
Bimetallic-ion co-intercalation to stabilize vanadium–oxygen bonds towards high-performance aqueous zinc-ion storage†
Yulin
Jiang
,
Xia
Wen
,
Yinuo
Li
,
Yuhang
Li
,
Yanan
Peng
,
Wang
Feng
,
Xiaohui
Li
,
Junbo
Yang
,
Luying
Song
,
Ling
Huang
,
Hang
Sun
and
Jianping
Shi
 *
*
The Institute for Advanced Studies, Wuhan University, Wuhan 430072, China. E-mail: jianpingshi@whu.edu.cn
First published on 21st November 2024
Abstract
Aqueous zinc-ion batteries (AZIBs) have received increasing attention in large-scale energy storage systems because of their appealing features with respect to safety, cost, and scalability. Although vanadium oxides with different compositions demonstrate promising potential as cathodes for AZIBs, the narrow interlayer spacing, inferior electronic conductivity, and high dissolution in electrolyte seriously restrict their practical applications. Here we design an ingenious bimetallic-ion (Mg2+ and Al3+) co-intercalation strategy to boost the performance of AZIBs using V6O13·1.31H2O (VOH). The bimetallic-ion intercalation expands the interlayer spacing, increases electronic conductivity, and more importantly stabilizes the vanadium–oxygen bond in VOH, thus promoting ion/electron transport kinetics and restraining vanadium oxide dissolution. As expected, MgAl-VOH cathodes deliver ultrahigh specific capacities of 524.9 and 275.6 mA h g−1 at current densities of 0.1 and 5 A g−1, respectively, comparable to the highest value reported for vanadium oxides. The underlying zinc-ion storage mechanism is unambiguously clarified with the aid of density functional theory calculations and in situ structural characterization. This work opens up a new avenue for boosting the performance of AZIBs by designing bimetallic-ion co-intercalated cathodes.
1. Introduction
Due to their high energy density, long cyclability, and scalability, lithium-ion batteries (LIBs) have achieved unprecedented success in portable electronic devices and electric vehicles.1,2 However, the scarcity and uneven geographical distribution of lithium resources, as well as the safety concerns arising from the employment of flammable carbonate electrolytes have seriously limited their application scenarios.3–5 Aqueous zinc-ion batteries (AZIBs) are considered as a promising alternative to LIBs due to their cost-effectiveness, environmental friendliness, and intrinsic safety.6–9 In particular, the high theoretical capacity (820 mA h![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) g−1 and 5851 mA h cm−3), low redox potential (−0.763 V vs. standard hydrogen electrode), and excellent electrochemical stability of the Zn metal anode in water can overcome the inferior energy density resulting from limited potential windows of aqueous media.10,11 Nevertheless, the commercialization of AZIBs is still hindered by some obstacles including (1) the narrow interlayer spacing and low electronic conductivity of cathodes, which limit the ion/electron transport;12 (2) the high dissolution of vanadium oxides in electrolyte, leading to unsatisfactory cycling stability;13 (3) the high polarization of Zn2+, which reduces the capacity and lifetime.14 In this regard, developing suitable cathode materials to match the Zn metal anode of AZIBs is of great significance.
g−1 and 5851 mA h cm−3), low redox potential (−0.763 V vs. standard hydrogen electrode), and excellent electrochemical stability of the Zn metal anode in water can overcome the inferior energy density resulting from limited potential windows of aqueous media.10,11 Nevertheless, the commercialization of AZIBs is still hindered by some obstacles including (1) the narrow interlayer spacing and low electronic conductivity of cathodes, which limit the ion/electron transport;12 (2) the high dissolution of vanadium oxides in electrolyte, leading to unsatisfactory cycling stability;13 (3) the high polarization of Zn2+, which reduces the capacity and lifetime.14 In this regard, developing suitable cathode materials to match the Zn metal anode of AZIBs is of great significance.
The cathode exploration mainly focuses on Prussian blue analogues, manganese/vanadium oxides, and organic compounds.11,15–18 Among them, vanadium oxides with unique layered structures are regarded as one of the promising cathodes for AZIBs because of the high natural abundance, low cost, and multiple oxidation states of vanadium.19–22 Even so, the narrow interlayer spacing and irreversible phase transition in vanadium oxides, and high dissolution in electrolyte inevitably result in low ion/electron transport kinetics and rapid electrode structure degradation.23,24 To address these issues, intensive efforts have been made including introducing defects, constructing heterostructures, and intercalating cations/molecules.25–29 Thereinto, the intercalation of exotic species should increase the interlayer spacing of cathodes, thereby improving ion/electron transport and enhancing cycling stability.21 Given their large hydrated ionic radii and electronegativities, as well as moderate atomic weights and robust metal–O bonds, Mg2+ and Al3+ occupy distinct places among the intercalated ions.30 For example, intercalating Mg2+ ions into NH4V4O10 can reduce the migration energy barriers of Zn2+ ions and inhibit amine dissolution, which contribute to improving the AZIB performance.23 After introducing Al3+ ions into the H11Al2V6O23.2 cathode, the lamellar structures are stabilized and the interlayer spacings are enlarged accordingly.31 Besides, the co-intercalation of K+, Mg2+, and Al3+ ions has also been proven to be an effective method to improve the AZIB performance;32 however, the underlying physical mechanism should be further clarified.
To tackle such intractable challenges, we designed a one-step solvothermal method to synthesize Mg2+ and Al3+ bimetallic-ion co-intercalated V6O13·1.31H2O (MgAl-VOH). The advantages of MgAl-VOH as the cathode of AZIBs can be summarized as follows: (1) the intercalation of Mg2+ and Al3+ ions expands the interlayer spacing (from 0.98 to 1.31 nm) of the VOH cathode, which should accelerate Zn2+ ion transport; (2) the bimetallic-ion co-intercalation stabilizes the vanadium–oxygen (V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) bonds and restrains VOH cathode dissolution; (3) the doping of Mg2+ and Al3+ ions modifies the electronic structure of VOH and increases its conductivity. Accordingly, remarkable specific capacity, good rate performance, and excellent cycling stability are achieved in AZIBs composed of MgAl-VOH cathodes. This work offers a new strategy for designing high-performance AZIB cathodes through bimetallic-ion co-intercalation.
O) bonds and restrains VOH cathode dissolution; (3) the doping of Mg2+ and Al3+ ions modifies the electronic structure of VOH and increases its conductivity. Accordingly, remarkable specific capacity, good rate performance, and excellent cycling stability are achieved in AZIBs composed of MgAl-VOH cathodes. This work offers a new strategy for designing high-performance AZIB cathodes through bimetallic-ion co-intercalation.
