
Structural rule of heteroatom-modified single-atom catalysts for the CO2 electroreduction reaction†
Xinyuan
Sui
 ,
Haiyang
Yuan
* and
Yu
Hou
*
,
Haiyang
Yuan
* and
Yu
Hou
*
Key Laboratory for Ultrafine Materials of Ministry of Education, Shanghai Engineering Research Center of Hierarchical Nanomaterials, School of Materials Science and Engineering, East China University of Science and Technology, Shanghai 200237, China. E-mail: hyyuan@ecust.edu.cn; yhou@ecust.edu.cn
First published on 20th November 2024
Abstract
The carbon dioxide electroreduction reaction (CO2RR) has emerged as a viable strategy to address pressing energy and environmental challenges. Single-atom catalysts (SACs) are of particular interest for the CO2RR due to their maximized atom utilization. The incorporation of heteroatoms as ligands is a common strategy to modify the geometric and electronic structures of metal centers to enhance performance. Here, we employed density functional theory study to investigate nitrogen-coordinated SACs with various heteroatom ligands and elucidated the structural rule of SACs on the CO2RR. The results show that the stability of SACs exhibits a volcano-shaped trend as a function of the ligand radius, with both excessively large and small radius compromising stability, and the planar structural SACs exhibit relatively better stabilities than the raised ones. Although the raised structural SACs have better ability to activate CO2 for the tip effect, they also hinder CO desorption and facilitate H+ adsorption, leading to relatively poor CO2RR activity and selectivity (vs. the HER). In contrast, planar-structured SACs generally show better activity and CO2RR selectivity, where promoting the CO2 activation step is necessary. This work provides fundamental insights into the structure-dependence of SACs and offers guidance for designing SACs for the CO2RR or other reactions.
1. Introduction
The electrochemical CO2 reduction reaction (CO2RR) presents a promising pathway for converting CO2 into valuable fuels and chemical feedstocks under ambient conditions.1–5 The CO2RR is a multiple electron-transfer process, which can generate a range of products, such as carbon monoxide (CO),6,7 formic acid,8 ethylene,9 ethanol,10 and others. Among them, CO, which has high energy efficiency and selectivity in the CO2RR, serves as one of the most promising products in the CO2RR.11 Numerous catalysts have been developed for CO2RR to CO, such as Ag (or Au, Cu, and Ni)-based catalysts.12–16 Over the last decade, single-atom catalysts (SACs) have garnered significant attention due to their maximum atom-utilization efficiency by downsizing active sites to the atomic level, which can exhibit impressive efficiency for CO2RR to CO.17 Nevertheless, SACs still face great challenges in terms of stability and activity during the CO2RR. How to design effective SACs for the CO2RR has become a prominent topic in the field of electrocatalysis.The coordination environment of metal center of SACs is generally considered to play a key role in determining their catalytic performance.18–22 In SACs, nitrogen (N) atoms are commonly employed as ligands to anchor the metal center, due to their ability to hybridize the 2p orbital with the d orbital of the metal.23,24 The unique configuration of SACs, where a metal center is coordinated by four N atoms within the carbon matrix (denoted as M-N4), has been demonstrated to be effective for the CO2RR.25 Recently, numerous studies have focused on modifying the coordination environment of SACs by incorporating heteroatoms [e.g., boron (B), oxygen (O), and phosphorus (P)] to manipulate the electronic structure of the active metal center, with the aim of enhancing the stability and catalytic activity of SACs for CO2RR to CO.26–29 The substitution of the N-ligands of M-N4 SACs with other heteroatoms can also change the geometric structure due to the space effect; especially when the ligand atoms have a large radius, this can elevate the metal center, facilitating the adsorption of small molecules or key intermediates and thus modulating CO2RR performance.30 For instance, the larger size of the S atom protrudes the Fe center from the first shell coordination plane Fe-S1N3, optimizing the charge transfer between the Fe center and intermediates independently and thereby lowering the energy barrier of the CO2RR.31 Although some evidence has suggested that the introduction of other heteroatom ligands into N-coordinate SACs can effectively regulate the electronic or geometric structure of the metal center, helping to improve CO2RR performance, there is no general consensus on the structure–performance relation of the metal center of these SACs for the CO2RR at the atomic level,32 which is important for the design of superior SACs. It remains a significant challenge to clearly identify the atomic level structures of various SACs with different metal centers and heteroatom ligands, using advanced experimental characterization technologies.
In this study, we explored the CO2RR to CO on SACs by virtue of the density functional theory (DFT) calculations, where the metal center is anchored on the XN3-coordinated graphene (denoted as M-XN3, where X represents the heteroatom ligand, X = B, C, N, O and P). The results show that the planar and raised metal centers are typical geometric structures for SACs, which can result in the extremely different chemical properties of metal centers. Based on these M-XN3 SACs, we explored the structure dependence on their stability, CO2RR activity and selectivity and provided some fundamental understanding of the regularity of M-XN3 SACs for the CO2RR.
