
Efficient caustic and hydrogen production using a pressurized flow-through cathode†
Fan
Yang
 a,
Minhao
Xiao
a,
Sangsuk
Lee
a,
Javier Alan
Quezada Renteria
a,
Xinyi
Wang
a,
Minju
Cha
a,
Anya
Dickinson-Cove
a,
Sungsoon
Kim
a,
Guy Z.
Ramon
d,
Gaurav N.
Sant
abc,
Eric M. V.
Hoek
ae and
David
Jassby
*ab
a,
Minhao
Xiao
a,
Sangsuk
Lee
a,
Javier Alan
Quezada Renteria
a,
Xinyi
Wang
a,
Minju
Cha
a,
Anya
Dickinson-Cove
a,
Sungsoon
Kim
a,
Guy Z.
Ramon
d,
Gaurav N.
Sant
abc,
Eric M. V.
Hoek
ae and
David
Jassby
*ab
aDepartment of Civil & Environmental Engineering, University of California, Los Angeles (UCLA), Los Angeles, 90095, USA. E-mail: jassby@ucla.edu
bInstitute for Carbon Management, University of California, Los Angeles (UCLA), Los Angeles, California 90095, USA
cMaterials Science and Engineering, University of California, Los Angeles (UCLA), Los Angeles, California 90095, USA
dDepartment of Civil & Environmental Engineering, Technion – Israel Institute of Technology, Haifa, 32000, Israel
eEnergy Storage & Distributed Resources, Lawrence Berkeley National Lab, Berkeley, California 94270, USA
First published on 18th November 2024
Abstract
The emerging process of CO2 capture and sequestration will likely require large volumes of caustic. The fossil fuel demand and carbon footprint of transporting liquid caustic is self-defeating, and hence, there is a need for energy-efficient, on-site caustic production for carbon capture projects. Caustic production is dominated by the well-established “chlor-alkali” process. This process requires highly concentrated (∼25% w/v) and pure (>99.5 wt%) NaCl feed brines, uses high-cost ion-exchange membranes and high operating temperatures (90 °C), and generates a highly-concentrated (>33% w/v) caustic stream that can be further concentrated using thermal evaporation. This highly concentrated caustic is then shipped to customers, where it is typically diluted to the required level. We have developed a flow-through membrane/cathode electrolysis process that produces a caustic solution (pH 10.22–12.26) at a specific energy consumption (SEC) of 1.71 kW he kg−1 NaOH at room temperature using a 3.5% w/v NaCl solution as feed, while achieving pure H2 generation without the use of ion exchange membranes. We demonstrate that the SEC is strongly dependent on the flow rate through the cathode, reaching a minimum at a high rate of 1200 L m−2 h−1. Electrochemical impedance spectroscopy, confocal microscopy, and finite element modeling show that the SEC is lowered through a combination of enhanced mass transport (of H+ and OH− ions) to and from the cathode surface and H2 gas stripping, both facilitated by the high flow rates. This technology offers the opportunity for the on-site production of dilute caustic streams (potentially from softened seawater) at a significantly reduced energy cost (compared to conventional chlor-alkali processes that consume >2.1 kW he kg−1 NaOH).
1. Introduction
Efficient production of caustic (NaOH) is a critical requirement for many industrial processes, including chemical manufacturing, food processing, and water treatment.1,2 In recent years, because of the increasing number of abnormal climate events,3 there has been an increasing focus on the use of caustic in the transition toward carbon-free industrial processes, such as CO2-free cement manufacturing4 and CO2 capture and sequestration.5,6 In these applications, caustic is used to increase the alkalinity of water to either enhance the concentrations of bicarbonate (e.g., in ocean alkalinity enhancement and direct air capture), or to drive the formation of stable minerals such as CaCO3 (which offer a permanent storage solution for CO2).7–10 The chlor-alkali process is the traditional method used for the production of caustic, with several technology iterations developed over the decades (e.g., membrane, diaphragm, and oxygen depolarized cathode electrolysis).11,12 In all of these process configurations, the anode and cathode are separated by a hydraulic separator, which can be either a diaphragm or an ion exchange membrane.13,14 The electrodes are then placed in a high-salinity environment, typically a 25% w/v NaCl aqueous solution as anolyte, and a ∼30% w/v NaOH solution is fed to the cathodic compartment as catholyte. When the electrodes are polarized, chloride ions are oxidized on the anode to produce chlorine, while protons and water are reduced on the cathode to produce hydroxide ions and hydrogen gas.12 The typical NaOH concentrations produced by traditional chlor-alkali reactors range between 30% w/v and 35% w/v, and further concentration can be achieved through water evaporation and NaOH precipitation.12Due to the high cost and high resistance introduced by ion exchange membranes, which are widely used in many electrochemical applications, researchers began to consider the development of membraneless water electrolysis cells. The laminar flow approach, demonstrated by Hashemi et al.15 and Jiang et al.,16 uses fluid flow to keep hydrogen and oxygen gas bubbles in separate streams. However, this method suffers from high resistance due to gas bubbles, leading to low overall energy efficiency, making it unsuitable for practical applications. The capillary action approach, highlighted by Li et al.17 and Tiwari et al.,18 used hydrophobic gas diffusion electrodes to directly extract gases, eliminating bubbles and achieving high energy efficiency. However, this approach does not produce alkalinity. The convective flow approach, explored by Gillespie et al.19 and Swiegers et al.,20 involves pumping an electrolyte through porous electrodes, achieving higher energy efficiency and effective gas separation. However, the work does not fully address the effects of fluid flow rate (flux) through the electrode on bubble formation, polarization layers, and charge transfer resistance, and does not explore the impact of flow conditions on the SEC of caustic production.
