
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThermochemical and chemo-biological molecular recycling of plastic waste and plastic-biomass waste mixtures: an updated review
Paula S.
Mateos
,
Sofía
Sampaolesi
,
María Victoria
Toledo
and
Laura E.
Briand
 *
*
Centro de Investigación y Desarrollo en Ciencias Aplicadas “Dr Jorge J. Ronco” CINDECA, CCT La Plata-CONICET, UNLP, CICpBA, Calle 47 No. 257, B1900AJK La Plata, Buenos Aires, Argentina. E-mail: briand@quimica.unlp.edu.ar
First published on 8th January 2025
Abstract
Massive amounts of plastic and biomass waste are mismanaged worldwide, causing detrimental consequences to human health and the environment. In fact, the disposal of residues through landfills without further processing and burning for household heating and cooking are common practices. Thermochemical processing, such as pyrolysis, chemical depolymerization and bioprocessing, has proven feasible for recovering valuable building block molecules from plastic residues. The main goal of pyrolysis is to obtain aliphatic hydrocarbons useful as fuel, while chemical processing generates constitutive molecules of plastic (i.e., monomers and polyols) that can be repolymerized and reintroduced in the market. Alternatively, the bioprocessing of plastic waste requires prior chemical depolymerization in order to unleash the building blocks. Chemo-enzymatic treatment of waste plastic-biomass mixtures is an open challenge due to the diverse composition of their residues, along with the presence of additives and contaminants. The few reports found in the literature regarding the bioprocessing of plastic residues with lignocellulosic biomass and paper indicate that chemical pretreatment cannot be avoided and that some substances present in the residues can act as fermentation inhibitors that affect waste bioprocessing.
Sustainability spotlightThe depletion of fossil feedstocks and CO2 emissions have urged governments and the research community towards upgrading industrial processes to more eco-friendly chemo-biological based technologies. The use of plastic and biomass wastes as biorefinery feedstocks represents an unlimited and ubiquitous alternative that can be adapted to each country, region and climate's availability of renewable resources. In particular, the bio-based valorization of mixed wastes, such as plastic combined with textile residues, biomass or food waste, is an emerging research field that needs further development for industrial application. |
1. Motivation and outline of the review
The development of novel technology towards the conversion of wastes into valuable substances is at the cutting edge of the scientific community's interest. Nevertheless, attempts to process complex mixtures of wastes have been assessed only recently. The most recent advances in the valorization of residues will be addressed in this review, with special attention on the chemical, catalytic and biological treatment of mixtures of various types of plastics and mixtures of plastic and biomass wastes. This overview of cutting-edge processes for mixed waste valorization provides avenues and opportunities for further advancement of research devoted to solving the billions of tons of waste dumped around the world.Before discussing the more recent investigations concerning the valorization of plastics and plastic-biomass waste mixtures, it is important to present fundamental aspects regarding the magnitude of the problem in terms of the quantity of plastic waste, end-of-life management and emission of greenhouse gases (GHG) as carbon dioxide equivalent (CO2e).
2. Global plastic residue generation, disposal and environmental impact
According to the reports of the International Energy Agency and the World Bank, 44% of the worldwide waste is composed of biodegradable type of residues, such as food leftovers, food industry residues (i.e., potato peel, waste cooking oil, etc.) and green waste that includes tree pruning, grass clippings, branches, wood chips, bark, wood, palm trees and branches, and weeds.1,2 Moreover, 17% of the waste is paper and cardboard, and 12% is of plastic origin. Nowadays, plastic residues get worldwide attention owing to the debris found in the ocean and the detection of microplastics in water streams. The Organization for Economic Cooperation and Development (OECD) estimated that this year, around 23.5 million tons of macro- and microplastic waste leaked into the environment around the world.3Recently, Cottom et al. published a global macroplastic pollution emission data analysis.4 Interestingly, the authors defined the term “pollution emission” as materials that have moved from the managed or mismanaged system (controlled or contained state) to the uncontrolled or uncontained state, which is the environment. This is important because the analysis is focused on plastic waste management rather than the amount of plastic produced. The findings of Cottom et al. demonstrated that 52.1 million metric tons per year (Mt per year) of plastic debris are not adequately managed worldwide, which would have an impact on the carbon and environmental footprints. India generates the largest amount of plastic pollution, accounting for 9.3 Mt per year, followed by Nigeria (3.5 Mt per year), Indonesia (3.4 Mt per year) and China (2.8 Mt per year).
Going deeply into the relevance of proper management of plastic waste, Fig. 1 shows the amount of plastic produced per capita and recycled in various countries; the percentage of recycled plastic based on the total amount of plastic waste is indicated above each column.5 In this context, South Korea possesses the highest percentage of plastic waste recycling (60%), followed by various European countries, such as Germany, Denmark, Belgium and Norway (48–35%). The United States, one of the largest waste producers per capita (811 kg), recycles only 23%.
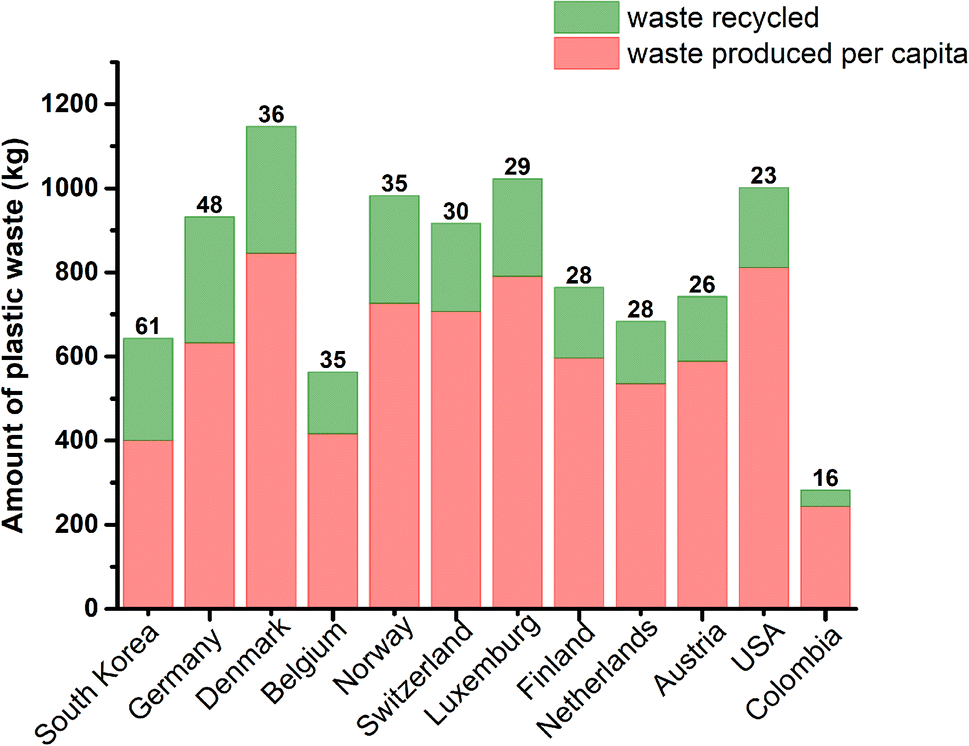 | ||
| Fig. 1 Amount of plastic produced per capita and recycled (in kg) in various countries and the percentage recycled indicated above each column. | ||
Landfills, even though it leads to long-term environmental contamination, are the end disposal of 40% of the global plastic waste, as depicted in Fig. 2. In addition, 32% goes directly to open non-regulated dump sites, and only 8% is disposed of in sanitary landfills with gas collection systems.2,6 In fact, plastic management is directly related to the socio-economic profile since low-income countries dump 93% of their plastic (solid) waste. Non-regulated dumping causes the pollution of waterways, which in turn generates marine litter as microplastics, accounting for 11.6–21.1 Mt in the Atlantic Ocean in 2020.6 In addition, non-regulated dumping goes along with the open burning of plastics within all types of solid garbage and the emission of harmful gases and ashes.7 For instance, bottles made of polyethylene terephthalate (PET) release CO2, methane, formaldehyde and polycyclic aromatic hydrocarbons; grocery bags made of high-density polyethylene (HDPE) produce olefins, aldehydes, CO and aromatic compounds; foam cups of polystyrene (PS) generate styrene gas, acrolein, hydrogen cyanide; and curtains, made of polyurethane (PU), release phosgene, among others.7
 | ||
| Fig. 2 Carbon footprint of plastic production and environmental impact of end-of-life management. Percentage accounts for landfilling (72%), combustion (14%) and recycling (9%) of plastic residues. | ||
Waste-to-energy incineration (WtE) is the end disposal of 14% of the plastic waste. This method involves CO2 emissions unless a technology for carbon capture and storage (CCS) or carbon capture and utilization (CCU) is applied downwards. Currently, only the Netherlands has three operational CCU facilities, one large-scale and two pilot plants.8 The former can process 360.635 tons per year of waste with 60 kt per year of CO2 capture through absorption. Norway and Japan also have operational pilot WtE-CCU plants.
Rubio-Domingo and Halevi gathered and analyzed various reports of the GHG emissions generated by the plastic end-of-life management option.9 The authors concluded that landfilling and mechanical recycling have the lowest GHG among the disposal methods. On the other hand, incineration possesses the highest emissions, with 1–2.5 kg CO2e per kg for WtE and 1.8–2.0 kg CO2e per kg (per kg of plastic) for incineration without an energy conversion-associated process. The investigation of Rubio-Domingo and Halevi also considered that gasification (0.2–1.8 kg CO2e per kg) and pyrolysis (almost zero emission) are low-emission methods. This last method will be further discussed in the following sections due to its low environmental impact and high potential to generate valuable substances.
The term “CO2e” means CO2 equivalent and is used to compare the emissions from various greenhouse gases GHG based on their global warming potential (GWP) by converting amounts of other gases to the equivalent amount of carbon dioxide with the same global warming potential. This concept is related to the carbon and environmental footprints of a product. The former is the total amount of GHG generated along the life cycle of a product. Moreover, the environmental footprint (also called the Life Cycle Assessment) involves not only the GHG emissions but also the environmental impact caused by particulate matter emission, human toxicity, ozone depletion, eutrophication, land use, resource depletion, among others.
Zheng and Suh10 calculated an emission of 1.8 Gt CO2e of fossil fuel-based plastics along their life cycle in 2015. By 2020, that number increased to 2.2 Gt CO2e and is projected to grow 31% by the year 2030 unless mitigation actions took place.11 The major contributors to GHG emissions at the resin production stage (the most polluting one) are polypropylene (PP), polyurethane (PU), low-density polyethylene (LDPE), high-density polyethylene (HDPE) and polyethylene terephthalate (PET).10 In this context, various strategies for the valorization of actual residues based on those types of plastics will be discussed in the following sections.
The carbon footprint is calculated considering that the life cycle of plastic involves coal combustion for the resin-production stage, which includes all activities from cradle to polymer-production factory gate, accounting for 61% of the total emission. In addition, the conversion stage covers the manufacturing processes that turn polymers into final plastic products (30% of the global emission), and the end-of-life stage refers to the treatment and disposal processes of plastic waste with 9% CO2e emission. Zheng and Suh pointed out that further efforts towards bio-based plastics and renewable energy (wind power and biogas) sources, lowering the demand, and recycling are the keys to diminishing the carbon footprint of plastics.10
Currently, only 9% of all the plastic waste is recycled. This observation is a driving force towards the development of valorization processes applicable to large amounts of plastic waste. In this context, various investigations of thermo-chemical-biological strategies of plastic waste recycling towards valuable platform molecules are discussed in the following sections.
