
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ruthenium-catalyzed dimerization of vanillin for the formation of a biobased epoxy thermoset resin†
Flavia
Ferrara
 a,
Iuliana
Ribca
b,
Namratha
Prabhu‡
a,
Josselin
Mante‡
a,
Maureen
Gumbo
a,
Andreas
Ekebergh
*a,
Mats
Johansson
*b and
Nina
Kann
*a
a,
Iuliana
Ribca
b,
Namratha
Prabhu‡
a,
Josselin
Mante‡
a,
Maureen
Gumbo
a,
Andreas
Ekebergh
*a,
Mats
Johansson
*b and
Nina
Kann
*a
aDepartment of Chemistry and Chemical Engineering, Chalmers University of Technology, SE-412 96 Göteborg, Sweden. E-mail: kann@chalmers.se
bKTH Royal Institute of Technology, Department of Fibre and Polymer Technology, Division of Coating Technology, SE-100 44 Stockholm, Sweden. E-mail: matskg@kth.se
First published on 11th April 2025
Abstract
Vanillin is one of few lignin platform chemicals that are currently available on industrial scale. Seeking to find biobased alternatives to the reprotoxic compound bisphenol A (BPA), we have successfully dimerized three different monomeric vanillin derivatives in a ruthenium-catalyzed Tischenko reaction. The resulting esters were characterized by NMR, FTIR, HRMS, and single crystal X-ray diffraction. The thermal behaviour of one of these derivatives, the epoxy divanillin ester EDVE, was studied further by DSC and TGA. EDVE was subsequently applied towards the preparation of an epoxy thermoset resin via curing with Jeffamine D-400. The thermoset formulation was thermally cured, monitoring the curing with DSC and FTIR. The final thermoset was then characterized with respect to physical and mechanical properties with DSC, TGA, and DMA. This catalytic approach provides a new strategy to access vanillin-based epoxides that could potentially replace bisphenol A.
Sustainability spotlightThe transition to a sustainable industry requires not only the replacement of precursors derived from petroleum with biomass alternatives, but also that current chemical processes adapt to accommodate the oxygenated structures of biobased molecules. Ideally, toxic chemicals should be exchanged for more benign alternatives, and catalytic, rather than stoichiometric, methods should be used. Vanillin is one of few biobased aromatic platform chemicals that is currently accessible on industrial scale. Striving to find a replacement for reprotoxic bisphenol A (BPA), we here report the catalytic dimerization of a vanillin-based monomer, as well as the application of the resulting ester in an epoxy thermoset. This work aligns with the UN Sustainable Development Goal 12 – Responsible consumption and production. |
Introduction
Bisphenol A (BPA, Fig. 1) is a common monomer used in polymeric materials such as polycarbonates and epoxy resins, and also in thermal papers and food packaging.1 However, BPA displays reproductive toxicity by causing endocrine disruption, and replacements for this monomer are thus urgently needed.2 In addition, current production of BPA affords toxic byproducts3 and the precursors are generally derived from fossil resources.4 Recent reports show that a methoxy substituent on one or both of the phenol rings of BPA can significantly reduce the estrogenic activity,5,6 making lignin-based aromatics well suited as precursors for BPA substitutes.3,7 | ||
| Fig. 1 Selected vanillin-based monomers for polymerization and their areas of application. | ||
One of the most important platform chemicals from lignin is vanillin, which is also one of very few aromatic biobased precursors that is accessible on an industrial scale.8 Caillol and co-workers have exploited this versatile precursor in the synthesis of a variety of vanillin-based monomers for use in biobased polymers.9Fig. 1 shows one such structure, the diglycidyl ether of vanillin alcohol (DGEVA),9,10 which was employed in the synthesis of biobased epoxy thermosets.10,11
DGEVA has also been used in conjunction with biobased anhydrides to form thermally stable epoxy resins for the aerospace sector, as reported by Mija.12 To mimic the diaryl structure of bisphenol A, the dimerization of vanillin has been implemented using different strategies. Pearl and colleagues used electrochemical coupling for this purpose as early as 1952,13,14 and this methodology was later optimized by Harvey et al., who incorporated the prepared vanillin bisphenols into polycyanurates and polycarbonates.15 Epoxy resins prepared via electrochemical vanillin dimerization have been reported by Amarasekara.16 Another approach is to oxidatively couple vanillin or vanillyl alcohol via the aromatic ring to form a dimeric structure (GEDVA or EDV, Fig. 1), a tactic applied by Cramail,17 as well as Fang and Nejad,18 to prepare biobased epoxy thermoset resins. The aldehyde functionality of vanillin can also be exploited for coupling reactions, for instance via reaction with a diol/polyol or with a diamine. A degradable diepoxy monomer (DGEVE, Fig. 1), with two vanillin units linked via a central bicycloacetal deriving from erythritol, was reported by Ma and colleagues.19DGEVE showed excellent degradability in acidic solutions after use, but was found to be stable under neutral conditions. Coupling a diamine to two equivalents of vanillin via imine formation is another approach to prepare bisphenol compounds, as described by Desnoes,20 as well as Li and Zeng.21 The corresponding vanillin-based diimines were subsequently applied towards the formation of biobased epoxy thermosets and vitrimers.
The base-catalyzed dimerization of aldehydes to produce an ester, originally reported by Claisen in 1887,22 but generally known as the Tishchenko reaction,23–25 provides another means for dimerizing vanillin. Chen and Xie applied this strategy with good results in the dimerization of allylated vanillin using sodium hydride as the catalyst, forming a divanillin ester (AMAM, Fig. 1).26AMAM was subsequently employed in thiol-ene click reactions and acyclic diene metathesis, to produce poly(thioether esters) and polyesters with high molecular weight. Thermoset thiol-ene networks were prepared by the same group,27 and Du and Zhu later applied AMAM towards polyurethane thermosets.28 Chen and Xie have also exploited the vanillin aldehyde functionality in another type of coupling reaction i.e. the aldol condensation between vanillin and platform chemical levulinic acid, forming the bio-based building block VLE (Fig. 1).29VLE was cured with an aromatic diamine, to form an imine-containing epoxy resin with improved thermal stability and mechanical properties compared to a bisphenol A epoxy resin. Xie has described the synthesis of a vanillin-based bisphenol carbonate and its application in the synthesis of polycarbonate esters and urethanes,30 while Llevot employed a divanillin carbonate in reactions with a myrcene-derived trithiol to prepare acid-degradable biobased polycarbonates via photopolymerization.31 Liu reported the synthesis of a bisguaiacol amide and ester and studied their material performances and cytotoxicity.32 Structures that more closely resemble BPA have also been prepared by Stanzione and colleagues, by coupling vanillyl alcohol with guaiacol under acidic conditions.33 The resulting bisguaicol structure was subsequently converted to a diglycidyl ether and applied in epoxy thermoset resins. Some recent reviews provide a comprehensive summary the use of vanillin building blocks in BPA replacements and in biobased polymers.34–36
While the base-catalyzed Tischenko reaction proceeds smoothly for dimerizing allylated vanillin into the corresponding ester, as reported by Chen and Xie,26 the basic reaction conditions used may be too harsh for more sensitive functionalities such as epoxides, that are commonly used in polymerization. In addition, sodium hydride is incompatible with certain solvents,37 and may be less suited for large-scale industrial scale applications due its pyrophoric nature.38 An alternative here could be to use transition metal catalysis for aldehyde dimerization under milder reaction conditions. Shvo has shown that catalytic dimerization of both aliphatic and aromatic aldehydes to form the corresponding esters can be catalyzed by a Shvo-type ruthenium complex.39 Darses has also reported that in situ generated ruthenium catalysts, prepared from [RuCl2(p-cymene)]2 and different phosphine ligands, can be applied for the same purpose.40 As part of studies in our respective groups, involving hydrogen transfer reactions using platform chemicals from biomass,41,42 as well as the preparation of polymers from renewable resources (vegetable oils,43 terpenes,44 suberin,45 and lignin46), we investigated the ruthenium-catalysed dimerization of vanillin and derivatives thereof to form biobased esters for polymer applications. We subsequently applied one of these compounds, the epoxy divanillin ester EDVE, towards the preparation of an epoxy thermoset resin. The results of these studies are disclosed herein.