While conventional caustic production processes produce highly concentrated NaOH, which is well-suited for specific applications like chemical manufacturing of various chemicals, including plastics, solvents, and synthetic fibers, many industries need non-pure, dilute caustic streams for simple acid neutralization.1,2 This is particularly applicable to the developing carbon capture and sequestration (CCS) technologies, including ocean alkalinity enhancement, direct air capture using caustic streams, and CO2 sequestration as mineral carbonates, where significant volumes of dilute caustic streams (pH > 10) are anticipated to be necessary for the process.21 As many CCS processes rely on seawater, the efficient electrochemical production of caustic using seawater as the feed is particularly attractive, as this reduces the need for external chemical addition.21 Recent work has demonstrated that increasing the pH of seawater above its ambient level (∼8.1, although this is a rapidly changing value) dramatically increases the carrying capacity of the water towards dissolved carbonate species.21 This increase results from the pH-dependent transformation of CO2(aq.) into HCO3− and CO32−, which reduces CO2(aq.) levels and enables additional dissolution of CO2(g) according to Henry's law. As a result, seawater can form cation-carbonate and cation–bicarbonate complexes, enhancing its carbon storage capacity.22,23 In solutions with a pH greater than 10, the solvation of CO2 through its reaction with OH− to form HCO3− occurs at a significantly faster rate (k = 8.5 × 103 M−1 s−1) compared to its reaction with H2O (k = 6.6 × 10−4 M−1 s−1), facilitating even more efficient carbon capture in alkaline environments.24,25
The electrochemical production of caustic, hydrogen, and chlorine using seawater as the feedstock involves specific reactions at both the cathode and anode (Fig. 1). The primary reactions at the cathode are proton reduction and water reduction. In seawater, water reduction predominates due to the limited availability of protons at the typical pH of seawater. The produced hydrogen gas and caustic can be further separated and purified to obtain the final products through gas–liquid separation processes.
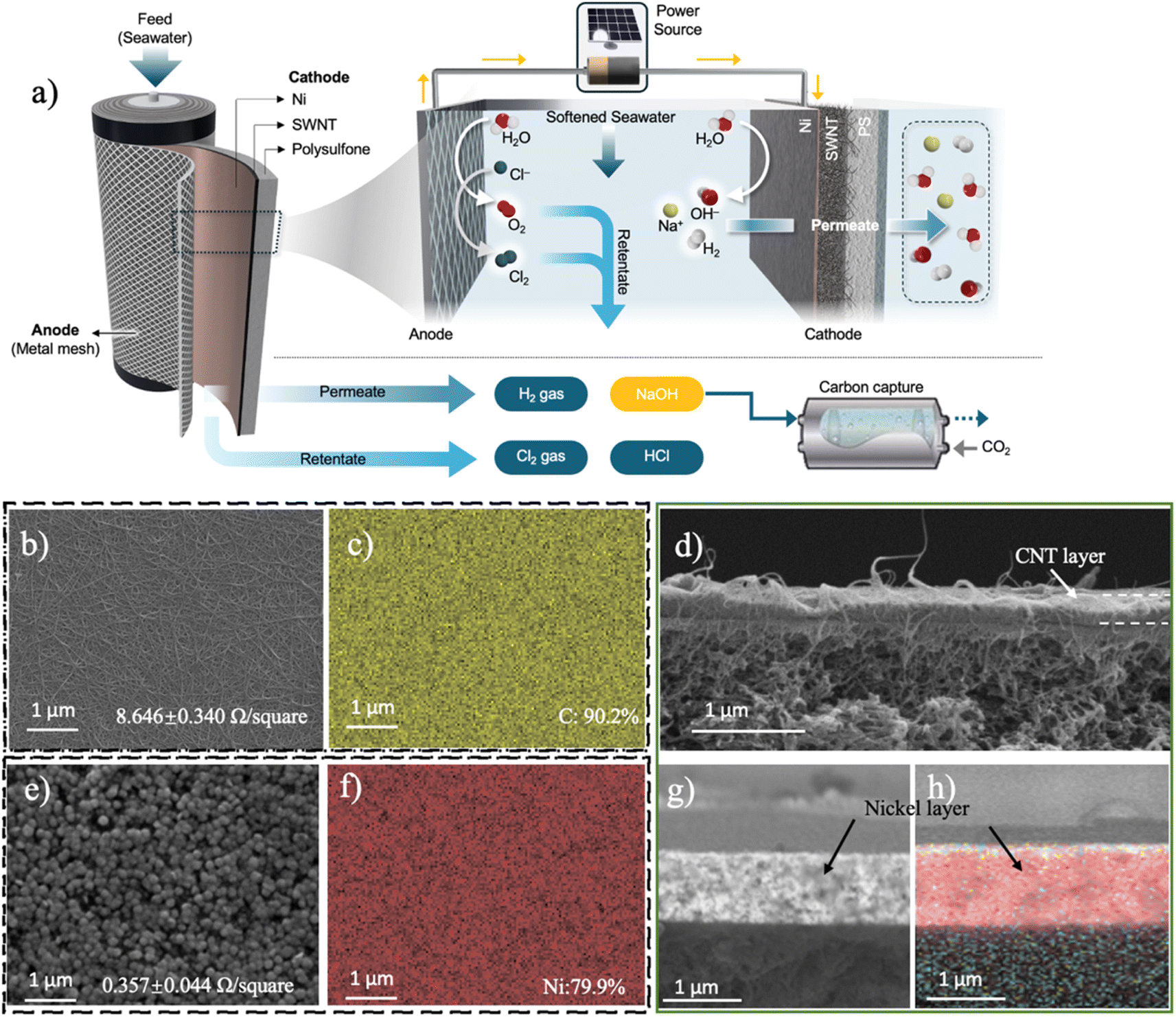 | ||
| Fig. 1 (a) Schematic diagram of a caustic production system composed of a porous Ni/SWNT cathode and an IrO2-coated Ti mesh anode. (b) SEM image of the CNT cathode surface, annotated with its measured sheet resistance. (c) EDS image of the CNT cathode surface, where yellow represents carbon. (d) Cross-sectional SEM image of the CNT-coated cathode showing the deposited CNT layer. (e) SEM image of the Ni-coated CNT coated cathode, with the associated sheet resistance of the material. (f) EDS image of the CNT/Ni cathode surface, where red represents nickel. (g) Cross-sectional SEM image of the Ni layer coating the CNT cathode. (h) Cross-sectional EDS image of the Ni/CNT cathode, where red represents Ni. | ||
At the anode, water molecules and chloride ions are oxidized, producing protons, oxygen gas, and chlorine gas. The Chlorine Evolution Reaction (CER) is thermodynamically less favorable compared to the Oxygen Evolution Reaction (OER).26 However, the CER is a two-electron reaction, whereas the OER involves four electrons, making OER kinetically less favorable. Despite progress in developing selective catalysts for these reactions in alkaline simulated seawater, both reactions still occur.27 The reactions can be summarized as follows:
Electrochemical reactions at the cathode:
| Proton reduction: 2H+ + 2e− → H2 | (1) |
| Water reduction: 2H2O + 2e− → H2(g) + 2OH− | (2) |
Electrochemical reactions at the anode
| OER: 2H2O → O2(g) + 4H+ + 4e− | (3) |
| CER: 2Cl− → Cl2 + 2e− | (4) |
The conventional chlor-alkali process has strict operational and material constraints such as the need for high-purity chemicals, deionized water, heat, and high-cost components (e.g., electrodes and ion exchange membranes in modern chlor-alkali reactors).28,29 While the theoretical minimum energy required to generate caustic is ∼1.6 kW he kg−1 NaOH, in practice, the actual energy requirement is higher (>2.1 kW he kg−1 NaOH), despite decades of process optimization.1 Chlor-alkali systems that rely on bipolar membrane electrodialysis have the potential to require significantly lower energy (as low as ∼0.7 kW he kg−1 NaOH), but in practice these systems require as much as 3.5 kW he kg−1 NaOH, as well as the use of high cost bipolar membranes.30 The use of oxygen depolarized cathodes can also reduce the energy demand of the chlor-alkali reaction, but at the cost of eliminating hydrogen production.31 In addition to electrical energy (needed to drive the electrolysis process), chlor-alkali reactors operate at elevated temperatures (90 °C), which requires additional thermal energy (0.128–0.196 kW ht kg−1 NaOH). Critically, these values do not include energy needed for pretreatment of the water (e.g., feed solutions are often made from distilled water), or any post-production concentration steps (often thermal-based evaporation processes). The departure from the minimum energy needed for the chlor-alkali reactions is influenced by various factors: (i) the use of ion exchange membranes increases the overall resistance of the system, leading to ohmic losses and higher voltage requirements and subsequent energy demand;32 (ii) H2 gas bubbles that evolve on the cathode can stick to the surface (known as gas masking), leading to current “hot spots” and lower faradaic efficiency;28,29 and (iii) concentration polarization of electrochemically-evolved species (e.g., OH−, Cl2) on the electrodes which increases the resistance to electron transfer.33 To minimize ohmic losses, chlor-alkali systems operate using extremely high solution concentrations (25% w/v NaCl as anolyte and 30% w/v NaOH as the catholyte).13 Also, chlor-alkali feed streams have strict purity requirements (in terms of Ca and Mg concentrations) so as to minimize cathode scaling.11 H2 gas, produced in large volumes on the cathode, is typically vented, as conventional chlor-alkali systems do not separate gasses well and the H2 is often mixed with chlorine, oxygen, and water vapor.34,35