2. Computational method
All spin-polarized density functional theory (DFT) calculations were executed utilizing the Vienna ab initio simulation package (VASP),33,34 with the project-augmented wave (PAW) method. The exchange-correlation functional was described by the Perdew–Burke–Ernzerhof (PBE) functional35 within the generalized gradient approximation (GGA).36 The cutoff energy for the plane wave basis of 450 eV was set. The graphene model size is 9.84 Å × 9.84 Å, in which a vacuum layer of 15 Å was applied to separate each periodic unit cell. During the structural optimization, all atoms in the model were relaxed and the Brillouin zone was sampled using 3 × 3 × 2 Monkhorst–Pack mesh k-points. As the force of each atom in the model is less than 0.05 eV Å−1, the optimization process is stopped. To more accurately describe the interactions between d orbital electrons, the DFT + U method was used.37 The specific U values for different metal elements are shown in Table S1.† To describe the van der Waals interactions, the empirical correction in Grimme's scheme was used. The adsorption energy of the adsorbate was calculated with the equations: Eads[X] = E[X/sur] − E[sur] − E[X], where E[X/sur], E[sur] and E[X] are the energies of the optimized surface with the adsorbed X molecule, the clean surface, and the gaseous X molecule, respectively. The more negative Eads[X] indicates the stronger adsorption. The Gibbs free energy change (ΔG) is calculated as: ΔG = ΔH + ΔEZPE − TΔS. ΔH is the enthalpy change of the reaction, resulting from DFT calculations; ΔEZPE and TΔS are the energy differences of the zero point energy and entropic contributions from the vibrational frequencies; for the gaseous molecules (CO2, CO and H2), the entropy contributions of gaseous molecules (TΔS) are obtained with the experimental values.3. Results and discussion
3.1 Model and stability of SACs
We focused on the representative N4-coordinated metal single-atom catalysts (M-N4) anchored on nitrogen-doped graphene as the basic model, which are popularly utilized in both experimental and theoretical studies. Several transition metal elements from different periods of the periodic table of elements experimentally used in the CO2RR, including Mn,38 Fe,39 and Co40 for 3d metals, Mo41 and Ru42 for 4d metals, and W43–45 for 5d metals, were selected as metal centers (M). Here, a heteroatom (X) was introduced into M-N4 to construct M-XN3 (X = B, C, N, O and P), as shown in Fig. 1a. The detailed radii of the corresponding metal atom and heteroatom are shown in Fig. 2a. Accordingly, we can obtain a series of M-XN3 catalyst models. After optimizing the structures of these M-XN3 catalyst models, we counted the height (denoted as H, see the calculation method in Fig. S1†) of the metal center above the graphene surface, as shown in Fig. 1b. The optimized structure of each M-XN3 is shown in Fig. S2.† It can be found that these M-XN3 catalysts can be structurally classified into two types (Fig. 1c): (i) the raised-structured M-XN3, where the metal center M has been pushed out of the graphene layer (H > ∼0.5 Å); (ii) the planar-structured M-XN3, where the metal center basically stays at the graphene surface with a small H (<∼0.5 Å). A metal center with a large atomic radius generally shows raised M-XN3 structures (Fig. 1b), such as M = W, Ru and Mo. To further illustrate the relationship between the protrusion of the metal center and the radius of the ligand X atom, we scaled the H value of different SACs with the radius of the corresponding ligand X atom; there are relatively good linear relationships between them, as shown in Fig. S3.† Their positive slopes illustrate that for a given metal center, with the increase of the radius of the ligand X atom, the protrusion of the metal center basically becomes more pronounced (i.e., larger H). In particular, in M-PN3, where the P atom has the largest radius, the protrusion of the metal center is most prominent and reaches its maximum height.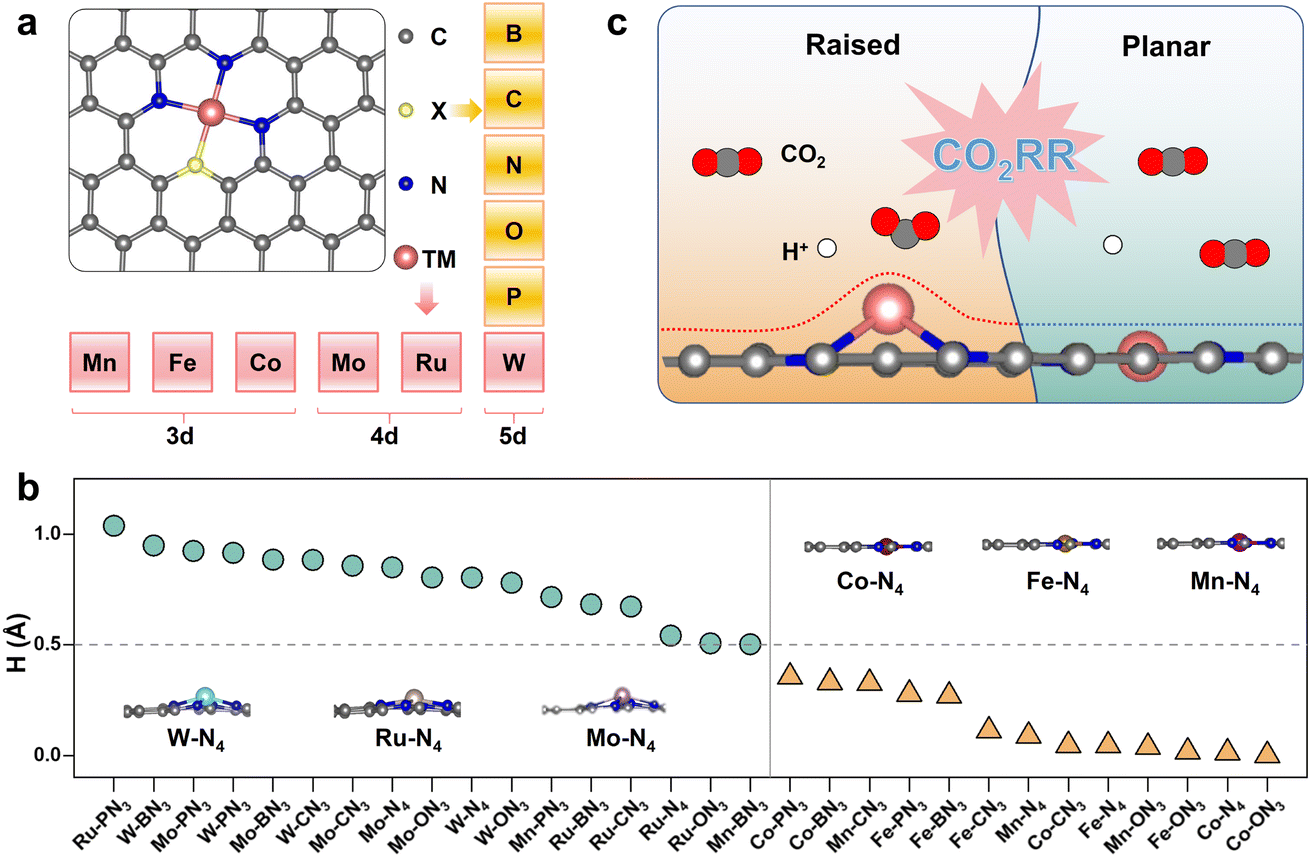 | ||
| Fig. 1 (a) Structural model of an M-XN3 catalyst, in which a heteroatom (X) is introduced into N4-coordinated SACs anchored on nitrogen-doped graphene. The blue, gray and pink balls represent nitrogen, carbon and metal atoms, respectively, and the yellow ball represents the heteroatom. Several metal centers (M = Mn, Fe, Co, Mo, Ru and W) and heteroatoms (X = B, C, N, O and P) were chosen to construct M-XN3 models. (b) Height (i.e., H) of the metal center above the graphene surface. (c) Schematic models of the raised and planar M-XN3. | ||
 | ||
| Fig. 2 (a) Distribution diagram of the atomic radius (Å) of the metal center and the ligand heteroatom.46 (b) Dissolution potentials (Udiss) of each M-XN3 as a function of the radius of the ligand heteroatom. (c) Integrated crystal orbital Hamilton population (ICOHP) of the M–X bond in M-XN3 SACs. | ||
The stability of SACs remains a primary challenge for their practical application in catalytic processes, as it directly determines the service life of SACs.47 In this position, the dissolution potentials (Udiss) of M-XN3 catalysts as an indicator of stability in the electrochemical environment were explored first (see the detailed calculation method in Note S1†). For each metal center, there is generally a volcano-shaped trend for Udiss as a function of the radius of the heteroatom (Fig. 2b). A higher Udiss means better stability of M-XN3 in the CO2RR. Specifically, as the radius of the heteroatom increases (from 0.66 Å for the O atom to 0.76 Å for the C atom), Udiss of each M-XN3 generally increases and reaches the highest value at the position of the C incorporation. Udiss decreases as the radius of the heteroatom further increases (from 0.76 Å for the C atom to 1.07 Å for the P atom). It is evident that the ligand heteroatom with either too small or too large a radius negatively affects the stability of SACs, and selecting the ligand heteroatom in SACs with an atomic radius close to that of the C atom may contribute to the stability of SACs.
