacement and reactions of dinitrogen pentoxide (N2O5), ozone (O3) and other species with Cl− containing aerosols (eqn (1)–(6)). The formation of atomic Cl has been demonstrated in NaCI aerosols in the presence of NOx and O3 gases.46 The photolytic precursor of atomic Cl was identified to be nitryl chloride (ClNO2), which is formed in the dark upon the reaction of N2O5 and NaCI (eqn (3)). Atmospheric measurements have shown that ClNO2 is produced via heterogeneous reactions on sea-salt particles at night, resulting in a peak of atomic Cl in the early morning with an estimated formation rate of 0.04 ppt s−1 (1 × 106 atom cm−3 s−1) upon its photolysis under sunlight.47 This atomic Cl source may represent a major oxidant of the troposphere in industrialized coastal areas.38–40
| O3(g) + 2NaCl(aq) → Cl2(g) + products | (1) |
| ˙OH(g) + NaCl(aq) → 1/2Cl2(g) + NaOH(aq) | (2) |
| N2O5(g) + NaCl(aq) → ClNO2(g) + NaNO3(aq) | (3) |
| ClNO2(g) + NaCl(aq) → Cl2(g) + NaNO3(aq) | (4) |
| HOCl(g) + NaCl(aq) → Cl2(g) + NaOH(aq) | (5) |
| ClONO2(g) + NaCl(aq) → Cl2(g) + NaNO3(aq) | (6) |
3.2 Methods for the artificial generation of atomic chlorine
The standard dissociation energy of Cl2 gas is 243.6 kJ mol−1 for atomic chlorine and related radicals. The dissociation of Cl2 gas can be artificially obtained by thermal decomposition , or via UV photolysis using appropriate light sources.48The thermal dissociation needs a high temperature. At 1500 °C the dissociation of Cl2 reaches 85% when the pressure is 17 torr and 95% when the pressure is 5 torr.49 Because of the very high activity of atomic chlorine at such high temperatures, inert reactors such as graphite ones are required. Moreover, such a process consumes more energy than the dissociation required to maintain the high temperature, resulting in quite low energy efficiency and therefore this method is not desirable for the purpose pursued.
The dissociation energy of Cl2 gas can be provided by light radiation and the dissociation can start in the visible region from 491 nm under ambient conditions, but is more efficient in the UV region of 330 nm.50 The UV photolysis of gaseous Cl2 can proceed rapidly with a quantum yield close to unity.51 The UV wavelengths of interest can be generated by traditional actinic lamps or by UV LEDs. Actinic lamps such as xenon and mercury ones form an arc discharge under high-frequency and high-voltage excitation. The xenon lamp radiates a continuous spectrum in a wide range of 250–2500 nm (Fig. 2a).
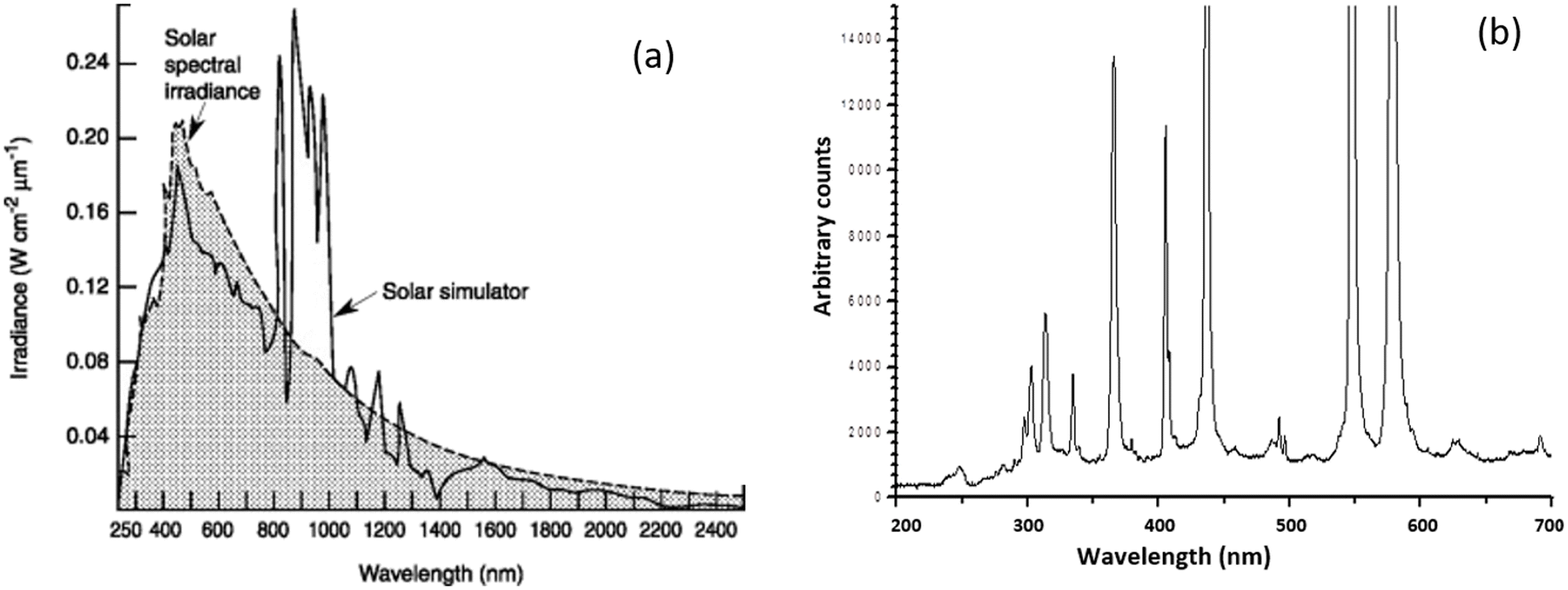 | ||
| Fig. 2 The light spectra of (a) xenon light lamps compared with that of solar spectral irradiance;52 and (b) mercury lamps.53 | ||
The spectrum distribution in the UV range of interest shadows that of the solar spectral irradiance, accounting for only a small fraction of the total irradiation. The mercury lamp radiates a powerful and stable UV, visible and IR spectrum with high energy density at several specified wavelengths as shown in Fig. 2b. Like xenon lamps, the irradiation in the wavelength range of interest only accounts for a small fraction. As point sources of light, these actinic lamps offer high luminance and radiance output for continuous operation, but they are not the best choice for the purpose pursued due to their too wide spectrum, high costs, short lifetime, and thermal runaway.
UV LED lights are distinctively characterised by monochromaticity and high energy efficiency at specified wavelengths, which cannot be matched by other light sources. UV light irradiation in the wavelength range of 300–350 nm has been effectively applied to generate atomic Cl in aqueous solutions and gas phases, as well as under aerobic or anaerobic conditions.54 Compared with other light sources with the same light flux, LED energy consumption is reduced by 80%.53
When it comes to the photolysis of Cl2 gas, the choice between UV LED lamps and traditional actinic UV lamps can have a significant impact on the process efficiency and effectiveness. Factors to consider include wavelength, energy efficiency (cost and energy consumption), lifetime, engineering control and maintenance as well as health and safety on top of all these factors.
Traditional UV lamps typically emit a wide spectrum including UV-C radiation, which has a shorter wavelength (lower than 254 nm) compared to most UV LED lamps. UV-C radiation is known for its germicidal properties and can break down certain chemical bonds. UV LED lamps, on the other hand, can be easily adjusted to emit a specific wavelength and intensity, allowing for more precise targeting of a desired wavelength, which will be 3–4 times more efficient than traditional UV lamps in terms of energy consumption. Due to the less energy damped as heat, the UV LED bulbs can be arranged in arrays for engineering applications. UV LED lamps typically have a significantly longer lifespan than traditional UV lamps, which reduces periodic bulb replacement for less downtime during operation.
In summary, UV LED lamps can be a suitable choice for the photolysis of Cl2 gas for GHG remediation. In practice, UV LEDs can be powered by solar or wind energy and can be flexibly combined with natural light for this conversion.