Flow-through electrodes have been explored in multiple applications such as batteries, metal recovery from industrial waste, and oxidation of organic pollutants.36,37 In these systems, the electrolyte is forced through a porous electrode through the application of hydraulic pressure.36 This advective flow through the electrode can reduce the thickness of the diffusive boundary layer at the electrode/electrolyte interface, which can lower mass transfer limitations in the system, as well as strip evolved gas bubbles.36,37 In addition, the porous structure of the electrode provides a larger surface area for electrochemical reactions.36,37 The improved mass transfer and large interfacial reaction area make flow-through electrodes highly suitable for applications with diluted feed solutions.37,38 While flow-through electrodes have been previously explored, few studies have systematically investigated their use and energy efficiency in caustic production. Specifically, key factors such as flux, flow rate, and their impact on critical phenomena like gas bubble formation, concentration polarization, and charge transfer resistance have not been extensively examined. Understanding these relationships is essential to optimizing the efficiency of caustic production and reducing the SEC of the process. To address these gaps, we have developed a novel flow-through cathode system and investigated the impact of flux and applied potential on key performance metrics such as the SEC, the pH of the catholyte, current density, and current efficiency. Through a combination of experimental measurements and simulations, we determine that the observed reduction in energy consumption is due to the minimization of bubble formation, concentration polarization, and charge transfer resistance.
2. Results and discussion
2.1 Process configuration and flow-through cathode characterization
Our flow-through system was composed of two parallel electrodes, with a pressurized 3.5% w/v NaCl stream (pH 7.02) flowing in between (Fig. 1). In this system, the anode is an IrO2-coated Ti plate, while the cathode is constructed from a porous carbon nanotube (CNT) network deposited on a porous polymeric support, and coated with a thin layer of nickel (Fig. 1b–j). The IrO2-coated Ti anodes used in this study have been demonstrated to be highly efficient materials that can drive both the CER and OER, with minimal overpotentials.39,40 Therefore, to isolate potential impact of anodic reactions on overall efficiency from our research, we selected IrO2 as the anode. The hydraulic pressure in the system pushes a portion of the water through the cathode (the permeate), while a portion of the water exits the system (the retentate). When the electrodes are polarized, electrochemical reactions on the cathode lead to the formation of OH− ions and H2 gas, while anodic reactions result in oxygen and chlorine evolution (eqn (1)–(4)). Because of the advective transport through the cathode, the permeate exits the cell at a higher pH (i.e., caustic), along with H2 gas, while the retentate becomes acidic and contains dissolved chlorine species.Detailed characterization of the porous cathode's microstructure and composition was performed using scanning electron microscopy (SEM) and energy dispersive X-ray spectroscopy (EDS) (Fig. 1b–h). SEM images of the cathode before it was coated with Ni show a porous CNT network with an average pore size of 22.22 nm, which is composed solely of carbon (90%) and oxygen (10%) (Fig. 1b and c). Cross-sectional image analysis shows that the CNT layer has a thickness of 324 nm (Fig. 1d). The CNT layer exhibited a sheet resistance of 8.65 Ω □−1. Once the Ni coating was added to the CNT layer, the sheet resistance dropped by more than an order of magnitude, to 0.36 Ω □−1 (Fig. 1e). This Ni layer formed a rough, nodule-like coating on the surface of the CNTs, with the layer showing a composition of 80% Ni and 14% C (Fig. 1f). Cross-sectional SEM images of the Ni-coated CNTs show a uniform layer of Ni with a thickness of 0.93 μm coating the surface evenly (Fig. 1g and h). In this coated material, the CNTs are no longer visible, suggesting that the Ni grows throughout the CNT network. In terms of its hydraulic properties, the porous cathode with only the CNT layer had a permeability of 783.01 L m−2 h−1 bar−1 (LMH/bar), with the permeability dropping to 262 LMH/bar once the Ni layer was added. The contact angle of the porous cathode when only CNTs are present was 70.5°, and when the Ni coating was added, the contact angle increased to 104.2°, probably due to the formation of larger air gaps within bigger pores of nickel.41
2.2 Caustic generation and process efficiency