2. Results and discussion
2.1. Synthesis of MgAl-VOH
The synthesis process of MgAl-VOH is illustrated in Fig. 1a with V2O5, Al(NO3)3, and Mg(NO3)2 as the precursors. The chemical compositions and valence states of MgAl-VOH, Mg-VOH, Al-VOH, and pure VOH are analysed by X-ray photoelectron spectroscopy (XPS), with the results shown in Fig. 1b, c and S1,† respectively. The coexistence of Mg and Al signals indicates the successful preparation of MgAl-VOH using this bimetallic-ion co-intercalation strategy. The binding energies at 1303.9 and 74.2 eV are attributed to Mg2+ and Al3+, respectively. In addition, the O 1s XPS spectra in Fig. S2† show that the Al–O and Mg–O peaks are not observed in MgAl-VOH, suggesting that Mg2+ and Al3+ ions are co-intercalated rather than co-doped in VOH. The weight fractions of Mg and Al are calculated to be 1.0 and 0.8 wt%, according to XPS and inductively coupled plasma optical emission spectroscopy (ICP-OES) results (Table S1, ESI†). Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) measurements were thus performed to characterize the morphologies of the obtained samples (Fig. 1d, e and S3, ESI†). The nanobelt structures are clearly observed for MgAl-VOH, consistent with Mg-VOH, Al-VOH, and pure VOH, indicating that the macrostructure of VOH is retained even after intercalating Mg2+ and Al3+ ions. Energy dispersive spectroscopy (EDS) maps of MgAl-VOH reveal the homogeneous distribution of V, O, Mg, and Al elements (Fig. 1f–i), confirming the intercalations of Mg2+ and Al3+ ions in VOH.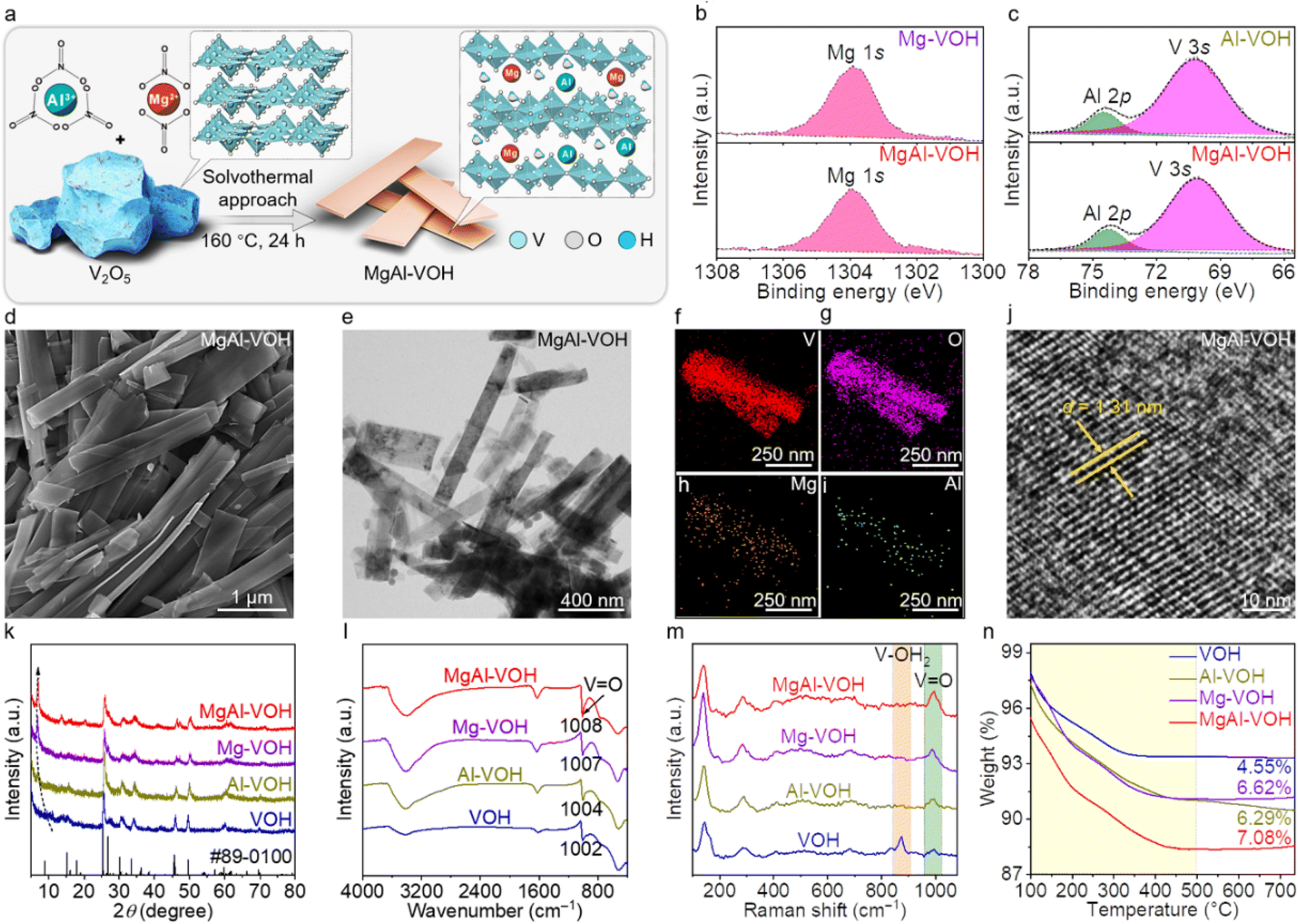 | ||
| Fig. 1 Synthesis of MgAl-VOH cathodes. (a) Schematic diagram for the preparation of MgAl-VOH. (b and c) XPS spectra of MgAl-VOH, Mg-VOH, and Al-VOH. The coexistence of Mg and Al signals indicates the successful synthesis of MgAl-VOH. (d and e) SEM and low-magnification TEM images of MgAl-VOH, revealing its nanobelt morphology. (f–i) EDS mapping images of MgAl-VOH, showing the uniform distribution of V, O, Mg, and Al elements. (j) Atomic resolution TEM image of MgAl-VOH. The large interlayer spacing is observed due to the co-intercalation of Mg2+ and Al3+ ions. (k) XRD patterns of MgAl-VOH, Mg-VOH, Al-VOH, and pure VOH. The characteristic peak of (001) plane shift reconfirms the increased interlayer spacing. (l) FTIR results of MgAl-VOH, Mg-VOH, Al-VOH, and pure VOH. The characteristic peaks of VO bonds are shifted to high wavenumbers after intercalating the metal ions. (m and n) Raman spectra and TGA results of MgAl-VOH, Mg-VOH, Al-VOH, and pure VOH. | ||
The atomic resolution TEM image in Fig. 1j shows an interlayer spacing of 1.31 nm for MgAl-VOH, much larger than those of Mg-VOH (1.29 nm), Al-VOH (1.26 nm), and pure VOH (0.98 nm), indicating that the co-intercalation of Mg2+ and Al3+ ions expands the interlayer spacings, which should accelerate the Zn2+ ion transport. X-ray diffraction (XRD) patterns in Fig. 1k reconfirm the intercalation of Mg2+ and Al3+ ions in VOH, as indicated by the characteristic peak shift of the (001) plane. To further determine the influence of Mg2+ and Al3+ ion intercalation on the atomic structure of VOH, Fourier transform infrared spectroscopy (FTIR) measurements were performed on MgAl-VOH, Mg-VOH, Al-VOH, and pure VOH, respectively, with the results shown in Fig. 1l. Notably, after intercalating Mg2+ and Al3+ ions, the characteristic peaks of VO bonds shift to high wavenumbers and the corresponding full width at half maxima (FWHM) decreases (Table S2, ESI†), suggesting that the VO bonds are stabilized after metal ion intercalation.33,34Fig. 1m shows the Raman spectra of these four samples; the weak or absent characteristic peak at 874 cm−1 (corresponding to the tensile vibration of V–OH2) indicates the high rotational freedom of water in the interlayers of MgAl-VOH, Mg-VOH, and Al-VOH, which contributes to the diffusion of Zn2+ ions.35 In addition, the tensile vibration peak of VO bonds at 995 cm−1 becomes sharper after intercalating metal ions, which suggests that the VO bonds are more stable, consistent with the FTIR results. The crystal water contents of MgAl-VOH (7.08%), Mg-VOH (6.62%), Al-VOH (6.29%), and pure VOH (4.55%) are also obtained according to thermogravimetric analysis (TGA) results shown in Fig. 1n. The specific surface areas of MgAl-VOH, Mg-VOH, Al-VOH, and pure VOH are calculated to be 15.20, 13.68, 12.26, and 6.36 m2 g−1, respectively (Fig. S4, ESI†). Electron paramagnetic resonance (EPR) spectroscopy was further performed on MgAl-VOH and pure VOH (Fig. S5, ESI†); the oxygen vacancies were not observed even after introducing Al3+ and Mg2+ ions. In short, the co-intercalation of Mg2+ and Al3+ ions expands the interlayer spacings of VOH and stabilizes the VO bonds, which is beneficial for accelerating the Zn2+ ion transport and restraining the VOH cathode dissolution during charging and discharging processes. Notably, it is the first report regarding the bimetallic-ion co-intercalation to stabilize VO bonds of the VOH electrode.