Structural analysis indicates that this difference in stability for a given M-XN3 incorporated by heteroatoms with different atomic radii may be related to the compatibility between the ligand atom and the carbon graphene substrate. The radius of the P atom is 1.07 Å, which is much larger than that of the C atom in the graphene substrate of r = 0.76 Å, making P incompatible with the lattice of the graphene substrate. The incorporation of the ligand atom with a large radius (e.g., P atom) can strengthen the M–X bonds significantly, as demonstrated by the integrated crystal orbital Hamilton population (ICOHP) in Fig. 2c. A more negative ICOHP means stronger bond strength.48 Interestingly, although M-PN3 generally has a stronger M–P bond than other M–X bonds, the M-PN3 structures have a decreased Udiss, implying poor stability. This instability arises because the heteroatom with a large radius could push the metal center out of the graphene plane, causing the metal center to detach from the anchoring site and potentially form clusters.49 Therefore, the geometric structure of the metal center (i.e., raised or planar) on SACs could be an important factor in determining their stability.
In contrast, the incorporation of a heteroatom with a smaller radius (e.g., the O atom) weakens the M–X bond, as shown by the relatively more positive ICOHP values in Fig. 2c, and thus decreases the stability of M-XN3. For example, for the O atom (r = 0.66 Å), which is smaller than the C atom in the graphene substrate, its incorporation results in the weakest M–X bond in all M-ON3 SACs. In general, a ligand heteroatom with too large or too small a radius can lead to the large structural change of the metal center (i.e., raised one) or weaken the M–X bond, thereby affecting the stability of SACs. The N atom and C atoms with a similar atomic radius could be the most effective ligands for stabilizing SACs at the carbon graphene substrate, which have been widely used in experiments.50
3.2 Activity and selectivity of SACs for the CO2RR
In addition to the stability, the catalytic activity of SACs is another measure of their effectiveness in the CO2RR. The adsorption and activation of CO2 is a key elementary process in the CO2RR, because the CO2 molecule is chemically inert with a linear structure and a strong C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond.51 Here, we first examined the adsorption energy of CO2 (denoted as Eads[CO2]) on different M-XN3 SACs and presented them in Fig. 3a. The more negative Eads[CO2] means the better activation of CO2. The bending of the O–C–O bond angle and the stretching of the CO bond serve as important indicators for CO2 activation. From Fig. 3a, we can find that Eads[CO2] on M-XN3 with a raised metal center is basically more negative than that on the planar M-XN3, implying the relatively good adsorption and activation capacities of the raised SACs for CO2. From the adsorption structures of CO2 on different M-XN3SACs, the linear CO2 molecule becomes more bent and the CO bonds are elongated more obviously on the raised metal center of SACs (Fig. S5†). On some M-XN3 SACs with a planar metal center, the CO2 molecule even remains linear, indicating their very poor ability to activate CO2. Therefore, it can be expected that the CO2 adsorption and activation can be better facilitated on the raised SACs compared with the planar ones.
O bond.51 Here, we first examined the adsorption energy of CO2 (denoted as Eads[CO2]) on different M-XN3 SACs and presented them in Fig. 3a. The more negative Eads[CO2] means the better activation of CO2. The bending of the O–C–O bond angle and the stretching of the CO bond serve as important indicators for CO2 activation. From Fig. 3a, we can find that Eads[CO2] on M-XN3 with a raised metal center is basically more negative than that on the planar M-XN3, implying the relatively good adsorption and activation capacities of the raised SACs for CO2. From the adsorption structures of CO2 on different M-XN3SACs, the linear CO2 molecule becomes more bent and the CO bonds are elongated more obviously on the raised metal center of SACs (Fig. S5†). On some M-XN3 SACs with a planar metal center, the CO2 molecule even remains linear, indicating their very poor ability to activate CO2. Therefore, it can be expected that the CO2 adsorption and activation can be better facilitated on the raised SACs compared with the planar ones.