Results and discussion
Synthesis of vanillin epoxy dimers via ruthenium-catalyzed coupling
Several different vanillin derivatives were investigated in the ruthenium-catalyzed coupling reaction to form vanillin epoxy dimers. In addition to vanillin itself, two derivatives where the phenol functionality was protected as an acetate or a silyl ether, were also prepared in case the free phenol functionality should interfere with the catalyst activity. Vanillin (1a, Scheme 1) was reacted with acetic anhydride, using 4-dimethylaminopyridine (DMAP) as a catalyst,47 to produce acetylated vanillin 1b in 98% yield. To form the silyl-protected derivative 1c, vanillin was instead treated with tert-butyldimethylsilyl chloride in the presence of imidazole,48 to afford the desired compound in 89% yield. Given the sensitivity of the epoxy group, we initially planned to perform the ruthenium-catalyzed coupling reaction using either unprotected vanillin, or one of the derivatives 1b or 1c, to form a diester, and subsequently introduce the epoxide functionality after dimerization. A different strategy, however, could be to investigate the direct dimerization of derivative 1d, where the epoxy functionality is present already at the start. To form the precursor needed for such a reaction, vanillin was reacted with epichlorohydrin in the presence of tetrabutylammonium bromide, under solventless conditions at 80 °C,49,50 affording 1d as a white solid in 60% yield. | ||
| Scheme 1 Preparation of vanillin monomer building blocks 1b–1d. | ||
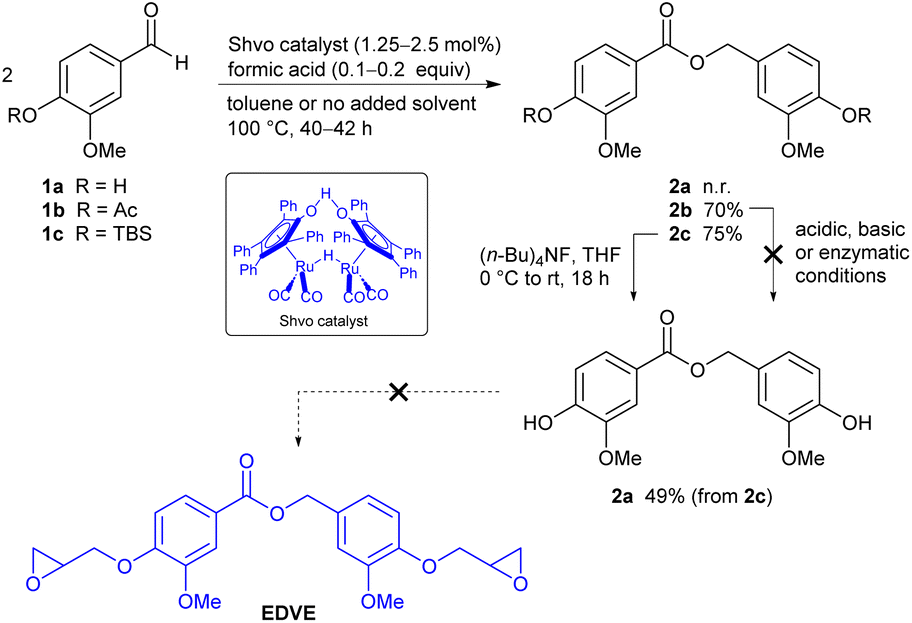
Direct coupling vanillin itself using 1.25 mol% of the Shvo catalyst 3 in the presence of formic acid under solventless conditions did not afford the desired coupling product 2a, despite heating for several days (Scheme 2). Deactivation of the catalyst via coordination to the phenolic group may be involved here. To see if this was indeed the case, we instead turned to the acetyl-protected derivative 1b. Initial attempts to dimerize 1b with the Shvo catalyst under solventless conditions, using a reaction time of 20 hours at 100 °C under argon, did indeed afford the desired ester 2b. However, the conversion seemed to plateau at around 50–60%. A similar trend was reported by Shvo in the case of electron rich aldehydes such as p-methoxybenzaldehyde.39 Postulating that catalyst deactivation or degradation was occurring, we added one portion of catalyst at the start of the reaction, together with a small amount of toluene to solubilize the starting material, and subsequently a second portion of catalyst after 20 hours, using 2.5 mol% catalyst in total. The reaction was then heated for an additional 20 hours. To our delight, ester 2b could be isolated in up to 70% yield following purification by automated flash chromatography on silica gel, using a gradient of ethyl acetate in pentane as the solvent. The structure of ester 2b was verified by 1H and 13C NMR, IR, HRMS (see Experimental section), and X-ray crystallography (Fig. 2).
 | ||
| Scheme 2 Initial strategy to prepare EDVE. | ||
 | ||
| Fig. 2 Solid state structure of compound 2b. Ellipsoids are represented at 50% probability. H atoms are shown as spheres of arbitrary radius. CCDC 2371862. | ||
The next step would be to deprotect the acetate in order to install the epoxy functionality. However, selective cleavage of the acyl ester in 2b, in the presence of the newly installed central ester functionality, proved to be challenging, despite the investigation of basic, acidic and enzymatic (porcine lipase) cleavage conditions, resulting in either disproportionation of the benzyl ester or low conversion to product. Reasoning that a silyl ether might be more facile to cleave in the presence of the central ester functionality, the dimerization of TBS-protected vanillin 1c was performed. As the precursor 1c is an oil rather than a solid in this case, no toluene was added and the reaction was performed under solventless conditions. In addition, a lower amount of catalyst (1.25 mol%) was needed in comparison to the earlier experiment. The resulting ester 2c was found to be difficult to separate from small amounts of residual 1c on a silica column, but switching to a reversed phase column (C18), afforded pure 2c in 75% yield. Cleavage of the silyl protecting group in 2c using tetrabutylammonium fluoride (TBAF) in THF afforded unprotected dimer 2a in a moderate yield (49%, non-optimized). However, subjecting 2a to the basic conditions needed for derivatizing the phenolic groups with epichlorohydrin again resulted in decomposition, and the initial strategy was thus abandoned.
