
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA new synthetic approach for high surface area mesoporous silica and its use towards sustainable antifouling materials †
Paola
Marzullo
ab,
Alessandro
Presentato
a,
Francesca
D'Anna
 ab,
Vincenzo
Campisciano
a,
Rosa
Alduina
ab,
Enrico
Tornatore
a,
Francesco
Giacalone
a,
Leonarda Francesca
Liotta
*c and
Michelangelo
Gruttadauria
*ab
ab,
Vincenzo
Campisciano
a,
Rosa
Alduina
ab,
Enrico
Tornatore
a,
Francesco
Giacalone
a,
Leonarda Francesca
Liotta
*c and
Michelangelo
Gruttadauria
*ab
aDepartment of Biological, Chemical and Pharmaceutical Sciences and Technologies (STEBICEF), University of Palermo, Viale delle Scienze, 90128 Palermo, Italy. E-mail: michelangelo.gruttadauria@unipa.it
bSustainable Mobility Center (Centro Nazionale per la Mobilità Sostenibile—CNMS), Via Durando 39, 20158 Milano, Italy
cInstitute for the Study of Nanostructured Materials (ISMN), National Research Council (CNR), Via Ugo La Malfa 153, 90146 Palermo, Italy. E-mail: leonardafrancesca.liotta@cnr.it
First published on 2nd April 2025
Abstract
For the first time, a silica gel was obtained by hydrolysis of tetraethyl orthosilicate in the presence of resorcinol. This simple method gave a nano-silica with high specific surface area (1000 m2 g−1) and narrow pore size distribution. Resorcinol cannot form a self-assembled structure such as a micelle. Therefore, the formation mechanism of this new silica differs from that of mesostructured silica. A possible explanation for the role played by resorcinol was given. Then, as an application of this new silica gel, a set of quaternary ammonium salt (QAS)-based silanes were synthesized and tested for their antibacterial activity against the Pseudomonas delhiensis PS27 strain earlier described for its resilience towards adverse and stressful environmental conditions. Therefore, the novel nano-silica alongside the most active QAS-based silane was successfully prepared and further incorporated into a polydimethylpolysiloxane (PDMS) polymer matrix. The resulting film exhibited significant antibiofilm activity, inhibiting bacterial cell attachment onto the QAS-silica/PDMS composite surface without killing planktonic cells. In contrast, the composite material prepared using commercially available silica gel did not show antibiofilm activity. This finding suggests a different role in activity when the QAS-based silane is covalently attached to very high surface area silica.
Sustainability spotlightEfficient and non-toxic marine antifouling materials are of fundamental interest considering that most trade routes are by sea. In this work, we have developed new mesoporous silica gels with high surface area through a simple and sustainable synthesis under mild conditions using tetraethyl orthosilicate and a cheap molecule, resorcinol, as template. Resorcinol can be removed by a simple filtration step without more expensive procedures such as extraction or calcination. This silica, modified with a simple quaternary ammonium salt embedded into a non-toxic PDMS film showed an excellent antibiofilm activity while no killing of planktonic cells was observed in solution. Our work emphasizes the importance of the following UN sustainable development goals: ensure sustainable consumption and production patterns (SDG 12) and life below water (SDG 14). |
Introduction
Amorphous silica nanoparticle materials are of great interest due to their potential use in catalysis and biomedicine, such as cancer treatment and drug delivery applications.1,2 Their wide range of applications is due to the possibility of tuning the pore volume, specific surface area, easy surface modification, biocompatibility, and degradation characteristics.3–5Silica nanoparticles are one of the five most widely used nanomaterials on the Woodrow Wilson International Center for Scholars' list of consumer goods.6 However, some concerns about toxicity have been raised.7–9
Amorphous silica materials can be obtained by the conventional sol–gel route starting from tetraethyl orthosilicate, (TEOS) via acid or base-catalysed hydrolysis and condensation reactions.10
The conventional sol–gel approach can give silica with a wide pore size distribution. However, the pores can be tuned by an organic template-assisted sol–gel process, in which the silica gel structure is trapped with organic molecules. The latter can be removed by calcination or extraction, resulting in porous silicate networks with pores affected by the template molecules. After the removal of the template, the porous structure may collapse if the silicate network is not mechanically strong enough. The different nature of interactions (electrostatic, van der Waals, and hydrogen bonding or combinations thereof) between the template and the silica may play a role. Equally important is the structural arrangement, as the volume fraction of the template can control the volume fraction of the silicate network.
Several methods reported in the literature involve the addition of organic molecules for surface area modulation and pore size distribution control.
A remarkably narrow pore size distribution was obtained by doping a sol–gel-derived from tetramethoxysilane with a small amount of octaphenylcyclotetrasiloxane. In this case, diphenylsiloxane units were incorporated into the material.11 Mesoporous silica was prepared by removing 1-dodecanol trapped in a gel formed by the reaction of TEOS and formic acid in the presence of 1-dodecanol. The silica samples were highly porous having up to the pore volume of about 0.74 cm3 g−1 and a high BET surface area of about 725 m2 g−1.12 Porous silica gels with high surface area (700–1000 m2 g−1) were prepared by calcination of silica/poly-(tetramethylene oxide) hybrid network materials.13 Silica with a controlled pore size distribution and a high surface area was obtained after calcination by the sol–gel process starting from TEOS in the presence of uncharged polymers (polyvinyl alcohol and polyethylene glycol), charged polymers (the anionic polyacrylic acid and the cationic quaternary salt of polyethylene imine), and proteins (lipase and albumin).14 An amorphous porous silica gel was prepared by pyrolysis of a composite of citric acid and silica obtained by the sol–gel reaction of TEOS. The silica gel had a high specific surface area, ca. 1000 m2 g−1, and its pore volume increased linearly as the citric acid content increased.15 Strongly basic anion-exchange gel resins were used as shape-directing macro-templates to obtain amorphous silica spherical macroparticles. The resin was removed by combustion, leaving silica particles with surface areas exceeding 1000 m2 g−1.16 The sol–gel process, followed by the calcination, in the presence of basic amino acids as catalysts and methanol/water, in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 volume ratio, as a co-solvent gave amorphous silica nanoparticles with an average particle size of 6.4 nm, a size distribution ranging between 2−11 nm, and a specific surface area of 386 m2 g−1.17
:1 volume ratio, as a co-solvent gave amorphous silica nanoparticles with an average particle size of 6.4 nm, a size distribution ranging between 2−11 nm, and a specific surface area of 386 m2 g−1.17
Given these premises and considering our research interests in hybrid materials for catalytic applications18–22 and, more recently, for antifouling applications,23,24 in the present study we focused on the development of new hybrid materials for this purpose.
Thus, we aimed to develop a straightforward method for producing silica gel with a high specific surface area and a narrow pore size distribution. Specifically, in a previous work, we mimicked the polydopamine chemistry preparing a set of hybrid organic-inorganic materials in a one-pot procedure under mild conditions.25 This procedure involved reacting several aminopropyl-trimethoxysilanes with catechol, which oxidizes to o-quinone, resulting in a complex mixture of products that led to the synthesis of a set of amine-functionalized silica-based materials with very low surface areas. Here, we replaced catechol with resorcinol to avoid the formation of such a complex product mixture from oxidation reactions of the former. Also, instead of using aminopropyl-trimethoxysilanes, we employed TEOS and performed a post-modification via grafting. The rationale of this approach relies on the hypothesis that resorcinol may facilitate the hydrolysis of TEOS, potentially yielding silica with a higher surface area than the previous method.
Further, resorcinol was earlier used to synthesize hollow silica nanoparticles with various morphologies; its combination with formaldehyde generated a resorcinol-formaldehyde resin, which served as a nanoplatform.26 Resorcinol was also used as an additive to prevent the polycondensation of silicate anions at early synthesis stages.27 Resorcinol was also used to prepare dioxybenzene-bridged hydrophobic silica aerogels by reaction with TEOS in the presence of trifluoroacetic acid (10 mol%) and refluxing the mixture at 160 °C for 4 hours.28 Resorcinol acts also as useful hydrogen bond donor molecule in deep eutectic solvents as structure-directing agents in the synthesis of hierarchical porous carbon monoliths.29 Resorcinol can interact with silanol groups on the silica surface30 with an effective H-bond network with the two types of silanol groups.31 Here, we successfully obtained amorphous silica with a high surface area; then, it was modified with a suitable QAS, and such modified silica was used to prepare a PDMS film which revealed excellent antibiofilm properties.