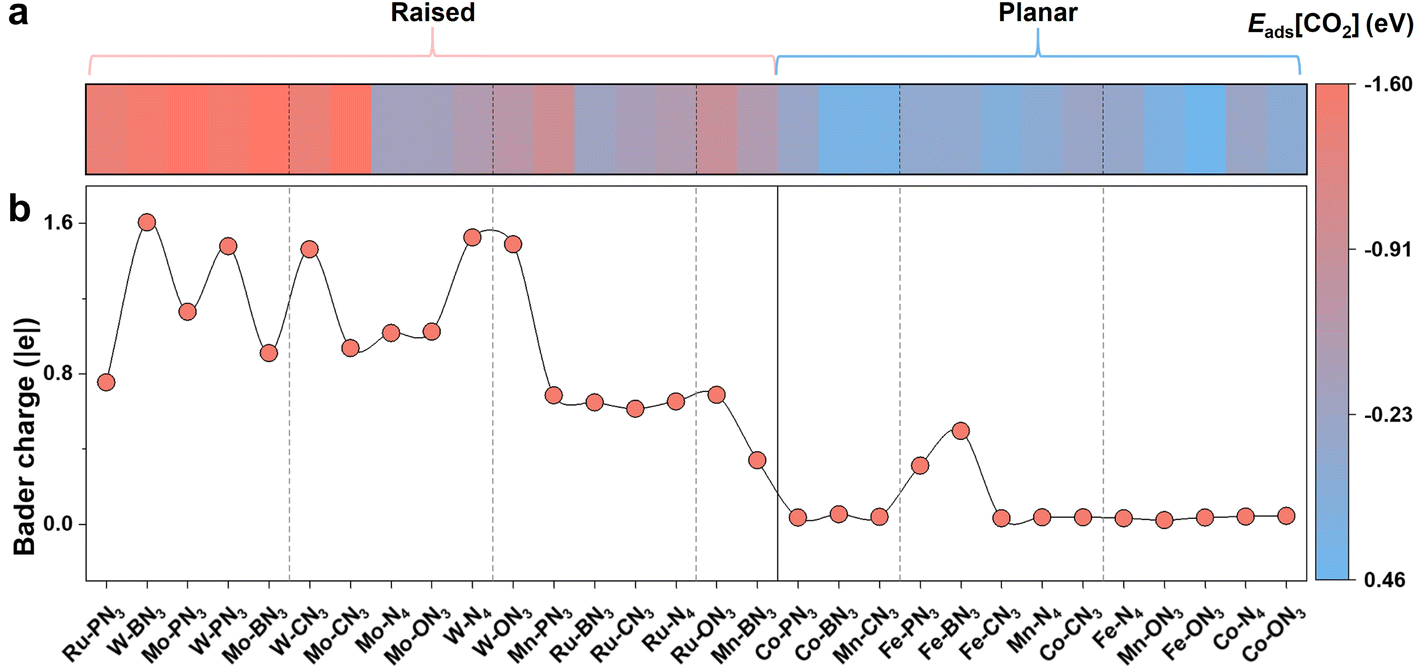 | ||
| Fig. 3 (a) Adsorption energy of the CO2 molecule (Eads[CO2]) on different M-XN3 SACs. (b) Electrons transferred from the metal center to the CO2 molecule upon CO2 adsorbing on different M-XN3 SACs. | ||
To gain a deeper insight into the different activation capacities of the raised and planar M-XN3 SACs for CO2, we calculated the charge density difference (CDD) upon CO2 adsorption on different M-XN3 structures. As shown in Fig. 3b, one can see that the raised metal center of M-XN3 can generally donate more electrons to the CO2 molecule via the C atom and activate the C–O bonds, leading to the remarkable bending of the O–C–O bond angle and stretching of the C–O bonds, while, on the planar M-XN3 catalysts, the metal center usually contributes few electrons to CO2, implying a limited ability to activate CO2. Furthermore, the projected crystal orbital Hamilton population (pCOHP) analysis was performed to describe the interaction between the CO2 molecule and the commonly used Fe center on different Fe-XN3 SACs as examples, aiming to further understand the underlying mechanism of CO2 activation on the raised or planar M-XN3. From Fig. 4, it can be seen that the greater electron-donating capacity of the raised metal center can contribute more electrons to the filling of the bonding state of the M–CO2 bond, strengthening the M–CO2 bond and thereby promoting the CO2 activation. This phenomenon can be attributed to the tip effect,52 where the directional aggregation of electrons in the raised metal center is promoted, thus facilitating more effective CO2 activation.
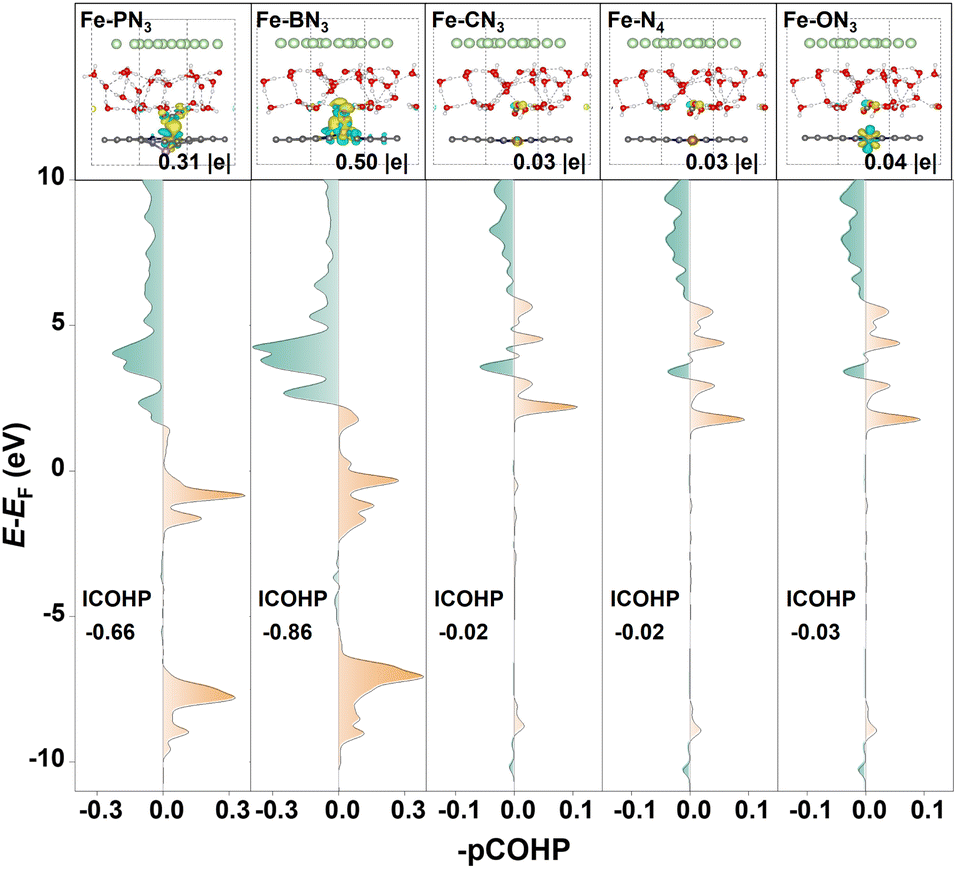 | ||
| Fig. 4 Charge density difference (CDD) for the CO2 adsorption on the commonly used Fe center of different Fe-XN3 SACs, and the corresponding projected crystal orbital Hamilton population (pCOHP) between the CO2 molecule and the Fe center. The yellow and blue regions in the CDD denote the charge accumulation and depletion with an isosurface level of 0.003 e bohr−3. | ||
Therefore, the raised M-XN3 SACs are anticipated to be more effective substrates for CO2 adsorption and activation compared to the planar one, on which the metal center exhibits the tip effect, accumulating more electrons and thereby facilitating the adsorption and activation of the CO2 molecule. Of course, this tip effect can also promote the adsorption of other key intermediates (e.g., *COOH and *CO) in CO2RR to CO; as shown in Fig. S6,† both *COOH and *CO on raised M-XN3 generally exhibit a stronger adsorption energy than that on planar ones, which also have important effects on the CO2RR (see the detailed discussion in the latter).