We had earlier refrained from using vanillin derivatized with an epoxy group already at the start, due to the sensitivity of this functionality. However, we now decided to investigate if vanillin epoxide derivative 1d (Scheme 1) was compatible with the ruthenium-catalyzed coupling, as this would provide a more direct route to the targeted epoxy divanillin ester EDVE. The results from this investigation are summarized in Table 1. While 1d is a solid, its relatively low melting point (80 °C) prompted us to initially investigate solventless conditions. The conversion was determined by 1H NMR of the crude product by comparing the aldehyde signal of 1d to the benzyl peak in 2d, after filtering the reaction mixture through neutral alumina to remove the catalyst. Using 2.5 mol% catalyst, added in two portions over 48 h, a promising 70% conversion of vanillin epoxide 1d to EDVE was obtained (Table 1, entry 1). However, we noticed that some of the aldehyde 1d sublimated upon heating and resolidified onto the walls of the vial, thus being removed from the reaction mixture. Adding a small amount of toluene proved to solve this issue, resulting in 81% conversion to dimer, still employing 2.5% mol of catalyst with a reaction time of 48 h (Table 1, entry 2). To see if the conversion could be improved even further, we added additional portions of catalyst and experimented with the reaction time. With 3.75 mol% catalyst, added in three portions, essentially the same conversion (88–89%) was obtained after 36 h and after 72 h (entries 3 and 4). A slightly higher conversion (92%) could be obtained using 5 mol% catalyst with a reaction time of 96 h. However, the conditions used in entry 3 provide a more optimized balance of catalyst amount and reaction time.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalysta (mol%) | HCOOHb or HCOONac (mol%) | Solvent (mL per mmol per vanillin) | Time (h) | Conv.d (%) |
| a Catalyst added in 1.25 mol% portions, see Experimental section for details. b HCOOH used in entries 1–5. c HCOONa used in entries 6–7. d Conversion determined by 1H NMR of the crude product. e 5 mol% CyPPh2 added. | |||||
| 1 | Shvo (2.5) | 20 | No solvent | 48 | 70 |
| 2 | Shvo (2.5) | 20 | Toluene (0.6) | 48 | 81 |
| 3 | Shvo (3.75) | 20 | Toluene (0.4) | 36 | 89 |
| 4 | Shvo (3.75) | 20 | Toluene (0.4) | 72 | 88 |
| 5 | Shvo (5) | 20 | Toluene (0.4) | 96 | 92 |
| 6e | [RuCl2(p-cym)]2 (2.5) | 15 | 1,4-Dioxane (1) | 16 | <5 |
| 7e | [RuCl2(p-cym)]2 (2.5) | 15 | 1,4-Dioxane (0.2) | 16 | <5 |
We also briefly investigated dimerization conditions reported by Simon and Darses,40 where a different ruthenium catalyst, [RuCl2(p-cym)]2, together with an added ligand (CyPPh2), were used to generate an active catalytic species in situ. While the authors report a high conversion of aromatic aldehydes to dimerized product, this methodology was not successful for the epoxy-functionalized substrate 1d, leaving the starting material essentially untouched (Table 1, entries 5 and 6). It may be that the high oxygen content of this particular substrate interferes with the catalyst under these reaction conditions in some way.
After obtaining a satisfactory conversion in the formation of EDVE, we then focused on purification of the crude product, where the main contaminant is unreacted 1d. Automated flash chromatography was found to be inefficient, as the majority of the product was trapped on the silica gel and only a small amount of product (<10%) could be eluted using ethyl acetate in pentane, albeit in high purity. By switching to a C18-column, using a gradient of H2O–MeOH as the eluent, the product was successfully eluted, initially in yields of 40–52%. However, with some optimization of the eluent, a 65% yield of pure EDVE could be achieved. For characterization purposes, we have aimed to isolate pure EDVE, resulting in some material loss on the C18 column. However, related studies on vanillin-containing epoxy dimers in many cases employ the crude product directly in the preparation of epoxy resins,18,20,51 without further purification. As a conversion in the range of 89–92% (Table 1) can be attained under the optimized conditions, with the main contaminant being unreacted 1d, an alternative for larger scale reactions could be to use the crude material directly in the epoxy resin synthesis. Overall, this direct coupling method provides a shorter route to EDVE than our initial strategy, by removing the protection/deprotection steps.