2.2. Bimetallic-ion co-intercalation to the stabilize V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) O bond
O bond
Density functional theory (DFT) calculations and detailed experimental characterization were carried out to clarify the effect of Mg2+ and Al3+ ion intercalation on the structural stability of VOH. The MgAl-VOH, Mg-VOH, Al-VOH, and pure VOH electrodes were constructed and then soaked into electrolytes, with the corresponding photographs shown in Fig. 2a. Interestingly, after 7 and 30 days, the electrolytes containing Mg-VOH, Al-VOH and pure VOH electrodes changed from colourless to yellow, indicative of the dissolution of vanadium oxides. Nevertheless, the electrolyte containing the MgAl-VOH electrode remained unchanged, even after 60 days, with its transparent feature still clearly visible. Such results manifest that the co-intercalation of Mg2+ and Al3+ ions slows down the vanadium oxide dissolution rate in electrolytes, which contributes to improving the capacity and cycling stability of AZIBs. Additionally, contrastive FTIR measurements of these four samples at different states were also performed, with the results shown in Fig. 2b. The characteristic peak of the VO bond in VOH disappeared after 60 days; nevertheless, these peaks are still clearly observed in the other three samples, strongly suggesting the robust structures of MgAl-VOH, Mg-VOH, and Al-VOH electrodes. To quantitatively understand this interesting phenomenon, ICP-OES analyses were carried out on the soaked electrolytes (Fig. 2c). The V ion concentration in the electrolyte containing the MgAl-VOH electrode is calculated to be 0.124 mg L−1, much smaller than those of Mg-VOH (0.412 mg L−1), Al-VOH (1.684 mg L−1), and pure VOH (12.061 mg L−1), which indicates that the vanadium oxide dissolution is significantly inhibited by the co-intercalated Mg2+ and Al3+ ions.
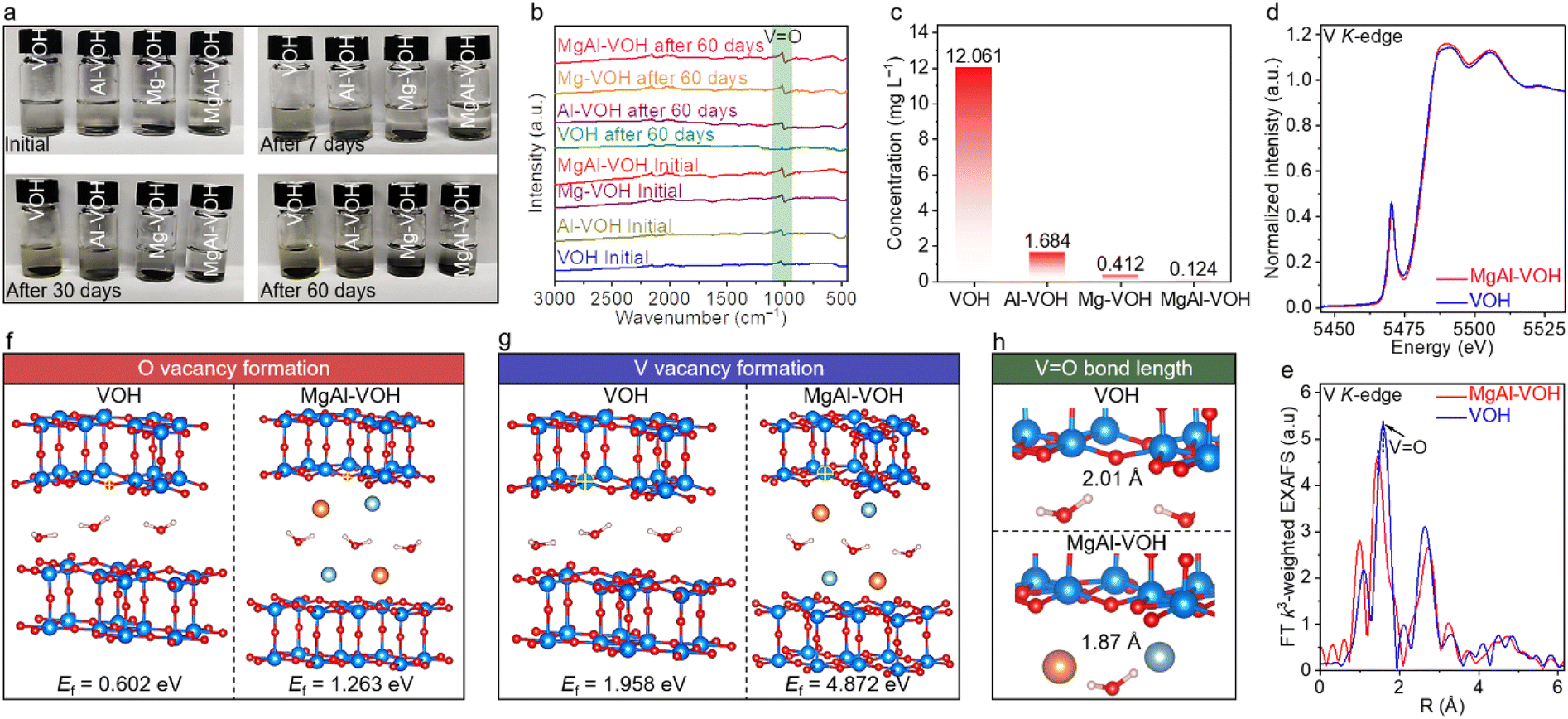 | ||
| Fig. 2 The influence of bimetallic-ion co-intercalation on the structure stability of VOH. (a) Photographs of electrolytes containing MgAl-VOH, Mg-VOH, Al-VOH, and pure VOH electrodes, respectively. (b) Corresponding FTIR spectra of such electrolytes at different states. The unchanged VO bond characteristic peaks in MgAl-VOH, Mg-VOH, and Al-VOH indicating their robust structures. (c) V ion concentrations in different electrolytes containing MgAl-VOH, Mg-VOH, Al-VOH, and pure VOH electrodes, respectively. (d and e) XANES and EXAFS spectra of MgAl-VOH and VOH, respectively, showing the smaller VO bond length in MgAl-VOH than in pure VOH. (f and g) Formation energies of O and V vacancies in MgAl-VOH and pure VOH. (h) Calculated VO bond lengths in MgAl-VOH and pure VOH. | ||
X-ray absorption near-edge structure (XANES) spectra present the existence of a low-strength pre-edge in MgAl-VOH, compared toVO2 and V2O5, indicating the abundance of 3d electrons in the V species of MgAl-VOH (Fig. 2d and S6, ESI†). Besides, the extended X-ray absorption fine structure (EXAFS) measurement results reveal a smaller VO bond length in MgAl-VOH (1.90 Å) than in VOH (1.92 Å), which should stabilize the VO bonds significantly (Fig. 2e, S7, and Table S3, ESI†). DFT calculations were performed to further expound the internal physical mechanism. The formation energies of O and V vacancies are calculated to be 1.263 and 4.872 eV for MgAl-VOH, much larger than those of pure VOH (0.602 and 1.958 eV), as shown in Fig. 2f and g, indicating that the VO bond is more stable in MgAl-VOH than in VOH. In addition, the VO bond lengths are also determined for MgAl-VOH (1.87 Å) and pure VOH (2.01 Å); the shorter bond length suggests the more stable structure of MgAl-VOH than pure VOH (Fig. 2h), which should prevent the electrode collapse during the charging and discharging processes.