Experimental
Materials and methods
All solvents and reagents were obtained from commercial sources and were used without purification. (N,N-Dimethyl-3-aminopropyl)trimethoxysilane, (3-chloropropyl)dimethoxy(methyl)silane, triacetoxymethylsilane, PDMS hydroxy terminated (cst. 700–800), N,N-dimethyloctylamine, N,N-dimethyldodecylamine, 1-iodooctane, tetraethyl orthosilicate (TEOS), resorcinol (99%), tetrabutylammonium fluoride (1 M in THF containing ca. 5 wt% of water), 1-butylimidazole, 1-hexylimidazole, 1-octylimidazole were purchased from Fluorochem (Hadfield, UK); 1-heptanol was purchased from Sigma-Aldrich (Burlington, MA, US); 1-dibutyltin dilaurate was purchased from TCI (Zwijndrecht, Belgium). All solvents (ACS grade) were purchased from VWR International (Milan, IT). Silica gel 60 (0.040–0.063 mm) (Merck, Milan, IT) with a surface area of 480–540 m2 g−1 (BET) was dried under vacuum for 5 h at 120 °C and used directly.Thermogravimetric analysis (TGA) measurements were carried out under oxygen flow from 100 to 1000 °C with a heating rate of 10 °C min−1 in a Mettler Toledo TGA STAR (Milan, IT). Combustion chemical analysis was performed on a PerkinElmer (Waltham, MA, US) 2400 Series II Elemental Analyzer System. Nitrogen adsorption–desorption analysis was performed at 77 K by using a volumetric adsorption analyzer (Micromeritics (Norcross, GA, US) ASAP 2020 Plus). Before the analysis, the sample was pre-treated at 150 °C for 16 h under reduced pressure (0.1 mbar). The Brunauer–Emmett–Teller (BET) method was applied in the p/p0 = 0.05–0.30 range to calculate the specific surface area (SSA). The mean pore size distribution was determined by the Barrett–Joyner–Halenda (BJH) method applied to the desorption curve.32 The average size of silica particles (d) was calculated by the equation: d(nm) = n/ρ × SSA,33 where n is the shape factor of the particles and it is assumed to be 6 for a sphere, ρ is the theoretical density of amorphous silica equal to 2.2 g cm−3.
1H and 13C-NMR liquid-state spectra were recorded at room temperature on Bruker (Billerica, MA, US) Advance II 400 MHz (9.4 T) spectrometer. 29Si Cross-Polarization Magic Angle Spinning (CP-MAS) NMR spectra were acquired on Bruker (Billerica, MA, US) Advance II 400 MHz (9.4 T) spectrometer operating at 79.5 MHz with a MAS rate of 6 kHz, 90° pulse on 1H of 5.1 μs, a delay time of 2 s, and a contact time of 8 ms. 13C Cross-Polarization Magic Angle Spinning (CP-MAS) NMR spectra were acquired on a Bruker (Billerica, MA, US) Advance II 400 MHz (9.4 T) spectrometer operating at 100.6 MHz with a MAS rate of 6 kHz, a delay time of 3 s, and a contact time of 2 ms. The Hartman–Hahn condition was optimized using adamantane as the standard.
General procedure for the synthesis of silica SIL2
Tetraethyl orthosilicate (11.25 g, 54 mmol) was dissolved in methanol (60 mL) then followed by TBAF (1.08 mL, 1 M in THF). The solution was stirred at 30 °C for about 15 hours. After this time the solution was concentrated under reduced pressure (200–150 mbar) at 30 °C to remove the methanol (see Table 1 for the amount of solvent removed), then water was added (30 mL). A precipitate was formed. The suspension was stirred at 50 °C for 3.5 h and at 80 °C for 1 h. After cooling at room temperature, the solid was filtered and washed with water (10 mL), ethanol (50 mL), and methanol (50 mL). The solid was dried at 60 °C under reduced pressure to give a white powder (1.40 g). Calcination was then carried out at 400 °C with a ramp of 2 °C min−1 up to 400 °C, then kept for 4 hours and cooled at room temperature overnight.| Entry | Silica | TEOS (mmol) | Resorcinol (mmol) | SiO2 (g) | SSAa (m2 g−1) | SSAb (m2 g−1) | da,c (nm) | db,c (nm) | Mean pore size BJHd (nm) |
|---|---|---|---|---|---|---|---|---|---|
| a Pre-calcination. b Post calcination. c Silica particles dimension; d(nm) = n/ρ × SSA where n = 6 and ρ is 2.2 g cm−3. d Calculated by desorption curve; calcined samples. e Solvent almost completely removed under reduced pressure. f Solvent removed under reduced pressure: 50 mL. g Not measured. h Same reaction condition of SIL4. | |||||||||
| 1e | SIL1 | 54 | 13.5 | 2.00 | 472 | 617 | 5.8 | 4.4 | 7.39 |
| 2e | SIL2 | 54 | — | 1.40 | 173 | 297 | 15.8 | 9.2 | 9.35 |
| 3e | SIL3 | 108 | 13.5 | 4.42 | 310 | 478 | 8.8 | 5.7 | 11.64 |
| 4f | SIL4 | 54 | 13.5 | 2.13 | n.mg | 1000 | n.mg | 2.7 | 9.32 |
| 5f,h | SIL5 | 54 | 13.5 | 2.10 | 776 | 1100 | 3.5 | 2.5 | 9.32 |
| 6 | SIL6 | 54 | 6.75 | 2.13 | n.mg | 850 | n.mg | 3.2 | 13.10 |
General procedure for the synthesis of silica SIL5 in the presence of resorcinol
Resorcinol (1.485 g, 13.5 mmol) was dissolved in methanol (60 mL) then tetraethyl orthosilicate (TEOS) was added (11.25 g, 54 mmol) followed by TBAF (1.08 mL, 1 M in THF). The solution was stirred at 30 °C for about 15 hours. After this time the pale pink solution was concentrated under reduced pressure (200–150 mbar) at 30 °C to remove the methanol (ca. 50 mL) and then water was added slowly (30 mL). A pale pink precipitate was formed. The suspension was stirred at 50 °C for 30 min and at 80 °C for 3 h. After cooling at room temperature, the solid was filtered and washed with water (10 mL) giving a pale pink solution containing the resorcinol residue, then the solid was washed with ethanol (50 mL), and methanol (50 mL) until the solid and the filtrate were colourless. The solid was dried at 60 °C under reduced pressure to give a white powder (2.00 g). Calcination was then carried out at 400 °C with a ramp of 2 °C min−1 up to 400 °C, then kept for 4 hours and cooled at room temperature overnight.General procedure for the synthesis of silica 3@SIL5
To a solution of N,N-dimethyl-N-(3-(dimethoxy(methyl)silyl)propyl)octan-1-aminium chloride 3 (150 mg, 0.44 mmol) in MeOH (3 mL) dry silica gel SIL5 (300 mg) was added. The mixture was stirred for 7 hours under reduced pressure (200 mbar) at rotavapor, setting the temperature of the bath to 45 °C. After this time, the silica was washed with ethanol and diethyl ether on a gooch filter to remove the excess silane. The obtained material was dried in an oven to give 397 mg of functionalized silica 3@SIL5.General procedure for the synthesis of silica 3@SiO2
To a solution of N,N-dimethyl-N-(3-(dimethoxy(methyl)silyl)propyl)octan-1-aminium chloride 3 (3 g, 9.9 mmol) in MeOH (25 mL) dry silica gel 0.060–0.040 mm, Merck (6 g) was added. The mixture was stirred for 7 hours under reduced pressure (200 mbar) at rotavapor, setting the temperature of the bath to 45 °C. After this time, the silica was washed with ethanol and diethyl ether on a gooch filter to remove the excess silane. The obtained material was dried in an oven to give 6.96 g of functionalized silica 3@SiO2.General procedure for the synthesis of silica 3@SIL5(tol)
To a solution of N,N-dimethyl-N-(3-(dimethoxy(methyl)silyl)propyl)octan-1-aminium chloride 3 (1.8 g, 5.3 mmol) in toluene (20 mL) dry silica gel SIL5 (3.6 g) was added. The mixture was stirred at 110 °C for 18 hours under argon. After cooling at room temperature, silica was filtered through a gooch filter and washed with ethanol and diethyl ether. The obtained material was dried in an oven to give 3.83 g of functionalized silica 3@SIL5(tol).General procedure for the synthesis of compound 1
To a solution of (N,N-dimethyl-3-aminopropyl)trimethoxysilane (1.036 g, 5 mmol) in toluene (4 mL), 1 eq of 1-bromooctane (5 mmol, 966 mg) was added. The mixture was stirred at 110 °C for 18 hours under argon. After this time the solvent was removed under reduced pressure and the obtained product was washed with diethyl ether to give 1.93 g of yellow oil (yield: 97%). 1H-NMR (CDCl3) δ: 0.63 (t, 2H, CH2–Si), 0.82 (t, 3H, CH3), 1.21–1.30 (m, 10H, N+–CH2–CH2–(CH2)5CH3), 1.66–1.74 (m, 4H, 2 × CH2–CH2–N+), 3.43–3.47 (m, 4H, 2 × CH2–N+), 3.28–3.53 (CH3–N+ and OCH3); 13C-NMR (CDCl3) δ: 5.49 (CH2–CH2–Si), 13.96 (CH2–CH3), 16.44 (CH2–Si), 22.48 (CH2–CH3), 22.64 (CH2), 26.19 (CH2), 28.95 (CH2), 29.11 (CH2), 31 (CH2), 50.70 (OCH3), 51.26 (CH3–N+), 63.84 (CH2–N+), 65.18 (CH2–N+).General procedure for the synthesis of compound 2