3. Strategies of valorization of plastic residues towards valuable products
Nowadays, mechanical processing is the main route for recycling waste plastics from various sources.1,12 This methodology involves the classification of the collected waste according to the polymers' nature and color. Then, it is washed and mechanically ground into a secondary raw material in the form of plastic flakes.12 Then, the flakes are melted (extrusion stage) and filtered to remove impurities. This recyclate that is ready to be reused in new plastic products is generally of a lower quality than starting virgin-grade plastics, mainly due to the changes in the polymer structure during the melting process.In contrast, chemical and biological recycling pursues the breaking down of the polymer into valuable molecules suitable for conversion into new materials. The so-called tertiary recycling of plastics comprises the pyrolysis and hydrolysis of the wastes.13,14 Those processes often involve a sequence of procedures that might begin with the mechanical treatment, followed by a chemical (catalytic or not) process and further biotransformation of the obtained molecules.
The biological upgradation of those building block molecules uses biocatalysts based on enzymes or microorganisms. Microbial bioprocessing of plastics involves the assimilation and mineralization of carbon degradation products to build more complex molecules. In contrast, the enzymatic treatment produces substances that can be further valorized into second-generation products.
Efficient recycling and valorization of plastic waste are challenging since there is a large number of different plastics, many of them consisting of a combination of different polymers as well as the presence of additives, such as plasticizers, fillers and reinforcements, thermal stabilizers and antioxidants, colorants, metals, among others. The variety and complexity of their composition is a drawback that traditionally involves multiple processing steps to be overcome.
3.1. Thermochemical (chemical and pyrolytic) based treatments: an overview
Tables 1–4 show a compilation of the latest reports on thermochemical and chemical methods that use plastic residues as feedstocks. In particular, the nature of the process, operative conditions, yield, recovery, purification and valorization have been addressed in the treatment of polyethylene terephthalate,14–22 polyethylene,23–30 polypropylene31–38 and polyurethane39–44 based wastes.| Feedstocks | Type of process | Catalyst | Operative conditions | Products and yield | Recovery and purification | Downstream valorization | Reference | |
|---|---|---|---|---|---|---|---|---|
| a PET, polyethylene terephthalate; TPA, terephthalic acid; EG, ethylene glycol; DMT, dimethyl terephthalate; BHET, 2-hydroxyethyl terephthalate. | ||||||||
| 1 | Carpet waste | Steam catalytic slow pyrolysis | CaO, ZSM-5 | Slow heating up to 750 °C, 5 °C min−1, 0.11 sccm steam, Ar flow | Gaseous phase (CO), 54% benzene | Condensable vapor collected in an impinger with methanol in a dry ice bath | None | |
| 2 | Waste bottles | Acidic hydrolysis | Concentrated acids | 3 M H2SO4, 150 °C, 10 h | 95% TPA and EG | Ethylene glycol is difficult to recover due to carbonization in strong acid solutions | None | 14 |
| 3 | Powder | Alkaline hydrolysis | Aqueous solution of NaOH | 1,3-Dimethyl-2-imidazolidinone (80 v%)-EG-NaOH (1.41 g), 70 °C, 30 min | 100% TPA and EG | Precipitated disodium TPA is filtered and decomposed with water to recover pure TPA | None | 15 |
| 4 | Waste bottles | Neutral hydrolysis | None | Water (liquid, compressed liquid, supercritical, vapor), 200–400 °C, 30 min–2 h | >80% TPA in 60 s fast hydrolysis mode | Precipitated TPA is dissolved in dimethyl sulfoxide | None | 16 |
| 5 | Waste bottles and PET-cotton fabrics | Transesterification with methanol (methanolysis) | K2CO3 | 70 °C, 300 rpm, 20 h, dichloromethane | 71% DMT (bottles) 42% DMT (fabric) | Filtration of residuals, cake and liquor containing DMT | None | 17 |
| 6 | Waste bottles | Methanolysis | Bamboo leaf ash | 200 °C, 2 h, autoclave | 78% DMT, 76% EG | Crystallization of DMT at 2 °C, 4 h | None | 18 |
| 7 | Waste bottles | Aminolysis | None | 1,2-Diaminopropane, 130 °C, 24 h, addition of methanol at the end of the reaction | N,N-bis-(2-aminopropyl)-terephthalamide | Removal of solvent, drying under vacuum | Synthesis of a Schiff-base with 25% yield | 19 |
| 8 | Waste bottles | Glycolysis | SO42−/Nb2O5 | EG, 195 °C, 3 h | 85% BHET, oligomers | Filtration and crystallization at 5 °C, 16 h | None | 20 |
| 9 | Bottle chips | Glycolysis | Deep eutectic solvent catalyst ChCl/Zn(AcO)2 | EG, 90 °C, pretreatment of PET with acetonitrile | 90% BHET | Cooling to −10 °C, 10 h; filtration to recover BHET, oligomers, unreacted PET. Dissolution in H2O at 60 °C and filtration. BHET crystallization at 5 °C | Polymerization of BHET to PET with recycled EG | 21 |
| 10 | Bottle, shirt, pillow | Hydrogenolysis | Hf(OTf)4, Pd/C | H2, 265 °C, 24 h | 95–97% TPA | None | None | 22 |
| Feedstocks | Type of process | Catalyst | Operative conditions | Products and yield | Recovery and purification | Downstream valorization | Reference | |
|---|---|---|---|---|---|---|---|---|
| a PE, polyethylene; LDPE, low density polyethylene; HDPE, high density polyethylene; FCC, fluid catalytic cracking. | ||||||||
| 1 | Low density LDPE scrap | Pyrolysis | None | N2, 500–900 °C | Propylene, propane, ethylene; C18–C35 and hydrocarbons | Two stage condensation at −1 °C and −40 °C | Products used as fuels without further modification | 23 |
| HZSM-11 | Propylene, propane, ethylene; C18–C35 and C8–C21 | |||||||
| 2 | LDPE waste carry bags | Pyrolysis | ZnO | N2, 30 mL min−1, 300 °C | 67 wt% oil with 40% aromatics, 41% aliphatic and cyclic hydrocarbons | Condensation at low temperature | 24 | |
| 3 | Post-consumer mixed polyolefinic wastes (75 wt% PE. 16 wt% PP) | Non-catalytic pyrolysis followed by catalytic cracking | HZSM-5 FCC catalyst | He, 60 mL min−1, pyrolysis at 550 °C, upgrading at 600–700 °C | 31 wt% propylene, 18 wt% ethylene | None | Conversion of pyrolysis products to C2–C4 olefins and aromatics | 25 |
| 4 | LDPE and HDPE waste | Microwave-assisted pyrolysis | ZSM-5, zeolites, MgO, SiC | 450–550 °C, 10–90 min | 24–57% oil (hydrocarbons in the gasoline and diesel fractions) | None | Products used as fuels without further modification | 26 |
| 5 | LDPE and HDPE waste; mixtures of PE and biomass | Pyrolysis and catalytic steam cracking of pyrolytic oil | None | 550–1000 °C, N2, fluidized bed, batch and conical spouted bed reactors | 15–48% Ethylene, other hydrocarbons | Distillation, adsorptive separation on molecular sieve | Polymerization to virgin polyethylene | 27 |
| Zeolites (Y, HY, ZSM-5, HZSM11), spent FCC, Al2O3, Al(OH)3 | 480–700 °C, N2, fluidized bed, batch and conical spouted bed reactors | 28% ethylene after steam cracking of pyrolytic oil | ||||||
| 6 | Mixture HDPE and poplar sawdust | Co-pyrolysis | MgCl2, ZSM-5 | Biomass/HDPE ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 500–600 °C, N2 :1, 500–600 °C, N2 |
55% Bio-oil enriched in toluene and xylene | Condensation of pyrolytic vapors | None | 29 |
| 7 | Spherical pellet | Hydrocracking | Pt/USY zeolite | H2, 350 °C, 1 h | 65.9% oil, 16.6% gas | Dissolution in CH2Cl2, filtration | None | 29 |
| 8 | Bottle | Hydrocracking | Pt/WO3/ZrO2, HY(30) zeolite | H2, 250 °C, 1 h | 20–30% C8–C12, 40–50% C5–C7, 10% C1–C4 | Filtration | None | 30 |
| Feedstocks | Type of process | Catalyst | Operative conditions | Products and yield | Recovery and purification | Downstream valorization | Reference | |
|---|---|---|---|---|---|---|---|---|
| a PP, polypropylene; PET, polyethylene terephthalate; LDPE, low density polyethylene; HDPE, high density polyethylene; FCC, fluid catalytic cracking. | ||||||||
| 1 | Personal protective equipment (face masks) | Flash, fast, slow pyrolysis | 5 wt% Ni/SiO2 | 450–600 °C, 15–180 °C min, 30–60 min, N2, CO2 | 33% char (slow pyrolysis); H2 and CH4 (catalytic pyrolysis) | None | None | 31 |
| 2 | PP plastic waste | Pyrolysis | Spent FCC catalyst | 700 °C, 10 °C min | 60% oil, 30% gas; 34% unsaturated aliphatics | Condensation at 10 °C | None | 32 |
| 3 | PP plastic waste | Pyrolysis | FCC catalyst, natural zeolite | 450–550 °C | 82–92 wt% oil | None | None | 33 |
| 4 | PP plastic waste; LDPE, HDPE waste | Pyrolysis | ZSM5, activated alumina, FCC catalyst, halloysite clay | N2, 30 mL min−1, 410–490 °C, 4 h, 2 wt% catalyst | 70% oil from PP; 70–80% olefins, 20% paraffins and cycloalkanes | Condensation | None | 34 |
| 5 | Packaging from ice cream | Oxidative thermolysis in aqueous media | None | 30% H2O2, 150 °C, 14 bar O2, 327.6 bar CO2, autoclave, 24 h | 74% acetic acid, 17% methanol, 7% propionic acid | None | None | 35 |
| 6 | Colorless and colored PP waste | Hydrothermal degradiation in supercritical water | None | 425–450 °C, 15–240 min, batch Parr reactor, 5 mL H2O per 1 g PP, 290–400 bar | 95% oil, 20% gas; 33% alkanes, 29% alcohols; 55% C2–C4, 20–32% propane | Removal of non-degraded plastic | None | 36 |
| 7 | PP and PET waste | Hydrothermal degradiation in sub/supercritical water | None | 400–415 °C, autoclave, N2, 30–120 min, water/plastic ratio from 1:5 to 1:40 |
17% H2 (PET), 25% H2(PP); 80 mg g−1 benzoic acid (PET); 130 mg g−1 4,4,5-trimethyl-2-hexene | Removal of non-degraded plastic through filtration | One | 37 |
| 8 | PP waste | Hydrocracking | NiMo/Al2O3 | H2, 450 °C | 86 wt% liquid hydrocarbons | Condensation | None | 38 |
| Pt/Al2O3 | ||||||||
| Feedstocks | Type of process | Catalyst | Operative conditions | Products and yield | Recovery and purification | Downstream valorization | Reference | |
|---|---|---|---|---|---|---|---|---|
| a PU, polyurethane; EG, ethylene glycol; DEG, diethylene glycol; MDA, methylenedianiline; DCA, dicarboxylic acid. | ||||||||
| 1 | Seat foam of end of life vehicle | CO2 assisted pyrolysis | 5 wt% Ni/SiO2 | 30–700 °C, 10 °C min, N2, CO2 | Syngas (5.65 mol% CO, 1.65 mol% H2) | None | Oxidation of aromatics with CO2 towards syngas | 39 |
| 2 | Retired wind turbine blades | Co-pyrolysis of PU and epoxy resin | None | 400–800 °C, 10 °C min | Aldehydes, ether, alcohols, epoxy compounds | None | None | 40 |
| 3 | PU scraps | Glycolysis and deamination | Potassium acetate | 2-Ethylhexyl glycidyl ether, acetic anhydride, diethylene glycol, 200 °C, 2 h | 100% conversion of MDA towards EG at 120 °C, 2 h | None | Rigid foams synthesis with recovered EG | 41 |
| 4 | PU wastes | Acidolysis | AlCl3, ZrO2, WO3 | HCl (60 °C), dicarboxylic acids (190–210 °C), inert gas, 5 h | Amine salt (with HCl), amide (DCA), 90% polyol, CO2 | Not informed | Adhesives for polymerization, synthesis PU foam | 42 |
| Hydrolysis | NaOH, NH3 | Pressurized water at 150–200 °C, steam 200–450 °C, inert gas or 80 bar CO2 | Diamine and polyol in aqueous phase, oligomers in oil phase, CO2 | Separation of the aqueous phase | ||||
| 5 | PU wastes | Aminolysis | NaOH, Al(OH)3, CH3ONa | Aliphatic and aromatic mono and polyamines, cyclo-aliphatic and heterocyclic amines, alkanolamines, NH3, NH4OH, 80–190 °C, inert gas, solvents | Non-miscible phases of polyol and disubstituted ureas | Separation of phases for product recovery | None | 42 |
| Glycolysis | NaOH, NaOAc, FeCl3, ionic liquids | EG, DEG, inert gas, 220 °C, 15 min–2 h | Non-miscible phases of polyol and ether polyol (90% conversion) | Direct reusability of products | Synthesis of rigid PU foam with propylene oxide | |||
| 6 | Rigid PU foam waste | Microwave-assisted glycolysis | Potassium acetate, stannous octoate, monoethanol amine | DEG, 200 °C, 15 min, 3–50 mmol catalyst/100 g PU | 0.4–2.5 wt% MDA; 475–550 mgKOH per g hydroxyl value for polyols | Direct use of polyols mixture in polymerization | Synthesis of PU | 43 |
| 7 | Upholstery foam from an office chair | Hydrogenolysis | Mn-complex, t-BuOK | H2, toluene, THF, 200 °C, 48 h | 81% conversion towards MDA, formate and polyol | None | None | 44 |
In general, pyrolysis is a thermochemical process carried out under an inert gaseous environment (non-oxidative atmosphere) provided by argon or nitrogen. More recently, the use of carbon dioxide has also been investigated, as will be discussed later on. The process involves the decomposition of a substrate through heating carried out either in a slow or fast mode, with or without a catalyst, in a batch, fluidized or spouted bed reactor.