4. Industrial synthesis of chlorine gas through sodium chloride electrolysis
Most industrial chlorine is produced by the chlor-alkali process, a process in which an electric current is passed through a saturated NaCl aqueous solution with sodium hydroxide (NaOH) and hydrogen gas (H2) as co-products. The global annual production of Cl2 by the chlor-alkali industry in 2021 was about 90 Mt.55 It was about 77 Mt in 201256 and is expected to reach 97 Mt by 2026.55 There are three variant technologies of the chlor-alkali process, respectively known as mercury, diaphragm and membrane methods (Fig. 3a–c). Table 1 summarises the key operational features of the three processes.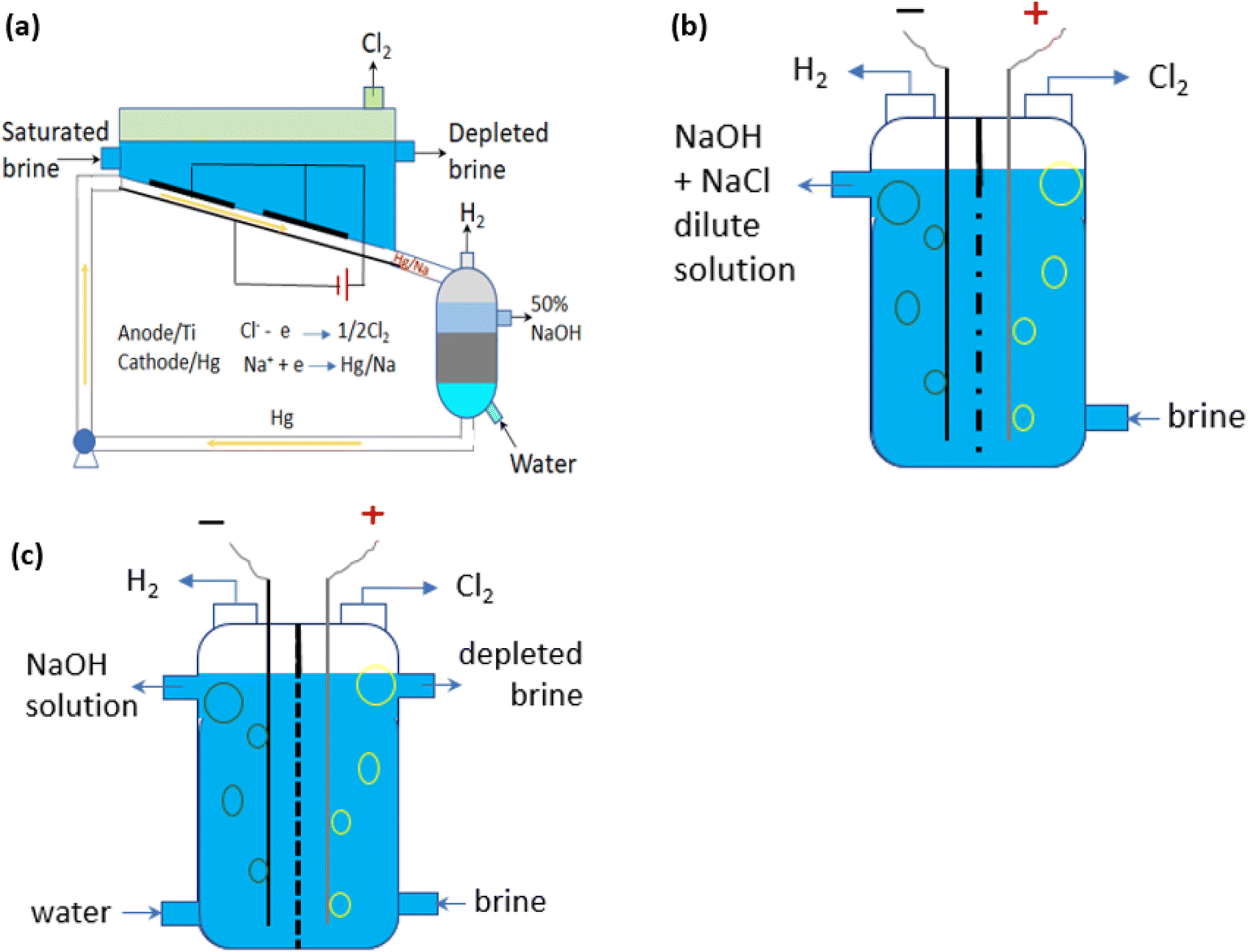 | ||
| Fig. 3 Schematic methods for the industrial chlor-alkali process to produce Cl2: (a) mercury-based; (b) diaphragm; and (c) membrane methods. Adapted from.57 | ||
| Feed | Voltage V(V) | Energy cons. E (kW h per tonne Cl2) | Efficiency Eef (%) | Drawbacks | |
|---|---|---|---|---|---|
| Mercury | Saturated NaCl | >3.25 | 3100 | 56.2 | Toxic emissions of Hg |
| Diaphragm | Saturated NaCl | >2.31 | 2300 | 75.8 | Low con. of NaOH, use of asbestos |
| Membrane | Ultra-pure saturated NaCl | >2.31 | 2010–2025 | 86.7 | Ultra-pure feed needed |
The mercury method is the least energy-efficient.58 It poses severe environmental and health risks due to the emission of highly toxic mercury,59 and therefore this type of plant has been phased out in the EU.60 The diaphragm process operates at a lower voltage compared to the mercury method;61 however, the dilute alkali by-product at ca. 12% requires a large amount of steam to bring it into the commercial concentration of 50%. In the diaphragm process (Fig. 3b), an asbestos (or polymer-fibre) diaphragm separates the cathode and anode, preventing the Cl2 generated at the anode from re-mixing with the NaOH and H2 formed at the cathode.62 The use of asbestos represents the other main drawback in terms of health and safety aspects and the diaphragm method represents only about 10% of the installed capacity in Europe. The membrane method (Fig. 3c) is currently the most energy efficient and has largely replaced the other two processes in the last decade, reaching 85% of Cl2 production in Europe in 2020.63 In the membrane electrolysis cell, a permeable membrane acting as a Na+-cation exchanger separates the two electrodes. To avoid membrane fouling or blockage for a long life and low-maintenance operation, the saturated NaCl aqueous solution needs to be filtered as an ultra-pure solution before being passed through the anode compartment.64 A diluted NaOH solution circulates through the cathode compartment, exiting in a concentrated solution. A fraction of the concentrated NaOH solution leaving the cell is diverted as a by-product at a specified concentration in the range of 30–35 wt%65 while the remainder of the solution is diluted again with deionized water and recycled back into the cell.
5. Proposed deployment of artificial Cl sinks
5.1 Proposed deployment methods
A two-step operation is proposed to enhance the atomic Cl sink for atmospheric methane removal at point sources as well as in the troposphere: the electrolysis of aqueous NaCl followed by the photolysis of Cl2 gas by using UV LED lights.At point sources, where methane is at much higher concentrations than the tropospheric one, such as landfills (where methane concentrations can be up to 45 to 60% but with low and discontinuous airflows66) and ventilation exits of underground coal mines (with typically less than 5% methane, as the dilution by ventilation prevents the explosive safety hazard to miners67), small devices could be designed and locally installed to generate chlorine radicals at a safe formation rate to meet the requirement. In this case, the Cl species may be released into a semi-closed system, such as the downstream of existing ventilation systems without reducing the exhaust flow rate or affecting the emergency escape. The chlorinated species and gas-phase by-products can be captured or neutralised using low-pressure drop alkaline filters or activated carbon, thus avoiding their release into the atmosphere. The unreacted Cl2 can be recycled to promote its utilisation efficiency.
At a larger scale for the removal of atmospheric methane of 1.9 ppm, the electrolysis-photolysis deployment method could be engineered as an integrated system in current chlor-alkali industries, which account for over 650 facilities worldwide. New chlor-alkali facilities could be built in remote coastal areas and be powered using offshore windmills or solar photovoltaic (PV) farms to maximise the negative carbon emissions. It is worth emphasising that all products of these large-scale chlor-alkali facilities will contribute to greenhouse gas removal (GGR): Cl species will oxidise CH4; NaOH can be used to capture CO2 forming NaHCO3 or Na2CO3; and H2 gas may be used as a carbon-free energy resource or for industrial hydrogenation processes.
The deployment of the large-scale system must envision the short- and long-term impacts of chlorine gas and its derived products (i.e. Cl2, Cl˙, HCl, HOCl, O3, etc.) on health, safety and the environment. The impacts assessments can be done on controlled release of Cl species at selected locations, i.e. in remote regions far from populated areas and sufficiently above local workers but still under the marine boundary layer to limit exposure.
Under ideal conditions, chlorine atom release would be made only under the planetary boundary layer, on wetland regions whose soils are calcareous or with large amounts of ultramafic rocks such as olivine or serpentine which are alkaline and can neutralize the deposition of hydrochloric acid resulting from the hydrogen abstraction from methane by the chlorine atom. Above the ocean under the marine boundary layer is also a possible region to perform the methane remediation. The buffering power of seawater can neutralize it in some regions where limestone is abundant and not too deep. The ocean acidification is a real and serious issue, due to the fact that the ocean absorbs about 25 to 30% of all CO2 emissions each year. It is worth pointing out that the possible acidification from HCl formation of oxidizing all tropospheric methane (about 576 Tg per year10) by Cl atoms would be smaller than that caused by absorbing new CO2 emissions in a year which are in the order of 10 Pg per year or 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Tg per year. Also, if all the NaOH co-produced with Cl2 is released (in a diluted form) in the ocean, globally the pH will remain unchanged (in that case the possible capture of CO2 by NaOH as Na2CO3 or as NaHCO3 is not accounted for in the negative emissions balance sheet). Several marine-CDR (carbon dioxide removal) methods are based on a similar principle:68 electrochemical production of HCl and NaOH, acidification of a volume of sea-water, capture of the CO2 released, neutralisation of the acidity of the same volume of sea-water and release of that volume of sea-water in the open ocean surface, with the end result of removing CO2 from surface waters, making them slightly more alkaline and able to reabsorb some atmospheric CO2.
000 Tg per year. Also, if all the NaOH co-produced with Cl2 is released (in a diluted form) in the ocean, globally the pH will remain unchanged (in that case the possible capture of CO2 by NaOH as Na2CO3 or as NaHCO3 is not accounted for in the negative emissions balance sheet). Several marine-CDR (carbon dioxide removal) methods are based on a similar principle:68 electrochemical production of HCl and NaOH, acidification of a volume of sea-water, capture of the CO2 released, neutralisation of the acidity of the same volume of sea-water and release of that volume of sea-water in the open ocean surface, with the end result of removing CO2 from surface waters, making them slightly more alkaline and able to reabsorb some atmospheric CO2.