We further explored the Gibbs free energy change of the following CO2 protonation and CO desorption steps to assess the CO2RR activity on different M-XN3 SACs using the computational hydrogen electrode (CHE) model53 (see the calculation details in Note S2†). The maximum Gibbs free energy (ΔGmax[CO2RR]) of the reaction steps was used to evaluate the intrinsic activities of catalysts for the CO2RR, which is an important indicator to identify the rate-determining step. By comparing the Gibbs free energy change of each reaction step in the CO2RR to obtain ΔGmax[CO2RR], as shown in Fig. 5a, it is obvious that the primary rate-determining step of the CO2RR on the raised M-XN3 is basically located at the CO desorption step (i.e., *CO → * + CO). This mainly results from the stronger binding strength of the raised metal center compared to the planar one, which significantly hinders the CO desorption. Conversely, for planar M-XN3, the rate-determining step basically changes to the *CO2 adsorption and activation (i.e., * + CO2 → *CO2). The planar metal center provides weaker binding ability, thus limiting the CO2 activation and protonation process. Comparing ΔGmax[CO2RR] on different M-XN3SACs, we can find that the planar M-XN3 SACs basically exhibit a lower ΔGmax[CO2RR] than the raised ones (see the blue line in Fig. 5b), implying the relatively better catalytic activity of the planar M-XN3 SACs for CO2RR to CO.
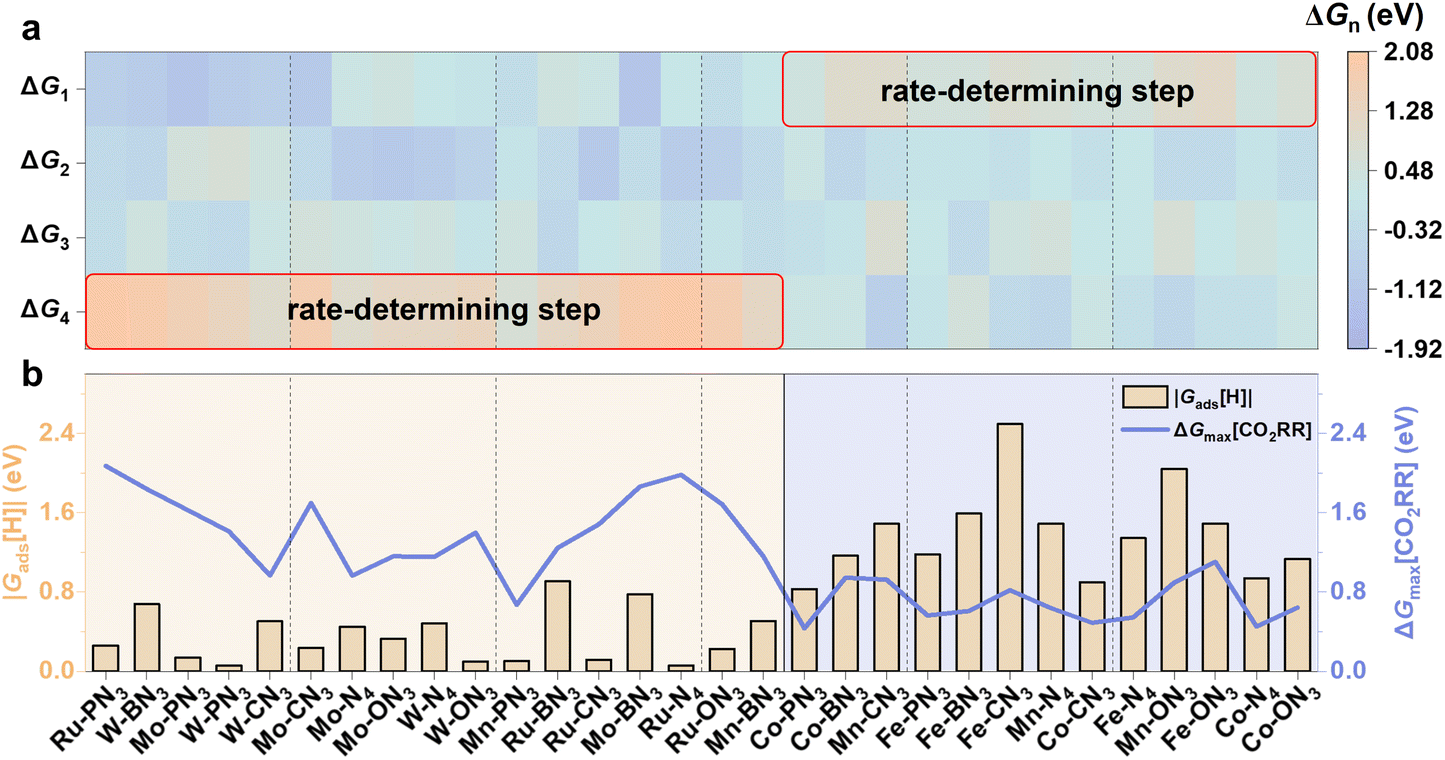 | ||
| Fig. 5 (a) Gibbs free energies (ΔGn) of the elementary steps in the CO2RR. ΔG1: CO2 + * → *CO2, ΔG2: *CO2 + H+/e− → *COOH, ΔG3: *COOH + H+/e− → *CO + H2O, and ΔG4: *CO → * + CO. (b) Distribution of ΔGmax[CO2RR] and |Gads[H]| on different M-XN3 SACs. | ||
During the CO2RR, the hydrogen evolution reaction (HER) serves as a common competitive side reaction, which significantly influences the efficiency of the CO2RR.54 In SACs, which have only one active site, once the metal center is covered by the H species to proceed with the HER, it is poisoned, thus hindering the CO2RR. Gads[H] is a good indicator to evaluate the activity of catalysts for the HER. As |Gads[H]| is closer to 0 eV, catalysts could have good activity for the HER to generate H2, which is undesirable for the CO2RR.55,56 To further illustrate the catalytic selectivity of the CO2RR (vs. the HER) on different M-XN3 SACs, we compared the absolute value of Gads[H] (i.e., |Gads[H]|) and ΔGmax[CO2RR] on different M-XN3 SACs (Fig. 5b). When the |Gads[H]| value is larger than ΔGmax[CO2RR], the catalyst is more likely to favor the CO2RR over the HER. From Fig. 5b, it can be found that |Gads[H]| on the raised M-XN3 is generally lower than ΔGmax[CO2RR], indicating the easier HER process compared to the CO2RR, i.e., the poor CO2RR selectivity of the raised M-XN3. A similar result was also reported in previous study.27 Hence, on most of the raised M-XN3 SACs, the chance of the HER or hydrogen poisoning is more. In contrast, on the relatively planar M-XN3 SACs, ΔGmax[CO2RR] is generally lower than |Gads[H]|, meaning their better adjustability for CO2RR selectivity, which can be realized by modifying the ligand.57 Therefore, from this perspective of the CO2RR selectivity, planar structures are preferable for designing SACs.
Combined with the stability and activity analysis above, it can be found that SACs exhibit a strong structure dependence in terms of stability, CO2RR activity and selectivity (vs. the HER). Raised or planar metal centers serve as typical active sites in SACs, in which the stability of a given metal center follows a volcano-shaped trend as a function of the radius of the ligand heteroatom. The metal center with a large atomic radius and the ligand atom with either too large or too small a radius are not conducive to stabilizing SACs. Too large a radius of the metal center or the ligand atoms tends