In a round bottom flask, (3-chloropropyl)trimethoxysilane (1 g, 5 mmol) reacted with 1 eq of N,N-dimethyloctylamine (5 mmol, 758 mg) at 110 °C for 18 hours under argon. After this time, the obtained product was washed with diethyl ether to give 1.73 g of yellow oil (yield: 95%). 1H-NMR (CDCl3) δ: 0.63–0.74 (m, 2H, –CH2–Si), 0.82 (t, 3H, CH3), 1.22–1.30 (m, 10H, N+–CH2–CH2–(CH2)5CH3), 1.67–1.87 (m, 4H, 2 × CH2–CH2–N+), 3.32–3.53 (m, 2 × CH2–N+, 2 × CH3–N+, 2 × –OCH3); 13C-NMR (CDCl3) δ: 5.51 (CH2–CH2–Si), 13.99 (CH2–CH3), 16.43 (CH2–Si), 22.51 (CH2–CH3), 22.76 (CH2), 26.23 (CH2), 28.99 (CH2), 29.13 (CH2), 31.59 (CH2), 50.70 (OCH3), 51.22 (CH3–N+), 63.68 (CH2–N+), 65.06 (CH2–N+).General procedure for the synthesis of compound 3
In a round bottom flask, (3-chloropropyl)dimethoxy(methyl)silane (1 g, 5.5 mmol) reacted with 1 eq of N,N-dimethyloctylamine (5.5 mmol, 860 mg) at 110 °C for 18 hours under argon. After this time, the obtained product was washed with diethyl ether to give 1.61 g of brown oil (yield: 86%). 1H-NMR (CDCl3) δ: 0.09 (s, 3H, CH3–Si), 0.55–0.60 (m, 2H, –CH2–Si), 0.80 (t, 3H, CH3), 1.18–1.28 (m, 10H, N+–CH2–CH2–(CH2)5CH3), 1.60–1.77 (m, 4H, 2 × CH2–CH2– N+), 3.21–3.45 (m, 2 × CH2–N+, 2 × CH3–N+, 2 × –OCH3); 13C-NMR (CDCl3) δ: −3.38 (CH3–Si), 9.65 (CH2–CH2–Si), 13.96 (CH2–CH3), 16.44 (CH2–Si), 22.46 (CH2–CH3), 22.69 (CH2), 26.24 (CH2), 28.92 (CH2), 29.09 (CH2), 31.54 (CH2), 50.32 (OCH3), 51.12 (CH3–N+), 63.58 (CH2–N+), 65.39 (CH2–N+).General procedure for the synthesis of compound 4
In a round bottom flask, (3-chloropropyl)dimethoxy(methyl)silane (2 g, 11 mmol) reacted with 1 eq of N,N-dimethyldodecylamine (11 mmol, 2.35 g) at 110 °C for 18 hours under argon. After this time, the obtained product was washed with diethyl ether to give 3.90 g of brown oil (yield: 90%). 1H-NMR (CDCl3) δ: 0.07 (s, 3H, CH3–Si), 0.57–0.64 (m, 2H, –CH2-Si), 0.82 (t, 3H, CH3), 1.19–1.29 (m, 18H, N+–CH2–CH2–(CH2)9CH3), 1.60–1.77 (m, 4H, 2 × CH2–CH2–N+), 3.21–3.45 (m, 2 × CH2–N+, 2 × CH3–N+, 2 × –OCH3); 13C-NMR (CDCl3) δ: −3.36 (CH3–Si), 12.68 (CH2–CH2–Si), 14.04 (CH2–CH3), 16.69 (CH2–Si), 22.60 (CH2–CH3), 22.76 (CH2), 26.29 (CH2), 27.08 (CH2), 29.24 (CH2), 29.33 (CH2), 29.40 (CH2), 29.51 (CH2), 31.50 (CH2), 31.82 (CH2), 50.34(OCH3), 59.04 (CH3–N+), 63.58 (CH2–N+), 64.23 (CH2–N+).General procedure for the synthesis of compound 5
In a round bottom flask, (3-chloropropyl)dimethoxy(methyl)silane (500 mg, 2.74 mmol) reacted with 1 eq of 1-butyl-1H-imidazole (2.74 mmol, 340.3 mg) at 110 °C for 18 hours under argon. After this time, the obtained product was washed with diethyl ether to give 813 mg of yellow oil (yield: 97%). 1H-NMR (CDCl3) δ: 0.05 (s, 3H, CH3–Si), 0.51–0.56 (m, 2H, CH2–Si), 0.90 (t, 3H, CH3), 1.27–1.36 (m, 2H, N+–CH2–CH2–(CH2)CH3), 1.82–1.85 (m, 2H, N+–CH2–CH2–CH2CH3), 2.00–3.07 (m, 2H, SiCH2CH2CH2–N+), 3.36–3.45 (OCH3), 4.23–4.32 (m, 4H, 2 × CH2N+), 7.41 (s, 1H, CH imidazole), 7.65 (d, 1H, CH imidazole), 9.96 (d, 1H, CH imidazole); 13C-NMR (CDCl3) δ: −1.08(CH3–Si), 13.05 (CH2–CH3 and CH2–CH2–Si), 19.32 (CH2–Si), 24.16 (CH2CH3), 32.09 (CH2CH2CH3), 49.65 (CH2–N+), 50.14 (OCH3), 52.02 (CH2–N+), 121.73 (CH imidazole), 122.90 (CH imidazole), 136.63 (CH imidazole).General procedure for the synthesis of compound 6
In a round bottom flask, (3-chloropropyl)dimethoxy(methyl)silane (500 mg, 2.74 mmol) reacted with 1 eq of 1-hexyl-1H-imidazole (2.74 mmol, 417.13 mg) at 110 °C for 18 hours under argon. After this time, the obtained product was washed with diethyl ether to give 813 mg of yellow oil (yield: 100%). 1H-NMR (CDCl3) δ: 0.09 (s, 3H, CH3–Si), 0.54–0.58 (m, 2H, CH2–Si), 0.82 (t, 3H, CH3), 1.24–1.27 (m, 6H, N+–CH2–CH2–(CH2)3CH3), 1.85–1.97 (m, 4H, N+–CH2–CH2–CH2CH3 and –SiCH2CH2CH2–N+), 3.36–3.45 (OCH3), 4.24–4.37 (m, 4H, 2 × CH2N+), 7.39 (1H, CH imidazole), 7.64 (1H, CH imidazole), 10.4 (1H, CH imidazole); 13C-NMR (CDCl3) δ: – 3.65(CH3–Si), 11.30 (CH2–CH2–Si), 13.65 (CH2–CH3 and CH2–Si), 22.31 (CH2CH3), 24.12 (CH2), 25.46 (CH2), 29.96 (CH2), 31.02 (CH2), 49.92 (CH2–N+), 50.06 (OCH3), 52.04 (CH2–N+), 121.55 (CH imidazole), 122.86 (CH imidazole), 136.49 (CH imidazole).General procedure for the synthesis of compound 7
In a round bottom flask, (3-chloropropyl)dimethoxy(methyl)silane (1 g, 5.5 mmol) reacted with 1 eq of 1-octyl-1H-imidazole (5.5 mmol, 992 mg) at 110 °C for 18 hours under argon. After this time, the obtained product was washed with diethyl ether to give 1.85 g of brown oil (yield: 86%). 1H-NMR (CDCl3) δ: 0.08 (s, 3H, CH3–Si), 0.49–0.61 (m, 2H, –CH2–Si), 0.82 (t, 3H, CH3), 1.19–1.28 (m, 10H, N+–CH2–CH2–(CH2)5CH3), 1.83–1.97 (m, 4H, 2 × CH2–CH2–N+), 3.38–3.45 (OCH3), 4.28–4.32 (m, 4H, 2 × CH2N+), 7.42 (1H, CH, imidazole), 10.53 (1H, CH, imidazole); 13C-NMR (CDCl3) δ: −3.38 (CH3–Si), 12.02 (CH2–CH2–Si), 13.79 (CH2–CH3 and CH2–Si), 22.50 (CH2–CH3), 24.46 (CH2), 26.21 (CH2), 28.96 (CH2), 30.27 (CH2), 31.60 (CH2), 50.31 (OCH3), 52.06 (CH2–N+), 49.95 (CH2–N+), 121.43(CH imidazole), 122.54 (CH imidazole), 136.87 (CH imidazole).General procedure for the synthesis of composite PDMS/SiO2 films
Heptane (1 mL) was added to a weighted amount of silica material and the resulting dispersion was sonicated for 15 minutes. After this time, the silica dispersion was added to 1 g of polydimethylsiloxane hydride-terminated (PDMS-OH) in a beaker and stirred at room temperature for 30 minutes. To promote cross-linking, triacetoxymethylsilane (133 μL) and 1-dibutyltin dilaurate (40 μL) were added as cross-linker and catalyst, respectively. The cross-linked films were obtained by casting the polymeric mixture in a Petri dish and allowing the solvent to evaporate in an oven at 50 °C for 24 hours.General procedure for the synthesis of silica SIL7
To a solution of (N,N-dimethyl-3-aminopropyl)trimethoxysilane (2.2 mmol, 456 mg) in heptanol (7 mL) dry silica SIL1 was added (1.1 g) and the mixture stirred at 110 °C, under argon atmosphere for 24 hours. After cooling at room temperature, the silica was filtered through a gooch filter and washed with ethanol and diethyl ether. The obtained material was dried in an oven to give 1.385 g of modified silica.General procedure for synthesis of silica SIL8
A mixture of silica SIL7 (500 mg) and 1-iodooctane (2 mmol, 480.26 mg) in 1-heptanol (4 mL) was refluxed at 110 °C, under an argon atmosphere for 18 hours. After cooling at room temperature, the silica was filtered through a gooch filter and washed with ethanol and diethyl ether. The obtained material was dried in an oven to give 600 mg of modified silica.Minimal inhibitory concentration of antifouling biocides
To determine the minimal inhibitory concentration (MIC) of antifouling compounds 1–7 against the Gram-negative bacterium Pseudomonas delhiensis PS27, a robust environmental isolate known for its resilience to stressors and unique metabolic capabilities,34 the experimental approach was adapted from a previously described method23 with minor modifications. Briefly, a pre-culture of P. delhiensis PS27 was initiated by inoculating a single colony into a liquid-rich medium, Tryptic Soy Broth (TSB; Merck Life Science S.r.l., Milan, Italy), followed by incubation at 37 °C under shaking conditions (180 rpm) for approximately 16 hours. Then, 5 × 105 colony-forming units per milliliter (CFU mL−1) were inoculated in fresh TSB medium amended with increasing concentrations (25, 50, 75, and 100 μg mL−1) of the antifouling biocides. The bacterial cells were exposed to the biocides for 24 hours under the specified conditions. Subsequent measurements of bacterial growth were obtained by recording the optical density at 600 nm (OD600nm) using a UV-vis spectrophotometer (Agilent Technologies, Singapore). Data represent mean OD600nm values from three independent experiments, with standard deviations (SD) adjusted by subtracting absorbance readings from uninoculated TSB medium containing corresponding biocide concentrations. For positive controls (untreated bacterial cultures), net absorbance values were determined by correcting for the absorbance of the TSB medium alone at 600 nm.Biofilm formation inhibition assay