More importantly, there is a safe window for the enhancement of chlorine radical sinks in locations which are not sensitive to acid rain. The current near-surface concentration of HCl, for example, ranges from 100 to 300 ppt in remote ocean regions, with concentrations below 50 ppt in altitude above the boundary layer.69,70 Nevertheless, above urban continental areas, peak HCl concentrations of up to 1–3 ppb have been reported70 due to anthropogenic sources. These concentrations are 10–20 times higher than those observed in natural environments and can cumulate other acid sources such as nitrates and sulphates from fossil fuel combustion pollution. Tropospheric peak HCl concentrations typically occur in the afternoon, coinciding with the peak concentrations of nitric acid (HNO3) and photochemical smog, which are attributed to the volatilisation of chloride (Cl−) from aerosol particles containing nitrates.71–74 For environment engineering, these factors can be used not only for the safe enhancement design of chlorine radical sinks, but also for making the operation more efficient and viable in terms of GHG emission and economics.
5.2 Cost estimation based on commercial pure Cl2 and NaOH
In this sub-section, we take the hypothesis of atmospheric methane removal by Cl2 photolysis by sunlight at low altitudes (well below the top of the marine boundary layer) above the ocean, far from populated areas and forests. It could also be above deserts, or abandoned open pit coal mines still leaking methane, but it can also be using semi-closed systems as described by the start-up Ambient Carbon,75 for instance the ventilation exhaust of underground coal mines in operation. The cost estimates made are based not only on the current commercial costs of Cl2 (g) and NaOH (solid or in solution), but also on the capital, infrastructure, and operational costs.The cost for a completely new membrane cell plant is estimated at ca. $1050 per tonne annual Cl2 capacity.76 The market price of commodities varies daily and by geographical market zones, but it was in average ca. $193 per tonne Cl2 and $760 per tonne NaOH in 2020.77 Our initial calculations are based on Cl2 gas at this market price and the production of atomic Cl using natural, free sunlight. Sunlight can provide irradiation of up to ca. 6.5 mW cm−2 for Cl2 gas UV photolysis, based on the UV flux in sunlight shown in Fig. 2a and their dissociation absorption efficiency at different wavelengths.50 The Cl2 gas can be released at a selected location as suggested above in an optimised manner in sunlight considering the 7 minute lifetime of Cl2 molecules in the atmosphere to allow its dissociation. Recent experimental measurement in our laboratory shows that the CH4 is dominantly converted into CO and HCl, and the molar ratio of CO:HCl is lower and close to ∼1:4.78 If the released Cl2 gas could be completely dissociated under sunlight and 1 kmol of molecular Cl2 would oxidise 0.5 kmol of methane, 1.00 tonne of Cl2 gas will be able to remove 9.46 tonnes of CO2-eq and the Cl2 gas cost for the removal will be in the range of $20.28 per tonne CO2-eq considering the molecular masses of Cl2 and CH4 (71 and 16 kg kmol−1, respectively) with 84 as the warming potential factor of methane over 20 years. Assuming the NaOH produced as a by-product (1.1 tonne per tonne of Cl2) is used directly for the neutralisation of atmospheric CO2, then the NaOH from the production of 1 tonne of Cl2 will also indirectly remove 1.21 tonnes of CO2 with a total cost of $691 per tonne CO2. On average, the production of 1 tonne of Cl2 and 1.1 tonnes of NaOH from 1.65 tonnes of NaCl can remove 10.67 tonnes of CO2-eq with an average cost of $96.4 per tonne CO2-eq. The cost contribution shows that the use of NaOH accounts for 78.96% of the overall cost, and the Cl radical approach provides a significant space for its development in an economic manner. These cost calculations are summarised in Table 2.
| Cl2(l) | NaOH(s) | |||
|---|---|---|---|---|
| a Calculation based on the formation of NaHCO3. | ||||
| Price ($ per tonne) | 193 | 760 | ||
| Cl2 dissociation conversion | 100% | 50% | 25% | |
| CH4 removal, tonne per tonne Cl2 | −0.11 | −0.056 | −0.028 | — |
| CO2-eq removal, tonne CO2-eq per tonne Cl2 | −9.46 | −4.73 | −2.36 | −1.21a |
| GHG removal cost, $ per tonne CO2-eq | 20.28 | 40.57 | 81.14 | 691 |
| Overall cost (CH4 and CO2), $ per tonne CO2-eq | 96.40 | 173.16 | 287.74 | |
However, as shown in the report of Johnson's group79 the dissociation of Cl2 is approximately >50% under irradiation of LED UV 365 nm at a Cl2 concentration of ∼100 ppm and a residence time of 3.4 minutes. Considering the lifetime of Cl2 is about 7 minutes the Cl2 dissociation of 50% and 25% is applied in the calculation. Under these conditions the cost of Cl2 gas required for the GHG removal will increase to $40.57 and $81.14 per tonne CO2-eq, respectively, as shown in Table 2. The cost calculation results show that using commercially available pure Cl2 gas to remove CH4 can be competitive to capturing CO2 from releasing points.80 It can be competitive to that of the direct air capture (DAC) technologies under development even when the commercial NaOH to capture atmospheric CO2 and sequestrate it into sea is included. The target cost of DAC is set to be less than $100 per tonne of CO2 by 2050.81 The DAC system developed by Climeworks in Hinwil, Switzerland,8 has an estimated capture cost of $500–600 per tonne of CO2. Carbon engineering estimates the cost of their process can reach between $94–232 per tonne of CO2 captured.82
Comparing with DAC costs, the following conclusions can be drawn: (a) even at the market prices of Cl2 and NaOH, artificial Cl radical sinks can be developed to remove atmospheric methane for a fast reduction of greenhouse gases contributing to reaching the mid-century goal of net-zero emissions; (b) there is a good cost space to achieve high Cl2 photolysis and recycle unreacted Cl2 gas and safe handling. Little cost data is available from the open literature. This will be discussed in more detail in Section 5.3.
5.3 Cost estimation based on an integrated chlor-alkali process
For large-scale facilities dedicated to the removal of methane and CO2 from the atmosphere, a Cl2 gas photolysis unit can be integrated with a chlor-alkali process as shown in Fig. 4. The formed Cl2 and NaOH can be directly used for methane and CO2 removal, respectively, without the complicated purification and pressurisation for pure Cl2(l) production, and concentration and crystallisation for pure NaOH(s) production as commercial products. These changes will significantly lower the cost of chlorine gas and NaOH to be used in the removal of methane and CO2 from the atmosphere.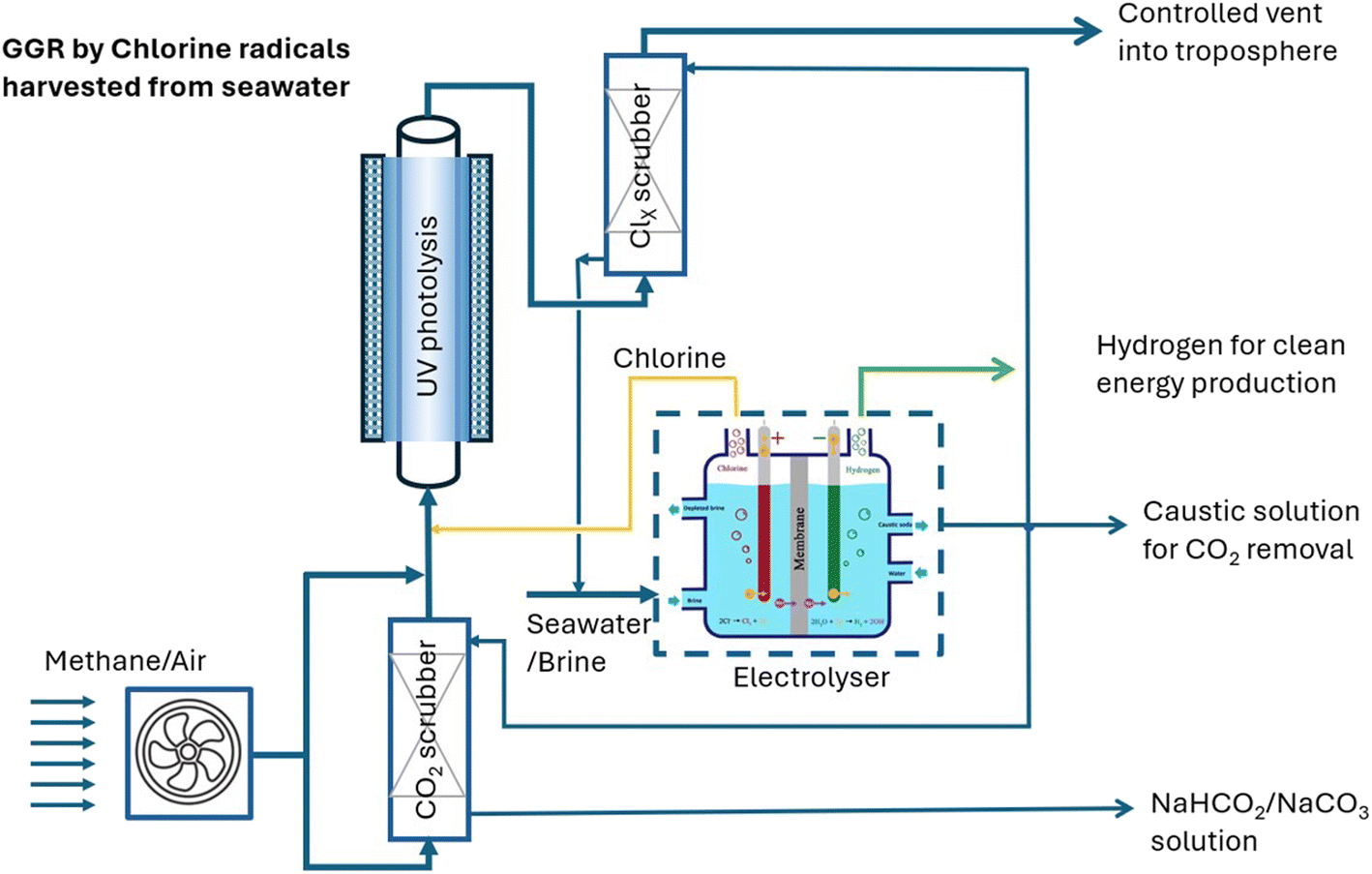 | ||
| Fig. 4 The proposed integration of brine/seawater electrolysis and photolysis of Cl2 gas for methane and CO2 removal. | ||
Fig. 5 shows the overall material flow and energy consumption distribution of current representative commercial chlor-alkali processes.65 For the proposed application of removing atmospheric methane and CO2, there will be no need to treat and pressurise Cl2 gas or concentrate NaOH after electrolysis. Hence, the post-treatment units after electrolysis may be excluded from the current commercial chlor-alkali systems. This will significantly reduce energy consumption (288–7308 MJ per tonneCl2) and the capital and operational costs for Cl2 generation. The reductions could be for covering the capital and operational costs and energy needs of the photolysis of Cl2 gas, especially when energy-efficient UV LED lights are used.