to push the metal center out of the anchoring graphene plane, easily leading to cluster formation and thus poor stability; too small a radius of the ligand atom can weaken the bond between the metal center and ligand heteroatom, diminishing the stability. Although raised M-XN3 SACs generally exhibit worse stability than the planar ones, they show a superior ability to activate CO2, due to the tip effect resulting from the directional aggregation of electrons in the raised metal center. However, this also strengthens the CO desorption and H+/e− adsorption, resulting in relatively worse CO2RR activity and selectivity (vs. the HER) compared to the planar M-XN3 SACs. In a comprehensive comparison, the planar M-XN3 could be more suitable for designing SACs for CO2RR to CO, where however promoting the rate-determining CO2 activation step is necessary.
4. Conclusions
In summary, we conducted a theoretical study toward the structure–performance relation of SACs after introducing different heteroatom ligands. We discovered that SACs generally exhibit two types of geometric structures: raised and planar metal centers. Interestingly, the stabilities of all these SACs follow a volcano-shaped trend with the change of the radius of the ligand. The ligands with too large or too small atomic radii are not conducive to the stability of SACs. The planar SACs exhibit a better stability compared with the raised ones. In terms of catalytic activity, although the raised SACs possess superior ability to activate CO2 for the tip effect, they limit the CO desorption and strengthen H+/e− adsorption, thus leading to their worse CO2RR activity and selectivity (vs. the HER). In comparison, the planar SACs generally exhibit better CO2RR activity and selectivity. Planar M-XN3 could be more suitably used to design SACs for CO2RR to CO, on which the rate-determining CO2 activation is necessarily promoted. We believe that this work can provide a fundamental theoretical understanding of the structure dependence of SACs and give some guidance on the design or modification of M-XN3 SACs for the CO2RR or even other reactions.Data availability
All relevant data are within the manuscript and its ESI†Author contributions
X. Y. S. performed the DFT calculations and data analysis and wrote the manuscript. Y. H. and H. Y. directed the whole research. All the authors participated in editing the manuscript and contributed their efforts to the discussion.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Ten Thousand Talent Program for Young Top-notch Talent, the National Natural Science Fund for Excellent Young Scholars (52022030), the National Natural Science Foundation of China (22472053 and 51972111), the Shanghai Municipal Natural Science Foundation (23ZR1416800 and 22ZR1418000), the “Dawn” Program of the Shanghai Education Commission (22SG28), Shanghai Fundamental Research Special Zone Projects (22TQ1400100-5), the Innovation Program of Shanghai Municipal Education Commission (E00014), the Fundamental Research Funds for the Central Universities (JKM01221621 and JKM01221678) and the Shanghai Engineering Research Center of Hierarchical Nanomaterials (18DZ2252400).Notes and references
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- W. Liu, P. Zhai, A. Li, B. Wei, K. Si, Y. Wei, X. Wang, G. Zhu, Q. Chen, X. Gu, R. Zhang, W. Zhou and Y. Gong, Nat. Commun., 2022, 13, 1877 CrossRef CAS PubMed.
- P. Wang, S. Meng, B. Zhang, M. He, P. Li, C. Yang, G. Li and Z. Li, J. Am. Chem. Soc., 2023, 145, 26133–26143 CrossRef CAS PubMed.
- C. Xu, Y. Dong, H. Zhao and Y. Lei, Adv. Funct. Mater., 2023, 33, 2300926 CrossRef CAS.
- N. Han, P. Ding, L. He, Y. Li and Y. Li, Adv. Energy Mater., 2020, 10, 1902338 CrossRef CAS.
- A. S. Varela, N. R. Sahraie, J. Steinberg, W. Ju, H.-S. Oh and P. Strasser, Angew. Chem., Int. Ed., 2015, 54, 10758–10762 CrossRef CAS PubMed.