Characterization and thermal behaviour of EDVE
EDVE was characterized by 1H NMR, 13C NMR, HRMS, ATR FTIR and single crystal X-ray diffraction. 1H NMR clearly shows the transition from an aldehyde proton at 9.86 ppm in 1d (Fig. 3a, peak h) to a benzylic ester peak at 5.27 ppm in EDVE (peak i), with new peaks appearing in both the aromatic (6.9–7.7 ppm) and aliphatic (2.7–4.4 ppm) area in EDVE, corresponding to an additional aromatic ring and epoxide functionality compared to 1d. In addition, the spectra for EDVE displays two -OMe signals at 3.91 and 3.89 ppm, compared to only one -OMe peak at 3.93 ppm in the case of 1d. In 13C NMR (Fig. 3b), the carbonyl signal for the aldehyde at 191 ppm disappears and is replaced by the ester carbonyl at 166 ppm, also verifying the formation of EDVE. HRMS analysis also corresponds to the correct mass for EDVE (see Experimental section).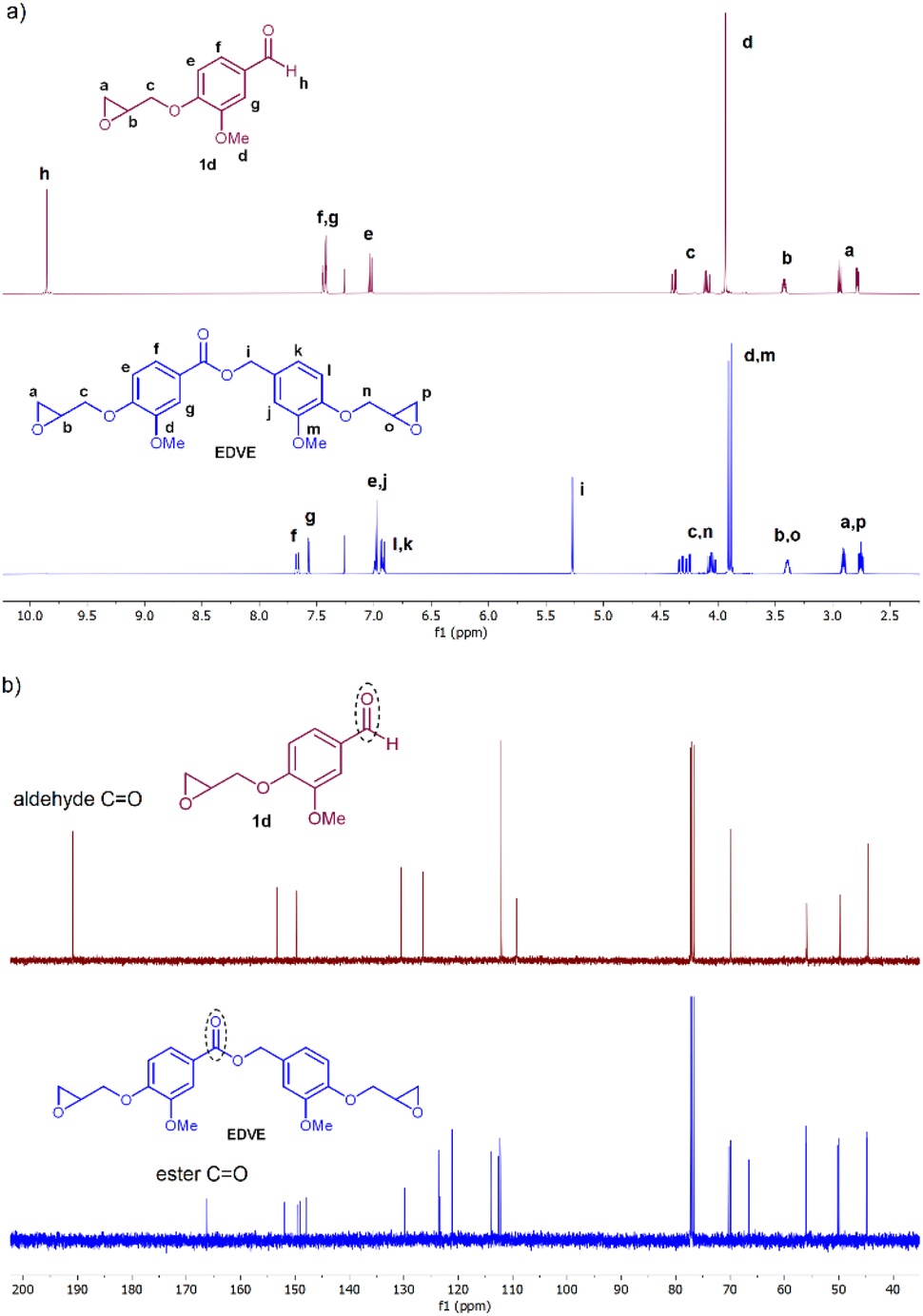 | ||
| Fig. 3 (a) 1H NMR spectrum of vanillin epoxide 1d (top) and EDVE (bottom) in CDCl3; (b) 13C NMR spectra of vanillin epoxide 1d (top) and EDVE (bottom) in CDCl3. | ||
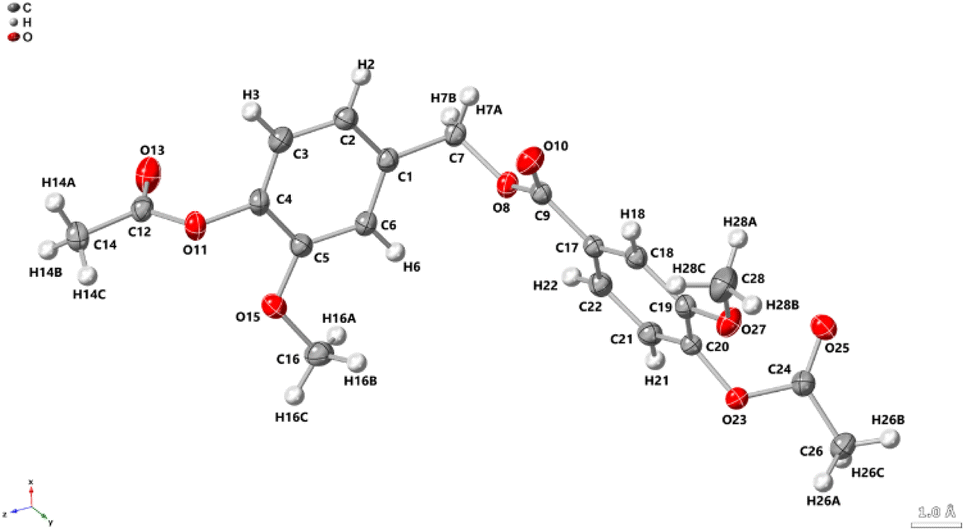
The ATR FTIR spectrum of EDVE is shown in Fig. 4, where the absorption signals at 917 and 845 cm−1 are assigned to the oxirane ring,52 while the signals at 1596 and 1517 cm−1 represent the aromatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibration.53 The signals at 1024 and 1237 cm−1 arise from the methoxy group,54 while the absorption band at 1713 cm−1 can be attributed to the stretching vibration of the ester carbonyl group.55 In comparison, the aldehyde carbonyl of 1d appears at 1683 cm−1.56 The shift of this carbonyl signal to a higher wavelength is consistent with the conversion of an aromatic aldehyde, typically with its CO stretch in the 1685–1710 cm−1 region, to an aromatic ester, which generally appears in the 1705–1725 cm−1 range.57 Single crystal X-ray diffraction (Fig. 5) provides additional proof of the EDVE structure.
C stretching vibration.53 The signals at 1024 and 1237 cm−1 arise from the methoxy group,54 while the absorption band at 1713 cm−1 can be attributed to the stretching vibration of the ester carbonyl group.55 In comparison, the aldehyde carbonyl of 1d appears at 1683 cm−1.56 The shift of this carbonyl signal to a higher wavelength is consistent with the conversion of an aromatic aldehyde, typically with its CO stretch in the 1685–1710 cm−1 region, to an aromatic ester, which generally appears in the 1705–1725 cm−1 range.57 Single crystal X-ray diffraction (Fig. 5) provides additional proof of the EDVE structure.
 | ||
| Fig. 4 ATR FTIR spectrum of EDVE. | ||
 | ||
| Fig. 5 X-ray crystallographic structure of EDVE with thermal ellipsoids at 50% probability. H atoms are shown as spheres of arbitrary radius. CCDC 2423529. | ||
The thermal behaviour of EDVE was studied by DSC and TGA The DSC analysis shown in Fig. 6a revealed a semi-crystalline structure, with the first heating curve presenting the endotherm corresponding to the melting point of EDVE with a maximum at 108 °C. During cooling from the melt, no recrystallization event was observed. The absence of recrystallization at lower temperatures is proposed to be due to limited molecular mobility and time for recrystallization during cooling. There is a small transition at 11 °C seen on the second heating corresponding to a Tg transition meaning that below this temperature is the system in the vitrified state where no crystallization can occur. The thermal decomposition of EDVE was studied by TGA and derivative TGA analysis. Fig. 6b shows that the decomposition of EDVE takes place in two steps. The first step is located between 203–338 °C with 31% mass loss and the maximum degradation rate peak in the DTGA at 309 °C, while the second step can be found between 338–442 °C with 79% mass loss and the maximum at 397 °C. The results suggest that the first degradation is coupled to the aryl–alkyl ether linkage that first breaks to split off a glycidyl group and then the ester linkage is broken at higher temperatures.