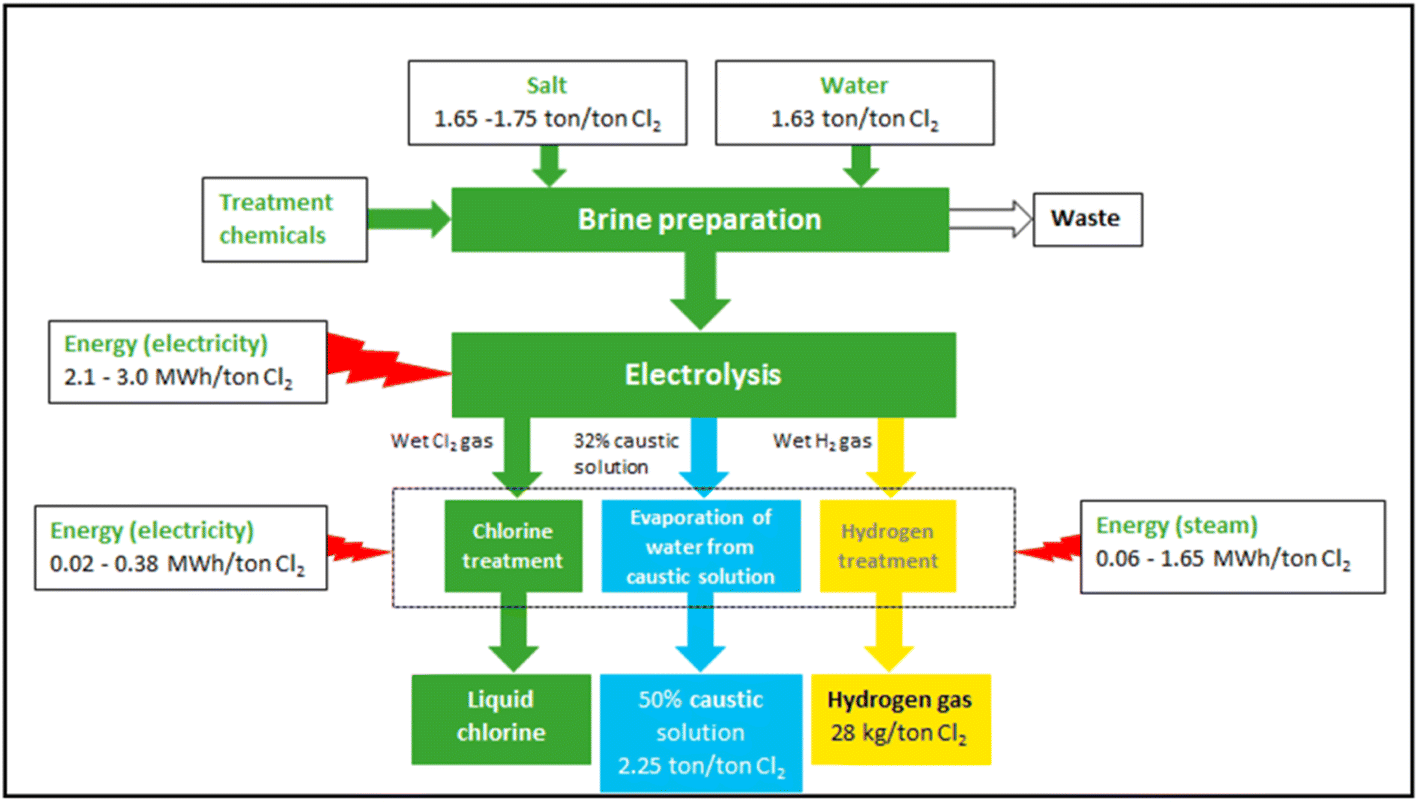 | ||
| Fig. 5 Overview of the main materials flows and process of the chlor-alkali production using the diaphragm process.65 | ||
The production cost of Cl2 varies within the range of $148–531 per tonne, which includes the associated electricity costs of $76–306.65 The techno-economic analysis to produce high-purity liquid Cl2 by the state-of-the-art chlor-alkali process has a carbon footprint of 0.96 kg CO2-eq per kgCl2 for the diaphragm and 0.92 kg CO2-eq per kgCl2 for membrane technologies. The corresponding energy consumption (including brine preparation and product concentration) is 19.52 MJ per kgCl2 for the diaphragm and 18.94 MJ per kgCl2 for membrane methods, as provided by the carbon foot-printing tool83 of the “Ecoinvent” life cycle inventory database.84
Considering the standard dissociation energy of Cl2 gas of 243.6 kJ mol−1, the energy consumption of converting Cl2 gas to atomic chlorine and related radicals, will be larger than 3.43 MJ kgCl−1. At an energy efficiency of the dissociation of 10%, the energy consumption will be 34.31 MJ kgCl−1. By using the carbon footprint value of 0.115 kgCO2 per MJ electricity from the combined heat and power production,84 the carbon footprint of dissociating Cl2 gas into Cl atoms is estimated at 3.95 kg CO2-eq kgCl−1. Considering the molecular masses of Cl2 and CH4 and the GWP of methane over 20 years, 1.00 tonne of Cl2 will be able to remove 9.46 tonnes of CO2-eq, while producing 0.31 kgCO2 per kgCl2reacted. The derived NaOH of 1.1 kg kgCl2−1 reacts with CO2 to form NaHCO3. The H2 gas obtained as a by-product can be used as a carbon-free energy source. These data are summarised in Table 3.
| Diaphragm | Membrane | Comments | |
|---|---|---|---|
| Chlor-alkali emission | |||
| kgCO2-eq kgCl2(l)−1 | 0.96 | 0.92 | EcoInvent83,84 |
|
|||
| Emission of Cl 2 photolysis | |||
| kgCO2-eq kgClradicals−1 |
3.95 | 3.95 | Quantum efficiency of 10% |
|
|||
| Negative emission | |||
| kgCH4 kgCl2−1 | −0.11 | −0.11 | Unit reaction |
| kgCO2-eq kgCl2−1 | −9.46 | −9.46 | CH4 GWP20 = 84 |
| kgCO2 kgCl2−1 | +0.31 | +0.31 | CO2 formation of CH4 oxidation |
| kgCO2-eq kgCl2viaNaOH−1 |
−1.24 | −1.24 | |
| kgCO2-eq kgCl2viaH2−1 |
−1.02 | −1.02 | EcoInvent83,84 |
| Net kgCO2-eq kgCl2(+NaOH+H2)−1 | −6.50 | −6.55 | |
|
|||
| Net GHG removal cost (with full recovery and recycling of unreacted Cl 2 gas) | |||
| $ per tonneCO2-eq | 22.74 | 22.59 | $148 per tonne Cl2 |
| 81.58 | 81.08 | $531 per tonne Cl2 | |
The calculated overall net-GHG emission is negative at 6.50 kgCO2-eq kgCl2−1 for the diaphragm technology and slightly higher at 6.55 kgCO2-eq kgCl2−1 for the membrane technology (Table 3). With full recovery and recycling of unreacted Cl2 gas, the corresponding cost (including CH4 and CO2) is estimated at $22.74 or 22.59 per tonne CO2-eq when the Cl2 production cost is $148 per tonneCl2 and $81.58 or 81.08 per tonne CO2-eq when the Cl2 production cost is $531 per tonneCl2. The costs calculated in both cases are well below the aimed cost of $100 per tonne CO2-eq for DAC, and comparable to that of CDR/CCS technologies from less concentrated point CO2 sources, such as cement production and power generation at $40–120 per tonne CO2.85
The calculation has been carried out also for the net GHG removal when the single path dissociation efficiency of Cl2 gas ranges from 1% to 100%. The corresponding costs have been estimated for the two scenarios i.e. (1) with the unreacted Cl2 being fully recycled and (2) without it being recycled. The result shown in Fig. 6 suggests that the GHG emission of the operation can be significantly negative even when the single path dissociation energy efficiency of Cl2 gas is 5% by using the electrical energy from the combined heat and power (CHP) production with a carbon footprint of 0.115 kgCO2-eq MJ−1.84 At a low single path dissociation energy efficiency of 5%, the cost of GHG removal is calculated to be $57.73 per tonne CO2-eq (well below $100 per tonne CO2-eq) when the Cl2 production cost is at $148 per tonne and with full recycling of the unreacted Cl2 gas; or £227.37 per tonne CO2-eq for the case without Cl2 gas recycling. Alkaline solutions generated in the chlor-alkali process can be used for the capture and recycling of Cl2 gas.