- T. Möller, W. Ju, A. Bagger, X. Wang, F. Luo, T. N. Thanh, A. S. Varela, J. Rossmeisl and P. Strasser, Energy Environ. Sci., 2019, 12, 640–647 RSC.
- K. R. Phillips, Y. Katayama, J. Hwang and Y. Shao-Horn, J. Phys. Chem. Lett., 2018, 9, 4407–4412 CrossRef CAS PubMed.
- Y. Chen, Z. Fan, J. Wang, C. Ling, W. Niu, Z. Huang, G. Liu, B. Chen, Z. Lai, X. Liu, B. Li, Y. Zong, L. Gu, J. Wang, X. Wang and H. Zhang, J. Am. Chem. Soc., 2020, 142, 12760–12766 CrossRef CAS PubMed.
- W. Xia, Y. Xie, S. Jia, S. Han, R. Qi, T. Chen, X. Xing, T. Yao, D. Zhou, X. Dong, J. Zhai, J. Li, J. He, D. Jiang, Y. Yamauchi, M. He, H. Wu and B. Han, J. Am. Chem. Soc., 2023, 145, 17253–17264 CrossRef CAS PubMed.
- H.-Y. Jeong, M. Balamurugan, V. S. K. Choutipalli, E.-s. Jeong, V. Subramanian, U. Sim and K. T. Nam, J. Mater. Chem. A, 2019, 7, 10651–10661 RSC.
- N. Zhang, X. Zhang, L. Tao, P. Jiang, C. Ye, R. Lin, Z. Huang, A. Li, D. Pang, H. Yan, Y. Wang, P. Xu, S. An, Q. Zhang, L. Liu, S. Du, X. Han, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2021, 60, 6170–6176 CrossRef CAS PubMed.
- X. Wang, P. Ou, J. Wicks, Y. Xie, Y. Wang, J. Li, J. Tam, D. Ren, J. Y. Howe, Z. Wang, A. Ozden, Y. Z. Finfrock, Y. Xu, Y. Li, A. S. Rasouli, K. Bertens, A. H. Ip, M. Graetzel, D. Sinton and E. H. Sargent, Nat. Commun., 2021, 12, 3387 CrossRef CAS PubMed.
- Q. Lei, L. Huang, J. Yin, B. Davaasuren, Y. Yuan, X. Dong, Z.-P. Wu, X. Wang, K. X. Yao, X. Lu and Y. Han, Nat. Commun., 2022, 13, 4857 CrossRef CAS PubMed.
- K. Jiang, S. Siahrostami, T. Zheng, Y. Hu, S. Hwang, E. Stavitski, Y. Peng, J. Dynes, M. Gangisetty, D. Su, K. Attenkofer and H. Wang, Energy Environ. Sci., 2018, 11, 893–903 RSC.
- H. B. Yang, S.-F. Hung, S. Liu, K. Yuan, S. Miao, L. Zhang, X. Huang, H.-Y. Wang, W. Cai, R. Chen, J. Gao, X. Yang, W. Chen, Y. Huang, H. M. Chen, C. M. Li, T. Zhang and B. Liu, Nat. Energy, 2018, 3, 140–147 CrossRef CAS.
- M. Li, H. Wang, W. Luo, P. C. Sherrell, J. Chen and J. Yang, Adv. Mater., 2020, 32, 2001848 CrossRef CAS PubMed.
- J. Leverett, R. Daiyan, L. Gong, K. Iputera, Z. Tong, J. Qu, Z. Ma, Q. Zhang, S. Cheong, J. Cairney, R. S. Liu, X. Lu, Z. Xia, L. Dai and R. Amal, ACS Nano, 2021, 15, 12006–12018 CrossRef CAS PubMed.
- L. Jiao, J. Li, L. L. Richard, Q. Sun, T. Stracensky, E. Liu, M. T. Sougrati, Z. Zhao, F. Yang, S. Zhong, H. Xu, S. Mukerjee, Y. Huang, D. A. Cullen, J. H. Park, M. Ferrandon, D. J. Myers, F. Jaouen and Q. Jia, Nat. Mater., 2021, 20, 1385–1391 CrossRef CAS PubMed.
- J. N. Tiwari, S. Sultan, C. W. Myung, T. Yoon, N. Li, M. Ha, A. M. Harzandi, H. J. Park, D. Y. Kim, S. S. Chandrasekaran, W. G. Lee, V. Vij, H. Kang, T. J. Shin, H. S. Shin, G. Lee, Z. Lee and K. S. Kim, Nat. Energy, 2018, 3, 773–782 CrossRef CAS.
- J. Su, L. Yu, B. Han, F. Li, Z. Chen and X. C. Zeng, J. Phys. Chem. Lett., 2024, 15, 8600–8607 CrossRef CAS PubMed.
- L. Yu, F. Li, J. Huang, B. G. Sumpter, W. E. Mustain and Z. Chen, ACS Catal., 2023, 13, 9616–9628 CrossRef CAS.
- Z. Li, R. Wu, S. Xiao, Y. Yang, L. Lai, J. S. Chen and Y. Chen, Chem. Eng. J., 2022, 430, 132882 CrossRef CAS.
- S. Liu, H. B. Yang, S.-F. Hung, J. Ding, W. Cai, L. Liu, J. Gao, X. Li, X. Ren, Z. Kuang, Y. Huang, T. Zhang and B. Liu, Angew. Chem., Int. Ed., 2020, 59, 798–803 CrossRef CAS PubMed.
- A. S. Varela, W. Ju, A. Bagger, P. Franco, J. Rossmeisl and P. Strasser, ACS Catal., 2019, 9, 7270–7284 CrossRef CAS.
- Y. Dai, H. Li, C. Wang, W. Xue, M. Zhang, D. Zhao, J. Xue, J. Li, L. Luo, C. Liu, X. Li, P. Cui, Q. Jiang, T. Zheng, S. Gu, Y. Zhang, J. Xiao, C. Xia and J. Zeng, Nat. Commun., 2023, 14, 3382 CrossRef CAS PubMed.
- Z. Chang, W. Zhao, M. Liu, M. Fan, B. Wang and R. Zhang, Appl. Surf. Sci., 2022, 597, 153755 CrossRef CAS.