2.3. Electrochemical performances of MgAl-VOH
The zinc-ion storage performances of MgAl-VOH, Mg-VOH, Al-VOH, and pure VOH were evaluated by assembling AZIBs. Fig. 3a and S8a–c† show the cyclic voltammetry (CV) curves of these four electrodes for the initial three cycles at a scan rate of 0.1 mV s−1 in the voltage range of 0.2–1.4 V. Two pairs of redox peaks with similar shapes are clearly observed for all the cathodes, indicating their similar electrochemical properties. For MgAl-VOH, these two pairs of redox peaks are located at 0.42/0.55 V and 0.96/0.99 V, respectively, suggesting the multistep Zn2+ ion de/intercalation processes.36 Particularly, the CV curves of the subsequent two cycles remain stable and overlap well, indicating the high reversibility of MgAl-VOH. Fig. 3b, c and S8d–f† reveal the galvanostatic charge/discharge (GCD) profiles of the four cathodes at a current density of 0.1 A g−1. In the first cycle, the discharge and charge capacities of MgAl-VOH are obtained to be 529.1 and 524.9 mA h g−1, much larger than those of Mg-VOH (481.1 and 455.7 mA h g−1), Al-VOH (434.0 and 424.1 mA h g−1), and pure VOH (296.2 and 286.5 mA h g−1). The rate capabilities of all the test samples were further evaluated at different current densities (Fig. 3d). As the current density increases from 0.1 to 5 A g−1, the average reversible capacities of 514.2 and 275.6 mA h g−1 are achieved for MgAl-VOH, much higher than those of Mg-VOH (457.7 and 228.9 mA h g−1), Al-VOH (436.8 and 197.3 mA h g−1), and pure VOH (321.6 and 160.3 mA h g−1). Notably, the MgAl-VOH cathode achieves a high specific capacity of 485.5 mA h g−1 when the current density returns to 0.1 A g−1, indicating its excellent reversibility. To clarify the influence of Mg2+ and Al3+ ion concentrations on AZIB performances, the rate capabilities of MgAl-VOH with different Mg2+ and Al3+ ion concentrations are shown in Fig. S9 and Table S4.† The rate capabilities of MgAl-VOH with different mass loadings (1.2, 1.5, 1.8, and 2.0 mg cm−2) were also obtained and are shown in Fig. S10.† Notably, a further increase in the mass loading of MgAl-VOH leads a reduction in charge transfer efficiency, which weakens the performance of AZIBs. The capacity comparison of MgAl-VOH with the other vanadium oxides at different current densities is shown in Fig. 3e and Table S5,† manifesting its distinguished application prospect in zinc-ion storage.23,27–29,35,37–44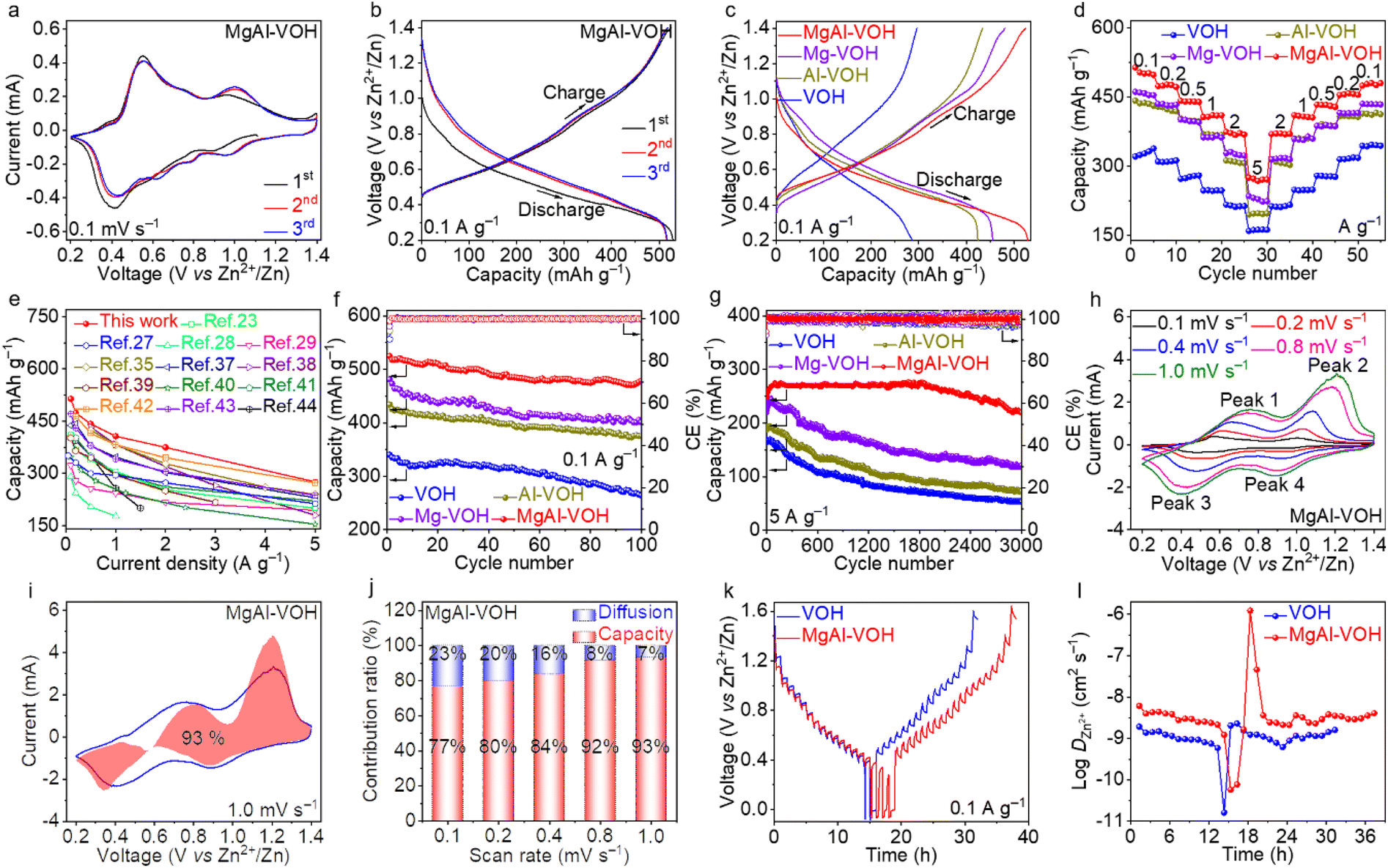 | ||
| Fig. 3 Electrochemical performances of MgAl-VOH, Mg-VOH, Al-VOH, and pure VOH. (a) CV curves of MgAl-VOH in the first three cycles with a scan rate of 0.1 mV s−1. (b) Charging and discharging curves of MgAl-VOH in the first three cycles at a current density of 0.1 A g−1. (c) Charging and discharging curves of MgAl-VOH, Mg-VOH, Al-VOH, and pure VOH at a current density of 0.1 A g−1. (d) Rate capabilities of MgAl-VOH, Mg-VOH, Al-VOH, and pure VOH at different current densities. (e) Capacity comparison of MgAl-VOH with the other vanadium oxides at different current densities. (f and g) Cycling performances of MgAl-VOH, Mg-VOH, Al-VOH, and pure VOH at the current densities of 0.1 and 5 A g−1, respectively. (h) CV curves of MgAl-VOH at different scan rates. (i) CV profile of MgAl-VOH with the capacitive contribution at a scan rate of 1.0 mV s−1. (j) Capacitive contributions in MgAl-VOH at different scan rates. (k) GITT potential profiles of MgAl-VOH and pure VOH. (l) Zn2+ ion diffusion coefficients in MgAl-VOH and pure VOH during the charging and discharging processes. | ||