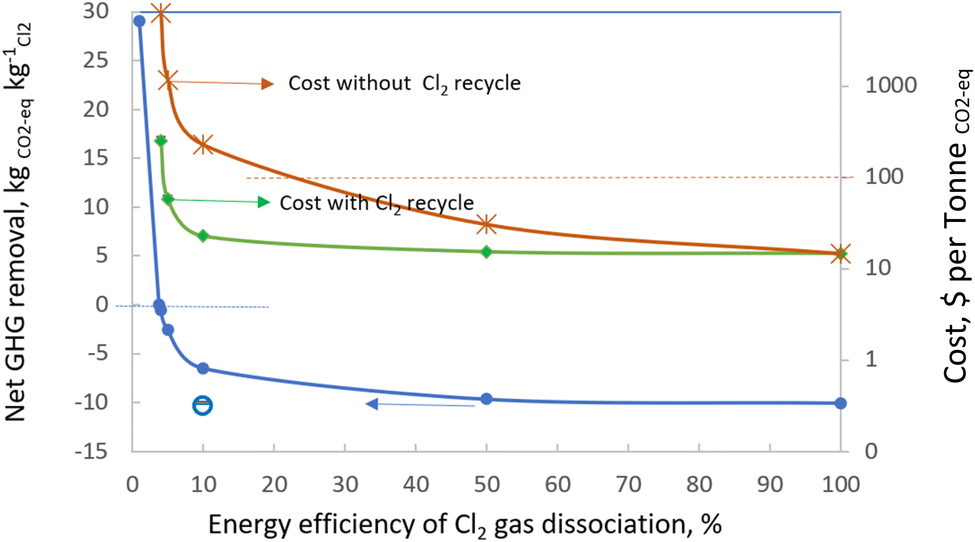 | ||
| Fig. 6 Estimation of net GHG removal and cost with energy efficiency in Cl2 gas dissociation. The hollow circle and the solid bar at the left bottom corner represent the net GHG removal when using clean energy from biomass, wind or nuclear sources. | ||
Furthermore, using clean energy from biomass, wind or nuclear sources will reduce the energy carbon footprint to 0.0111, 0.00311 or 0.00173 kgCO2-eq MJ−1, respectively. The net GHG removal will increase to 10.05–10.40 kgCO2-eq kgCl2(+NaOH+H2)−1 when the Cl2 dissociation energy efficiency is 10%, as shown by the hollow circle and the solid bar labelled at the left bottom corner in Fig. 5. This will lower the cost to less than $15 kgCO2-eq−1 before including the deployment cost. These calculations, therefore, demonstrate the potential of developing atomic chlorine technology by incorporating chlorine gas photolysis with brine electrolysis for atmospheric methane remediation.
6. Relevance to climate change at scale and complementary with DAC
The global annual production of Cl2 by the chlor-alkali industry in 2021 was about 90 Mt (with nearly the same amount of NaOH production), and is expected to reach 97 Mt by 2026.55 If the same amount of all the produced Cl2 is successfully used for tropospheric methane removal and a 50% conversion is achievable, it could be possible to directly remove ∼20 Mt of methane per year. This represents half of the current annual growth of methane in the troposphere, corresponding to 1.7 Gt CO2-eq per year based on the methane GWP of 84 over 20 years. This estimation does not consider the tropospheric recycling of HCl that is produced by the reaction of atomic Cl with methane. One mole of Cl2 will produce 2 moles of atomic Cl and consequently two moles of HCl, which can be recycled to 2 moles or more of Cl˙, following eqn (7)–(12) (where RH represents methane or other hydrocarbons):C![[l with combining dot above]](https://www.rsc.org/images/entities/char_006c_0307.gif) + RH → R˙ + HCl + RH → R˙ + HCl | (7) |
| HCl + ˙OH → C + H2O | (8) |
| N2O5 + HCl → ClNO2 + HNO3 | (9) |
| ClNO2 + hv → C + H2O | (10) |
2Cl− + O3 + H2O → Cl2 + ![[2 with combining dot above]](https://www.rsc.org/images/entities/char_0032_0307.gif) OH + O2 OH + O2 | (11) |
| Cl2 + hv → 2C | (12) |
To reach direct air capture of CO2 (DAC) capacity of 30 Gt CO2 per years based on aqueous hydroxide solutions (NaOH, KOH, Ca(OH)2) some authors envision the need to scale up the current chlor-alkali production with a production of about 5.1–8.7 Gt per years of NaOH86,87 and about 4.6 to 7.9 Gt per years of Cl2 by salt electrolysis. This Cl2 production level is more than 65 times the current capacity of about 90 Mt per years.
The methane removal method proposed in this work can be complementary to these DAC processes and can use excess Cl2 to remove more than 1.0–1.8 Gt of methane per years.86 Since the tropospheric stock of methane is about 4.6 Gt,39 thus in theory such stock of methane could be cleared within 3–5 years, with significant implications in the control of global warming. Of course, risks for the environment with acid rain or stratospheric ozone layer damage need to be thoroughly studied and assessed before any large-scale implementation is attempted. A more reasonable assumption would be that the target removal is obtained progressively over 20 or 30 years, which has the advantage of much lower new chlor-alkali infrastructure needs and after the target has been reached, to avoid production overcapacity.
If the global capacity of the chlor-alkali industry is to increase by 20% each year (its current growth rate is between 3 and 5% but for other purposes88), it would be tripled in 7 years. The cumulative reduction of the methane tropospheric stock will significantly reduce the methane global warming burden (0.5 °C currently) and also lessen the radiative impact of O3 and some fluorocarbons (HCFCs, HFCs).
Apparently, these calculations do not consider the steep evolution of the DAC industry. According to current status, for instance, a DAC capacity of 3 Gt per years of CO2 through NaOH absorption will need about 47 years to remove the amount of CO2 that is equivalent to the warming of the current methane tropospheric stock of 5.6 Gt.
Along with methane neutralization, the Cl2 produced with NaOH devoted to the DAC industry could be used to generate HCl, which can be used to convert globally abundant serpentine, olivine or other silicate minerals or ultramafic rocks to benign metal salts and/or silica as useful resources. On the other hand, the re-generation technology of the spent alkali from the DAC hydroxide solution will be developed for recycling, to reduce the requirement of fresh NaOH.89
The proposed method might also help in dealing with localised methane leaks due for example to pipe bursting in operation, as well as for dealing with more progressive but also more diffuse thawing permafrost or destabilization of submarine methane hydrates. Even with a very low probability,90 methane bursts represent a real threat: for instance, several gigatons of methane could have been released during the Storegga submarine landslide that took place on the continental slope west of Norway in the early-Holocene period.91
Furthermore, the chlorine sink can also have an impact on the reduction of tropospheric ozone (O3), another critical GHG.92 Oceanic emissions of bromine, iodine and chlorine have been shown to improve the atmospheric oxidation capacity,93,94 with an estimated average decrease in O3 concentration of ∼15%.95 The GEOS-Chem model96 has also estimated a halogen-driven O3 reduction of up to 46% with respect to the typical hourly O3 concentration (above 50 nmol mol−1) in European air quality, except for highly NOx-polluted areas where the amount of O3 increased slightly. Similar observations have been reported globally, for example in Los Angeles, California, where O3 levels were reported to decrease by 5 ppb due to marine halogen emissions.97
7. Future research
Enhancing the atomic Cl concentration in the atmosphere by the method proposed in this article involves (a) volatilisation of Cl2, HOCl and other chlorine species from NaCl aerosols/solution or other sources; (b) photolysis of the volatilised species to produce Cl radicals; (c) Cl-driven oxidation of methane as well as other hydrocarbons with the production of hydrogen chloride (HCl); (d) removal of HCl upon reaction with hydroxyl radicals, wet/dry deposition or scavenging by aerosols. To develop applicable environment engineering technologies, more scientific and technology development research is urgently required to achieve high efficiencies of material and energy utilisation as well as low impacts on the environment for high negative emissions and avoiding harm.Recent pioneer work79 showed that the energy efficiency of diffuse methane removal by Cl2 (100 ppm) photolysis is at ∼0.83% in the single-pass gas phase photoreactor developed. Advances to improve the energy efficiency to above 5% as calculated, shown in Fig. 5, or above 9%98 are the major engineering requirements for any chlorine radical technology economically applicable. In combination with reactor design effective photocatalysis is possible to provide a solution to achieve the desired photolysis and methane oxidation under milder irradiation conditions or using sunlight only. On the other hand, one mole of Cl2 produces 2 moles of atomic Cl and consequently two moles of HCl, which can be recycled according to eqn (8)–(12) to reduce fresh Cl2 feed and HCl capture and to increase the overall energy efficiency of methane removal.