 | ||
| Fig. 6 Thermal behaviour of EDVE studied by (a) DSC and (b) TGA/DTGA. | ||
Synthesis of a vanillin epoxy thermoset resin from EDVE and Jeffamine D-400
The curing process of EDVE and Jeffamine D-400 (Scheme 3) was studied by DSC in order to choose the optimum curing temperature. Fig. 6a and 7a show the DSC thermograms of EDVE and Jeffamine D-400 respectively, where it is seen that EDVE is semi-crystalline with a melting point at 108 °C, while the Jeffamine is amorphous. In Fig. 7b, the heat-flow data as a function of temperature for a sample which was mixed 45 minutes before the measurement, using a small amount of acetone as a co-solvent, is displayed. This waiting period was necessary to allow for a partial evaporation of acetone and ensure minimum impact on the thermal behavior of the curing process. The first heating curve presents an endothermic maximum at 82 °C (melting point of EDVE = 108 °C) and two exothermic maxima at 129 °C and 168 °C. Two maxima can indicate the two main stages of the reaction. The second heating curve shows a Tg of 45 °C and a completely cured thermoset (no visible exotherm). This curing behaviour is in line with the corresponding system based on a BPA-based epoxy previously studied.58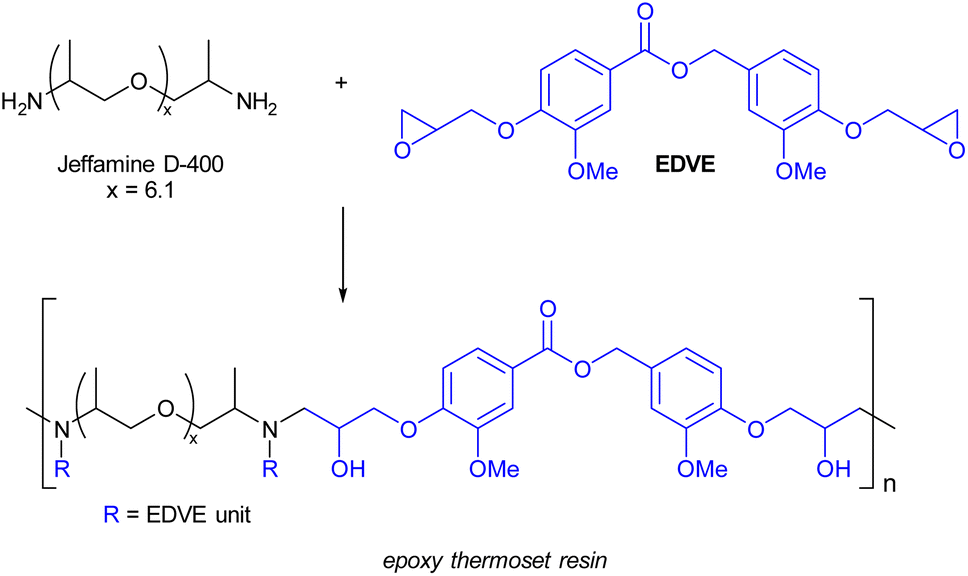 | ||
| Scheme 3 Epoxy thermoset prepared from EDVE and Jeffamine D-400. | ||
 | ||
| Fig. 7 DSC thermogram of (a) Jeffamine D-400, and (b) EDVE and Jeffamine D-400 during the curing process. See Fig. 5a for a DSC thermogram of EDVE only. | ||
The spectral evolution at 140 °C of the curing process of EDVE with Jeffamine D-400 was also studied by real-time ATR FTIR, as seen in Fig. 8. A curing temperature of 140 °C was selected to prevent degradation or evaporation of the monomers. In situ DSC measurements of the reaction between EDVE and Jeffamine D-400 revealed two exothermic maxima at 129 °C and 168 °C, indicating that a higher curing temperature, such as 170 °C, could accelerate the reaction. Thermogravimetric analysis (TGA) of Jeffamine D-400 showed a 5 wt% weight loss at 155 °C, and considering the vapor pressure of 1 mm Hg per °C at 165 °C, temperatures around 170 °C could lead to monomer evaporation/degradation. While a higher curing temperature could have accelerated the reaction, 140 °C was chosen as an optimal compromise between curing efficiency and avoiding monomer evaporation/degradation.
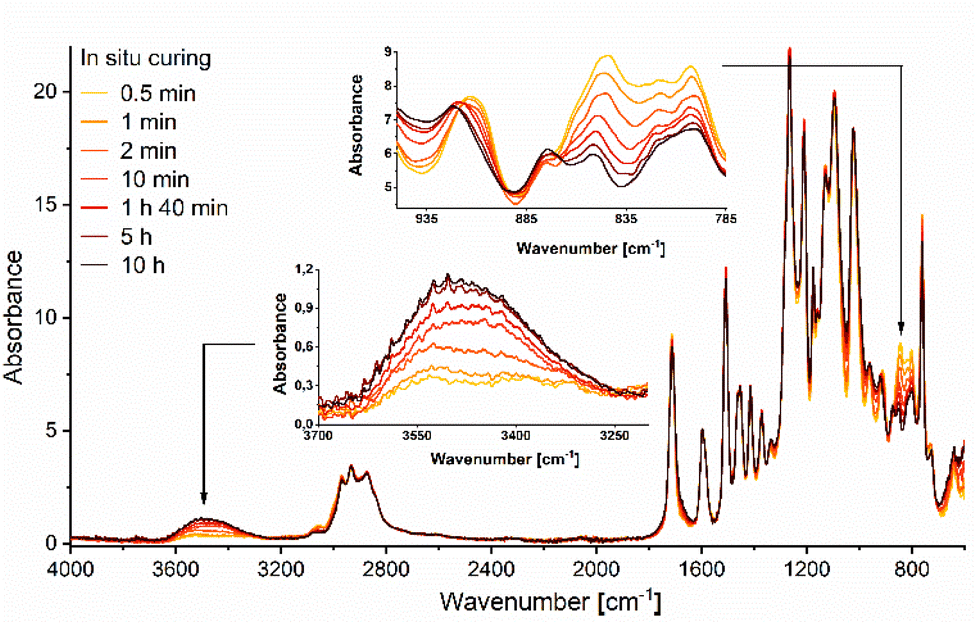 | ||
| Fig. 8 Real-time ATR FTIR analysis of the curing of EDVE with Jeffamine D-400 at 140 °C. | ||
The signals at 917 and 845 cm−1 are drastically reduced already after 10 minutes, and continue to decrease over several hours, with significant changes observed between 10 minutes and 5 h. The changes between 5 h and 10 h are minimal, which indicated that the reaction is almost completed and the epoxy groups consumed. In order to accelerate the reaction, the curing temperature can be further increased. Simultaneously, the signal corresponding to the OH vibrational stretching at ∼3500 cm−1 is increasing, indicating that a ring-opening reaction of the oxirane ring is occurring during the curing process.52 The combined results from the FTIR and DSC measurements confirm that EDVE works well as an alternative to the corresponding BPA-based epoxy.