The caustic generation process efficiency was determined by evaluating key metrics such as the pH of the permeate and the retentate, the specific energy consumption (SEC) of the process, and the system's current efficiency. The pH of the permeate served as an indicator of the hydroxide generation at the cathode. The SEC of the process was used as the primary metric of process efficiency. The current efficiency, calculated using Faraday's law, quantifies the effectiveness of electron use for hydroxide production in the permeate. It is important to note that in our system some hydroxide ions may remain in the retentate. Only the hydroxide ions captured in the permeate were used to calculate the current efficiency and SEC. In this study, we explore how the flux of water through the porous cathode impacted the system's SEC and current efficiency, and explore the mechanistic reasons behind these observations. Here, we discuss the SEC of the electrochemical process alone. A more comprehensive evaluation of the process' SEC that includes the energy consumption associated with pumping is presented in a later section.The pH of the permeate was measured as a function of the applied cell potential (2–3.5 V) and flux through the cathode (Fig. 2a). As expected, the pH of the permeate was strongly impacted by both the applied cell potential and flux. Higher potentials resulted in higher currents, which translates to a higher rate of electrochemical reactions that generate OH− and higher permeate pH values (Fig. 2b). In these experiments, the ratio between the volume of the permeate and the volume of the feed was 1%, and therefore, the retentate pH change was far less significant. The measured currents fall in the typical range observed by previous studies that use IrO2 anodes and Ni-coated cathodes for seawater electrolysis.39 The flux through the cathode had a more complex relationship with pH and current. In terms of current, higher fluxes resulted in an increase in current for all applied voltages (Fig. 2b). For example, increasing the flux from 300 LMH to 1800 LMH increased the current from 70 A m−2 to 73.3 A m−2, a 4.76% increase, when 2.5 V was applied. Although the measured current increases slightly with increasing flux at various potentials, the overall increase is marginal. This is due to two factors: first, bubble coverage on the electrode surface is already very low, starting at around 2%, and decreases further with increasing pressure and flux. Second, the reduction in overpotential from concentration polarization mitigation is small (∼0.022 V), which contributes only a modest increase in current density. Together, these factors explain the limited effect of flux on current density. A detailed discussion can be found in later sections. However, the pH of the permeate declined when the system was operated at higher fluxes, likely due to dilution of the stream (Fig. 2a). For example, at an applied potential of 3.5 V, the pH of the permeate declined from 12.26 to 11.69 when the flux increased from 300 to 1800 LMH, despite the increase in OH− flux with increased flux (Fig. S2†). A detailed exploration as to why the current increases with increasing flux is provided in the following sections, but can be summarized by (i) enhanced transport of easily reduced species towards the cathode, (ii) reduced pH along the cathode, and (iii) enhanced gas (H2) stripping from the cathode surface.
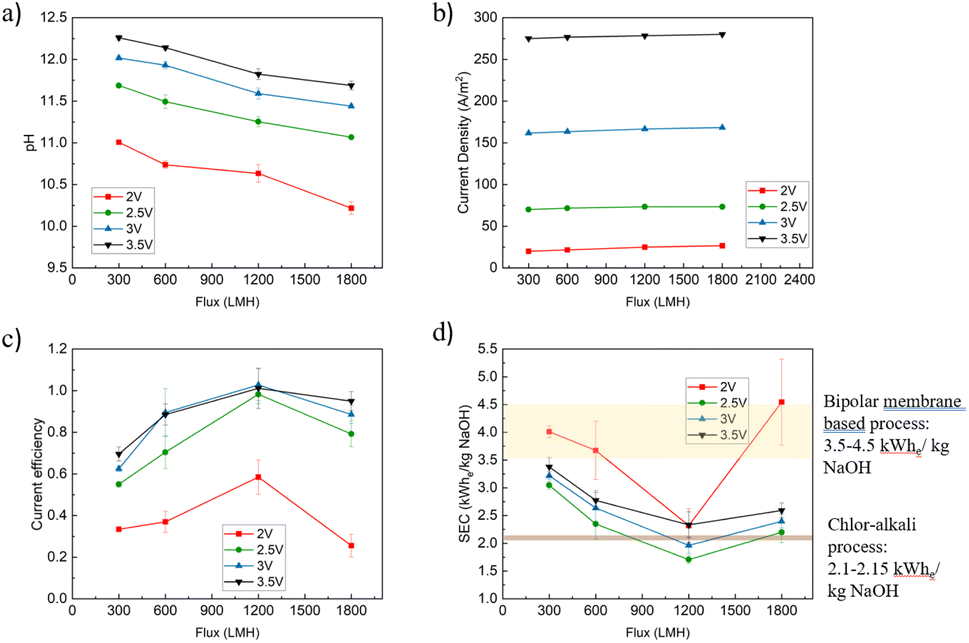 | ||
| Fig. 2 Variation of key performance metrics of the flow-through cathode system operating with a 3.5% w/v NaCl feed solution under different electrical and hydraulic conditions: (a) pH of permeate under different cell potentials. (b) Current density as a function of flux and cell potential. (c) Current efficiency of the electrolysis process as a function of flux and cell potential, (d) SEC of the process as a function of flux and cell potentials, where the SEC of the conventional chlor-alkali process is represented by the narrow brown band, and the SEC of a bipolar membrane electrodialysis system is represented by a wide yellow band. | ||
The system exhibits an interesting non-monotonic relationship between flux and current efficiency (Fig. 2c). Across all applied potentials, the current efficiency increases as the flux rises from 300 LMH, reaching the highest efficiency at 1200 LMH. At this flux, the current efficiency is 98.26%, 102.78%, and 101.13%, for 2.5 V, 3 V, and 3.5 V, respectively, while it is significantly lower at 2 V (58.46%). At the lowest applied potential (2 V) the electrochemical driving force is too low to drive efficient OH− production (as evidenced by electrochemical impedance spectroscopy (EIS) data below), which leads to the observed low current efficiency. Interestingly, the current efficiency drops at higher fluxes under all applied potential conditions (Fig. 1c). This is likely due to enhanced proton transport (generated at the anode) that neutralizes some of the caustic generated at the cathode when more rapid water transport through the cathode is imposed on the system. Therefore, maximizing the current efficiency in the system is dependent on balancing the advantages and disadvantages of rapid mass transfer to/from the cathode, with optimal conditions identified to exist at 1200 LMH in our system, a flux that facilitates gas bubble stripping and minimizes concentration polarization, while balancing dilution and anode-generated proton transport towards the cathode.