The pyrolysis generates a liquid fraction called oil or biooil (if coming from biomass), composed of organic molecules that are lighter than the ones of the feedstock, a non-condensable gaseous fraction and a solid phase composed of a carbonaceous material (char or biochar). It is obvious that the nature of the pyrolysis products is related primarily to the starving oxygen environment that suppresses gasification and combustion, preserving the integrity of the organic molecules and avoiding the generation of non-condensable gases. Secondly, the abundance of the liquid, gaseous and solid fractions and their composition depends on the heating rate and the temperature. Slow heating (slow pyrolysis), high residence time of the inert gas with the substrate (5–60 min) and temperatures from 300 °C to 650 °C yield a higher proportion of the solid fraction. Fast heating (fast and flash pyrolysis), low residence time (0.5–1.0 s) and temperatures in the range of 450–600 °C improve the yield of oil.33
In turn, the pyrolysis might use a catalytic material in order to direct the decomposition towards targeted reactions and products. For instance, the non-catalyzed pyrolysis of PET typically yields terephthalic acid, benzoic acid vinyl ester and acetophenone. However, the pyrolysis of PET waste carpet catalyzed with basic material such as CaO with steam co-feeding directs the deoxygenation of PET's oligomers towards benzene14 (see Table 1, row 1).
The pyrolysis of low-density polyethylene (LDPE) waste in the presence of HZSM-11 zeolite yields light aliphatic hydrocarbons in the C8–C21 range rather than the C18–C35 obtained in the non-catalyzed process.23 The acidic material catalyzes not only the cracking of hydrocarbons but also the dehydrogenation of propane towards propylene (see Table 2, row 1).
In this context, the typical heterogeneous catalysts used in pyrolysis are aluminosilicate materials such as zeolites (ZSM-5, ZSM-11, Y, HY)14,23,25–27,33,34 and the commercial catalyst used in the fluid catalytic cracking (FCC) process of petroleum refinery25,27,32,33 (see Table 2, rows 3 and 5; and Table 3, rows 2, 3 and 4).
Recently, Kanattukara et al. published a detailed investigation of the influence of various catalysts, such as ZSM5, activated alumina, FCC catalyst and halloysite nanotube clay, in the pyrolysis of wastes containing polyethylene (HDPE and LDPE) and polypropylene (PP)34 (see Table 3, row 4). The catalysts allowed the lowering of the pyrolysis temperature from 470 °C to 450 °C, reaction time from 5 h to 4 h, and improved the yield of the oil fraction compared with the non-catalyzed pyrolysis. These observations were attributed to the acidic property of the materials that catalyze the cracking of fragments initially produced in the pyrolysis to even lower molecular weight hydrocarbons. In general, 70–80% of the oil was composed of olefins, followed by 20% of paraffins and cycloalkanes and a minor content of aromatic compounds.
Overall, catalytic pyrolysis is intended to improve the amount of the liquid fraction towards suitable hydrocarbons to be applied as fuels. More precisely, C5–C15 hydrocarbons containing olefins and aromatic compounds are key pyrolytic products. This goal is achieved primarily in the catalytic pyrolysis of polyethylene wastes, as shown in Table 2.23–27 In some cases, a tandem pyrolysis, that is, two successive pyrolysis and tandem pyrolysis-steam cracking, was applied to further tune the desired products25,27 (Table 2, rows 3 and 5).
Typically, pyrolysis is carried out through conventional conduction heating; that is, the feedstock is heated up at the surface and then the energy is transferred to the inner part of the particles. Microwave-assisted pyrolysis uses radiation that directly penetrates plastic material (of insulating nature) without absorption or is absorbed by dielectric materials (i.e., biomass) that are heated from the inside out. In this context, Table 2 (see row 4) shows that microwave pyrolysis of polyethylene (LDPE and HDPE) generates a high proportion of oil enriched in hydrocarbons that can be used as fuels.26
Another non-conventional heating is plasma pyrolysis. This one provides extreme heat and high temperature (around 1200 °C) in a short period of time, leading to the generation of gases (i.e., CO, H2 and hydrocarbons) and a low proportion of residues. This process is particularly indicated for treating infectious medical plastic waste31 (see Table 3, row 1).
The pyrolysis of polyurethane (PU) gives rise to harmful aromatic compounds, such as benzene, toluene, aniline, styrene, p-xylene, methylenedianiline (MDA), among others (Table 4, rows 1 and 2). Jung et al. reported the pyrolysis of PU waste catalyzed with 5 wt% Ni/SiO2 in an N2/CO2 environment to convert those chemicals into H2 and CO (this gas mixture is known as syngas).39 The process was performed in a tandem mode through a pyrolytic reactor, followed by a second one containing the catalyst. The slow pyrolysis was carried out between 100 °C to 700 °C at 10 °C min−1, while the second reactor was set at 600 °C.
A similar approach was used to obtain syngas from the pyrolysis of disposable facemasks composed of polypropylene, polyethylene and nylon31 (see Table 3, row 1).
The non-catalyzed co-pyrolysis of PU with an epoxy resin also suppresses the decomposition of the methylene diphenyl diisocyanate monomer of PU towards HCN and aromatics (Table 4, row 2). In fact, the investigation of Wu et al. suggested that the acid sites of the epoxy resin catalyze the secondary cracking of those substances towards aliphatic hydrocarbons, alcohols, ethers and epoxides at temperatures above 500 °C.40
As observed in Tables 1–4, the pyrolytic process is a commonality in plastic waste treatment since it is suitable for application regardless of the nature of the polymeric matrix. Nevertheless, less harsh processes for plastic waste depolymerization, such as hydrolysis, glycolysis, methanolysis, aminolysis, hydrothermal degradation, hydrogenolysis and hydrocracking, have also been investigated.
Hydrolysis is the reaction with water at high temperatures performed under acidic, alkaline or neutral conditions with or without a catalyst.14 Hydrolysis depolymerizes the plastic waste into the terephthalic acid (TPA) monomer of PET and polyols in the case of PU. Table 1 (rows 2, 3 and 4) shows that the hydrolysis of waste PET bottles towards the monomer is highly effective in recovering up to 100% of TPA.14–16 Similarly, the acidolysis of PU with dicarboxylic acids produces polyol, amine and esters (see Table 4, row 4).42
Polyethylene is based on the polymerization of ethylene C2H2; therefore, the linear alkyl chains of the polymer (C2H4)n do not have polar functions. In turn, PE is rather inert and not suitable for hydrolysis. In the case of polypropylene, hydrothermal degradation with water in sub and supercritical conditions, at about 450 °C, in an inert gas at high pressure, proved effective in degrading the PP waste to an oil containing alkanes, alkenes, and alcohols, among others36,37 (see Table 3, rows 6 and 7). The hydrolysis of ice cream packaging under oxidative conditions, provided by hydrogen peroxide and CO2, yields mostly acetic acid at a lower temperature (150 °C vs. 450 °C) than the process described before35 (Table 3, row 5).
Glycolysis comprises the cleavage of the ester bonds of PET with ethylene glycol to release oligomers, dimers and finally, 2-hydroxyethyl terephthalate (BHET)20,21 (see Table 1, rows 8 and 9). Typically, heterogeneous catalysts, such as metal (Zn, Mn, Co and Pb) salts, sulfated niobia, ZnMn2O4, g-Fe2O3, zeolites and silica nanoparticles, are used.13,20 More recently, deep eutectic solvent catalysts have been successfully used in the glycolysis of PET waste with an essential reduction of the reaction temperature compared with the heterogeneous catalyzed process21 (Table 1, row 9). PU also undergoes glycolysis through the reaction of the urethane group NHCOO with diethylene glycol, releasing polyol and carbamate compounds, R1NHCOOR2. The nature of the latter depends on the isocyanate that was originally used for synthesizing the polymer. Most frequently, the isocyanate is diphenylmethane-4,4′-diisocianate, which provides the carcinogenic amine 4,4′-methylendianiline (MDA) upon PU glycolysis.41,43,44 In this context, Donadini et al. studied the reaction of MDA with 2-ethylhexylglycidyl ether, acetic anhydride and ethylene carbonate in order to lower its concentration in the reaction media.41 The deaminated solution was then successfully used to synthesize new rigid PU foam (see Table 4, row 3). Microwave-assisted glycolysis of rigid foams made of PU catalyzed with potassium acetate and stannous octoate proved far less time-consuming and energy-saving than conventional heating (Table 4, row 5). The combination of the catalysts and diethylene glycol provided a dielectric media for efficient heating that led to PU depolymerization towards polyols and a low content of the harmful MDA.43
The depolymerization of PET waste through methanolysis and ethanolysis implies the transesterification with methanol or ethanol from 70 °C to 200 °C catalyzed with zinc acetate, potassium carbonate or biomass ashes.14,17–19 The reaction yields dimethyl terephthalate and diethyl terephthalate, among other substituted monomers containing the terephthalate backbone (see Table 1, rows 5 and 6).