The resulting epoxy network was also characterized by TGA and DSC, as shown in Fig. 9. The resulting thermoset has a thermal stability up to 300 °C where the degradation starts. This is very similar to the thermal stability of the corresponding BPA-based epoxy system.58 Comparing the TGA results for the monomers (Fig. 6b and 9a) and the final thermoset (Fig. 9b), one can see that the thermal stability is improved for the thermoset, being at least 60 °C higher compared to the thermal stability of the monomers, which is in line with the previous discussion. Additionally, the Tg of the epoxy thermosets is around 47 ± 2.6 °C, which aligns well with the DSC data obtained during the in situ curing process. This significant increase in Tg from around 11 °C for the EDVE monomer to 47 °C for the thermoset further confirms the formation of a crosslinked network during curing, with the final Tg in the typical range for a system based on Jeffamine D-400. BPA-based epoxy systems combined with Jeffamine D-400 normally have a Tg around 50 ± 5 °C and an elastic modulus around 2 GPa.58,59
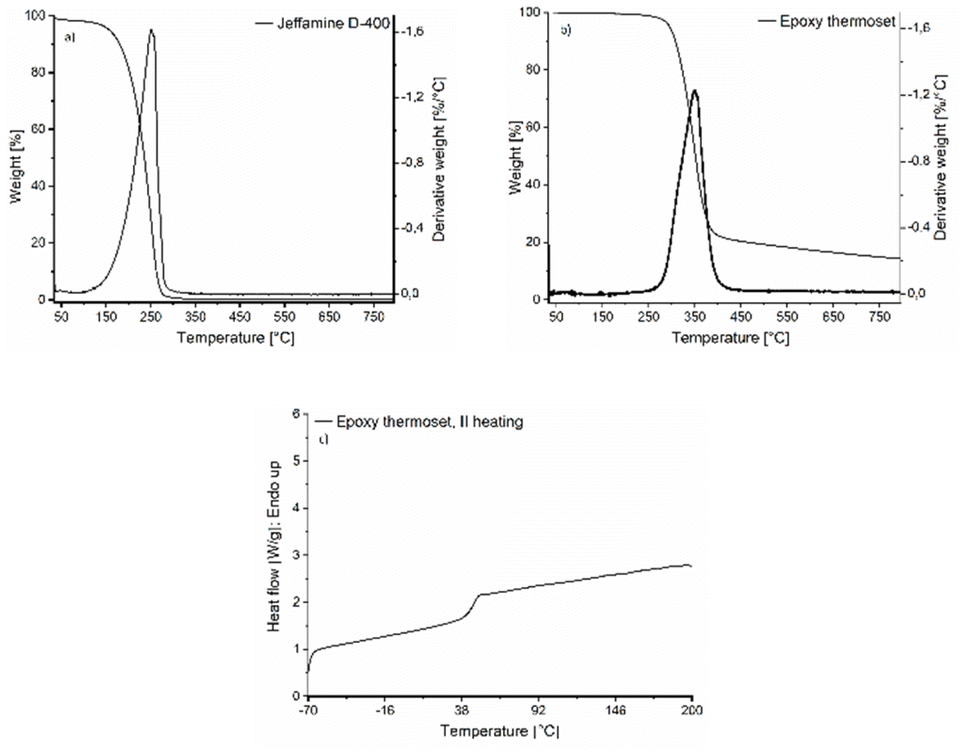 | ||
| Fig. 9 TGA of (a) Jeffamine D-400, and (b) epoxy thermoset prepared from EDVE and Jeffamine D-400. (c) DSC of the epoxy thermoset. | ||
In Fig. 10, the DMA data of the epoxy thermoset is shown. The storage modulus at −50 °C is around 1502 ± 185 and drops significantly around the glass transition temperature which is around 49 ± 1.5 °C, providing a good correlation between the Tg measured by DSC and DMA.
 | ||
| Fig. 10 DMA of epoxy thermoset prepared from EDVE and Jeffamine D-400. | ||
Conclusions
In summary, we have developed a convenient ruthenium-catalyzed synthetic route to the epoxy divanillin ester EDVE, where the target compound is formed in a single step from the corresponding vanillin epoxide derivative. The reaction is highly atom economic, with all atoms in the precursor vanillin epoxide being incorporated into the product. EDVE was characterized by NMR, FTIR, HRMS and single crystal X-ray diffraction, and subsequently applied in the synthesis of an epoxy thermoset resin via curing with Jeffamine D-400. The properties of the resulting resin were investigated using DSC, TGA and DMA, showing that the thermoset exhibited good thermal stability and physical and mechanical properties suitable for thermoset applications, in par with corresponding BPA-epoxy system. Our initial results indicate that biobased EDVE could potentially function as a replacement to BPA-based epoxy. In addition, the introduction of an ester moiety into the epoxy resin structure could facilitate recycling of the polymer by chemical or enzymatic hydrolysis after use. We envisage that EDVE can be a valuable addition to the current collection of biobased building blocks for polymer applications.Experimental section
Materials
Vanillin (1a, 99%) was purchased from Sigma Aldrich. The Shvo catalyst (1-hydroxytetraphenylcyclopentadienyl-(tetraphenyl-2,4-cyclopentadien-1-one)-μ-hydrotetracarbonyl-diruthenium-(II)) was purchased from Strem. Jeffamine D-400 polyetheramine was purchased from Huntsman. All other solvents and reagents were purchased from commercial suppliers and used without purification or drying, unless otherwise stated. Microwave reaction vials with caps were purchased from Biotage. Reactions were heated using a Radleys Heat-On Block in conjunction with an IKA magnetic stirrer/heater.Analytical methods
C stretching vibration, and baseline corrected by using Spectrum software.52 The curing reaction was followed by monitoring the decrease in RT-FTIR absorption intensity of the oxirane ring absorption (917 and 845 cm−1)42 add the increase in hydroxyl (–OH) absorption around 3500 cm−1.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ were continuously recorded as a function of temperature. The Tg of the thermosets was reported as maximum of the tanδ curve.
:H2O:EtOAc (1:0.5:0.5). Suitable crystals were selected and placed on a mounted cryo loop and mounted on an XtaLAB Synergy R, HyPix diffractometer. The crystal was kept at a steady T = 145.07(10) K for 2b and T = 153(5) K for EDVE during data collection. The structures were solved with the ShelXT46 (ref. 60) structure solution program using the Intrinsic Phasing solution method and by using Olex247 (ref. 61) as the graphical interface. The model was refined with version 2016/6 of ShelXL 2016/646 (ref. 61) using Least Squares minimisation. The data was submitted to the CSD with deposition numbers CCDC 2371862 (2b) and CCDC 2423529 (EDVE).
δ were continuously recorded as a function of temperature. The Tg of the thermosets was reported as maximum of the tanδ curve.
:H2O:EtOAc (1:0.5:0.5). Suitable crystals were selected and placed on a mounted cryo loop and mounted on an XtaLAB Synergy R, HyPix diffractometer. The crystal was kept at a steady T = 145.07(10) K for 2b and T = 153(5) K for EDVE during data collection. The structures were solved with the ShelXT46 (ref. 60) structure solution program using the Intrinsic Phasing solution method and by using Olex247 (ref. 61) as the graphical interface. The model was refined with version 2016/6 of ShelXL 2016/646 (ref. 61) using Least Squares minimisation. The data was submitted to the CSD with deposition numbers CCDC 2371862 (2b) and CCDC 2423529 (EDVE).
Synthetic procedures and polymerization methods
Automated flash chromatography was performed on a Biotage Isolera Spektra One with SNAP KP-sil or KP-C18-HS columns, or on a Biotage Selekt with Sfär Silica D or Sfär C18 D columns, eluting with EtOAc–pentane (silica) or H2O–MeOH (C18). Conventional flash chromatography using glass columns may also be used for purification of the products.O), 1717 (CO), 1191 (C–O); 1H NMR (400 MHz, CDCl3) δ 7.70 (dd, J = 8.1, 1.8 Hz, 1H), 7.66 (app d, J = 1.8 Hz, 1H), 7.10 (app d, J = 8.1 Hz, 1H), 7.06–7.00 (m, 3H), 5.32 (s, 2H), 3.88 (s, 3H), 3.84 (s, 3H), 2.32 (s, 3H), 2.31 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 169.0, 168.5, 165.7, 151.1 (2C), 143.7, 139.7, 134.8, 128.6, 122.9, 122.8, 122.7, 120.7, 113.5, 112.4, 66.6, 56.1, 55.9, 20.6 (2C); HRMS (ESI+) m/z: [M + Na]+ calcd for C20H20NaO8 411.1056; found 411.1054.