- K. Li, S. Zhang, X. Zhang, S. Liu, H. Jiang, T. Jiang, C. Shen, Y. Yu and W. Chen, Nano Lett., 2022, 22, 1557–1565 CrossRef CAS PubMed.
- Z. Qiao, R. Jiang, H. Xu, D. Cao and X. C. Zeng, Angew. Chem., Int. Ed., 2024, 40, e202407812 Search PubMed.
- Z. Lou, W. Li, H. Yuan, Y. Hou, H. Yang and H. Wang, J. Mater. Chem. A, 2022, 10, 3585–3594 RSC.
- Z. Jin, D. Jiao, Y. Dong, L. Liu, J. Fan, M. Gong, X. Ma, Y. Wang, W. Zhang, L. Zhang, Z. Gen Yu, D. Voiry, W. Zheng and X. Cui, Angew. Chem., Int. Ed., 2024, 63, e202318246 CrossRef CAS PubMed.
- T. Tang, Z. Wang and J. Guan, Adv. Funct. Mater., 2022, 32, 2111504 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B:Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B:Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS PubMed.
- G. Kresse and J. Furthmiiller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- Q. Fan, P. Hou, C. Choi, T. S. Wu, S. Hong, F. Li, Y. L. Soo, P. Kang, Y. Jung and Z. Sun, Adv. Energy Mater., 2019, 10, 1903068 CrossRef.
- H.-Y. Tan, S.-C. Lin, J. Wang, C.-J. Chang, S.-C. Haw, K.-H. Lin, L. D. Tsai, H.-C. Chen and H. M. Chen, ACS Appl. Mater. Interfaces, 2021, 13, 52134–52143 CrossRef CAS PubMed.
- W. Ni, Z. Liu, Y. Zhang, C. Ma, H. Deng, S. Zhang and S. Wang, Adv. Mater., 2021, 33, 2003238 CrossRef CAS PubMed.
- P. Yin, T. Yao, Y. Wu, L. Zheng, Y. Lin, W. Liu, H. Ju, J. Zhu, X. Hong, Z. Deng, G. Zhou, S. Wei and Y. Li, Angew. Chem., Int. Ed., 2016, 55, 10800–10805 CrossRef CAS PubMed.
- Y. Liu, D. Wang, B. Yang, Z. Li, X. Peng, Z. Liu, L. Zeng, T. Zhang, R. D. Rodriguez, L. Lei and Y. Hou, Chem. Eng. Sci., 2023, 273, 118638 CrossRef CAS.
- Z. Lian, Y. Lu, C. Wang, X. Zhu, S. Ma, Z. Li, Q. Liu and S. Zang, Adv. Sci., 2021, 8, 2102550 CrossRef CAS PubMed.
- Z. Chen, W. Gong, Z. Liu, S. Cong, Z. Zheng, Z. Wang, W. Zhang, J. Ma, H. Yu, G. Li, W. Lu, W. Ren and Z. Zhao, Nanomater. Energy, 2019, 60, 394–403 CrossRef CAS.
- S.-L. Li, Y. Song, G. Tian, Q. Liu, L. Qiao, Y. Zhao and L.-Y. Gan, Fuel Process. Technol., 2024, 261, 108106 CrossRef CAS.
- Y. Feng, C. Wang, P. Cui, C. Li, B. Zhang, L. Gan, S. Zhang, X. Zhang, X. Zhou, Z. Sun, K. Wang, Y. Duan, H. Li, K. Zhou, H. Huang, A. Li, C. Zhuang, L. Wang, Z. Zhang and X. Han, Adv. Mater., 2022, 34, 2109074 CrossRef CAS PubMed.
- B. Cordero, V. Gómez, A. E. Platero-Prats, M. Revés, J. Echeverría, E. Cremades, F. Barragán and S. Alvarez, Dalton Trans., 2008, 2832–2838 RSC.
- J. Wan, W. Chen, C. Jia, L. Zheng, J. Dong, X. Zheng, Y. Wang, W. Yan, C. Chen, Q. Peng, D. Wang and Y. Li, Adv. Mater., 2018, 30, 1705369 CrossRef PubMed.
- R. Dronskowski and P. E. Bloechl, J. Phys. Chem. C, 1993, 97, 8617–8624 CrossRef CAS.
- H. Hu, J. Wang, P. Tao, C. Song, W. Shang, T. Deng and J. Wu, J. Mater. Chem. A, 2022, 10, 5835–5849 RSC.
- F. Pan, H. Zhang, K. Liu, D. Cullen, K. More, M. Wang, Z. Feng, G. Wang, G. Wu and Y. Li, ACS Catal., 2018, 8, 3116–3122 CrossRef CAS.
- A. Álvarez, M. Borges, J. J. Corral-Pérez, J. G. Olcina, L. Hu, D. Cornu, R. Huang, D. Stoian and A. Urakawa, ChemPhysChem, 2017, 18, 3135–3141 CrossRef PubMed.
- B. Yang, K. Liu, H. Li, C. Liu, J. Fu, H. Li, J. E. Huang, P. Ou, T. Alkayyali, C. Cai, Y. Duan, H. Liu, P. An, N. Zhang, W. Li, X. Qiu, C. Jia, J. Hu, L. Chai, Z. Lin, Y. Gao, M. Miyauchi, E. Cortes, S. A. Maier and M. Liu, J. Am. Chem. Soc., 2022, 144, 3039–3049 CrossRef CAS PubMed.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- S. Jin, Z. Hao, K. Zhang, Z. Yan and J. Chen, Angew. Chem., Int. Ed., 2021, 60, 20627–20648 CrossRef CAS PubMed.
- J. Greeley, T. F. Jaramillo, J. Bonde, I. B. Chorkendorff and J. K. Nørskov, Nat. Mater., 2006, 5, 909–913 CrossRef CAS PubMed.
- J. K. Nørskov, T. Bligaard, A. Logadottir, J. R. Kitchin, J. G. Chen, S. Pandelov and U. Stimming, J. Electrochem. Soc., 2005, 152, J23 CrossRef.
- A. Tripathi and R. Thapa, Carbon, 2023, 208, 330–337 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta06604a |
| This journal is © The Royal Society of Chemistry 2025 |