To assess the stability of MgAl-VOH, Mg-VOH, Al-VOH, and pure VOH electrodes, cycling tests at different current densities were performed. MgAl-VOH displays a high reversible capacity of 524.9 mA h g−1 at 0.1 A g−1 with a coulombic efficiency (CE) of 100%, and the capacity is maintained at 478.3 mA h g−1 even after 100 cycles, corresponding to a capacity retention of 91.1% (Fig. 3f). In contrast, Mg-VOH, Al-VOH and pure VOH cathodes show low reversible capacities (400.9, 375.2, and 267.2 mA h g−1) and small capacity retentions (83.2%, 86.4%, and 90.9%) after 100 cycles. At a high current density of 5 A g−1, MgAl-VOH maintains an excellent cycling performance and a reversible capacity of 210.6 mA h g−1 is achieved even after 3000 cycles with the capacity retention of 88.3%, much higher than those of Mg-VOH (54.5%), Al-VOH (38.4%) and pure VOH (31.2%), as shown in Fig. 3g. The better electrochemical performance of MgAl-VOH compared to Mg-VOH and Al-VOH is attributed to its larger interlayer spacing, more stable VO bonds, and the synergistic effect. Notably, the small difference in the intercalated metal content of MgAl-VOH (1.8%), Mg-VOH (1.2%), and Al-VOH (0.8%) possibly results in insignificant influence on the zinc-ion storage performances, as has been demonstrated in Mg2+ doped NH4V4O10.45 These results indicate that the co-intercalation of Mg2+ and Al3+ ions accelerates the Zn2+ ion transport, restrains cathode dissolution, and then improves the reversible capacity and cycling performances.
The electrochemical kinetics of MgAl-VOH, Mg-VOH, Al-VOH, and pure VOH were evaluated via CV, galvanostatic intermittent titration technique (GITT), and electrochemical impedance spectroscopy (EIS) measurements. To determine the capacitive contribution, CV curves of the four electrodes were examined at various scan rates from 0.1 to 1.0 mV s−1 (Fig. 3h and S11a–c, ESI†). The capacitive contribution can be obtained using the equation i = k1v + k2v1/2, where k1v and k2v1/2 represent the capacitive contribution and ion diffusion, respectively (Fig. S12, ESI†). As shown in Fig. 3i and S11d–f,† the capacitive contribution of MgAl-VOH is calculated to be 93% at a scan rate of 1.0 mV s−1, much larger than those of Mg-VOH (84%), Al-VOH (82%) and pure VOH (68%). In addition, MgAl-VOH also shows higher capacitive contribution (Fig. 3j) than the other three cathodes at different scan rates (Fig. S11g–i, ESI†). To explore the electrochemical reaction kinetics, GITT measurements were performed and similar zinc-ion storage mechanisms for MgAl-VOH, Mg-VOH, Al-VOH, and pure VOH were observed (Fig. 3k and S13, ESI†). Particularly, MgAl-VOH possesses a higher Zn2+ ion diffusion coefficient (DZn2+) (10−6–10−10 cm2 s−1) than pure VOH (10−9–10−11 cm2 s−1), suggesting its outstanding Zn2+ ion diffusion kinetics (Fig. 3l). Moreover, the charge transfer resistance extracted from the EIS of MgAl-VOH is smaller than those of Mg-VOH, Al-VOH, and pure VOH, indicating the fast charge transfer (Fig. S14, ESI†).