The SEC of the system exhibited an inverse relationship with the current efficiency, with the conditions that lead to the highest current efficiencies leading to the lowest SEC values (Fig. 2d). At 2 V, the SEC was highest among the four voltages tested, decreasing to its lowest point at 2.5 V before gradually increasing with further voltage increments. This pattern is attributed to the insufficient half-cell potentials for water splitting at a 2 V cell potential (the detailed half cell potential conversion can be found in Table 1): the cathode half-cell potential was −0.90 V vs. Ag/AgCl, where water electrolysis begins at −1.02 V vs. Ag/AgCl wire in 3.5% w/v NaCl solution (see EIS data below). Raising the voltage to 2.5 V provided sufficient energy to drive water electrolysis (the half-cell potential of the cathode was −1.36 V vs. Ag/AgCl). As the voltage increased further, concentration polarization and gas masking increased, leading to increased ohmic losses and higher SEC. At all applied potentials, the lowest SEC was measured at a flux of 1200 LMH, with SEC values of 2.32, 1.71, 1.96, and 2.33 kW he kg−1 NaOH at 2 V, 2.5 V, 3 V, and 3.5 V, respectively. Compared to the SEC (electrical) of conventional (membrane) and bipolar-based chlor-alkali processes (i.e., 3.5–4.5 kW h kg−1 NaOH),2 the SEC of our process is significantly lower, representing a 51.14% and 44.00% reduction when the applied potential was 2.5 V and 3 V, respectively. The hydrogen production rate under conditions with the lowest SEC was calculated using Faraday's law:
| Hydrogen production rate (mol s−1) = I/(2F) | (5) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1). Substituting the values of the current density and the surface area, the hydrogen production rate can be calculated to be 3.8 × 10−4 mol s−1 m−2. At standard temperature and pressure (STP), this corresponds to 8.5 × 10−3 L s−1 m−2. At the highest flux (1800 LMH), the SEC of the process increases, likely due to dilution and neutralization of the OH− ions from anode-generated protons.
485 C mol−1). Substituting the values of the current density and the surface area, the hydrogen production rate can be calculated to be 3.8 × 10−4 mol s−1 m−2. At standard temperature and pressure (STP), this corresponds to 8.5 × 10−3 L s−1 m−2. At the highest flux (1800 LMH), the SEC of the process increases, likely due to dilution and neutralization of the OH− ions from anode-generated protons.
| Cell potentials (V) | Cathode half-cell potentials (vs. Ag/AgCl in 3.5% NaCl, V) | Cathode half-cell potentials (RHE, V) |
|---|---|---|
| 2 | −0.90 | −0.29 |
| 2.5 | −1.36 | −0.75 |
| 3 | −1.79 | −1.18 |
| 3.5 | −2.22 | −1.61 |
Any evaluation of the overall SEC of the process must consider the energy associated with pressurizing the water to induce flow through the porous cathode. For this, we assume that the permeability of the cathode is similar to that of a conducting CNT membrane (2900 LMH/bar).42 The energy needed to achieve a certain flux at a given pressure can be derived using:43
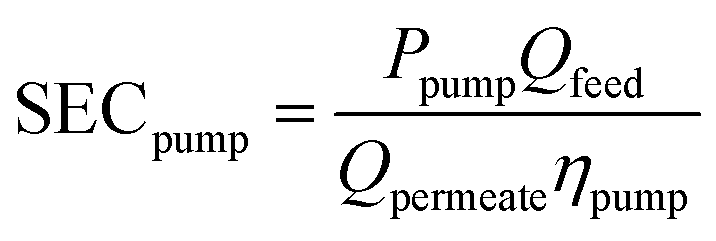 | (6) |
To investigate the charge transfer resistance on the cathode under different applied potentials and fluxes, high-voltage EIS measurements were conducted using a 3-electrode system, with a Ag/AgCl wire placed in between the anode and cathode serving as a reference electrode. In these experiments, a constant DC potential was applied to the electrode pair, while an oscillating potential of 300 mV at varying frequencies (20 mHz to 100 kHz) was overlain on top of it. A Randles circuit was used to fit the EIS data, with RS being the electrolyte/electrode/circuit resistance, RCT being the charge transfer resistance, and CPE being a constant phase element. In all cases, the left-side intercept of the curve with the X-axis corresponds to the RS, determined to be 2.06 ± 0.12 Ω (Fig. 3a–d). When a constant DC potential of 2 V was applied to the electrode (−0.90 V vs. Ag/AgCl on the cathode), plotting the real vs. imaginary impedance (on a Nyquist plot) and fitting an equivalent circuit to the curve clearly showed the impact of flux on the RCT (Fig. 3a). In this case, RCT declines from 113.0 Ω at 300 LMH to 37.8 Ω at 1800 LMH. When the constant potential was increased beyond 2 V, the resistance to charge transfer declined to 10.7 Ω, 1.2 Ω, and 0.5 Ω for the 2.5 V, 3 V, and 3.5 V, respectively (Fig. 3b–d). However, at these higher applied potentials, no relationship between flux and the RCT was observed.
 | ||
| Fig. 3 Electrochemical characterization of the reaction process at different fluxes and voltages. (a) EIS at −0.90 V vs. Ag/AgCl wire in 3.5% w/v NaCl (2 V cell potential) for 4 different fluxes; (b) EIS at −1.36 V vs. Ag/AgCl wire in 3.5% w/v NaCl (2.5 V cell potential) for 4 different fluxes; (c) EIS at −1.79 V vs. Ag/AgCl wire in 3.5% w/v NaCl (3 V cell potential) for 4 different fluxes; (d) EIS at −2.22 V vs. Ag/AgCl wire in 3.5% w/v NaCl (3.5 V cell potential) for 4 different fluxes; (e) LSV scanning from −0.3 V to −1.2 V vs. Ag/AgCl with a 5 mV s−1 scan rate in 3.5% w/v NaCl for 4 different fluxes. | ||
The difference between the 2 V conditions and the higher applied potentials (in terms of the observed relationship between flux and RCT) can be explained through the different electrochemical reactions taking place on the cathode surface, which we explored using linear sweep voltammetry (LSV) (Fig. 3e). As the potential moves toward more cathodic values, before the onset of the reduction reaction, the current is primarily due to capacitive current and non-faradaic processes. Once the electrode potential exceeds the critical potential for the reaction, the reduction reaction begins to occur, and a noticeable inflection point can be observed. There are two principal electrochemical reactions taking place on the cathode: (i) the proton reduction reaction (at −0.77 V vs. Ag/AgCl in 3.5% w/v NaCl) (eqn (1)) can be seen on the LSV curve as the first plateau, and (ii) water reduction (at −1.02 V vs. Ag/AgCl in 3.5% w/v NaCl) (eqn (2)) that can be seen as the current inflection curve on the LSV curve (Fig. 3e). When the applied potential was 2 V (−0.90 V vs. Ag/AgCl on the cathode), only proton reduction can take place (−0.77 V vs. Ag/AgCl). Increasing the flux through the cathode increases the transport of protons from the feed stream (also potentially protons generated on the anode), which lowers the pH along the cathode and reduces the resistance to charge transfer, as observed from the EIS data (Fig. 3a and e). While the RCT declines under higher fluxes when the 2 V cell potential is applied (from 113 Ω to 37.8 Ω), current measurements do not show a corresponding increase – the current density increases from 20.00 A m−2 at 300 LMH to 26.67 A m−2 at 1800 LMH (Fig. 2b). The overall reaction and current are influenced by both the anode and cathode, so a decrease in charge transfer resistance at one electrode does not necessarily lead to a proportional increase in current. Additionally, EIS measurements isolate the charge transfer resistance by using high-frequency signals to minimize capacitive and diffusive components. Under real operating conditions, diffusion terms also contribute to the overall current, making the relationship non-linear. Furthermore, according to the Butler–Volmer equation, the relationship between overpotential and current is logarithmic. These factors collectively explain why the current density does not increase proportionally to the decrease in charge transfer resistance on the cathode observed under higher flux conditions. At higher potentials, the dominant cathodic electrochemical reaction is the water reduction reaction (at −1.02 V vs. Ag/AgCl), evidenced by the far higher currents measured using LSV at these potentials (Fig. 3e). Since increasing flux through the cathode does not impact the concentration of water at the cathode surface, there is no change in the RCT at these higher voltages.
2.3 Gas masking and bubble stripping
The mitigation of gas masking on electrode surfaces was systematically investigated through the application of changing hydraulic pressure, flux, and cross-flow velocities using confocal microscopy (Fig. 4, S3 and S4†). All three physical mechanisms were found to reduce gas masking on the cathode surface. Not surprisingly, the application of higher hydraulic pressure reduces the number of H2 gas bubbles (Fig. 4a and S5†). When the pressure was increased (in a static system with no flow), the average bubble coverage reduced from 2.10% ± 0.12% at atmospheric pressure to 0.84% ± 0.04%. When water flux through the cathode was encouraged, a further reduction in bubble coverage occurred, with the average coverage reducing to 0.18% ± 0.03% at the highest pressure and flux, an 79% decline compared to coverage at the same pressure (10 psi) with no flux (Fig. 4b). Water flux through the cathode strips gas bubbles from the cathode surface, and carries these bubbles into the permeate stream, where they can be recovered as pure H2 gas. The H2 gas generated at the cathode was collected from the permeate line, and its concentration was determined to be 79.9% ± 0.03%, with the remainder being air, based on the composition of this fraction (N2:O2 = 3.7). The presence of air is likely from leaks during the gas collection process. The cross-flow velocity across the cathode also had a large impact on bubble coverage (Fig. 4c). When the pressure and flux were fixed at 0 psi and 0 LMH, respectively, increasing the cross-flow velocity across the cathode significantly reduced bubble coverage from 4.83% ± 0.61% at 0.031 m s−1 to 0.18% ± 0.03% at 0.138 m s−1, a 96.3% decline. At higher cross-flow velocities, the exertion of horizontal drag and longitudinal shear-induced lift forces effectively displaced the gas bubbles from the cathode surface, resulting in a substantial reduction in bubble coverage. And the reduction of overpotentials by mitigated bubble coverage is shown in Fig. S6 and S7.† Together, these data suggest that the hydrodynamic conditions in the system have a large impact on gas masking, and through that, on the energy efficiency of the process.
 | ||
| Fig. 4 Bubble generation on a cathode surface during the electrochemical reaction at (a) different pressures without cathode fluxes (0 psi, 3 psi, 10 psi), where the cross flow velocity was fixed at 0.056 m s−1; (b) different pressures with permeate flux (0 LMH at 0 psi, 160 LMH at 3 psi, 540 LMH at 10 psi), where cross flow velocity was fixed at 0.056 m s−1; (c) different cross flow velocities (0.031 m s−1, 0.056 m s−1, 0.138 m s−1), where the pressure was fixed at 0 psi. The dimensions of the characterized cathode are 8.45 mm × 7.66 mm, where the total number of bubbles (No.) and the bubble coverage area (%) are annotated in each figure. | ||
Finite-element simulations (using COMSOL) were used to examine the concentration polarization layer of electrochemically-generated hydroxide ions on the cathode surface under various operational scenarios, at a fixed cell potential of 2.5 V. Water flows in two directions in the vicinity of the cathode: a lateral cross-flow and a longitudinal permeate flux (Fig. 5a). These simulations revealed significant changes in the concentration of hydroxide ions on the cathode surface upon modulating the permeate flux (Fig. 5b) and cross flow (Fig. S8†). Under constant cross-flow conditions, an increase in flux led to a decrease in the thickness of the hydroxide ion concentration polarization layer, indicating enhanced transport of ions away from the cathode surface. This effect is evident in Fig. 5b, where the intensity of the red color, representing higher concentrations of hydroxide ions, decreases as the flux increases from 300 LMH to 1800 LMH, implying a mitigation of the concentration polarization layer (assuming pH 10 as the boundary value, the thickness of this boundary layer contracts from 29.8 μm to 16.9 μm). When considering the concentration of hydroxide ions on either side of the electrode (i.e., in the feed stream and in the permeate stream), an increase in flux was observed to reduce the pH on the sides of the electrode surface (Fig. 5c). Comparisons under different flux conditions showed that the simulated pH at the electrode's bottom surface (in contact with the permeate) matches well with the actual measured pH values in the permeate – both exhibiting a decrease in pH with increasing flux. The pH changes on the cathode surface were further analyzed using the Nernst equation to determine the shift in the hydroxide ion generation potentials under varying pH conditions. Our calculations suggest that a decrease in pH from 10.86 to 10.48 would lead to a reduction in the overpotential of 0.022 V. This result also demonstrates that in addition to the gas masking minimization, increasing water flux through the cathode reduces the process's energy consumption by minimizing the overpotential associated with the presence of a concentration polarization layer (composed of electrochemically generated OH− ions), another important reason, besides bubble mask removal, that increasing flux helps to improve current efficiency. This comprehensive analysis sheds light on the dynamic changes occurring at the cathode surface, offering valuable insights for optimizing electrochemical processes.
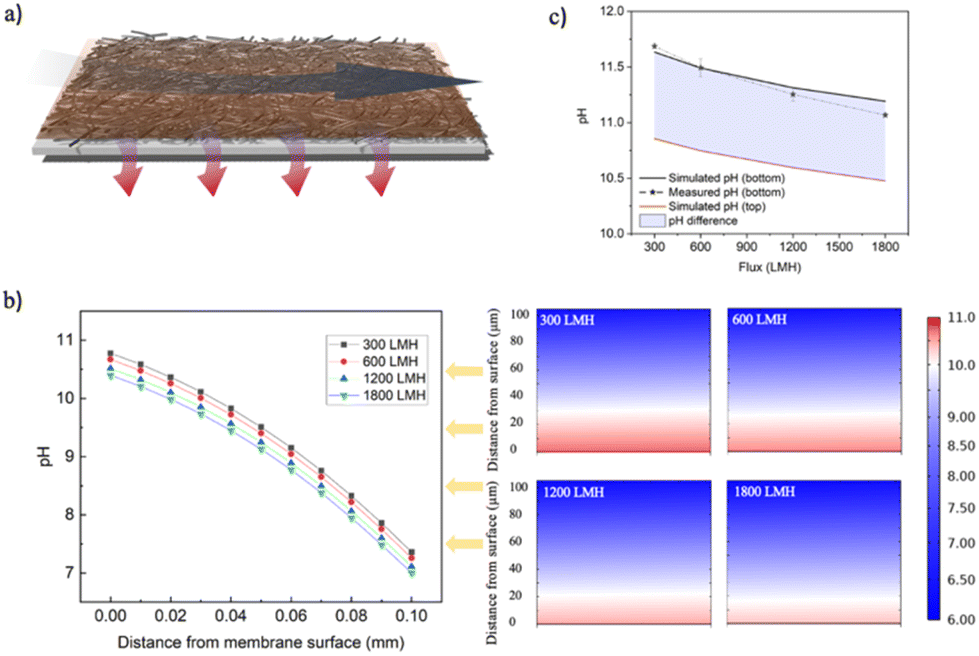 | ||
| Fig. 5 Simulation of hydroxide layer mitigation using COMSOL multiphysics. (a) Schematic of the cathode surface with cross flow and permeate flux effects; a 30 mm long cathode surface with a 2 mm channel height is simulated. (b) Changes in hydroxide ion concentration as a function of distance from the cathode surface at different fluxes (300 LMH, 600 LMH, 1200 LMH, 1800 LMH). (c) Simulated pH on the top and bottom surfaces of the porous cathode, and comparison with actual measurement values. | ||
3. Conclusions
The research presented herein represents a step forward for energy-efficient, on-site caustic generation that could be relevant for carbon sequestration applications. By forcing a NaCl stream through a porous cathode, mass transfer limitations and gas masking are minimized, while gas separation is achieved without the need for an ion exchange membrane. Through optimizing hydraulic and electrochemical conditions, we achieved a SEC of 1.71 kW he kg−1 NaOH, surpassing conventional chlor-alkali processes, without the need for heating the solution. This process innovation holds significant promise for large-scale clean caustic production that could enable efficient CO2 capture, for example through ocean alkalinity enhancement.Data availability
Data for this article are available at Open Society Framework (OSF) at https://osf.io/9aqxw/?view_only=df8c5b42950a430faab4dd1d211ff30b.Author contributions
F. Y. contributed to conceptualization, investigation, and writing of the original manuscript. M. X. was responsible for image analysis and SEC characterization. S. L. conducted the COMSOL multi-physics simulation. J. A. Q. R. performed electrochemical characterization. X. W. and M. C. both contributed to material characterization. A. D.-C. and S. K. worked on data visualization. G. N. S. and G. Z. R. provided supervision. E. M. V. H. and D. J. contributed to supervision and manuscript writing, including review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from the National Science Foundation (1926360). The author(s) would also like to acknowledge the assistance on confocal microscope usage from UCLA CNSI.References
- A. Kumar, F. Du and J. H. Lienhard, ACS Energy Lett., 2021, 6, 3563–3566 CrossRef CAS.
- G. P. Thiel, A. Kumar, A. Gómez-González and J. H. Lienhard, ACS Sustain. Chem. Eng., 2017, 5, 11147–11162 CrossRef CAS.
- Q. Xu, W. Chen and L. Song, Geophys. Res. Lett., 2022, 49, e2021GL095431 CrossRef.
- P. Badjatya, A. H. Akca, D. V. Fraga Alvarez, B. Chang, S. Ma, X. Pang, E. Wang, Q. Van Hinsberg, D. V. Esposito and S. Kawashima, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2114680119 CrossRef CAS PubMed.
- S. Chu, Science, 2009, 325(5948), 1599 CrossRef CAS PubMed.
- H. Pilorgé, N. McQueen, D. Maynard, P. Psarras, J. He, T. Rufael and J. Wilcox, Environ. Sci. Technol., 2020, 54, 7524–7532 CrossRef PubMed.
- C. Breyer, M. Fasihi, C. Bajamundi and F. Creutzig, Joule, 2019, 3, 2053–2057 CrossRef.
- S. Nawaz, J. Lezaun, J. M. Valenzuela and P. Renforth, Environ. Sci. Technol., 2023, 57, 8863–8869 CrossRef CAS PubMed.
- F. Lucile, P. Cézac, F. Contamine, J.-P. Serin, D. Houssin and P. Arpentinier, J. Chem. Eng. Data, 2012, 57, 784–789 CrossRef CAS.
- E. C. La Plante, X. Chen, S. Bustillos, A. Bouissonnie, T. Traynor, D. Jassby, L. Corsini, D. A. Simonetti and G. N. Sant, ACS ES&T Engineering Journal, 2023, 3, 955–968 Search PubMed.
- S. Madaeni and V. Kazemi, Sep. Purif. Technol., 2008, 61, 68–74 CrossRef CAS.
- P. Millet, in Handbook of Membrane Reactors, Elsevier, 2013, pp. 384–415 Search PubMed.
- J.-H. Shim, J.-Y. Jeong and J.-Y. Park, Int. J. Electrochem. Sci., 2015, 10, 6338–6347 CrossRef CAS.
- C. Huang and T. Xu, Environ. Sci. Technol., 2006, 40, 5233–5243 CrossRef CAS PubMed.
- S. M. H. Hashemi, M. A. Modestino and D. Psaltis, Energy Environ. Sci., 2015, 8, 2003–2009 RSC.
- L. Jiang, B. Myer, K. Tellefsen and S. Pau, J. Power Sources, 2009, 188, 256–260 CrossRef CAS.
- J. Li, Y. Zhu, W. Chen, Z. Lu, J. Xu, A. Pei, Y. Peng, X. Zheng, Z. Zhang and S. Chu, Joule, 2019, 3, 557–569 CrossRef CAS.
- P. Tiwari, G. Tsekouras, K. Wagner, G. F. Swiegers and G. G. Wallace, Int. J. Hydrogen Energy, 2019, 44, 23568–23579 CrossRef CAS.
- M. Gillespie, F. Van Der Merwe and R. Kriek, J. Power Sources, 2015, 293, 228–235 CrossRef CAS.
- G. F. Swiegers, A. L. Hoang, A. Hodges, G. Tsekouras, C.-Y. Lee, K. Wagner and G. Wallace, Curr. Opin. Electrochem., 2022, 32, 100881 CrossRef CAS.
- E. C. La Plante, D. A. Simonetti, J. Wang, A. Al-Turki, X. Chen, D. Jassby and G. N. Sant, ACS Sustain. Chem. Eng., 2021, 9, 1073–1089 CrossRef CAS.
- F. J. Millero and R. N. Roy, Croat. Chem. Acta, 1997, 70, 1–38 CAS.
- F. J. Millero, T. B. Graham, F. Huang, H. Bustos-Serrano and D. Pierrot, Mar. Chem., 2006, 100, 80–94 CrossRef CAS.
- W. Stumm and J. J. Morgan, Aquatic chemistry: chemical equilibria and rates in natural waters, John Wiley & Sons, 2012 Search PubMed.
- X. Wang, W. Conway, R. Burns, N. McCann and M. Maeder, J. Phys. Chem. A, 2010, 114, 1734–1740 CrossRef CAS.
- L. Yu, J. Xiao, C. Huang, J. Zhou, M. Qiu, Y. Yu, Z. Ren, C.-W. Chu and J. C. Yu, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2202382119 CrossRef CAS.
- J. S. Ko, J. K. Johnson, P. I. Johnson and Z. Xia, ChemCatChem, 2020, 12, 4526–4532 CrossRef CAS.
- M. T. de Groot and A. W. Vreman, Electrochim. Acta, 2021, 369, 137684 CrossRef CAS.
- A. Angulo, P. van der Linde, H. Gardeniers, M. Modestino and D. F. Rivas, Joule, 2020, 4, 555–579 CrossRef CAS.
- M. Reig, S. Casas, C. Valderrama, O. Gibert and J. Cortina, Desalination, 2016, 398, 87–97 CrossRef CAS.
- I. Moussallem, J. Jörissen, U. Kunz, S. Pinnow and T. Turek, J. Appl. Electrochem., 2008, 38, 1177–1194 CrossRef CAS.
- M. Seko, Ind. Eng. Chem. Prod. Res. Dev., 1976, 15, 286–292 CrossRef CAS.
- Y. Guo, X. Li, H. Guo, Q. Qin, Z. Wang, J. Wang and G. Yan, Energy Storage Mater., 2022, 51, 476–485 CrossRef.
- M. Erden and M. Karakilcik, Int. J. Hydrogen Energy, 2024, 52, 546–560 CrossRef CAS.
- K. Li, Q. Fan, H. Chuai, H. Liu, S. Zhang and X. Ma, Trans. Tianjin Univ., 2021, 27, 202–216 CrossRef CAS.
- T. Doherty, J. Sunderland, E. Roberts and D. Pickett, Electrochim. Acta, 1996, 41, 519–526 CrossRef CAS.
- J. Newman and W. Tiedemann, AIChE J., 1975, 21, 25–41 CrossRef CAS.
- M.-A. Goulet, M. Eikerling and E. Kjeang, Electrochem. Commun., 2015, 57, 14–17 CrossRef CAS.
- A. El-Moneim and M. Mohamed, Int. J. Electrochem. Sci., 2012, 7, 671–685 CrossRef CAS.
- T. Naito, T. Shinagawa, T. Nishimoto and K. Takanabe, Inorg. Chem. Front., 2021, 8, 2900–2917 RSC.
- M. Xiao, F. Yang, S. Im, D. S. Dlamini, D. Jassby, S. Mahendra, R. Honda and E. M. Hoek, Journal of Membrane Science Letters, 2022, 2, 100022 CrossRef.
- M. J. Larocque, D. R. Latulippe and C.-F. de Lannoy, J. Membr. Sci., 2021, 620, 118859 CrossRef CAS.
- A. Zhu, P. D. Christofides and Y. Cohen, J. Membr. Sci., 2009, 339, 126–137 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section and supplementary data. See DOI: https://doi.org/10.1039/d4ta04680c |
| This journal is © The Royal Society of Chemistry 2025 |