Furthermore, significant yields of Cl2 and Br2 gases can be produced upon high-energy photon absorption above the deliquescence point of their respective salts (NaCl, NaBr) in the presence of O3.99,100 It is known, for example, that the efficiencies of the former process could be increased up to 6 times upon substitution of NaCl by an NH4Cl aerosol.101 Likewise, halogen salts such as KCl deposited onto standard photocatalytic materials, such as titanium dioxide (TiO2), can form Cl2 gas as well as other chlorinated compounds (ClO, HOCl) under illumination.102 The amount of Cl2 produced in that case depends on the light intensity, relative humidity and amount of salt on the photocatalyst. The role of these materials can have a major impact at least on the photolysis share of the deployment method proposed, with an anticipated reward in overall costs and energy consumption.
Before any field tests or deployment of the method proposed here, safety assessments, as well as full environmental impact assessments, are essential at all locations where it might be envisioned, acid rain being one of the possibly worst drawbacks in regions where soil alkalinity is reduced.
8. Concluding remarks
Reducing CO2 and CH4 emissions is an absolute priority; however, based on the current progress of methane reduction strategies, alternative mitigation and preventive measures as well as remediation methods need to be explored to achieve staying below the 2 °C global warming goal settled in the Paris Agreement. Consequently, both mitigation and remediation measures are necessary and should be implemented together with any emission cut-down strategies.Even if anthropogenic emissions of CO2 were drastically reduced, global warming will continue rising, causing an acceleration in the generation and release of methane from natural systems – those biogenic emissions indirectly related to man-made actions cannot be ignored. Large-scale methane remediation methods are particularly encouraged in this sense, preventing the impact of any potential methane burst or feedback loop from melting permafrost as well as dealing with the acceleration of biogenic methane release from wetlands, lakes, rice paddies, and hydroelectric reservoirs.
Chlorinated species – such as HCl, Cl2, HOCl and atomic Cl, derived from sea brine and sea salt, sunlight and natural processes-represent a natural sink for ca. 1–4% of the tropospheric methane.28–30 Even if some authors103 consider the contribution of Cl˙ to the tropospheric removal of methane to be probably much lower than currently assumed, others consider the chlorine atmospheric chemistry into NaCl droplets as much more important.104 Enhancing this natural sink using Cl2 gas produced by the existing chlor-alkali industrial process at a market price could have a cost of about $34 per tonne of CO2-eq, allowing for the removal of ca. 20 Mt per years of methane. This method can be cost-competitive and complementary to DAC technologies.85 Importantly, costs can be significantly lowered to less than $10 per tonne CO2-eq if efficient chlor-alkali plants were installed and dedicated to the removal of diffuse GHG directly from the air. Further economic and environmental benefits will come from installations using sustainable energy sources such as offshore windmills or solar PV farms. In addition, environmental co-benefits include the reduction of tropospheric O3 with human health and agricultural co-benefits.105 Simulations using an Earth System Model92 have shown a mean global surface O3 reduction of 1.0 ± 0.2 ppb and a mean global surface temperature reduction of 0.21 ± 0.04 °C per gigaton of methane removed.
Data availability
Data for this article are from the literature as referenced in the paper. The data for Table 3 and Fig. 5 include chlorine production cost by commercial electrolysis and corresponding carbon footprints are available at https://www.eurochlor.org/wp-content/uploads/2021/04/12-Electrolysis-production-costs.pdf; http://www.ccalc.org.uk/ and https://ecoinvent.org/, respectively.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to all reviewers for their constructive comments and suggestions for this manuscript revision. This research was supported by a CO2RE Pathfinders fund (2022) for QY and BX. RQC would like to thank MEFP (Beatriz Galindo Program) and ACIISI (ProID2021010047), Spain, for funding this work.References
- IPCC, Summary for Policymakers, in Climate Change 2014: Mitigation of Climate Change. Contribution of Working Group III to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, ed. Edenhofer , O., R. Pichs-Madruga, Y. Sokona, E. Farahani, S. Kadner, K. Seyboth, A. Adler, I. Baum, S. Brunner, P. Eickemeier, B. Kr iemann, J. Savolainen, S. Schlömer, C. von Stechow, T. Zwickel and J. C. Minx, Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA, 2014, https://www.ipcc.ch/site/assets/uploads/2018/02/ipcc_wg3_ar5_summary-for-policymakers.pdf, Accessed 20- February 2022 Search PubMed.
- The NOAA, Annual Greenhouse Gas Index (AGGI), https://gml.noaa.gov/aggi/, accessed 15 August 2024 Search PubMed.
- IPCC, AR6 WG1, Climate Change 2021, The Physical Science Basis - Summary for Policymakers, 2014, https://www.ipcc.ch/report/ar6/wg1/downloads/report/IPCC_AR6_WGI_SPM.pdf, accessed 11 August. 2021 Search PubMed.
- IPCC, AR6 WG1 Climate Change 2021: The Physical Science Basis, https://www.ipcc.ch/report/ar6/wg1/downloads/report/IPCC_AR6_WGI_Full_Report.pdf, accessed 4 May 2022. 2021 Search PubMed.
- N. McQueen, et al., A review of direct air capture (DAC): scaling up commercial technologies and innovating for the future, Prog. Energy, 2021, 3, 032001 CrossRef CAS.
- GMI, Methane's Role in Global Warming, 2021, https://www.globalmethane.org/methane/index.aspx, Accessed 20 February 2022 Search PubMed.
- NOAA. Global Monitoring Laboratory - Eath System Research laboratories, 2019, https://gml.noaa.gov/ccgg/trends_ch4/, accessed 20 December 2021 Search PubMed.
- IPCC, Climate Change 2013, in The Physical Science Basis, Working Group I Contribution to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, ed. Stocker, T. F., D. Qin, G.-K. Plattner, M. Tignor, S. K. Allen, J. Boschung, A. Nauels, Y. Xia, V. Bex and P. M. Midgley, Cambridge University Press, Cambridge, UK and New York, NY, USA, 2013, p. 1535, https://www.ipcc.ch/report/ar5/wg1/ Search PubMed.
- I. B. Ocko, et al., Acting rapidly to deploy readily available methane mitigation measures by sector can immediately slow global warming, Environ. Res. Lett., 2021, 16(5), 054042 CrossRef CAS.
- M. Saunois, et al., The global methane budget 2000–2017, Earth Syst. Sci. Data, 2020, 12(3), 1561–1623 CrossRef.
- R. Atkinson and J. andArey, Atmospheric degradation of volatile organic compounds, Chem. Rev., 2003, 103(12), 4605–4638 CrossRef CAS PubMed.
- Y. Wang, et al., Atmospheric Removal of Methane by enhancing the natural hydroxyl radical sink, Greenhouse Gases:Sci. Technol., 2022, 12(6), 784–795 CrossRef CAS.
- T. Ming, et al., Perspectives on removal of atmospheric methane, Adv. Appl. Energy, 2022, 100085 CrossRef CAS.
- R. de Richter, et al., Removal of non-CO2 greenhouse gases by large-scale atmospheric solar photocatalysis, Prog. Energy Combust. Sci., 2017, 60, 68–96 CrossRef.
- (a) R. J. Brenneis, et al., Atmospheric-and Low-Level Methane Abatement via an Earth-Abundant Catalyst, ACS Environ. Au, 2021, 2(3), 223–231 CrossRef PubMed; (b) Y. Wang and W. Li, et. al., Removing low-concentration methane via thermo-catalytic oxidation on CuOx/zeolite, Appl. Surf. Sci., 2025, 682, 161691 CrossRef CAS.
- R. B. Jackson, et al., Methane removal and atmospheric restoration, Nat Sustainability, 2019, 2(6), 436–438 CrossRef.
- Y. Wang, S. Xu, H. Zhang, Y. Wang, E. Wang and Z. He, et al., Highly dispersed CuOx decorated ZnO photocatalyst for low-concentration methane removal, J. Environ. Chem. Eng., 2024, 12(6), 114954 CrossRef CAS.
- F. D. Oeste, et al., Climate engineering by mimicking natural dust climate control: the iron salt aerosol method, Earth Syst. Dynam., 2017, 8(1), 1–54 CrossRef.
- T. Ming, et al., A nature-based negative emissions technology able to remove atmospheric methane and other greenhouse gases, Atmos. Pollut. Res., 2021, 12(5), 101035 CrossRef CAS.
- https://www.konokogs.com/systems/resources/what-is-a-regenerative-thermal-oxidizer. retrieved on 31 July 2024.
- A. M. Tsopelakou, et al. , Exploring the bounds of methane catalysis in the context of atmospheric methane removal, Environ. Res. Lett., 2024, 19, 054020 CrossRef CAS.
- K. S. Lackner, Practical constraints on atmospheric methane removal, Nat Sustainability, 2020, 3(5), 357 CrossRef.
- X. Li, C. Wang and J. Tang, Methane transformation by photocatalysis, Nat. Rev. Mater., 2022, 1–16 Search PubMed.
- X. Chen, Y. Li and X. Pan, et al. , Photocatalytic oxidation of methane over silver decorated zinc oxide nanocatalysts, Nat. Commun., 2016, 7, 12273, DOI:10.1038/ncomms12273.
- H. Xiong and T. Ming, et. al., Experimental and kinetic studies on the photocatalysis of UV–vis light irradiation for low concentrations of the methane, Appl. Energy, 2024, 377(part A), 124388 Search PubMed.
- D. R. Crosley, et al., Gas-phase photolytic production of hydroxyl radicals in an ultraviolet purifier for air and surfaces, J. Air Waste Manage. Assoc., 2017, 67(2), 231–240 CrossRef CAS PubMed.
- T. Minamikawa, et al., Quantitative evaluation of SARS-CoV-2 inactivation using a deep ultraviolet light-emitting diode, Sci. Rep., 2021, 11(1), 1–9 CrossRef PubMed.
- Z. Peng, S. L. MillerJose and J. L. Jimenez, Evaluation of Secondary Chemistry due to Disinfection of Indoor Air with Germicidal Ultraviolet Lamps, Environ. Sci. Technol. Lett., 2023, 10(1), 6–13 CrossRef CAS.
- L. Pennacchio, M. K. Mikkelsen, M. Krogsbøll, M. van Herpen and M. S. Johnson, Physical and practical constraints on atmospheric methane removal technologies, Environ. Res. Lett., 2024, 19(10), 104058 CrossRef CAS.
- J. Wittmer and C. Zetzsch, Photochemical activation of chlorine by iron-oxide aerosol, J. Atmos. Chem., 2017, 74(2), 187–204 CrossRef CAS.
- J. Wittmer, et al., Iron (III)-induced activation of chloride from artificial sea-salt aerosol, Environ. Chem., 2015, 12(4), 461–475 CrossRef CAS.
- M. K. Mikkelsen, J. B. Liisberg, M. M. van Herpen, K. V. Mikkelsen and M. S. Johnson, Photocatalytic chloride-to-chlorine conversion by ionic iron in aqueous aerosols: a combined experimental, quantum chemical, and chemical equilibrium model study, Aerosol Res., 2024, 2(1), 31–47 CrossRef.
- C. Zetzsch, S. Bleicher and J. Wittmer, Smog Chamber Investigation on the Iron-Catalyzed Activation of Chloride from Seasalt for a Depletion of Tropospheric Methane, in AGU Fall Meeting Abstracts, 2013 Search PubMed.
- M. M. van Herpen, Q. Li, A. Saiz-Lopez and J. B. Liisberg, et al., Photocatalytic chlorine atom production on mineral dust–sea spray aerosols over the North Atlantic, Proc. Natl. Acad. Sci. U. S. A., 2023, 120(31), e2303974120 CrossRef CAS PubMed.
- G. Chen, L. Xu, S. Yu, L. Xue and Z. Lin, et al., Increasing Contribution of Chlorine Chemistry to Wintertime Ozone Formation Promoted by Enhanced Nitrogen Chemistry, Environ. Sci. Technol., 2024, 58(51), 22714–22721 CrossRef CAS PubMed.
- D. Meidan, Q. Li, C. A. Cuevas and S. C. Doney, et al., Evaluating the potential of iron-based interventions in methane reduction and climate mitigation, Environ. Res. Lett., 2024, 19(5), 054023 CrossRef CAS.
- Q. Li, D. Meidan and P. Hess, et al., Global environmental implications of atmospheric methane removal through chlorine-mediated chemistry-climate interactions, Nat. Commun., 2023, 14(1), 4045 CrossRef CAS PubMed.
- X. Wang, et al., The role of chlorine in global tropospheric chemistry, Atmos. Chem. Phys., 2019, 19(6), 3981–4003 CrossRef CAS.
- R. Hossaini, et al., A global model of tropospheric chlorine chemistry: Organic versus inorganic sources and impact on methane oxidation, J. Geophys. Res.:Atmos., 2016, 121(23), 14271–14297 Search PubMed.
- W. Allan, H. Struthers and D. Lowe, Methane carbon isotope effects caused by atomic chlorine in the marine boundary layer: Global model results compared with Southern Hemisphere measurements, J. Geophys. Res.:Atmos., 2007, 112, D04306 CrossRef.
- M. Mendez, et al., Reactivity of chlorine radical with submicron palmitic acid particles: kinetic measurements and product identification, Atmos. Chem. Phys., 2013, 13(23), 11661–11673 CrossRef.
- M. Khalil, Reactive chlorine compounds in the atmosphere, in Reactive Halogen Compounds in the Atmosphere, 1999, pp. 45–79 Search PubMed.
- X. Peng, et al., Unraveling the daytime source of molecular chlorine in the extra-polar atmosphere,Earth Space Science Open Archive ESSOAr, 2021 Search PubMed.
- G. McFiggans, et al., Active chlorine release from marine aerosols: Roles for reactive iodine and nitrogen species, J. Geophys. Res.:Atmos., 2002, 107(D15), ACH10 CrossRef.
- E. K. Frinak and J. P. Abbatt, Br2 production from the heterogeneous reaction of gas-phase OH with aqueous salt solutions: Impacts of acidity, halide concentration, and organic surfactants, J. Phys. Chem. A, 2006, 110(35), 10456–10464 CrossRef CAS PubMed.
- W. Behnke and C. Zetzsch, Heterogeneous photochemical formation of Cl atoms from NaCl aerosol, NOx and ozone, J. Aerosol Sci., 1990, 21, S229–S232 CrossRef CAS.
- H. D. Osthoff, et al., High levels of nitryl chloride in the polluted subtropical marine boundary layer, Nat. Geosci., 2008, 1(5), 324–328 CrossRef CAS.
- Y.-H. Chuang, et al., Photolysis of Chlorine Dioxide under UVA Irradiation: Radical Formation, Application in Treating Micropollutants, Formation of Disinfection Byproducts, and Toxicity under Scenarios Relevant to Potable Reuse and Drinking Water, Environ. Sci. Technol., 2022, 56(4), 2593–2604 CrossRef CAS PubMed.
- C. Pan, et al., Chlorine-activated diamond chemical vapor deposition, J. Electrochem. Soc., 1994, 141(11), 3246 CrossRef CAS.
- J. Ganske, H. Berko and B. Finlayson-Pitts, Absorption cross sections for gaseous ClNO2 and Cl2 at 298 K: Potential organic oxidant source in the marine troposphere, J. Geophys. Res.:Atmos., 1992, 97(D7), 7651–7656 CrossRef.
- B. Finlayson-Pitts, Chlorine atoms as a potential tropospheric oxidant in the marine boundary layer, Res. Chem. Intermed., 1993, 19(3), 235–249 CrossRef CAS.
- Zeiss. Education in Microscopy and Digital Imaging, https://zeiss-campus.magnet.fsu.edu/articles/lightsources/xenonarc.html, accessed 25 May 2023 Search PubMed.
- BCE, Beijing China Education Au-light Technology Co., Ltd. (in Chinese), http://www.bjaulight.com/ParentList-1047937.html, Accessed on 25 May 2023.
- Y. Lei, et al., Reactivity of chlorine radicals (Cl˙ and Cl2˙−) with dissolved organic matter and the formation of chlorinated byproducts, Environ. Sci. Technol., 2020, 55(1), 689–699 CrossRef PubMed.
- Globaldata. Chlorine Industry Capacity And Capital Expenditure Forecasts With Details Of All Active And Planned Plants, 2021-2026; Report Code: GDCH01224ICR, 2022, https://www.globaldata.com/, accessed 12 November 2022 Search PubMed.
- T. Brinkmann, et al., Best available techniques (BAT) reference document for the production of chlor-alkali. Joint Research Centre, Institute for Prospective Technological Studies. Industrial Emissions Directive 2010/75/EU (integrated pollution prevention and control), Publications Office, 2014, https://data.europa.eu/doi/10.2791/13138, accesed 12 November 2022 Search PubMed.
- S. Lakshmanan and T. Murugesan, The chlor-alkali process: work in progress, Clean Technol. Environ. Policy, 2014, 16(2), 225–234 CrossRef CAS.
- Eurochlor, Mercury cell process, https://www.eurochlor.org/about-chlor-alkali/how-are-chlorine-and-caustic-soda-made/mercury-cell-process/, accessed 20 February 2022 Search PubMed.
- I. Garcia-Herrero, et al., Life Cycle Assessment model for the chlor-alkali process: A comprehensive review of resources and available technologies, Sustain. Prod. Consum., 2017, 12, 44–58 CrossRef.
- A. Scott, EU's chlorine makers end mercury-based production, 2017, https://cen.acs.org/articles/95/web/2017/12/EUs-chlorine-makers-end-mercury.html, accessed 31 May 2022 Search PubMed.
- D. M. Kiefer, When the industry charged ahead.Today’s Chemist at Work, 2002, vol. 11, 3, p. 9 Search PubMed.
- Eurochlor, Diaphragm cell process, 2007, https://www.eurochlor.org/about-chlor-alkali/how-are-chlorine-and-caustic-soda-made/diaphragm-cell-process/, accessed 20 February 2022 Search PubMed.
- Eurochlor. Chlorine & caustic Soda Production, 2021, https://www.eurochlor.org/about-chlor-alkali/how-are-chlorine-and-caustic-soda-made/, accessed 20 February 2022 Search PubMed.
- Eurochlor. Membrane cell process, 2007. https://www.eurochlor.org/about-chlor-alkali/how-are-chlorine-and-caustic-soda-made/membrane-cell-process/, accessed 20 February 2022 Search PubMed.
- Eurochlor. The Electrolysis process and the real costs of production. Eurochlo communication, 2018, https://www.eurochlor.org/wp-content/uploads/2021/04/12-Electrolysis-production-costs.pdf, accessed 20 February 2022 Search PubMed.
- EPA, Basic Information about Landfill Gas, https://www.epa.gov/lmop/basic-information-about-landfill-gas, accessed 18 June 2023 Search PubMed.
- EPA, Ventilation Air Methane (VAM) Utilization Technologies, 2019, https://www.epa.gov/sites/default/files/2019-11/documents/vam_technologies.pdf, Accessed 18 June 2023 Search PubMed.
- M. D. Eisaman, Pathways for marine carbon dioxide removal using electrochemical acid-base generation, Front. Clim., 2024, 6, 1349604 CrossRef.
- W. C. Keene, et al., The geochemical cycling of reactive chlorine through the marine troposphere, Global Biogeochem. Cycles, 1990, 4(4), 407–430 CrossRef CAS.
- T. Graedel and W. Keene, Tropospheric budget of reactive chlorine, Global Biogeochem. Cycles, 1995, 9(1), 47–77 CrossRef CAS.
- R. Griffin, et al., Final Report, Surface Measurements and One-Dimensional Modeling Related to Ozone Formation in the Suburban Dallas-Fort Worth Area AQRP Project UTA10-000875-RICE-RP24-TO2, 2011, http://aqrp.ceer.utexas.edu/projectinfo/10-024/10-024FinalReport.pdf, accessed 12 March 2022 Search PubMed.
- B. Appel, et al., The measurement of atmospheric hydrochloric acid in Southern California, Atmospheric Environment. Part A. General Topics, 1991, 25(2), 525–527 CrossRef.
- R. M. Harrison and A. Allen, Measurements of atmospheric HNO3, HCl and associated species on a small network in eastern England, Atmospheric Environment. Part A. General Topics, 1990, 24(2), 369–376 CrossRef.
- C. Faxon and D. Allen, Chlorine chemistry in urban atmospheres: a review, Environ. Chem., 2013, 10(3), 221–233 CrossRef CAS.
- Ambient Carbon, https://ambientcarbon.com/, accessed 18 June 2023.
- T. Brinkmann, et al., Best Available Techniques (BAT) Reference Document for the Production of Chlor-alkali, https://publications.jrc.ec.europa.eu/repository/handle/JRC91156, accessed 31 May 2022. 2014, Report. ISBN 978-92-79-40945-5 Search PubMed.
- Westlake, Average Quarterly Industry Prices, 2021, https://www.westlake.com/industry-product-pricing, accessed 12 March 2022 Search PubMed.
- Q. Yuan and B. Xiao, CO2RE pathfinder project report (2023) methane removal and remediation using chlorine radicals sink, https://co2re.org/pathfinders-blog-chlorine-radicals-methane/ Search PubMed.
- M. Krogsbøll, H. S. Russell and M. S. Johnson, A high efficiency gas phase photoreactor for eradication of methane from low-concentration sources, Environ. Res. Lett., 2024, 19(1), 014017 CrossRef.
- W. J. Schmelz, G. Hochman and K. G. Miller, Total cost of carbon capture and storage implemented at a regional scale: northeastern and midwestern United States, Interface Focus, 2020, 10(5), 20190065 CrossRef PubMed.
- N. McQueen, et al., Cost analysis of direct air capture and sequestration coupled to low-carbon thermal energy in the United States, Environ. Sci. Technol., 2020, 54(12), 7542–7551 CrossRef CAS PubMed.
- D. W. Keith, et al., A process for capturing CO2 from the atmosphere, Joule, 2018, 2(8), 1573–1594 CrossRef CAS.
- CCaLC2, Second generation of the carbon footprinting tool, 2022, http://www.ccalc.org.uk/, accssessed 12 january 2022 Search PubMed.
- Ecoinvent-database, Life cycle inventory database, https://ecoinvent.org/, accessed 12 January 2022 Search PubMed.
- IEA, Is carbon capture too expensive?, IEA, Paris, 2021, https://www.iea.org/commentaries/is-carbon-capture-tooexpensive, accessed 20 February 2022 Search PubMed.
- S. Chatterjee and K.-W. Huang, Unrealistic energy and materials requirement for direct air capture in deep mitigation pathways, Nat. Commun., 2020, 11(1), 1–3 CrossRef PubMed.
- G. Realmonte, et al., An inter-model assessment of the role of direct air capture in deep mitigation pathways, Nat. Commun., 2019, 10(1), 1–12 CrossRef PubMed.
- Mordor, Chlor-alkali Market Size & Share Analysis - Growth Trends & Forecasts (2023–2028), https://www.mordorintelligence.com/industry-reports/chlor-alkali-market, 2023, accessed 18 June 2023 Search PubMed.
- E. Medina-Martos, et al., Environmental and economic performance of carbon capture with sodium hydroxide, J. CO2 Util., 2022, 60, 101991 CrossRef CAS.
- E. G. Nisbet and D. J. Piper, Giant submarine landslides, Nature, 1998, 392(6674), 329–330 CrossRef CAS.
- C. Paull, W. Ussler and W. Holbrook, Assessing methane release from the colossal Storegga submarine landslide, Geophys. Res. Lett., 2007, 34(4), L04601 CrossRef.
- S. Abernethy, et al., Methane removal and the proportional reductions in surface temperature and ozone, Philos. Trans. R. Soc., A, 2021, 379(2210), 20210104 CrossRef CAS PubMed.
- Q. Li, et al.Potential Effect of Halogens on Atmospheric Oxidation in China. EGU General Assembly 4–8 May 2020, 2020 Search PubMed.
- Q. Li, et al., Potential effect of halogens on atmospheric oxidation and air quality in china, J. Geophys. Res.:Atmos., 2020, 125(9), e2019JD032058 CrossRef CAS PubMed.
- Q. Li, et al., Chemical interactions between ship-originated air pollutants and ocean-emitted halogens, J. Geophys. Res.:Atmos., 2021, 126(4), e2020JD034175 CrossRef CAS PubMed.
- T. Sherwen, et al., Effects of halogens on European air-quality, Faraday Discuss., 2017, 200, 75–100 RSC.
- M. Muñiz-Unamunzaga, et al., The influence of ocean halogen and sulfur emissions in the air quality of a coastal megacity: The case of Los Angeles, Sci. Total Environ., 2018, 610, 1536–1545 CrossRef PubMed.
- S. Abernethy, M. Kessler and R. Jackson, Assessing the potential benefits of methane oxidation technologies using a concentration-based framework, Environ. Res. Lett., 2023, 18, 094064 CrossRef CAS.
- E. M. Knipping and D. Dabdub, Modeling Cl2 formation from aqueous NaCl particles: Evidence for interfacial reactions and importance of Cl2 decomposition in alkaline solution, J. Geophys. Res.:Atmos., 2002, 107(D18), ACH8 CrossRef.
- P. Nissenson, et al., Rapid formation of molecular bromine from deliquesced NaBr aerosol in the presence of ozone and UV light, Atmos. Environ., 2014, 89, 491–506 CrossRef CAS.
- C. B. Faxon, et al., Heterogeneous production of Cl2 from particulate chloride: Effects of composition and relative humidity, AIChE J., 2018, 64(8), 3151–3158 CrossRef CAS.
- Y. Li, et al., Photoinduced production of chlorine molecules from titanium dioxide surfaces containing chloride, Environ. Sci. Technol. Lett., 2020, 7(2), 70–75 CrossRef CAS.
- S. Gromov, C. A. Brenninkmeijer and P. Jöckel, A very limited role of tropospheric chlorine as a sink of the greenhouse gas methane, Atmos. Chem. Phys., 2018, 18(13), 9831–9843 CrossRef CAS.
- R. Zhang and C. K. Chan, Simultaneous formation of sulfate and nitrate via co-uptake of SO2 and NO2 by aqueous NaCl droplets: combined effect of nitrate photolysis and chlorine chemistry, Atmos. Chem. Phys., 2023, 23(11), 6113–6126 CrossRef CAS.
- UNEP, Global Methane Assessment: Benefits and Costs of Mitigating Methane Emissions, https://www.unep.org/resources/report/global-methane-assessment-benefits-and-costs-mitigating-methane-emissions, Accessed 22 June 2021. ISBN: 978-92-807-3854-4. 2021 Search PubMed.
| This journal is © The Royal Society of Chemistry 2025 |