2.4. Zinc-ion storage mechanism of MgAl-VOH
The charge/discharge curves of AZIBs composed of MgAl-VOH cathodes is shown in Fig. 4a. As displayed in Fig. 4b and S15,†in situ XRD measurements were performed to clarify the structure evolution of the MgAl-VOH cathode during the charging and discharging processes. The (001) characteristic peak of MgAl-VOH first shifts to a higher angle and then returns to its initial position, corresponding to the de/intercalation of Zn2+ ions and electrostatic adsorption with the vanadium oxides. However, the (110) characteristic peak remains unchanged throughout the charging and discharging processes, strongly indicating the excellent structure stability of MgAl-VOH. To further analyse the structure evolution of MgAl-VOH, ex situ Raman characterization was also carried out, with the results shown in Fig. 4c. Two Raman characteristic peaks of VOH gradually disappeared during the discharging process; nevertheless, after charging to 1.4 V, these two peaks reappeared, manifesting the high reversibility of the MgAl-VOH electrode. Additionally, ex situ FTIR and EDS mapping results in Fig. S16 and S17† show the formation of Znx(OH)y(CF3SO3)2x−y·nH2O by-products, which suggests the co-de/intercalation mechanism of Zn2+/H+ ions in the MgAl-VOH cathode.21 | ||
| Fig. 4 The zinc-ion storage mechanism of MgAl-VOH. (a) Charging and discharging curves of MgAl-VOH. (b) In situ XRD patterns of MgAl-VOH during the charging and discharging processes. The unchanged (110) characteristic peak indicates the high structure stability of MgAl-VOH. (c) Ex situ Raman spectra of MgAl-VOH during the charging and discharging processes. (d and e) XPS spectra of MgAl-VOH at different charging and discharging states. (f–h) SEM images of MgAl-VOH at different charging and discharging states, showing the high structural stability during the charging and discharging processes. (i) Schematic illustration of zinc-ion storage processes for MgAl-VOH and pure VOH. | ||
Ex situ XPS was employed to test the valence state and composition variation of MgAl-VOH, as displayed in Fig. 4d and e. Compared with the initial state, the proportions of V4+ and V3+ increase after discharging to 0.2 V, because the intercalation of Zn2+ ions results in the partial reduction of V5+. Nevertheless, after charging to 1.4 V, the valence state of V returns to its initial state, reconfirming the remarkable reversibility of MgAl-VOH. In addition, the appearance of Zn 2p1/2 and 2p3/2 characteristic peaks indicates the intercalation of Zn2+ ions. SEM and EDS measurements were then carried out to visualize the morphology evolution of MgAl-VOH (Fig. 4f–h and S17–S19, ESI†). The nanobelt feature of MgAl-VOH is clearly observed in the initial state and after charging to 1.4 V, indicating its robust atomic structure, which is also confirmed by the uniform distribution of Mg and Al elements at different charging/discharging states (Fig. 4f, h, S18 and S19, ESI†). However, after discharging to 0.2 V, the surfaces of MgAl-VOH nanobelts were covered with by-products (Znx(OH)y(CF3SO3)2x−y·nH2O), which possess nanosheet morphologies, as shown in Fig. 4g and S17.† These results reconfirm the co-de/intercalation mechanism of Zn2+/H+ ions in MgAl-VOH. As illustrated in Fig. 4i, the pure VOH cathode is prone to the irreversible process of Zn2+/H+ co-de/intercalation, ultimately leading to structural collapse and electrode dissolution. In contrast, the co-intercalation of Mg2+ and Al3+ ions expands the interlayer spacing of VOH and stabilizes the VO bonds, which improves the performance of AZIBs.
2.5. DFT calculations
DFT calculations were carried out to further uncover the internal physical mechanism of high-performance zinc-ion storage fin MgAl-VOH, with the atomic models shown in Fig. S20.† The electronic structures of MgAl-VOH and pure VOH are displayed in Fig. 5a and b, respectively, where a higher density of states (DOS) is observed in MgAl-VOH compared to pure VOH, which should enhance the conductivity and accelerate the charge transfer. Furthermore, new energy levels are also detected in the band gap of VOH after intercalating Mg2+ and Al3+ ions, contributing to the electron transport. Fig. 5c reveals the differential charge densities of Zn2+ ion-intercalated MgAl-VOH and pure VOH, respectively. The transferred charge numbers between Zn2+ ions and O atoms are calculated to be 0.37 and 0.24 e− for MgAl-VOH and pure VOH, respectively, the larger transferred charge number indicates the higher adsorption capacity of MgAl-VOH towards Zn2+ ions. In addition, the lower migration barriers of Zn2+ ions in MgAl-VOH compared to pure VOH are clearly observed, as shown in Fig. 5d and S21,† suggesting fast reaction kinetics. The adsorption energy of Zn2+ ions in MgAl-VOH is calculated to be −0.835 eV, much lower than that in pure VOH (−0.591 eV), indicating that Zn2+ ions are more easily anchored in MgAl-VOH than in pure VOH, which is favourable for zinc-ion storage, as demonstrated in Fig. 5e. | ||
| Fig. 5 DFT calculations for clarifying the high zinc-ion storage performances of MgAl-VOH. (a and b) Electronic structures of MgAl-VOH and pure VOH. The high DOS indicates the high conductivity of MgAl-VOH. (c) Differential charge densities of MgAl-VOH and pure VOH. The yellow and blue regions represent the accumulation and depletion of electrons, respectively. (d) Migration energies of Zn2+ ions in MgAl-VOH and pure VOH. (e) Adsorption energies of Zn2+ ions in MgAl-VOH and pure VOH. | ||
3. Conclusions
We designed a facile one-step solvothermal strategy to synthesize bimetallic-ion intercalated vanadium oxides. The unique co-intercalation of Mg2+ and Al3+ ions expands the interlayer spacing and improves the electronic conductivity of vanadium oxide cathodes, significantly enhancing the ion/electron transport kinetics. Particularly, the bimetallic-ion intercalation stabilizes the VO bonds and restrains the dissolution of vanadium oxides. Accordingly, excellent specific capacity, high rate performance, and long cycling stability are obtained in the AZIBs composed of MgAl-VOH cathodes. These findings represent significant advances in boosting the zinc-ion storage performance by constructing bimetallic-ion co-intercalated cathode materials and enable the further practical applications.