Aminolysis involves the reaction with an aqueous solution of a primary amine at 20–200 °C under an inert environment with and without a catalyst.13,14 This process was investigated in the depolymerization of PET13,14,19 and PU.42 The aminolysis of PET might be carried out with ethanolamine, ethylene diamine, allyl amine, hydrazine hydrate, hydroxylamine hydrochloride or alkyl amine, yielding bis(-2-hydroxyethyl) terephthalamide, bis(2-aminoethyl) terephthalamide; N,N′-diallyl terephthalamide, terephthalic dihydrazide, terephthalohydroxamic acid and N,N′-dialkyl terephthalamide, respectively.13 In particular, the non-catalyzed solvent-free aminolysis of PET bottle waste with 1,2-diaminopropane at 130 °C provided a water-soluble amide of the monomer. This product is suitable for condensation with salicylaldehyde towards a Schiff base19 (see Table 1, row 7).
Table 4 (row 4) shows that the aminolysis of PU generates a biphasic system containing an upper phase with a polyol and a bottom one with disubstituted ureas. In this context, the polyols can be easily recovered and reused to synthesize new PU. In addition, urea might be further valorized through decomposition in amines and CO2.42
Hydrogenolysis is the selective scission of C–C and C–O bonds through reaction with hydrogen. Typically, metal particles in acid or basic media are used as catalysts.45 Depolymerization of plastic through hydrogenolysis has gained attention because numerous studies have demonstrated that polyolefins are successfully converted to short-chain hydrocarbons.46 At 200–250 °C, liquid products are obtained from polyolefins, and the selective alkoxy C–O bond hydrogenolysis of polyesters drives high yields of the terephthalic acid (see Table 1, row 10).
Table 4 (row 6) shows that the hydrogenation of upholstery PU foam catalyzed with Mn-complex and t-BuOK generates methylenedianiline, the corresponding formate and the polyol.44
Hydrocracking is a process that converts heavy plastic molecules into lighter molecules by breaking the long polymer chains in the presence of hydrogen with a bifunctional metal/acid catalyst.46,47 Zeolites are often used to catalyze these reactions due to their strong acidity, high thermal and hydrothermal stability and regeneration capacity. Furthermore, the porosity of zeolites allows accessibility of certain reactants to the reaction sites, leading to high selectivity.48 Efficient hydrogen transfer from the hydrogen donors to the polyolefins is crucial to this process. Typically, the reaction temperature is 150–450 °C, and the hydrogen pressure is between 20 and 100 bar. Hydrocracking of PE or PP with a bifunctional metal/acid catalyst can achieve a liquid yield of over 60% and the distribution of hydrocarbons in liquid fuels will depend on the metal and acid sites of the catalyst49,50 (see Table 2, rows 7 and 8 and Table 3, rows 8 and 9).
3.2. Chemo-biological valorization of plastics residues
The section above presented the investigations regarding the chemical and thermochemical methods for the valorization of plastic wastes reported in the past 5 years. In comparison, biodegradation is a more environmentally friendly approach since bioprocesses use milder reaction conditions, such as room temperatures and mid-range pH. More interestingly, they avoid or minimize the use of biologically incompatible-toxic-reagents. Interaction of bacteria and fungi with plastic waste has been successfully applied to degrade the polymers into shorter low-molecular weight chains under aerobic or anaerobic conditions.51,52 Till today, three different strategies have been explored, including fungal, bacterial (isolates and consortia) and enzymatic biodegradation (native and bio-engineered isolated enzymes). Each of these is best suited to depolymerize different post-consumer plastics, as we will review in this section.Combined strategies that valorize plastic waste through both chemical and biotechnological methods were designed to overcome the challenges of deconstructing highly crystalline polymers, such as PET or PU. Table 5 gathers reported tandem processes involved in the treatment of plastic wastes to recover valuable building block molecules. In addition, the biological and enzymatic-based technologies developed for further valorization of the recovered substances are summarized and illustrated in Fig. 3A and B. Chemo-enzymatic treatment of textile waste based on polyesters was investigated by Quartinello et al.53 As a first step, the plastic waste was hydrolyzed in an aqueous environment at 250 °C and 40 bar (neutral hydrolysis), which depolymerized 85% of the PET fibers into terephthalic acid and oligomers (see Table 5, row 1, Fig. 3A). This energy-consuming pre-treatment was necessary to degrade highly crystalline fractions of PET into oligomers that enzymes can catabolize. The oligomers were further hydrolyzed with 1–2 mg mL−1 of Humicola insolens cutinase, an enzyme from the α/β hydrolase family, at pH 7, 50 °C, for 6 h. The overall process provides 97% of terephthalic acid (TPA).
| Feedstock | Chemical treatment | Downstream bio-valorization | Recovery and purification | Reference | |||
|---|---|---|---|---|---|---|---|
| Process | Product yield | Process | Product yield | ||||
| a PET, polyethylene terephthalate; PS, polystyrene; TPA, terephthalic acid; EG, ethylene glycol; DEG, diethylene glycol; BHET, 2-hydroxyethyl terephthalate; MHET, mono(2-hydroxyethyl) terephthalate; LDPE, low density polyethylene; LLDPE, linear low density polyethylene; HDPE, high density polyethylene; BH medium, Bushnell Haas medium. | |||||||
| 1 | Polyester-composed waste textiles | Neutral hydrolysis, 250 °C, 39 bar, 1/10 PET/water ratio, 90 min | 85% TPA, oligomers | Enzymatic hydrolysis of PET oligomers, 1–2 mg mL−1Humicola insolens cutinase, pH 7, 50 °C, 6 h | 97% TPA | None | 53 |
| 2 | Waste PET bottles | Catalytic glycolysis, EG, 190 °C, 5 h, orange peel ash catalyst | 92% BHET | Bacterial degradation of BHET, 30 °C, 5 bacteria strains | 35% degradation of BHET to TPA and EG after 10 weeks | None | 54 and 55 |
| 3 | Waste PET cups | Catalytic glycolysis and enzymatic hydrolysis, K2CO3 catalyst, commercial esterases, 30 °C, pH 7.5 | 97.7% TPA | Whole-cell bioconversion of TPA with a catechol biosynthetic strain in Escherichia coli, 30 °C, 20 h | 99.5% Catechol | BHET, MHET, oligomers and EG directly used in the bioconversion | 56 |
| 4 | Post-consumer plastic waste (HDPE, PE, and PS) | Catalyzed chemical oxidation with 11.3% N-hydroxyphthalimide, 8 bar O2, 72 bar N2, 210 °C, 5.5 h, Co–Mn catalysts | 59–65% benzoic acid, 20% C2–C22 dicarboxylic acids, 60% TPA | Bioconversion of oxygenates with engineered Pseudomonas putida, 36 h | 66% 3-hydroxydodecanoic and 34% 3-hydroxydecanoic acids; 57% β-ketoadipate | Metal-catalysts recovery, mixed products used directly in the bioconversion | 59 |
| 5 | LDPE and HDPE waste from soil and sludge of a cooling tank | Catalytic hydrogenolysis, 5 wt% Ru/C, 30 bar H2, 250 °C, 3 h | C4–C35 liquid alkane mixture | Bioconversion with a microbial consortia growth as a biofilm on LDPE, Rhodococcus aetherivorans strains, BH medium, 30 °C, 14 days | 71–85% alkane conversion, 36 mg g−1 cetyl palmitate, 9.7 mg g−1 1-hexadecanol, 3.6 mg g−1 myristyl palmitate | Condensation of alkanes and removal of catalyst | 62 |
| 6 | Post-consumer PU foam from a pillow | Catalyzed glycolysis, DEG, 200 °C, tin(II)-2-ethylhexanoate catalyst, 2 h | Polyols; 2,4 and 2,6-toluene diamine | Enzymatic hydrolysis of dicarbamates, metagenomic urethanases, pH 10, 30–70 °C, 48 h | 65% Conversion, aromatic diamine, DEG, CO2 | Recovery of the top with polyols and bottom layer with dicarbamates | 69 |
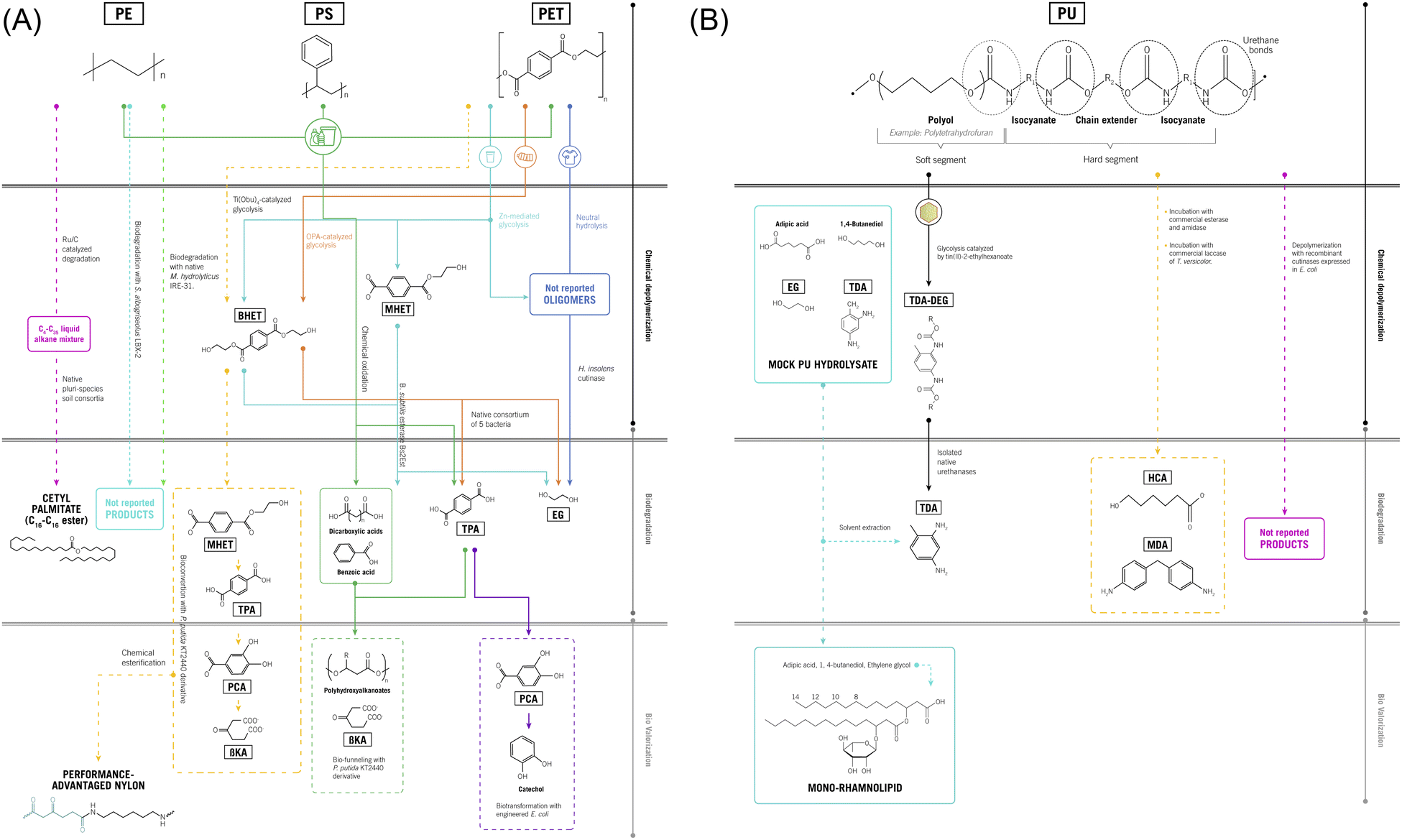 | ||
| Fig. 3 (A) Combination of chemical, biological and enzymatic based technologies for recovery and valorization of platform molecules from PET, PS, PE and blends of those plastics. PET: polyethylene terephthalate; PE: polyethylene; PS: polystyrene; EG: ethylene glycol; MHET: mono(2-hydroxyethyl) terephthalate; BHET: bis(2-hydroxyethyl) terephthalate; TPA: terephthalic acid; βKA: β-ketoadipic acid; PCA: protocatechuate. --→ Research using plastic wastes as feedstock. -.-.→ Research using model polymers. (B) Combination of chemical, biological and enzymatic based technologies for recovery and valorization of platform molecules from PU. PU: polyurethane; TDA: 2,4-toluenediamine; MDA: 4,4′-methylendianiline; DEG: diethylene glycol; HCA: 6-hydroxycaproic acid. --→ Research using plastic wastes as feedstock. -.-.→ Research using model polymers. | ||
Going even further in the biorefinery concept, Shingwekar et al. developed a two-step process for the depolymerization of post-consumer PET bottles to 92 wt% of bis(2-hydroxyethyl) terephthalate (BHET).54 The glycolysis performed at 190 °C for 1.5 h was catalyzed with ashes obtained from orange peel (Table 5, row 2, Fig. 3A), yielding a biocompatible mixture rich in BHET suitable for biological degradation since the crystalline fraction of PET was eliminated. A native consortium of five bacteria strains, investigated previously by León-Zayas et al., was isolated from soils polluted with petroleum products, taking advantage of the rapid adaptation and evolution capacities of bacteria.55 Composed of Bacillus thuringiensis C15, Bacillus albus, Pseudomonas sp. B10, Pseudomonas sp. SWI36 and Pseudomonas sp. PFYNo1, the consortium uses BHET more readily than PET, synergistically degrading the glycolysis products by 62.63% in 2 weeks towards TPA and ethylene glycol (EG). The overall 2-step process stands as a sustainable methodology to valorize plastic waste without using toxic or costly reagents.
In a similar approach, Kim et al. investigated the chemical glycolysis and enzymatic hydrolysis of PET cup waste, followed by the biological transformation of TPA towards catechol.56 Altogether, the cascade-type process comprises the glycolysis of the polymer to BHET and mono(2-hydroxyethyl) terephthalate (MHET) with potassium carbonate K2CO3 catalyst, followed by the enzymatic degradation of BHET, MHET and oligomers to TPA (Table 5, row 3, Fig. 3A). Four commercial esterases from Bacillus subtilis, Paenibacillus barcinonesis Rhizopus oryzae, and Methylobacterium populi, were assayed. The latter catalyzed a complete depolymerization of BHET at a loading of 41.8 μg mL−1, 30 °C in 10 h. Finally, the biotransformation of terephthalic acid to catechol was achieved with a bioengineered strain of Escherichia coli at 30 °C for 20 h. Addressing the complete biorefinery concept, the authors proposed a PET upcycling using catechol as a coating agent directly from the previous step by removing E. coli cells without further purification. The catechol coating provided the scaffolding to further functionalize different materials with a broad range of applications.
Previous work of Yoshida et al., reviewed by Blank et al., supply the basis for more recent studies that engineer bacteria with a set of genes codifying PET-degrading enzymes.57 The authors isolated the bacteria Ideonella sakaiensis 201-F6, capable of degrading PET and assimilating its monomers, and characterized the specific enzymes involved with unusual features, namely PETase and MHETase. The enzymes have the potential for improvement by genetic manipulation and to achieve PET depolymerization at milder temperatures and biologically relevant conditions.
A catalyzed glycolysis of model PET (not from a waste source) coupled with biological upgrading of BHET towards β-ketoadipic acid (β-KA, monomer of nylon 66) was reported by Werner et al.58 In this case, degrading enzymes from I. sakaiensis were used to transform Pseudomonas putida KT2440, obtaining a strain capable of harnessing EG by constitutive expression of native genes and BHET by heterologous expression of PETase, MHETase, TPA transporters and enzymes for TPA conversion to protocatechuate (PCA). The strain further converts BHET into β-ketoadipic acid with a molar yield of 76% at 30 °C, pH 7 in 96 h of fermentation. This scheme of chemical depolymerization coupled with biodegradation proved to be yield-efficient, but improving the biocompatibility of the glycolysis products is necessary to achieve process fluency and scaling. Valorizing the EG obtained as a by-product remains a future challenge since β-KA represses EG utilization by bacteria.58
The performance of P. putida KT2440 and engineered derivatives on post-consumer PET feedstocks is reviewed next. In this context, Sullivan et al. reported the chemical and biological processing of a mixture of high-density polyethylene, polystyrene, and poly(ethylene terephthalate), which are regular components of post-consumer plastics waste.59 The authors used expanded polystyrene cups, milk containers made of polyethylene, and single-use beverage bottles (Table 5, row 4, Fig. 3A). In the first step, the mixture was subjected to auto-oxidization and depolymerization through a catalyzed process, which leads to a random type of chain scissoring. The process, typically carried out with manganese/copper-containing catalysts and N-hydroxyphthalimide as an oxidation promoter, was performed at 180–200 °C for 5.5 h. As a result, benzoic acid, dicarboxylic acids and terephthalic acids were produced, resulting in substantial energy consumption and need for wastewater treatment. An important observation highlighted by the authors was the fact that the catalytic treatment in an oxygen atmosphere, unlike typical pyrolysis, generates a mixture of products with enhanced water solubility suitable for biological fermentation. In this context, the authors used P. putida genetically engineered for the bioconversion of acetate, C4 to C17 dicarboxylates, benzoate, and terephthalate to polyhydroxyalkanoate, which is a natural polyester. In addition, these remarkable strains were designed to use acetate and dicarboxylates as a carbon source for cellular growth while converting benzoate and terephthalate to β-ketoadipate, a polymer monomer. The authors called downstream valorization a “biological funneling” since the various molecules produced in the chemical treatment were bio-transformed to only two building block molecules. This approach enables the treatment of blends of plastic residues without previous sorting, which makes it cost-effective.
In the case of polyethylene (PE), it is interesting to discuss the investigation of Li et al., who isolated the marine bacteria Microbulbifer hydrolyticus IRE-31, capable of degrading the recalcitrant low-density polyethylene (LDPE).60 The bacteria, found in the wastewater of a lignin-rich pulp mill, were able to oxidize the surface of linear LDPE, monitored by scanning electron microscopy after 30 days of incubation. FTIR analysis disclosed the unknown metabolic pathways of biodegradation of PE, revealing the formation of additional hydroxyl and carbonyl functional groups at the polymer surface, implying that oxidative reactions may be the initial step for depolymerization. Nevertheless, the products of degradation were not reported.
A study by Shao et al. reports the biodegradation of untreated PE with the native strain Streptomyces albogriseolus LBX-2 isolated from soil.61 The authors highlight the importance of using microorganisms that can degrade virgin PE, avoiding photo- and thermal pre-treatments. It has been proposed that the alkane hydrolase system, particularly alkane monooxygenase, is involved in PE degradation, which is supported by the similarities in the chemical structures of these substances. Arguing this hypothesis, the authors found 21 monooxygenase genes in the genome of S. albogriseolus LBX-2, while other bacterial genomes commonly harbor a few. Further genomic, transcriptomic and metabolic studies of the novel strain are needed to design rational biodegradation processes.
Gregory et al. reported the catalytic hydrogenolysis of waste polyolefins followed by biotransformation towards an ester wax and alcohols62 (see Table 5, row 5, Fig. 3A). Interestingly, the authors isolated a consortium of bacteria (composed of two Rhodococcus aetherivorans strains) from LDPE debris found in the soil of a plastic recycling plant. Those bacteria were cultivated in a C10–C40 alkane mixture as a sole carbon source in order to enhance the bioconversion of the PE deconstruction mixture.
The investigations discussed above prove that most of the research efforts are devoted to the treatment and valorization of PET-based wastes. Nevertheless, this kind of plastics represents 10.2% of the global plastic production. But what about the chemo-enzymatic treatment of more recalcitrant and by far less recycled plastics, such as polyurethane (PU)?
The diverse composition and variety of monomers of PU hinder the implementation of chemical depolymerization processes and require the design of new recycling strategies. As described by Rossignolo et al.,63 degradation of PU takes place in three steps: breaking the polymer chains into oligomers, deeper depolymerization towards low molecular weight species and conversion to carbon dioxide and water (aerobic conditions) or, alternatively, to methane (anaerobic conditions).
In this context, an early investigation by Schmidt et al. demonstrated that enzymatic hydrolysis of model PU and thermoplastic polyester PU (TPU) with various strains of cutinases was achievable.64 The authors cloned synthetic gene constructs corresponding to polyester hydrolases LC cutinase (LCC), TfCut2, Tcur1278 and Tcur0390 in E. coli, expressed and purified the recombinant enzymes that allowed the hydrolysis of solid polyurethane plastic. The degradation, performed under incubation at 70 °C for 200 h, was concluded from surface depletions and weight loss of PU, but the products of degradation were not reported.
Biological funneling for the upcycling of PU hydrolysates was proposed by Catur Utomo et al.65 The researchers applied a defined microbial mixed culture composed of microorganisms trained to use specific PU monomers and genetically engineered to yield rhamnolipids. The advantage of this strategy relies on saving time and effort by developing various strains with different metabolic capacities rather than a single strain with multiple biotransformation events or multiple targeted features achieved by directed evolution. While three P. putida KT2440 derivatives harboring different genetic optimizations enable the utilization of adipic acid, 1,4-butanediol and EG, the addition of a fourth Pseudomonas sp. strain that degrades 2,4-toluene diamine (TDA) was not enough to reduce the inhibition caused by this isocyanate by-product. To overcome the drawback, the authors proposed a chemical removal of TDA from the PU hydrolysate prior to incubation with the microbial consortium. The scheme allows the recovery of valuable TDA and the full utilization of other PU monomers originating from a variety of PU wastes, but some issues regarding the biocompatibility of the extractants remain.
The review by Magnin et al. pointed out that there is only one investigation about the use of oxidoreductases, such as laccase and horseradish peroxidase, in the enzymatic degradation of PU.66 That review dates from the year 2021, and to our knowledge, there have been no additional publications on that matter. Magnin et al. discovered that the combination of an amidase (E4143) and an esterase (E3576) was capable of hydrolyzing model PU films towards 6-hydroxycaproic acid (HCA) and 4,4′-methylene dianiline (MDA).67 The researchers developed an interesting strategy using specifically designed TPU that allows us to understand the molecular mechanisms of enzymatic catalysis further. A synergistic two-discrete step degradation was postulated, in which esterase first attacks the polymer and releases water-soluble oligomers containing urethane bonds, allowing amidase to better access and exert its urethanase (hydrolase) activity. Finally, the efficient enzymatic system yielded 1 g L−1 of 6-hydroxycaproic acid and 0.3 to 3 mg L−1 of MDA and MDA linked to caprolactone. As a limitation, this time-consuming process requires boosting/replacement of the lost enzymatic activity every two to three days during 51 days of incubation.
More recently, the same research group demonstrated that a commercial laccase from Trametes versicolor fungi was active in the degradation of model foams, thermoplastic, polyester and polyether-based PU incubated in 1-hydroxybenzotriazole at 37 °C for 18 days.68 Further investigation is needed to test if the combination of the previously studied esterase and amidase with the novel laccase could create an efficient enzymatic system suitable for different kinds of PU. Moreover, the development of coupled schemes with physical (grinding towards PU powder) or chemical (glycolysis) pre-treatments could enable full degradation of the plastic.
It is worth noting that the investigations discussed above used model materials, which denotes the difficulty of the biological recycling of the actual PU waste towards substances suitable for valorization, except for those motivated by basic research. In fact, a very recent review by Rossignolo et al. pointed out that biodegradation is limited by the number of microorganisms and enzymes able to degrade polyurethanes. In addition, the various structures (polyester PU, polyether PU, among others) and forms (flexible and rigid foams, elastomers, thermoplastic, etc.) would demand a prior PU waste separation to enable an effective upcycling.63 In this context, more research is needed to obtain engineered microorganisms harboring the gene constructs necessary to overproduce genetically optimized enzymes. The use of the metagenomic approach to find new enzymatic activities among microorganisms belonging to degrading communities of PU and petroleum derivatives seems to be the next step to overcome these issues.
Such a strategy was used by Branson et al. in the only investigation that, to our knowledge, reports the chemo-enzymatic recycling of an actual PU waste69 (see Table 5, row 6, Fig. 3B). The authors isolated DNA from soil largely exposed to PU residues and developed a metagenome library. The screening for urethanase activity led to the discovery of three new enzymes, identified as UMG-SP-1 to UMG-SP-3, with GenBank accession codes OP972509, OP972510, and OP972511. These enzymes converted 65% of the dicarbamates generated in the glycolysis of polyether-polyurethane foam waste towards aromatic diamines under mild conditions and at room temperature.
The development of tandem processes for plastic recycling is an ongoing effort that requires a critical analysis of the technical feasibility and economic and environmental factors before going towards a scaling stage. In this sense, the need for waste sorting before treatment is a bottleneck. Even though some bio-funneling strategies have been proposed to overcome this drawback, developing a more integral waste processing approach to achieve circular economy goals is still challenging. The few operative technologies that reached that stage of maturity will be further addressed in Section 4 of this review.
3.3. Valorization of plastic and biomass waste mixtures: Does a synergic effect exist?
The previous sections described the valorization of plastic waste of various sorts. This section dives into the chemo-biological treatment of mixtures of plastic and biomass wastes and the effect of combining those major streams of residues. A detailed analysis has been published by Seah et al. regarding the synergistic effect of the co-pyrolysis of biomass and plastics to improve the yield and quality of biofuels.70 In this context, the up-to-date reports (published in the past 5 years) on the treatment of mixtures composed of plastic waste with textile fabrics (cotton-based and synthetic),71–76 waste food77–79 and paper wastes80 are discussed.Table 6 summarizes the feedstocks, characteristics of the chemical pre-treatment, bioprocessing of the waste mixture, products obtained and further bioprocessing, if applied.
| Feedstocks | Pretreatment | Type of bioprocess | Biocatalyst | pH, temperature, enzyme conc., time | Products and downstream valorization | Reference | |
|---|---|---|---|---|---|---|---|
| a PET, polyethylene terephthalate; PE, polyethylene; PP, polypropylene; PS, polystyrene; TPA, terephthalic acid; FPU, filter paper unit assay; PBAT, polybutylene adipate terephthalate; PLA, polylactic acid. | |||||||
| 1 | Cotton/PET waste colored textiles | Grinding and soaking in NaOH 6 M, 1 h | Enzymatic hydrolysis of cotton | Commercial cellulase cocktail | Buffered pH 5, 55 °C, 5 mLenzyme L−1, 24 h | PET and glucose | 71 |
| Steam explosion, 150 °C, 5 bar, 15 min, alkaline pretreatment | Enzymatic hydrolysis of cotton | Cellulase cocktail | Buffered pH 4.8, 50 °C, 7 days | Glucose fermented with Saccharamyces cerevisiae to bioethanol | 72 | ||
| 5 min milling | Enzymatic hydrolysis of cotton and PET simultaneously | Humicola insolens cutinase, cellulases CTec2® | Sodium phosphate 100 mM, pH 7.3, 55 °C | 30% TPA yield; 83% glucose yield | 73 | ||
| Autoclavage 121 °C, 15 psi; NaOH 15%, 121 °C, 15 min | Enzymatic hydrolysis of cotton | Commercial cellulases CTec2® | Buffered pH = 4.8, 50 °C, 25 FPU g−1 enzyme, 96 h | 66.7% glucose yield | 74 | ||
| Acid/alkaline pre-treatment | Enzymatic hydrolysis of cotton | Commercial cellulases CTec2® | 50 °C, 32 FPU g−1 enzyme, 19 h | 99 g L−1 glucose | 75 | ||
| 2 | Wool/cotton/PET waste textiles | Grinding, boiling in water, dried | Enzymatic hydrolysis of wool amino-acids and cotton | Proteases, cellulases | 8 Um L−1 protease, 50 °C, 2 days; buffered pH 4.8, 2750 Um L−1 cellulase, 50 °C, 5 days | 0.6 g L−1 glucose fermented with S. cerevisiae to bioethanol; peptides, pure PET | 76 |
| 3 | Rice husks-waste food/PE-PP-PS | Hydrothermal carbonization, 204 °C, 30–60 min, subcritical water | Anaerobic co-digestion | Mesophilic bacteria | 38 °C, 30 days | Methane CH4 | 77 |
| 4 | Sewage sludge-waste food/PET microplastics | None | Anaerobic co-digestion | Bacteroides vadin HA17, Clostridium Sphaerochaeta | 37 °C, 35 days | >40 mL g−1 CH4 acetic, valeric, propionic and butyric acids | 78 |
| 5 | Waste food/PBAT-PLA-starch plastic | Mechanical processing | Anaerobic co-digestion | Thermophilic, mesophilic bacteria and archaea | 35–55 °C, 35 days | 550 mL CH4 | 79 |
| 6 | Paper waste/plastic waste | Shredding, milling, NaOH 7 wt%, 85 °C, 2 h | Enzymatic hydrolysis | Commercial cellulases CTec2® | 4 mg g−1, 50 °C, 5 days | 70% Glucose 85% xylose | 80 |
In the case of textiles containing PET (with the exception of those containing wool), grinding and a chemical pre-treatment (i.e., alkaline hydrolysis) are required to increase the available sites for the biocatalytic saccharification with cellulases. Table 6 (see rows 1 and 2) shows that saccharification of cotton-based wastes is performed through a commercial cocktail of cellulases. This biocatalyst contains endoglucanases that randomly cut cotton cellulose chains, exoglucanases, which act at the ends of the cellulose chain yielding cellobiose; and β-glucosidases, which degrade cellobiose towards glucose.71,72 Only one research study presents the simultaneous depolymerization of PET and cotton biocatalyzed with cutinases and cellulases, which gives rise to terephthalic acid and glucose73 (Table 6, row 1). Unlike other methods, the work reported by Kaabel et al. uses the minimum amount of liquid to provide enough moisture for the bioprocessing, but no pre-treatment is performed. Bioethanol is also produced through fermentation with Saccharomyces cerevisiae of carbohydrates obtained from textiles72,76 (Table 6, rows 1 and 2).
Various types of plastics have also been recently addressed. These include: PET microplastics, polyethylene, polypropylene and polystyrene found in films, plastics from disposable bags and food containers, and also polylactic acid from biodegradable bags. Particularly, complex mixtures of plastics with waste food and sewage sludge have been treated through anaerobic co-digestion both in mesophilic and thermophilic fashions with bacteria and archaea microorganisms.77–79 In this process, organic matter is degraded to form biogas by the action of anaerobic bacteria at temperatures of 30 to 50 °C. The first stage involves acid-forming bacteria that use carbohydrates as raw material. A second step implicates the generation of acetic acid. The last stage comprises the bioconversion of acetic acid, carbon monoxide, and hydrogen in biogas composed mainly of methane.
The mixture of wastes is frequently pretreated before anerobic digestion. In this context, Farghali et al. reported the pre-treatment of 2 m3 of a mixture of plastic films, food waste and rice husks under subcritical water in a large-scale reactor at high temperature and pressure77 (see Table 6, row 3). Pretreatment was required to reduce the wastes to liquid and solid fractions containing solubilized lignocellulose. In fact, volatile fatty acids released from biomass through the hydrothermal process served as a carbon source for bacteria, improving biogas production.
In contrast with the positive effect of the biomass-plastic mixture described above, the investigations of Wang et al. regarding the co-digestion of PET microplastics with sewage sludge and waste food were not synergistic.78 In this case, the decomposition of PET into diisobutyl phthalate and dibutyl phthalate was harmful to the microbial community of the anerobic digestor (see Table 6, row 4). Indeed, those substances caused a decrease in the amount of hydrolytic bacteria Bacteroides vadin HA17 and the acidification performed by Clostridium and Sphaerochaeta.
A similar outcome was obtained by Yu et al. in the co-digestion of food waste and biodegradable plastic bags made of polybutylene adipate terephthalate (PBAT)/polylactic acid (PLA)/starch.79 The authors detected a decline in biogas production at a bioplastic/waste food ratio above 30% due to a detrimental effect on bacterial community diversity (see Table 6, row 5). The less effective anaerobic digestion under mesophilic conditions and high plastic loading was attributed to the decrease of the Synergistota phylum type of bacteria. Similarly, a decrease in the abundance of bacteria related to the hydrolysis and acidification of organic substances (i.e., Firmicutes and Bacteroidota) was observed under thermophilic conditions and high plastic loading. On the other hand, the composition of the archaeal community (i.e., Methanosarcina, Methanospirillum, Methanothermobacter and unclassified_k_norank_d_Archaea) was not influenced by the proportion of bioplastics mixed with food waste.
Finally, Brown et al. investigated the co-fermentation of various plastics (PET, polypropylene, low-density and high-density polyethylene, polystyrene, and polyvinyl chloride) and paper waste through enzymatic hydrolysis with the commercial cellulase CTec2®.80 The authors demonstrated that the presence of acetic acid in the paper acts as an enzymatic inhibitor of cellulases (Table 6, row 6). In this context, an alkaline pre-treatment proved to be effective in removing contaminants and increasing the surface area of the cellulose fibers, which in turn, enhances the substrate-enzyme interaction. However, the presence of plastic, ink, and stickies within the mixed paper stream did not have an impact on the enzymatic hydrolysis of cellulose towards glucose and xylose.
Fig. 4 summarizes the synergistic and non-synergistic effects of mixing plastic and biomass wastes. PET is one of the most common plastic wastes polluting the planet, and it is also the most deeply studied in this section. Better yields in glucose and bioethanol production were achieved by employing cotton or wool combined with PET as a biomass feedstock. Nevertheless, the presence of PET was detrimental when methane production from sewage sludge-waste food or glucose generation from paper waste was studied. Poly-ethylene, polypropylene and polystyrene were analyzed combined with paper and food waste, giving only a synergistic effect in the anaerobic digestion of rice husks. Finally, plastic residues from biodegradable bags did not present a beneficial effect in the anaerobic digestion, studied with waste food as raw material.
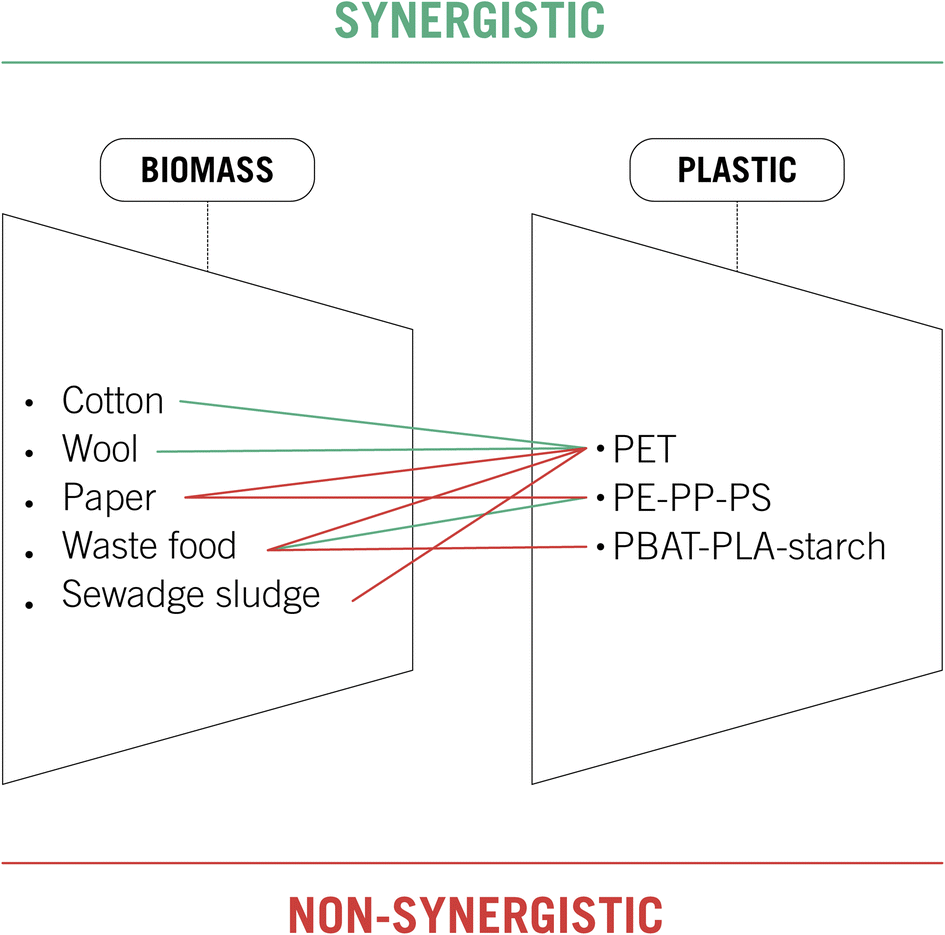 | ||
| Fig. 4 Synergistic and non-synergistic effects of mixing plastic and biomass in waste processing technologies. PET: polyethylene terephthalate; PE: polyethylene; PP, polypropylene; PS: polystyrene; PBAT: polybutylene adipate terephthalate, PLA: polylactic acid. | ||
4. From the lab bench to industry: actual large-scale processes
Previous sections discussed a variety of investigations dealing with thermochemical and chemo-biological methods to treat plastic waste and mixtures of plastic-biomass waste in order to obtain valuable substances. This section presents the technologically mature and cost-effective processes that have scaled up towards industrial application and are effectively applied to this day.The report published by the capital investment firm Closed Loop Partners and our own search on the World Wide Web performed in January 2024 showed that pyrolysis is the first choice for plastic recycling in 36 companies worldwide. Chemical depolymerization occupies the second place with 19 companies.81 Only one company, Carbios (located in France), applies chemo-biological methods. Carbios depolymerizes PET and other polyesters from urban plastic and textile wastes through enzymatic hydrolysis. The process uses an engineered cutinase where a divalent-metal-binding (formed by the side chains of three acidic amino acid residues) site was replaced by a disulfide bridge to increase the thermal stability of the hydrolase. The obtained TPA monomer is recycled to produce new plastic.81–83
In addition, it is worth noticing that the startups, Scindo (London, UK), the University of Portsmouth (USA) and Xampla (Cambridge, UK), developed biological-based recycling technologies that are currently at the lab-scale and are moving towards large-scale application.81
5. Conclusions and future perspectives
This review provides insights into the up-to-date research regarding the valorization of the most common plastic-type residues and plastic-biomass mixed wastes that account for the major quantity of residues generated worldwide.The valorization of waste-derived platform molecules for the production of refined chemicals and commodities has been extensively explored in the last decades. Most of these compounds originate from petroleum-based raw materials. However, the depletion of fossil feedstocks and the GHG emissions associated with those energy sources is turning the governments and research community into the obtention of bio-based building blocks, upgrading the industrial processes to more eco-friendly ways.
Using waste as biorefinery feedstocks represents an unlimited and ubiquitous alternative that can be adapted to each country, region and climate's availability of renewable resources. Nevertheless, molecular recycling of mixtures of plastic and biomass residues is not an easy task due to the variety of compositions and properties of those wastes, which in turn might be either an advantage, as in the case of the pyrolysis of mixed plastic-biomass residues, or a problem to overcome with an appropriate pre-treatment. A remarkable fact is that always chemical or physical, or both treatments, are required for further application of either an enzymatic or biological process in order to obtain valuable substances from the waste. In fact, multiple approaches to pre-treatment have been investigated, and novel, less harsh ones are part of the ongoing research.
The use of microorganisms, as microscopic bioreactors, for the production of commodities has been largely exploited by humanity since early times. They have the complex metabolic pathways and biological machinery to synthesize all kinds of biomolecules, including biopolymers and their constituent building blocks. By this approach, with the appropriate selection of the bacteria or fungi to be cultured, bulk mixtures enriched in target biomolecules can be obtained.
This review clearly shows that significant research is needed in order to develop reliable chemo-enzymatic bioprocesses to treat and valorize mixtures of waste. In particular, the bio-based valorization of mixed wastes, such as plastic and textiles, plastic and biomass, and food wastes and plastics, is an emerging research field that needs further development for industrial application. In fact, the cutting-edge investigations outlined in this contribution show a variety of shortcomings and, therefore, opportunities for advancement in the field.
Data availability
No primary research results, software or code have been included and no new data were generated or analyzed as part of this review.Author contributions
The manuscript was written with contributions from all authors.Conflicts of interest
The authors do not declare conflicts of interest.Acknowledgements
This research was funded by the Consejo Nacional de Investigaciones Científicas y Técnicas CONICET of Argentina (project PIP 11220200102016CO and project PIBAA 28720210100040CO) and Universidad Nacional de La Plata (project 11X-898).References
- I. Johansson, M. E. Giménez, D. Roberts, B. Hoffman, M. Becidan, G. Ciceri, F. Murphy, C. Trois, T. P. Curran and D. Stapf. Material and energy valorization of waste as part of a circular model. Bioenergy Annual Report, Technology Collaboration Programme [internet], International Energy Agency IEA, 2023, https://www.ieabioenergy.com/wp-content/uploads/2023/05/Colour-Feature-Article-Annual-Report-2022.pdf (accessed September 2024) Search PubMed.
- Worldbank.org, What a waste 2.0. A global snapshot of solid waste management to 2050, Trends in Solid Waste Management, World Bank, Washington (DC), 2023, https://datatopics.worldbank.org/what-a-waste/trends_in_solid_waste_management.html (accessed September 2024) Search PubMed.
- Organization for Economic Co-operation and Development [internet], Plastic leakage to the environment, OECD.Stat, OECD environment statistics, 2023, https://stats.oecd.org/viewhtml.aspx?datasetcode=PLASTIC_LEAKAGE_V2_3%26lang=en# (accessed September 2024) Search PubMed.
- J. W. Cottom, E. Cook and C. A. Velis, Nature, 2024, 633, 101–108 CrossRef CAS PubMed.
- Global waste index 2022: These are biggest waste producers in the world[internet], https://sensoneo.com/global-waste-index/(accessed September 2024).
- A. F. J. Tang, S. Yu, Ch. Wang, G. H. Yeoh, W. Y. Teoh and A. C. K. Yip, npj Mater. Sustain., 2024, 3, 2 Search PubMed.
- G. Pathak, M. Nichter, A. Hardon, E. Moyer, A. Latkar, J. Simbaya, D. Pakasi, E. Taqueban and J. Love, Global Environ. Change, 2023, 80, 102648 CrossRef.
- M. Bertone, L. Stabile and G. Buonanno, Sustainability, 2024, 16, 4117 CrossRef CAS.
- G. Rubio-Domingo and A. Halevi, Making plastics emissions transparent, Coalition on Materials Emissions Transparency eds., published in February 2022, https://ccsi.columbia.edu/sites/default/files/content/COMET-making-plastics-emissions-transparent.pdf (accessed September 2024) Search PubMed.
- J. Zheng and S. Suh, Nat. Clim. Change, 2019, 9, 374–378 CrossRef.
- L. Cabernard, S. Pfister, C. Oberschelp and S. Hellweg, Nature, 2022, 5, 139–148 Search PubMed.
- H. Jeswani, C. Krüger, M. Russ, M. Horlacher, F. Antony, S. Hann and A. Azapagic, Sci. Total Environ., 2021, 769, 144483 CrossRef CAS PubMed.
- K. Ghosal and C. Nayak, Mater. Adv., 2022, 3, 1974–1992 RSC.
- T. Thiounn and R. C. Smith, J. Polym. Sci., 2020, 58, 1347–1364 CrossRef CAS.
- H. Chen and H. Hu, Ind. Eng. Chem. Res., 2023, 62, 12925–12934 CrossRef CAS.
- P. Pereira, P. E. Savage and C. W. Pester, ACS Sustain. Chem. Eng., 2023, 11, 7203–7209 CrossRef CAS.
- F. Tollini, L. Brivio, P. Innocenti, M. Sponchioni and D. Moscatelli, Chem. Eng. Sci., 2022, 260, 117875 CrossRef CAS.
- Z. T. Laldinpuii, V. Khiangte, S. Lalhmangaihzuala, C. Lalmuanpuia, Z. Pachuau, C. Lalhriatpuia and K. Vanlaldinpuia, J. Polym. Environ., 2022, 30, 1600–1614 CrossRef CAS.
- A. A. Al Otaibi, A. K. D. Alsukaibi, M. A. Rahman, M. Mushtaque and A. Hauqe, Polymers, 2022, 14, 1861 CrossRef PubMed.
- S. Shirazimoghaddam, I. Amin, J. A. Faria Albanese and N. R. Shiju, ACS Eng. Au, 2023, 23, 37–44 CrossRef PubMed.
- J. Huang, D. Yan, Q. Zhu, X. Cheng, J. Tang, X. Lu and J. Xin, Polym. Degrad. Stab., 2023, 208, 110245 CrossRef CAS.
- A. A. Y. Kratish and T. J. Marks, Angew. Chem., Int., 2022, 61, e202112576 CrossRef PubMed.
- N. Lee, J. Joo, K. A. Lin and J. Lee, Polymers, 2021, 13, 1198 CrossRef CAS PubMed.
- K. P. Rajan, I. Mustafa, A. Gopanna and S. P. Thomas, Recycling, 2023, 8, 63 CrossRef.
- A. Eschenbacher, R. J. Varghese, M. S. Abbas-Abadi and K. M. Van Geem, Chem. Eng. J., 2022, 428, 132087 CrossRef CAS.
- X. Hu, D. Ma, G. Zhang, M. Ling, Q. Hu, K. Liang, J. Lu and Y. Zheng, Carbon Resour. Convers., 2023, 6, 215–228 CrossRef CAS.
- S. W. Kim, Y. T. Kim, Y. F. Tsang and J. Lee, Sci. Total Environ., 2023, 903, 166789 CrossRef CAS PubMed.
- X. Xue, J. Mi, F. Huang, J. Wang, L. Chen and J. Liang, J. Anal. Appl. Pyrolysis, 2023, 170, 105909 CrossRef CAS.
- Z. Chen, B. J. Erwin and L. Che, J. Cleaner Prod., 2023, 424, 138861 CrossRef CAS.
- S. Liu, P. A. Kots, B. C. Vance, A. Danielson and D. G. Vlachos, Sci. Adv., 2021, 7, eabf8283 CrossRef CAS PubMed.
- M. M. Harussani, S. M. Sapuan, U. Rashid, A. Khalina and R. A. Ilyas, Sci. Total Environ., 2022, 803, 149911 CrossRef CAS PubMed.
- P. Palmay, C. Medina, C. Donoso, D. Barzallo and J. C. Bruno, Clean Technol. Environ., 2023, 25, 1539–1549 CrossRef CAS.
- A. Zabaniotou and I. Vaskalis, Energies, 2023, 16, 593 CrossRef CAS.
- B. V. Kanattukara, J. Singh, P. Sarkar, A. Chopra, D. Singh, S. Mondal, G. Singh Kapur and S. S. V. Ramakumar, Environ. Sci. Pollut. Res., 2023, 30, 64994–65010 CrossRef CAS PubMed.
- V. V. Zefirov, I. V. Elmanovich, A. I. Stakhanov, A. A. Pavlov, S. V. Stakhanova, E. P. Kharitonova and M. O. Gallyamov, Polymers, 2022, 14, 744 CrossRef CAS PubMed.
- M. Čolnik, P. Kotnik, Ž. Knez and M. Škerget, Polymers, 2022, 20, 4415 CrossRef PubMed.
- Z. Fu, Y. S. Zhang, G. Ji and A. Li, Chemosphere, 2024, 350, 141045 CrossRef CAS PubMed.
- M. S. Al-Iessa, B. Y. Al-Zaidi, R. S. Almukhtar, Z. M. Shakor and I. Hamawand, Energies, 2023, 16, 4871 CrossRef CAS.
- J. M. Jung, T. Lee, S. Jung, Y. F. Tsang, A. Bhatnagar, S. S. Lee, H. Song, W.-K. Park and E. E. Kwon, Chem. Eng. J., 2022, 442, 136358 CrossRef CAS.
- Z. Wu, C. Li, R. Shan and J. Zhang, Waste Biomass Valorization, 2024, 15, 1603–1614 CrossRef CAS.
- R. Donadini, R. Boaretti, L. Scopel, A. Lorenzetti and M. Modesti, Chem.–Eur. J., 2023, e202301919 Search PubMed.
- B. Liu, Z. Westman, K. Richardson, D. Lim, A. L. Stottlemyer, T. Farmer, P. Gillis, V. Vlcek, P. Christopher and M. M. Abu-Omar, ACS Sustain. Chem. Eng., 2023, 11, 6114–6128 CrossRef CAS.
- R. Donadini, R. Boaretti, A. Lorenzetti, M. Roso, D. Penzo, E. Dal Lago and M. Modesti, ACS Omega, 2023, 8, 4655–4666 CrossRef CAS PubMed.
- V. Zubar, A. T. Haedler, M. Shütte, A. S. K. Hashmi and T. Schaub, ChemSusChem, 2022, 15, e202101606 CrossRef CAS PubMed.
- C. M. Osmundsen, K. Egeblad and E. Taarning, in New and Future Developments in Catalysis, ed. S. L. Suib, Elsevier, Amsterdam, 2013, vol. 4, pp. 73–89 Search PubMed.
- X. B. Wu, K.-H. Lin, W.-T. Lee, R. C. Turnell-Ritson, P. J. Dyson and P. C. L. Delannoi, Nat. Commun., 2023, 14, 6524 CrossRef CAS PubMed.
- D. Munir, M. F. Irfan and M. F. Usman, Renewable Sustainable Energy Rev., 2018, 90, 490–515 CrossRef CAS.
- R. Saab, K. Polychronopoulou, L. Zheng, S. Kumar and A. Schiffer, J. Ind. Eng. Chem., 2020, 89, 83–103 CrossRef CAS.
- P. A. Kolts, B. C. Vance and D. G. Vlachos, React. Chem. Eng., 2022, 7, 41–54 RSC.
- M. S. Al-Iessa, B. Y. Al-Zaidi, R. S. Almukhtar, Z. M. Shakor and I. Hamawand, Energies, 2023, 16, 4871 CrossRef CAS.
- M. Farghali, A. Shimahata, I. M. A. Mohamed, M. Iwasaki, J. Lu, I. Ihara and K. Umetsu, Biochem. Eng. J., 2022, 186, 108546 CrossRef CAS.
- L. M. Blank, T. Narancic, J. Mampel, T. Tiso and K. O'Connor, Curr. Opin. Biotechnol., 2020, 62, 212–219 CrossRef CAS PubMed.
- F. Quartinello, S. Vajnhandl, J. Volmajer Valh, T. J. Farmer, B. Vončina, A. Lobnik, E. H. Acero, A. Pellis and G. M. Guebitz, Microb. Biotechnol., 2017, 10, 1376–1383 CrossRef CAS PubMed.
- D. Shingwekar, H. Laster, H. Kemp and J. L. Mellies, Bioengineering, 2023, 10, 1253 CrossRef CAS PubMed.
- R. León-Zayas, C. Roberts, M. Vague and J. L. Mellies, Microbiol. Resour. Announce., 2019, 8(25), 002377–e319 Search PubMed.
- H. T. Kim, M. H. Ryu, Y. J. Jung, S. Lim, H. M. Song, J. Park, S. Y. Hwang, H.-S. Lee, Y. J. Yeon, B. H. Sung, U. T. Bornscheuer, S. J. Park, J. Chan Joo and D. X. Oh, ChemSusChem, 2021, 14, 4251–4259 CrossRef CAS PubMed.
- L. M. Blank, T. Narancic, J. Mampel, T. Tiso and K. O'Connor, Curr. Opin. Biotechnol., 2020, 62, 212–219 CrossRef CAS PubMed.
- A. Z. Werner, R. Clare, T. D. Mand, I. Pardo, K. J. Ramirez, S. J. Hauge, F. Bratti, G. N. Dexter, J. R. Elmore, J. D. Huenemann, V. G. L. Peabody, C. W. Johnson, N. A. Rorrer, D. Salvachúa, A. M. Guss and G. T. Beckman, Metab. Eng., 2021, 67, 250–261 CrossRef CAS PubMed.
- K. P. Sullivan, A. Z. Werner, K. J. Ramirez, L. D. Ellis, J. R. Bussard, B. A. Black, D. G. Brandner, F. Bratti, B. L. Buss, X. Dong, S. J. Haugen, M. A. Ingraham, M. O. Konev, W. E. Michener, J. Miscall, I. Pardo, S. P. Woodworth, A. M. Guss, Y. Román-Leshkov, S. S. Stahl, T. Gregg and G. T. Beckham, Science, 2022, 378, 207–211 CrossRef CAS PubMed.
- Z. Li, R. Wei, M. Gao, Y. Ren, B. Yu, K. Nie, H. Xu and L. Liu, J. Environ. Manage., 2020, 263, 110402 CrossRef CAS PubMed.
- H. H. Shao, M. J. Chen, X. T. Fei, R. L. Zhang, Y. Zhong, W. M. Ni, X. Tao, X. Y. He, E. L. Zhang, B. Yong and X. M. Tan, Microorganisms, 2019, 7, 1–13 Search PubMed.
- G. J. Gregory, C. Wang, S. Sadula, S. Koval, R. F. Lobo, D. G. Vlachos and E. T. Papoutsakis, ACS Sustain. Chem. Eng., 2023, 11, 3494–3505 CrossRef CAS.
- G. Rossignolo, G. Malucelli and A. Lorenzetti, Green Chem., 2024, 26, 1132–1152 RSC.
- J. Schmidt, W. Ren, T. Oeser, L. A. D. e Silva, D. Breite, A. Schulze and W. Zimmermann, Polymers, 2017, 9, 65 CrossRef PubMed.
- R. N. Catur Utomo, W.-J. Li, T. Tiso, C. Eberlein, M. Doeker, H. J. Heipieper, A. Jupke, N. Wierckx and L. M. Blank, ACS Sustain. Chem. Eng., 2020, 8(47), 17466–17474 CrossRef CAS.
- A. Magnin, E. Pollet and L. Avérous, Methods Enzymol., 2021, 648, 317–336 CAS.
- A. Magnin, E. Pollet, R. Perrin, C. Ullman, C. Persillon, V. Phalip and L. Avérous, Waste Manag., 2019, 85, 141–150 CrossRef CAS PubMed.
- A. Magnin, L. Entzmann, E. Pollet and L. Avérous, Waste Manag., 2021, 132, 23–30 CrossRef CAS PubMed.
- Y. Branson, S. Söltl, C. Buchmann, R. Wei, L. Schaffert, C. P. S. Badenhorst, L. Reisky, G. Jäger and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2023, 62, e202216220 CrossRef CAS PubMed.
- C. C. Seah, C. H. Tan, N. A. Arifin, R. S. R. M. Hafriz, A. Salmiaton, S. Nomanbhay and A. H. Shamsuddin, Results Eng., 2023, 17, 100989 CrossRef CAS.
- B. Piribauer, A. Bartl and W. Ipsmiller, Waste Manag. Res., 2021, 39, 1277–1290 CrossRef CAS PubMed.
- S. M. Gritsch, S. Mihalyi, A. Bartl, W. Ipsmiller, U. Jenull-Halver, R. F. Putz, F. Quartinello and G. M. Guebitz, Resour. Conserv. Recycl., 2023, 188, 106701 CrossRef CAS.
- S. Kaabel, J. Arciszewski, T. H. Borchers, J. P. D. Therien, T. Friščić and K. Auclair, ChemSusChem, 2023, 16, e202201613 CrossRef CAS PubMed.
- A. Boondaeng, J. Keabpimai, P. Srichola, P. Vaithanomsat, C. Trakunjae and N. Niyomvong, Polymers, 2023, 15, 1964 CrossRef CAS PubMed.
- J. Egan, S. Wang, J. Shen, O. Baars, G. Moxley and S. Salmon, Resour. Environ. Sustain., 2023, 13, 100118 Search PubMed.
- F. Quartinello, S. Vecchiato, S. Weinberger, K. Kremenser, L. Skopek, A. Pellis and G. M. Guebitz, Polymers, 2018, 10, 1107 CrossRef PubMed.
- M. Farghali, A. Shimahata, I. M. A. Mohamed, M. Iwasaki, J. Lu, I. Ihara and K. Umetsu, Biochem. Eng. J., 2022, 186, 108546 CrossRef CAS.
- P. Wang, Y. Guo, M. Yu, S. Riya, Y. Zheng and L. Ren, Biochem. Eng. J., 2023, 198, 109012 CrossRef CAS.
- C. Yu, B. Dongsu, Z. Tao, K. Zhe, J. Xintong, W. Siqi, C. Ming, S. Zheng and Z. Yalei, Biochem. Eng. J., 2023, 199, 109072 CrossRef CAS.
- R. M. Brown, A. N. Hoover, J. L. Klinger, B. D. Wahlen, D. Hartley, H. Lee and V. S. Thompson, Front. Energy Res., 2022, 10, 834832 CrossRef.
- Closed Loop Partners-Investors in the Circular Economy [internet]. B. Miñana and E. Mills. Global Directory of Molecular Recycling Technologies. Supplemental Resource from Transitioning to a Circular System for Plastics: Assessing Molecular Recycling Technologies in the United States and Canada. New York (US): Closed Loop partners; cited 2021 Nov 13. https://www.closedlooppartners.com/wp-content/uploads/2021/11/CLP_Molecular-Recycling-Directory-2021.pdf (assessed October, 2024).
- V. Tournier, C. M. Topham, A. Gilles, B. David, C. Folgoas, A. Moya-Leclair, E. Kamionka, M.-L. Desrousseaux, H. Texier, S. Gavalda, M. Cot, E. Guémard, M. Dalibey, J. Nomme, G. Cioci, S. Barbe, M. Chateau, I. André, S. Duquesne and A. Marty, Nature, 2020, 580, 216–219 CrossRef CAS PubMed.
- Carbios website [internet], Carbios Produces First Clear Plastic Bottles from Enzymatically Recycled Textile Waste, Carbios industries, Clermont-Ferrand (France), 2020, press release 2020 Nov 19, https://www.carbios.com/en/enzymatic-recycling/(assessed November, 2024) Search PubMed.
| This journal is © The Royal Society of Chemistry 2025 |