:MeOH (40:60 → 0:100). The last fraction afforded the product as a colourless viscous oil (312 mg, 0.59 mmol, 75%); νmax/cm−1 (neat) 1713 (CO), 1277 (C–O); 1H NMR (400 MHz, CDCl3) δ 7.60 (dd, J = 8.2, 2.0 Hz, 1H), 7.56 (app d, J = 2.0 Hz, 1H), 6.94–6.88 (m, 2H), 6.85 (app t, J = 8.2 Hz, 2H), 5.25 (s, 2H), 3.85 (s, 3H), 3.82 (s, 3H), 0.994 (s 9H), 0.988 (s, 9H), 0.16 (s, 6H), 0.15 (s, 6H); 13C NMR (101 MHz, CDCl3) δ 166.4, 150.9, 150.7, 149.7, 145.1, 129.6, 123.7, 123.4, 121.2, 120.8, 120.4, 113.1, 112.5, 66.6, 55.5 (2C), 25.7 (3C), 25.6 (3C), 18.5 (2C), 18.4 (2C), −4.6 (2C); HRMS (ESI+) m/z: [M + Na]+ calcd for C28H44NaO6Si2 555.2574; found 555.2573. This reaction has also been performed using 11.3 mmol of 1c, affording 2c in 71% yield.
:MeOH (0 → 70% MeOH). The second fraction afforded the product as a white solid (270 mg, 65%); mp 108 °C; νmax/cm−1 (neat) 1708 (CO), 1270 (C–O); 1H NMR (400 MHz, CDCl3) δ 7.67 (dd, J = 8.4, 2.1 Hz, 1H), 7.57 (app d, J = 2.0 Hz, 1H), 7.01–6.96 (m, 2H), 6.94–6.90 (m, 2H), 5.27 (s, 2H), 4.32 (dd, J = 11.4, 3.2 Hz, 1H), 4.26 (dd, J = 11.4, 3.4 Hz, 1H), 4.07 (dd, J = 11.4, 5.6 Hz, 1H), 4.04 (dd, J = 11.5, 5.6 Hz, 1H), 3.91 (s, 3H), 3.89 (s, 3H), 3.42–3.37 (m, 2H), 2.91 (ddd, J = 6.7, 4.9, 4.1 Hz, 2H), 2.75 (ddd, J = 7.3, 4.9, 2.6 Hz, 2H); 13C NMR (101 MHz, CDCl3) δ 166.2, 152.0, 149.6, 149.0, 148.0, 129.8, 123.5, 123.4, 121.1, 113.9, 112.6, 112.3, 112.2, 70.2, 69.8, 66.6, 56.0 (2C), 50.1, 50.0, 44.9, 44.8; HRMS (ESI+) m/z: [M + H]+ found 417.1548; calcd for C22H25O8 417.1549. Some decomposition of EDVE was seen in halogenated solvents and longer term storage in such solvents is thus not recommended.
Data availability
The 1H and 13C NMR data supporting this article has been included as part of the ESI.† Crystallographic data for 2b and EDVE has been deposited at the CCDC under 2371862 (for 2b) and 2423529 (for EDVE) and can be obtained from https://www.ccdc.cam.ac.uk/structures/.Author contributions
F. F. and I. R.: investigation, validation, writing – review & editing. N. P. and J. M.: investigation. M. G.: investigation, formal analysis, writing – review & editing. A. E., M. J. and N. K.: conceptualization, methodology, supervision, funding acquisition, resources, writing – original draft.Conflicts of interest
A. E. is an employee of AstraZeneca and may or may not own stock options. There are no other conflicts of interest to declare.Acknowledgements
Financial support from the Swedish Research Council Formas (NK, grant no. 2015-01106), the C. F. Lundström Foundation (NK, grant no. CF2021-0027), the Adlerbertska Foundation (NK) and the Knut and Alice Wallenberg Foundation (KAW) through the Wallenberg Wood Science Center (MJ) is gratefully acknowledged.References
- J. Michalowicz, Environ. Toxicol. Pharmacol., 2014, 37, 738–758 CrossRef CAS PubMed
.
- J. S. Siracusa, L. Yin, E. Measel, S. X. Liang and X. Z. Yu, Reprod. Toxicol., 2018, 79, 96–123 CrossRef CAS PubMed
- F. Liguori, C. Moreno-Marrodan and P. Barbaro, Chem. Soc. Rev., 2020, 49, 6329–6363 RSC
- Y. Jiang, J. Li, D. Li, Y. K. Ma, S. C. Zhou, Y. Wang and D. H. Zhang, Chem. Soc. Rev., 2024, 53, 624–655 RSC
- Y. Peng, K. H. Nicastro, T. H. Epps and C. Q. Wu, Food Chem., 2021, 338, 127656 CrossRef CAS PubMed
- A. Amitrano, J. S. Mahajan, L. T. J. Korley and T. H. I. Epps, RSC Adv., 2021, 11, 22149–22158 RSC
- R. Tavernier, M. Semsarilar and S. Caillol, Green Mater., 2023, 12, 121–167 CrossRef
- M. Fache, B. Boutevin and S. Caillol, ACS Sustain. Chem. Eng., 2016, 4, 35–46 CrossRef CAS
- M. Fache, E. Darroman, V. Besse, R. Auvergne, S. Caillol and B. Boutevin, Green Chem., 2014, 16, 1987–1998 RSC
- M. Fache, R. Auvergne, B. Boutevin and S. Caillol, Eur. Polym. J., 2015, 67, 527–538 CrossRef CAS
- A. S. Mora, R. Tayouo, B. Boutevin, G. David and S. Caillol, Green Chem., 2018, 20, 4075–4084 RSC
- R. Dinu, U. Lafont, O. Damiano and A. Mija, ACS Appl. Polym. Mater., 2022, 4, 3636–3646 CrossRef CAS
- I. A. Pearl, J. Am. Chem. Soc., 1952, 74, 4593–4594 CrossRef CAS
- I. A. Pearl, J. Am. Chem. Soc., 1952, 74, 4260–4262 CrossRef CAS
- B. G. Harvey, A. J. Guenthner, H. A. Meylemans, S. R. L. Haines, K. R. Lamison, T. J. Groshens, L. R. Cambrea, M. C. Davis and W. W. Lai, Green Chem., 2015, 17, 1249–1258 RSC
- A. S. Amarasekara, R. Garcia-Obergon and A. K. Thompson, J. Appl. Polym. Sci., 2019, 136, 47000 CrossRef
- E. Savonnet, E. Grau, S. Grelier, B. Defoort and H. Cramail, ACS Sustain. Chem. Eng., 2018, 6, 11008–11017 CrossRef CAS
- Z. Fang, S. Nikafshar, E. L. Hegg and M. Nejad, ACS Sustain. Chem. Eng., 2020, 8, 9095–9103 CrossRef CAS
- W. C. Yuan, S. Q. Ma, S. Wang, Q. Li, B. B. Wang, X. W. Xu, K. F. Huang, J. Chen, S. S. You and J. Zhu, Eur. Polym. J., 2019, 117, 200–207 CrossRef CAS
- E. Desnoes, L. Toubal, A. H. Bouazza and D. Montplaisir, Polym. Eng. Sci., 2020, 60, 2593–2605 CrossRef CAS