4. Experimental section
4.1. Synthesis of MgAl-VOH nanobelts
0.5 g of V2O5 powder was added into 25 ml of anhydrous ethanol and then stirred to obtain the solution A. 0.05 g of Al(NO3)3 and 0.05 g of Mg(NO3)2 were added into 20 ml of deionized water and then stirred to achieve the solution B. Subsequently, solution B was added into solution A and then stirred for 30 minutes. The mixed solution was transferred into a 100 mL autoclave and kept for 24 hours at 160 °C. After the reaction was completed and cooled down to room-temperature, the products were filtered and washed with deionized water and anhydrous ethanol. Finally, the obtained MgAl-VOH samples were dried overnight at 60 °C. Using the same method, Mg-VOH, Al-VOH, and pure VOH were also achieved.4.2. Material characterization
XRD (Rigaku, using Cu Kα radiation in the 2θ range of 5–80°) was performed to investigate the phase structure evolution and interlayer spacing of MgAl-VOH. The chemical compositions of MgAl-VOH were analysed by Raman spectroscopy (Horiba, with 532 nm excitation), FTIR (Nicolet iS50), and XPS (ESCALAB250Xi, with an Al Kα excitation source). The morphologies and atomic structures of MgAl-VOH were characterized by SEM (Hitachi S-4800, with an acceleration voltage of 5 kV) and TEM (JEOL JEM-F200 and JEM-NEOARM, with an acceleration voltage of 200 kV). The crystal water and Mg/Al contents in MgAl-VOH were determined by TGA (Mettler-Toledo TGA/DSC 3+) and ICP-OES (Agilent 5110). The EXAFS measurements were carried out on the sample at the 5S1 X-ray absorption beamline of the Aichi Synchrotron Radiation Center. This beamline utilized a double-bounce channel-cut Si (111) monochromator for mono-beam X-ray absorption spectroscopy. The end-station was equipped with three ionization chambers and a seven-element SDD detector positioned after the sample for transmission and fluorescence mode X-ray absorption spectroscopy. The photon flux on the sample ranged from 3 × 1010–4 × 1010 photons per sec for X-ray energy from 5–9 keV in low energy mode. The photon flux on the sample ranged from 1 × 1011–2.2 × 1010 photons per sec for X-ray energy from 7–18 keV in normal energy mode. The photon flux on the sample ranged from 2.3 × 1010–5 × 109 photons per sec for X-ray energy from 17–22 keV in high energy mode.4.3. Electrochemical measurements
All the electrodes were prepared by mixing active materials (70 wt%), acetylene black (20 wt%), and polyvinylidene fluoride binder (10 wt%), with the mass loading set to 1.2 mg cm−2. The CR2025 coin cells were assembled using zinc metal, GF/D-Whatman glass fibre, and Ti foil as the anode, separator, and collector, respectively. 3.0 M Zn(CF3SO3)2 was used as the electrolyte. The galvanostatic charge and discharge cycling, GITT, and rate capability measurements were performed on a LAND CT2001A multichannel battery testing system with a voltage window of 0.2 to 1.4 V. The CV and EIS curves were recorded on a CHI 660D electrochemical workstation.4.4. Computational methods
The Vienna Ab Initio Package (VASP) was employed to perform DFT calculations46 within the generalized gradient approximation (GGA) using the Perdew, Burke, and Ernzerhof (PBE) formulation.47 The projected augmented wave (PAW) potentials were applied to describe the ionic cores,48 and the valence electrons were considered using a plane wave basis set with a kinetic energy cutoff of 450 eV. The partial occupancies of Kohn–Sham orbitals were treated using the Gaussian smearing method with a width of 0.1 eV. The electronic energy was considered self-consistent when the energy change was smaller than 10−5 eV. A geometry optimization was considered convergent when the force change was smaller than 0.05 eV Å−1. Grimme's DFT-D3 methodology was used to describe the dispersion interactions. The Brillouin zone integral was performed utilizing a 2 × 2 × 1 Monkhorst–Pack k point sampling. During structure optimization, the gamma point in the Brillouin zone was used for k-point sampling, and all atoms were allowed to relax. The migration of Zn2+ ions was determined by the nudged elastic band (NEB) method.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Author contributions
Yulin Jiang: conceptualization, investigation, data curation, visualization, writing – original draft, writing – review & editing. Xia Wen: investigation, formal analysis. Yinuo Li: investigation, formal analysis. Yuhang Li: investigation, formal analysis. Yanan Peng: investigation, formal analysis. Wang Feng: investigation, formal analysis. Xiaohui Li: investigation, formal analysis. Junbo Yang: investigation, formal analysis. Luying Song: investigation, formal analysis. Hang Sun: investigation, formal analysis. Ling Huang: investigation, formal analysis. Jianping Shi: conceptualization, methodology, supervision, validation, writing – review & editing. All authors agreed to the publication of the article.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (92164103), the National Key R&D Program of China (2021YFA1200800), the Natural Science Foundation of Hubei Province (2024AFA052), and the Fundamental Research Funds for the Central Universities (2042023kf0187). The authors would like to acknowledge the Center for Electron Microscopy at Wuhan University for their substantial support of the JEM-F200 and JEM-NEOARM. The authors would like to thank the Shiyanjia Lab (https://www.shiyanjia.com) for conducting the XPS analysis.References
- B. Dunn, H. Kamath and J.-M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS PubMed.
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- Y. L. Liang, H. Dong, D. Aurbach and Y. Yao, Nat. Energy, 2020, 5, 646–656 CrossRef CAS.
- Y. M. Sun, N. Liu and Y. Cui, Nat. Energy, 2016, 1, 16071 CrossRef CAS.
- Y. L. Liang and Y. Yao, Nat. Rev. Mater., 2023, 8, 109–122 CrossRef.
- S. W. D. Gourley, R. Brown, B. D. Adams and D. Higgins, Joule, 2023, 7, 1415–1436 CrossRef CAS.
- J. F. Parker, C. N. Chervin, I. R. Pala, M. Machler, M. F. Burz, J. W. Long and D. R. Rolison, Science, 2017, 356, 415–418 CrossRef CAS PubMed.
- L. Ma, M. A. Schroeder, O. Borodin, T. P. Pollard, M. S. Ding, C. S. Wang and K. Xu, Nat. Energy, 2020, 5, 743–749 CrossRef CAS.
- D. Kundu, B. D. Adams, V. Duffort, S. H. Vajargah and L. F. Nazar, Nat. Energy, 2016, 1, 16119 CrossRef CAS.
- F. Wang, O. Borodin, T. Gao, X. L. Fan, W. Sun, F. D. Han, A. Faraone, J. A. Dura, K. Xu and C. S. Wang, Nat. Mater., 2018, 17, 543–549 CrossRef CAS PubMed.
- X. X. Jia, C. F. Liu, Z. G. Neale, J. H. Yang and G. Z. Cao, Chem. Rev., 2020, 120, 7795–7866 CrossRef CAS PubMed.
- G. Z. Fang, J. Zhou, A. Q. Pan and S. Q. Liang, ACS Energy Lett., 2018, 3, 2480–2501 CrossRef CAS.
- F. Wan and Z. Q. Niu, Angew. Chem., Int. Ed., 2019, 131, 16508–16517 CrossRef.
- H. L. Pan, Y. Y. Shao, P. F. Yan, Y. W. Cheng, K. S. Han, Z. M. Nie, C. M. Wang, J. H. Yang, X. L. Li, P. Bhattacharya, K. T. Mueller and J. Liu, Nat. Energy, 2016, 1, 16039 CrossRef CAS.
- Q. Zhao, W. W. Huang, Z. Q. Luo, L. J. Liu, Y. Lu, Y. X. Li, L. Li, J. Y. Hu, H. Ma and J. Chen, Sci. Adv., 2018, 4, eaao1761 CrossRef PubMed.
- Y. L. Liang, Y. Jing, S. Gheytani, K.-Y. Lee, P. Liu, A. Facchetti and Y. Yao, Nat. Mater., 2017, 16, 841–848 CrossRef CAS PubMed.
- J. P. Xie, L. Ma, J. L. Li, X. Q. Yin, Z. R. Wen, Y. L. Zhong, C. W. Li, Y. Liu, Z. X. Shen, W. J. Mai, G. Hong and W. J. Zhang, Adv. Mater., 2022, 34, 2205625 CrossRef CAS PubMed.
- C. Xia, J. Guo, P. Li, X. X. Zhang and H. N. Alshareef, Angew. Chem., Int. Ed., 2018, 130, 4007–4012 CrossRef.
- L. L. Wang, K.-W. Huang, J. T. Chen and J. R. Zheng, Sci. Adv., 2019, 5, eaax4279 CrossRef CAS PubMed.
- Y. Kim, Y. Park, M. Kim, J. Lee, K. J. Kim and J. W. Choi, Nat. Commun., 2022, 13, 2371 CrossRef CAS PubMed.
- T. T. Lv, G. Y. Zhu, S. Y. Dong, Q. Q. Kong, Y. Peng, S. Jiang, G. X. Zhang, Z. L. Yang, S. Y. Yang, X. C. Dong, H. Pang and Y. Z. Zhang, Angew. Chem., Int. Ed., 2023, 135, e202216089 CrossRef.
- Z. H. Wang, Y. Song, J. Wang, Y. L. Lin, J. M. Meng, W. B. Cui and X.-X. Liu, Angew. Chem., Int. Ed., 2023, 135, e202216290 CrossRef.
- X. R. Wang, Y. L. Wang, A. Naveed, G. T. Li, H. W. Zhang, Y. Zhou, A. C. Dou, M. R. Su, Y. J. Liu, R. Q. Guo and C. C. Li, Adv. Funct. Mater., 2023, 33, 2306205 CrossRef CAS.
- N. Zhang, Y. Dong, M. Jia, X. Bian, Y. Y. Wang, M. D. Qiu, J. Z. Xu, Y. C. Liu, L. F. Jiao and F. Y. Cheng, ACS Energy Lett., 2018, 3, 1366–1372 CrossRef CAS.
- M. Liao, J. W. Wang, L. Ye, H. Sun, Y. Z. Wen, C. Wang, X. M. Sun, B. J. Wang and H. S. Peng, Angew. Chem., Int. Ed., 2020, 132, 2293–2298 CrossRef.
- Y. Xu, G. L. Fan, P. X. Sun, Y. Guo, Y. Y. Wang, X. J. Gu, L. M. Wu and L. Yu, Angew. Chem., Int. Ed., 2023, 62, e202303529 CrossRef CAS PubMed.
- D. Bin, W. C. Huo, Y. B. Yuan, J. H. Huang, Y. Liu, Y. X. Zhang, F. Dong, Y. G. Wang and Y. Y. Xia, Chem, 2020, 6, 968–984 CAS.
- Y. Liu, C. J. Lu, Y. T. Yang, W. S. Chen, F. Ye, H. L. Dong, Y. P. Wu, R. Z. Ma and L. F. Hu, Adv. Mater., 2024, 36, 2312982 CrossRef CAS PubMed.
- N. N. Liu, X. Wu, L. S. Fan, S. Gong, Z. K. Guo, A. S. Chen, C. Y. Zhao, Y. C. Mao, N. Q. Zhang and K. N. Sun, Adv. Mater., 2020, 32, 1908420 CrossRef CAS PubMed.
- M. Clites and E. Pomerantseva, Energy Storage Mater., 2018, 11, 30–37 CrossRef.
- L. L. Xing, C. Y. Zhang, M. Li, P. Hu, X. Y. Zhang, Y. H. Dai, X. L. Pan, W. Y. Sun, S. L. Li, J. X. Xue, Q. Y. An and L. Q. Mai, Energy Storage Mater., 2022, 52, 291–298 CrossRef.
- M.-Y. Ma, Y. Liu, J.-L. Yang, S.-Y. Li, M. Du, D.-H. Liu, Z.-L. Hao, J.-Z. Guo and X.-L. Wu, J. Colloid. Interf. Sci., 2024, 670, 174–181 CrossRef CAS PubMed.
- E. H. Wang, W. Xiang, R. Rajagopalan, Z. G. Wu, J. Yang, M. Z. Chen, B. H. Zhong, S. X. Dou, S. L. Chou, X. D. Guo and Y.-M. Kang, J. Mater. Chem. A, 2017, 5, 9833–9841 RSC.
- C. L. Xu, J. M. Zhao, Y.-A. Wang, W. B. Hua, Q. Fu, X. M. Liang, X. H. Rong, Q. Q. Zhang, X. D. Guo, C. Yang, H. Z. Liu, B. H. Zhong and Y.-S. Hu, Adv. Energy Mater., 2022, 12, 2200966 CrossRef CAS.
- Q. Zong, Y. L. Zhuang, C. F. Liu, Q. L. Kang, Y. Z. Wu, J. J. Zhang, J. Y. Wang, D. W. Tao, Q. L. Zhang and G. Z. Cao, Adv. Energy Mater., 2023, 13, 2301480 CrossRef CAS.
- C. F. Liu, Z. Neale, J. Q. Zheng, X. X. Jia, J. J. Huang, M. Y. Yan, M. Tian, M. S. Wang, J. H. Yang and G. Z. Cao, Energy Environ. Sci., 2019, 12, 2273–2285 RSC.
- M. H. Wu, C. Shi, J. W. Yang, Y. Zong, Y. Chen, Z. G. Ren, Y. X. Zhao, Z. Li, W. Zhang, L. Y. Wang, X. L. Huang, W. Wen, X. L. Li, X. Ning, X. C. Ren and D. M. Zhu, Adv. Mater., 2024, 36, 2310434 CrossRef CAS PubMed.
- M. Li, M. Z. Liu, Y. Y. Lu, G. D. Zhang, Y. Zhang, Z. Y. Li, Q. J. Xu, H. M. Liu and Y. G. Wang, Adv. Funct. Mater., 2024, 34, 2312789 CrossRef CAS.
- J.-J. Ye, P.-H. Li, H.-R. Zhang, Z.-Y. Song, T. J. Fan, W. Q. Zhang, J. Tian, T. Huang, Y. T. Qian, Z. G. Hou, N. Shpigel, L.-F. Chen and S. X. Dou, Adv. Funct. Mater., 2023, 33, 2305659 CrossRef CAS.
- K. Y. Zhu, T. Wu and K. Huang, Adv. Energy Mater., 2019, 9, 1901968 CrossRef CAS.
- F. Wan, Z. M. Hao, S. Wang, Y. X. Ni, J. C. Zhu, Z. W. Tie, S. S. Bi, Z. Q. Niu and J. Chen, Adv. Mater., 2021, 33, 2102701 CrossRef CAS PubMed.
- W. Shi, B. S. Yin, Y. Yang, M. B. Sullivan, J. Wang, Y.-W. Zhang, Z. G. Yu, W. S. V. Lee and J. M. Xue, ACS Nano, 2021, 15, 1273–1281 CrossRef CAS PubMed.
- J. D. Guo, J. X. Liu, W. B. Ma, Z. Y. Sang, L. C. Yin, X. Q. Zhang, H. Chen, J. Liang and D. A. Yang, Adv. Funct. Mater., 2023, 33, 2302659 CrossRef CAS.
- L. Yang, Y.-J. Zhu, F. L. Zeng, H.-P. Yu, L.-Y. Dong, J. C. Tao, G. He and H. Li, Energy Storage Mater., 2024, 65, 103162 CrossRef.
- X. R. Wang, Y. L. Wang, A. Naveed, G. T. Li, H. W. Zhang, Y. Zhou, A. C. Dou, M. R. Su, Y. J. Liu, R. Q. Guo and C. C. Li, Adv. Funct. Mater., 2023, 33, 2306205 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B:Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B:Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta05938g |
| This journal is © The Royal Society of Chemistry 2025 |