The ability of composite PDMS-based film formulations (i.e., PDMS@SIL5<, PDMS@SIL5, PDMS@3@SIL5<, PDMS@3@SIL5, PDMS@3@SiO2, PDMS@3@SiO2<, PDMS@SiO2, and PDMS@SiO2<) to prevent P. delhiensis PS27 biofilm formation was carried out as described elsewhere35 with slight modifications. Pseudomonas cells were pre-cultivated as described earlier and inoculated (200 μL) at an initial biomass load corresponding to 1 × 107 CFU mL−1 in a 96-well microtiter plate in which submerged circular disks of the above-mentioned composite films. Thus, bacterial cultures were incubated at 37 °C for 24 hours under shaking conditions (180 rpm) to allow bacterial cells to grow as a biofilm. Then, planktonic cells were serially diluted in base ten in a sterile saline solution (NaCl 0.9% w/v) to estimate the viable cell count. Disks of composite films were instead harvested, washed thrice to remove traces of loosely adherent cells, resuspended in saline solution (200 μL), and sonicated for 30 minutes at a frequency of 35 kHz, using a bath sonicator filled with iced water, to remove the microbial biomass that grew as a biofilm onto the disk surface. The microbial titer of bacterial cells within the biofilm was evaluated by serially diluting the sonication product. All assays were conducted at least three times, and data are reported as the average value of the CFU per disk in the logarithmic scale with SD.Results and discussion
Synthesis of silica gel with high surface area
Hydrolysis of TEOS was carried out in the presence of fluoride ions36 and resorcinol. In a first attempt, resorcinol was dissolved in methanol, then TEOS was added followed by tetrabutylammonium fluoride (TBAF). The solution was stirred at 30 °C for about 15 h. After this time the pale pink solution was concentrated under reduced pressure (200–150 mbar) at 30 °C to remove almost all solvent, then water was slowly added. A pale pink precipitate was obtained. The suspension was stirred at 50 °C for 30 min and at 80 °C for 3 hours. After cooling at r.t., the solid was filtered and washed with water, ethanol, and methanol until the solid and the filtrate were colourless. The solid was dried at 60 °C under reduced pressure to give silica SIL1 (Table 1). Measurement of the specific surface area (SSA) using the BET method gave a value of 472 m2 g−1 (Fig. S1†). This silica was calcined at 400 °C for 4 hours. After this treatment, SSA increased up to 617 m2 g−1 (Table 1, entry 1)To understand whether resorcinol played a role in the formation of the silica with high surface area, the same procedure was carried out without adding resorcinol (SIL2, Table 1, entry 2). A much lower value of the specific surface area was obtained for both the not-calcined and the calcined silica. By doubling the concentration of TEOS and TBAF and using the same amount of resorcinol, the obtained silica (SIL3) featured a higher surface than that obtained without resorcinol (entry 3) (Fig. S2 and S3†).
The procedure of entry 1 was repeated with a slight modification, the pale pink solution was concentrated under reduced pressure (200–150 mbar) at 30 °C to remove 50 mL of solvent, and then water was slowly added (entry 4, SIL4). Silica was calcined as earlier described and a material with a very high SSA was obtained (1000 m2 g−1). This procedure was duplicated (entry 5, SIL5). The SSA was determined before and after calcination (Fig. 1a).
 | ||
| Fig. 1 (a) N2 adsorption/desorption isotherms of SIL5 before (black line) and after calcination (red line) and (b) pore size distribution as dV/dw (cm3 g−1·nm−1) vs. pore width (nm) calculated by the Barrett–Joyner–Halenda (BJH) model of SIL5 before (black line) and after calcination (red line). | ||
Again, a sharp increase in SSA was observed, as well as a very high SSA of the calcined silica. A further experiment was conducted by halving the amount of resorcinol (entry 6, SIL6). Calcined silica still showed a high SSA.
Pore size distribution in SIL2 before and after calcination are alike showing a large range up to ca. 20 nm (Fig. S2†). The use of resorcinol (SIL1 and SIL3, Fig. S1 and S3†) gave a more uniform distribution and in the case of SIL5, the distribution was very sharp (Fig. 1b).
13C-CPMAS-NMR of silica SIL5 (CPMAS = cross-polarization magic angle spinning) before calcination showed the absence of signals due to resorcinol and the presence of ethoxy groups (Fig. 2a). Indeed, the hydrolysis reaction may lead to silica-containing unreacted SiOEt groups.3729Si-CPMAS-NMR of silica SIL5 (Fig. 2b) showed signals at ca. −110 and −101 ppm that can be assigned to Q4 [Si(OSi)4], Q3 [Si(OSi)3OH] systems, and Q2 [Si(OSi)2(OH)2] as a small shoulder of the Q3 peak. 29Si-CPMAS-NMR after calcination agreed with a different nature of the silica material. Indeed, the ratios Q4/Q3 and Q4/Q2 decreased, which is in line with the loss of the ethoxy groups to give more silanol groups as well as the transformation of siloxane bridges into silanols (Fig. 2b and 5h–i). This finding is in agreement with the observed increase in SSA after calcination. Fig. 3 reports TGA before and after calcination of SIL5.
 | ||
| Fig. 2 (a) 13C CPMAS NMR of non-calcined SIL5; (b) 29Si CPMAS NMR of non-calcined and calcined SIL5. | ||
 | ||
| Fig. 3 TGA under air flow of SIL5 before (black line) and after (red line) calcination and ion current vs. temperature (blue dotted line) for CO2 evolution relative to TGA of SIL5 before calcination. | ||
This analysis showed an appreciable loss of organic substance attributable to the presence of non-hydrolyzed ethoxy groups. Moreover, mass analysis coupled with TGA clearly indicated the evolution of CO2 during TGA (Fig. 3) as well as water (Fig. S4†). Transmission electron microscopy (TEM) analysis showed the uniform and small size of the silica nanoparticles (Fig. 4) in agreement with data obtained by nitrogen adsorption/desorption (Table 1).33
 | ||
| Fig. 4 TEM images of SIL5. | ||
The results demonstrated that resorcinol facilitated both hydrolysis and condensation processes, as well as influenced the uniform morphology of the material. Additionally, the quantity of solvent plays a crucial role, as its near-complete evaporation enabled the synthesis of small-sized amorphous silica with a narrow pore size distribution and a very high specific surface area. The mechanism by which resorcinol achieves this effect may depend on its acidity, which enhances hydrolysis and subsequent condensation. Yet, understanding its role in achieving a high specific surface area is more complex. A plausible hypothesis is that the combination of fluoride ions and resorcinol facilitates hydrolysis, also through intermediate hypervalent species (Scheme 1).36,38
 | ||
| Scheme 1 Hypervalent silicon structures and hydrolysis. | ||
Fig. 5a schematically represents what could be present in the solution (TEOS, resorcinol, hypervalent silicon species). Removing some solvent leads to a concentrated solution where resorcinol could form a hydrogen bonds network (Fig. 5b), whereas, water addition can further cause rapid hydrolysis and condensation, leading to silica particles (Fig. 5c).
 | ||
| Fig. 5 Schematic representation of silica formation in the presence of resorcinol. (a–f) Steps of silica gel formation. (g–i) Structural representation of steps d–f (see text for explanation). | ||
Resorcinol may aid this process by “solvating” silane molecules in certain regions (Fig. 5d), effectively acting as a “shield” toward other regions. The subsequent sample filtration removed unreacted silane and resorcinol (Fig. 5e). Moreover, controlled calcination with a gradual temperature ramp allowed the carbonization of ethoxy groups, as evidenced by CO2 and H2O formation during TGA, without collapsing the silica structure (Fig. 5f). Fig. 5d can be represented as in Fig. 5g in which the resulting molecules from hydrolysis are joined together to build larger structures but the presence of the resorcinol controls such process avoiding further aggregation towards bigger structures. Filtration and subsequent calcination (Fig. 5h–i) allow the obtainment of silica with very high SSA. Water evolution could also play a role in the formation of silanol groups through rehydroxylation of thermally dehydroxylated silica (Q4 → Q3 see Fig. 5h–i).39
Synthesis of quaternary ammonium salts (QASs)-based silane and antimicrobial evaluation
To exploit the novel high surface area silica gel, we decided to use it as a platform for antifouling materials upon the grafting of suitably designed alkoxysilanes. To do this, we first synthesized a set of quaternary ammonium salts (QASs)-based silanes by reaction of (3-chloropropyl)dimethoxy(methyl)silane, and (3-chloropropyl)– or (3-bromopropyl)trimethoxysilane with the corresponding N,N-dimethyl-alkyl amine or 1-alkyl-1H-imidazole (Scheme 2), and in turn, the antibacterial activity of these molecules was tested against the highly resilient Pseudomonas delhiensis PS27 strain to environmental stresses.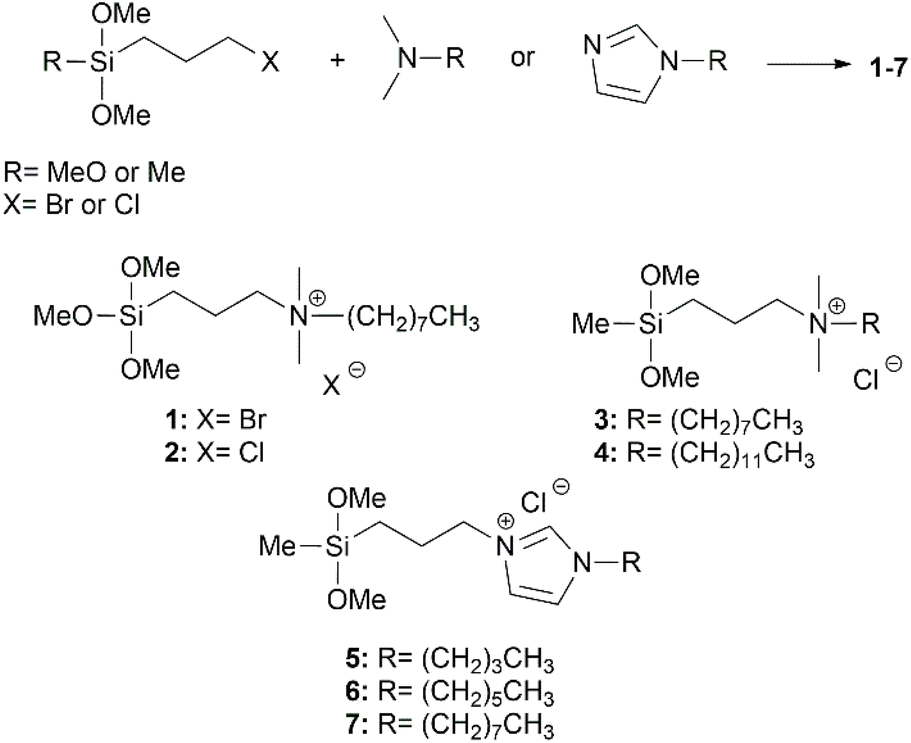 | ||
| Scheme 2 Preparation of quaternary ammonium salts (QASs)-based silanes 1–7. | ||
Trimethoxysilanes 1 and 2 did not exhibit antibacterial activity within the tested range of concentrations (Fig. 6). A comparative analysis of the antibacterial properties of 1–3 underscored the critical role of the silicon-bonded methyl group in enhancing the molecule biocidal effect. Indeed, silanes 2 and 3, differing only for the methyl groups on silicon, show significant differences in terms of biocidal activity (Fig. 6). Instead, the dimethoxy(methyl)silane QAS with a longer alkyl chain (i.e., 4) displayed a slightly reduced antibacterial activity (50 μg mL−1) as compared to 3 (25 μg mL−1). This could be due to the increase in conformational freedom, which can make more difficult the interaction with cell membranes.40–42 Moreover, compounds 5, 6, and 7, which featured the identical dimethoxy(methyl)silane group as 3, and the replacement of the aliphatic nitrogen with an imidazolium ring, resulted in diminished antibacterial effectiveness, except for 7. Specifically, reducing the alkyl chain length to six carbons, as in the case of 6, caused the worsening of its biocidal power, which was only improved by increasing the concentration of QAS 6. Instead, shortening the alkyl chain to four carbon atoms with the compound 5 completely abolished the antibacterial activity.43
 | ||
| Fig. 6 Minimal inhibitory concentration of antifouling compounds against the P. delhiensis PS27 strain. * p-value ≤ 0.05 based on the T-test comparing data deriving from the biological trials (n = 3) performed for antifouling biocides 3 and 7. | ||
Thus, these findings highlight the importance of the moieties (type of silane, ammonium, and alkyl chain length) in mediating the antibacterial properties among the biocides tested. Hence, based on these data, QAS 3 emerged as the most suitable candidate for grafting onto silica.
Grafting of silica with antimicrobial quaternary ammonium salts (QASs)-based silane
Dimethoxy(methyl)silane QAS 3 was grafted on silica by gentle heating of a methanolic solution under reduced pressure (Scheme 3). | ||
| Scheme 3 Preparation of quaternary ammonium-based silica 3@SIL5 from silica precursor SIL5. | ||
In Fig. 7a the 29Si CPMAS NMR spectra of the modified silica gel 3@SIL5 and the pristine calcined silica (SIL5) are reported. From this comparison, it is possible to observe in the former the increase of the Q4 signal due to [Si(OSi)4] systems at ca. −108 ppm with respect to the Q3 one. In the modified silica gel spectrum, there are also present D signals due to R2Si(SiO)2 at ca. −21 ppm and RSi(OSi)OH and RSi(OSi)OMe as shoulder at ca. −13 ppm. These findings are indicative of the success of the grafting process.
 | ||
| Fig. 7 (a) 29Si CPMAS-NMR spectra of 3@SIL5 material overlapped with pristine silica SIL5; (b) 13C CPMAS-NMR spectra of 3@SIL5. | ||
In the 13C CPMAS-NMR spectra of 3@SIL5 (Fig. 7b) methylene groups bound to nitrogen resonate at ca. 65 ppm. The signal at ca. 51 ppm can be ascribed to N,N-dimethyl group. Signals between 32 and 14 ppm can be assigned to the alkyl chain together with the methylene of the aminopropyl chain (–CH2CH2CH2N+–). Signals at lower chemical shifts ca. 12 ppm and −2 ppm can be ascribed to the methylene group and methyl group bound to the silicon atom, respectively.
Thermogravimetric analysis of the 3@SIL5 sample is shown in Fig. S5.† The organic content is 26.3%, which corresponds to an estimated loading of ca. 1.05 mmol g−1. Grafting was also performed by refluxing dry SIL5 in the presence of silane 3 in toluene under an argon atmosphere to give the silica material 3@SIL5(tol). TGA showed a slightly lower loading of ca. 0.86 mmol g−1. For comparison purposes, the grafting procedure reported in Scheme 3 was applied to functionalize commercial Silica gel (0.060–0.040 mm, Merck). 29Si CPMAS-NMR (Fig. 8) and 13C CPMAS-NMR (Fig. S6†) spectra of obtained material 3@SiO2 show the successful functionalization with the characteristic signals previously discussed for 3@SIL5.
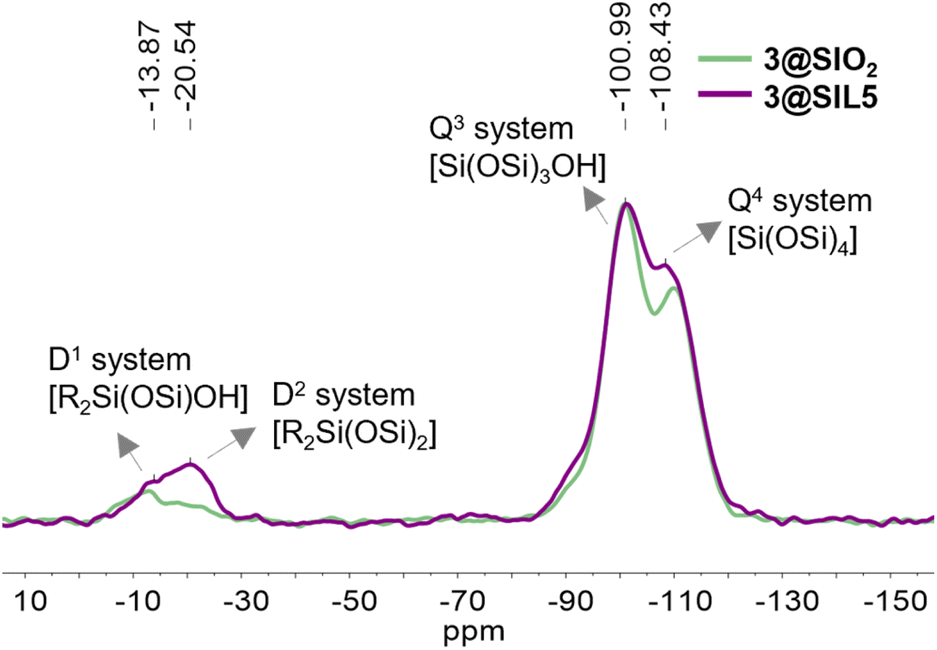 | ||
| Fig. 8 29Si CPMAS-NMR spectra of 3@SIL5 material overlapped with 3@SiO2. | ||
In the case of 3@SiO2, it is worth noting in the 29Si CPMAS-NMR spectrum (Fig. 8) that the strongest D signal is found at ca. −13 ppm due to the RSi(OSi)OH and/or RSi(OSi)OMe group. In 3@SIL5 silica material, the high SSA of silica precursor together with the higher availability of superficial silanol groups, explain the major intensity of peak at ca. −20 ppm due to R2Si(SiO)2 systems.
TGA shows a different degradation profile than that acquired for 3@SIL5 material (Fig. S7†). Indeed, a lower organic loading is observed (0.45 mmol g−1 for 3@SiO2 compared to 1.05 mmol g−1 for 3@SIL5). Again, this finding can be related to the very large SSA observed for allowing a better functionalization via grafting. As an ancillary investigation on silica SIL5, we applied our recent procedure for the grafting of trimethoxysilanes in the presence of alcohol.44 Silica SIL5 was heated with (N,N-dimethyl-3-aminopropyl)trimethoxysilane in 1-heptanol (Scheme 4a).
 | ||
| Scheme 4 (a) Synthesis of silica SIL7; (b) synthesis of silica SIL8. | ||
The 29Si CPMAS-NMR spectrum of SIL7 (Fig. S8a†) shows signals at ca. −111 ppm which is characteristic of the Q4 system [Si(OSi)4]. The peak at ca. −103 ppm assigned to the Q3 system (Si(OSi)3OH) is less noticeable. This is indicative of a lower presence of free silanol groups. Signal at ca. −67 ppm characterizes the T3 system [RSi(OSi)3] of silanol portion. The peak at ca. −59 ppm can be ascribed to T2 systems [RSi(OSi)2OH]. Fig. S8a† reports also the 29Si CPMAS-NMR spectrum of the same material obtained using commercial silica.44 In the present case, the main difference is in the T3 signal.
The 13C CPMAS-NMR spectrum in Fig. S8b† shows peaks of the heptyl moiety of the heptanol alcohol used in the synthesis. The peak at 10 ppm is due to the methylene group linked to the silicon atom. The peak at 12 ppm can be assigned to the methyl group of the heptyl chain. The deshielded signals at ca. 62 and 63 ppm can be assigned to –OCH2– or –NCH2– groups while the peak at ca. 44 ppm could be ascribed to methyl bound to a nitrogen atom. Signals between 21 and 32 ppm can be assigned to the alkyl chain together with the methylene of the aminopropyl chain. In this case, an almost perfect superimposition is observed with material reported previously (Fig. S8b†).44
Thermogravimetric analysis of SIL7 (Fig. S9†) shows a loading of organic content of 21.7%, which resulted higher than that obtained previously (12.5%)44 due to the high SSA of the new silica. Material SIL7 was alkylated with 1-iodo-octanol in 1-heptanol to give material SIL8 (Scheme 4b). 13C CPMAS NMR spectrum (Fig. S10†) showed the complete superimposition with the corresponding material prepared from commercial silica while TGA showed a higher organic content (33.5% vs. 24%) (Fig. S11†).
Composite polydimethylsiloxane/SiO2 films and their antibiofilm activity
To investigate the possible antifouling properties of the modified silica, we prepared polydimethylsiloxane (PDMS) films containing dispersed silica 3@SIL5 (samples PDSM@3@SIL5 and PDSM@3@SIL5< containing 0.18 and 0.09 mmol g−1 of QAS respectively, see Table 2) or 3@SiO2 (samples PDMS@3@SiO2 and PDMS@3@SiO2<) (Table 2). For comparison, a PDMS film without silica and a PDMS containing unmodified silica (samples PDMS@SiO2, PDMS@SiO2<, PDMS@SIL5, and PDMS@SIL5<) were prepared as blank tests. Table 2 reports all investigated samples with their weight composition in terms of silica and quaternary ammonium compound content.| Entry | Sample name | Silica (mg) | QAS (mmol g−1) |
|---|---|---|---|
| 1 | PDMS@SIL5< (control) | 100 | — |
| 2 | PDMS@3@SIL5< | 100 | 0.09 |
| 3 | PDMS@SIL5 (control) | 200 | — |
| 4 | PDMS@3@SIL5 | 200 | 0.18 |
| 5 | PDMS@3@SiO 2 | 360 | 0.18 |
| 6 | PDMS@SiO 2 (control) | 309 | — |
| 7 | PDMS@3@SiO 2 < | 180 | 0.09 |
| 8 | PDMS@SiO 2 < (control) | 154.5 | — |
These PDMS-based films were prepared by a reaction of hydroxy-terminated polydimethylsiloxane (MW 18 K) (1 g) with triacetoxymethylsilane (133 μL) as a cross-linker (Scheme 5).
 | ||
| Scheme 5 Preparation of PDMS/silica composite films. | ||
The PDMS film containing silica 3@SIL5 was characterized by 29Si and 13C CPMAS NMR (Fig. 9). Fig. 9a reports 29Si CPMAS NMR of PDMS@3@SIL5 overlapped with 3@SIL5 for comparison. In particular, the T3 peak assigned to RSi(OSi)3 system and the small peak at ca. −55 ppm due to the T2 system [RSi(OSi)2OH] confirm the cross-linked nature of the film. The sharp peak of the D2 system [R2Si(OSi)2] at −22 ppm is due to the PDMS structure. The small shoulder, as well as the Q4 peak, are due to the presence of silica 3@SIL5, as also confirmed by peaks in the 13C CPMAS NMR spectrum (Fig. 9b). Furthermore, in the 29Si CPMAS NMR, the less pronounced signal for the Q3 system [Si(OSi)3OH] at ca. −105 ppm could be indicative of a lower number of free silanol groups and hence the occurrence of some cross-linking with silanol groups.
 | ||
| Fig. 9 (a) 29Si CPMAS-NMR spectra of 3@SIL5 material overlapped with PDSM@3@SIL5 film; (b) 13C CPMAS-NMR spectra of PDMS@3@SIL5 film. | ||
Bacterial species can form complex structural communities known as biofilms, which process follows microbial cells' adhesion and aggregation to a given surface, encapsulating them-selves within an extracellular polymeric substance. In this context, bacterial cells acquire unique biological traits, heightened tolerance and resistance to antimicrobials, and resilience to external stresses.45,46 Bacterial biofilms contribute significantly to biofouling in aquaculture, affecting cultured species and infrastructure, and leading to substantial economic losses.47,48 Therefore, early intervention in biofilm formation employing antifouling compounds is critical, mostly when it comes to Pseudomonas species, which are of particular concern due to their opportunistic behavior, proficiency in biofilm production, and their involvement in spoilage within aquatic environments.35,47,49 Thus, PDMS-based composite films were tested in vitro to ascertain their suitability as antifouling agents against P. delhiensis PS27 (Fig. 10a). The PDMS film had notable efficiency in inhibiting bacterial adhesion, resulting in approximately a three-log reduction in viable colony-forming unit (CFU) counts. This antifouling effect likely arises from the hydrophobic nature of the PDMS polymer. Incorporating the biocide 3 grafted to silica into the PDMS film, producing the antifouling variant PDMS@3@SIL5, completely prevented bacterial adhesion, as no viable CFUs were detected compared to the unchallenged cells. However, a modified film containing half the biocide amount (PDMS@3@SIL5<) exhibited reduced antifouling performance compared to PDMS@3@SIL5, showing only a marginal improvement over the PDMS film.
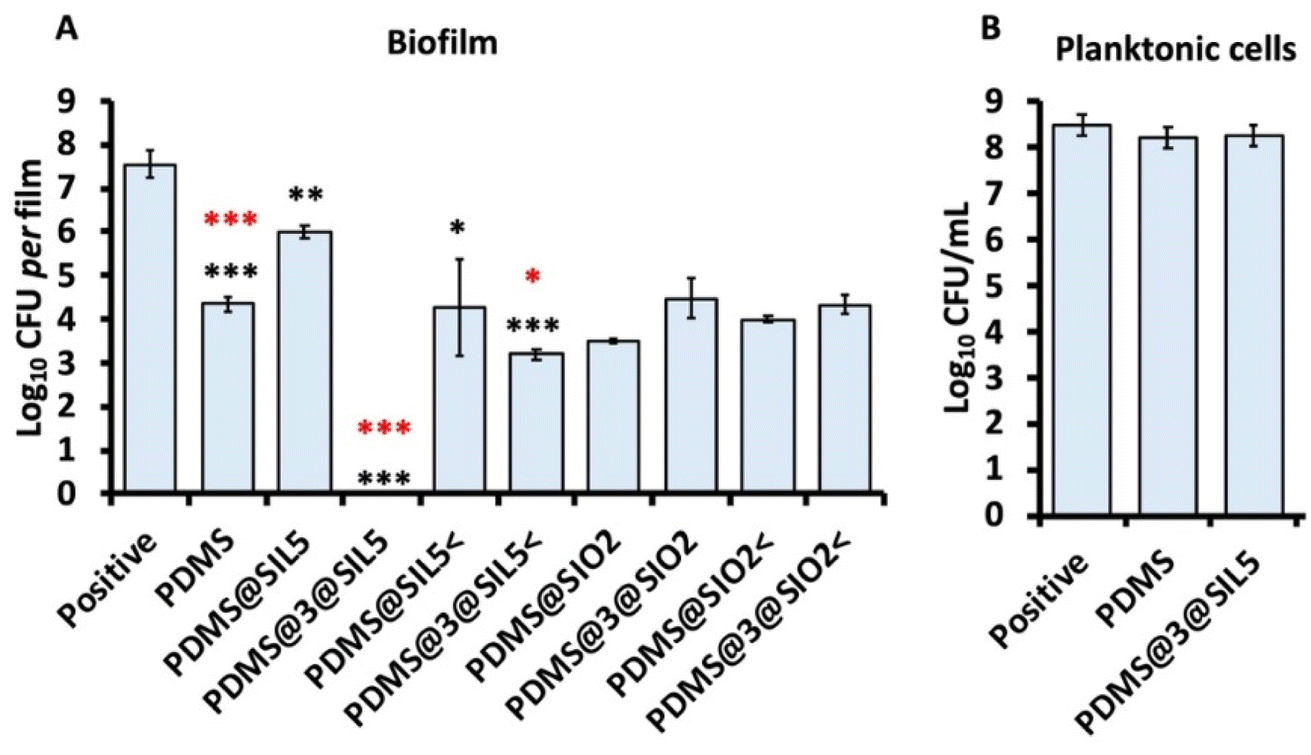 | ||
| Fig. 10 (a) Biofilm inhibition efficacy of PDMS-based composite films against the Pseudomonas delhiensis PS27 strain. Black asterisks refer to p-values obtained comparing microbial titers of the positive control with those of each film tested. Red asterisks refer instead to p-values considering the microbial titer of the PDMS film compared to that obtained for films containing different amounts of biocide (i.e., 3@SIL5 and 3@SIL5<), as well as that containing not grafted silica with the biocide (i.e., PDMS@SiO2; *p-value ≤ 0.05, **p-value ≤ 0.01, ***p-value ≤ 0.001). (b) Bar graph showing the unaltered physiological fitness of planktonic cells experiencing the presence of either PDMS or PDMS@3@SIL5 films. | ||
Further analysis with PDMS films containing silica without the biocide aimed to assess whether silica alone could contribute to antifouling properties.
Results indicated that silica alone in PDMS@SIL5 decreased antifouling efficacy probably due to the exposition of numerous silanol groups and a consequent reduction of PDMS hydrophobicity. Instead, PDMS@SIL5< showed performance comparable to PDMS within the standard deviation range. Collectively, these findings emphasize that the biocide, at an optimal concentration, is essential for achieving full antifouling potential.
Simultaneously, the antifouling effects of PDMS and PDMS@3@SIL5 films did not compromise the physiological fitness of planktonic Pseudomonas cells (Fig. 10b), as microbial titers remained statistically similar between challenged and unchallenged cells. This finding is particularly significant, suggesting minimal or no biocide release from the polymeric matrix into the extracellular environment. Consequently, this antifouling film offers additional applicative advantages, as the biocide embedded within the film is unlikely to threaten aquatic wildlife within environmental niches. Finally, we also evaluated the antifouling efficacy of PDMS-based films containing commercial silica grafted with QAS-based silane, namely samples PDMS@3@SIO2 and PDMS@3@SIO2< (Fig. 10a). Despite matching the biocide content of PDMS@3@SIL5 and PDMS@3@SIL5<, these films exhibited different behavior. Compared to their controls (i.e., samples PDMS@SIO2 and PDMS@SIO2<) these films did not significantly enhance bacterial adhesion inhibition, demonstrating antifouling effects comparable to the PDMS film or their respective controls. These results suggest that the previously used silica (3@SIL5), synthesized in-house with a very high SSA, plays a crucial role in enhancing the performance of these antifouling materials.
Conclusion
In conclusion, we have described a new simple method for preparing a silica gel with a very narrow pore size distribution and high surface area through a simple and sustainable synthesis under mild conditions using tetraethyl orthosilicate and a cheap molecule, resorcinol, as a template. Resorcinol can be removed by a simple filtration step without the need for expensive procedures such as extraction or calcination.Then, as an application of this new silica gel, a set of QAS-based silanes was synthesized, and their antibacterial activity was tested against P. delhiensis PS27.
Using the more active QAS-based silane, the corresponding QAS-based silica was successfully prepared. The incorporation of this QAS-based silica into a PDMS film results in a material with excellent fouling release properties. No bacterial biofouling occurred on the surface of the QAS-silica/PDMS composite.
It is also important to stress that, although the QAS-based silane used was the one that presented the higher antibacterial activity, no killing of planktonic cells was observed. This indicated a different role in the activity when the QAS-based silane is covalently linked to silica with very high SSA.
Indeed, comparison with the corresponding material having the same QAS moiety but linked to commercial silica clearly showed the complete loss in antibiofilm activity, then indicating the usefulness of the reported strategy.
Our recent strategy for trimethoxysilane grafting in heptanol and subsequent transformation into QAS-based silica was also applied and further investigations using this approach will be developed.
Finally, it is worth to spend some consideration about the role of resorcinol in the synthesis of silica gel. Resorcinol cannot form a self-assembled structure such as a micelle. Therefore, the formation mechanism of the new silica differs from those in mesostructured silica such as MCM-41. Furthermore, the pore formation mechanism in the resorcinol-silica system is also different from that of other organic-inorganic systems such as PEG-silica, although both resorcinol-silica and PEG-silica systems form amorphous nanocomposites.
Here, one resorcinol molecule per four TEOS molecules forms hydrogen bonding, first with partially hydrolysed TEOS and secondly, after condensation reactions, with surface silanols. The strength of hydrogen bonding between silanol and the hydroxyl group of resorcinol in the concentrated phase could be sufficiently strong (at 30 °C) to restrict the condensation of silanol groups. Since the silica gel network cannot form additional Si–O–Si bonds due to the existence of resorcinol, the space occupied with bulk resorcinol remains as pores after filtration. Nevertheless, the silica gel gains rigidity high enough to maintain the bulky structure up to 400 °C. The resorcinol forming hydrogen bonding with silanol contributes to the increase in the specific surface area, while all resorcinol contributes to the narrow pore size distribution.
Also, it is noteworthy that the silica gel network does not shrink when heated. Indeed, the template molecule usually remains in the structure, being removed after calcination. In the present case, the template is removed with filtration, then no “protection” to shrinkage can be operative.
It is interesting how a simple molecule like resorcinol can be so useful in the sol–gel process. Of course, both its solubility in the system and its ability to form good interactions with silanols are important: not too strong to remain adsorbed in the composite after filtration, not too weak to allow complete hydrolysis and condensation towards large siliceous agglomerates.
These synthetic approaches represent interesting sustainable ways for developing new hybrid materials possessing antifouling properties.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research has been partially supported by the European Union—NextGenerationEU—National Sustainable Mobility Center CN00000023, Italian Ministry of University and Research Decree n. 1033—17/06/2022, spoke 3, CUP B73C22000760001. The authors are thankful to Nunzio Gallì (ISMN-CNR) for performing surface area and pore size analyses.References
- A. Watermann and J. Brieger, Nanomaterials, 2017, 7, 189 CrossRef PubMed.
- F. Tang, L. Li and D. Chen, Adv. Mater., 2012, 24, 1504–1534 CrossRef CAS PubMed.
- Q. Liang, M. Sun, Y. Ma, F. Wang, Z. Sun and J. Duan, Chemosphere, 2023, 311, 136955 CrossRef CAS PubMed.
- A. A. Nayl, A. I. Abd-Elhamid, A. A. Aly and S. Bräse, RSC Adv., 2022, 12, 13706–13726 RSC.
- F. Rizzi, R. Castaldo, T. Latronico, P. Lasala, G. Gentile, M. Lavorgna, M. Striccoli, A. Agostiano, R. Comparelli, N. Depalo, M. L. Curri and E. Fanizza, Molecules, 2021, 26, 4247 CrossRef CAS PubMed.
- M. E. Vance, T. Kuiken, E. P. Vejerano, S. P. McGinnis, M. F. Hochella, Jr., D. Rejeski and M. S. Hull, Beilstein J. Nanotechnol., 2015, 6, 1769–1780 CrossRef CAS PubMed.
- J. G. Croissant, K. S. Butler, J. I. Zink and C. J. Brinker, Nat. Rev. Mater., 2020, 5, 886–909 CrossRef CAS.
- M. Yazdimamaghani, P. J. Moos, M. A. Dobrovolskaia and H. Ghandehari, Nanomedicine, 2019, 16, 106–125 CrossRef CAS PubMed.
- G.-H. Lee, Y.-S. Kim, E. Kwon, J.-W. Yun and B.-C. Kang, Pharmaceutics, 2020, 12, 826 CrossRef CAS PubMed.
- C. J. Brinker and G. W. Scherer, Hydrolysis and Condensation II: Silicates, in Sol-Gel Science, ed. C. J. Brinker and G. W. Scherer, Academic Press, San Diego, 1990, ch. 3, pp. 96–233 Search PubMed.
- N. A. J. M. Sommerdijk, E. R. H. van Eck and J. D. Wright, Chem. Commun., 1997, 159–160 RSC.
- P. S. Singh, J. Colloid Interface Sci., 2008, 325, 207–214 CrossRef CAS PubMed.
- J. Wen, B. Dhandapani, S. T. Oyama and G. L. Wilkes, Chem. Mater., 1997, 9, 1968–1971 CrossRef CAS.
- S. Sato, T. Murakata, T. Suzuki and T. Ohgawara, J. Mater. Sci., 1990, 25, 4880–4885 CrossRef CAS.
- R. Takahashi, S. Sato, T. Sodesawa, M. Kawakita and K. Ogura, J. Phys. Chem. B, 2000, 104, 12184–12191 CrossRef CAS.
- L. Tosheva, V. Valtchev and J. Sterte, J. Mater. Chem., 2000, 10, 2330–2337 RSC.
- D. Yang, G. Yang, G. Liang, Q. Guo, Y. Li and J. Li, Colloids Surf., A, 2021, 610, 125700 CrossRef CAS.
- F. Giacalone, V. Campisciano, C. Calabrese, V. La Parola, L. F. Liotta, C. Aprile and M. Gruttadauria, J. Mater. Chem. A, 2016, 4, 17193–17206 RSC.
- V. Campisciano, F. Giacalone and M. Gruttadauria, Chem. Rec., 2017, 17, 918–938 CrossRef CAS PubMed.
- V. Campisciano, M. Gruttadauria and F. Giacalone, ChemCatChem, 2019, 11, 90–133 CrossRef CAS.
- C. Calabrese, V. Campisciano, F. Siragusa, L. F. Liotta, C. Aprile, M. Gruttadauria and F. Giacalone, Adv. Synth. Catal., 2019, 361, 3758–3767 CrossRef CAS.
- I. Ziccarelli, R. Mancuso, M. Novello, A. De Salvo, C. Calabrese, L. Valentino, A. Pettignano, M. Gruttadauria, F. Giacalone and B. Gabriele, ChemCatChem, 2025, 17, e202401841 CrossRef CAS.
- A. Presentato, E. La Greca, L. Consentino, R. Alduina, L. F. Liotta and M. Gruttadauria, Nanomaterials, 2023, 13, 1291 CrossRef CAS PubMed.
- P. Marzullo, M. Gruttadauria and F. D’Anna, Biomol, 2024, 14, 957 CAS.
- C. Calabrese, L. F. Liotta, L. Soumoy, C. Aprile, F. Giacalone and M. Gruttadauria, Asian J. Org. Chem., 2021, 10, 2932–2943 CrossRef CAS.
- D. Ren, N. Chen, J. Xu, Z. Ye, X. Li, Q. Chen and S. Ma, Colloids Surf., A, 2021, 625, 126508 CrossRef CAS.
- N. I. Maliavski, O. V. Dushkin and G. Scarinci, Ceram. Silik., 2001, 45, 48–54 CAS.
- Q. Wang, D. B. Mahadik, P. Meti, Y.-D. Gong, K.-Y. Lee and H.-H. Park, Microporous Mesoporous Mater., 2020, 294, 109863 CrossRef CAS.
- D. Carriazo, M. C. Gutiérrez, M. L. Ferrer and F. del Monte, Chem. Mater., 2010, 22, 6146–6152 CrossRef CAS.
- M. Djebbar, F. Djafri, M. Bouchekara and A. Djafri, Appl. Water Sci., 2012, 2, 77–86 CrossRef CAS.
- M. Sulpizi, M.-P. Gaigeot and M. Sprik, J. Chem. Theory Comput., 2012, 8, 1037–1047 CrossRef CAS PubMed.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CrossRef CAS.
- M. Dell'Edera, F. Petronella, A. Truppi, L. F. Liotta, N. Gallì, T. Sibillano, C. Giannini, R. Brescia, F. Milano, M. Striccoli, A. Agostiano, M. L. Curri and R. Comparelli, Catalysts, 2020, 10, 893 CrossRef.
- A. Presentato, S. Lampis, A. Vantini, F. Manea, F. Daprà, S. Zuccoli and G. Vallini, Microorganisms, 2020, 8, 92 CrossRef CAS PubMed.
- E. Piacenza, A. Presentato, E. Zonaro, J. A. Lemire, M. Demeter, G. Vallini, R. J. Turner and S. Lampis, Microb. Biotechnol., 2017, 10, 804–818 CrossRef CAS PubMed.
- E. Reale, A. Leyva, A. Corma, C. Martínez, H. García and F. Rey, J. Mater. Chem., 2005, 15, 1742–1754 RSC.
- J. Cihlář, Colloids Surf., A, 1993, 70, 239–251 CrossRef.
- M. Cypryk, B. Gostyński and M. Pokora, New J. Chem., 2019, 43, 15222–15232 RSC.
- V. V. Potapov and L. T. Zhuravlev, Glass Phys. Chem., 2005, 31, 661–670 CrossRef CAS.
- A. Carangio, S. Srinivasan, C. McGuigan, G. Andrei, R. Snoeck, E. De Clercq and J. Balzarini, Antiviral Chem. Chemother., 2002, 13, 263–271 CrossRef CAS PubMed.
- C. McGuigan, A. Brancale, H. Barucki, S. Srinivasan, G. Jones, R. Pathirana, A. Carangio, S. Blewett, G. Luoni, O. Bidet, A. Jukes, C. Jarvis, G. Andrei, R. Snoeck, E. De Clercq and J. Balzarini, Antiviral Chem. Chemother., 2001, 12, 77–89 CrossRef CAS PubMed.
- C. Rizzo, P. Cancemi, L. Mattiello, S. Marullo and F. D'Anna, ACS Appl. Mater. Interfaces, 2020, 12, 48442–48457 CrossRef CAS PubMed.
- P. Cancemi, M. Buttacavoli, F. D'Anna, S. Feo, R. M. Fontana, R. Noto, A. Sutera, P. Vitale and G. Gallo, New J. Chem., 2017, 41, 3574–3585 RSC.
- P. Marzullo, V. Campisciano, L. F. Liotta, F. D'Anna, F. Giacalone and M. Gruttadauria, Molecules, 2024, 29, 4730 CrossRef CAS PubMed.
- K. D. Xu, P. S. Stewart, F. Xia, C. T. Huang and G. A. McFeters, Appl. Environ. Microbiol., 1998, 64, 4035–4039 CrossRef CAS PubMed.
- B. Purevdorj-Gage, W. J. Costerton and P. Stoodley, Microbiology, 2005, 151, 1569–1576 CrossRef CAS PubMed.
- I. Fitridge, T. Dempster, J. Guenther and R. de Nys, Biofouling, 2012, 28, 649–669 CrossRef PubMed.
- S. Wu, G. Liu, W. Jin, P. Xiu and C. Sun, Front. Microbiol., 2016, 7, 102 Search PubMed.
- F. C. Capri, E. Prazzi, G. Casamento, D. Gambino, G. Cassata and R. Alduina, Microb. Ecol., 2023, 86, 1923–1933 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5su00047e |
| This journal is © The Royal Society of Chemistry 2025 |