- P. X. Tian, Y. D. Li, Y. X. Weng, Z. Hu and J. B. Zeng, Eur. Polym. J., 2023, 193, 112078 CrossRef CAS
- L. Claisen, Ber. Dtsch. Chem. Ges., 1887, 20, 646–650 CrossRef
- V. E. Tishchenko, J. Russ. Phys.-Chem. Soc., 1906, 38, 355–418 Search PubMed
- V. E. Tischenko, Chem. Zentralbl., 1906, 77, 1309–1311 Search PubMed
- T. Seki, T. Nakajo and M. Onaka, Chem. Lett., 2006, 35, 824–829 CrossRef CAS
- T. H. Ren, Q. Chen, C. B. Zhao, Q. Zheng, H. B. Xie and M. North, Green Chem., 2020, 22, 1542–1547 RSC
- Z. J. Gao, Y. You, Q. Chen, M. North and H. B. Xie, Green Chem., 2023, 25, 172–182 RSC
- D. Zhao, X. Liang, J. W. Wang, J. Z. Du and Y. Q. Zhu, ACS Appl. Polym. Mater., 2023, 5, 4536–4545 CrossRef CAS
- Z. T. Cao, Y. You, Y. Q. Li, C. J. Huang, Y. Z. Tian, S. H. Zhao, Q. Chen and H. B. Xie, Polym. Chem., 2023, 14, 3978–3988 RSC
- D. Bai, Q. Chen, Y. Chai, T. H. Ren, C. J. Huang, I. D. V. Ingram, M. North, Q. Zheng and H. B. Xie, RSC Adv., 2018, 8, 34297–34303 RSC
- P. S. Löser, A. Lamouroux, M. A. R. Meier and A. Llevot, Polym. Chem., 2024, 15, 2240–2252 RSC
- H. R. Sun, J. Y. Jiang, Y. L. Zheng, S. F. Xiang, S. J. Zhao, F. Y. Fu and X. D. Liu, Polym. Chem., 2023, 14, 1613–1621 RSC
- E. D. Hernandez, A. W. Bassett, J. M. Sadler, J. J. La Scala and J. F. Stanzione, ACS Sustain. Chem. Eng., 2016, 4, 4328–4339 CrossRef CAS
- H. R. Qiang, J. W. Wang, H. X. Liu and Y. Q. Zhu, Polym. Chem., 2023, 14, 4255–4274 RSC
- M. A. Rashid, M. N. Hasan, M. A. R. Dayan, M. S. I. Jamal and M. K. Patoary, Reactions, 2023, 4, 66–91 CrossRef CAS
- W. J. Yang, H. Ding, D. Puglia, J. M. Kenny, T. X. Liu, J. Q. Guo, Q. W. Wang, R. X. Ou, P. W. Xu, P. M. Ma and P. J. Lemstra, SusMat, 2022, 2, 535–568 CrossRef CAS
- Q. Yang, M. Sheng, J. J. Henkelis, S. Y. Tu, E. Wiensch, H. L. Zhang, Y. Q. Zhang, C. Tucker and D. E. Ejeh, Org. Process Res. Dev., 2019, 23, 2210–2217 CrossRef CAS
- C. Fluegeman, T. Hilton, K. P. Moder and R. Stankovich, Process Saf. Prog., 2005, 24, 86–90 CrossRef CAS
- N. Menashe and Y. Shvo, Organometallics, 1991, 10, 3885–3891 CrossRef CAS
- M. O. Simon and S. Darses, Adv. Synth. Catal., 2010, 352, 305–308 CrossRef CAS
- A. Said Stålsmeden, J. L. B. Vázquez, K. van Weerdenburg, R. Rae, P. O. Norrby and N. Kann, ACS Sustain. Chem. Eng., 2016, 4, 5730–5736 CrossRef
- A. Ekebergh, R. Begon and N. Kann, J. Org. Chem., 2020, 85, 2966–2975 CrossRef CAS PubMed
- K. Ohlsson, T. Bergman, P. E. Sundell, T. Deltin, I. Tran, M. Svensson and M. Johansson, Prog. Org. Coat., 2012, 73, 291–293 CrossRef CAS
- M. Claudino, J. M. Mathevet, M. Jonsson and M. Johansson, Polym. Chem., 2014, 5, 3245–3260 RSC
- S. Nameer and M. Johansson, J. Coat. Technol. Res., 2017, 14, 757–765 CrossRef CAS
- M. Jawerth, M. Lawoko, S. Lundmark, C. Perez-Berumen and M. Johansson, RSC Adv., 2016, 6, 96281–96288 RSC
- S. Nagasawa, S. Fujiki, Y. Sasano and Y. Iwabuchi, J. Org. Chem., 2021, 86, 6952–6968 CrossRef CAS PubMed
- T. Schneider, V. Kubyshkin and N. Budisa, Eur. J. Org Chem., 2018, 2018, 2053–2063 CrossRef CAS
- S. Zhao and M. M. Abu-Omar, Macromolecules, 2018, 51, 9816–9824 CrossRef CAS
- S. Wang, S. Q. Ma, Q. Li, X. W. Xu, B. B. Wang, W. C. Yuan, S. H. Zhou, S. S. You and J. Zhu, Green Chem., 2019, 21, 1484–1497 RSC
- M. Fache, B. Boutevin and S. Caillol, Green Chem., 2016, 18, 712–725 RSC
-
M. G. González, J. C. Cabanelas and J. Baselga, in Infrared Spectroscopy - Materials Science, Engineering and Technology, ed. T. Theophanides, InTech, Rijeka, 2012, ch. 13, pp. 261–284 Search PubMed
- Z. Y. Wang, P. Gnanasekar, S. S. Nair, R. Farnood, S. L. Yi and N. Yan, ACS Sustain. Chem. Eng., 2020, 8, 11215–11223 CrossRef CAS
- V. Balachandran and K. Parimala, Spectrochim. Acta, Part A, 2012, 95, 354–368 CrossRef CAS PubMed
- C. C. Pang, J. Zhang, Q. F. Zhang, G. L. Wu, Y. N. Wang and J. B. Ma, Polym. Chem., 2015, 6, 797–804 RSC
- X. M. Ding, L. Chen, Y. J. Xu, X. H. Shi, X. Luo, X. Song and Y. Z. Wang, ACS Sustain. Chem. Eng., 2023, 11, 14445–14456 CrossRef CAS
-
P. Larkin, in Infrared and Raman Spectroscopy : Principles and Spectral Interpretation, Elsevier, Waltham, UK, 2011, ch. 6, pp. 73–116 Search PubMed
- D. Ratna, S. B. Jagtap, R. Nimje and B. C. Chakraborty, J. Therm. Anal. Calorim., 2024, 149, 1073–1087 CrossRef CAS
-
B. Burton, D. Alexander, H. Klein, A. Garibay-Vasquez, A. Pekarik and C. Henkee, Epoxy formulations using Jeffamine polyether-amines, Technical Report, Huntsman, 2005 Search PubMed
- G. M. Sheldrick, Acta Crystallogr., Sect. C:Struct. Chem., 2015, 71, 3–8 CrossRef PubMed
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS
- K. Sommer and R. M. Williams, Tetrahedron, 2009, 65, 3246–3260 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2371862 and 2423529. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5su00165j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |