
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Inorganic solid electrolytes for all-solid-state lithium/sodium-ion batteries: recent developments and applications
Muhammad
Muzakir
 ab,
Karnan
Manickavasakam
b,
Eric Jianfeng
Cheng
*c,
Fangling
Yang
c,
Ziyun
Wang
d,
Hao
Li
c,
Xinyu
Zhang
e and
Jiaqian
Qin
*ab
ab,
Karnan
Manickavasakam
b,
Eric Jianfeng
Cheng
*c,
Fangling
Yang
c,
Ziyun
Wang
d,
Hao
Li
c,
Xinyu
Zhang
e and
Jiaqian
Qin
*ab
aInternational Graduate Program of Nanoscience and Technology, Graduate School, Chulalongkorn University, Bangkok 10330, Thailand. E-mail: Jiaqian.Q@chula.ac.th
bCenter of Excellence on Advanced Materials for Energy Storage, Department of Materials Science, Faculty of Science, Chulalongkorn University, Bangkok 10330, Thailand
cAdvanced Institute for Materials Research (WPI-AIMR), Tohoku University, Sendai 980–8577, Japan. E-mail: ericonium@tohoku.ac.jp
dSchool of Chemical Sciences, University of Auckland, Auckland, New Zealand
eState Key Laboratory of Metastable Materials Science and Technology, Yanshan University, Qinhuangdao 066004, P. R. China
First published on 14th November 2024
Abstract
The rapid evolution in electrolyte engineering has significantly propelled the development of synthesis and the precise tailoring of the properties of inorganic solid electrolytes (ISEs). These advancements are crucial to meeting the stringent performance requirements of high-performance all-solid-state batteries (ASSBs). This review comprehensively summarizes recent progress in the synthesis techniques and electrochemical characteristics of various ISEs, including oxides, sulfides, hydroborates, antiperovskites, and halides, highlighting their applications in ASSBs. Additionally, we review key challenges in ASSB development, such as the limited compatibility between ISEs and electrodes, and the detrimental interfacial reactions. Strategies to overcome these challenges, including the use of composite cathodes and solid interface layers, are discussed. Finally, we present current ASSB models and propose emerging approaches driving the future development of ASSBs for the next generation of energy storage solutions.
 Muhammad Muzakir | Muhammad Muzakir is currently a PhD student at the Department of Nanoscience and Technology, Graduate School, Chulalongkorn University, Thailand. He received his M. Eng. in Engineering Technology Program from Sirindhorn International Institute of Technology (SIIT), Thammasat University, Thailand in cooperation with Thailand National Science & Technology Development Agency (NSTDA) and Tokyo Institute of Technology, Japan (TAIST-Tokyo Tech Joint Degree Program: Sustainable Energy & Resources Engineering Program) in 2023. His research interests focus on supercapacitors and solid-state batteries. |
 Karnan Manickavasakam | Karnan Manickavasakam is presently employed as a C2F post-doctoral fellow at the Metallurgy and Materials Science Research Institute, located at Chulalongkorn University in Thailand. He obtained his Doctorate in Chemical Sciences from the CSIR-Central Electrochemical Research Institute, India, in 2021. He possesses both a master's and a bachelor's degree in chemistry from Bharathiar University, India. From 2021 to 2023, he held the position of a postdoctoral researcher at IIT Hyderabad, India, and UMPSA Pahang, Malaysia. His research work mainly focused on the synthesis, characterization, and electrochemical measurements of novel materials for energy storage devices, especially on supercapacitors, Li-ion, Na-ion, and Zn-ion batteries. |
 Eric Jianfeng Cheng | Eric Jianfeng Cheng is an Associate Professor at the Advanced Institute for Materials Research (AIMR), Tohoku University, Japan. He earned his PhDs in Materials Engineering from Tohoku University and Energy Chemistry from Kyoto University, respectively. His research primarily focuses on ceramic solid electrolytes for Li metal batteries. He pioneered the direct observation of Li dendrite growth along grain boundaries in garnet-type solid electrolytes. |
 Fangling Yang | Fangling Yang is a PhD candidate at Sichuan University. He is studying at Tohoku University in Japan under the supervision of Dr Hao Li. His research focuses on optimizing and designing novel solid-state electrolytes based on machine learning and theoretical calculations. |
 Ziyun Wang | Ziyun Wang is an emerging researcher specializing in computational chemistry and theoretical catalysis. He conducted his doctoral research in theoretical catalysis at the Queen's University of Belfast under the guidance of Prof. Peijun Hu and Prof. Chris Hardacre. This was followed by postdoctoral research with Prof. Jens Nørskov and Prof. Thomas Bligaard at Stanford University, and with Prof. Edward Sargent at the University of Toronto. In 2021, Dr Wang joined the School of Chemical Sciences at the University of Auckland, where he is currently a Senior Lecturer. |
 Hao Li | Hao Li is an Associate Professor and Group Leader at the Advanced Institute for Materials Research (WPI-AIMR), Tohoku University, Japan. He received his PhD degree at The University of Texas at Austin in 2019. His research interests include catalysis theory, AI for materials, and computational methodology development. |
 Xinyu Zhang | Xinyu Zhang is a Professor of Materials Science and Engineering at the State Key Laboratory of Metastable Materials Science and Technology, Yanshan University. He received his PhD in materials science in 2009 from Yanshan University. Afterwards, he worked in Swiss Federal Laboratories for Materials Testing and Research (EMPA) (2009–2011) as a visiting research scientist. His research involves design and development of novel materials with unique properties and structures including energy conversion and storage materials, and high strength materials. |
 Jiaqian Qin | Jiaqian Qin obtained his PhD in physics from Sichuan University in 2010, and subsequently worked as a JSPS Post doctoral Researcher at Ehime University, Japan and joined the Metallurgy and Materials Science Research Institute, Chulalongkorn University and established the energy conversion and storage Lab in 2012. He was promoted to Senior Expert Researcher (Professor) in 2022. His research group mainly works on key materials and technologies for Li-ion batteries and next-generation rechargeable batteries. |
1. Introduction
Battery storage technology plays an essential role in cutting-edge technologies in information and communication technology, transportation, healthcare, defense, security, and other related industries.1–4 Batteries with high energy density, the ability to operate safely at extreme temperatures, and long-term stability are essential for these applications.5–7 We can achieve the aforementioned objectives by concentrating on enhancing battery components with exceptional performance.8–15 Recent research interest in battery storage technology has centered on the production of solid-state electrolytes with high conductivity.16–20 Liquid electrolytes in LIBs and SIBs are less reliable at low temperatures, the viscosity of the solution increases and the ionic mobility of the liquid electrolyte decreases. Overcharging also raises the risk of explosions and fires because of the flammable electrolytes and solvents present, as well as the release of harmful gases and pollution into the environment.21–24 Hence, switching liquid electrolytes to solid-state electrolytes and broadening the utilization of all-solid-state batteries are capable of solving these problems.All-solid-state batteries that incorporate ISEs are considered to be at the forefront of battery storage technology. These batteries provide both high energy density and dependable security systems.25,26 Inorganic solid electrolytes demonstrate numerous advantages compared to liquid electrolytes. These include a broad electrochemical window, excellent air stability, high ionic conductivity at room temperature, superb electrochemical stability, good deformability, compatibility with electrode materials, and being environmentally friendly.1,27–29 To construct ASSBs, it is necessary to use electrolytes based on solid-state ion conductors, which must fulfill the previously mentioned requirements. Nevertheless, there is currently no solid-state ion conductor that exhibits the capability to fulfill all of these specified criteria.30–32 The solid-state ion conductors are constructed via a combination of metal and non-metal ions, generating a polyhedral network. Polyhedral structures exhibit patterns based on the angular orientation of their positions. The crystal structure and the variety of defects formed within it determine the conductivity of the crystal.33 Some examples of solid-state ion conductors are Garnet,34–38 NASICON,39–41 halide,2,30,42–46 sulfide,47–49 hydroborate,50–52 and antiperovskite.24,31,53 High conductivity in solid-state crystals is achieved when the number of vacancies for mobile ions exceeds the number of mobile ions, the activation energy between adjacent vacancy sites is sufficiently low to allow ion diffusion, and the channels for ion diffusion are linked.17,51,54 The challenges associated with the applications of ISEs in ASSBs include insufficient wettability and inadequate interfacial compatibility between the electrode and solid electrolytes, issues with electronic and ionic conductivities, fluctuations in the electrochemical window of solid electrolytes that do not meet the cathode potential, and issues related to fabrication time and cost.55
Today's goal in solid-state electrolyte research for ASSBs applications is to create solid-state electrolytes with superior performance compared to conventional liquid electrolytes.2,56–58 Improved electrochemical performance, such as the electrochemical stability window and ionic conductivity of inorganic solid electrolytes, may be obtained by modifying the crystal structure framework, doping with cations and anions, and improving synthesis techniques and conditions.16,37,42,59–64 Liang et al.56 presented the compositions and crystal structures of a ternary halide solid electrolyte, Li3−3xM1+xCl6 (−0.14 < x ≤ 0.5, M = Tb, Dy, Ho, Y, Er, Tm). For example, the orthorhombic structure of Li2.73Ho1.09Cl6 achieves a conductivity higher than that of the trigonal structure of Li3HoCl6. The change from a trigonal to an orthorhombic structure makes it much easier for Li-ions to move around and lowers the activation energy in different L–M–Cl systems. The electrochemical window for Li2.73Ho1.09Cl6 and other inorganic solid electrolytes is seen in Fig. 1.
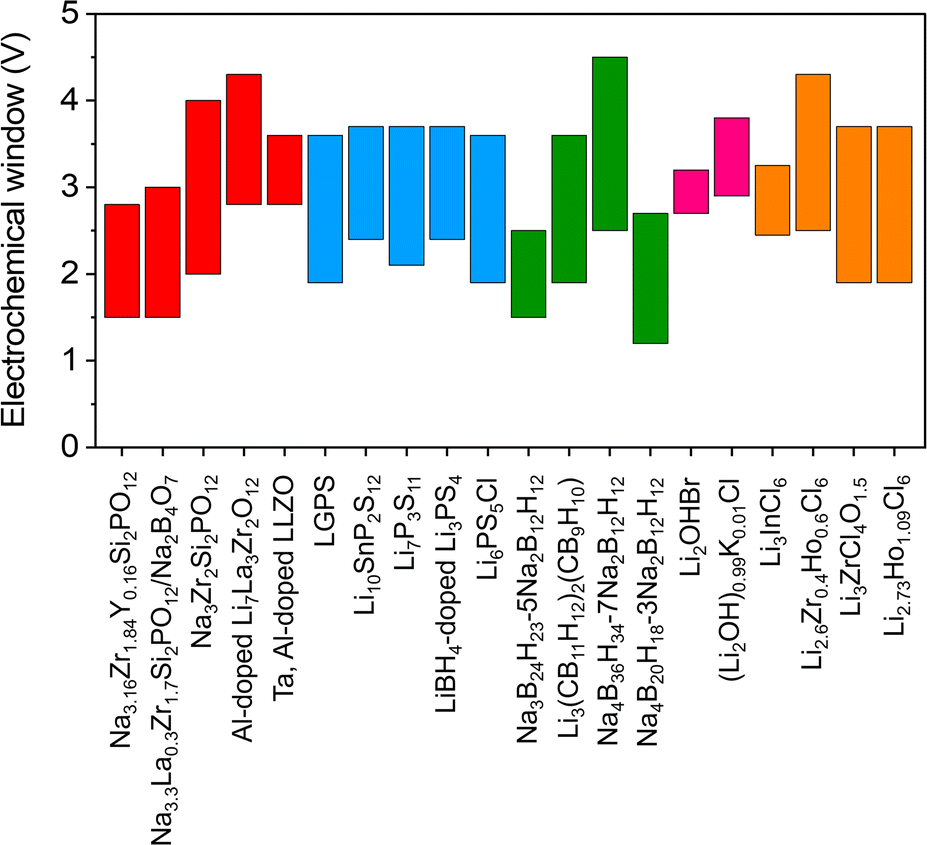 | ||
| Fig. 1 Summary of electrochemical windows for the selected ISEs in ASSBs.6,39,41,56,57,59,65–78 | ||
The selected method of synthesis has a significant impact on several characteristics of the ISEs, including particle size, material density, lattice parameter, and crystal defect. Furthermore, it also affects the mechanical and electrochemical properties of the solid electrolyte.19,79,80 Ma et al.81 utilized freeze-drying technology and heat treatment to produce Li3InCl6 electrolytes. This method resulted in 80% of the particles having a size smaller than 200 nm, a uniform distribution of particle sizes, and the elimination of solvents during the solidification of the electrolyte solution. Additionally, this approach successfully solved the issue of particle agglomeration caused by the growth of particles and high temperatures during the thermal evaporation process of the solutions. It is believed that particle size has a significant effect on the charge transport capabilities of composite cathodes and interface contacts in ASSBs.82,83 The growth of lithium dendrites within the pores and cracks of electrodes is a significant challenge for ASSBs and can even impair the integrity of solid electrolytes.84,85 To solve this issue, increased density and homogenization of the solid electrolyte are essential to suppressing lithium dendrite growth.23,40,86–88 Li et al.23 proposed a two-stage sintering technique to enhance the homogeneity and density of a sulfide-type solid electrolyte (Li7P3S11). The initial powder is subjected to annealing and subsequently densely packed into pellets. The Li7P3S11 undergoes re-annealing by controlled modifications of the annealing temperature and time. The treatment had significant impacts on the conductivity of Li7P3S11, leading it to increase to 8.04 mS cm−1 at room temperature. Furthermore, optimal heat treatment reduces the resistance at the grain boundaries, enhances the durability from cracking, and increases the ionic conductivity of Li7P3S11. One-step synthesis and two-step synthesis that have been applied to date include ultrafast high-temperature sintering,39 calcination + cold sintering process + annealing,83 high-pressure low-temperature,89 spark plasma sintering + annealing,90 sol–gel + calcination,91 liquid-phase method + wet milling + sintering,7 liquid-phase method + annealing,21 ion exchange method,69 wet chemical + low-temperature heating,16,92 radio frequency magnetron sputtering,53 ball-milling + annealing/sintering,22,24,52 two-step milling,71 two-step annealing,93 melt-quenching + high-energy ball-milling,94 solid–gas reaction,95etc.
Understanding the behavior of the electrode-ISE interface is crucial for achieving high-performance ASSBs. This includes investigating the diffusion of Li-ions at the interface, chemical reactions that occur at the interface, changes in the contact area between the electrode and electrolyte, changes in the volume of the electrode material, and concerns about poor compatibility leading to the formation of a high-resistance passive layer during the charge–discharge cycle.76,96,97 Completely eliminating the electrode/ISE interface reaction is not possible. However, the interface reaction can be controlled by employing coating layers and modifying the composition of composite electrodes.98–100 For instance, sulfide solid electrolytes exhibit excellent conductivity within the ISE class.25,79,101 However, when utilized in ASSBs, they meet challenges such as decomposition at high voltages, interfacial reactions, the formation of a space-charge layer between the ISEs and the electrode, and a reduction in capacity and voltage due to structural deterioration on the surface and grain boundaries of cathode particles.68,102–104 The purpose of applying a coating to the electrode surface is to prevent the spontaneous reaction between ISEs and electrodes. Additionally, the coating helps to minimize the oxidative decomposition of the ISE, prevent significant changes in the contact area and reduce the formation of cracks, maintain strong adhesion between the electrode and the ISEs, and suppress the growth of lithium dendrites.42,73,91,97,105
The interface layers should possess excellent lithium conductivity, electron-blocking capabilities to hinder redox reactions in the ISEs, high surface energy to inhibit the formation of lithium dendrites, and compatibility with both the electrode and ISEs.1,66 The electrode/ISE interface layers often employed include LiNbO3,42,103 Li3PO4,56,106 SnO2,107 ZrO2,108 Li3N–LiF,97 ZnO–LiF,1 carbon,91etc. ISEs that have been applied as an interface layer, such as Li7.5La3Zr1.5Co0.5O12,76 Li6PS5Cl,22,42,67,75 and Li3YCl6.57 The coating method is essential to achieving a uniform coating distribution. The coating material must possess plastic properties if it covers the entire surface of the cathode and ISEs. When fabricating the electrodes using mechanical compression tools, this is necessary to prevent the coating from becoming brittle. Additional research is required to investigate the characteristics of coatings and the techniques used to apply them to electrodes, as well as their substantial influence on ASSBs.105
In ASSBs, the composite cathode is usually made up of conductive carbon, CAM, and catholyte (ISEs).50,54 This composition will have a highly intricate interface system, specifically CAM/carbon, carbon/catholyte, and CAM/catholyte. The utilization of single cathode active materials like LCO, LTO, LFP, NCM622, NCM811, NCM83, NCM85, NCM88, and NCM90 is deemed disadvantageous due to the occurrence of volume changes and structural degradation during the charge–discharge process. These factors result in a reduction in the capacity of the cathode materials.103,109 The purpose of incorporating conductive carbon is to enhance the electrical conductivity of the electrode, improve the contact between the CAM and ISEs, and preserve the structural integrity of the electrode during the charge–discharge process.39,41,49,78,110,111 On the other hand, the presence of ISEs in the composite cathode acts to decrease the resistance at the interface of the composite cathode, enhance Li-ion transport, and enhance the performance of the cathode.75,112 To decrease the resistance at the anode/ISEs interface in the case of lithium or sodium anodes, a protective layer can be applied, or the metal can be modified into an alloy such as Li–In,25,65,66,71,80 Li–Si,75 and Na–Sn.30
In recent years, numerous review articles have focused on inorganic solid electrolytes for ASSBs.113–116 However, a scarcity of articles exists that provide a comprehensive review of ISEs, present designs of ASSBs, and prospective strategies therein. Through reviewing the most recent developments in the study of inorganic solid electrolytes. Herein, recent findings in the field of inorganic solid electrolytes such as oxide, sulfide, hydroborate, antiperovskite, and halide are described. This section discusses current synthesis and modification techniques that affect the crystal structure of inorganic solid electrolytes, as well as the impact of doping on electrochemical stability, ionic conductivity, and other characteristics. The stability of each ISEs toward the anode and cathode is then explained, along with several of the current techniques to reduce the shortcomings produced. In discussing the application of ISEs in all-solid-state batteries, especially for Li-ion batteries and sodium-ion batteries. Furthermore, this review summarizes the potential of ISE classes implemented as catholytes and solid interfacial layers to enhance the electrochemical stability of ASSBs and minimize electrode instability. Finally, we provide an overview of the various configuration models for ASSBs and discuss the future progress of ASSBs.
2. Overview of inorganic solid electrolytes (ISEs)
ISEs are regularly updated to fulfill the specifications of next-generation ASSBs due to their tremendous potential. This chapter discusses 5 groups of ISEs, including oxides (Garnet and NASICON), sulfides (lithium thiophosphate, thio-LISICON, and argyrodite), halides, hydroborates, and antiperovskites. The following topics will be included: an overview of ISEs, the basic structures, the impact of metal and additive doping, synthesis techniques, the chemical and electrochemical stability of ISEs, and their compatibility with anodes and cathodes.2.1. Oxide-based ISEs
Inorganic solid-state electrolytes based on oxide materials are commonly classified into several structural types, including LISICON-like, NASICON-like, perovskite, garnet, and β-Al2O3.62,117,118 These oxide-based ISEs are renowned for their wide electrochemical stability, good chemical stability, and high ionic conductivity which make them attractive candidates for advanced energy storage applications.35,36,98,119–125 However, despite these advantageous properties, they suffer from significant limitations that hinder their overall performance. Notably, the inherent brittleness of materials, while the challenge of achieving intimate solid–solid contact at the electrolyte–electrode interface further exacerbates their performance deficiencies.83,126 These issues are critical obstacles that must be addressed to fully realize the potential of oxide-based ISEs in practical applications.The crystal structure of cubic LLZO (Fig. 2a1), exemplifies the garnet-type framework, characterized by its intricate arrangement of dodecahedral LaO8 and octahedral ZrO6 units. These units serve as the backbone of the structure, establishing a robust and stable matrix. Within the cubic Ia![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) d space group, Li-ions are distributed across two distinct crystallographic sites: the tetrahedral (24d sites, referred to as Li1 sites) and octahedral site and off-centered octahedral site (48g/96h sites, referred to as Li2 sites). Meanwhile, La, Zr, and O occupy sites 24c, 16a, and 96h respectively. The Li1 sites demonstrate a remarkably high occupancy rate of 0.94, the highest recorded among garnet-type oxides, indicating a strong preference for Li-ions to reside within these tetrahedral sites. In contrast, the Li2 sites exhibit partial occupancy, with an occupancy value of 0.35, and are associated with significant positional disorder. This pronounced disorder at the Li2 sites is posited to play a critical role in the high ionic conductivity characteristic of cubic LLZO. Specifically, the partial occupancy and the dynamic nature of the Li2 sites likely facilitate the fast Li-ion transport within the structure, thereby enhancing the material's performance as a solid electrolyte.36,37,138
d space group, Li-ions are distributed across two distinct crystallographic sites: the tetrahedral (24d sites, referred to as Li1 sites) and octahedral site and off-centered octahedral site (48g/96h sites, referred to as Li2 sites). Meanwhile, La, Zr, and O occupy sites 24c, 16a, and 96h respectively. The Li1 sites demonstrate a remarkably high occupancy rate of 0.94, the highest recorded among garnet-type oxides, indicating a strong preference for Li-ions to reside within these tetrahedral sites. In contrast, the Li2 sites exhibit partial occupancy, with an occupancy value of 0.35, and are associated with significant positional disorder. This pronounced disorder at the Li2 sites is posited to play a critical role in the high ionic conductivity characteristic of cubic LLZO. Specifically, the partial occupancy and the dynamic nature of the Li2 sites likely facilitate the fast Li-ion transport within the structure, thereby enhancing the material's performance as a solid electrolyte.36,37,138
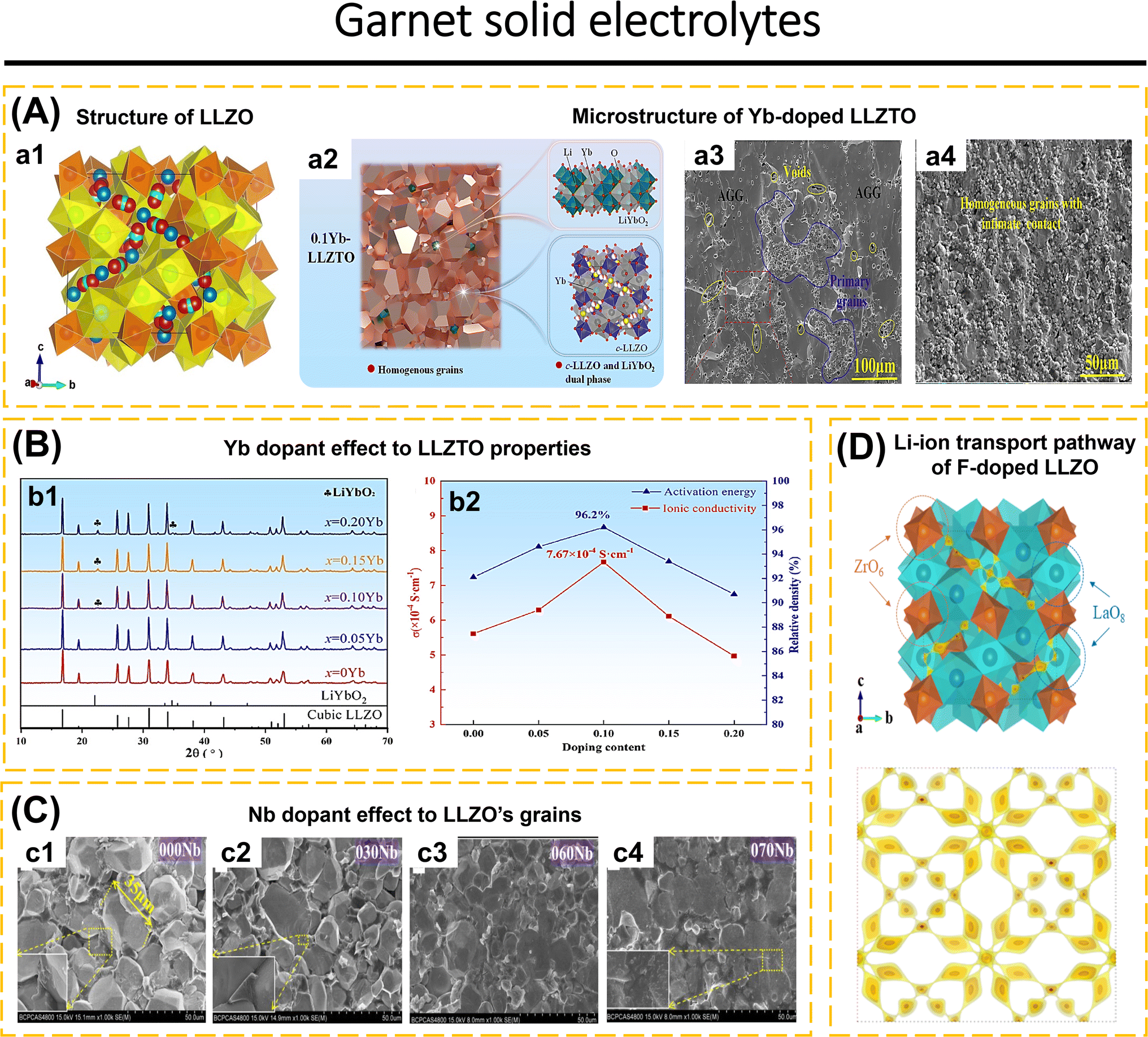 | ||
| Fig. 2 (A) (a1) Crystal structure of cubic LLZO. The yellow is dodecahedrally coordinated La3+ (at the Wyckoff position 24c) and the orange is octahedrally coordinated Zr4+ (16a). The blue spheres correspond to tetrahedrally coordinated (24d) Li+, the green spheres to octahedrally coordinated (48g) Li+, and the red ones to distorted 4-fold coordinated (96h) Li+. Reproduced with permission from ref. 135. Copyright 2014, American Chemical Society. (a2) Microstructural schematic analysis of further Yb-doped LLZTO. SEM images of the cross-sectional grain boundary microstructure of xYb-LLZTO electrolytes: 0Yb-LLZTO (a3) and 0.10Yb-LLZTO (a4). Reproduced with permission from ref. 86. Copyright 2024, Elsevier. (B) (b1) XRD patterns and peak shift of xYb-LLZTO (x = 0, 0.05, 0.10, 0.15, 0.20) in the range of 25–32°. (b2) Ion conductivity (σ) and relative density of xYb-LLZTO (x = 0, 0.05, 0.10, 0.15, 0.20) at 25 °C. Reproduced with permission from ref. 86. Copyright 2024, Elsevier. (C) SEM images of the cross-sections of the Nb-LLZO pellets. Reproduced with permission from ref. 136. Copyright 2018, Elsevier. (D) BVSE analysis of Li-ion transport pathway in the garnet structure: LLZO-F0.2. Reproduced with permission from ref. 137. Copyright 2024, American Chemical Society. | ||
Enhancing the stability of the cubic structure, density, ionic conductivity, and other characteristics of LLZO may be accomplished by cation or anion doping. Several studies have been conducted on doping and its impact on their properties:
(1) Cation substitution on Li sites such as Al(III), Ga(III), Fe(III),139 Ge(IV).140
(2) Cation substitution on La sites such as Yb(III)-doped LLZTO.86
(3) Cation substitution on Zr sites such as Ta(V),141 Nb(V),136 W(VI),121 Sc(III), Mg(II),142 Sb(V), Mo(VI),139 W(VI).143
(4) Double cation substitution on Zr sites such as Y(III), Nb(V).119
(5) Double cation substitution on La and Zr sites such as Sr(II), Sb(V) co-doped LLZO,36 Ca(II), Ta(V) co-doped LLZO,35 Sr(II), Mo(VI) co-doped LLZO,144 Ce(III), Bi(III) co-doped LLZO.145
(6) Double cation substitution on Li and Zr sites such as Ga(III), Mg(II) co-doped LLZO.146
(7) Anion substitution on O site such as F.147
Li7−xLa3Zr2−xTaxO12 (LLZTO) is a Ta-substituted LLZO derivative on the Zr site with high conductivity making it a frequently investigated SE. When x = 0 and x = 0.6, the LLZO pattern shows a single tetragonal and cubic phase, respectively. Unlike the LLZO pattern observed when (0.2 ≤ x ≤ 0.5), which exhibits a combination of tetragonal and cubic phases. Li6.5La3Zr1.5Ta0.5O12 (with x = 0.5) shows the highest ionic conductivity. The presence of Ta content in the range of x = 0.3 to 0.5 leads to a displacement of the Li2 atom at the 96h site, resulting in a reduction of the Li–Li distance from 1.73 Å to 1.50 Å. The precise location of the Li2 atom plays a crucial role in the significant ionic conductivity observed in the LLZO structure.64 The conductivity of Li6.5La3Zr1.5Ta0.5O12 will vary according to the production process used, although having the same composition.64,86,148 The improvisation of LLZTO was carried out due to the low ionic conductivity, low lithium dendrite inhibition, and AGG during preparation which was still difficult to avoid. Lu et al.86 incorporated a Yb2O3-based dopant into the LLZTO structure to manipulate the microstructure of LLZTO (Fig. 2a2). This dopant plays a crucial role in suppressing AGG and enhancing lattice parameters by forming YbO8 in the LaO8 site. The Yb–O bond distance in this area is greater than that of La–O, facilitating the migration of Li-ions. Additionally, the formation of a second phase (LiYbO2) in the grain boundary region enables efficient Li-ion transport, resulting in a significant increase in the conductivity of the LLZO electrolyte. The relative density reached 96.2% and the conductivity was measured at 7.67 × 10−4 S cm−1 when the Yb doping concentration was increased to 0.1. The microstructure noticed a significant drop in both density and conductivity when the Yb concentration reached x = 0.2. SEM images of 0Yb-LLZTO reveal that AGG is widespread and comprises disconnected grains, together with gaps between the grains, resulting in decreased density and conductivity (Fig. 2a3). Elevating Yb concentration to 1.0 results in the suppression of voids and aggregation, leading to a uniform morphological condition (Fig. 2a4). XRD elucidates the appearance of diffraction peaks corresponding to LiYbO2 for Yb concentrations of 0.1 and 0.15 at 25°, and for Yb = 0.2 at 25° and 32° (Fig. 2b1). The rise in Yb concentration correlates with the elevation of LiYbO2 phase concentration at the grain boundary, which reduces intergranular contact, leading to a reduction in density and ionic conductivity (Fig. 2b2).
The single and double doping of cations at Zr4+ sites in LLZO significantly affects its conductivity. Zhao et al.136 explained that the substitution of Zr4+ (0.79 Å) with Nb5+ (0.70 Å) resulted in a reduction of the crystal lattice from 12.9673 Å (0.1 Nb-doped LLZO) to 12.9136 Å (0.7 Nb-doped LLZO) when Nb concentration increased. The SEM photograph indicates an escalation in transgranular fractures from 000Nb to 070Nb, characterized by variation particle sizes that are interconnected without discernible voids (Fig. 2c). Elevating Nb concentration reduces the average particle size of LLZO. The magnification in Fig. 2c1–c3 clarifies the transition from amorphous intergranular fracture to a smooth transgranular fracture. It is inferred that elevated Nb concentration affects particle borders, enhancing Li-ion transport inside the grain boundary area. For 0.25 Nb-doped LLZO (Li6.75La3Zr1.75Nb0.25O12) shows an ionic conductivity of 8.07 × 10−5 S cm−1 at 30 °C, while 0.6 Nb-doped LLZO shows an ionic conductivity of 5.22 × 10−4 S cm−1 at 30 °C. Gai et al.119 substituted Zr4+ sites by double doping with Nb5+ and Y3+ in Li7La3Zr2−2xNbxYxO12. This finding indicates that Nb5+ doping facilitates the formation of Li vacancies, while Y3+ enhances the concentration of Li-ions. The SEM figures indicate that the grains of undoped LLZO exceed 20 μm. Upon the introduction of Nb, the particle size of 0.25 Nb-doped LLZO (Li6.75La3Zr1.75Nb0.25O12) ranges from 5 to 20 μm. The ionic conductivity of Li6.75La3Zr1.75Nb0.25O12 is measured at 4.82 × 10−4 S cm−1 at 30 °C, surpassing the findings of Zhao et al.,136 indicating that the preparation method influences the properties of Nb-doped LLZO. This topic is further discussed in Section 3.1. In the case of 0.5 Nb/0.5 Y co-doped LLZO (Li7La3Zr1.5Nb0.25Y0.25O12), all the grains measure less than 10 μm. It is determined that Nb and Y co-doping influences the reduction of grain size, while simultaneously impacting an increase of grain boundaries. Elevating the doping concentration of 0.5 Nb/0.5 Y co-doped LLZO (Li7La3ZrNb0.5Y0.5O12) indicates that, despite the grain size being less than 10 μm, the grains exhibit interconnectivity. Li7La3ZrNb0.5Y0.5O12 has a conductivity of 8.29 × 10−4 S cm−1 at 30 °C.
Anion replacement at the oxygen position in the garnet structure has been examined. Lu et al.147 replaced the oxygen anion with fluorine in Li6.25Ga0.25La3Zr2O12 (LGLZO). Fluorine doping results in a stiffer LGLZO structure with decreased atomic displacement. Fluorine substitution in the anionic sublattice enhances the strength of La–(O,F) and Zr–(O,F) bonds. The Li-ion conductivity of F-LGLZO is superior to that of LGLZO, measuring 1.28 mS cm−1 compared to 0.543 mS cm−1, respectively. The Li|SPE-F-LGLZO-SPE|Li symmetric cell can operate for up to 650 hours with consistent lithium plating and stripping. NPD analysis elucidates the lack of additional phases in F-doped and undoped LGLZO. Rietveld analysis verifies that La is located at 24c, Zr at 16a, and O1 at 96h. In F-doped LGLZO, F atoms are randomly allocated at the 96h site, in conjunction with O1 atoms. Li1 atoms occupy the 24d position, whereas Li2 atoms are located in the 96h position. Ga is randomly distributed with Li1 atoms. There are just 0.2 F per formula among the 12 anionic sites, which does not significantly influence the bond distance. Each atom has a small displacement, so the atoms are more tightly bound in the F-LGLZO structure. This affects the lithium transport in the F-LGLZO structure due to the smaller displacement of the anionic sublattice. Liu et al.137 employed BSVE to investigate the influence of Fluorine on the transport pathway of LLZO (Fig. 2d). Generally, lithium atoms occupy Li(1)O4 and Li(2)O6 sites. Fluorine doping at oxygen sites leads to the rearrangement of lithium atoms between Li1 and Li2, with Li(1)O4 tetrahedrons linked to distorted Li(2)O6 octahedrons. This process leads to a decrease in the Li-ion transport barrier energy and an enhancement in ionic conductivity. The high sintering temperature of 1200 °C for 12 hours required for the production of Li6.3La3Zr1.65W0.35O12 (LLZWO) poses a challenge for the commercialization of garnet. The incorporation of CuO additive effectively reduced the sintering temperature while enhancing the properties compared to pristine LLZWO. Zhang et al.143 incorporated varying amounts of CuO into LLZWO, specifically LLZWO-x wt% CuO where x = 0, 1, 2, 5. The diffraction pattern of LLZWO pellets sintered at 1180 °C for 6 hours exhibited signals corresponding to La2Zr2O7. No impurity phase was observed in the 1 and 2 wt% CuO–LLZWO pellets sintered at 1120 °C for 6 hours. Once increasing CuO to 5 wt%, signals for Li3Cu2O4 and CuO were observed. The maximum relative density attained for 2 wt%-LLZWO was 97.6%.
Zhang et al.149 clarified the diffusion mechanism of Na-ions in NZSP-type NASICON, incorporating insights from prior research on four Na sites.152 They introduced a novel Na5 site that significantly increases the diffusion mechanism, utilizing a combination of four methodologies: BVEL, DFT, MEM, and NPD. In the rhombohedral structure, sodium is predominantly located at the Na1 (6b) and Na2 (18e) sites, with a minor presence at the Na3 (36f) sites. The Na3 site is adjacent to Na1, preventing simultaneous occupation of both sites (Fig. 3a1). In the monoclinic structure, the Na2 (18e) site is subdivided into Na2 (4e) and Na3 (8f) sites, while the Na3 (36f) site is further divided into Na4 (8f) and Na5 (8f) sites. In the monoclinic 3D network of NZSP, Na+ diffusion is observed at all five sites; however, probability density analysis indicates a limited diffusion pathway along the [101] direction that connects adjacent bc planes (red lines in Fig. 3a3). The additional site Na5 functions as a crossover site, facilitating rapid Na+ mobility. In the rhombohedral NZSP structure, two correlated three-dimensional networks have been identified: Path 1 (Na2–Na3–Na1–Na3–Na2) and Path 2 (Na2–Na3–Na3–Na2). AIMD and CI-NED calculations indicate that the correlated migration mechanism predominates over the single ion jumping mechanism. Consequently, an increased Na-ion concentration leads to a higher percentage of correlated jumping, attributed to the coulombic repulsion among Na-ions. Enhancing ionic conductivity is more effectively achieved by increasing the Na-ion concentration than by expanding the NASICON framework while keeping a constant Na+ concentration.
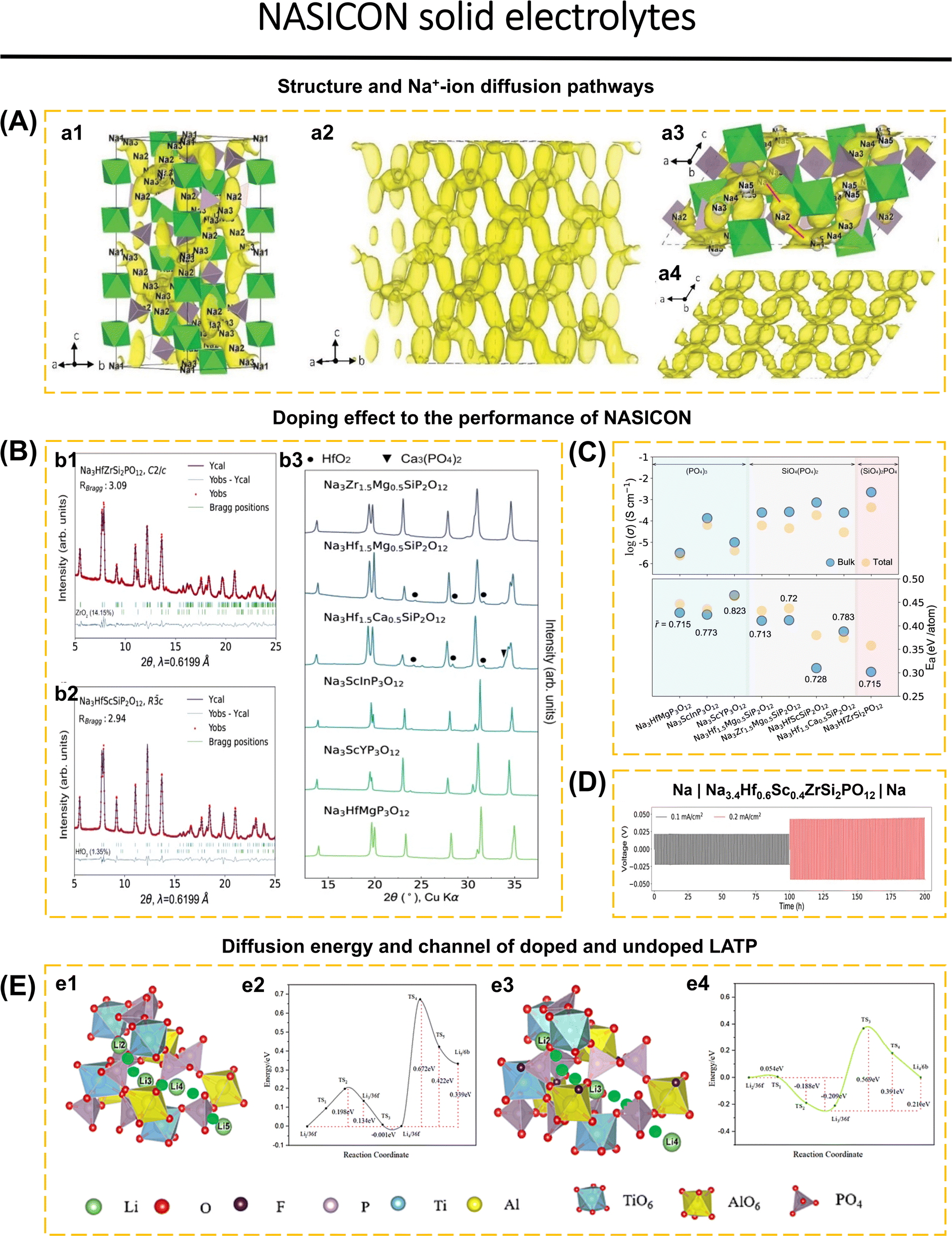 | ||
| Fig. 3 (A) Na-ion diffusion pathways in monoclinic and rhombohedral Na3Zr2Si2PO12 from AIMD and MEM. (a1 and a2) The Na-ion probability density isosurface (yellow) of rhombohedral Na3Zr2Si2PO12 at 1400 K from AIMD simulations; all pathways follow the Na2–Na3–Na1–Na3–Na2 trajectory, and no Na2–Na2 diffusion is observed. The polyhedra are not shown in (a2). (a3 and a4) Na-ion probability density isosurfaces (yellow) of monoclinic Na3Zr2Si2PO12 at 1400 K from AIMD simulation. The red lines in (a3) indicate the [101] direction, which is the rate-limiting step in the Na+-ion migration process. The polyhedra are not shown in (a4) (green: ZrO6 octahedra, light purple: Si(P)O4 tetrahedra, yellow: Na atoms). Reproduced with permission from ref. 149. Copyright 2019, Wiley-VCH. (B) XRD patterns of the as-synthesized NASICONs. Rietveld refinement results of synchrotron XRD data of Na3HfZrSi2PO12 with monoclinic C2/c symmetry (b1) and Na3HfScSiP2O12 with rhombohedral Rc symmetry (b2). (b3) Lab XRD patterns of other as-synthesized NASICONs. (C) Measured ionic conductivity of as-synthesized NASICONs at room temperature (∼25 °C). The bulk properties are indicated by blue circles, whereas the total properties are indicated by orange ones. (D) Na stripping and plating tests in a temperature chamber at 25 °C at various current rates of a Na|Na3.4Hf0.6Sc0.4ZrSi2PO12|Na symmetric cell. Reproduced with permission from ref. 40. Copyright 2023, Springer Nature. (E) (e1) Li-ion diffusion channel model in LATP and corresponding (e2) diffusion energy barrier and (e3) Li ion diffusion channel model in LATP-0.2F and corresponding (e4) diffusion energy barrier. Reproduced with permission from ref. 150. Copyright 2024, Elsevier. | ||
The current versions of NASICON, such as LiSn2(PO4)3,153 LiTi2(PO4)3,83,123 LiGe2(PO4)3,154 NaTi2(PO4)3,78 Na3V2(PO4)3,5etc. Doping occurs when a metal cation with a radius comparable to that of the NASICON metal cation and the ability to form octahedral coordination is introduced, regardless of whether the metal cation is divalent (Mg2+, Ca2+, Cu2+),125,155,156 trivalent (Yb3+, Gd3+, Sc3+, Al3+, Fe3+),157–162 tetravalent (Ge4+, Zr4+, Ce4+),157,158,163–165 or pentavalent (Nb5+).124 For example, doping LiGe2(PO4)3 with divalent or pentavalent cations will impact the concentration of Li species and its ionic conductivity. By depositing divalent cations of varying ionic radius sizes into LiGe2(PO4)3, the ionic conductivity will differ due to the hindrance of Li diffusion caused by the larger size of the doping metal cation.
The selection of doping has a significant role in expanding the diffusion channel of Na+ or Li+, enhancing the density of the solid electrolytes, inhibiting the impurities formation at high temperatures, lowering the sintering temperature, and increasing the ionic conductivity. However, not all doping agents and methods comply with NASICON compliance standards due to their inability to increase conductivity, contaminant growth, and lack of economic feasibility.60,125,155 Wang et al.40 examined the electrochemical characteristics of eight recently discovered NASICON compounds. The ionic conductivity of these compounds can increase up to a certain point with the average size of the metal. Sodium concentration, activation barrier, and substitution of the polyanion PO43− with SiO44−, these factors collectively contribute to the enhancement of ionic conductivity. Out of the eight recently discovered NASICON compounds, seven compounds exhibit a rhombohedral structure (Rc), while the remaining one has a monoclinic structure (C2/c). Fig. 3b1–b3 displays the Rietveld refinement results for Na3HfZrSi2PO12 (monoclinic) and Na3HfScSiP2O12 (rhombohedral), together with XRD patterns of additional NASICON compounds. The value of the lattice parameter a is influenced by the species of metal present, whereas the value of the lattice parameter c is determined by the type of polyanion present. The compounds Na3ScInP3O12 and Na3Hf1.5Ca0.5SiP2O12 exhibit a relative density of ∼87%, and the remaining compounds are in the range of ∼93% to ∼95%. Electrochemical impedance spectroscopy analysis performed at room temperature revealed that the ionic conductivity of NASICON increased as the concentration of silicate increased, irrespective of the cation type (Fig. 3c). NASICON exhibits conductivity values ranging from 10−6 to 10−5 S cm−1 for the pure phosphate group, from 10−5 to 10−4 S cm−1 for the (SiO4)(PO4)2 group, and a maximum conductivity of 4.4 × 10−4 S cm−1 for the (SiO2)2PO4 group. The ionic conductivity of Na3HfZrSi2PO12 was enhanced by doping it with Sc3+, which increased the average cation radius and modification of the composition. Na3.4Hf0.6Sc0.4ZrSi2PO12 exhibits an increase in the average cation size from 0.715 Å to 0.722 Å. It has a completely rhombohedral structure and achieves an ionic conductivity of 1.2 mS cm−1, which is higher than that of the pristine Na3HfZrSi2PO12 compound. Na3.4Hf0.6Sc0.4ZrSi2PO12 demonstrated stability in Na stripping and plating for 200 h (Fig. 3d).
Yin et al.150 assessed the impact of partially substituting fluorine for oxygen at the 36f1 and 36f2 sites within the LATP-type NASICON structure. The fluorine atom exhibits a propensity to substitute the oxygen atom in the Al–O–P configuration, shifting closer to the phosphorus atom (P–F, 1.60 Å) and away from the aluminum atom (Al–F, 2.49 Å). Doping leads to the elongation of the Al–O and P–O bonds, accompanied by the distortion of the octahedron (from AlO6 to AlO5F) and tetrahedron. The doping effect enlarges the crystal lattice while preserving the Rc crystal structure. The analysis of Li vacancies elucidates the impact of F doping. The incorporation of a single fluorine atom (LATP-0.1F) reduces the vacancy energy of lithium ions in Li4, while simultaneously increasing the vacancy energy in Li1. In LATP-0.3F, the vacancy energy in Li3 and Li4 is reduced. LATP-0.2F demonstrates the ability to maintain vacancy energy in Li1, Li2, and Li3 at significantly lower energy levels compared to LATP-0.1F and LATP-0.3F. The vacancies-assisted diffusion mechanism of LATP and LATP-0.2F is represented by the following pathways: Li2 → Li3 → Li4 → Li5 (36f → 36f → 36f → 6b) for LATP (Fig. 3e(1)) and Li2 → Li3 → Li4 (36f → 36f → 6b) for LATP-0.2F (Fig. 3e(3)). The graph indicates that the initial energy barriers for both are minimal. LATP requires crossing multiple barriers, specifically from the octahedral [TiO6] to the tetrahedral [PO4], then to the octahedral [AlO6], and finally back to the tetrahedral [PO4]. This outcome is attributable to LATP containing four lithium ions. Lithium migrates from 36f to 36f with minimal energy consumption (0.001 eV). The diffusion of 36f → 36f in LATP-0.2F exhibits a low energy level of 0.054 eV. The transition from the [AlO6] octahedron to [AlO5F] results in a modification of the Al–O bond length (1.93 Å) to the Al–F bond length (2.49 Å), thereby facilitating the passage of the initial diffusion barrier with minimal required energy (Fig. 3e2). The 36f → 6b diffusion barrier for LATP-0.2F (0.578 eV) is lower than that for LATP (0.672 eV) due to the shorter Al–O bond length of 1.85 Å in the [AlO5F] octahedron compared to 1.96 Å in the [AlO6] octahedron (Fig. 3e4). F doping is concluded to facilitate the opening of diffusion channels and to reduce the diffusion energy barrier. The ionic conductivities of LATP and LATP-0.2F are measured at 0.142 mS cm−1 and 0.358 mS cm−1, respectively. Li et al.122 presented another study related to Cl doping in LATP. Chlorine atoms are distributed randomly between two oxygen positions (36f), with occupancy rates of 12.3% for position 36f1 and 7.2% for position 36f2. This suggests that chlorine predominates in occupying the O site (36f1). This influences the asymmetry of the metal–oxygen bond length (M = Li, Al, or Ti). The bond length of Li (36f)–O/Cl (36f1) increases from 2.145 Å to 2.304 Å, whereas the Li (36f)–O/Cl (36f2) decreases from 2.069 Å to 1.874 Å. The variation in Li–O bond distance influences the diffusion rate of Li-ions. The conductivity of LATP-0.4Cl is approximately 0.423 mS cm−1, whereas pristine LATP exhibits a conductivity of 0.139 mS cm−1.
Wu et al.166 enhanced the stability and mobility of Na-ions in Na3V2(PO4)2F3 (NVPF) by the doping of K+ cations, which partially substitute Na+ in the NVPF structure. The unique structure of NVPF consists of the interconnection of octahedra [VO4F2] via F atoms and the interconnection of tetrahedra [PO4] via O atoms, resulting in a three-dimensional NASICON framework with Na+ migration channels oriented in the [110] direction. NVPF has three pairs of redox potentials (about 3.4, 3.7, and 4.2 V) that influence structural deformation, modify the diffusion pathway, and reduce the mobility of Na+ during the discharging process compared to the charging process. Cation doping at the V site of NVPF effectively enlarges the crystal lattice parameters when the doping cation's size exceeds that of V, thereby enhancing Na+ migration and improving the structural and physicochemical stability of the crystal, as the doping has no impact on the electrochemical process. K+ doping at the Na+ site results in a reduction of bandgap energy from 3.31 eV to 2.70 eV, hence enhancing electrical conductivity from 7.95 × 10−6 S cm−1 (NVPF/C) to 3.78 × 10−5 S cm−1 (NKVPF/C). The optimization of K+ doping (x = 0.08, N0.92K0.08VPF/C) promotes a reduction in particle size and enhances crystallinity, resulting in a lattice spacing of 0.322 nm. K+ doping does not influence the V3+ valence state in NKVPF. The CV profiles and dQ/dV plot indicate that NKVPF/C significantly reduces polarization and voltage hysteresis throughout the charge–discharge operation. N0.92K0.08VPF/C has a highly reversible phase shift during sodiation and desodiation. EIS study indicates that K doping enhances Na+ transport, leading to improved cell performance.
Wang et al.41 demonstrated that the addition of 3 wt% Na2B4O7 (NBO3) to Na3.3La0.3Zr1.7Si2PO12 (NLZSP) can solve deficiencies in the synthesis and properties of NASICON. This addition facilitates a reduction in sintering temperature to 950 °C, enhances NLZSP grain connection, decreases grain boundary resistance (Rgb) from 135 Ω cm2 for NLZSP to 8 Ω cm2 for NLZSP (NBO3), and increases ionic conductivity to 1.81 mS cm−1 with an activation energy of 0.22 eV. Fig. 4a1–a8 shows the SEM of NLZSP (NBO3) sintered from 850 to 1150 °C. At 850 °C, NLZSP (NBO3) has many tiny holes and cracks, but at 950 °C, it exhibits small grains with different morphologies and a higher density. These variables are shown to impact the differences in Rg, Rgb, and Rt values in NLZSP (NBO), Fig. 4b. In another case, Li1.5Al0.5Ge0.5P3O12 (LAGP) with 0.5 wt% LiBF4 improved conductivity to 3.21 × 10−4 S cm−1, reduced grain boundary impedance from 162.2 Ω to 35.2 Ω, raised relative density from 93.6% to 96.5%, eliminated the impurity phase, and reduced the sintering temperature from 800 to 700 °C. The Li|LAGP-LiBF4|LFP cell has a discharge capacity of 164.2 mA h g−1 at 0.2C and 117.5 mA h g−1 after 100 cycles.167 The decrease in ionic conductivity can be caused by the increase in temperature during the synthesis of NASICON because it has an impact on the decrease in the concentration of mobile ions (Li+ and Na+). This is followed by grain cracking, which disrupts the ionic transport pathway, the formation of by-products that inhibit ionic transport, and the formation of unfavorable pores and becomes a place for the growth of Li dendrites during the cycle. Modification in sintering techniques is the solution to obtain high-density materials, without additional phases, with high conductivity, and efficiency in energy consumption and cost. Common synthesis methods of NASICON that have been applied to date include solid-state reaction,40,41,60,78,125 sol–gel,168,169 spark plasma sintering,90 stereolithography,170 melt-casting technique,123 CSP followed by post-annealing,83 and ultrafast high-temperature sintering.39
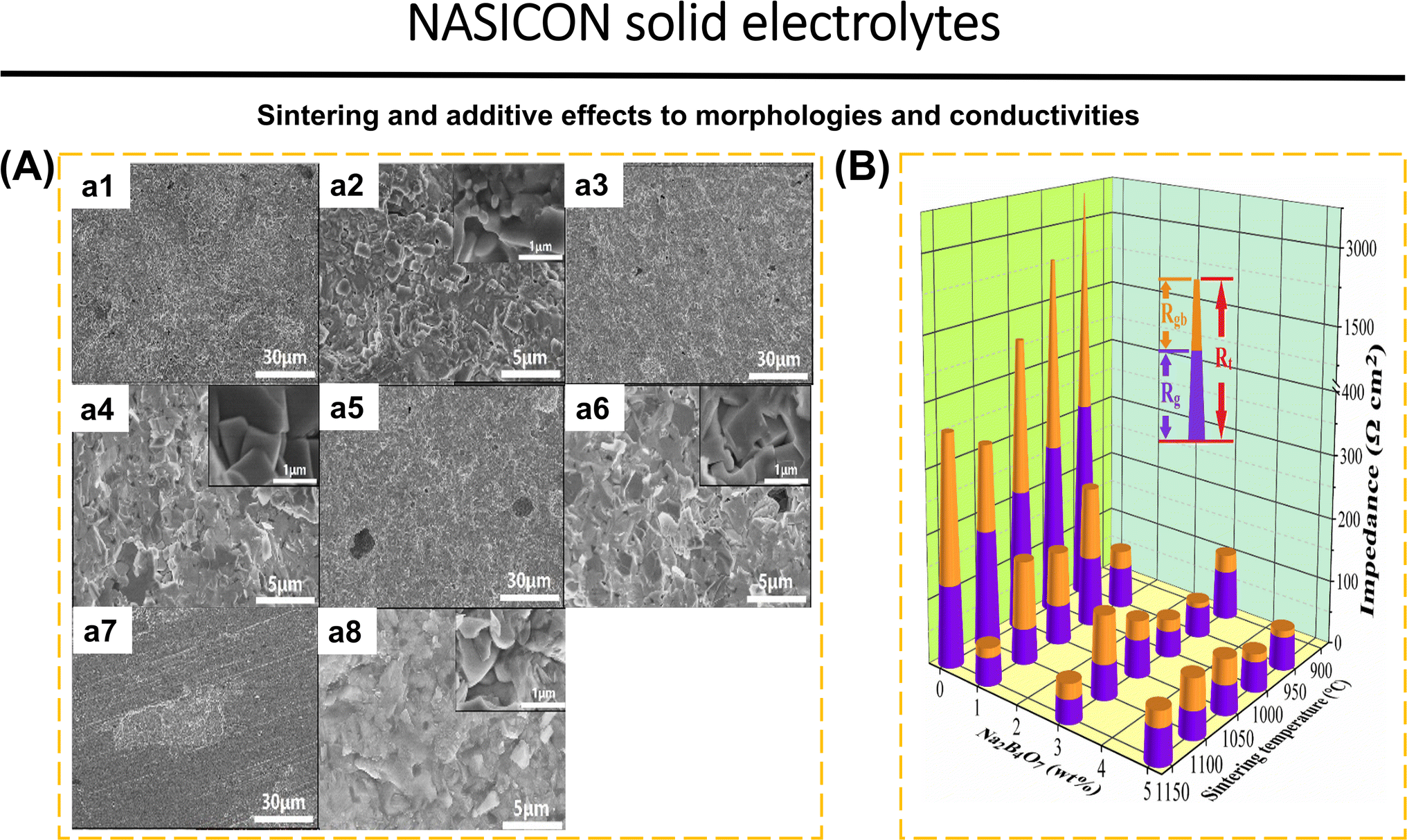 | ||
| Fig. 4 (A) Cross-sectional SEM images of NLZSP (NBO3) ceramic electrolyte sheets sintered at (a1 and a2) 850 °C, (a3 and a4) 950 °C, (a5 and a6) 1050 °C and (a7 and a8) 1150 °C. (B) Rg, Rgb, and Rt of NLZSP and NLZSP (NBO) ceramic electrolyte. Reproduced with permission from ref. 41. Copyright 2023, Elsevier. | ||
2.2. Sulfide-based ISEs
In this section, sulfides are divided into several groups: lithium thiophosphate, thio-LISICON, and argyrodite. In the ISE class, sulfides show good ductility and high ionic conductivity.47,80,82,88,171–173 The properties of each class are described further.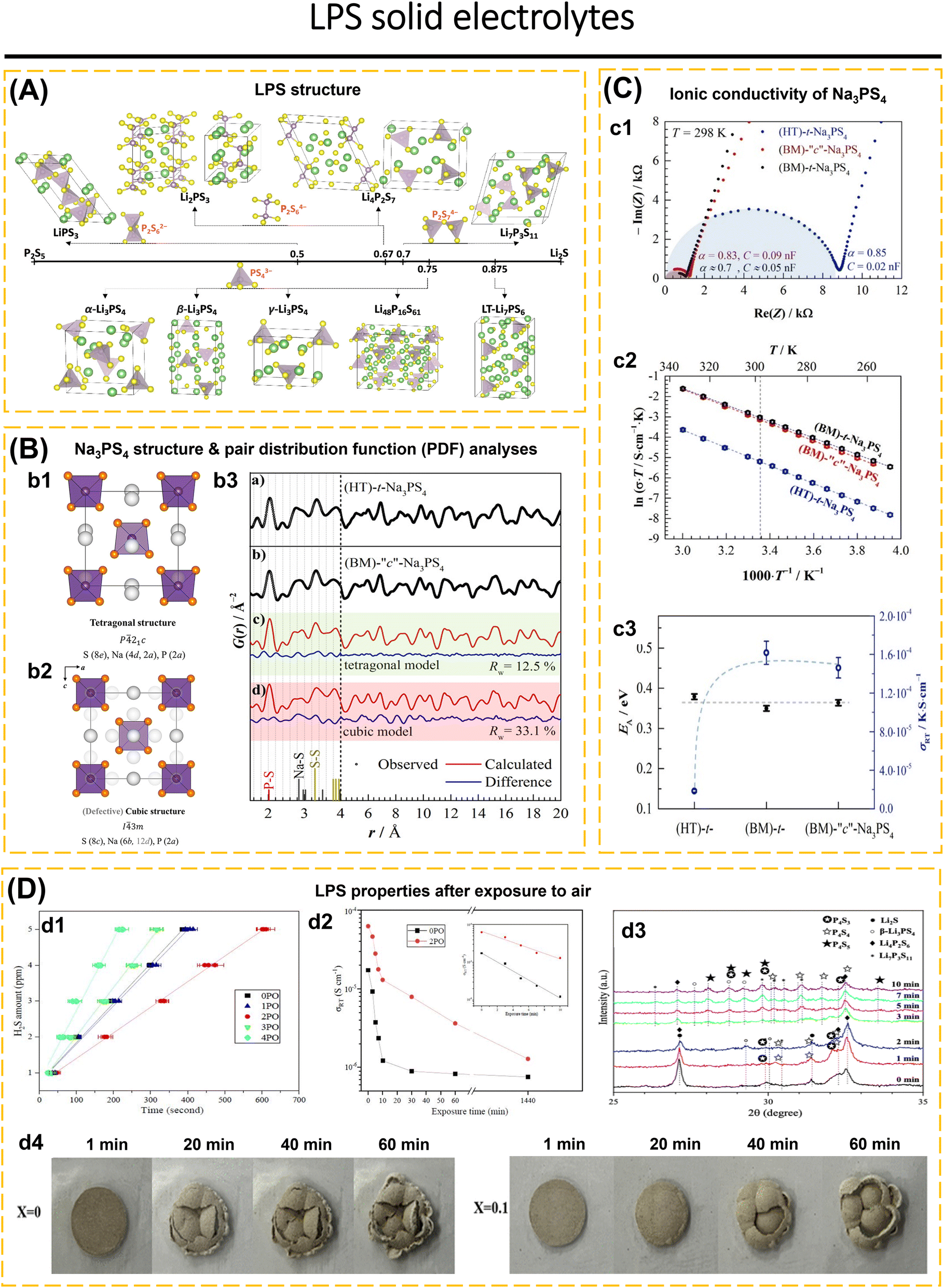 | ||
| Fig. 5 (A) Crystal structures of LPS compositions on and near the Li2S–P2S5 composition line (Li: green; S: yellow; P: purple). Reproduced with permission from ref. 174. Copyright 2022, American Chemical Society. (B) Crystal structure of (b1) tetragonal and (b2) cubic Na3PS4 projected in the (010) plane. The perfectly cubic phase (i.e., no occupancy of the 12d positions) shows PS43− tetrahedra in a body-centered lattice. In the tetragonal modification, a minor rotation of the tetrahedra leads to a splitting of the Na positions and an elongation of the c-lattice parameter. (b3) Experimentally obtained G(r) data for (a) HT-t-Na3PS4 and (b) BM-“c”-Na3PS4 showing that there is no significant difference in the local structure. BM-“c”-Na3PS4 was fitted using a (c) tetragonal model, shaded in green and a (d) cubic model shaded in red. Experimental data are shown as black points. The red line denotes the calculated pattern, and the difference profile is shown in blue. A fit to the low-r range (1.5–4 Å) was independently performed and leads to larger scale factors, thereby indicating an additional fraction of amorphous content or phase with a low coherence length. (C) (c1) Nyquist plots of t-Na3PS4 and “c”-Na3PS4 synthesized using the different synthetic approaches. Data points are only displayed up to 2 MHz. (c2) Arrhenius plots for all compounds. (c3) Activation energy and room temperature ionic conductivity for each of the Na3PS4 compounds, showing that while the activation barrier for ionic motion remains unchanged the ball-milling procedure leads to much higher conductivities irrespective of the crystal structure. Reproduced with permission from ref. 176. Copyright 2018, American Chemical Society (D) LPS properties after exposure to air: (d1) amounts of H2S released from 0PO, 1PO, 2PO, 3PO, and 4PO after exposure to air (left). (d2) The ionic conductivity (RT) of 0PO and 2PO electrolytes at various exposure times (middle). (d3) XRD patterns of 2PO exposed to air for 1 min, 2 min, 3 min, 7 min, and 10 min in the 2θ of 25° to 35° range. Reproduced with permission from ref. 177. Copyright 2023, Royal Society of Chemistry. (d4) Optical images of Li7P3S11 and Li7P2.9Ge0.05S10.75O0.1 electrolyte at 1, 20, 40, and 60 min exposure in the open air at RT. Reproduced with permission from ref. 65. Copyright 2022, American Chemical Society. | ||
Sodium-based thiophosphate, Na3PS4, exhibits two types of crystal structures: cubic and tetragonal. The cubic structure exhibits higher conductivity compared to the tetragonal structure (Fig. 5b1 and b2). In the cubic structure, PS43− polyhedra are arranged within a body-centered cubic lattice, with one Na+ occupying the octahedral sites (6b). The tetragonal structure results from the rotation of the PS43− polyhedra towards the Na+ positions (4d and 2a), resulting in an increased ratio of the lattice parameters c/a. Krauskopf et al.176 examined the influence of the crystal lattice of Na3PS4, which was prepared through ball-milling (BM-“c”-Na3PS4) and high temperature (HT-t-Na3PS4). The cubic structure of (BM)-“c”-Na3PS4 determined by Bragg diffraction exhibited no significant change when compared to the local tetragonal structure derived from PDF analysis. Despite the brief annealing of BM-“c”-Na3PS4, the resulting product, (BM)-t-Na3PS4, exhibited structural parameters comparable to those of HT-t-Na3PS4 (Fig. 5b3). This indicates that varying preparation conditions lead to distinct crystal structures, and the ionic conductivity of Na3PS4 is independent of the crystal structure. Despite the sintering of the HT-t-Na3PS4 (500 °C for 10 hours) to decrease grain boundary resistance, SEM results indicated a smooth and dense surface; however, the conductivity exhibited no significant change. The conclusion indicates that conductivity is influenced by variations in defect concentration rather than by the grain boundary (Fig. 5c1–c3). The milling conditions result in a reduced grain size, which enhances conductivity, while simultaneously increasing defect concentration.
Park et al.179 demonstrated the hydrolysis stability of sulfides (Li3PS4, LGPS, and Li6PS5Cl) in the presence of moisture. The reaction energies for Li3PS4, LGPS, and Li6PS5Cl were measured at 49, 50, and 92 meV per atom, respectively. This suggests that sulfides easily interact with H2O, resulting in the formation of H2S,177,179 as demonstrated by the reaction:
| 0.9412H2O + 0.1176Li10GeP2S12 → 0.2353Li3PO4 + 0.1176Li4GeS4 + 0.9412H2S |
| 0.5H2O + 0.5Li6PS5Cl → 0.125Li3PO4 + 0.375Li3PS4 + LiHS + 0.5LiCl |
| 0.9412H2O + 0.2353Li3PS4 → 0.9412H2S + 0.2353Li3PO4 |
Research and development focus mainly on synthesis and modification using addictive materials to reduce metal and moisture instability, inhibit lithium dendrites, and boost conductivity.49,65,180,181 Mi and Hall177 developed a glass-ceramic electrolyte containing (100 − x)(0.75Li2S·0.25P2S5)·xP2O5 (mol%) (x = 0, 1, 2, 3 and 4). A glass-ceramic based on 98(0.75Li2S·0.25P2S5)·2P2O5 (2PO) has strong air stability, producing 5 ppm of H2S gas after 10 minutes of exposure at 20–25 °C and 70% humidity (Fig. 5d1). 2PO has 3.7 times better ionic conductivity than undoped P2O5 and is stable in air (Fig. 5d2). The ss-NMR data revealed that phases with high conductivity in 2PO (β-Li3PS4 and Li7P3S11) were sustained, resulting in high conductivity. 2PO EDS analyzes the stability of 2PO in terms of element composition. At 10 minutes of exposure, the conductivity of 2PO decreases more slowly than that of 0PO and serves several levels higher. The XRD investigation demonstrates that 2PO only underwent minor changes after being exposed to air for several days; nonetheless, an S8 layer was produced, which preserves the material's stability against air and delays deterioration (Fig. 5d3).
Lv et al.65 developed Li7P3−xGe0.5xS11−2.5xOx (x = 0, 0.1, 0.2, and 0.3) glass-ceramic electrolytes. Under 30% humidity at 30 °C for 60 minutes, Li7P3S11 and Li7P2.9Ge0.05S10.75O0.1 produced 0.5472 cm3 g−1 and 0.3168 cm3 g−1 of H2S, respectively. XRD analysis before and after air exposure shows that there is no structural change in Li7P2.9Ge0.05S10.75O0.1. Fig. 5d4 depicts the conditions of Li7P3S11 and Li7P2.9Ge0.05S10.75O0.1 solid electrolytes exposed to open air. In electrochemical stability tests, Li7P3S11 exhibited anodic fluctuations in the range of 3 to 5 V (vs. Li/Li+), whereas Li7P2.9Ge0.05S10.75O0.1 continues to be stable up to 5 V. The electrochemical stability of Li/Li7P3S11/Li symmetric cell shows an overpotential of ∼48 mV followed by a voltage drop after the 7th cycle caused by short circuit due to the growth of lithium dendrites. While the Li/Li7P2.9Ge0.05S10.75O0.1/Li cell shows a small and stable overpotential (∼5 mV) for 100 h @ 0.1 mA cm−2/0.1 mA h cm−2 at room temperature condition. The presence of GeO2 to Li7P3S11 structure improves the ionic conductivity, stability in the air, electrochemical stability, and compatibility with Li anodes.
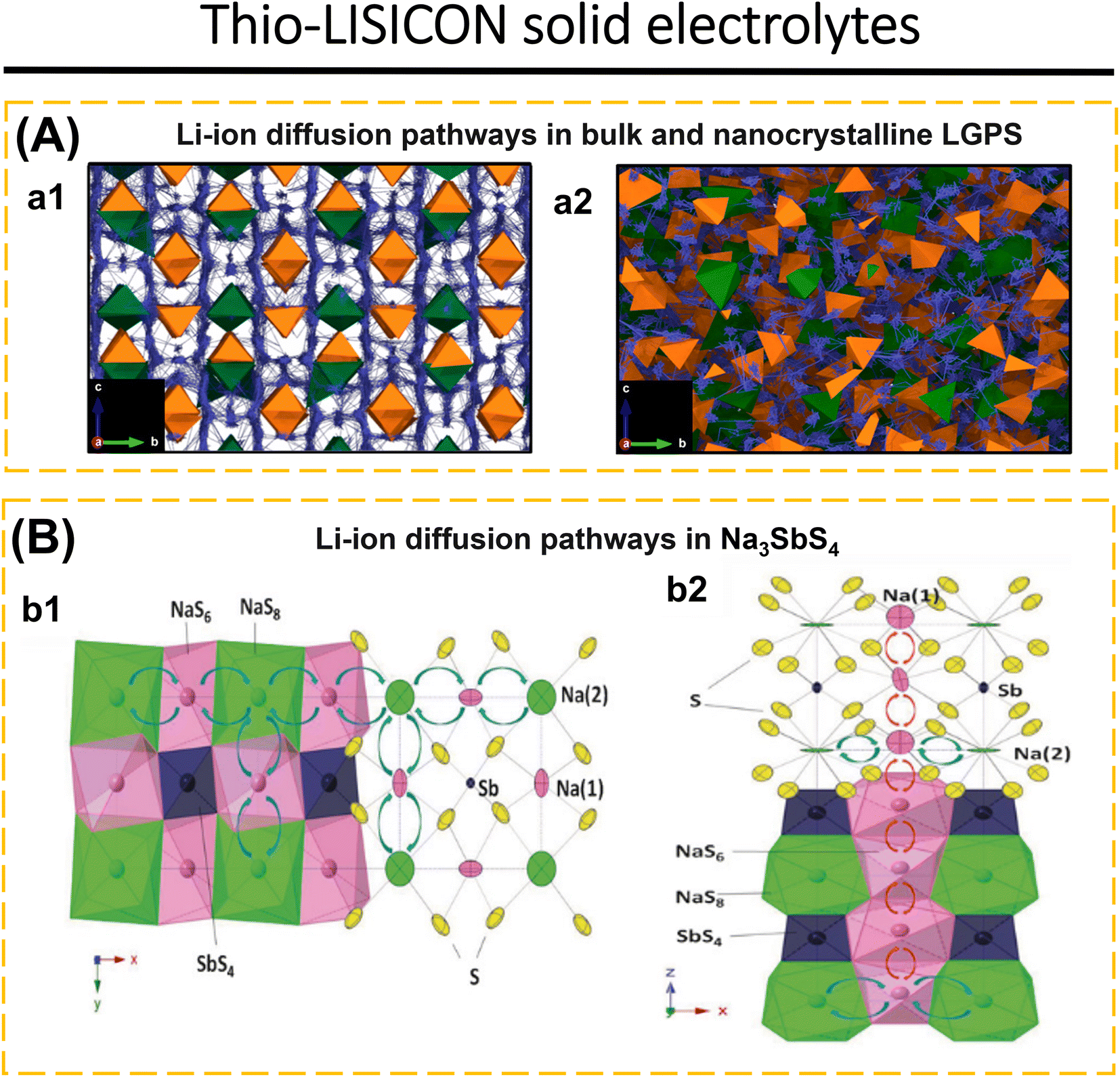 | ||
| Fig. 6 (A) Li-ion diffusion pathways in bulk and nanocrystalline LGPS. Diffusion density plots of Li ions (blue) overlaid on GeS4 (green) and PS4 (orange) tetrahedra in (a1) bulk and (a2) nanocrystalline LGPS with a particle volume of 10 nm3 at 300 K (2.4 × 3.5 nm cross-section). Reproduced with permission from ref. 183. Copyright 2023, American Chemical Society. (B) Li-ion diffusion pathways in LGPS: (b1) [001] and (b2) [010] views of the framework structure of Na3SbS4. The 2D planar Na+ transport tunnels in the xy-plane are formed by alternatively arranged NaS6 and NaS8 polyhedra, which share common faces. Planar tunnels are connected along the z-axis through chains formed by NaS6 octahedra, which share common edges. Reproduced with permission from ref. 184. Copyright 2016, Wiley-VCH. | ||
Lu et al.3 fabricated a glass-ceramic LGPS using Li2S, GeS2, and P2S5. This was achieved by subjecting the mixture to high-energy ball milling for a duration of 40 to 520 minutes. The formation of glassy-ceramic LGPS may be elucidated by considering three distinct stages: the vitrification zone refers to the stage when the mixture of Li2S, GeS2, and P2S5 becomes uniform and transitions into an amorphous state. In the intermediary zone, intermediate substances such as Li4GeS4 and Li3PS4 are formed. Finally, in the product stage, glassy-ceramic LGPS is generated. Nevertheless, the EIS investigation revealed that LGPS–HBM 520 min had the greatest ionic conductivity, 1.07 mS cm−1. The percentage of LGPS is 40%. The ionic conductivity of LGPS can be increased by post-heating at 575, 600, and 625 °C for 8 h. The highest ionic conductivity at room temperature was attained by LGPS 600 °C, 3.27 mS cm−1, with an LGPS concentration of 91.1%. Luo et al.66 replaced Ge with Sn to reduce costs and synthesized a solid electrolyte based on Li10SnP2S12. Li10SnP2S12 has an ionic conductivity of 4.79 mS cm−1 and electronic conductivity of 2.6 × 10−8 S cm−1 at room temperature. LiNi0.6Mn0.2Co0.2O2‖Li–In cells exhibit limited discharge capacity and poor SE compatibility with the electrode. The use of Li3InCl6 as a catholyte and a cathode protective layer results in an initial discharge capacity of 186.9 mA h g−1 at 60 °C. The combination of Li3InCl6 may separate the direct contact between ISE and cathode, reducing space charge effects and side reactions.
The incorporation of additive Al2O3 into the LGPS structure has been examined by Yabuzaki et al.80 The XRD study indicated no significant alterations in lattice parameters and Ge/P ratio for LGPS and LGPS–Al2O3. Increase the concentration of Al2O3 (3, 10, and 30 wt%) did not considerably increase the Al2O3 particles inside LGPS grains; nevertheless, when Al2O3 reached the maximum level in LGPS, the particles occupied the voids in LGPS. The ionic conductivity remained almost unchanged with varying Al2O3 concentrations: LGPS (5.4 mS cm−1); LGPS-3 wt% Al2O3 (5.7 mS cm−1); LGPS-10 wt% Al2O3 (4.8 mS cm−1); and LGPS-30 wt% Al2O3 (4.9 mS cm−1). Section 2.2.1 discusses the reactivity of sulfide to moisture.179 According to HSAB theory, oxygen is harder base than sulfur, binds with hard acid phosphorous and releases H2S. The ionic conductivity retention of LGPS–Al2O3 following air exposure surpasses that of LGPS. The substitution of oxide in LGPS enhances moisture stability. The moisture stability of LGPS–Al2O3 is attributed to the Al2O3 filling the voids of LGPS, which inhibits moisture penetration at the grain boundaries. Additionally, the reactivity of Al2O3 with H2O forms a passivation layer of Al(OH)3, inhibiting water residue and reactions at the LGPS interface. The electrolyte composite approach has the potential to serve as an option for preserving moisture stability while maintaining good LGPS conductivity.
![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 3m structure (Fig. 7b2). In most argyrodites, Li+ atoms occupy disordered T5 (48h) and/or T5a (24g) sites. Recent research suggests that partial occupancy of T2, T3, or T4 sites might improve ionic conductivity by providing shorter and more favorable pathways for Li+ diffusion, as occurs when P5+ is partly substituted by other cations.
3m structure (Fig. 7b2). In most argyrodites, Li+ atoms occupy disordered T5 (48h) and/or T5a (24g) sites. Recent research suggests that partial occupancy of T2, T3, or T4 sites might improve ionic conductivity by providing shorter and more favorable pathways for Li+ diffusion, as occurs when P5+ is partly substituted by other cations.
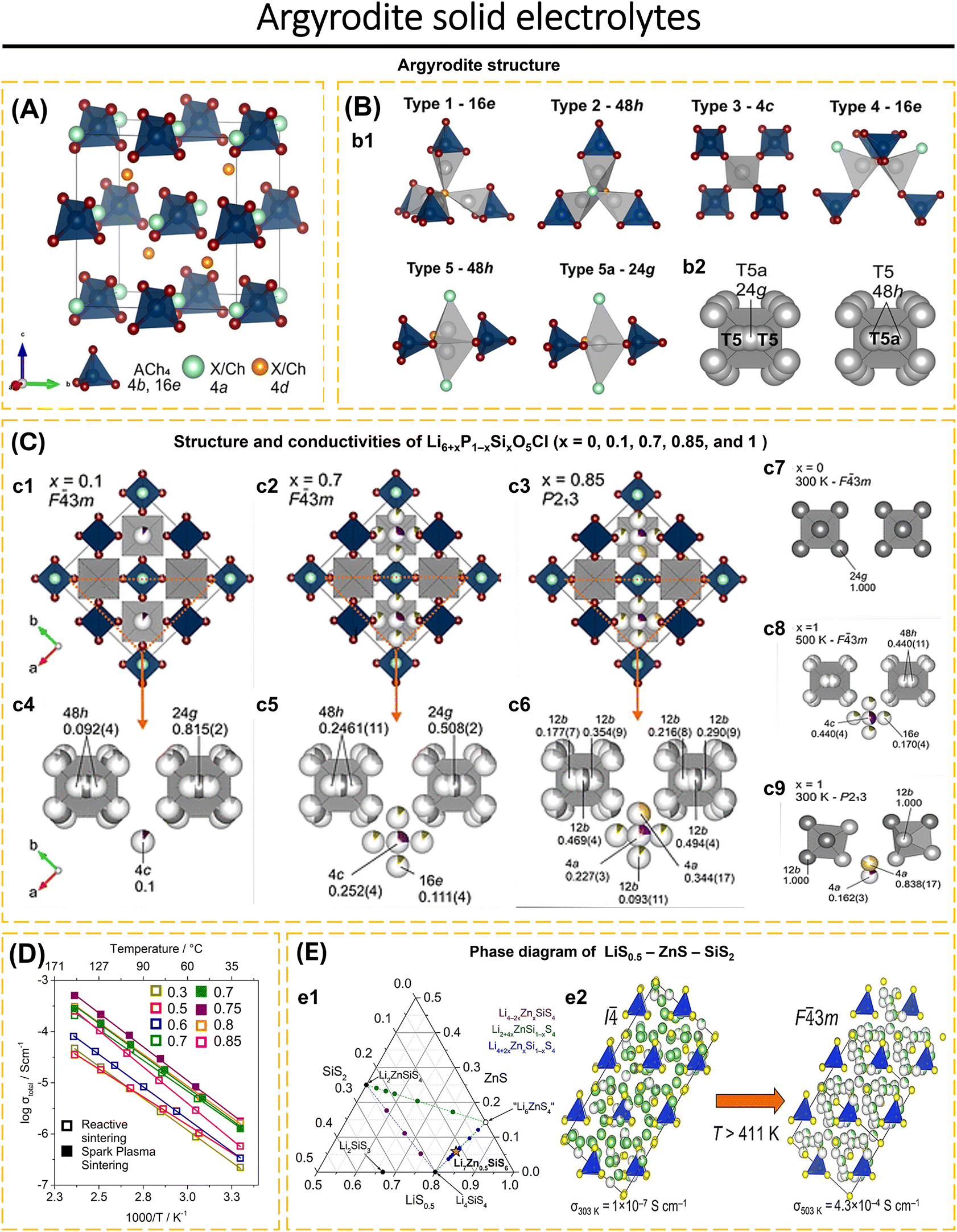 | ||
| Fig. 7 (A) Unit cell of Li6ACh5X (A = P, Si, and Al; Ch = O, S, and Se; X = Cl, Br, and I); Ch/X anions are tetrahedrally close-packed on Wyckoff site (4a, 4d, and 16e) forming 136 tetrahedral voids; four are occupied by A cations on the 4b site defining Ach4 tetrahedra (Ch: 16e). Lithium atoms are not shown. (B) (b1) Panels showing five types (T1, T2, T3, T4, and T5) of possible interstitial tetrahedral sites available for lithium occupancy and the trigonal bipyramidal T5a site. Lithium atoms are shown in gray. (b2) Octahedral Li+-ion cages consisting of T5 and T5a sites surrounding central anions on the 4d site; due to the proximity of T5 and T5a sites, the total occupancy over the two sites is constrained to unity. Reproduced with permission from ref. 88. Copyright 2022, American Chemical Society. (C) Structures of x = 0, 0.1, 0.7, 0.85, and 1 in Li6+xP1−xSixO5Cl highlighting the different Li sites occupied and associated symmetry settings as a function of composition and temperature in Li7SiO5Cl; atom and polyhedra colors: SiO4 tetrahedra (dark blue), O (dark red), Cl (light blue), Li-T5a site (dark gray), Li-T5 site (light gray), Li-T3 site (purple), Li-T4 site [green (16e/12b), and yellow (4a)]. Unit cell for (c1) x = 0.1, (c2) x = 0.7, and (c3) x = 0.85 (T5 and T5a Li atoms are omitted for clarity with octahedral cages shown in gray); orange triangle highlights the structural subsection shown in panels (c4–c6); (c7–c9) octahedral Li ion cages (T5 and T5a) surrounding central oxide anions and interstitial T3 and T4 sites with their site occupancy factor for (c4) x = 0.1, (c5) x = 0.7, (c6) x = 0.85, (c7) x = 0, (c8) x = 1, (500 K), and (c9) x = 1 (300 K). (D) Arrhenius plots for Li6+xP1−xSixO5Cl for samples prepared via reactive sintering (RS) (x = 0.3, 0.5, 0.6, 0.7, 0.8, 0.85) and SPS (x = 0.7, 0.75); reproduced with permission from ref. 88. Copyright 2022, American Chemical Society. (E) (e1) Pseudoternary LiS0.5–ZnS–SiS2 phase field was explored by synthesizing the compositions shown. Known phases are shown as black circles and are labeled. Purple, green, and blue circles show compositions explored on the Li4−2xZnxSiS4, Li2+4xZnSi1−xS4, and Li4+2xZnxSi1−xS4 tie lines, respectively. The orange star indicates the new Li7Zn0.5SiS6. (e2) High-temperature (T = 448 K) and room-temperature structures of Li7Zn0.5SiS6 show cubic close-packed layers of SiS44− polyanions stacked along the [111] and [011] directions, respectively. Atom colors: Li, green; Zn, gray; S, yellow; SiS4 tetrahedra, blue. Reproduced with permission from ref. 185. Copyright 2022, American Chemical Society. | ||
Morscher et al.88 synthesized a Li-rich oxide argyrodite, Li7SiO5Cl, with more than 6 Li atoms per formula unit. By substituting the P5+ by Si4+ in Li6+xP1−xSixO5Cl, to understand the distribution behavior of Li+ on the T5 (48h), T5a (24g), T3 (4c), and T4 (16e) sites which resembles the behavior of Li7SiO5Cl. The oxide was produced by ball milling a mixture of Li4SiO4, Li3PO4, Li2O, and LiCl under an argon environment, followed by heat treatment at 823 K for 3 hours. In addition, Li6+xP1−xSixO5Cl (x = 0.7, 0.75, and 0.8) was prepared by SPS. The cubic argyrodite structure is adopted by all compositions in Li6+xP1−xSixO5Cl. In Li6+xP1−xSixO5Cl, the O and Cl anions are arranged in a regular pattern without mixing between the O2− (4d) and Cl− (4a) anions, which is different from the commonly reported arrangement of S2− and Cl− anions in argyrodite sulfide. It is known that Li6+xP1−xSixO5Cl has 4 Li sites (48h, 24g, 4c, and 16e) compared to argyrodite sulfide. For x = 0.1 (F3m), the Li+ atom occupies three separate sites, 4c, 48h, and 24g (Fig. 7c1). Most of the Li+ atoms occupy the 24g and 48h sites, the remaining Li+ atoms occupy the 4c site (Fig. 7c4). While for x = 0, Li6PO5Cl, has only one site, 24g (Fig. 7c7). When x = 0.3–0.75, the occupancy of 24g and 48h sites increases, resulting in the disorder of Li+ sites in the octahedral cage and an increase in ionic conductivity in argyrodite. Li6+xP1−xSixO5Cl was the first to investigate the disorder of Li+ sites in argyrodite oxide at room temperature. Additional Li+ atoms introduced by cation substitution will occupy the 16e position sporadically (Fig. 7c2 and c5). This composition remains at 500 K with the crystal structure F3m (Fig. 7c8). At x = 0.8, the transition from F3m to P213 occurs (Fig. 7c3). For x = 0.8 to x = 0.85, site 4a (T4) shows a stronger occupancy tendency than site 12b (T4) (Fig. 7c6). The Li+ site arrangement for x = 0.8 and 0.85 (Fig. 7c3 and c6) is similar to that of Li7SiO5Cl at 300 K (Fig. 7c9), where only site 4a (T4) is occupied. Thus, controlling the composition of Li6+xP1−xSixO5Cl may explain the behavior of Li7SiO5Cl. Based on EIS analysis (Fig. 7d and Table 1), the Li6.75P0.25Si0.75O5Cl sample prepared via SPS shows the highest conductivity of 1.82 × 10−6 S cm−1 at 303 K with an activation energy of 0.522 eV. The Li6.75P0.25Si0.75O5Cl–SPS sample demonstrates electrochemical stability against Li metal, showing its capacity to transport Li-ions at a rate of 20 mA cm−2 (298 K) for 60 hours. Additionally, the sample exhibits stability at the Li/electrolyte interface. Li6PS5Cl shows instability after exposure to air for 1 hour. In contrast to Li6.7P0.3Si0.7O5Cl which does not show significant changes after contact with air for 1 hour. After exposure for 60 hours, Li6.7P0.3Si0.7O5Cl diffraction data indicates the presence of Li2CO3. The superior air stability of oxide argyrodite opens up opportunities for commercialization in ASSBs devices and the replacement of sulfide argyrodite electrolytes.
| Composition | Sintering method | Relative density (%) | Ionic conductivity (S cm−1) | Activation energy (eV) |
|---|---|---|---|---|
| 0.1 | RS | 73.4 | ∼10−9 | 0.687(13) |
| 0.3 | RS | 79.0 | 2.23(1) × 10−7 | 0.496(4) |
| 0.5 | RS | 74.5 | 3.53(4) × 10−7 | 0.433(4) |
| 0.6 | RS | 77.1 | 4.72(3) × 10−7 | 0.502(3) |
| 0.7 | RS | 79.7 | 1.16(4) × 10−6 | 0.478(11) |
| 0.7 | SPS | 96.0 | 1.63(4) × 10−6 | 0.50(1) |
| 0.75 | SPS | 95.1 | 1.82(1) × 10−6 | 0.522(5) |
| 0.8 | RS | 85.0 | 1.54(2) × 10−6 | 0.491(7) |
| 0.8 | SPS | 96.5 | 1.42(5) × 10−6 | — |
| 0.85 | RS | 78.4 | 4.93(4) × 10−7 | 0.564(8) |
Leube et al.185 first reported the argyrodite type of LiS0.5–ZnS–SiS2 phase mixture via solid-state reaction. According to the pseudoternary phase diagram of LiS0.5–ZnS–SiS2 (Fig. 7e1), the synthesis of Li4−2xZnxSiS4 (purple line) does not result in the formation of a new phase. Instead, only Li4SiS4 and Li2ZnSiS4 are formed. Likewise, the Li2+4xZnSi1−xS4 line (green line) only consists of a mixture of Li2ZnSiS4, Li4SiS4, Li2S, and ZnS. A new phase can be observed in Li4+2xZnxSi1−xS4 (blue line) at x = 0.267, despite the presence of certain impurities of Li2S and Li4SiS4. By increasing the composition to x = 0.33, a pure Li4.67Zn0.33S0.67S4 phase is obtained, without the presence of Li2S and Li4SiS4. The composition may be expressed as Li7Zn0.5SiS6, which represents a novel argyrodite phase with an I4 crystal structure (Fig. 7e2). This crystal structure is tetragonal under room temperature conditions. At a temperature of 411.1 K, the crystal structure transforms into an F3m structure (cubic). In structure I4, five Li sites (T5, T2, T4, T1, and T2a) are observed. In structure F3m, four Li sites (T5, T5a, T2, and T4) are observed. The substitution of Zn2+ in the lattice creates a jumping variation. At high temperatures, Zn2+ occupies the T5-48h position in the F3m structure, whereas at room temperature, it only partially fills the T5 position in the I4 structure. The transition from the ordered I4 structure to the disordered F3m is associated with a significant decrease in the activation energy to 0.34(1) eV above 411 K, while the conductivity increases from 1.0(2) × 10−7 S cm−1 (RT) to 4.3(4) × 10−4 S cm−1 (503 K).
Cation and anion doping of argyrodite increase ionic conductivity while also improving argyrodite stability with Li anodes and moisture stability.187,188 Arnold et al.16 reported F-doped Li5+yPS5Fy argyrodite and double dopants (F−/Cl− and F−/Br−) synthesized using a solution-based method. Li5+yPS5Fy argyrodites with various F levels (y = 0.1, 0.2, 0.5, 1.0, and 2.0) exhibit diffraction at 2θ = 25.5°, 30°, and 31.2°, indicating a cubic crystal phase (F3m). For y > 0.5, diffraction at 2θ = 38.6° and 65.6° indicates the existence of LiF. The Raman spectra of Li7PS6 and Li5+yPS5Fy (y = 0.1, 0.2, 0.5, 1.0, and 2.0) show a strong peak at 421–426 cm−1 indicates the stretching vibration of the P–S bond in PS43− (ortho-thiophosphate), a shift slightly owing to the replacement of the F− anion at the 16e site. The introduction of 9.5 wt% LiF in Li6PS5F (y = 1) shows the conductivity of 2.3 × 10−4 S cm−1 (Fig. 8a1). Li6PS5F0.5X0.5 with double halogen (X = Cl, Br) were synthesized using the same method. XRD analysis of Li6PS5F0.5X0.5 (X = Cl, Br) samples revealed 7.9 wt% LiF in Li6PS5F0.5Cl0.5. At room temperature, Li6PS5F0.5Cl0.5 and Li6PS5F0.5Br0.5 possessed ionic conductivities of 3.5 × 10−4 S cm−1 and 3.2 × 10−4 S cm−1, respectively. It concluded that double doping of Li6PS5F0.5X0.5 increased the ionic transport rate more than single doping (Fig. 8a2). Li6PS5F0.5Cl0.5 is stable to Li metal for up to 300 cycles (Fig. 8a3). Fig. 8b1 shows the ionic conductivity of Li6PS5I and hybrid-doped Li6PS5FxI1−x argyrodites at RT. Li6PS5F0.25I0.75 displays the highest ionic conductivity, 40% higher than that of Li6PS5I. The increase in ionic conductivity is due to the anion disorder caused by the double dopants F− and I−. The stability test of Li6PS5I, Li6PS5F0.75I0.25, and Li6PS5F0.25I0.75 with lithium anode at 0.05 mA cm−2 showed good cycle stability. The interfacial reaction between Li6PS5I and Li metal caused an increase in polarization-voltage up to the 100th cycle. While Li6PS5F0.75I0.25 and Li6PS5F0.25I0.75 had stable polarization-voltage. The concentration of F dopant impacts the stability of Li6PS5F0.75I0.25 in Li symmetric cells, demonstrating stability for up to 1100 hours (Fig. 8b2).92
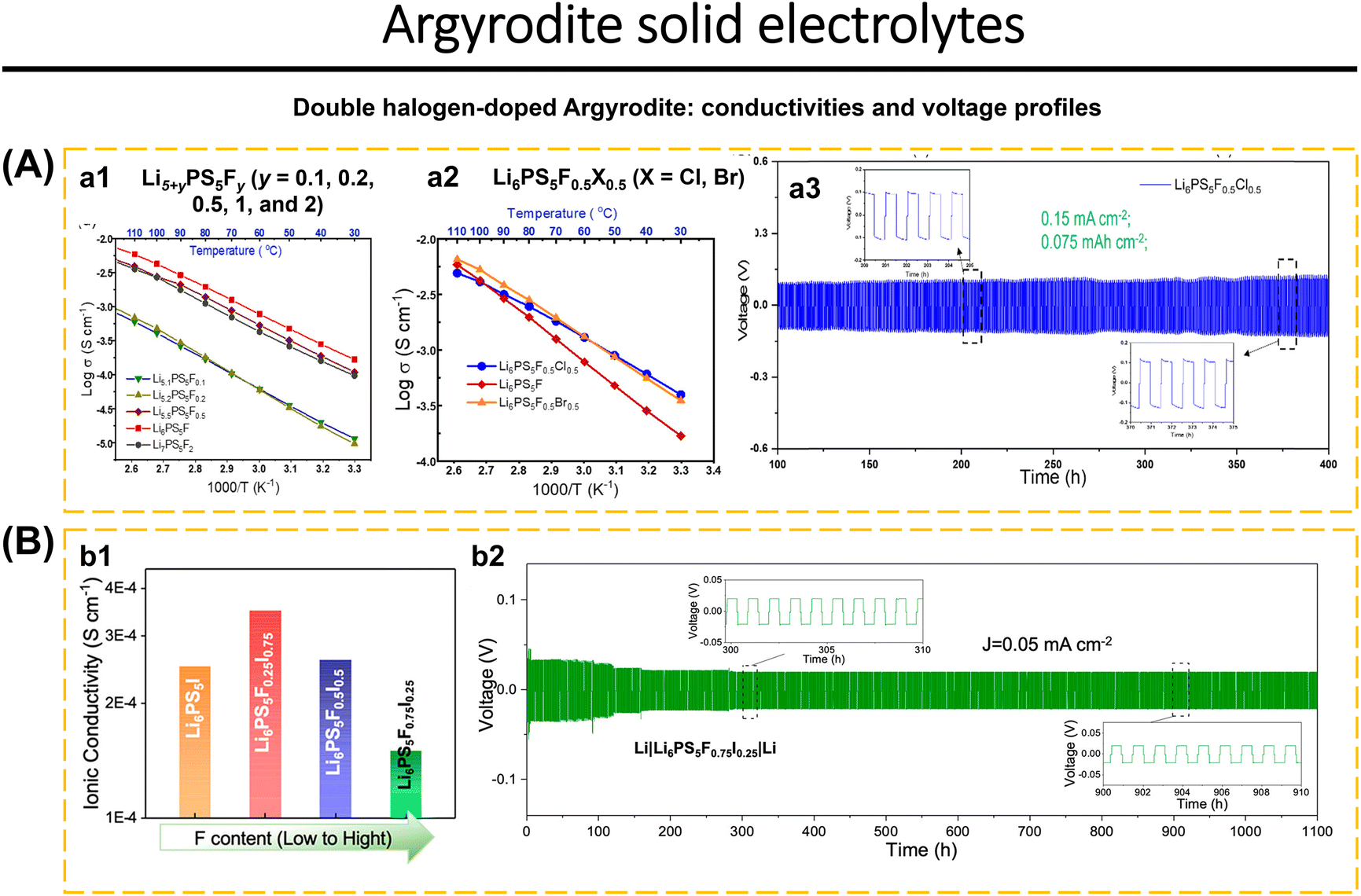 | ||
| Fig. 8 Double halogen-doped argyrodite. (A) (a1) Arrhenius plots of F-incorporated lithium argyrodites Li5+yPS5Fy (y = 0.1, 0.2, 0.5, 1, and 2) that were synthesized from a solvent-based method. (a2) Arrhenius plots of Li6PS5F0.5X0.5 samples (X = Cl, Br), in comparison with Li6PS5F. (a3) Voltage profiles of Li|Li6PS5F0.5Cl0.5|Li symmetric cell cycling under current density of 0.15 mA cm−2 (0.075 mA h cm−2). Reproduced with permission from ref. 16. Copyright 2022, American Chemical Society. (B) (b1) Composition dependent ionic conductivity of Li6PS5I and hybrid-doped Li6PS5FxI1−x argyrodites at RT. (b2) Voltage profiles of Li6PS5F0.75I0.25-based symmetric cell cycling up to 1100 h (current density of 0.05 mA cm−2). Reproduced with permission from ref. 92. Copyright 2023, American Chemical Society. | ||
2.3. Hydroborate-based ISEs
Hydroborate-type solid electrolytes provide attractive properties such as outstanding stability when combined with sodium and lithium metals, high thermal and chemical stability, soft mechanical properties, low crystallographic density, ease of processing, and low toxicity. In addition, hydroborates have an electrochemical stability window of around 4.0 V.6,51,52,59,69,70,84 Jin et al.70 presented a novel conjuncto-hydroborate compound, Na3B24H23, which exhibits a wide electrochemical stability window. The [B24H23]3− anion is produced by oxidizing [B12H12]2− using the ion-exchange technique. Na3B24H23–xNa2B12H12 was synthesized using the ball-milling method under the N2 atmosphere. The Na3B24H23–5Na2B12H12 composite, consisting of a double anion hydroborate, has an excess conductivity of 1.42 mS cm−1 at room temperature. It also demonstrates a Na-ion transference amount of 0.97 and a wide electrochemical window of 5.8 V vs. Na/Na+. These properties make it a promising candidate for use in high-voltage all-solid-state batteries. Braun et al.59 conducted an investigation that mixed LiCB11H12 and LiCB9H10 and examined how different ratios of these compounds affected the oxidative stability of the electrolyte. Li3(CB11H12)2(CB9H10) with a ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 shows a stability window of ∼3.9 V vs. Li/Li+ is lower than LiCB9H10-rich 1:2 ratio, with stability windows of ∼3.6 V vs. Li/Li+. An appropriate electrolyte can be fully integrated with a high-voltage NCM811 cathode. Meanwhile, Li-ion conductivity reaches 1.5 mS cm−1 at RT and 29.4 mS cm−1 at 60 °C. Electrochemical analyses of the NCM811‖LiIn cell show an initial discharge capacity of 145 mA h g−1 (RT) and 175 mA h g−1 (60 °C) at C/10. This cell showed capacity retention of 98% (RT) and 90% (60 °C) after 100 cycles at C/5. Excellent cycle stability was achieved using Li–In and graphite-based anodes. In addition, the formation of anion hydroborate dimers and TiO2 coating on the surface of NCM811 also contribute to the stability of the electrode–electrolyte interface (Fig. 9A and B).
:1 shows a stability window of ∼3.9 V vs. Li/Li+ is lower than LiCB9H10-rich 1:2 ratio, with stability windows of ∼3.6 V vs. Li/Li+. An appropriate electrolyte can be fully integrated with a high-voltage NCM811 cathode. Meanwhile, Li-ion conductivity reaches 1.5 mS cm−1 at RT and 29.4 mS cm−1 at 60 °C. Electrochemical analyses of the NCM811‖LiIn cell show an initial discharge capacity of 145 mA h g−1 (RT) and 175 mA h g−1 (60 °C) at C/10. This cell showed capacity retention of 98% (RT) and 90% (60 °C) after 100 cycles at C/5. Excellent cycle stability was achieved using Li–In and graphite-based anodes. In addition, the formation of anion hydroborate dimers and TiO2 coating on the surface of NCM811 also contribute to the stability of the electrode–electrolyte interface (Fig. 9A and B).
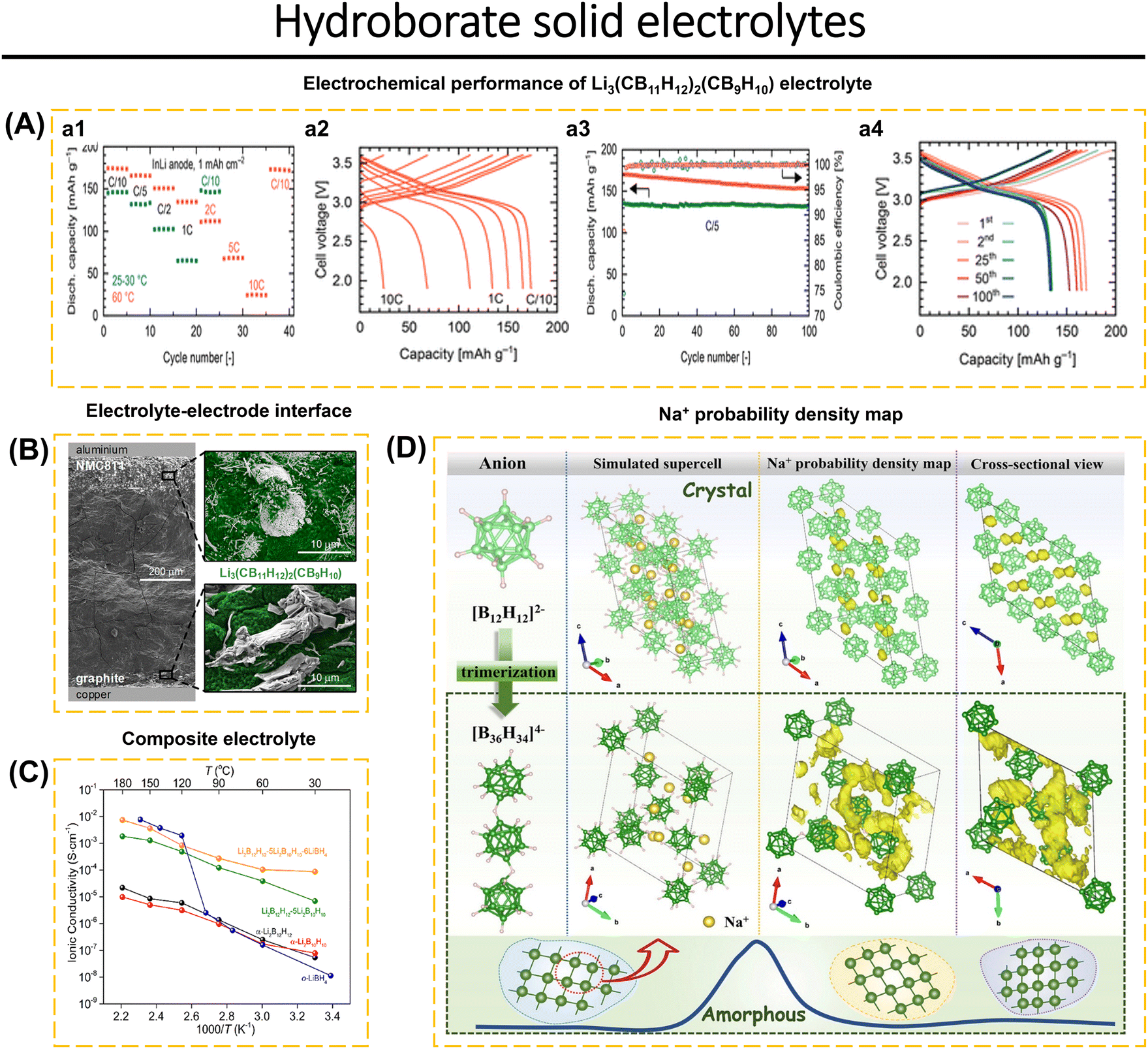 | ||
| Fig. 9 (A) Solid-state cell performance at room temperature (blue) and at 60 °C (red) with Li3(CB11H12)2(CB9H10) solid electrolyte, NCM811 composite cathode, and In–Li anode, in terms of (a1 and a2) rate capability, (a3 and a4) cycling stability at C/5. (B) SEM images of the cell cross-section with schematically added current collectors: aluminum current collector, cathode composite with NCM811 and solid electrolyte, solid electrolyte separator, anode composite with graphite and solid electrolyte, and copper current collector (top to bottom). Enlarged views show SEM images of NCM811 particles and vapor-grown carbon fibers (top) and graphite flakes (bottom) embedded in Li3(CB11H12)2(CB9H10) solid electrolyte (false-colored green). Current collectors are not drawn to scale. Reproduced with permission from ref. 59. Copyright 2024, American Chemical Society. (C) Lithium ionic conductivities σ(Li+) as a function of the temperature of Li2B12H12–5Li2B10H10–6LiBH4, compared to the Li+ conductivity of pristine α-Li2B12H12, α-Li2B10H10, o-LiBH4, and Li2B12H12–5Li2B10H10. Reproduced with permission from ref. 52. Copyright 2023, American Chemical Society. (D) Proposed structural models of Na4B36H34 and Na2B12H12 ([B36H34]4−, green; [B12H12]2−, light green; Na+, yellow) and the corresponding Na-ion probability density maps (yellow isosurface) from AIMD simulations at 500 K. All H atoms are omitted for clarity except for the B–H–B bridges. Reproduced with permission from ref. 69. Copyright 2024, American Chemical Society. | ||
Li2B12H12 and Li2B10H10 in the closo-borates class show relatively low conductivity of 10−8 S cm−1 at RT. However, after compositing, Li2B12H12–5Li2B10H10, its conductivity is still relatively low (10−5 S cm−1). In addition, fast decay and low coulombic efficiency are the disadvantages of the closo-borates class. Zhou et al.52 solved the ionic conductivity and instability problem of the Li2B12H12–5Li2B10H10 type closo-borate composite by introducing LiBH4. This modification increases the Li-ion conductivity to 1.0 × 10−4 S cm−1 at RT (Fig. 9C) and the electrochemical window reaches ∼3.0 V. This is supported by 7Li MAS NMR spectra data showing that the full-width at half maximum of Li2B12H12–5Li2B10H10–6LiBH4 (2.4 ppm) is smaller than Li2B12H12–5Li2B10H10 (5.2 ppm), which demonstrates the rapid diffusion of Li-ion in solid-state electrolytes based on Li2B12H12–5Li2B10H10–6LiBH4. In a recent study by Jin et al.69 a nonflammable compound called Na4B36H34 was produced. This compound was synthesized by oxidizing [B12H12]. At high temperatures, the compound forms the trimeric anion [B36H34]4− exhibiting an electrochemical stability range of up to 6.9 V. Density functional theory (DFT) investigation verified that the polyanion [B36H34]4− may lead to the expansion of interstitial sites, creating a favorable channel for the diffusion of sodium ions. This was further corroborated by the paddle-wheel effect of the polyanion [B36H34]4− (Fig. 9D). The Na4B36H34–7Na2B12H12 solid electrolyte exhibits numerous benefits within the hydroborate-SE class. These include low electronic conductivity (9.3 × 10−12 S cm−1), Na+ transference number of 0.97, ionic conductivity of 1.02 × 10−3 S cm−1, and an electrochemical stability window of 5.5 V. In a symmetric cell configuration (Na|Na4B36H34–7Na2B12H12|Na), it can maintain stability for over 200 hours at 0.1 mA cm−2. Additionally, it possesses favorable properties such as good compressibility, non-flammability, and decent mechanical properties. The application of Na4B36H34 as solid-state electrolytes is advantageous for high-voltage all-solid-state batteries.
2.4. Antiperovskite-based ISEs
Antiperovskite, reverse-charged versions of perovskite, with the general structure (X3AB, where X is an alkali cation, A and B are anions), is a highly potential solid electrolyte because it can be processed at low temperatures, has a wide electrochemical stability window, high ionic conductivity, good reduction stability, and low melting point.189–192 Lithium-rich antiperovskite-type solid electrolytes have garnered significant interest due to their large ion vacancy area and low activation energy barrier.32 Li-rich antiperovskite (LiRAP) materials demonstrate excellent ionic conductivity and high electrochemical stability when used with Li metal anodes.24,74,94 Antiperovskite has features that make it suitable for use as a protective layer for Li metal anodes, solid electrolyte films in thin film batteries, and solid electrolytes in all-solid-state batteries.189 Li3−x(OHx)Cl is an example of LiRAPs that exhibit instability in the presence of air and undergo decomposition to form LiOH and LiCl.24 Li3−x(OHx)Cl has a tetragonal structure at room temperature but transitions to a cubic form at 40 °C, this structure arises from the presence of lithium vacancies in the absence of cation or anion doping (Fig. 10a5). The Li3−x(OHx)Cl compound has a conductivity of around 7.1 × 10−6 S cm−1 when not exposed to air (Fig. 10a1). After being subjected to ambient air conditions (25 °C, 25 ± 5% RH) for 1 minute, the conductivity rises by 1.2 × 10−4 S cm−1 (Ea = 0.45 eV) and reaches 3.3 × 10−3 S cm−1 after 60 minutes (Fig. 10a2–a4). However, non-Arrhenius behavior occurs as the temperature goes over 60 °C, causing a reduction in the ionic conductivity of Li3−x(OHx)Cl. Fig. 10a6 indicates that prior to being exposed to open air, Li3−x(OHx)Cl exhibits significant resistance in both its bulk and grain boundaries. By extending the duration of exposure, moisture may be absorbed via the grain borders, resulting in a reduction in grain boundary resistance. After 60 minutes, all grain boundaries are fully wetted, and the Nyquist plot only shows the Warburg resistance, while the bulk resistance is attributed to ionic migration in the aqueous solution. Li3−x(OHx)Cl was determined to be unstable in the presence of air. The hygroscopic features can be decreased by introducing 0.1 molar F on the Cl site. This weakens the hydrogen bonds between LiRAPs and H2O, resulting in the formation of Li3−x(OHx)Cl0.9F0.1. This compound exhibits resistance to moisture, with a water content of 0.27 wt% for pellets and 0.93 wt% for powder. Unlike Li3−x(OHx)Cl, Li3−x(OHx)Cl0.9F0.1 exhibits an enhancement in conductivity until a duration of 60 minutes (2.9 × 10−5 S cm−1).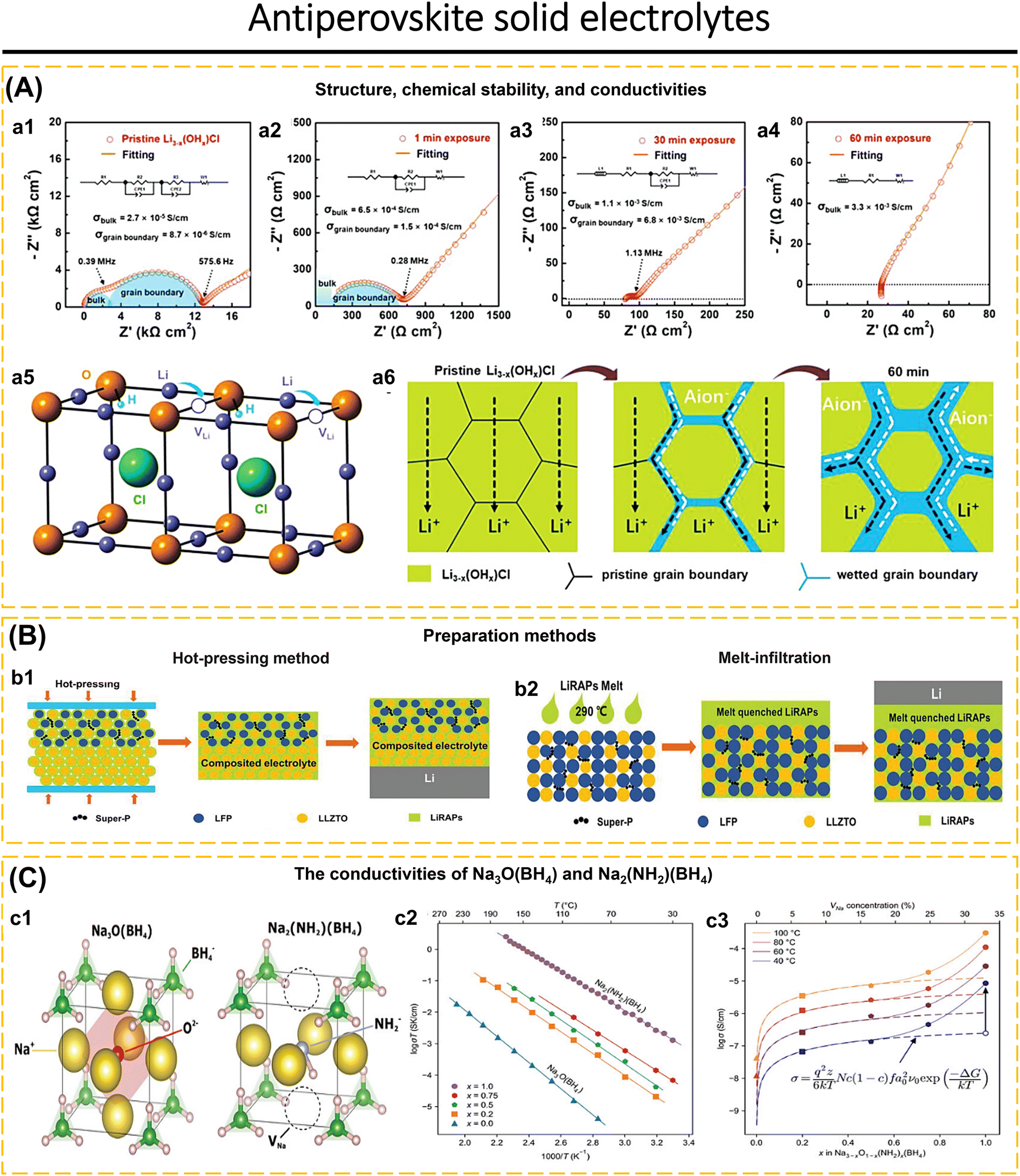 | ||
| Fig. 10 (A) The effects of hygroscopicity on ionic conductivity. (a1–a4) EIS profiles of Li3−x(OHx)Cl with different expose durations: (a1) pristine, (a2) 1 min, (a3) 30 min, (a4) 60 min. (a5) Crystal structure of Li3−x(OHx)Cl. (a6) Temporal evolution of Li-ion conducting mechanisms in Li3−x(OHx)Cl. (B) Schematics of the battery assembling through hot-pressing (b1) and melt-infiltration (b2). Reproduced with permission from ref. 24. Copyright 2023, Wiley-VCH. (C) Sodium ion transport in Na3−xO1−x(NH2)x(BH4) (x = 0–1) upon systematic substitution of O2− by NH2−, with corresponding increase in Na+ vacancy concentration. (c1) Powder X-ray diffraction (PXRD) shows that the whole series of compositions Na3−xO1−x(NH2)x(BH4) form cubic antiperovskites with space group Pmm at room temperature. Between the endmember compositions Na3O(BH4) and Na2(NH2)(BH4), the Na+ vacancy concentration increases from zero to one-third at the full substitution of O2− by NH2−. In the cubic unit cell Na3O(BH4) (c1, left), Na-ions at the face-centered sites comprise the vertices of the Na6O octahedra, and (BH4−) cluster anions occupy the corner sites. In the cubic unit cell Na2(NH2)(BH4) (c1, right), two of the six Na-ions are missing, and the O2− ion residing at the center of the pseudo-octahedron is replaced with an NH2− anion. (c2) Temperature-dependent ionic conductivity of Na3−xO1−x(NH2)x(BH4) (where x = 0, 0.2, 0.5, 0.75, and 1.0) exhibits Arrhenius behavior above the room temperature region and ionic conductivity increases with the substitution of O2− by NH2−. Activation energies are 0.358, 0.299, 0.301, 0.278, and 0.268 eV, respectively. The endmember composition Na2(NH2)(BH4) with the highest Na+ vacancy and NH2− cluster anion concentration shows 104 higher ionic conductivity than that of the endmember Na3O(BH4) without the substitution of NH2−. (c3) The dependence of ionic conductivity on the concentration of Na+ vacancy and NH2− cluster ion concentration at several temperatures between 40 °C and 100 °C is fitted as the dashed line using the vacancy-dependent ionic conductivity model (inset equation). At low x (≤0.75), the ionic conductivity can be fit well by the classical vacancy-concentration model. At high x (>0.75), the ionic conductivity of Na3−xO1−x(NH2)x(BH4) exhibits a large positive deviation from the expected classical vacancy-dependent conductivity. It is correlated with not only high vacancy concentration but also the NH2− cluster anion concentration, suggesting a paddle-wheel effect. Reproduced with permission from ref. 26. Copyright 2022, Wiley-VCH. | ||
The mechanical softness and low melting point (274 °C) of Li3−x(OHx)Cl0.9F0.1 antiperovskite make it applicable as a composite material for ASSBs electrodes in two ways. The first technique used is the hot-pressing method, where a layer of antiperovskite material is applied onto the surfaces of LFP and LLZTO (Li6.75La3Zr1.75Ta0.25O12). Antiperovskite may function as an adhesive, enhancing the contact between LFP and LLZTO, and reducing strain on the cathode. The cathode and electrolyte are combined using the hot-pressing technique at a temperature of 290 °C. This is followed by the addition of Li metal on the opposite side (Fig. 10b1). The second technique involves the melt-infiltration method, in which the antiperovskite is heated to 290 °C and then poured in excess onto the cathode composite mixture to fill the cathode cavity. This results in the formation of a solid antiperovskite membrane over the cathode composite. The membrane is subsequently collected along with Li metal (Fig. 10b2). The initial discharge capacity of the two manufacturing processes is competitive, with 1st-type ASSBs exhibiting a capacity of about 83.6 mA h g−1 and 2nd-type ASSBs exhibiting a capacity of approximately 70.8 mA h g−1 at a rate of 0.05C.
The ionic conductivity of Li2OHCl1−xBrx (x = 0, 0.1, 0.3, 0.5, 0.7, 0.9, and 1) antiperovskite electrolyte was investigated by Lee et al.31 The crystal structure of Li2OHCl1−xBrx is significantly influenced by the Br to Cl ratio, as proven by X-ray diffraction studies. This ratio directly affects the conductivity of Li ions inside the grains. It can be deduced that both the Li2OHCl and Li2OHCl0.9Br0.1 structures are defect-rich and have a distorted structure, as shown by the reduction in the Li–O distance around the Schottky defect. In the Li2OHCl structure, the Li–O distance decreases from 1.926 Å to 1.814 Å (a decrease of 0.112 Å), while in the Li2OHCl0.9Br0.1 structure, the Li–O distance decreases from 1.929 Å to 1.839 Å (a decrease of 0.09 Å). The Li2OHCl0.9Br0.1 compound exhibited a higher ionic conductivity (2.52 × 10−3 mS cm−1) compared to Li2OHCl (1.93 × 10−3 mS cm−1). While the replacement of Br may maintain the cubic crystal structure under room temperature conditions, a high ratio of Br to Cl might hinder the migration of Li-ions owing to the larger size of the Br ion. After 7 days, EIS data indicates that the interfacial resistance between the solid electrolyte and Li metal in the Li|Li2OHCl0.9Br0.1|Li cell has increased to 705 Ω cm2 at 80 °C. After 200 hours at 80 °C, the voltage profile of the Li–Li symmetric cell remains constant and does not show any increase in polarization.
Tsai et al.,26 Ahiavi et al.,193 and Sun et al.194 elucidate the “paddle-wheel” effects, which enhance the movement of cations in solid electrolytes. Structures that include cluster anions, also known as polyanions, complex anions, or rotor groups, such as sulfate (SO42−), nitrite (NO2−), borohydride (BH4−), phosphate (PO43−), aluminum hydride (AlH4−), thiophosphates (PS43−), closo-borates and carba-closo-borates, etc.195 These structures have a larger ion radius and create a “paddle-wheel” effect, where the rotation of the anion group facilitates the movement of cations with a lower energy barrier for migration, resulting in enhanced cation mobility. Cluster anions can enhance the conductivity of cations compared to configurations containing single atoms that are similar in nature. Systems containing numerous types of anion clusters are believed to exhibit various ways of motion using multiple paddle wheels and cations. Antiperovskite is a suitable framework for investigating the “paddle-wheel” phenomenon due to its ability to include various ionic substitutions, including cluster anions. The research published by Tsai et al.26 showcased the utilization of sodium oxide borohydride (Na3O(BH4)) with a unique antiperovskite structure consisting of a single umbrella wheel. In this structure, the oxide anion fraction (O2−) was substituted with an amide cluster anion (NH2−) (Fig. 10c1). The compound Na3−xO1−x(NH2)x(BH4) (where x = 0, 0.2, 0.5, 0.75, 1.0) was synthesized using a solid-state reaction method. The PXRD analysis reveals that the primary phases of Na3O(BH4) and Na3−xO1−x(NH2)x(BH4) exhibit a cubic crystal structure (Pmm). Furthermore, when the value of x rises, the lattice parameters likewise increase. Borohydride cluster anions are located at the corners of a cube, whereas amide/oxide anions are located in the center of the cube. Sodium cations are positioned at the center of each face of the cube, forming an octahedron. At x = 1, there are two vacant octahedral sites, and all body center sites are occupied by amide cluster anions. An increase in the concentration of the amino group anion (NH2−) leads to a corresponding rise in both the ionic conductivity and the concentration of cation vacancies. The NH2− and BH4− species have a Na-ion conductance that is 100 times greater when x = 1 (Fig. 10c2). Despite the conductivity values plotted with the sodium vacancy concentration exhibiting no correlation between the two (Fig. 10c3). Extensive investigation and comprehension of the interactions between cluster anions and cations are crucial for the advancement of solid-state electrolytes in the future.
2.5. Halide-based ISEs
Halides exhibit high conductivity (>1 mS cm−1 @ RT), oxidation stability (∼4 V), better performance under high voltage, and temperature processability.43,196 Several articles have described the division of halide electrolytes. Based on the type of metal element, halides are divided into three categories, i.e., LiaMXb halide with group 3 elements (Sc, Y, La–Lu), LiaMXb halide with group 13 elements (M = Al, Ga, In), and LiaMXb halide with divalent metal elements (M = Ti, V, Cr, Mn, Fe, Co, Ni, Cu, Zn, Cd, Mg, Pb).45 Huang et al.43 describes the division of halide division, which is Li3M3+X6 (M3+ = In, Sc, Ti, Y, Ho, Er, Yb and X = Cl, Br, I), Li2M4+X6 (M4+ = Zr, Hf and X = Cl, Br, I), and LiM5+Cl4O (M5+ = Nb, Ta). Based on the sublattice structure of halogen anions for ternary halides: CCP (cubic Fdm symmetry and monoclinic C2/m symmetry) and HCP (trigonal Pm1 symmetry and orthorhombic Pnma symmetry).197 Park and co-worker179 investigated the Li-ion transport pathways in Li3MCl6 (where M = Bi, Dy, Er, Ho, In, Lu, Sc, Sm, Tb, Tl, Tm, and Y) utilizing BSVE and AIMD methods. The analysis revealed that the monoclinic structure exhibited the lowest migration energy barrier, approximately 0.5 eV, compared to the orthorhombic and trigonal structures. AIMD simulations indicated that the orthorhombic and trigonal structures exhibited slow ion migration within the 2D plane, leading to low ionic diffusion and increased activation energy. The monoclinic structure shows an Oct–Tet–Oct transport mechanism along both the 2D (ab-plane) and 3D (cross-layer) pathways. In the trigonal and orthorhombic structures, an Oct–Oct transport mechanism is observed along the 1D path, while an Oct–Tet–Oct mechanism is present along the 2D path (Fig. 11a1–a3).
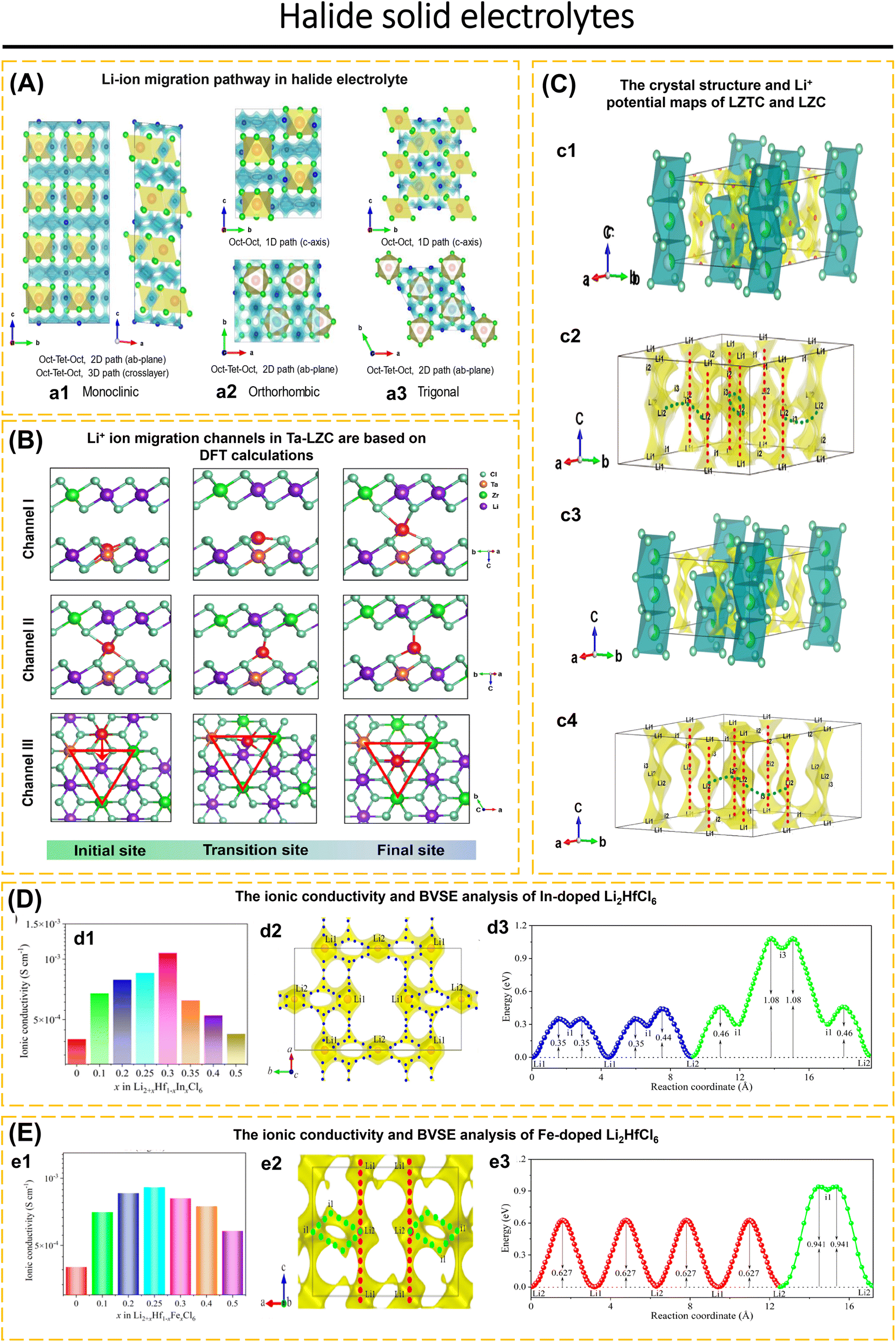 | ||
| Fig. 11 (A) Li-ion migration pathway in (a1) monoclinic, (a2) orthorhombic, and (a3) trigonal Li3MCl6 obtained using the BVSE method. Blue atom and yellow polyhedral correspond to Li and MCl6, respectively, while the isosurface with light blue corresponds to the ionic migration path. The monoclinic phase (CCP anion sublattice) exhibits two-dimensional intra-layer and three-dimensional cross-layer paths between octahedral sites via tetrahedral interstitial sites (Oct–Tet–Oct), while trigonal and orthorhombic phase (HCP anion sublattice) exhibit anisotropic paths including one dimensional path along c-axis between octahedral sites (Oct–Oct) and two-dimensional migration path along ab plane between octahedral sites via interstitial tetrahedral sites (Oct–Tet–Oct). Reproduced with permission from ref. 179. Copyright 2020, American Chemical Society. (B) The key local environment fragments of three Li+ ion migration channels in Ta-LZC are based on DFT calculations. The red ball is highlighted to illustrate the migrations of Li+ ions in three different channels. Concisely, channel I for Li+ ion conduction pathway in c direction. Channel II for Li+ ion conduction pathway along with a and b directions in layer B. Channel III for Li+ ion conduction pathway in a and b directions of layer A. (C) (c1) The crystal structure of LZTC superimposed with the Li+ potential map. (c2) Li+ migration pathways of LZTC. (c3) The crystal structure of LZC is superimposed with the Li+ potential map. (c4) Li+ migration pathways of LZC. Reproduced with permission from ref. 46. Copyright 2024, Elsevier. (D) (d1) The Li-ion conductivities of Li2+xHf1−xInxCl6 (0 ≤ x ≤ 0.5). (d2) Li-ions migration pathways within the LHIC crystal structure along with the ab plane. (d3) Energy barrier profile of various migration pathways within the LHIC crystal structure, and i1 and i3 represent the different interstitial tetrahedra (Tet) sites. Reproduced with permission from ref. 198. Copyright 2023, Elsevier. (E) (e1) the Li-ion conductivities Li2+xHf1−xFexCl6 (0 ≤ x ≤ 0.5). (e2) Diffusion pathways of the Li-ion within LHFC crystalline structure through the face-sharing lithium-centered octahedron along the c axis. (e3) Energy barrier profile of different Li-ion diffusion pathways within the LHFC crystalline structure. Reproduced with permission from ref. 199. Copyright 2024, American Chemical Society. | ||
The ionic conductivity enhancement of halide-based ISEs is affected by the types of substituted aliovalent cations and their respective substitution ratios.43,45,56,198–200 A novel halide structure, Li2−xZr1−xTaxCl6 (0 ≤ x ≤ 0.7), was synthesized through high-energy ball-milling.46 The LZC structure consists of two layers: layer A (LiA), which is occupied by ZrCl62− and LiCl65− octahedra, and layer B (LiB), positioned between layer A. The migration route of Li-ions can be explained through three primary channels: channel I represents the conduction path of Li-ions in the c direction; channel II denotes the conduction path along the a and b directions within layer B; and channel III indicates the conduction path in the a and b directions of layer A. The integration of the three channels establishes the three-dimensional migration pathway in LZTC (Fig. 11b). The [Li1–Li2–Li1] transport pathway in the c direction is the most favorable for LZC and LZTC. Nonetheless, the migration barriers will vary as a result of Ta doping. Ta5+ doping in LZC decreases the migration barriers for Li-ions along the [Li1–Li2–Li1] pathway in the c direction and the [Li2–Li3–Li2] pathway in the ab plane. The migration barriers of LZTC are lower than those of LZC by 0.849 eV and 0.988 eV, respectively. The substitution of Ta5+ is concluded to decrease the migration barriers of Li+ and enhance the transport of Li+ within the crystal lattice (Fig. 11c1–c4).
Li2HfCl6 type halides were synthesized through mechanical milling, with partial substitution of Hf4+ atoms by M3+ ions (Fe3+, V3+, Cr3+, and In3+).198,199 The variation in atomic radius significantly influences the structure of the bare LHC, specifically Hf4+ (71 pm), Fe3+ (65 pm), Cr3+ (61.5 pm), V3+ (64 pm), and In3+ (94 pm). The optimal percentage and ionic conductivity of M3+ substituted Li2+xHf1−xMxCl6 are as follows: Cr3+ (x = 0.2, 0.81 mS cm−1), V3+ (x = 0.2, 0.64 mS cm−1), In3+ (x = 0.3, 1.05 mS cm−1) (Fig. 11d1) and Fe3+ (x = 0.25, 0.91 mS cm−1) (Fig. 11e1). The substitution of Hf4+ with M3+ (where rHf4+ < rM3+) will result in an expansion of the Li-ion diffusion barrier. Conversely, if rHf4+ > rM3+, the pure LHC crystal lattice will contract, leading to a disruption in the distribution of Li-ions. Optimization of the concentration of substituted M3+ is essential; excessive amounts can reduce ionic conductivity due to a deficiency of vacancies in the LHC structure. The diffraction pattern of LHC is identical to that of Li3YCl6 (P3m1). Substituting Fe (0 ≤ x ≤ 0.5) into LHC results in trigonal Li3YCl6. In the case of In3+ substitution in LHC, if In3+ (x > 0.3) leads to a monoclinic Li3InCl6 structure. The In3+-substituted LHC exhibits a diffusion path of [Li1–Tet.1–Li1] in the ab plane with an energy barrier of 0.35 eV. Furthermore, an additional alternative path [Li1–Tet.1–Li2] is linked with the [Li2–Tet.1–Tet.3–Tet.1–Li2] chain to establish a three-dimensional network (1.08 eV) (Fig. 11d2 and d3). In the case of LHC, an energy barrier of 1.31 eV exists between two adjacent ab planes. The Fe3+-substituted LHC exhibits a diffusion path of [Li2–Li1–Li2] along the c direction, characterized by an energy barrier of 0.627 eV. This path is interconnected with the [Li2–Tet.1–Li2] path, resulting in a three-dimensional diffusion network with an energy barrier of 0.941 eV (Fig. 11e2 and e3). The energy remains lower than that of pure LHC and In3+-doped LHC. Aliovalent substitution induces a rearrangement of Li-ions and an anisotropic distortion of the local structure, resulting in a reduction of migration barrier energy. This process facilitates the formation of efficient Li-ion diffusion pathways and enhances ionic conductivity.
Li3−xY1−xHfxCl6 (0 ≤ x < 1) were synthesized through mechanical milling.200 Pristine LYC and Hf-doped LYC exhibited a uniform and interconnected particle size of less than 10 μm, characterized by a porous structure. The diffraction pattern of LYC closely resembled the space group P3m1. The soft nature and low crystallinity of LYHC facilitated its processing into dense pellets through cold pressing. LYHC exhibited an ionic conductivity of 1.49 mS cm−1, surpassing that of pristine LYC, which measured 0.139 mS cm−1. In LYHC, the radius of the doping ion Hf4+ (71 pm) is smaller than that of Y3+ (90 pm). The substitution of Hf4+ (x > 0.6) in LYC results in a narrowing of the crystal lattice and ionic migration channels, leading to a reduced concentration of Li-ions. Analysis of the Bond Valence Energy Landscape indicates Li+ migration LYHC. The transition from Li1 (6h) to Li2 (6g) may occur along the c-axis. The Li1 site exhibits six-fold coordination perpendicular to the z-axis and is situated between the Y1–Cl6 octahedra. Li2 atoms are linked to other Li2 atoms in the a or b direction. The percolation energy calculated along the three axes is uniform at 2.74 eV. Hf-doped LYC exhibits a percolation energy of approximately 2.8 eV, facilitating the three-dimensional diffusion of Li-ions.
The combination of good deformability, high Ionic conductivity, and electrochemical stability allows for the synthesis of a novel group of halides with heterogeneous structures by combining high- and low-coordinated halide frameworks. Fu et al.30 synthesized a compound by combining the UCl3-type halide framework, which is based on a high-coordination framework with a coordination number of more than 6, such as Na3xM2−xCl6 (M = La, Sm), and a low-coordination framework represented by scattered [MCl6]x− octahedra, such as NaTaCl6 (LCF-Ta). The arrangement of Na0.75Sm1.75Cl6 (HCF-Sm) and Na0.75La1.75Cl6 (HCF–La) is shown as a tricapped trigonal prism, with the Na atom occupying the octahedral site and the La or Sm atoms occupying the other sites (Fig. 12a1). The LCF-Ta crystal consists of an octahedral framework [TaCl6]− positioned at the corner of the unit cell (P21/c). This framework shares an edge with two [NaCl7]6− closed trigonal prisms. The mixing of HCF and LCF leads to the formation of new heterogeneous structures, the study revealed that the optimal ionic conductivity was 2.7 mS cm−1 for the combination of 0.62[HCF-Sm]·0.38[LCF-Ta], and 1.8 mS cm−1 for the combination of 0.57[HCF-La]·0.43[LCF-Ta], both at a temperature of 25 °C.
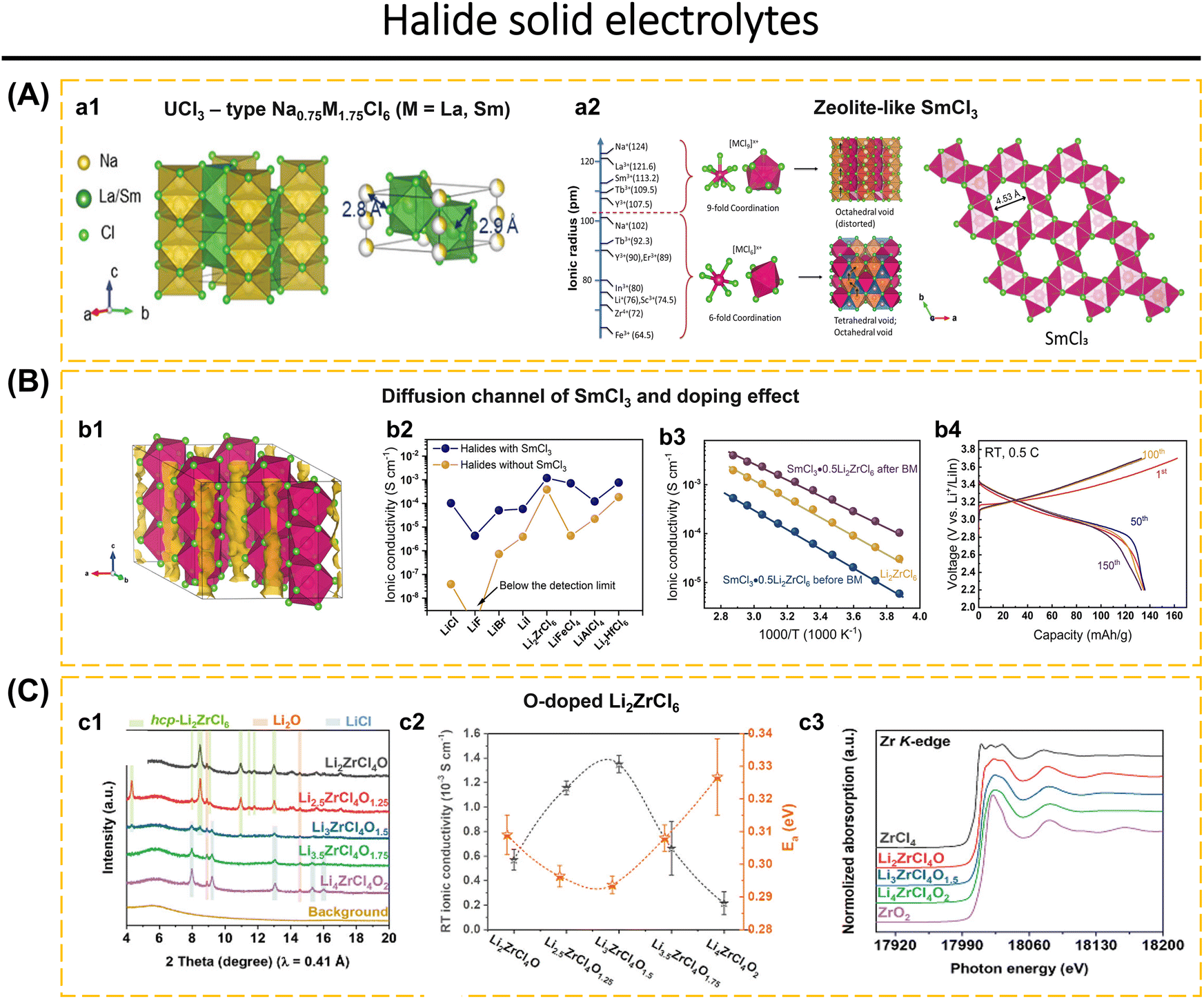 | ||
| Fig. 12 (A) (a1) The crystal structure of UCl3-type Na0.75M1.75Cl6 (M = La, Sm). Reproduced with permission from ref. 30. Copyright 2023, Wiley-VCH. (a2) Porous framework structure based on halides. Relationship between the ionic radius of M and the coordination of [MClx] polyhedra. The black arrows on the crystal structures indicate the Li+ diffusion pathway. Top view of the SmCl3 lattice along the c axis to show the existing plentiful channels with an inner diameter of 4.53 Å. Reproduced with permission from ref. 2. Copyright 2023, American Chemical Society. (B) (b1) Crystal structure of SmCl3 superimposed with Li+ probability density (yellow iso-surface) from AIMD simulations of Li0.17SmCl3 at 300 K. Reproduced with permission from ref. 2. Copyright 2023, American Chemical Society. (b2) Ionic conductivities of the SmCl3 framework coupled with different halide adsorbents at 30 °C. (b3) Arrhenius-plots of Li2ZrCl6 and the SmCl3·0.5Li2ZrCl6 (before and after ball-milling). (b4) Charge and discharge voltage profiles of the ASSLIB using LiNi0.83Mn0.06Co0.11O2 (NCM83) as the cathode and SmCl3·0.5Li2ZrCl6 as the ISE layer at different cycles (1st, 50th, 100th, and 150th). Reproduced with permission from ref. 2. Copyright 2023, American Chemical Society. (C) (c1) SXRD patterns of the Li2+2xZrCl4O1+x (x = 0, 0.25, 0.5, 0.75, and 1). (c2) Comparison of ionic conductivities and activation energies of the Li2+2xZrCl4O1+x. (c3) XANES spectra of the Li2ZrCl4O, Li3ZrCl4O1.5, and Li4ZrCl4O2 at the Zr K-edge, respectively. ZrCl4 and ZrO2 are reference samples. Reproduced with permission from ref. 57. Copyright 2024, American Chemical Society. | ||
A novel class of zeolite-like halide framework called SmCl3 was introduced by Fu et al.2 The ionic radius of metal atoms in Li-M-Cl halides, such as Li3InCl6, Li3YCl6, and Li2ZrCl6, are in close proximity. Specifically, Zr4+ (72 pm), Li+ (76 pm), In3+ (80 pm), and Y3+ (90 pm) form an octahedral framework [MCl6]x−. When the ionic radius of the atom M is raised, specifically for M = Sm3+ (113.2 pm) and La3+ (121.6 pm), the halide structure undergoes a transition from 6-fold coordination to either 8-fold or 9-fold coordination. This transition occurs in a P63/m crystal lattice and is accompanied by the formation of 1D vacancy channels along the c-axis. The obtained pore size is comparable to the pore size of zeolite (Fig. 12a2). Zeolite-like SmCl3 framework has a one-dimensional channel that is surrounded by [SmCl9]6− structures, allowing for a small distance of 2.08 Å between two octahedral structures, enabling the transit of Li-ions. AIMD simulations indicate that the Li+ diffusion is facilitated by the presence of several tunnels (between distorted octahedral) promoting efficient diffusion pathways for mobile ions (Fig. 12b1). BM-SmCl3, produced through the ball-milling method, can be combined with halide species like LiCl, LiF, LiBr, LiI, Li2ZrCl6, LiFeCl4, LiAlCl4, and Li2HfCl6 to improve the ionic conductivity (Fig. 12b2). The solid electrolyte BM-SmCl3·0.5Li2ZrCl6, which exhibits the highest ionic conductivity, is suitable for use as a solid electrolyte in ASSBs and this solid electrolyte demonstrates stability for up to 150 cycles (Fig. 12b3 and b4).
Zhang et al.57 investigated the impact of the amorphous structure on the mechanical deformability and Li+ conduction of the quasi-crystalline Li2ZrCl6 structure, which was partly replaced by O at the Cl site. The Li2+2xZrCl4O1+x ISE (where x = 0, 0.25, 0.5, 0.75, and 1) is generated by adjusting the proportion of ZrCl4 and Li2O in the synthesis process. Raising the quantity of Li2O decreases the formation of crystalline hcp-Li2ZrCl6. When the value of x is 0.5, the level of amorphization is 89.5%, and there are minor impurities, including hcp-Li2ZrCl6, LiCl, and Li2O. The substitution of O reaches 42.9%. The Li3ZrCl4O1.5 solid electrolyte has the highest ionic conductivity, measured at (1.35 ± 0.07) × 10−3 S cm−1, and the lowest activation energy of 0.294 ± 0.003 eV (Fig. 12c2). Li3ZrCl4O1.5 ISE has a much lower electronic conductivity of 7.10 × 10−10 S cm−1 compared to Li2ZrCl6, which has an electronic conductivity of 5.61 × 10−9 S cm−1. Zr K-edge FT-EXAFS spectroscopy is utilized to quantitatively determine the coordination system around the Zr atom in Li2+2xZrCl4O1+x ISEs (x = 0, 0.5, and 1). Increasing the value of x leads to an increase in Zr–O intensity, indicating a higher degree of anion replacement of Cl by the O anion (Fig. 12c3). Consequently, the Cl anion combines with the Li cation to form a LiCl compound, which is in agreement with the SXRD results (Fig. 12c1).
Li et al.54 reported a crystal structure called ternary halide, which is formed by combining LaCl3 and CeCl3. This crystal structure follows the UCl3 type structure with space group P63/m. It has one-dimensional hexagonal channels that may hold monovalent cations such as Li+, Na+, K+, Cu+, and Ag+. The UCl3-type superionic chloride SE is composed of LiCl, LaCl3, CeCl3, AlCl3, TaCl5, ZrCl4, and HfCl4. The ionic conductivity values for different combinations of halides are as follows: Li–LaCeHfAlTa–Cl (1.3 mS cm−1), Li–LaCeZrHfTa–Cl (1.8 mS cm−1), Li–LaCeZrAlTa–Cl (1.0 mS cm−1), Li–LaCeZrHfAlTa–Cl (1.1 mS cm−1). The activation energy of certain UCl3-type chloride solid-state electrolytes is quite low. Examples include Li–LaCeZrHfTa–Cl (0.318 eV), Li–LaCeZrAlTa–Cl (0.336 eV), Li–LaCeHfAlTa–Cl (0.364 eV), Li–LaCeZrHfAlTa–Cl (0.383 eV), Li–LaZr–Cl (0.404 eV), and Li–La–Cl (0.511 eV). The modification is achieved by substituting LiCl with Li2O and introducing monovalent cations, namely Na+, K+, Cu+, and Ag+. These cations are denoted as Li–LaCeZrHfTa–OCl, Na–LaCeZrHfTa–Cl, K–LaCeZrHfTa–Cl, Cu–LaCeZrHfTa–Cl, and Ag–LaCeZrHfTa–Cl. The Li–LaCeZrHfTa–OCl sample exhibits an ionic conductivity of approximately 1.02 mS cm−1 at 25 °C. In comparison, the Na–LaCeZrAlTa–Cl sample has a conductivity of 0.88 mS cm−1 at the same temperature and an activation energy of 0.326 eV. The K–LaCeZrAlTa–Cl sample, on the other hand, has a conductivity of 1.32 × 10−3 mS cm−1 at a temperature of 55 °C and an activation energy of 0.552 eV. The Cu–LaCeZrAlTa–Cl sample demonstrates a conductivity of 7.13 mS cm−1 at 25 °C with an activation energy of 0.283 eV. Lastly, the Ag–LaCeZrAlTa–Cl sample displays a conductivity of 7.75 mS cm−1 at 25 °C and an activation energy of 0.264 eV.
Wang et al.87 investigated the air stability characteristics of Li3InCl6-based halides. Pelletized halides exhibit a greater relative density compared to their powdered forms. Applying pressure ranging from 100 to 400 MPa during pelletizing enhances relative density and stability in air. The water absorption rate of halides in air correlates with their relative density. Li3InCl6 absorbs water and will form hydrate crystals and partially decompose into InCl3 and LiCl. Subsequent hydrolysis of InCl3 results in the formation of corrosive compounds. The pH value of LiCl indicates no significant change; however, the hydrolysis of In3+ results in the formation of H+, as illustrated by the reaction: In3+ + 3H2O ↔ In(OH)3 + 3H+. The stability of halides in air can be enhanced by applying a coating of aluminum oxide (Al2O3) to their surface. The water absorption rate of Li3InCl6@Al2O3 is 25% that of Li3InCl6. The stability duration of Li3InCl6@Al2O3 is sevenfold superior to that of Li3InCl6. Overall, Tables 2 and 3 summarize several examples of Li/Na-based ISEs and their characteristics.
| Solid electrolyte | Synthesis method | Ionic conductivity | E a | Ref. |
|---|---|---|---|---|
| Na3HfZrSi2PO12 | Solid-state reaction | 4.4 × 10−4 S cm−1 @ RT | 0.358 eV | 40 |
| Na3.4Hf0.6Sc0.4ZrSi2PO12 | Solid-state reaction | 1.2 × 10−3 S cm−1 @ RT | — | 40 |
| NZSP | UHS (20 A, 60 s) | 2.62 × 10−4 S cm−1 @ RT | 0.28 eV | 39 |
| NZSP | Sol–gel method + calcinating (950 °C, 10 h) | 4.43 × 10−3 S cm−1 @ 50 °C | — | 201 |
| Al-doped NZSP | Sol–gel method + calcinating (950 °C, 10 h) | 1.06 × 10−3 S cm−1 @ 50 °C | — | 201 |
| Na3.2Zr1.9Mg0.1Si2PO12 | Solid-state reaction | 1.16 × 10−3 S cm−1 @ RT | 0.25 eV | 202 |
| Ca-doped NZSP | Sol–gel method + heat treatment (600 °C, 6h and 950 °C, 12 h) | 1.67 × 10−3 S cm−1 @ RT | 0.29 eV | 156 |
| NZSP–ATO | Calcinating (1100 °C, 9 h, air) + sintering (1100 °C, 6 h) | 1.43 × 10−3 S cm−1 | — | 60 |
| NLZSP–NBO3 | Solid-state reaction (950 °C, 6 h) | 1.81 × 10−3 S cm−1 @ RT | 0.22 eV | 41 |
| Mg-NZSP-0.128 | Solid-state reaction (1250 °C, 5 h, air) | 3.2 × 10−3 S cm−1 @ RT | 0.247 eV | 155 |
| Cu-doped NZSP | Solid-state reaction (1150 °C, 12 h) | 5.57 × 10−4 S cm−1 @ RT | — | 125 |
| 1.42 × 10−3 S cm−1 @ 60 °C | ||||
| Na3Zr2Si2PO12 | SPS (1050 °C, 20 min) + annealing (1100 °C, 10 h) | 8.2 × 10−4 S cm−1 @ 75 °C | — | 90 |
| Na5GaS4 | Gas passing | 4.0 × 10−7 S cm−1 @ RT | 0.28 eV | 203 |
| Na2.9Sb0.9W0.1S4 | Solid-state reaction (823 K, 20 h) | 41 × 10−3 S cm−1 | 0.19 eV | 79 |
| Na3SbS4 | Solid-state reaction (823 K, 20 h) | 1.3 × 10−5 S cm−1 | 0.25 eV | 79 |
| Ca-doped c-Na3PS4 | Mechanochemical milling (500 rpm, 3–5 h) + heat treatment (700 °C, 12 h) | 0.94 × 10−3 S cm−1 @ 25 °C | 0.49 eV | 101 |
| NaB3H8·xNH3@NaB3H8 | Mechanical milling | 0.84 × 10−3 S cm−1 @ RT | 1.46 eV @ RT | 50 |
| 20.64 × 10−3 S cm−1 @ 318 K | 0.18 eV @ 318 K | |||
| Na2B20H18 | Wet chemical method | 2.5 × 10−6 S cm−1 @ 25 °C | — | 51 |
| Na2B10H10 | Thermolysis (185 °C) + wet chemical method | 1.7 × 10−7 S cm−1 @ 25 °C | — | 51 |
| Na2B20H18–4Na2B12H12 | Ball-milling (400 rpm, 3 h, N2 atmosphere) | 2.8 × 10−4 S cm−1 @ 25 °C | — | 51 |
| >10−3 S cm−1 @ 50 °C | ||||
| Na4B20H18–3Na2B12H12 | Ball-milling (400 rpm, 3 h, N2 atmosphere) | 2.2 × 10−4 S cm−1 | 0.449 eV | 6 |
| Na4B20H18 | Wet chemical method | 3.8 × 10−6 S cm−1 | — | 6 |
| Na3B24H23–5Na2B12H12 | Ball-milling (400 rpm, 3 h, N2 atmosphere) | 1.42 × 10−3 S cm−1 @ 25 °C | 0.385 eV | 70 |
| Na3B24H23 | Ion-exchange method | 2.3 × 10−5 S cm−1 @ 25 °C | 0.59 eV | 70 |
| NaBH4@Na2B12H12 | Solid–gas reaction (10 MPa of H2, 150 °C, 4 h) | 4 × 10−6 S cm−1 @ RT | 0.57 eV | 95 |
| Na4B36H34–7Na2B12H12 | Ball-milling (400 rpm, 3 h, Ar atmosphere) | 1.02 × 10−3 S cm−1 | — | 69 |
| Na4B36H34 | Ion exchange method | — | 0.55 eV | 69 |
| NaNbCl6 | Ball-milling (500 rpm) + heat treatment (200 °C, 24 h) | 3.1 × 10−6 S cm−1 @ 30 °C | 0.48 eV | 43 |
| Na1.5Nb0.5Zr0.5Cl6 | Ball-milling (500 rpm) + heat treatment (200 °C, 24 h) | 2.3 × 10−5 S cm−1 @ 30 °C | ∼0.40 eV | 43 |
| NaTaCl6 | Ball-milling (500 rpm) + heat treatment (200 °C, 24 h) | 5.2 × 10−5 S cm−1 @ 30 °C | 0.39 eV | 43 |
| Na1.5Ta0.5Zr0.5Cl6 | Ball-milling (500 rpm) + heat treatment (200 °C, 24 h) | ∼7 × 10−5 S cm−1 @ 30 °C | ∼0.40 eV | 43 |
| Na3YCl6 | Ball-milling (500 rpm) + heat treatment (200 °C, 24 h) | 5.7 × 10−8 S cm−1 @ 30 °C | — | 43 |
| Solid electrolyte | Synthesis method | Ionic conductivity | E a | Ref. |
|---|---|---|---|---|
| Yb-doped Li6.5La3Zr1.5Ta0.5O12 | Solid-state reaction and ceramic pressure-less sintering | 7.67 × 10−4 S cm−1 @ 25 °C | — | 86 |
| Li7La3Zr2O12 | Solid-state, cold pressing, 1100 °C, 24 h | 9.14 × 10−7 S cm−1 | 0.431 eV | 144 |
| Li6.6La3Zr1.8Mo0.2O12 | Solid-state, cold pressing, 1100 °C, 24 h | 3.17 × 10−4 S cm−1 | 0.26 eV | 144 |
| Li6.65La2.95Sr0.05Zr1.8Mo0.2O12 | Solid-state, cold pressing, 1100 °C, 24 h | 6.43 × 10−4 S cm−1 | 0.25 eV | 144 |
| Li6.25Al0.25La3Zr2O12 | Solid-state reaction (950 °C, 12 h + 1200 °C, 12 h) | 3.72 × 10−4 S cm−1 | 0.31 eV | 139 |
| Li6.25Ga0.25La3Zr2O12 | Solid-state reaction (950 °C, 12 h + 1200 °C, 12 h) | 13.0 × 10−4 S cm−1 | 0.26 eV | 139 |
| Li6.25Fe0.25La3Zr2O12 | Solid-state reaction (950 °C, 12 h + 1200 °C, 12 h) | 11.2 × 10−4 S cm−1 | 0.22 eV | 139 |
| Li6.5La3Zr1.5Ta0.5O12 | Solid-state reaction (950 °C, 12 h + 1200 °C, 12 h) | 2.38 × 10−4 S cm−1 | 0.37 eV | 139 |
| Li6.5La3Zr1.5Nb0.5O12 | Solid-state reaction (950 °C, 12 h + 1200 °C, 12 h) | 1.91 × 10−4 S cm−1 | 0.44 eV | 139 |
| Li6.5La3Zr1.5Sb0.5O12 | Solid-state reaction (950 °C, 12 h + 1200 °C, 12 h) | 3.41 × 10−4 S cm−1 | 0.41 eV | 139 |
| Li6.5La3Zr1.75W0.25O12 | Solid-state reaction (950 °C, 12 h + 1200 °C, 12 h) | 5.43 × 10−4 S cm−1 | 0.37 eV | 139 |
| Li6.5La3Zr1.75Mo0.25O12 | Solid-state reaction (950 °C, 12 h + 1200 °C, 12 h) | 3.03 × 10−4 S cm−1 | 0.36 eV | 139 |
| Li7La2.5Ce0.5Zr1.625Bi0.3O12 | Sol–gel method and heat treatment (1150 °C, 6 h) | 5.12 × 10−4 S cm−1 @ RT | 0.1083 eV | 145 |
| Li7La3Zr1.625Bi0.3O12 | Sol–gel method and heat treatment (1150 °C, 6 h) | 8.04 × 10−5 S cm−1 @ RT | 0.1750 eV | 145 |
| Li7La3Zr1.75Ce0.25O12 | Solid-state reaction | 2.2 × 10−4 S cm−1 @ RT | 0.30 eV | 34 |
| Li6.55Ga0.15La3Zr2O12 | Solid-state reaction | 0.13 × 10−3 S cm−1 @ RT | — | 146 |
| Li6.65Ga0.15La3Zr1.95Mg0.05O12 | Solid-state reaction | 1.13 × 10−3 S cm−1 @ RT | — | 146 |
| CuO-doped Li6.3La3Zr1.65W0.35O12 | Solid-state reaction (1120 °C, 6 h) | 1.88 × 10−4 S cm−1 @ 25 °C | 0.34 eV | 143 |
| Li1.3Al0.3Ti1.7(PO4)3 | Calcinating (800 °C, 2 h) + CSP (250 MPa, 250 °C, 1 h) + annealing (900 °C, 1 h) | 4.29 × 10−4 S cm−1 @ RT | — | 83 |
| LATP | HPLT (200 °C, 2 min, 1 GPa) | 6.15 × 10−5 S cm−1 | — | 89 |
| Te-doped LAGP | Sol–gel pre-sintered (500 °C, 4 h) + calcinated (850 °C, 5 h) | 6.33 × 10−4 S cm−1 | 0.29 eV | 91 |
| LAGP | Sol–gel pre-sintered (500 °C, 4 h) + calcinated (850 °C, 5 h) | 1.94 × 10−4 S cm−1 | 0.35 eV | 91 |
| Li7P3S11 glass-ceramic | Mechanical milling (270 °C, 3 h) | 1.27 × 10−3 S cm−1 @ RT | 0.22 eV | 48 |
| GeO2-doped L7P3S11 (Li7P2.9Ge0.05S10.75O0.1) | Ball-milling (510 rpm, 40 h @ RT) + annealing (240–290 °C, 4 h) | 4.77 × 10−3 S cm−1 @ RT | 18.66 kJ mol−1 | 65 |
| P2O5-doped 75Li2S·25P2S5 | Ball-milling (370 rpm, 48 h) and heat treatment (260 °C, 3.5 h) | 6.3 × 10−5 S cm−1 @ RT | 20 kJ mol−1 | 177 |
| CaS-doped Li7P3S11 (Li6.72Ca0.14P3S11) | Liquid-phase synthesis (50 °C, 72 h + 80 °C, 6 h + 270 °C, 1 h) | 0.95 × 10−3 S cm−1 @ 30 °C | 34 kJ mol−1 | 171 |
| 0.98Li7P3S11–0.02CaI2 | Liquid-phase synthesis (50 °C, 72 h + 80 °C, 6 h + 270 °C, 1 h) | 0.78 × 10−3 S cm−1 @ 29 °C | 36 kJ mol−1 | 171 |
| 95Li7P3S11–5CaI2 | Liquid-phase synthesis (50 °C, 24 h + 270 °C, 2 h) | 1 × 10−3 S cm−1 @ RT | 30 kJ mol−1 | 181 |
| 10.1 × 10−3 S cm−1 @ 70 °C | ||||
| 90Li7P3S11–5CaI2–5LiI | Liquid-phase synthesis (50 °C, 72 h + 270 °C, 1 h) | 3.1 × 10−3 S cm−1 @ RT | 25 kJ mol−1 | 181 |
| 13 × 10−3 S cm−1 @ 70 °C | ||||
| (1 − x)Li3PS4–2xLiBH4 (x = 0.54) | Two-step milling (213 rpm, 45 h and 510 rpm, 15 h) | 11 × 10−3 S cm−1 @ RT | 34.4 kJ mol−1 | 71 |
| Li4PS4I | Solid-state reaction (500 °C, 24 h) | 2.2 × 10−5 S cm−1 @ RT | 0.45 eV | 204 |
| Li4.3P0.7Si0.3S4 | Solid-state reaction (700 °C, 5 h) | 1.46 × 10−3 S cm−1 @ RT | 0.32 eV | 205 |
| Li4PS4I | Solid-state reaction (700 °C, 5 h) | 4.6 × 10−5 S cm−1 @ RT | 0.44 eV | 205 |
| Li6PS5I | Mechanochemical process (510 rpm, 45 h) and heat treatment (550 °C, 2 h, Ar) | 3.8 × 10−7 S cm−1 @ RT | 43 kJ mol−1 | 47 |
| Li10GeP2S12 | Solid-state reaction (680 °C, 8 h) | 6.2 × 10−3 S cm−1 | 49 | |
| SbCl3-doped LGPS | Solid-state reaction (680 °C, 8 h) | 6.8 × 10−3 S cm−1 @ 25 °C | 0.196 eV | 49 |
| Li5GaS4 | Gas passing | 1.8 × 10−7 S cm−1 @ RT | 0.54 eV | 203 |
| Li4.025Sn0.975Bi0.025S4 | Two-step ball milling (180 rpm for 2 h + 550 rpm for 18 h) and annealing (300 °C, 1.5 h, N2) | 1.35 × 10−4 S cm−1 @ RT | 0.275 eV | 180 |
| Li10.2P2.95Sb0.05S12I | High-energy ball milling (220 °C, 2 h, Ar) | 5.9 × 10−3 S cm−1 @ 25 °C | 0.28–0.29 eV | 111 |
| Li10P3S12I | High-energy ball milling (220 °C, 2 h, Ar) | 5.06 × 10−3 S cm−1 @ 25 °C | 0.28–0.29 eV | 111 |
| Li3.68Ge0.69Sb0.31S4 | Solid-state reaction (773 K, 8 h) | 1.23 × 10−4 S cm−1 @ 303 K | ∼0.28 eV | 25 |
| Li4GeS4 | Solid-state reaction (973 K) | 3.98 × 10−6 S cm−1 @ 303 K | 25 | |
| Li10SnP2S12 | High-energy ball-milling (550 °C, 4 h) | 4.79 × 10−3 S cm−1 @ RT | 0.15 eV | 66 |
| LGPS–Al2O3 (97:3) |
Solid-state reaction (823 K, 8 h, Ar) | 5.7 × 10−3 S cm−1 @ RT | — | 80 |
| LGPS | Liquid-phase method (mix solvents, stirring time 30 min) + heat treatment (550 °C, 6 h) | 1.6 × 10−3 S cm−1 @ RT | — | 206 |
| LGPS | High-energy ball milling 520 min + heat treatment | 3.27 × 10−3 S cm−1 @ RT | 0.28 eV | 3 |
| Li10P2.8S10O2 | Liquid-phase method (2-propanol solvent) + annealing (300 °C, 2 h) | 1.6 × 10−4 S cm−1 @ 25 °C | 20 kJ mol−1 | 21 |
| LSiPSCl | Liquid-phase method (ACN solvent) + wet-milling (300 rpm, 20 h) + sintering (748 K, 8 h, Ar) | 6.6 × 10−3 S cm−1 @ RT | — | 7 |
| LSiPSCl | Ball-milling (380 rpm, 40 h) + sintering (748 K, 8h, Ar) | 8.8 × 10−3 S cm−1 @ RT | — | 7 |
| Li6.75P0.25Si0.75O5Cl | Spark plasma sintering | 1.82 × 10−6 S cm−1 @ 303 K | 0.522 eV | 88 |
| Li7Zn0.5SiS6 | Solid-state reaction (973 K, 24 h) | 1 × 10−7 S cm−1 @ RT | 0.66 eV (I4) | 185 |
| 4.3 × 10−4 S cm−1 @ 503 K | 0.34 eV (F3m) |
|||
| Li7GeS5Br | Ball-milling (1 h, 250 rpm + 10 h, 450 rpm) | 0.63 × 10−3 S cm−1 @ 298 K | 0.43 eV | 207 |
| Li6PS5Cl | Wet milling and post-annealing (500 °C, 4 h) | 1.9 × 10−3 S cm−1 | 0.245 eV | 68 |
| Li6PS5Cl | Solid-state reaction (550 °C, 6 h) | 3.19 × 10−3 S cm−1 | — | 102 |
| Li6SbS5I | Mechanochemical process (510 rpm, 45 h) and heat treatment (400–450 °C, 2 h, Ar atmosphere) | 2.1 × 10−6 Scm−1 @ 25 °C | 35 kJ mol−1 | 47 |
| Li6.5[P0.25Si0.25Ge0.25Sb0.25]S5I | High-energy milling and post-annealing at 500 °C | 13.2 × 10−3 S cm−1 @ RT | 0.19 eV | 208 |
| Li5.6PS4.6I1.4 glass–ceramic | Ultimate-energy mechanical alloying (1500 rpm, 1 h + 550 °C, 2 h) | 2.04 × 10−3 S cm−1 | 0.31 eV | 209 |
| Li6PS5F0.5Cl0.5 | Wet chemical (ethanol solvent, Ar atmosphere, and heating 200 °C, 1 h) | 3.5 × 10−4 S cm−1 | 0.32 eV | 16 |
| Li6PS5F | Wet chemical (ethanol solvent, Ar atmosphere, and heating 200 °C, 1 h) | 2.4 × 10−4 S cm−1 | 0.45 eV | 16 |
| Li6PS5F0.25I0.75 | Wet chemical (ethanol solvent, Ar atmosphere, and heating 200 °C, 1 h) | 3.5 × 10−4 S cm−1 | 0.34 eV | 92 |
| Li6PS5F0.75I0.25 | Wet chemical (ethanol solvent, Ar atmosphere, and heating 200 °C, 1 h) | 1.5 × 10−4 S cm−1 | 0.377 eV | 92 |
| Li6PS5I | Wet chemical (ethanol solvent, Ar atmosphere, and heating 200 °C, 1 h) | 2.5 × 10−4 S cm−1 | 0.387 eV | 92 |
| Li2B12H12–5Li2B10H10 – 6LiBH4 | Ball-milling (350 rpm, 30 min) + annealing (280 °C, 1 h, 20 MPa of H2) | ∼1.0 × 10−4 S cm−1 @ RT | 0.16 eV | 52 |
| Li2B12H12–5Li2B10H10 | Ball-milling (350 rpm, 30 min) + annealing (380 °C, 1 h) | 6.92 × 10−6 S cm−1 @ 30 °C | 0.51 eV | 52 |
| Li3(CB11H12)2(CB9H10) | Ball-milling (ratio of 2:1, Ar atmosphere) |
1.5 × 10−3 S cm−1 @ RT | — | 59 |
| 29.4 × 10−3 S cm−1 @ 60 °C | ||||
| Li3OCl | Melt-quenching and high-energy ball-milling (350 °C, 6 h) | 3.21 × 10−5 S cm−1 @ RT | 0.52 eV | 94 |
| Li2OHCl0.9Br0.1 | Solid-state reaction (350 °C, 30 min) | 2.52 × 10−6 S cm−1 @ RT | 0.52 eV | 31 |
| Li2OHCl | Solid-state reaction (350 °C, 30 min) | 1.93 × 10−6 S cm−1 @ RT | 0.83 eV | 31 |
| Li3SI | High-energy ball-milling (400 rpm, 24 h) + heat treatment (150–550 °C, 3 h) | 2.2 × 10−5 S cm−1 @ RT | 27.6 kJ mol−1 | 32 |
| Li3−x(OHx)Cl0.9F0.1 | Ball-milling (400 rpm, 24 h) + sintering (350 °C, 5 h) | 9.0 × 10−6 S cm−1 @ 25 °C | — | 24 |
| Li2OHBr | Mechanochemical method (700 rpm, 48 h) | 1.6 × 10−6 S cm−1 @ 25 °C | — | 74 |
| Li3OCl | Radio frequency magnetron sputtering (P = 1–2 × 10−3 mbar, RF power = 22 W, Ar atmosphere) | 1.27 × 10−8 S cm−1 @ 25 °C | 0.93 eV | 53 |
| Li2OHCl | Wet mechanical milling (hexane solvent, 400 rpm, 10 h) | 4.27 × 10−6 S cm−1 @ 25 °C | 1.03 eV | 210 |
| Li2OHCl | Solid-state reaction (360 °C, 2 h) | 2.37 × 10−7 S cm−1 @ 25 °C | 0.52 eV | 210 |
| (Li2OH)0.99K0.01Cl | Solid-state reaction (400 °C, 1 h, Ar atmosphere) | 4.5 × 10−6 S cm−1 @ 25 °C | 0.56 eV | 72 |
| LIC | Water removal route (100–200 °C) | 1.6 × 10−3 S cm−1 @ 298 K | — | 42 |
| Li2.73Ho1.09Cl6 | Solid-state reaction (650 °C, 4 h + annealing for 24 h) | 1.3 × 10−3 S cm−1 @ RT | — | 56 |
| SmCl3·0.5LiCl | Ball-milling (100–500 rpm, 4 h) | 1.2 × 10−4 S cm−1 @ 30 °C | <0.4 eV | 2 |
| SmCl3·0.5LiCl | Co-melting (650 °C, 10 h + 50 °C, 1 h) | 3.1 × 10−8 S cm−1 @ 30 °C | — | 2 |
| HCF-Sm | Ball-milling | 1.1 × 10−4 S cm−1 | — | 30 |
| HCF-La | Ball-milling | 5.5 × 10−5 S cm−1 | — | 30 |
| LCF-Ta | Ball-milling | 5.0 × 10−5 S cm−1 | 0.387 eV | 30 |
| LCF-Zr | Ball-milling | ∼1.0 × 10−5 S cm−1 | — | 30 |
| LCF-Hf | Ball-milling | ∼1.0 × 10−5 S cm−1 | — | 30 |
| 0.62[HCF-Sm]·0.38[LCF-Ta] | Ball-milling | 2.7 × 10−3 S cm−1 @ 298 K | 0.305 eV | 30 |
| 0.62[HCF-Sm]·0.38[LCF-Hf] | Ball-milling | ∼2.0 × 10−4 S cm−1 | — | 30 |
| 0.62[HCF-Sm]·0.38[LCF-Zr] | Ball-milling | ∼2.0 × 10−4 S cm−1 | — | 30 |
| 0.57[HCF-La]·0.43[LCF-Ta] | Ball-milling | 1.8 × 10−3 S cm−1 @ 298 K | 0.309 eV | 30 |
| 0.57[HCF-La]·0.43[LCF-Hf] | Ball-milling | ∼1.0 × 10−4 S cm−1 | — | 30 |
| 0.57[HCF-La]·0.43[LCF-Zr] | Ball-milling | ∼1.0 × 10−4 S cm−1 | — | 30 |
| Li3ScCl6 | Ball-milling (480 rpm, 12 h) + heat treatment (450 °C, 12 h) | 0.6 × 10−3 S cm−1 | 0.367 eV | 211 |
| Li2.6Sc0.6Zr0.4Cl6 | Ball-milling (480 rpm, 12 h) + heat treatment (450 °C, 12 h) | 1.61 × 10−3 S cm−1 | 0.326 eV | 211 |
| Li2.6Sc0.6Hf0.4Cl6 | Ball-milling (480 rpm, 12 h) + heat treatment (450 °C, 12 h) | 1.33 × 10−3 S cm−1 | 0.323 eV | 211 |
| Li2.7In0.7Zr0.3Cl6 | Two-step annealing (450 °C, 24 h + 450 °C, 8 h) | 2.02 × 10−3 S cm−1 @ RT | ∼0.28 eV | 93 |
| Li2.4Zr0.6Ho0.4Cl6 | Solid-state reaction (450 °C, 48 h) | ∼1.8 × 10−3 S cm−1 | ∼0.34 eV | 75 |
| Li2.4Zr0.6Lu0.4Cl6 | Solid-state reaction (450 °C, 48 h) | ∼1.8 × 10−3 S cm−1 | ∼0.34 eV | 75 |
| Li3HoCl6 | Solid-state reaction (650 °C, 48 h) | ∼0.1 × 10−3 S cm−1 | — | 75 |
| Li3LuCl6 | Solid-state reaction (650 °C, 48 h) | ∼0.1 × 10−3 S cm−1 | — | 75 |
| Li3ZrCl4O1.5 | Ball-milling (100 rpm, 2 h + 500 rpm, 10 h) | 1.35 × 10−3 S cm−1 @ 25 °C | 0.294 eV | 57 |
| Li2.375Sc0.375Zr 0.625Cl6 | Ball-milling (400 h, 10 h) + annealing (300 °C, 5 h) | 2.2 × 10−3 S cm−1 @ 25 °C | 0.31 eV | 22 |
| Li2.375Sc0.375Hf0.625Cl6 | Ball-milling (400 h, 10 h) + annealing (300 °C, 5 h) | 1.1 × 10−3 S cm−1 @ 25 °C | 0.33 eV | 22 |
| Li3ScCl6 | Ball-milling (400 h, 10 h) + annealing (300 °C, 5 h) | ∼4 × 10−4 S cm−1 @ 25 °C | 0.44 eV | 22 |
| Li2.1Zr0.83Ta0.27Cl6 | Ball-milling (550 rpm, 48 h) | 1.68 × 10−3 S cm−1 @ 25 °C | 0.28 eV | 46 |
| Li2.3Hf0.7In0.3Cl6 | Ball-milling | 1.05 × 10−3 S cm−1 @ 30 °C | 0.337 eV | 198 |
| Li2.4Y0.4Hf0.6Cl6 | Ball-milling | 1.49 × 10−3 S cm−1 @ RT | 0.331 eV | 200 |
| Li3YCl6 | Ball-milling | 1.39 × 10−4 S cm−1 @ RT | — | 200 |
| Li2ZrCl6 | Ball-milling (500 rpm, 45 h) | 8.08 × 10−4 S cm−1 at 25 °C | 0.35 eV | 212 |
| Li2.25Hf0.75Fe0.25Cl6 | Ball-milling | 0.91 × 10−3 S cm−1 @ 30 °C | 0.34 eV | 199 |
| Li2.2Hf0.8V0.2Cl6 | Ball-milling | 0.64 × 10−3 S cm−1 @ 30 °C | 0.351 eV | 199 |
| Li2.2Hf0.8Cr0.2Cl6 | Ball-milling | 0.81 × 10−3 S cm−1 @ 30 °C | 0.342 eV | 199 |
| Li2HfCl6 | Ball-milling | 0.39 × 10−3 S cm−1 @ 30 °C | — | 199 |
3. ISEs preparations and interface of ISEs/electrode
3.1. ISEs preparations
The method of preparation impacts the mechanical and electrochemical characteristics of solid electrolytes, which consequently impacts the commercialization of these electrolytes in the assembly of all-solid-state batteries.7,64,82,90,102,126,210,212–215 The following overview outlines various preparation methods that have been presented to date, including solid-state synthesis, liquid-phase synthesis, and other advanced techniques. Solid-state reactions can be categorized into three distinct methods: mechanochemical milling, mechanical milling combined with heat treatment, and direct solid-state sintering.216In oxide electrolytes, attention must be given to preparation conditions to achieve high density, ensure appropriate stoichiometry, and prevent the formation of impurity phases.39,40,64,89,126,133 In general, these electrolytes necessitate high sintering temperatures and extended holding time required to achieve high-density oxides. Conversely, high-temperature conditions can lead to the evaporation of Li2O, and prolonged exposure to severe conditions may result in the decomposition of the oxide.119 Lithium deficiency resulting from Li2O evaporation inhibits oxide electrolyte formation and promotes the development of secondary oxide phases. High-temperature sintering can create microstructural defects that block ion transport.83,89 Therefore, understanding the preparation conditions is invaluable for ISE development. Utilizing considerably reduced temperatures and durations may enhance manufacturing yields while minimizing energy consumption, thus promoting a more environmentally sustainable approach.
Garnet-type Nb-doped LLZO is generally synthesized via solid-state reaction.119,136,217,218 Li7−xLa3Zr2−xNbxO12 (Nb-LLZO, x = 0.25) was synthesized by Ohta and coworkers using a conventional solid-state reaction method that included ball-milling, calcination (950 °C, 12 hours), pressing, and sintering (1200 °C, 36 hours).218 All LLZO and Nb-LLZO-type garnets were synthesized in a cubic structure, achieving ionic conductivities of ∼0.8 mS cm−1 for 0.25Nb-LLZO and ∼0.2 mS cm−1 for LLZO at 25 °C, with relative densities between 89% and 92%. Zhao et al.136 synthesized 0.25Nb-LLZO via a self-consolidation method (Fig. 13a1), including ball-milling (400 rpm, 6 h), calcination (1150 °C, 10 h), and ball-milling (500 rpm, 6 h). The Nb-LLZO powder was manually compacted using vibration and sintered at 1150 °C for 15 hours. The material properties attained include relative density, crystal phase, and ionic conductivity for LLZO (93%, tetragonal phase, 0.0564 mS cm−1 at 30 °C) and 0.25Nb-LLZO (91%, cubic phase, 0.0807 mS cm−1 at 30 °C). The maximum relative density and ionic conductivity were attained by 0.6Nb-LLZO, 94% and 0.522 mS cm−1 at 30 °C. Gai et al.119 synthesized 0.25Nb-LLZO by solid-state reaction, which included ball-milling (8 h), pressing, calcination (900 °C, 8 h), further ball-milling (8 h), and sintering (1230 °C, 15 h). The material properties obtained from this approach include relative density, crystal phase, and ionic conductivity for LLZO (84.1%, tetragonal phase, 0.00734 mS cm−1 at 30 °C) and 0.25Nb-LLZO (90%, cubic phase, 0.482 mS cm−1 at 30 °C). The solid-state reaction exhibits a consistent sequence with modifying temperature, sintering duration, and pellet densification by the pressing process.
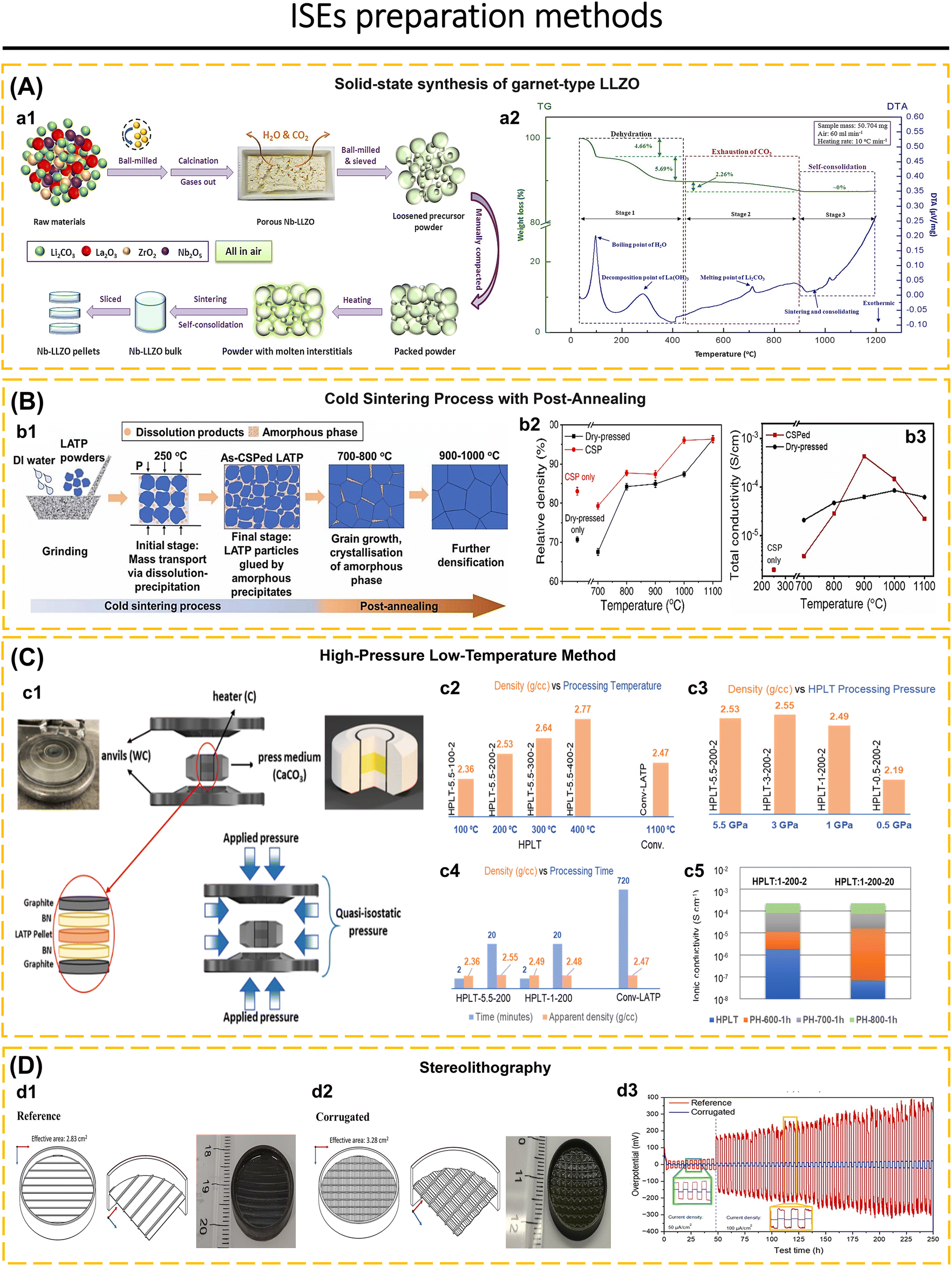 | ||
| Fig. 13 (A) (a1) Schematic illustration for the preparation of Nb-LLZO pellets by self-consolidation method. (a2) TG/DTA curves of the precursor 060Nb-LLZO powder before sintering. Reproduced with permission from ref. 136. Copyright 2018, Elsevier. (B) (b1) Schematic illustration of the microstructure evolution in LATP ceramics during CSP and subsequent post-annealing. (b2) Effect of annealing temperatures on the relative density of LATP samples produced by CSP and dry-pressing. All samples were annealed for 1 h. (b3) Total ionic conductivity (measured at room temperature) of the CSPed and dry-pressed LATP samples as a function of annealing temperature. Reproduced with permission from ref. 83. Copyright 2023, Elsevier. (C) (c1) The schematic representation of components for the HPLT technique. Density of LATP pellets densified by HPLT processing and conventional sintering. From top to down: (c2) LATP density vs. HPLT processing temperature for pellets processed at 5.5 GPa and 2 minutes compared to conventional sintering, (c3) LATP density vs. HPLT processing pressure for pellets processed at 200 °C and for 2 min, (c4) LATP density vs. HPLT processing time for pellets processed at 200 °C and under high (5.5 GPa) and low (1 GPa) processing pressures compared to conventional sintering conditions. (c5) Evolution of Li-ion conductivity with increase in PH temperature: HPLT-1-200-2 (b) pellet: HPLT-1-200-20. Reproduced with permission from ref. 89. Copyright 2024, Elsevier. (D) 3D CAD drawings, details of the cross-sections, and photos of the as-printed membranes: reference (d1) and corrugated (d2). (d3) Galvanostatic cycling of the printed electrolytes in a Li|Ge-LAGP-Ge|Li configuration. Reproduced with permission from ref. 170. Copyright 2023, Royal Society of Chemistry. | ||
The TGA/DTA analysis for the Nb-LLZO sample (Fig. 13a2)136 highlights the thermal gravimetry mechanism into three distinct stages. The first stage involves the dehydration of H2O, shown by an endothermic peak at 97 °C, and the decomposition of La(OH)3, seen at an endothermic peak of 280 °C. The second stage is the decomposition process of Li2CO3, characterized by an endothermic peak at 714 °C. The third stage is the sintering process. There is no weight loss within the temperature range of 880 to 1020 °C. This region has an exothermic peak. While weight loss is not evident in the third stage, heat treatment at temperatures up to 1200 °C results in volume shrinkage, yielding dense sintered bulk. Zhao et al.136 explain that significant shrinkage occurs during the sintering process, thereby necessitating careful adjustment of the heating rate. The heating rate from room temperature to 800 °C was set at 10 °C min−1 to eliminate remaining gas components, while the rate from 800 °C to 1150 °C was set at 1 °C min−1 to mitigate significant volume shrinkage. Subsequently, hold the sintering temperature to get a dense sintered bulk. LLZO sintered at 1150 °C for 15 hours achieved a relative density of 93%,136 while at 1230 °C for 15 hours, the relative density was 84.5%.119 The 0.25Nb-LLZO exhibits competing relative densities of 91% at 1150 °C (ref. 136) and 90% at 1230 °C.119 The elevated sintering temperature in the LLZO sintering process is likely the cause. The LLZO synthesized by Zhao et al.136 and Gai et al.119 has a tetragonal structure. Elevating the sintering temperature reduces the stability of the tetragonal structure, hence leading to material decomposition and/or a reduction in the volume of LLZO. Doping Nb into LLZO leads to a stable cubic structure. Ohta et al.218 achieved a relative density, 89–92% using a sintering temperature of 1200 °C for 36 hours. Despite using a longer sintering holding time that may lead to lithium evaporation or product degradation, the density of LLZO remains notably high.
Nonetheless, it is difficult to ascertain whether density correlates to ionic conductivity. In the case of Zhao et al.136 LLZO and 0.25Nb-LLZO exhibit comparable densities (93% and 91%) and ionic conductivities (0.05 and 0.08 mS cm−1). In the case of Gai et al.119 LLZO and 0.25Nb-LLZO exhibit significantly higher densities (84.5% and 90%) and ionic conductivities (0.00734 and 0.482 mS cm−1). SEM analysis from Zhao et al.136 indicate that the average particle size for LLZO and 0.25Nb-LLZO remains unchanged at around 12 μm, with a maximum particle size of 50 μm. In the case of Gai et al.,119 the reduction in particle size is significant for LLZO (>20 μm) and 0.25Nb-LLZO (5–20 μm). The reduction in grain size due to preparation conditions will impact the increase of grain boundaries and ionic conductivity of LLZO. Although the grain sizes vary but are interconnected, this has an impact on the stability of Li-ion transport in Nb-LLZO. Huge voids are seen in LLZO, affecting the ionic conductivity. Gai et al.119 clarified that the doping of Y and Nb in LLZO effectively enhanced the ionic conductivity. Li7La3Zr2−2xNbxYxO12 (x = 0.5) sintered at 1230 °C for 15 hours exhibited a particle size reduction to below 10 μm, demonstrating excellent inter-grain connectivity compared to 0.25Nb-LLZO. The sintering of 0.5Nb/0.5Y co-doped LLZO at 1230 °C resulted in the formation of a secondary phase, Y2O3, which infiltrated the grain boundaries, enhanced grain connectivity, reduced grain boundaries resistance, and enhanced Li-ion transport pathways.
Cai et al.83 reported that the combination of the cold sintering process (CSP) technique and annealing is highly successful in removing amorphous phase residues in NASICON. Additionally, this approach allows for the control of LATP growth and the decrease of impurity phases (Fig. 13b1). The proposed CSP presents initial densification facilitated by dissolution precipitation. This aimed to regulate grain growth. The CSP was optimized at 250 °C under 250 MPa for 1 hour. The density of the as-CSPed sample reached 83%, which is slightly lower than that achieved through conventional sintering, but superior to the 70% observed in dry-pressed samples. The as-CSPed sample exhibited a higher density compared to the dry-pressed sample, with a particle size range of 30 to 180 nm. The as-CSPed LATP samples were post-annealed at 700–1100 °C for 1 h. The as-CSPed sample achieved optimal density at 1000 °C (96%), with a particle size ranging from 0.4 to 1.7 μm, comparable to the dry-pressed samples, 0.3 to 1.8 μm (Fig. 13b2). Raising the temperature resulted in variable grain size formation, characterized by an inhomogeneous microstructure and the presence of micro defects. The best ionic conductivity of the as-CSPed sample after annealing at 900 °C for 1 h (4.29 × 10−4 S cm−1) which is 213 times higher than that of the CSP-only samples, 2.01 × 10−6 S cm−1. While the dry-pressed sample achieved the best conductivity at annealing at 1000 °C for 1 h (8.51 × 10−5 S cm−1). While the conductivity obtained remains slightly below that of traditional methods, it measures at 1.09 × 10−3 S cm−1 after 40 hours of mechanical milling followed by sintering at 900 °C for 6 hours (Fig. 13b3).214 The applied CSP method offers benefits related to reduced processing temperature and duration, regulation of microstructure, and enhanced energy efficiency.
Valiyaveettil-SobhanRaj and co-workers89 enhanced the density and ionic conductivity of NASICON-type LATP by applying the HPLT processing technique followed by post-heat treatment. The HPLT method involves positioning the pellet at the center of a toroid, which is a container made of CaCO3. This setup is sealed within a graphite tube that has been coated on the interior with hexagonal boron nitride (h-BN), effectively preventing any contact between the graphite and the sample. The graphite tube provides effective thermal conductivity. The toroid is situated between two anvils and functions to regulate the pressure (Fig. 13c1). Varying the temperature, pressure, and processing time will affect the microstructure and ionic transport processes. Sample nomenclature was used in the format: HPLT-(pressure)-(temperature)-(time). For example, in the case of the LATP pellet densified by HPLT at 5.5 GPa, 200 °C for 2 min, the nomenclature is HPLT-5.5-200-2. The density of conventional LATP (2.47 g cm−3) achieved at 1100 °C for 720 min can be compared with that of 2.77 g cm−3 (for HPLT-5.5-400-2) and 2.55 g cm−3 (for HPLT-3-200-2 and HPLT-5.5-200-20) (Fig. 13c2–c4). For HPLT-5.5 GPa-2 min under temperature 100–400 °C shows low conductivity (∼10−8 S cm−1). Lowering pressure for HPLT-200 °C-2 min increases conductivity 2 orders of magnitude higher (∼10−6 S cm−1). The effect of holding time (20 min) for HPLT-5.5 GPa-200 °C and HPLT-1 GPa-200 °C samples show conductivity at ∼10−7 S cm−1. The conductivity of HPLT pellets is improved through post-heat treatment (PH) in the range of 600 to 800 °C and a duration of between 1 minute to 1 hour. Samples HPLT-1 GPa-200 °C-2 min and HPLT-1 GPa-200 °C-20 min exhibited conductivity greater than 10−4 S cm−1 after PH treatment at 800 °C, a temperature lower than that used in conventional sintering at 1000 °C (Fig. 13c5). The duration of PH treatment significantly affects conductivity. Samples sintered at 800 °C with HPLT-3 GPa-200 °C for holding times of 2, 10, and 60 minutes exhibited conductivities of 1.9 × 10−5 S cm−1, 5.1 × 10−5 S cm−1, and 1.5 × 10−4 S cm−1, respectively. The study of HPLT-based solid electrolyte synthesis is still relatively new, and further improvisation is needed to increase ionic conductivity, but a significant increase in density is a promising achievement for low-temperature sintering.
Sabato et al.170 have researched enhancing the surface reactivity of glass-derived LAGP NASICON-type electrolytes by modifying their surface morphology. They employed a 3D printing process called stereolithography, which was helped by CAD. The LAGP ceramic powder was evenly dispersed in an acrylate-based resin that contained a monofax additive. The ratio of the components was 65 wt% LAGP ceramic powder, 34.35 wt% acrylate-based resin, and 0.65 wt% monofax additive. A temperature of at least 600 °C guarantees a structure that closely resembles the crystal structure of LiGe2(PO4)2 (PDF#1-080-1924), with no observable impurities such as AlPO4 or GeO2. By utilizing CAD technology, the solid electrolyte is constructed in two forms: reference-planar (Fig. 13d1) and corrugated (Fig. 13d2). The wrinkling effect enhances the active surface area of the membrane by 15%. The EIS results showed that the corrugated structures in symmetric cells, Au|LAGP|Au configuration, can decrease the total area-specific resistance by 15%. Fig. 13d3 shows the cycling test results of the Li|Ge-LAGP-Ge|Li associated corrugated structure, indicating a consistently low overpotential for up to 250 hours.
Solid-state methods for synthesizing sulfide-type solid electrolytes have drawbacks, including significant energy consumption and limited large-scale production capacity. Liquid-phase synthesis offers a more economical method for large-scale production. The solution state divides liquid-phase synthesis into two categories: solution synthesis and suspension synthesis.91,145,147,171,181,206 Hikima et al.206 described a simple suspension synthesis method to produce LGPS using liquid phase shaking. LGPS with ionic conductivity of 1.6 × 10−3 S cm−1, electrical conductivity of 9.8 × 10−9 S cm−1, and low impurities such as GeS2, Li3PO4, β-Li3PS4, and Li2S were obtained by improving the synthesis conditions utilizing a combination of solvents such as ACN, THF, and ethanol, a shaking temperature of 45 °C, a stirring time of 30 minutes, excess sulfur, and heat-treatment at 550 °C for 6 hours. Ito et al.7 reported the preparation of LGPS-type LSiPSCl using the liquid-phase method (Fig. 14a1). Solution 1 contains Li, P, and S and solution 2 contains Li, Si, and S, each prepared separately with ACN solvent. ACN as a solvent is an aprotic polar, which is not easily decomposed by precursors. The two solutions are mixed to produce a Li–Si–P–S solution containing Li2S:P2S5:SiS2 with a molar ratio of 2.1:1.1:2.0. This ratio is lower than the target molar ratio for LSiPSCl, Li2S:P2S5:SiS2 = 6.6:1.1:2.0. To meet the precursor target, fine-Li2S was added to the precursor slurry solution. During the drying process of the slurry at 453 K under vacuum conditions, a certain amount of sulfur evaporates, and to compensate for the lost sulfur, excess sulfur is added and adjusted to achieve optimal conditions following the ratio Li2S:P2S5:SiS2:LiCl:S = 6.6:1.1:2.0:0.4:X (X = 0, 1.0, and 2.9). The sample was sintered at 748 K for 8 hours under an Argon atmosphere. LSiPSCl is prepared by a liquid-phase synthetic route and a solid-state reaction is coded L-LSiPSCl and S-LSiPSCl, respectively. L-LSiPSCl-1.0 shows minimal impurity phases, while its ionic conductivity of 6.6 mS cm−1 at 298 K is higher than L-LSiPSCl-X (X = 0 and 2.9) and slightly lower than S-LSiPSCl of 8.8 mS cm−1. The electrochemical performance of ASSB was tested in LCO‖Li–In cells using L-LSiPSCl-1.0 and S-LSiPSCl electrolytes. The electrochemical stability of both electrolytes competes with each other (Fig. 14a2). The particle size factor of solid electrolytes has a significant impact on particle defect reduction, ionic conductivity, and long-term stability. Adjusting particle size can be accomplished by selecting a synthesis method; at this point, liquid-phase synthesis is considered to be capable of controlling particle size. Zhou et al.82 reported the synthesis of Li7P3S11 using ethyl acetate solvent (Fig. 14b1). Adjusting the precursor concentration (10 to 40 mg mL−1) and solvent evaporation temperature (80 to 150 °C) results in a particle size of ∼100 nm and a maximum ion conductivity of 1.05 mS cm−1. Fig. 14b2 shows the SEM photograph of the Li7P3S11 sample produced under optimum conditions. Compared to solvents like ACN, THF, and DME, ethyl acetate has superior Li2S and P2S5 dissolving systems. The selection of solvent is an important aspect in liquid-phase synthesis to prevent Li2S–P2S5 insolubility, side reactions, crystalline-amorphous phase mixing, and non-uniform particle size.221–224
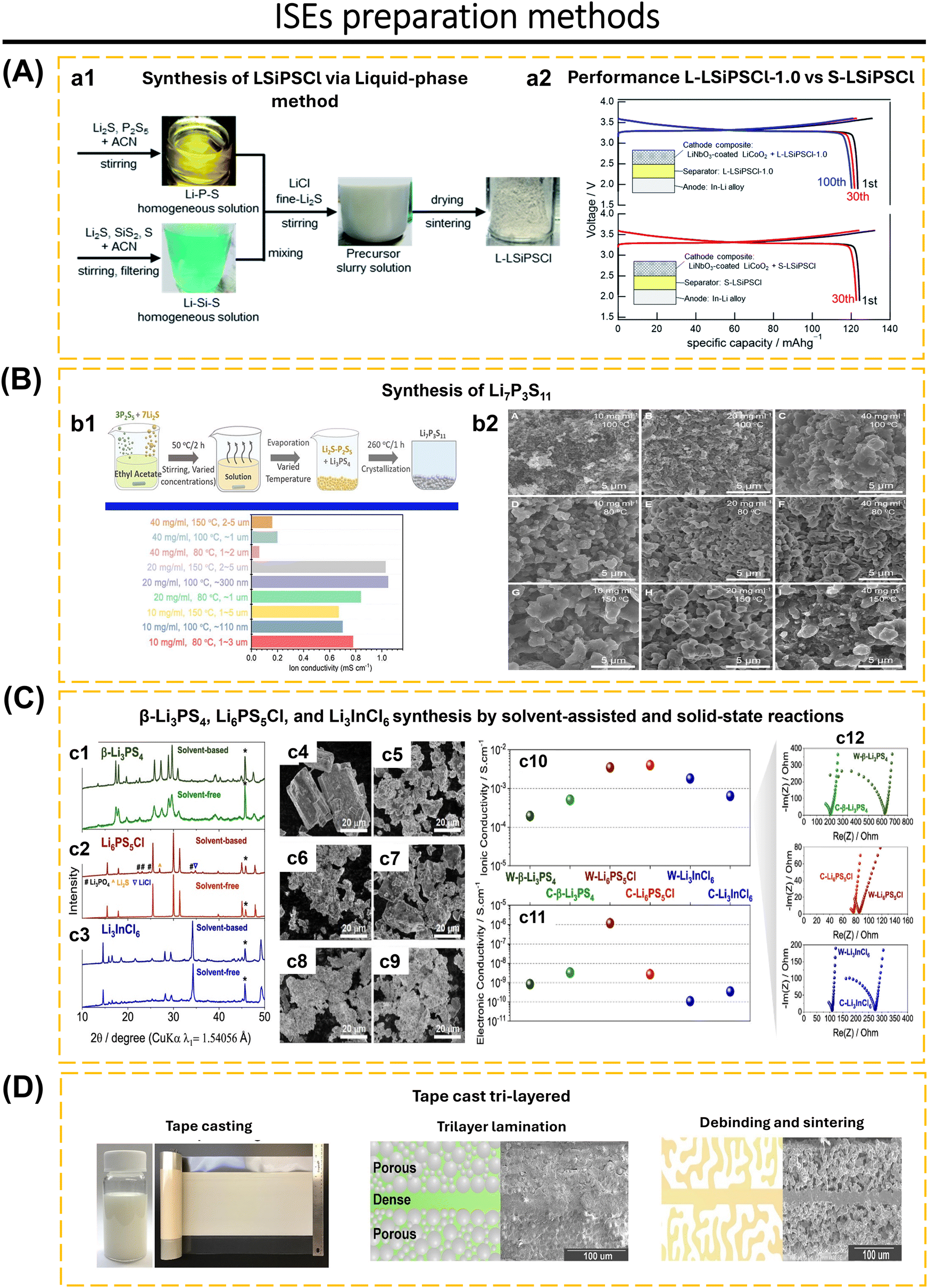 | ||
| Fig. 14 (A) (a1) Schematic representation of the LSiPSCl synthesized via a liquid-phase process. (a2) Charge–discharge curves of the all-solid-state cells prepared using L-LSiPSCl-1.0 or S-LSiPSCl as the solid electrolyte. The current density applied to the cell was 0.096 mA cm−2, which corresponds to a 0.2C rate. Reproduced with permission from ref. 7. Copyright 2022, Royal Society of Chemistry. (B) (b1) A schematic synthesis mechanism of Li7P3S11via ethyl acetate solvent. Ionic conductivity of samples prepared under varying conditions of precursor concentration (10 to 40 mg mL−1) and evaporation temperature (80 to 150 °C). (b2) The SEM images of Li7P3S11 samples synthesized at different concentrations and evaporation temperatures followed by heating at 260 °C: (A) 10–100, (B) 20–100, (C) 40–100, (D) 10–80, (E) 20–80, (F) 40–80, (G) 10–150, (H) 20–50, (I) 40–150. Reproduced with permission from ref. 82. Copyright 2022, Elsevier. (C) Powder X-ray diffraction patterns of (c1) W-β-Li3PS4 (dark green), C-β-Li3PS4 (green), (c2) W–Li6PS5Cl (wine), C–Li6PS5Cl (red), (c3) W–Li3InCl6 (navy), and C–Li3InCl6 (blue). Asterisks indicate Be reflections from the sample holder. SEM images of (c4) W-β-Li3PS4, (c5) C-β-Li3PS4, (c6) W–Li6PS5Cl, (c7) C–Li6PS5Cl, (c8) W–Li3InCl6, and (c9) C–Li3InCl6 as synthesized. Comparison of (c10) ionic and (c11) electronic conductivities of studied ISEs and (c12) Nyquist plots from up to down: β-Li3PS4, Li6PS5Cl, and Li3InCl6 prepared through solvent-engineered and solvent-free methods. Reproduced with permission from ref. 219. Copyright 2021, American Chemical Society. (D) (d1) Photographs of a tape-casting slurry in a glass vial and after casting/drying on a mylar sheet. (d2) Diagram and SEM cross-sectional image of the trilayer in the green unfired state after lamination of the porous and dense tapes. (d3) Diagram and cross-sectional SEM of the trilayer after debinding and sintering. Reproduced with permission from ref. 220. Copyright 2018, Elsevier. | ||
Koç et al.219 compared the properties of β-Li3PS4, Li6PS5Cl, and Li3InCl6 were prepared using solvent-assisted and solid-state reactions, indicated by the prefixes W- and C-, respectively. Both methods indicated that the XRD patterns of all samples exhibited pure phases. Solvent-assisted Li6PS5Cl exhibited impurities, including Li3PO4, Li2S, and LiCl (Fig. 14c1–c3). The ball-milling method facilitated the formation of nano-sized ISE particles and resulted in the formation of microstructural defects. For Li3InCl6, no morphological differences were observed between the two synthesis routes (Fig. 14c4–c9). The ionic and electronic conductivities at room temperature indicated distinct characteristics of the ISEs (Fig. 14c10–c12), with C-β-Li3PS4 exhibiting higher ionic conductivity compared to W-β-Li3PS4. Similar to prior research by Krauskopf et al.,176 the defect concentration influences the ionic conductivity of Na3PS4. The ionic conductivity of Li6PS5Cl remains consistent across both synthesis routes. The solvent-assisted synthesis of Li3InCl6 demonstrates superior ionic conductivity.
Balijapelly et al.203 successfully synthesized ternary alkali ion thiogallates, A5GaS4 (A = Li and Na), utilizing a gas-passing synthesis route. The synthesis of Li5GaS4 using a combination of Li2S, Ga, and S is hindered by the formation of a secondary phase, LiGaS2. Li5GaS5 was produced through the combination of Li2CO3 and Ga2O3 in a precisely controlled tube furnace. A carrier gas was used to flow CS2 gas into a furnace filled with oxides for sulfidation at high temperatures.
The electrolytes Na3B24H23 and Na4B36H34 were synthesized through the ion-exchange method.69,70,225 The anion [B24H23]3− was derived from the oxidation of [B12H12]2−. The precursor Na2B12H12 underwent treatment with a strong acid ion exchange column and was subsequently neutralized using Me4NCl, resulting in the formation of [Me4N]3B24H23. The product underwent processing via an H+ ion exchange column and was subsequently neutralized using NaOH to yield Na3B24H23. The hybrid Na3B24H23.xNa2B12H12 was synthesized by the mechanical ball-milling of Na3B24H23 and Na2B12H12.
Luo et al.95 developed a novel solid electrolyte with a core–shell structure using a gas–solid reaction method by controlling the concentration of B2H6 to form a Na2B12H12 layer on the surface of the NaBH4 particles. The NaBH4/B2H6 ratio of 2:5 results in NaBH4@Na2B12H12 with an ionic conductivity of 10−4 S cm−1 at 115 °C, which makes it highly suitable for ASSBs. Solid-state 11B NMR shows peaks of −42 ppm attributed to [BH4]− in NaBH4, −21 and −15 ppm attributed to [B9H9]2– and [B12H12]2–. Formation of [B9H9]2– consequence of partial decomposition of NaBH4. FTIR spectroscopic investigations revealed that the presence of OH vibrations in the 3600, 3200, and 1614 cm−1 regions was associated with the hygroscopic nature of Na2B12H12 and NaBH4. The XPS results of NaBH4@Na2B12H12 show two peaks in the B 1s spectrum region that shift to a higher energy level, indicated as an increase in boron in NaBH4@Na2B12H12.
Advanced fabrication techniques for LLZO membranes have also gained significant attention. For instance, the sintering of tape-cast trilayered LLZO frameworks represents a promising strategy to produce membranes with desirable properties.220 Another innovative method involves sintering a freeze-cast, vertically aligned LLZO scaffold, which offers potential improvements in the microstructural control of the electrolyte (Fig. 14d).226,227 These sintering techniques, while promising, face considerable challenges in scaling up, particularly in achieving high manufacturing yields of thin, high-performance LLZO-based electrolytes. Consequently, the widespread commercial adoption of these materials may be delayed for years, if not decades, until these production hurdles are overcome. Looking forward, research on LLZO must prioritize several key areas to accelerate the practical application of LLZO-based solid electrolytes. First, engineering the cathode–LLZO interface is critical to improving overall battery performance and longevity. Second, efforts should be directed toward scaling up the production of LLZO-based sheet electrolytes, ensuring that manufacturing processes are both cost-effective and capable of producing consistent, high-quality SEs. Finally, addressing the persistent issue of Li dendrite growth is essential to enhance the safety and reliability of solid-state batteries.
3.2. Interface of ISEs/electrode
The compatibility of interfaces between ISEs and electrodes is a critical factor in achieving high performance in ASSBs, alongside ionic conductivity and electrochemical stability.120 This section summarizes the behavior of ISEs at the anode and cathode interfaces, as well as the approach to controlling these interfaces through the implementation of artificial SEI and CEI layers, referred to as anode and cathode interface layers in this review. This may decrease interfacial resistance, mitigate ISEs decomposition, and enhance battery performance.Sulfide exhibits reactivity with Na and Li metals, leading to the spontaneous formation of an SEI layer. The stable formation of the solid electrolyte interphase at the anode/ionic solid electrolyte interface enhances solid–solid contact, minimizes interfacial resistance, and facilitates the migration of lithium ions.120 The phase diagram of lithium grand potential aids in identifying phase equilibria across various potentials and determining the thermodynamically dominant products influenced by these potentials (Table 4). The SEI layers, including LiF, Li2O, Li2S, Li3N, and Li3P, exhibit stability in relation to Li metal.235 Although NASICONs are superior for high voltage operation and are stable in air, they have poor chemical and mechanical compatibility with Li anodes, resulting in inhomogeneous Li+ transfer and reduction of the active material, such as the reduction of Ge4+ to Ge2+ in LAGP, which occurs frequently when in contact with Li metal.236,237 For garnet-type oxides, LLZO at 2.91 V undergoes reduction into Li2O2, Li6Zr2O7, and La2O3. The increase in voltage discovered for O2 and La2Zr2O7 resulted from the decomposition of Li2O2 and Li6Zr2O7, respectively. At a low voltage of 0.05 V, LLZO undergoes reduction to form Li2O, Zr3O, La2O3, and Zr3O. Zr3O undergoes reduction to Zr at potentials below 0.004 V. The electrochemical stability remains relatively unaltered for Al-doped LLZO (Li6.28La3Zr2Al0.24O12) and Ta-doped LLZO (Li6.75La3Zr1.75Ta0.25O12). As voltages near 0 V, Ta-doped LLZO undergoes reduction to Ta metal, while Al-doped LLZO is transformed into Al–Zr alloy.120 The expectation is that Ta/Al-doped materials can be reduced at slightly high potentials; however, the low concentration of dopants results in insufficient contribution to enhancing stability. Additional investigation is required regarding the Lithium grand potential phase diagram of Ta/Al-doped LLZO as the concentration of dopants increases. The stable SEI does not consistently prevail at the interface, as the interface layer also includes decomposition products that serve as ionic and electronic conductors. During charge–discharge cycles, interface products inevitably form, leading to the accumulation of unstable SEI. This accumulation deteriorates interface contact, elevates interfacial resistance, and promotes dendrite formation due to the presence of electronic conductors at the interface. The resulting dendrites compromise the ISE structure and ultimately reduce battery performance.238
| ISEs | Anode | Phase equilibria at low voltage | Phase equilibria at high voltage | Ref. |
|---|---|---|---|---|
| Li3PS4 | Li | P, Li2S @ 1.72 V | P2S5, S @ ∼2.4 V | 230 |
| Li3P, Li2S @ 0.5 V | ||||
| Li4GeS4 | Li | Ge, Li2S @ 1.62 V | Li2GeS3, S @ ∼2.4 V | 230 |
| Li15Ge4, Li2S @ 0 V | GeS2, S @ > 2.5 V | |||
| Li6PS5Cl | Li | P, Li2S, Li3P, LiCl @ 1.71 | S, P2S5, LiCl @ 2.01 | 172 and 231 |
| Li7P3S11 | Li | Li3PS4, P4S9 @ 2.28 V | S, P2S5 @ 2.31 | 231 |
| Li7P2S8I | Li | P, Li2S, Li3P, LiI @ 1.71 | S, P2S5, LiI @ 2.31 | 231 |
| LGPS | Li | Li4GeS4, P, Li2S @ 1.71 V | Li3PS4, S, GeS2 @ 2.14 V | 120 |
| Li2S, Li15Ge4, Li3P @ 0 V | P2S5, S, GeS2 @ 2.31 V | |||
| LLZO | Li | Li2O, Zr3O, La2O3 @ 0.05 V | Li2O2, Li6Zr2O7, La2O3 @ 2.91 V | 120 |
| Li2O, Zr, La2O3 @ 0.004 V | O2, Li6Zr2O7, La2O3 @ 3.3 V | |||
| Ta-doped LLZO | Li | Li2O, Zr3O, La2O3, Ta @ 0.05 V | Li2O2, Li6Zr2O7, Li5TaO5, La2O3 @ 2.91 V | 120 |
| Li2O, La2O3, Zr, Ta @ 0.004 V | O2, La3TaO7, La2Zr2O7, La2O3 @ 3.3 V | |||
| Al-doped LLZO | Li | Li2O, Zr3O, La2O3, Zr3Al @ 0.05 V | Li2O2, LiAlO2, Li6Zr2O7, La2O3 @ 3.04 V | 120 |
| Li2O, La2O3, Zr, Zr3Al@0.004 V | O2, La4Al2O9, La2Zr2O7, La2O3 @ 3.3 V | |||
| LLTO | Li | Li4Ti5O12, Li7/6Ti11/6O4, La2Ti2O7 @ 1.75 V | O2, TiO2, La2Ti2O7 @ 3.71 V | 231 |
| LATP | Li | P, LiTiPO5, AlPO4, Li3PO4 @ 2.17 V | O2, LiTi2(PO4)3, Li4P2O7, AlPO4 @ 4.21 V | 231 |
| LAGP | Li | Ge, GeO2, Li4P2O7, AlPO4 @ 2.70 V | O2, Ge5O(PO4)6, Li4P2O7, AlPO4 @ 4.27 V | 231 |
| Li3ScCl6 | Li | Sc5Cl8, LiCl @ <0.91 V | ScCl3, Cl2 @ >4.26 V | 232 |
| Sc, LiCl @ 0.V | ||||
| Na3SbS4 | Na | Sb, Na2S @ 0.66 V | Sb2S3, Na2S @ 2.19 V | 104 and 233 |
| Na3Sb, Na2S @ 0 V | Sb2S3, S @ >3.4 V | |||
| Na3PS4 | Na | Na2PS3, Na2S @ <1.55 V | Na2PS3, Na2S @ >2.25 V | 234 |
| Na3P, Na2S @ 0 V | P2S7, S @ 3.0 V | |||
| Na3PSe4 | Na | Na2PSe3, Na2Se @ <1.80 V | Na2PSe3, Se @ >2.15 V | 234 |
| Na3P, Na2Se @ 0 V | PSe, Se @ 3.0 V |
The Na3SbS4/Na interface has been evaluated by Hu et al.233 and Wu et al.104 The symmetric Na/Na3SbS4/Na cell's Nyquist plot indicates an increase in both bulk resistance and charge transfer as Na ions are progressively consumed by Na3SbS4 to form the solid electrolyte interphase (Fig. 15a1).104 The grand potential phase stability plot demonstrates that in the anodic region, Na3SbS4 transforms into Na3Sb and Na2S, whereas in the cathodic region, Sb2S3 and S are present (Fig. 15a3).104,233 The XPS S 2p spectra show that all conditions exhibit a peak at 159.4 eV, which is confirmed to be Na2S. The XPS Sb spectra exhibit a separation of 9.39 eV in the 3d orbital, specifically observed in the Sb 3d5/2 (529.5 and 528.5 eV) and Sb 3d3/2 (538.9 and 537.9 eV) regions (Fig. 15a2). The galvanostatic voltage profiles for 160 cycles at a current density of 0.1 mA cm−2, with 0.5 h allocated for Na plating and 0.5 h for Na stripping, indicate a significant increase in the overpotential of the cell (Fig. 15a4).233 In a related study, Wu et al.104 discussed how the presence of Cl dopant in Na3PS4 influences the stability of the Na/Na3PS4 interface. The ASR values of Na3PS4, Cl-doped Na3PS4 (6.25%), and Cl-doped Na3PS4 (12.5%) exhibited increases from 1986 to 3740 Ω cm2 (88.3%), 992 to 2139 Ω cm2 (115.6%), and 6372 to 7163 Ω cm2 (12.5%), respectively. The findings indicate that adjusting the dopant concentration from 6.25% to 12.5% results in enhanced cycling stability and ionic conductivity. Na2S and Na3P are both solid electrolyte interphases generated from Na3PS4 and Cl-doped Na3PS4. Furthermore, the NaCl peak is observed as a decomposition product of Cl-doped Na3PS4. The NaCl passivation layer serves as an electronic insulator, effectively inhibiting electronic percolation across ISEs and interfaces.
 | ||
| Fig. 15 (A) (a1) Nyquist plot of the Na|Na3SbS4|Na symmetric cell from 0 to 50 h, showing the increasing impedance with time. Inset: equivalent circuit used to fit the data with a corresponding fit plotted on the 50 h curve. Inset: the fitted impedance components with time. (a2) S 2p region scan of the pristine NAS, anode interface of the cell cycled to completion, the symmetric cell, and the Na2S precursor, overlaid on top of each other. O 1s/Sb 3d region scan of the Na|NAS SSEI from the cell cycled to completion. Reproduced with permission from ref. 104. Copyright 2018, American Chemical Society. (a3) Calculated equilibrium voltage profile and phase equilibria for sodiation and desodiation reactions of Na3SbS4. (a4) Galvanostatic voltage profile of the cell cycled at a current density of 0.1 mA cm−2 for 0.5 h Na deposition and 0.5 h Na stripping during each cycle at 60 °C after 1 h rest at the beginning of cycling. Reproduced with permission from ref. 233. Copyright 2019, American Chemical Society. (B) Moisture leading to the byproduct layer formed on the surface of NZSP and its thermal stability verified by the HT. Schematic illustration of the NZSP surface structure change in moisture (H2O + CO2) and further HT. First, a byproduct layer was formed on the NZSP surface; secondly, decomposition of the byproduct layer during HT; at last, a Na-deficient surface formed after decomposition of Na compounds contained in the byproduct layer. (C) (c1 and c2) Contact angle measurement of the molten metallic Na on NZSP SSE and Na-deficient NZSP. Reproduced with permission from ref. 229. Copyright 2020, American Chemical Society. (D) Effects of coating methods on Li wetting behavior with LLZO: stacking-coating (upper panel) and rub-coating (lower panel). Reproduced with permission from ref. 239. Copyright 2022, Wiley-VCH. (E) Li dendrite growth phenomenon and potential mechanisms. SEM images of intergranular Li dendrite growth along grain boundaries of polycrystalline LLZO. Reproduced with permission from ref. 38. Copyright 2016, Elsevier. | ||
Byproduct layers were also found to form periodically as a result of air exposure on the NZSP surface. Gao et al.229 studied the heat treatment (HT) technique that effectively eliminates surface contaminants, thereby enhancing the wettability of the NZSP surface (Fig. 15B). The contact angle of the as-prepared NZSP was measured at 122.5°, in contrast to the 72.5° observed for HT-NZSP. The reduction in contact angle following the elimination of the impurity layer led to a surface deficient in Na ions, thereby enhancing the contact power with the molten Na (Fig. 15C). HT did not produce any side effects by altering the structure of NZSP and facilitating the formation of the stable SEI layers (Na2O) to inhibit the further decomposition of NZSP. The total resistance measured of Na/NZSP/Na and Na/HT-NZSP/Na is about 13725 and 680 Ω cm2.
A major challenge with LLZO is its propensity to react with atmospheric moisture and CO2, resulting in the formation of surface contaminants such as LiOH and Li2CO3. These contaminants significantly hinder the material's ability to wet molten Li, which is critical for its application as a solid electrolyte in Li-metal batteries. Several mechanisms have been proposed to explain the interactions between LLZO and ambient air.240 The reactions leading to the formation of carbonate and hydroxide impurities in the presence of CO2 and H2O, respectively, can be described by the following equations:
| 2Li7La3M2O12 + 7H2O ⇌ 14LiOH + 3La2O3 + 4MO2 |
| 2Li7La3M2O12 + 7CO2 ⇌ 7Li2CO3 + 3La2O3 + 4MO2 |
Interfacial instability at the Li/LLZO interface is a critical challenge, primarily due to lithiophobicity and the propensity for Li dendrite formation.38,120,239,247 The poor wettability between Li and LLZO results in inadequate interfacial contact, leading to an uneven current distribution, which can facilitate the nucleation and growth of Li dendrites (Fig. 15E).248,249 While substantial experimental and theoretical research suggests that grain boundaries play a pivotal role in Li penetration behavior, it is important to recognize that this is not the sole mechanism by which Li penetration occurs. Evidence of Li penetration has been documented in both amorphous and single-crystalline solid electrolytes, which inherently lack grain boundaries.247,250,251 Recent studies have also proposed that residual stresses within the solid electrolyte may significantly contribute to Li penetration, particularly through mechanisms analogous to stress-corrosion cracking, where these stresses are amplified by chemical interactions with Li.252 These residual stresses may originate from grain boundaries, impurities, or processing-induced defects, and could potentially be correlated with the observed heterogeneous stresses associated with polymorphism in LLZO.253 Moreover, the role of electronic conductivity in Li penetration within ISEs has garnered considerable attention in recent literature. Several reports suggest that even trace levels of electronic conductivity, whether located in the grain interior or at grain boundaries, may facilitate the nucleation of metallic Li within the solid electrolyte matrix.254–256 This underscores the complexity of Li penetration phenomena, indicating that multiple, interrelated factors contribute to the interfacial instability at the Li/LLZO interface. Furthermore, previous research has focused on enhancing the compatibility between garnet and lithium by using coating layers such as SnO2,257 LIPON,258 and graphite.259
Fu et al.260 proposed a solution to the instability of the Na metal and NASICON interface by using Na–SiO2 as the anode, effectively decreasing the interfacial resistance from 1658 to 101 Ω cm2. At the Na/NASICON interface, the ability of molten Na to wet the NASICON electrolyte surface is limited. The molten Na–SiO2 completely covers the NASICON surface (Fig. 16a1–a4). The high surface tension and poor wettability of planar NZSP present challenges for the application of molten Na on the NZSP surface. The adoption of monolithic electrolyte architecture is attributed to its unique characteristics that integrate solid and porous layers (Fig. 16b1).156,201 NZSP electrolyte is composed of a solid layer (300 μm) sandwiched by a porous layer (100 μm) (Fig. 16b2). The SnO2 additive is applied to the NZSP surface to improve the sodiophilic properties and facilitate the wetting of the NZSP surface (Fig. 16b3). The integration of monolithic architecture with high sodiophilicity results in an extensive contact area, enhancing the solid–solid interaction of NZSP-Na (Fig. 16b4 and b5). This approach effectively reduces the surface resistance significantly, decreasing from 10000 Ω cm2 to 275 Ω cm2, outperforming planar NZSP.156
 | ||
| Fig. 16 (A) (a1) the intimate contact between Na–SiO2 composite and NASICON SE facilitates a close physical contact and a stabilized solid–solid interface. (a2) Digital photo of Na–SiO2|NASICON interface. (a3) SEM images of the cross-section of Na–SiO2|NASICON interface, which indicates drastically enhanced physical contact at the interface. (a4) EIS spectra of symmetric cells using Na metal and Na–SiO2 as electrodes. Reproduced with permission from ref. 260. Copyright 2020, American Chemical Society. (B) (b1) Schematic of the trilayer NZSP solid-state electrolyte. (b2) SEM images of the trilayer membrane at low magnification. (b3) SnO2 modified porous electrolyte surface. (b4) Photos of the SnO2 modified-trilayer membranes with melting sodium. (b5) SEM images of the Na-impregnated porous layer. Reproduced with permission from ref. 156. Copyright 2019, Wiley-VCH. (C) (c1) Schematic diagram of the preparation process of the Na15Sn4/Na composite foil. (c2) Voltage profiles of symmetric coin cells with Na15Sn4 alloys/Na with various atomic ratios of Na/Sn = 25:4, 45:4, and 75:4 and bare Na electrodes at 50 μA cm−2, and the inset shows the enlarged voltage profiles of the symmetric cells with Na15Sn4/Na electrode (Na/Sn = 45:4) during 200–202, 600–602, and 1200–1202 h, respectively. Reproduced with permission from ref. 261. Copyright 2024, American Chemical Society. | ||
The use of metal alloy reduces the reactivity of ISEs with metal anodes. The Na/Na12Sn4 electrode was fabricated using a mechanical folding and calendaring technique, as detailed by Wang et al.261 An alloy with a thickness of 300 μm was produced by spreading Sn granules on Na foil and subsequently rolling it repeatedly (Fig. 16c1). The electrochemical behavior of Na alloy (Na/Sn = 25:4, 45:4, and 75:4) and pure Na were tested in symmetrical cells Na15Sn4/Na|NZSPF3-PVDF-HFP|Na15Sn4/Na and Na|NZSPF3-PVDF-HFP|Na at 50 μA cm−2 (Fig. 16c2). Na/Sn alloy (Na/Sn = 45:4) showed low polarization voltage and cycle stability reaching 1500 hours. This indicates that Na alloy is more stable facilitating Na deposition and suppressing dendrite growth. Na/Sn alloy (Na:Sn = 25:4) experienced faster cell failure due to the lack of Na as a binder. The cell was subsequently damaged by the irregular sodium deposition of the Na/Sn alloy (Na:Sn = 75:4). The voltage fluctuations observed in the Na anode indicated that the discharging mechanism was unstable. The ratio of Na/Sn = 45:4 as a Na/Sn alloy with low reactivity, good sodiophilic properties, and stable precipitation. After the plating/stripping process, the Na metal anode exhibited morphological irregularities and cracks, as determined by SEM analysis. While the Na/Sn alloy demonstrated stability under the same measurement conditions. The Na/Sn alloy is electrochemically stable and devoid of dendrites due to the fact that the surface remains stable despite the presence of precipitation in certain regions.30,101
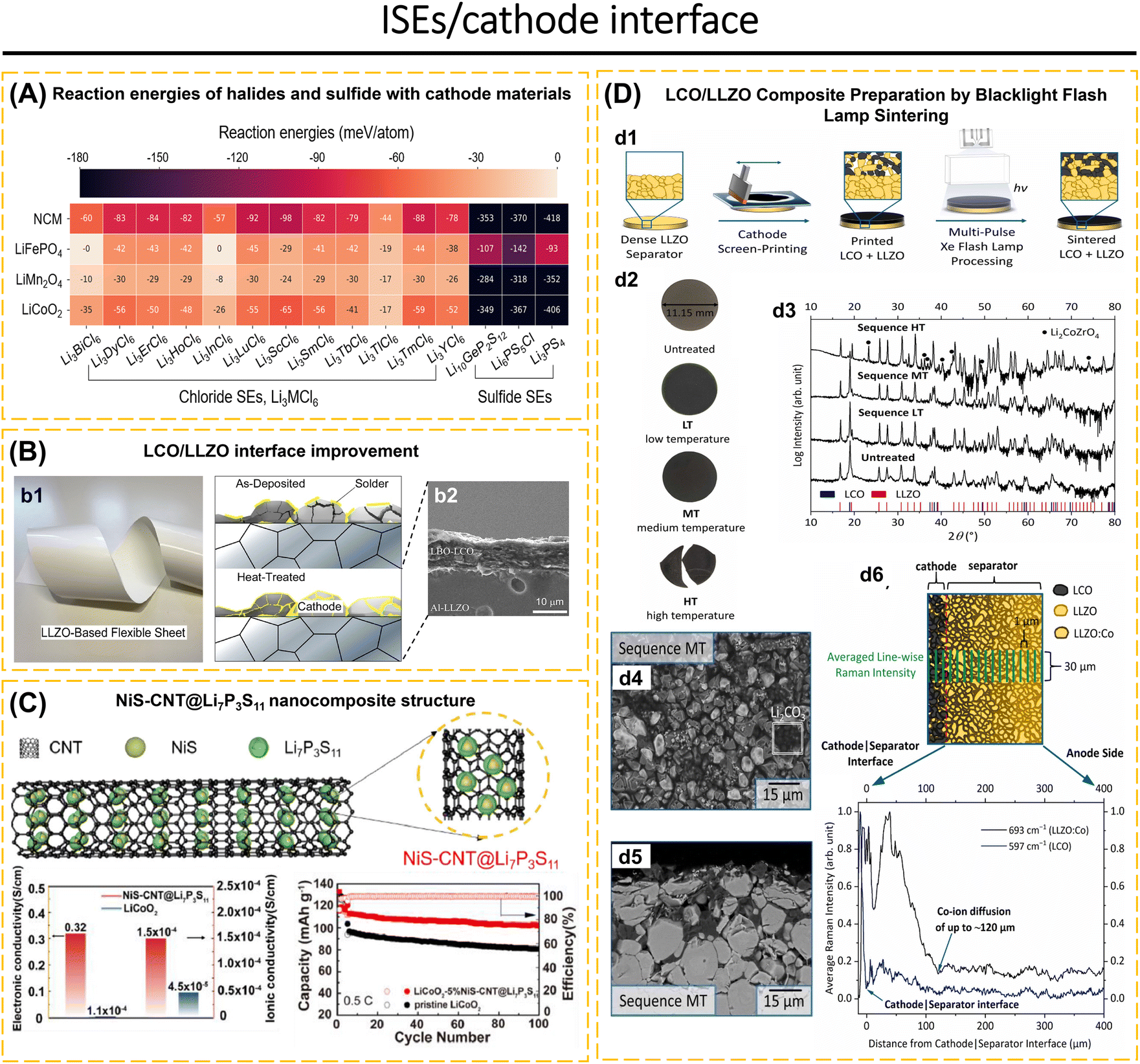 | ||
| Fig. 17 (A) Heat map of the maximum reaction energy between 12 lithium chloride and three lithium sulfide SEs and four cathode materials (LCO, LMO, LFP, and NMC). Reproduced with permission from ref. 179. Copyright 2020, American Chemical Society. (B) (b1) Strategies to overcome the contact issue between the electrode and LLZO solid electrolyte: LLZO-based flexible sheet electrolyte for improved contact between LLZO and cathode layer (left). Reproduced with permission from ref. 61. Copyright 2020, American Chemical Society. (b2) A solid electrolyte solder-coated cathode layer is deposited onto an LLZO substrate by aerosol deposition; the cracks and voids are filled by the solid electrolyte solder after heating. Reproduced with permission from ref. 73. Copyright 2022, American Chemical Society. (C) NiS-CNT@Li7P3S11 nanocomposite structure. Electronic and ionic conductivities of the LCO and NiS-CNT@Li7P3S11 at room temperature. Cycling stability of pristine LCO and LCO-5%NiS-CNT@Li7P3S11 at current densities of 0.5C. Reproduced with permission from ref. 110 Copyright 2021, Elsevier. (D) (d1 and d2) Schematic representation of LLZO-LCO preparation and images of the cathode surface in the untreated condition and after processing of 3 different pulse sequences. (d3) XRD patterns of the processed and untreated composite cathodes with the reference pattern of LLZO (red, ICSD 422259) and LCO (blue, ICDD 010702685). (d4) Top-view SEM images (BSE) of the flash lamp processed composite cathodes (sequence MT). (d5) Polished cross-sections of the flash lamp processed half-cells (sequence MT). (d6) Scheme of the Raman measurement of the sintered half-cell cross-section processed with sequence MT to detect Co-ion diffusion into the LLZO separator as a function of distance from the cathode|separator interface. The measurement was performed in a range of 30 μm over the whole half-cell in 1 μm steps (x and y) and the resulting signals were averaged line-wise and normalized; (b) Raman spectra with the LLZO:Co photoluminescence signal at 693 cm−1 (black) and the LCO phase signal at 597 cm−1 (blue) are shown as a function of distance from the cathode|separator interface (0 μm). Reproduced with permission from ref. 77. Copyright 2023, Elsevier. | ||
| ISEs | Cathode | Decomposition product | Ref. |
|---|---|---|---|
| Li3PS4 | NMC | Co(NiS2)2, Li3PO4, Li(MnS2)2, Co2NiS4, MnS2, Li2S | 179 |
| LFP | Li4P2O7, FePS, FeS2 | ||
| LMO | Li3PO4, Li(MnS2)2 | ||
| LCO | Co9S8, Li3PO4, Li2SO4, Li2S | ||
| Li6PS5Cl | NMC | Li3PO4, Co2NiS4, Li(MnS2)2, Li2S, Co(NiS2)2, MnS2, LiCl | 179 |
| LFP | P4S7, Li3PO4, FePS, FeS2, LiCl | ||
| LMO | Li3PO4, Li(MnS2)2, Li2S, LiCl | ||
| LCO | Li3PO4, Li2SO4, Li2S, Co9S8, LiCl | ||
| Li10GeP2S12 | NMC | Co(NiS2)2, MnO, Li2MnGeO4, Li3PO4, Li2SO4, Co2NiS4, Li2S | 179 |
| LFP | Li4P2O7, Li3PO4, FePS, GeS2, FeS2 | ||
| LMO | Li(MnS2)2, Li3PO4, Li2MnGeO4, MnS2, Li2S | ||
| LCO | Li4GeO4, Co9S8, Li3PO4, Li2SO4, Li2S | ||
| Li3YCl6 | NMC | Li2MnCo3O8, YClO, Li2Mn3NiO8, NiCl2, Co3O4, LiCl | 179 |
| LFP | Fe2PClO4, YPO4, LiCl | ||
| LMO | MnO2, Mn8Cl3O10, YMn2O5, LiCl | ||
| LCO | YClO, LiClO4, Co3O4, LiCl | ||
| LATP | LCO | Co3O4, CoAl2O4, Co2TiO4, and Li3PO4 | 262 |
| LLZO | LCO | La2O3, Li6Zr2O7, Li5CoO4 | 232 |
| Li3ScCl6 | LCO | LiCl, Co3O4, LiClO4, Sc2O3 | 232 |
The interface between cathode active materials and crystalline LLZO presents significant challenges during the fabrication and operation of solid-state batteries. A primary concern lies in the chemical instability at the cathode/LLZO interface, which critically impacts the overall performance and durability of these batteries. In typical solid-state Li-metal batteries, the cathode is often a composite, consisting of cathode active materials and ISEs. Achieving effective contact between these components frequently necessitates high-temperature co-sintering. However, this process can induce thermal decomposition or Li loss from the active materials,263 as well as promote undesirable elemental diffusion at the cathode/solid electrolyte interface, further exacerbating interfacial instability. The challenges associated with the cathode/LLZO interface can be broadly categorized into three key types: mechanical, chemical, and electrochemical instability. These issues are critical as they contribute to increased interfacial resistance, which hinders ion transport and degrades battery performance. To address these challenges, various strategies have been proposed. Cheng et al. have made significant advancements in this area by developing LLZO-based, castable, and flexible sheet electrolytes aimed at improving interfacial contact (Fig. 17b1).61,264 In addition, their work highlights the use of a buffer layer composed of low-melting-point Li3BO3 to enhance interfacial contact between LCO particles and the LLZO substrate. Upon heating, Li3BO3 melts and forms a Li-ion-conducting liquid phase, which infiltrates cracks and voids, thereby improving the connectivity and stability at the interface (Fig. 17b2).73 Such innovative approaches hold promise for overcoming the interfacial challenges in solid-state Li-metal batteries and should be further explored in future studies.
The integration of oxide-based cathodes in sulfide-based ASSBs presents challenges to their applications: (1) decomposition of sulfide electrolyte at high voltage; (2) interface reactions and detrimental decomposition products; (3) formation of space-charge layer; and (4) structural degradation of oxide cathodes.106,179 Transition-metal sulfide cathodes are the preferred cathode type because they provide strong interfacial compatibility with sulfide-type solid electrolytes while also providing superior ionic conductivity and specific capacity.12,48,109,110 The use of a single material as a cathode, such as amorphous niobium polysulfides (a-NbSx, x = 3, 4, 5), has drawbacks for ASSBs applications owing to a-NbSx's weak electronic and ionic conductivity, which affects battery cycle performance. The usage of carbon-based materials may increase the cathode material's electrical conductivity while protecting the cathode with sulfide electrolyte layers improves the solid–solid interface contact issue. Xie and co-workers109 investigated the performance of ASSBs using a-NbS4.5/20% Super P@15% Li7P3S11 cathodes produced via ball milling and annealing. Bilayer electrolytes consisted of Li10GeP2S12 (6.2 × 10−3 S cm−1) and 75% Li2S–24% P2S5–1% P2O5 (1.54 × 10−3 S cm−1). The results demonstrate an initial discharge capacity of 975.7 mA h g−1 at 0.1 A g−1 and a reversible capacity of 464.8 mA h g−1 after 2500 cycles at 0.5 A g−1. After 40 cycles of EIS measurement, the battery cathode containing a-NbS4.5/20% Super P@15% Li7P3S11 had lower Re and Rct values than the a-NbS4.5 and a-NbS4.5/20% Super P cathodes. It was concluded that using Li7P3S11 layer and Super P in the cathode might increase electrochemical performance as well as the electrolyte–electrode interface. Jiang et al.110 used the solvothermal technique to synthesize the NiS-CNT@Li7P3S11 nanocomposite (260 °C, 1 h). LCO-NiS-CNT@Li7P3S11 composite cathodes were produced by combining 2.5, 5.0, and 7.5 wt% NiS-CNT@Li7P3S11 with LCO. Another composite cathode consists of LCO-NiS-CNT@Li7P3S11 and Li10GeP2S12 in a 7:3 ratio. The solid electrolyte utilized is a combination of Li10GeP2S12 and 75% Li2S–24% P2S5–1% P2O5. In the Li10GeP2S12/NiS-CNT@Li7P3S11 nanocomposite/Li10GeP2S12 symmetric cell, NiS-CNT@Li7P3S11 achieves a higher ionic conductivity of 1.5 × 10−4 S cm−1 than LCO, which is 4.5 × 10−5 S cm−1. In ASSBs with LCO-5%NiS-CNT@Li7P3S11 cathode has a capacity of 118.6 mA h g−1, which is higher than the pristine LCO cathode's capacity of 115.1 mA h g−1 at 0.1C after 10 cycles. At 0.5C, LCO-5%NiS-CNT@Li7P3S11 achieves a reversible capacity of 99.6 mA h g−1 after 100 cycles, compared to pure LCO, which has a capacity of 81.9 mA h g−1 (Fig. 17c).
Scheld et al.77 used a blacklight flash lamp sintering approach using an Xe flash lamp to fabricate an LCO/LLZO composite cathode in 20 seconds, rivaling the speed of RTP and laser sintering methods. The three used sintering modes—low temperature (LT), medium temperature (MT), and high temperature (HT)—revealed that the HT mode induced cracks in the composite cathode, led to the decomposition of the LCO phase and resulted in the development of secondary phases such as Li2CoZrO4 (Fig. 17d1–d3). Composite cathodes sintered at medium-temperature conditions exhibited many linkages between LLZO and LCO particles. The upper cross-section exhibited cracks resulting from thermal shock induced by rapid and strong heating and cooling conditions. Nevertheless, cracks were only observable on the LCO surface, since this region experienced the most substantial heat contrast relative to the lower area (Fig. 17d4 and d5). In LLZO composites, the migration of Co-ions from LCO and LLZO must be prevented. Raman spectroscopy may be used to assess the contamination of LLZO by Co (Fig. 17d6). The contamination level was around 120 μm, lower than RTP sintering.213 The short sintering duration resulted in reduced Co ion migration into LLZO, or maybe none at all. The findings suggest that this sintering approach is viable for commercializing the manufacturing of composite cathodes by enhancing the solid–solid contact between the cathode and ISE, although further advancements in the production of LLZO composites are necessary to minimize surface cracks.
Koç et al.219 evaluated the stability of a core-shell-like cathode composite of ISEs-coated NMC622 using ISEs based on lithium thiophosphates (β-Li3PS4), argyrodite (Li6PS5Cl), and halide (Li3InCl6). In cases of ISEs using β-Li3PS4 and Li6PS5Cl, the battery cells were arranged as follows: NMC622/ISE/VGCF|ISE|Li0.5In/ISE. Specifically, Li3InCl6 was arranged as follows: NMC622/Li3InCl6/VGCF|Li6PS5Cl|Li0.5In/Li6PS5Cl. The NCM/Li6PS5Cl and NCM622/β-Li3PS4 cathode composites exhibited capacity retention rates of 97% and 90%, respectively, but the NCM622/Li3InCl6 cathode composite dropped to 50%. Galvanostatic profile testing of the composite cathode (NCM622/Li3InCl6) in a three-electrode cell configuration revealed a reduction in electrochemical performance, corroborating findings from two-electrode configuration cell testing, attributed to the highly resistive positive electrode interface and the incompatibility between the Li3InCl6/Li6PS5Cl and Li3InCl6/Li3PS4 interfaces. The EIS analysis reveals a considerable rise in positive electrode resistance until the 20th cycle.
The charge–discharge process will often induce volume fluctuations in CAM, resulting in the loss of contact force among CAM particles and leading to the formation of cracks in the composite cathode materials. The inherent characteristics of the ISE surface that are incompatible with certain cathodes, intact with the cathode–ISE interface reactions capable of generating unstable products, worsen the interface contact,268 and ultimately degrade battery performance. CAM protection layers and artificial interface layers will serve to enhance the structural and interfacial stability of the electrolyte–electrode and mitigate unstable side reactions during charging and discharging.262,269 The parameters for CAM coating materials and their interaction processes with cathode and electrolyte materials have been clarified.105 The choice of coating type and processes in the design of ASSBs must be meticulous to reduce performance deterioration.268 If CAM coating is the selected approach, the compression pressure during densification should not result in the CAM coating breaking and possibly contacting the ISE directly. Alternatively, the composite cathode may be densified first, followed by the compression or synthesis of the interface layer above the composite cathode layer. The variation in construction will influence battery performance. ISE-coated cathodes offer superior advantages over the CAM and ISE mixing methods due to a more uniform partial distribution between CAM and ISE, which minimizes voids between particles, thereby enhancing particle contact and increasing ionic conduction in the cathode and electrodes.270,271
Here we will explain the method of preparing coated CAM and coating the electrode-ISE interface. Several articles have described the method for depositing the interface layer including ALD,106,108,269,270,272 mechanical mixing,262,265 solution-processed coating,273 direct pressing process,274 magnetic sputtering,1 spin coating,98,266in situ synthesis.97 The electrode–electrolyte interface material applied can be a metal oxide such as SnO2,107 ZrO2,108 LiNbO3,273,275 Li3PO4,106,272,273 LiTaO3,269 Li4Ti5O12,273 graphite,91 Li3N–LiF,97 ZnO–LiF,1 and ISEs. Here, we will explain the progress of ISE application not only as electrolyte and catholyte but also as an artificial interface layer. Until now, ISE has been applied as an interface layer, for example LATP,262 Na3SbS4,271 Li7.5La3Zr1.5Co0.5O12,76 Li3OCl,98 Li6PS5Cl,22,42,67,75 and Li3YCl6.57
3.2.3.1. Mechanical mixing technique. Wang et al.262 applied a simple mechanical mixing technique to coat the surface of the LCO electrode with LATP at high-temperature conditions. The LATP and LCO powders were blended until homogeneous in a mixing machine. The composite samples were calcined at 400–900 °C. The adjustments of ball-milling and calcination conditions influenced the thickness and uniformity of LATP on the LCO surface (Fig. 18a1). The deposition of the LATP layer on the LCO surface leads to the formation of a passivation layer consists of spinel phases (Co3O4, CoAl2O4, and Co2TiO4) and Li3PO4 (Fig. 18a2 and a3), which exhibit stable properties at high voltages, suppress the decomposition reactions, enhance Li-ion transport, and extend the voltage range to 4.6 V. LATP-coated LCO heated at 700 °C has a capacity retention of 88.3% at 25 °C and 72.9% at 45 °C after 100 cycles.
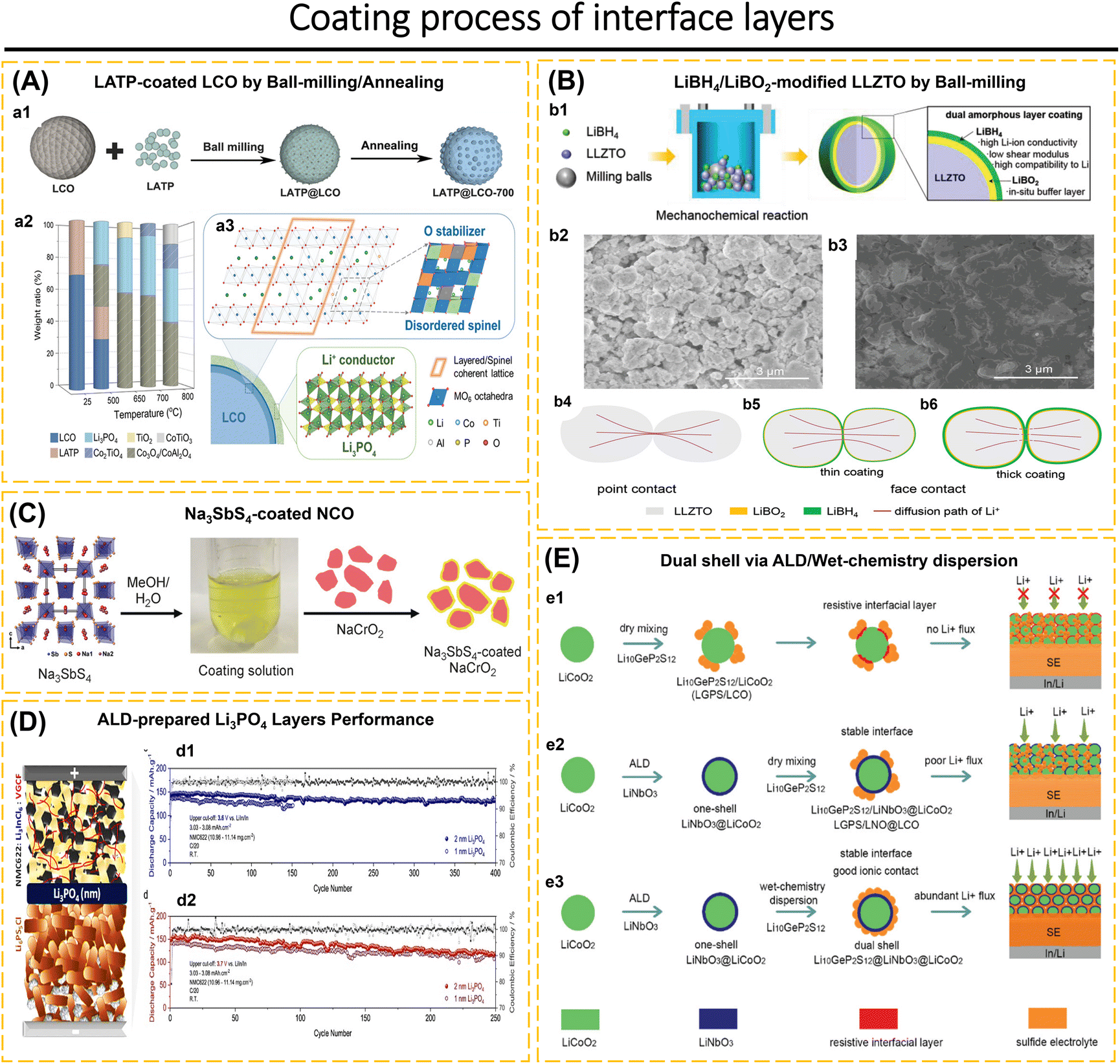 | ||
| Fig. 18 (A) (a1) Schematic illustrations of the surface engineering of LCO. (a2) Phase constitutions of reaction products of LiCoO2 and LATP heat-treated at different temperatures. (a3) Schematic illustration of the surface layer growth mechanism. The spinel phase is structurally more stable than the layered phase with weaker oxygen-anion oxidizing ability at high voltages. Reproduced with permission from ref. 262. Copyright 2020, Wiley-VCH. (B) (b1) Schematic of the preparation of LiBH4-modified LLZTO. SEM images of pellets of (b2) pristine LLZTO and (b3) LLZTO-4LiBH4. Schematic illustration of contact and the Li+ diffusion path between particles in the pellets of (b4) pristine LLZTO sample, with (b5) low and (b6) high LiBH4 content. Reproduced with permission from ref. 265. Copyright 2021, Wiley-VCH. (C) Schematic illustrations of Na3SbS4-coated NCO. Reproduced with permission from ref. 271. Copyright 2016, Wiley-VCH. (D) Long-term cycling performance of cells (NMC622:Li3InCl6:VGCF|Li3PO4-coated Li6PS5Cl|Li6PS5Cl:Li0.5In) with varied surface coating thicknesses at (d1) 3.6 V and (d2) 3.7 V cutoff voltages. Reproduced with permission from ref. 272. Copyright 2022, American Chemical Society. (E) Schematic diagram of ASSLIBs with various interfacial nanostructures. (e1) LiCoO2 directly mixed with Li10GeP2S12 without interfacial design for ASSLIBs. (e2) A one-shell LiNbO3@LiCoO2 cathode for ASSLIBs. (e3) A dual shell LGPS@LNO@LCO for ASSLIBs. Reproduced with permission from ref. 270. Copyright 2019, Wiley-VCH. | ||
Garnet-type LLZO is often manufactured using high-temperature sintering to optimize ionic conductivity; however, this process leads to inadequate solid–solid contact with the electrode and increased brittleness. Gao et al.265 addressed the issue by doping LiBH4 by the ball-milling technique. The interaction between LLZO and the LiBH4 layer results in the formation of a LiBO2 passivation layer, which stabilizes the LLZO–LiBH4 interface (Fig. 18b1). The cold pressing method resulted in a reduction in open porosity from 26.4% (LLZO) to 14.4% (LLZO–4LiBH4) (Fig. 18b2 and b3). This accomplishment elucidates that the coating outcomes function as fillers (enhancing the density of LLZO), adhesives (improving solid–solid contact and reducing interfacial resistance), and connections (efficient ionic conductors) (Fig. 18b4–b6). Ionic conductivity improved from 4.17 × 10−9 S cm−1 (LLZO) to 8.02 × 10−5 S cm−1. Hydroborate serves as an alternate option for a coating layer to enhance oxide-based ion-selective electrodes (ISEs).
3.2.3.2. Solution-processed coating. The technique offers a straightforward preparation procedure for coating, although the challenges of this method are the control of coating concentration, thickness, and homogeneity of the coating. Liu et al.273 synthesized solution-based LiNbO3/Li3PO4/Li4Ti5O12-coated LiNi0.5Mn1.5O4 (LNMO). LNMO and the precursors of Li3PO4 coating (LiOH, NH4H2PO4) were synthesized in aqueous solution and subsequently given heat treatment to produce Li3PO4-coated LNMO. HRTEM examination verified that the coating layer effectively encapsulated LNMO with a thickness of 10–20 nm. The cathode composite comprising 8 wt% LiNbO3-coated LNMO and Li7PS5Cl (70
:30) exhibited an initial discharge capacity of 115 mA h g−1 and a reversible discharge capacity of 80 mA h g−1 after 20 cycles. Banerjee et al.271 deposited a Na3SbS4 layer onto the NaCrO2 surface in a methanol solvent followed by low-temperature treatment (Fig. 18c). Na3SbS4 layers (13 wt%) applied on NaCrO2 ensured optimal ionic contact and strong adherence. High-Resolution Transmission Electron Microscopy (HRTEM) verified that the thickness of the Na3SbS4 layers on NaCrO2 was about 200 nm. NASICON could serve as a protective layer by using a 10 nm LAGP buffer layer (with a conductivity of 0.41 mS cm−1) as a solid electrolyte interface coating that covers the sulfide electrolyte infiltrating NCM622 cathode.18 The LAGP layer formed on the NCM622 surface using the sol–gel method may diminish interfacial side reactions while enhancing Li-ion transport. The dip-coating approach is then used, wherein the LGPS/NMP solution is permeated into the LAGP-coated NCM622 cathode sheet to improve solid–solid contact. NCM622, infused with LGPS and LAGP buffer layers on ASSLBs, had an initial discharge capacity of 141.5 mA h g−1 at 0.05C and maintained stability for 100 cycles at 0.1C.
3.2.3.3. Direct pressing process. The Li3N-based interface layer was applied on the Li/Li2ZrCl6 (LZC) interface. Commercial Li3N (c-Li3N) was converted into the α-phase and β-phase using ball-milling and annealing treatment, respectively. The EIS study of β-Li3N reveals an ionic conductivity of 2.46 × 10−4 S cm−1, and its deformability characteristics allow production of a pellet by direct pressing with the solid electrolyte. The conductivity of LZC with and without the β-Li3N layer is 2.69 × 10−4 S cm−1 and 2.95 × 10−4 S cm−1, respectively, indicating that the β-Li3N layer is stable on LZC. The stability of lithium plating/stripping in symmetrical Li|LZC|Li cells at a current density of 0.1 mA cm−2 exhibits an initial overpotential of 250 mV, which expands to around 1 V after 80 hours. The symmetrical Li|β-Li3N/LZC/β-Li3N|Li cells exhibit an initial overpotential of 100 mV. After reaching 300 hours of operation with an overpotential of around 50 mV. The Li3N layer effectively decreases interfacial resistance, enhances Li+ diffusion, and preserves the stability of the plating/stripping cycle.274
3.2.3.4. Atomic layer deposition. For halide ISE, alongside the use of metal alloys to mitigate the spontaneous interaction between halides and ISEs, the implementation of sulfide-based anode interface layers (i.e. Li6PS5Cl) presents an alternative strategy.46,199,212 Li6PS5Cl in contact with lithium will create SEI layers (Li2S, Li3P, and P2S5) that restrict the interaction between the halide ion-conducting electrolyte and lithium. The contact established between the halide and sulfide electrolytes is significant. Koç et al.272 elucidated that the SXRD investigation of the Li3InCl6/Li6PS5Cl interface revealed the decomposition products, LiCl. The assembly of dual ISEs using Li3InCl6 and Li6PS5Cl facilitates interfacial processes that impede the efficiency of ASSBs. A possible approach involves interface engineering using a protective layer, such as Li3PO4. Koç and co-workers used the ALD process as an outcome. ALD has difficulties in modifying layer thickness because of the rough surface of Li6PS5Cl. Nonetheless, the benefit of ALD is found in its ability to produce ultra-uniform and ultra-thin structures on a wide scale without damaging the basic crystal structure of the coated material.269 The thickness of the applied layer is determined by the number of cycles, 17 ALD cycles for a 2 nm thick Li3PO4. In the cell configuration NCM622(coated)/Li3InCl6/VGCF|Li3InCl6/Li3PO4-coated Li6PS5Cl|Li0.5In/Li6PS5Cl, the cell with a 2 nm thick Li3PO4 layer exhibits 92.3% capacity retention after 400 cycles, while the cell with a 1 nm thick Li3PO4 layer demonstrates 88% retention after 151 cycles. The voltage of 3.7 V exhibited comparable retention rates of 79.9% and 80.1% after 250 cycles for Li3PO4 films of 2 nm and 1 nm thickness, respectively (Fig. 18d1 and d2). Raising the cut-off voltage to 3.8 V results in diminished performance. The application of a Li3PO4 layer with a thickness of up to 10 nm exhibited no electrochemical activity.
Wang et al.270 integrated atomic layer deposition and wet-chemical techniques to create a dual shell design for lithium cobalt oxide cathodes. The direct interaction between LCO and LGPS electrolyte promotes the formation of a high-resistivity CEI layer (Fig. 18e1); therefore, the application of LiNbO3-based layer material via the ALD process results in a one-shell LiNbO3@LCO structure. LiNbO3 serves as an interfacial layer and stabilizes the LGPS–LCO contact. The LiNbO3@LCO/LGPS composite, synthesized by the dry mixing process, exhibits reduced aggregation; nevertheless, the ionic conductivity of the combination requires enhancement (Fig. 18e2). The second layer, LGPS, is formed on LiNbO3@LCO via a wet-chemistry dispersion technique, resulting in the dual shell LGPS@LiNbO3@LCO. This approach effectively produces LiNbO3@LCO particles uniformly coated with LGPS (Fig. 18e3). The GITT study indicates that the Li+ diffusion efficiency of the dual-shell design is four times higher than that of the single-shell design. ASSBs with a single-shell LiNbO3@LCO cathode exhibited a specific capacity of 20 mA h g−1 at 1C, while the dual-shell LGPS@LiNbO3@LCO electrode demonstrated a capacity of 87.7 mA h g−1 at the same rate. The advancement of dual-shell LGPS@LiNbO3@LCO using ALD/Wet-chemical techniques is expected to provide promising outcomes for high-performance ASSBs.
3.2.3.5. In situ synthesis process. According to Kato et al.,276 lithium superionic conductors such as Li9.6P3S12 and Li9.54Si1.74P1.44S11.7Cl0.3 have LGPS-like crystal structures, with Li9.54Si1.74P1.44S11.7Cl0.3 having the highest conductivity of 25 mS cm−1, which is twice the LGPS conductivity value (12 mS cm−1) reported by Kamaya et al.277 In ASSBs, the coulombic efficiency of cells with solid electrolytes based on Li9.6P3S12, Li10GeP2S12, and Li9.54Si1.74P1.44S11.7Cl0.3 is 90%, 61%, and 39%, respectively. The loss in efficiency is caused by a certain amount of lithium being consumed during the reaction, which forms an interfacial layer on the electrode/SE interface. To prevent the formation of lithium dendrites, an electron-insulating layer for the lithium anode must be introduced. Wu et al.97 developed an anode interface layer based on LiF–Li3N (Fig. 19a1). Li@LiF–Li3N was synthesized by an in situ reaction of C7H2F5NO in DMF solvent. The C7H2F5NO-DMF solution was dropped on lithium foil using spin coating methods and dried at 60 °C for 2 hours to produce Li@LiF–Li3N electrodes. Stability performance analysis of Li/Li10GeP2S12/Li symmetric cell shows the overpotential reaches 2 V after 403 hours @ 0.1 mA cm−2/0.1 mA h cm−2. The Li@LiF–Li3N/Li10GeP2S12/Li@LiF–Li3N symmetric cell can achieve stability of more than 9000 hours with an overpotential of 0.39 V. SEM was used to observe the anode morphology of Li and Li@LiF–Li3N anodes from all-solid-state batteries after 20 cycles (Fig. 19a2 and a3). The anode without a protective coating looks to have holes on its surface, as well as side reactions that damage the electrode/electrolyte contact. The anode's LiF–Li3N layer ensures robust surface integrity. Li3N, as a good ionic conductor, may prevent lithium side reactions that produce sulfide, but LiF, with its poor electronic conductivity and high interfacial energy, can reduce dendrite formations. The combination of both allows the electrode–electrolyte interface to remain stable while also providing superior electrochemical performance stability for ASSBs (Fig. 19a4 and a5).
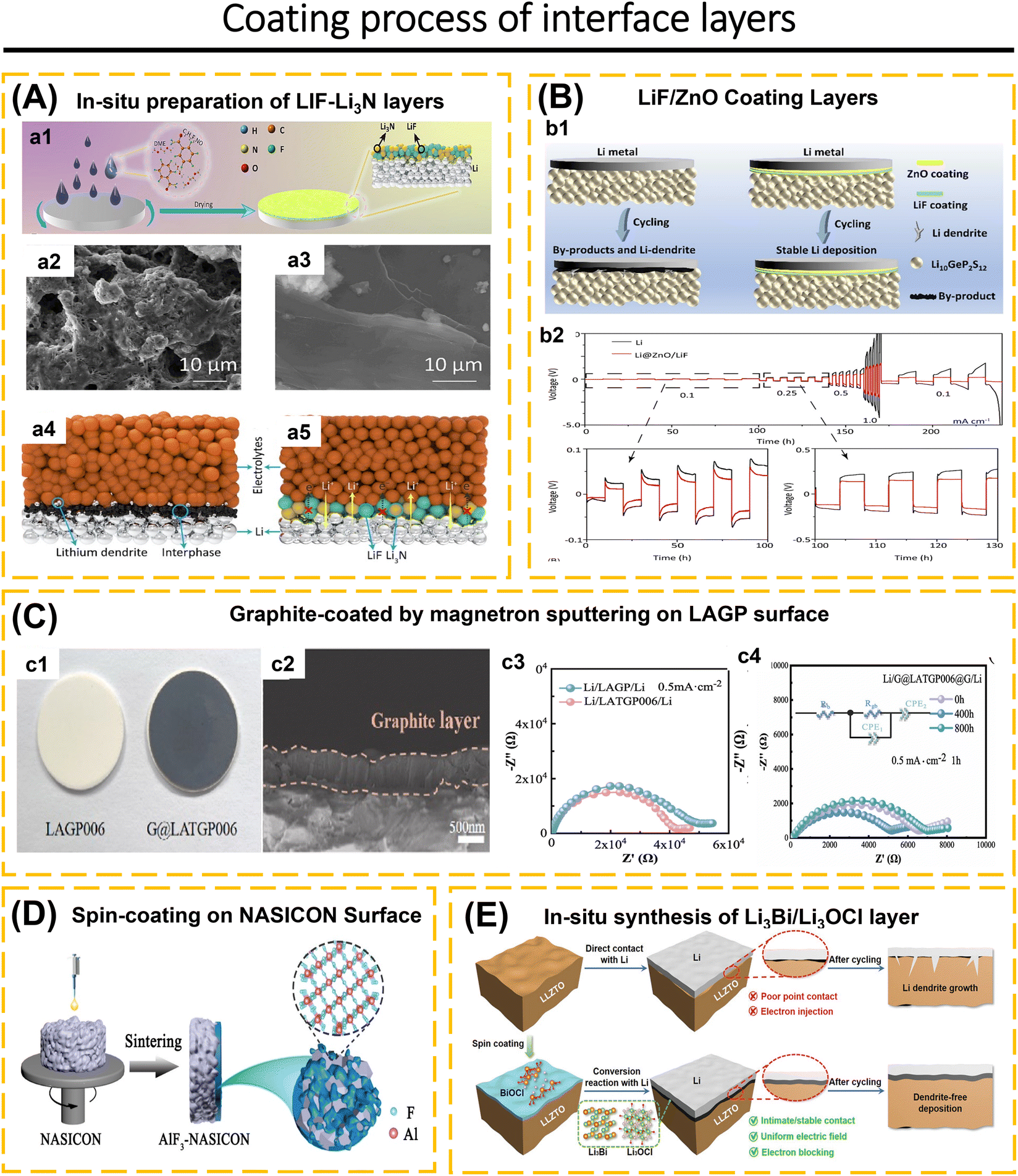 | ||
| Fig. 19 (A) (a1) The Li@LiF–Li3N anode is in situ formed via a manipulated reaction of C7H2F5NO with lithium metal, realizing highly stable all-solid-state lithium batteries. Surface SEM images of Li anodes obtained from (a2) LCO|LGPS|Li and (a3) LCO|LGPS|Li@LiF–Li3N batteries after 20 cycles at 0.1C. Lithium deposition behaviors of (a4) Li/electrolytes and (a5) Li@LiF–Li3N/electrolytes. Reproduced with permission from ref. 97. Copyright 2023, Elsevier. (B) (b1) Proposed interfacial evolution of Li and Li@ZnO/LiF negative electrodes with LGPS after deposition. (b2) Rate capability of the symmetric cells of Li@ZnO/LiF|LGPS|Li@ZnO/LiF, Li|LGPS|Li at 0.1, 0.25, 0.5, 1.0 mA cm−2. Reproduced with permission from ref. 1. Copyright 2023, Wiley-VCH. (C) (c1) Optical images of the polished LATGP006 pellet and the LATGP006 pellet with a graphite layer. (c2) SEM images of the LATGP006 pellet with a graphite layer: cross-sectional image. (c3) EIS spectra of the Li/LAGP/Li and Li/LATGP006/Li cells after cycling at 0.5 mA cm−2. (c4) Nyquist plots of the symmetric Li/G@LATGP006@G/Li cell before and after cycling at current densities of 0.5 mA cm−2. Reproduced with permission from ref. 91. Copyright 2023, Elsevier. (D) (d1) Schematic illustration of the synthesis process of AlF3-NASICON pellets. (d2) SEM images of the cross-sections of Na/AlF3-NASICON interfaces. Insets are the corresponding digital images showing the wetting behaviors of molten Na on NASICON and AlF3-NASICON, respectively. Reproduced with permission from ref. 266. Copyright 2020, Elsevier. (E) Schematic diagrams of the fabrication of alloy@antiperovskite hybrid layer by in situ conversion reaction between garnet and Li metal, and the comparison of dendrite suppression capability for the pristine and modified Li/garnet interfaces. Reproduced with permission from ref. 98. Copyright 2023, Wiley-VCH. | ||
3.2.3.6. Sputtering technique. Chang et al.1 applied a double layer of ZnO/LiF to prevent side reactions at the Li–LGPS metal contact. Magnetic sputtering provides benefits over ALD and CVD due to its high deposition rate, favorable environmental impact, ease of control, and high production efficiency on an industrial scale. ZnO was sprayed on the surface of the lithium metal disc using magnetic sputtering (RH450) at 50 W, 0.5 Pa, and a rotation speed of 20 rpm for 1 hour. Then, LiF in the same way for 6 hours to generate Li@ZnO/LiF. The lithiophilic ZnO layer limits direct contact between LGPS and lithium anode, while the lithiophobic LiF layer prevents lithium dendrite growth (Fig. 19b1). The Li@ZnO/LiF|LGPS|Li@ZnO/LiF symmetric cell demonstrates stability after 2000 hours of lithium plating/striping cycling, with a small overpotential of 200 mV at 0.1 mA cm−2 for 1 hour. Fig. 19b2 illustrates tiny overpotentials of 15, 153, 564, 1431, and 198 mV for current densities of 0.1, 0.25, 0.5, 1.0, and 0.1 mA cm−2. The conclusion is that the ZnO/LiF coating may improve the stability of the Li/LGPS interface.
Graphite exhibits good conductivity, superior mechanical properties, and electrochemical stability when used as the Li–LATGP interface layers. The graphite layer uniformly covers the LATGP surface with a thickness of 0.5 μm (Fig. 19c1 and c2). The Nyquist plots of LAGP, LAGTP006, and G@LAGTP006 in a symmetrical cell at 0.5 mA cm−2 indicate that the impedance of G@LAGTP006 is inferior to that of LAGP and LAGTP006 (Fig. 19c3 and c4). The graphite layer effectively enhances the stability of the Li–LAGTP006 contact. The initial discharge of the Li/LAGTP006/LFP cell reaches 142.1 mA h g−1, exhibiting a retention of 17.6% after 100 cycles. The Li/G@LAGTP/LFP cell has a retention rate of 87.8%. The graphite interface layer enhances cycling performance by preventing damage at the interface during charge–discharge cycles.91
3.2.3.7. Spin-coating with heat-treatment process. Miao et al.266 synthesized an AlF3-based protective layer at the Na/NASICON interface by a spin-coating technique. The AlF3 solution was prepared from Al(NO3)3.9H2O and NH4F. The NASICON pellet was coated with 60 μL of AlF3 at 4000 rpm for 30 seconds, then sintered at 450 °C for 5 hours (Fig. 19d). The AlF3 solution enhanced interfacial contact, exhibiting a conductivity of 2.415 × 10−4 S cm−1 at RT, and filled the gaps in the NASICON to inhibit dendrite growth. Under high voltage conditions, the AlF3-coated NASICON exhibited cycle stability, while the uncoated NASICON demonstrated significant voltage drop and short circuit. Furthermore, the propensity of AlF3 to react with Na during the first phase of activation led to the formation of stable SEI layers, which reduces electron distribution and increases the dendrite resistance ability. This significantly enhances the cycle stability of solid-state batteries. Bi et al.98 explained the formation of a hybrid layer consisting of Li3Bi alloy nanoparticles embedded inside an antiperovskite Li3OCl matrix (Li3Bi@Li3OCl) in situ at the interface of Li|Li6.75La3Zr1.75Ta0.25O12 (Fig. 19e). BiOCl is deposited onto the Li6.75La3Zr1.75Ta0.25O12 surface using spin-coating and undergoes a spontaneous reaction with Li, resulting in the formation of a hybrid Li3Bi@Li3OCl layer. The density of states (DOS) indicates that Li3OCl, possessing a bandgap of 5.06 eV, can inhibit electron tunneling from Li to LLZTO and serve as a good ionic conductor at the interface. On the other hand, Li3Bi alloy, with a bandgap of 0.44 eV, functions as an electron conductor, with Li3Bi uniformly dispersed within the Li3OCl matrix, thereby restricting electron conduction and preventing electron movement through the Li3OCl matrix. This functions to inhibit dendritic growth. Li3B exhibits superior lithiophilic capabilities due to its reduced surface energy in comparison to Li3OCl. The Li3Bi@Li3OCl hybrid layer effectively decreased the interface resistance from 349 Ω cm2 to 27 Ω cm2. In conclusion, BiOCl can spontaneously react with Li to form a Li3Bi@Li3OCl hybrid layer at the Li–LLZTO interface and exhibits good lithiophilicity, ionic conductivity, and electron-blocking properties. This also indicates that antiperovskite can act as an interfacial layer to improve the incompatibility of garnet to Li metal.
4. Current models of ASSBs and multi-phase ISEs-based ASSBs
4.1. Current models of all-solid-state lithium/sodium-ion batteries
The solid-state battery configuration model is an innovative improvement over liquid electrolyte-based batteries. It replaces the liquid electrolyte and separator with a solid electrolyte, such as ISE, which performs the functions of both.200 Chen et al.278 clarified the classification of batteries based on the presence or absence of LE. In general, solid-state batteries consist of an anode (Li or Na metal), ISE, and a cathode (containing cathode active materials such as LCO, LTO, NMC, and LFP). This setup forms a straightforward interface system between the anode/ISE and ISE/cathode, as shown in Fig. 20. This section has summarized some published model designs of lithium/sodium-based ASSBs. First, it is necessary to know the general configuration of both systems. Table 6 shows the comparison of components in both systems based on the information gathered. Some solid electrolytes may exhibit incompatibility with either the anode or the cathode or both. In conventional solid-state batteries, a certain quantity of liquid electrolytes is frequently applied to improve the wetting properties of the interface between the ISE and cathode.91,125,158,159,163,279 In order to eliminate the demand for flammable organic materials in solid-state batteries, the liquid electrolyte can be replaced with a solid interface layer made of metal oxide, or ISEs. A solid interface layer is typically applied to facilitate solid–solid contact interactions in ASSBs.91,97,270 Additionally, a composite cathode consisting of CAM, conductive carbon, and catholyte (ISEs) can be applied to enhance resistance and reduce side reactions at the ISE/cathode interface.77,103,109,110,165,196,269 This will complicate the interface system in ASSBs such as CAM–carbon, CAM–catholyte, carbon–catholyte, CAM–cathode interface layer, carbon–cathode interface layer, catholyte–cathode interface layer, ISE/cathode interface layer, ISE–anode interface layer, and anode–anode interface layer. In Fig. 21–23 present the existing 28 ASSBs models and categorize them based on ISEs type. (1) Antiperovskite: this group of ISEs remains stable when in contact with a lithium anode, even without the presence of an interface layer. The cathode typically consists of CAM, ISEs, and conductive carbons. (2) Sulfide: ASSBs that use sulfide electrolytes often employ Li/Na metal or alloy as the anode. Several models display an interface layer on either the anode, cathode, or both. The composition of the cathode varies. Hybrid sulfide-based electrolytes may be used either as a homogenous mixture or as stacked layers of electrolyte. (3) Oxide: this ISE not only serves as an electrolyte and a catholyte, but also functions as a CAM based on the model presented. Further development, single-class ASSBs in NASICON class can be collected, with the possibility of the material having the same or different chemical composition. This ISE exhibits stability when exposed to both the anode and cathode. In some cases, an interface layer may be added. (4) Halide: electrolytes exhibit such high reactivity towards lithium anodes that the use of anode interface layers or alloys is increasingly prevalent. For some cases with halide electrolyte, the utilization of interface layers is applied even though anodes (alloys) or composite cathodes have been used. (5) Hydroborate: in the ASSBs models do not include the usage of anode and cathode interface layers. This ISE has excellent stability when used with metal anodes, alloy anodes, and composite cathodes. Tables 7 and 8 summarize example configurations of each current ASSB model along with a comparison of their respective electrochemical performances.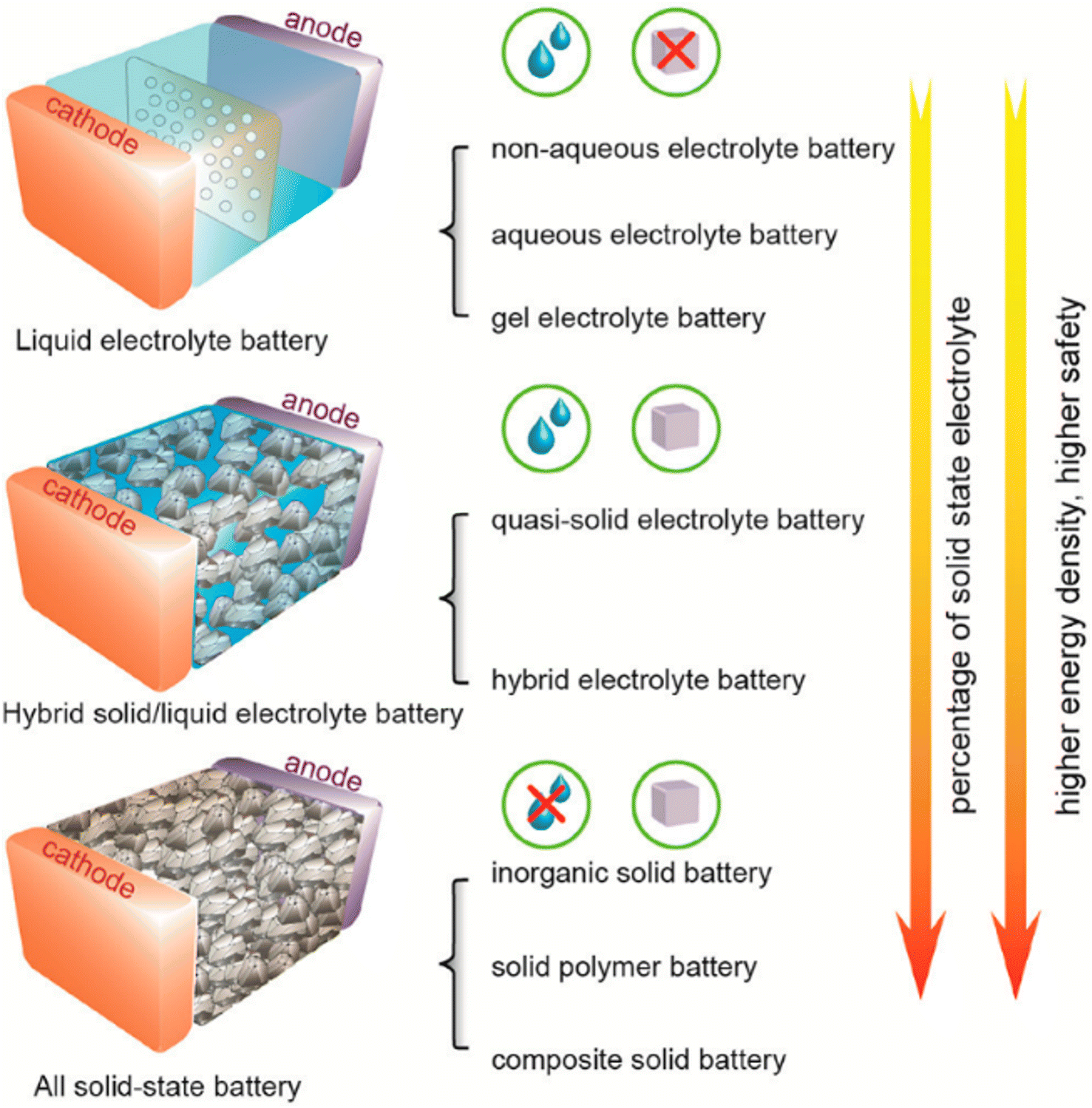 | ||
| Fig. 20 Classification of batteries based on the use of liquid electrolyte. Reproduced with permission from ref. 278. Copyright 2020, American Chemical Society. | ||
| Materials | ASSSIBs | ASSLIBs |
|---|---|---|
| Cathode active material | NaTi2(PO4)3 | LFP |
| Na3V2(PO4)3 | NCA | |
| Na0.67Mn0.47Ni0.33Ti0.2O2 | LMO | |
| Na[Ni1/3Fe1/3Mn1/3]O2 | LCO | |
| TiS2 | LTO | |
| Na3V2(PO4)2O2F | NCM811 | |
| Na0.85Mn0.5Ni0.4Fe0.1O2 | NCM622 | |
| NaNi0.68Mn0.22Co0.1O2 | NCM83 | |
| Na3V2(PO4)2F3 | NCM85 | |
| NaMO2 (M = Fe, Ni, Mn, Co) | NCM90 | |
| TiS2 | ||
| Catholyte | Na-based ISEs | Li-based ISEs |
| Conductive material | Carbon black, carbon nanofiber, MWCNT, RGO, etc. | Carbon black, carbon nanofiber, MWCNT, RGO, etc. |
| Cathode interface layer | — | Metal oxide-based layer |
| Oxide-based ISEs | ||
| Halide-based ISEs | ||
| Electrolyte | Na-based ISEs | Li-based ISEs |
| Anode interface layer | Metal oxide | Sulfide-based ISEs |
| Metal halide | Halide-based ISEs | |
| Metal halide-based layer | ||
| Metal oxide-based layer | ||
| Graphite-based layer | ||
| Nitride-based layer | ||
| Anode | Na | Li |
| Na–Sn | Li–In | |
| Na–SiO2 | Li–Si | |
| Hard carbon | Graphite |
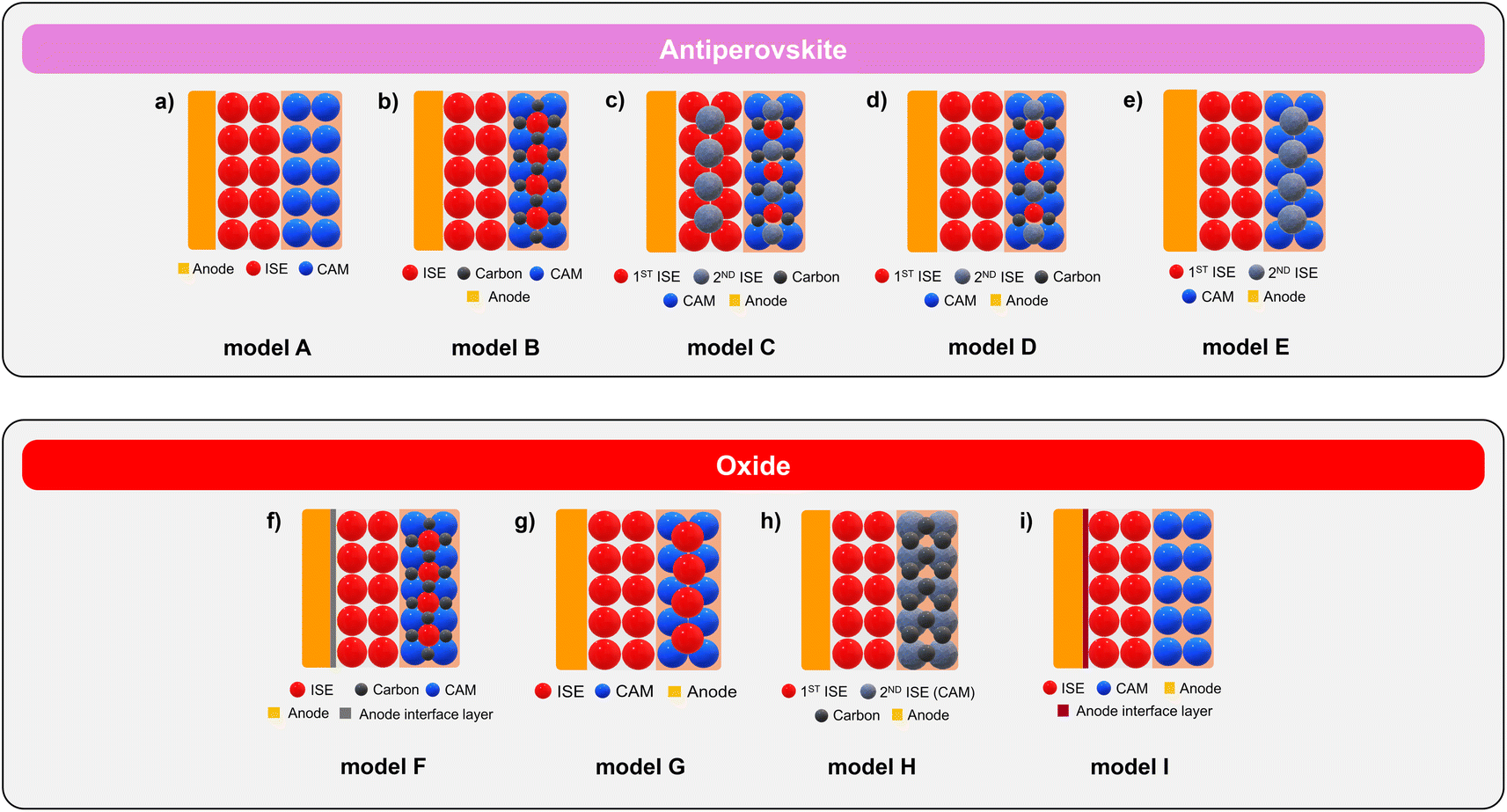 | ||
| Fig. 21 Current models of ASSBs assembly for antiperovskite-based ISEs: (a) model A, (b) model B, (c) model C, (d) model D, (e) model E, and oxide-based ISEs: (f) model F, (g) model G, (h) model H, (i) model I. | ||
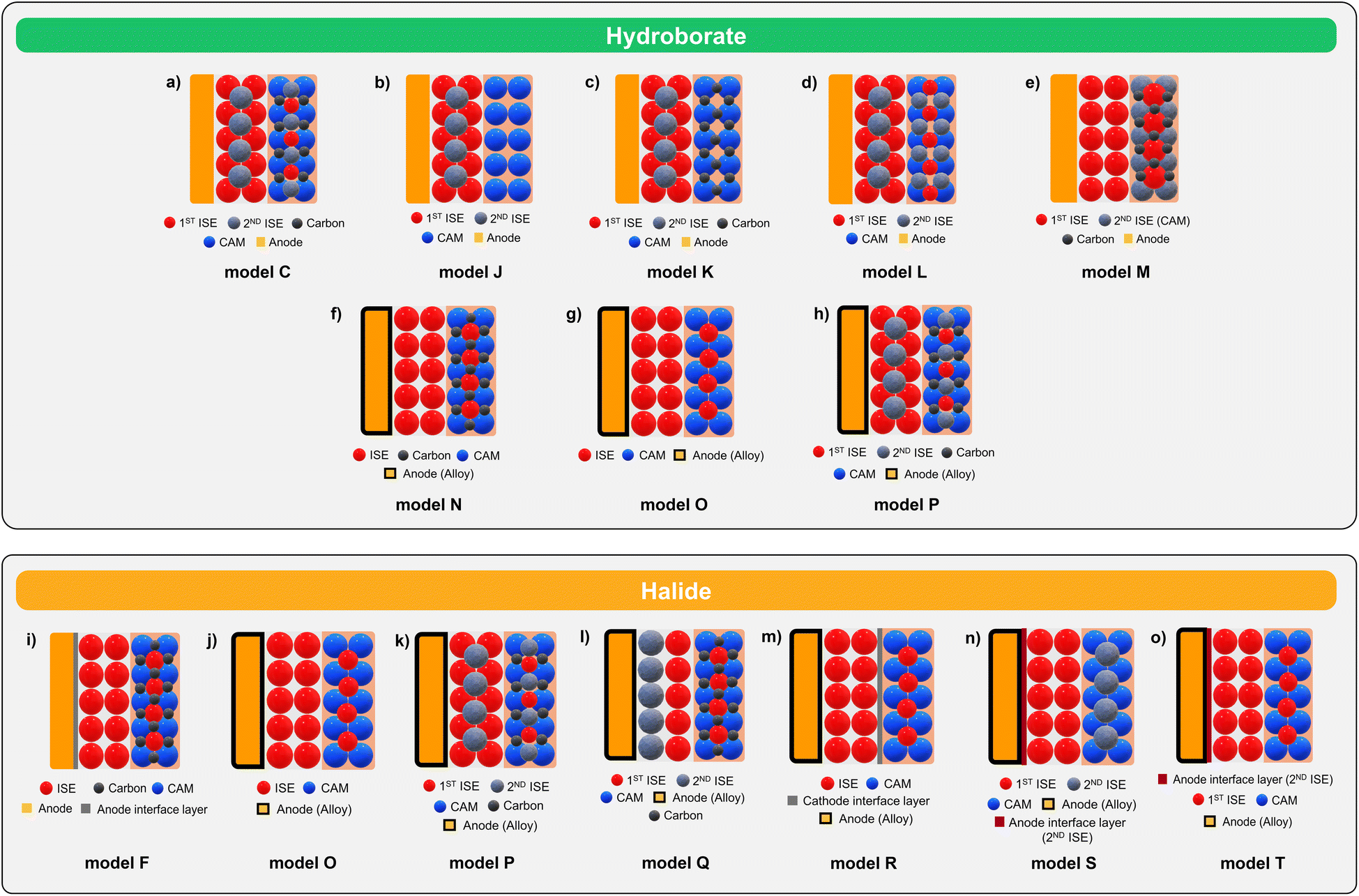 | ||
| Fig. 22 Current models of ASSBs assembly for hydroborate-based ISEs: (a) model C, (b) model J, (c) model K, (d) model L, (e) model M, (f) model N, (g) model O, (h) model P, and halide-based ISEs: (i) model F, (j) model O, (k) model P, (l) model Q, (m) model R, (n) model S, (o) model T. | ||
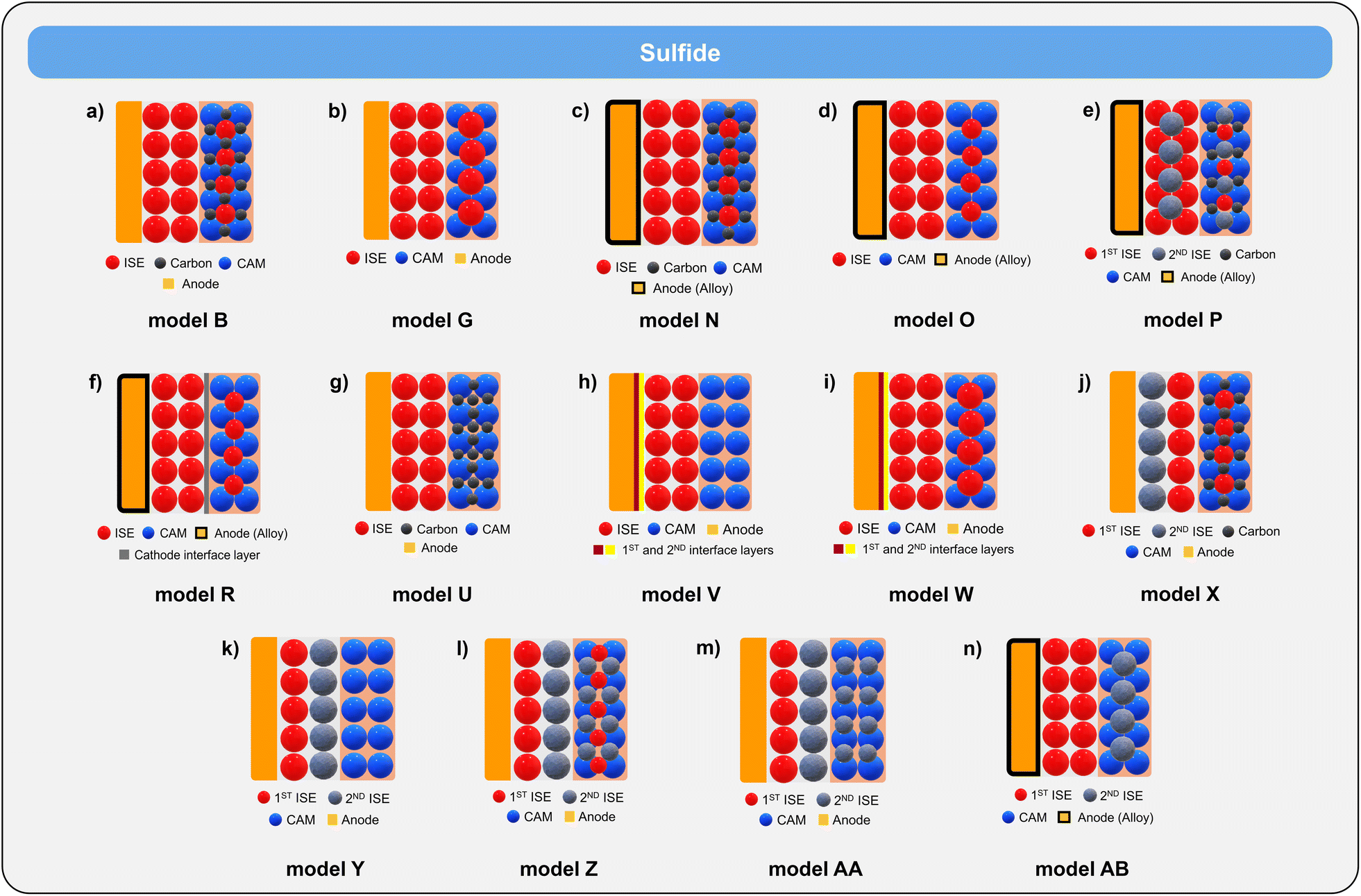 | ||
| Fig. 23 Current models of ASSBs assembly for sulfide-based ISEs: (a) model B, (b) model G, (c) model N, (d) model O, (e) model P, (f) model R, (g) model U, (h) model V, (i) model W, (j) model X, (k) model Y, (l) model Z, (m) model AA, (n) model AB. | ||
| Cathode | Solid electrolyte | Anode | ASSBs model | Capacity | Potential window | Capacity retention | Ref. |
|---|---|---|---|---|---|---|---|
| NaTi2(PO4)3 + Carbon | Na3.16Zr1.84Y0.16Si2PO12 | Na | H | Reversible capacity of 56.6 mA h g−1 at C/5 | 1.5–2.8 V vs. Na/Na+ | 28.3% capacity retention after 950 cycles at C/5 | 78 |
| Na3V2(PO4)3 + carbon black + PVDF | UHS-NZSP | Na | H | Initial discharge capacity of 102 mA h g−1 (NVP loading of ∼2 mg cm−2) | 2.0–4.0 V vs. Na/Na+ | 82% capacity retention after 850 cycles | 39 |
| NaTi2(PO4)3 + RGO | NLZSP-NBO3 | Na | H | Initial discharge capacity of 95.4 mA h g−1 | 1.5–3.0 V vs. Na/Na+ | 91.4% capacity retention after 200 cycles at 1C | 41 |
| Na2/3Ni1/3Mn2/3O2 | NZSP | Na | A | Initial discharge capacity of 92.5 mA h g−1 at 0.1C | 2.0–4.0 V vs. Na/Na+ | — | 229 |
| TiS2 + Na3SbS4 | Na3SbS4 | Na | G | Initial charge capacity of 115.9 mA h g−1 at 0.1C | 1.2–2.4 V vs. Na/Na+ | — | 104 |
| TiS2 + Ca-doped Na3PS4 | Ca-doped Na3PS4 | Na–Sn alloy | P | 2nd charge capacity of 198 mA h g−1 at 0.06C | 1.0–2.5 V vs. Na/Na+ | 91% capacity retention after 100 cycles | 101 |
| Na3V2(PO4)3 + carbon + t-Na3PS3.8Cl0.2 | t-Na3PS3.8Cl0.2 | Na | M | Initial discharge capacity of 100 mA h g−1 at 10 mA g−1 | 2.5–3.8 V vs. Na/Na+ | — | 63 |
| Na[Ni1/3Fe1/3Mn1/3]O2 + Na3B24H23–5Na2B12H12 + carbon black | Na3B24H23–5Na2B12H12 | Na | C | Initial discharge of 51.2 mA h g−1 at 0.1C | 2.0–4.2 V vs. Na/Na+ | 87% capacity retention after 50 cycles at 0.1C at 25 °C | 70 |
| TiS2 | Na3B24H23–5Na2B12H12 | Na | J | Initial discharge of 171.4 mA h g−1 at 0.1C | 1.5–2.5 V vs. Na/Na+ | ∼150 mA h g−1 after 100 cycles at 0.1C | 70 |
| TiS2 + acetylene black + PEG | NaB3H8·xNH3@NaB3H8 (x < 1) | Na | K | Initial discharge of 237.2 mA h g−1 at 1C | 1.40–2.35 V vs. Na/Na+ | 90.1% capacity retention after 240 cycles at 1C | 50 |
| Na3V2(PO4)2O2F + Na4B36H34–7Na2B12H12 + carbon black | Na4B36H34–7Na2B12H12 | Na | M | Initial discharge of 71.2 mA h g−1 at 0.1C | 2.5–4.5 V vs. Na/Na+ | 85.8% capacity retention at 25 °C after 100 cycles | 69 |
| TiS2 + Na2B20H18–4Na2B12H12 | Na2B20H18–4Na2B12H12 | Na | L | Initial discharge of 64.8 mA h g−1 | 1.5–2.7 V vs. Na/Na+ | 82% capacity retention after 100 cycles at 0.1C | 51 |
| TiS2 + Na4B20H18–3Na2B12H12 | Na4B20H18–3Na2B12H12 | Na | L | Initial discharge of 38.1 mA h g−1 at 0.1C | 1.2–2.7 V vs. Na/Na+ | 92% capacity retention after 50 cycles at 0.1C | 6 |
| NMNFO + CB + 0.62[HCF-Sm]·0.38[LCF-Ta] | 0.62[HCF-Sm] | Na–Sn alloy | P | Initial discharge of 117.9 mA h g−1 | 2.3–4.0 V vs. Na/Na+ | 88.3% after 100 cycles at 0.2C (25 °C) | 30 |
| 0.38[LCF-Ta] | |||||||
| NMNFO + CB + 0.57[HCF-La]·0.43[LCF-Ta] | 0.57[HCF-La] | Na–Sn alloy | P | Initial discharge of 95.1 mA h g−1 | 2.3–4.0 V vs. Na/Na+ | 91.0% after 100 cycles at 0.2C (25 °C) | 30 |
| 0.43[LCF-Ta] |
| Cathode | Solid electrolyte | Anode | ASSBs model | Capacity | Potential window | Capacity retention | Ref. |
|---|---|---|---|---|---|---|---|
| Li3BO3-coated LiCoO2 | Al-doped Li7La3Zr2O12 | Li (Au layer) | I | Initial discharge capacity of 128 mA h g−1 at 0.2C | 2.8–4.3 V | 87% after 30 cycles | 73 |
| LCO/Ta, Al-doped LLZO | Ta, Al-doped LLZO (Li6.45Al0.05La3Zr1.6Ta0.4O12) | In | G | Initial discharge capacity of 60 mA h g−1 | 2.8–3.6 V | — | 77 |
| LFP + LAGP + carbon black + PVDF | Te-doped LAGP | Li (graphite layer) | F | Initial discharge of 154.4 mA h g−1 at 0.1 mA cm−2 | 2.5–4.0 V vs. Li/Li+ | 135.6 mA h g−1 (87.8% capacity retention) after 100 cycles | 91 |
| LiNbO3@NCA + Li7P2.9Ge0.05S10.75O0.1 | Li7P2.9Ge0.05S10.75O0.1 | Li–In | O | Initial discharge capacity of 158.9 mA h g−1 at 0.064 mA cm−2, RT | 2.1–3.7 V vs. Li/Li+ | 89% capacity retention after 50 cycles | 65 |
| LiNbO3@NCA + Li7P3S11 | Li7P3S11 | Li–In | O | Initial discharge capacity of 118.4 mA h g−1 at 0.064 mA cm−2, RT | 2.1–3.7 V vs. Li/Li+ | 61.57% capacity retention after 50 cycles | 65 |
| LCO | Li10GeP2S12 + 75% Li2S − 24% P2S5 − 1% P2O5 | Li | Y | Initial discharge capacity of ∼124 mA h g−1 at 0.1C | 3.0–4.2 V vs. Li/Li+ | 115.1 mA h g−1 after 10 cycles at 0.1C | 110 |
| 93.4 mA h g−1 after 50 cycles at 0.1C | |||||||
| 70 wt% LCO + 30 wt% (5% NiS-CNT@Li7P3S11) | Li10GeP2S12 + 75% Li2S − 24% P2S5 − 1% P2O5 | Li | X | Initial discharge capacity of ∼124 mA h g−1 at 0.1C | 3.0–4.2 V vs. Li/Li+ | 118.6 mA h g−1 after 10 cycles at 0.1C | 110 |
| 114.3 mA h g−1 (91.9% capacity retention) after 50 cycles at 0.1C | |||||||
| 70 wt% LCO + 30 wt% Li10GeP2S12 | Li10GeP2S12 + 75% Li2S − 24% P2S5 − 1% P2O5 | Li | X | Initial discharge capacity of 135.1 mA h g−1 at 0.05C | 3.0–4.2 V vs. Li/Li+ | 118.5 mA h g−1 after 25 cycles at 0.05C | 110 |
| 80 wt% (LCO + 5% NiS-CNT@Li7P3S11) + 20 wt% Li10GeP2S12 | Li10GeP2S12 + 75% Li2S − 24% P2S5 − 1% P2O5 | Li | Z | Initial discharge capacity of 135.1 mA h g−1 at 0.05C | 3.0–4.2 V vs. Li/Li+ | 124 mA h g−1 after 25 cycles at 0.05C | 110 |
| LiNi0.7Mn0.15Co0.15O2 + Li3PS4–2.3LiBH4 + conductive carbon | Li3PS4–2.3LiBH4 | Li–In | P | Initial discharge of 177.5 mA h g−1 at 0.05C | 2.4–3.7 V vs. Li/Li+ | — | 71 |
| LCO + SbCl3-doped LGPS + VGCF + p(MMA/nBA) | SbCl3-doped LGPS | Li | B | Initial discharge capacity of 125.6 mA h g−1 | — | 86.3% capacity retention at 0.1C (60 °C) after 250 cycles | 49 |
| NCM 811 + Li10.2P2.95Sb0.05S12I + vapor-grown nanofiber carbon | Li10.2P2.95Sb0.05S12I | Li | B | Initial discharge of 147 mA h g−1 at 0.1C | 2.5–4.25 V vs. Li/Li+ | 141 mA h g−1, 96.6% capacity retention (200 cycles) | 111 |
| NCM 811 + Li10P3S12I + vapor-grown nanofiber carbon | Li10P3S12I | Li | B | Initial discharge of 126 mA h g−1 at 0.1C | 2.5–4.25 V vs. Li/Li+ | 95 mA h g−1, 75.4% capacity retention (200 cycles) | 111 |
| TiS2 + LGSbS-04 | LGSbS-04 | Li–In | O | Initial discharge of 221 mA h g−1 | 0.78–2.08 V vs. Li/Li+ | 157 mA h g−1 after 44 cycles | 25 |
| LCO + LGPS (LLZCO coating layer) | LGPS | Li–In | R | Initial discharge of 121.0 mA h g−1 at 0.2C | 1.9–3.6 V vs. Li/Li+ | 96% capacity retention after 100 cycles | 76 |
| LCO + LGPS | LGPS | Li–In | O | Initial discharge of 73.1 mA h g−1 at 0.2C | 1.9–3.6 V vs. Li/Li+ | 59% capacity retention after 100 cycles | 76 |
| NCM622 + Li10SnP2S12 | Li10SnP2S12 | Li–In | O | Initial discharge of 122.9 mA h g−1 at 0.1C | 2.4–3.7 V vs. Li/Li+ | — | 66 |
| NCM622 + Li3InCl6 (catholyte/coating layer) | Li10SnP2S12 | Li–In | AB | Initial discharge of 176.1 mA h g−1 at 0.1C | 2.4–3.7 V vs. Li/Li+ | 106.2 mA h g−1 after 100 cycles | 66 |
| LCO + LGPS | LGPS | Li (Li3N–LiF layer) | W | Initial discharge of 132 mA h g−1 at 0.1C | 3.0–4.2 V vs. Li/Li+ | 93.9% capacity retention after 50 cycles | 97 |
| Initial discharge 95 mA h g−1 at 1C | 93.5% capacity retention after 500 cycles at 1C | ||||||
| LCO + LGPS | LGPS | Li | G | Initial discharge of 126.5 mA h g−1 at 0.1C | 3.0–4.2 V vs. Li/Li+ | 14.6 mA h g−1 after 20 cycles at 0.1C (11.5% capacity retention) | 97 |
| LiNbO3-coated LCO + LGPS | LGPS–Al2O3 (90:10) |
Li–In | O | Initial discharge of 111 mA h g−1 at 0.2C (unexposed) | 1.9–3.6 V vs. vs. Li/Li+ | 90% capacity retention after 50 cycles at 0.2C (unexposed) | 80 |
| Initial discharge 108 mA h g−1 at 0.2C (exposed at −30 °C, 1 h) | 92.6% capacity retention after 50 cycles at 0.2C (exposed at a dew point of −30 °C, 1 h) | ||||||
| LiNbO3-coated LCO + L-LSiPSCl-1.0 | L-LSiPSCl-1.0 | Li–In | O | Initial discharge of 123 mA h g−1 at 0.2C | 1.9–3.6 V vs. Li/Li+ | >97% capacity retention after 100 cycles at 0.2C | 7 |
| LiNbO3-coated LCO + LGPS | LGPS | Li (ZnO–LiF layer) | V | 5th -discharge capacity of 80 mA h g−1 at 0.1C | 3.0–4.2 V vs. Li/Li+ | 97% capacity retention after 500 cycles at 0.1C | 1 |
| NCM811 + Li6PS5Cl + super P | Li6PS5Cl | Li–In | N | Initial discharge of 192.4 mA h g−1 | 1.9–3.6 V vs. vs. Li/Li+ | — | 68 |
| LCO + Li6PS5Cl | Li6PS5Cl + Li5.6PS4.6I1.4 | Li | AA | Initial discharge of 128.8 mA h g−1 at 0.1C | 2.5–4.2 V vs. Li/Li+ | 79.6% capacity after 50 cycles | 209 |
| LFP + PVDF + Super P | Li6PS5F0.5Cl0.5 | Li | U | Initial discharge of 122 mA h g−1 at 0.2C | 2.5–4.0 V vs. Li/Li+ | Discharge capacity above 105 mA h g−1 after 50 cycles | 16 |
| LTO + PVDF + Super P | Li6PS5F0.75I0.25 | Li | U | Initial discharge of 140 mA h g−1 | 1.0–2.8 V vs. Li/Li+ | 91.5% capacity retention after 50 cycles | 92 |
| TiS2 + Li2B12H12–5Li2B10H10–6LiBH4 | Li2B12H12–5Li2B10H10–6LiBH4 | In1.3Li0.3 | O | Initial discharge of 147.8 mA h g−1 | 2.0–2.7 V vs. Li/Li+ | 80% capacity retention after 120 cycles | 52 |
| NCM811 + Li3(CB11H12)2(CB9H10) + carbon fibers | Li3(CB11H12)2(CB9H10) | Li–In | P | Initial discharge of ∼145 mA h g−1 at C/10, RT | 1.9–3.6 V vs. Li/Li+ | 54% capacity retention after 2000 cycles at C/2, 60 °C | 59 |
| NCM811 + Li3(CB11H12)2(CB9H10) + carbon fibers | Li3(CB11H12)2(CB9H10) | Graphite | N | Initial discharge of ∼125 mA h g−1 at C/10, RT | 2.4–4.1 V vs. Li/Li+ | 75% capacity retention after 350 cycles at C/2, RT | 59 |
| Fe2(MoO4)3 | Li2OHBr | Li | A | Initial discharge of ∼30 μAh cm−2 μm−1 | 2.7 to 3.2 V vs. Li/Li+ | ∼35 μAh cm−2 μm−1 after 60 cycles | 74 |
| LFP + Li3InCl6 | (Li2OH)0.99K0.01Cl | Li | E | Initial discharge of ∼120 mA h g−1 at 80 mA g−1 | 2.9–3.8 V vs. Li/Li+ | 96.1% capacity retention after 150 cycles at 80 mA g−1 | 72 |
| LFP + LLZTO + Super-P + Li3−x(OHx)Cl0.9F0.1 | LLZTO + Li3−x(OHx)Cl0.9F0.1 | Li | C | Initial discharge of 83.6 mA h g−1 at 0.05C, 90 °C | 2.7–4.0 V vs. Li/Li+ | 70.1% capacity retention after 50 cycles | 24 |
| LFP + LLZTO + Super-P + Li3−x(OHx)Cl0.9F0.1 | Li3−x(OHx)Cl0.9F0.1 | Li | D | Initial discharge of 70.8 mA h g−1 at 0.05C, 90 °C | 2.7–4.0 V vs. Li/Li+ | 62.5% capacity retention after 50 cycles | 24 |
| NCM622 + Li3SI + Super P | Li3SI | In | B | Initial discharge of 96.8 mA h g−1 at 0.02C | — | 50% capacity retention after 50 cycles at 0.04C | 32 |
| VCl3 + LIC + AB | LIC | Li (Li6PS5Cl layer) | F | Initial discharge capacity of ∼90 mA h g−1 at 6C | 2.45–3.25 V vs. Li/Li+ | 85.7% retention after 200 cycles at 6C | 67 |
| NCM811 + Li2.73Ho1.09Cl6 (Li3PO4 coating layer) | Li2.73Ho1.09Cl6 | Li–In | R | Initial charge capacity of 209 mA h g−1 at 0.1C (25 °C) | 1.9–3.7 V vs. Li/Li+ | Specific capacity of 125.5 mA h g−1 after 180 cycles (25 °C) | 56 |
| LCO + LIC | LIC | Li–In (Li6PS5Cl layer) | T | Initial discharge of 102.9 mA h g−1 at 1C | 2.7–4.2 V vs. Li/Li+ | 89.1% retention after 150 cycles at 1C | 42 |
| NCM83 + Li–Y–Cl | BM-SmCl3·0.5Li2ZrCl6 | Li–In (Li–Y–Cl electrolyte layer) | S | Initial discharge capacity of ∼135 mA h g−1 at 0.5C | 2.2–3.7 V vs. Li/Li+ | 85% retention after 600 cycles | 2 |
| NCM88 + Li–LaCeZrHfTa–Cl + AB | Li–LaCeZrHfTa–Cl + Li6PS5Cl | Li–In alloy | Q | Initial discharge capacity of 217.2 mA h g−1 at 0.1C, 25 °C | 2.5–4.3 V vs. Li/Li+ | 80% retention after 3000 cycles at 4C | 54 |
| 80 wt% NCM85 + 20 wt% Li2.6Zr0.4Ho0.6Cl6 | Li2.6Zr0.4Ho0.6Cl6 | In/Li–In (Li6PS5Cl coating layer) | T | Initial discharge of 180 mA h g−1 at 0.5C | 2.5–4.3 V vs. Li/Li+ | 96.8% retention after 180 cycles at 0.5C | 75 |
| 80 wt% NCM85 + 20 wt% Li2.6Zr0.4Lu0.6Cl6 | Li2.6Zr0.4Lu0.6Cl6 | In/Li–In (Li6PS5Cl coating layer) | T | Initial discharge of 202 mA h g−1 at 0.5C | 2.5–4.6 V vs. Li/Li+ | 88.9% retention after 180 cycles at 0.5C | 75 |
| 80 wt% NCM85 + 20 wt% Li2.6Zr0.4Lu0.6Cl6 | Li2.6Zr0.4Lu0.6Cl6 | Li0.7Si | O | Initial discharge of 16.1 mA h cm−2 at 0.5 mA cm−2 | 1.8–4.3 V vs. Li/Li+ | — | 75 |
| NCM83 + Li3ZrCl4O1.5 | Li3ZrCl4O1.5 | Li–In (Li3YCl6 coating layer) | T | Initial discharge of ∼135 mA h g−1 at 1C | 1.9–3.7 V vs. Li/Li+ | 90.1% retention after 300 cycles at 1C | 57 |
| LCO + Li3ZrCl4O1.5 | Li3ZrCl4O1.5 | Li–In (Li3YCl6 coating layer) | T | Initial discharge of ∼115 mA h g−1 at 0.5C | 1.9–3.7 V vs. Li/Li+ | 90% retention after 240 cycles at 0.5C | 57 |
| NCM811 + LSZC | LSZC | Li–In (Li6PS5Cl coating layer) | T | Initial discharge of ∼200 mA h g−1 at 0.1C, ∼150 mA h g−1 at 1C | 2.8–4.3 V vs. Li/Li+ | 82.7% retention after 1000 cycles at 1C, 30 °C | 22 |
| NCM90 + LSZC | LSZC | Li–In (Li6PS5Cl coating layer) | T | Initial discharge of 219.4 mA h g−1 at 0.1C, 200.7 mA h g−1 at 0.5C | 2.8–4.3 V vs. Li/Li+ | 78.1% retention after >200 cycles at 0.5C, 30 °C | 22 |
| LCO + LZTC | LZTC | Li–In foil (Li6PS5Cl coating layer) | T | Initial discharge capacity of 129.9 mA h g−1 at 0.05C | 2.3–3.6 V vs. Li–In/Li+ | Discharge capacity of 76.3 mA h g−1 after 200 cycles | 46 |
| NCM811 + LZTC | LZTC | Li–In foil (Li6PS5Cl coating layer) | T | Initial discharge capacity of 187.8 mA h g−1 at 0.05C | 2.2–3.7 V vs. Li–In/Li+ | 85.7% retention after 600 cycles | 46 |
| LCO + Li2ZrCl6 | Li2ZrCl6 | Li–In foil (Li6PS5Cl coating layer) | T | Initial discharge capacity of 137 mA h g−1 | 1.9–3.6 V vs. Li–In/Li | Discharge capacity of 114 mA h g−1 after 100 cycles at 0.5C | 212 |
| NCM811 + Li2ZrCl6 | Li2ZrCl6 | Li–In foil (Li6PS5Cl coating layer) | T | Initial discharge capacity of 181 mA h g−1 | 2.2–3.8 V vs. Li–In/Li+ | Discharge capacity of 149 mA h g−1 after 200 cycles at 1C | 212 |
| LCO + Li2.25Hf0.75Fe0.25Cl6 | Li2.25Hf0.75Fe0.25Cl6 | Li–In foil (Li6PS5Cl coating layer) | T | Initial discharge capacity of 113.4 mA h g−1 | 2.5–4.2 V vs. Li–In/Li | 88% retention after 100 cycles | 199 |
4.2. Multi-phase ISEs-based ASSBs
The development and application of ISEs-based ASSBs in recent decades have yielded favorable outcomes across various fields, demonstrating energy densities comparable to liquid electrolyte-based batteries and exhibiting promising safety levels.216,274 The stability of ISE with respect to electrode materials is critical, as it influences battery performance. The stability of the ISE/cathode interface has been addressed through the development of composite cathodes with an ISE-based catholyte and/or a cathode interface layer.51,65,70,110 The stability of the ISE–anode interface is addressed through the utilization of Li/Na alloy and/or an anode interface layer.30,42,71,75,101 The recent advancement indicates that ISE can be utilized as an anode–cathode interface layer. This expands the potential applications of ISEs, which can function in three distinct roles within ASSBs: as an electrolyte, catholyte, and interface layer at both anode and cathode sites.The ongoing innovation has resulted in the advancement of multi-phase ISEs-based ASSBs, characterized by the incorporation of multiple types of ISEs in the latest ASSB assembly model. The application of a single ISE as an electrolyte encounters numerous challenges, including ionic conductivity, chemical and electrochemical stability, and potential window limitations. Hybrid ISEs combinations may serve as an alternate approach, with reported examples including hybrid LGPS–LPS (Li10GeP2S12 + 75% Li2S − 24% P2S5 − 1% P2O5),110 LPS-hydroborate (Li3PS4–2.3LiBH4),71 double-argyrodite (Li6PS5Cl + Li5.6PS4.6I1.4),209 double-hydroborate (Li3(CB11H12)2(CB9H10)),59 garnet-antiperovskite (LLZTO + Li3−x(OHx)Cl0.9F0.1),24 double-halide (BM-SmCl3·0.5Li2ZrCl6),2 halide–argyrodite (Li–LaCeZrHfTaCl + Li6PS5Cl).54 The hybrid combination of ISEs at the catholyte and electrolyte sites includes catholyte (halide)–ISE (antiperovskite),72 catholyte (garnet)–ISE (antiperovskite),24 catholyte (halide)–ISE (halide),2 catholyte (halide)–ISE (halide–sulfide),54 catholyte (halide)–ISE (sulfide),66 catholyte (sulfide)–hybrid ISE (sulfide–sulfide),110 and catholyte (sulfide)–ISE (halide).198,200 The integration of ISEs at both the electrolyte and interface layers includes ISE (halide)–anode interface layer (sulfide),22 ISE (halide)–anode interface layer (halide),57 ISE (sulfide)–cathode interface layer (garnet),76 and ISE (sulfide)–cathode interface layer (halide).66 The wide scope of ISE types developed requires more investigation into the integration of multiple ISE types and the analysis of hybrid ISE interfaces to advance the next generation of ASSBs.
5. Dynamic database of solid-state electrolyte (DDSE)
Given the significant progress of various ISE classes in recent decades, it is imperative for researchers to have an ISE performance database. This database serves as a valuable resource for comparing and referencing current findings with existing ones. It also serves as a motivation for researchers to carry on their commitment to innovation in ISE research. Yang et al.280 developed a database referred to as the DDSE, which utilizes Machine Learning techniques. The program facilitates researchers in assessing the performance of ISEs discovered and allows for comparison with existing datasets. The DDSE database includes data from many ISEs that include both mono- and divalent cations such as Li+, Na+, K+, Ag+, Ca2+, Mg2+, and Zn2+, as well as anions including halide, hydride, and sulfide. This database will undoubtedly be invaluable for the progress of the next generations of ISEs and SSBs.6. Conclusion and future perspectives
In this review, we summarize recent advances in ISEs in terms of new chemical compounds, doping types, crystal structures, preparation methods, electrochemical behavior, chemical reactivity, and alternative strategies to alleviate the limitations of ISEs. As demonstrated, halides exhibit relatively wide stability windows, highlighting the importance of further investigation into their structures and characteristics to fulfill the performance requirements of high-voltage electrodes. In addition, their ionic conductivity is comparable to other ISEs. Oxide electrolytes typically exhibit excellent chemical stability; nevertheless, their stiffness compromises the contact with the electrode materials, necessitating the use of liquid electrolytes in some instances. Regarding garnet, engineering the cathode–LLZO interface is critical to improving the overall battery performance and longevity. Moreover, efforts should be directed toward scaling up the production of LLZO-based sheet electrolytes. NASICON is an ISE class with various benefits, including good mechanical properties, non-toxic, air stability, and wide electrochemical window. NASICON's further development is going to focus on low-temperature synthesis and controlling impurities concentration during synthesis, as well as addressing poor interface contact with the electrode materials. Sulfide has the highest conductivity among ISEs and good mechanical deformability; however, sulfides are vulnerable to atmospheric moisture, leading to hydrolysis and a consequent reduction in conductivity. Antiperovskite is a new class of ISE that warrants additional investigation due to its good mechanical properties and ease of production, but it must be handled carefully since it is less stable to moisture and has poor ionic conductivity when compared to other ISEs. Finally, the hydroborate class has considerable promise due to its straightforward preparation, good chemical stability, and wide potential window for high-voltage electrodes. To boost the ionic conductivity, the chemical structure of the hydroborate group should be improved. ISE innovation has advanced rapidly providing promising features for high-performance ASSBs. Fig. 24 compares five classes of ISEs as considerations for the sustainable development of high-performance ASSBs.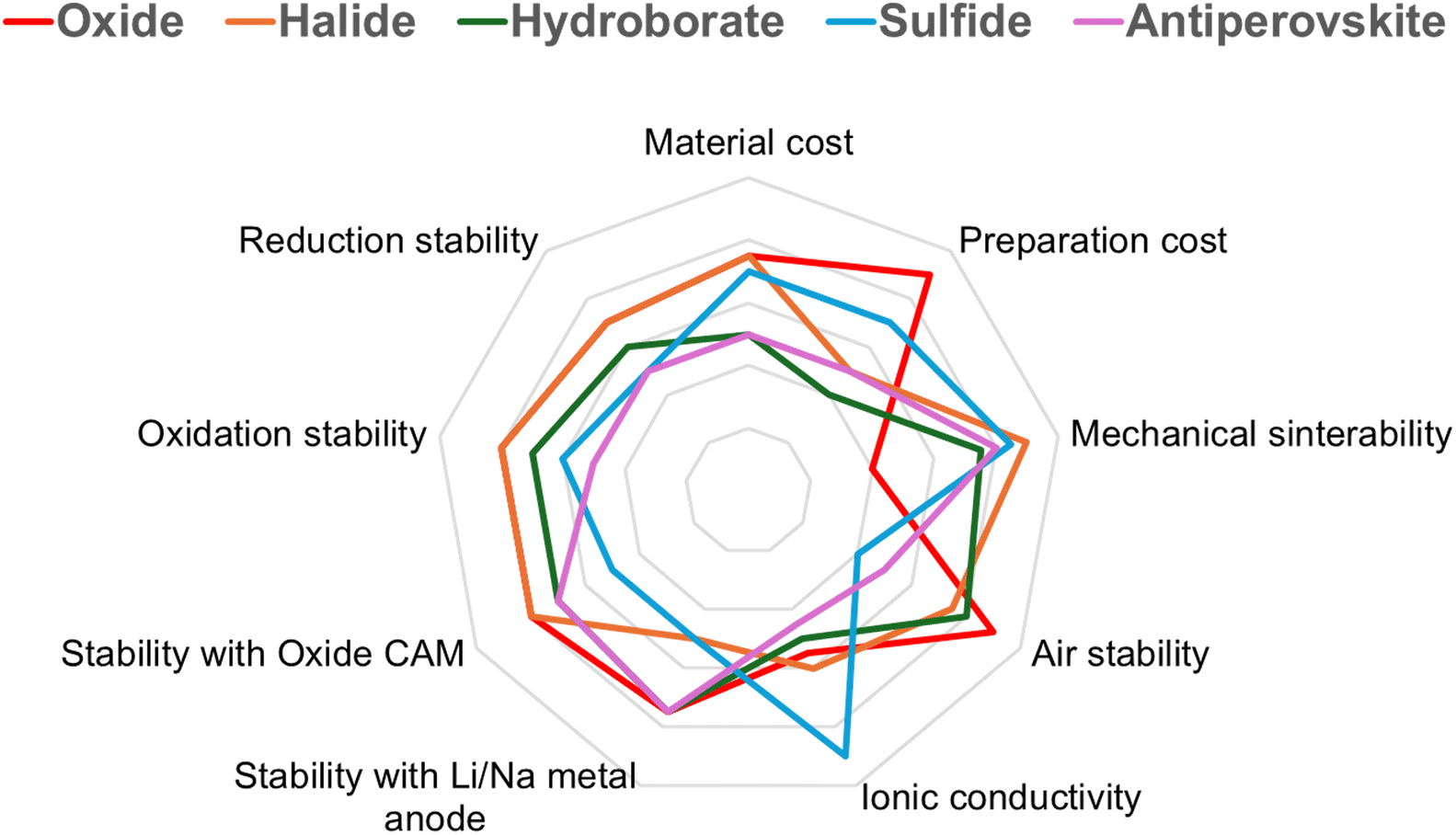 | ||
| Fig. 24 Comparison of the properties of Na/Li-based ISEs. | ||
Based on our analysis, the strategies, and recommendations to encourage the widespread application of ISEs and ASSBs are described as follows and presented in Fig. 25:
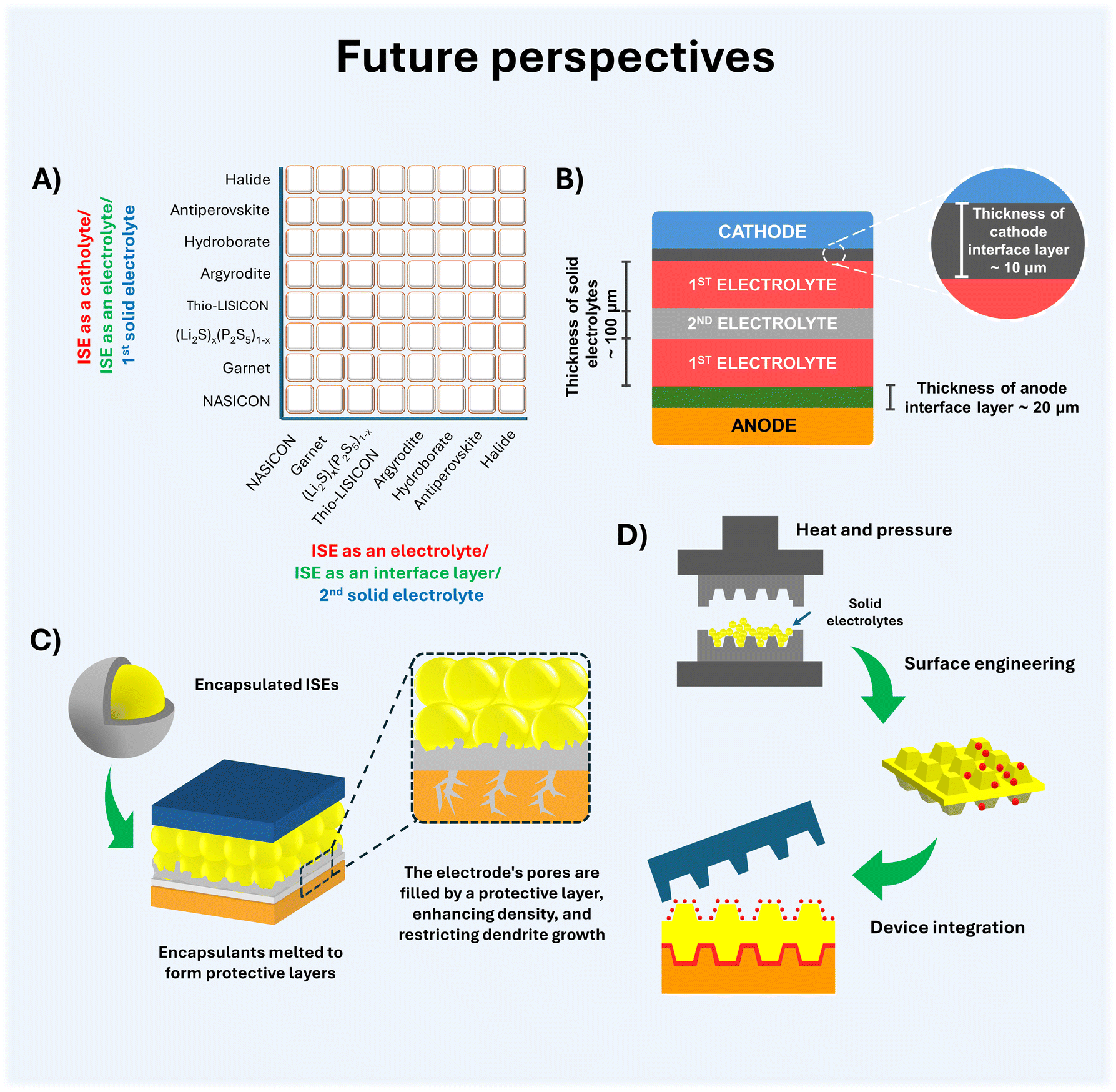 | ||
| Fig. 25 Schematic representation of the future perspectives on ASSBs based on inorganic solid electrolytes: (A) multi-phase ISEs in ASSBs, (B) effect of layer thickness on ASSBs performance, (C) encapsulated electrolytes, and (D) lock and key concept. | ||
(1) The incorporation of multiphase ISE into ASSBs. The ISEs group has the potential to function as an electrolyte, an interface layer, and a catholyte. Considering that there is a lack of knowledge about the combination of two different types of ISEs in the same class or different classes, it is necessary to conduct further investigation on this topic. When it comes to the application of ISEs in three different positions in ASSBs, we demonstrate 64 different combination arrangements between ISEs that can serve as a guide. These arrangements include (a) ISE as a catholyte–ISE as an electrolyte; (b) ISE as an electrolyte–ISE as an interface layer; and (c) utilization of the hybrid inorganic solid electrolytes.
(2) According to point No. 1, it is critical to investigate the impact of hybrid inorganic solid-state electrolyte thickness on ASSB device performance. In the case of hybrid ISEs, we divide them into two concepts: first, hybrid ISEs are manufactured by mixing two types of ISE in a specific ratio until they reach a homogeneous equilibrium and are implemented as a unified ISE electrolyte; and second, hybrid ISEs manufactured by stacking two layers of different ISEs while paying attention to their respective thickness ratios. We will focus on the second point in this section; the use of this concept in ASSBs necessitates further research, particularly when stacking the combination into several thin layers.
(3) Continuing from point No. 2, we are interested in the influence of the thickness of each component layer in an ASSBs system consisting of a composite cathode, a cathode interface layer, a solid electrolyte, an anode interface layer, and an anode. Developing thin-film batteries with a high energy density offers a huge opportunity. An interface layer is an alternate option for solid electrolytes that are reactive towards the anode and cathode in order to minimize side reactions. Furthermore, the solid electrolyte's reactivity on both sides varies, necessitating research into regulating the thickness of the interface layer. The application of interface layers at insufficient levels is likely to have a negative impact on ASSB performance and production costs.
(4) Develop ISE derivative compounds from the present ISE group. Each class of ISE has benefits and drawbacks that can be attributed to the type and coordination of cations and anions, respectively. As a result, using modern computational methods to design the chemical structure is critical for developing next-generation ISE electrolytes.
(5) Utilization of the encapsulation concept for solid electrolytes with poor air stability. How does this concept perform for ASSBs? Sulfide, antiperovskite, and other solid electrolytes are covered with encapsulants that exhibit strong ionic conductivity and low electron conductivity. The aim is to simplify the preparation and incorporation of electrolytes into ASSBs devices. The encapsulant (coating) will melt after a certain period of time when the encapsulated electrolyte is incorporated into the ASSB device, therefore releasing ISEs. The molten encapsulant will fill the gaps between ISE particles concurrently to improve the interaction between them. At the electrolyte–electrode interface, the molten encapsulant also generates a protective layer to control interfacial side reactions. Melted encapsulant is supposed to be an electronic insulator and an ionic conductor to improve ionic transport and suppress dendrite growth.
(6) Utilization of modified textured surfaces. Enhancing the contact area between two solid surfaces will enhance the transfer of mobile ions. The sophistication of computing systems can be utilized to design textured surfaces for electrodes and solid electrolytes. The touch-plane concept is derived from the mechanism of enzymes interacting with biological substrates via the “lock and key” principle. Enzymes exhibit the best performance when they encounter a substrate surface that precisely fits their structure. By using this principle, the contact surfaces of the two substances, the electrode, and solid electrolyte, are deliberately modified to have a textured structure that is coherent and compatible with each other. Furthermore, one of the surfaces is coated with a substance that enhances the adhesion between the two surfaces and serves as an interface layer.
(7) Comparative analysis of the effectiveness of developing hybrid ISEs vs. using electrolyte additives. Both exhibit the same objectives for ISEs, including reducing sintering temperature, inhibiting impurity growth, enhancing grain connectivity, decreasing grain boundary resistance, improving ionic conductivity, increasing density, rectifying mechanical issues, and enhancing chemical and moisture stability. Materials in ISE classes (NASICON, halide, sulfide, antiperovskite, hydroborate, etc.) and electrolyte additives such as oxides, nitrides, carbides, and solid polymer electrolytes can be alternatives and tailored to match the specific substrate properties and affordability for fabricating high-performance ISEs.
List of formulas
| Cu-doped NZSP | Na3.12Zr1.94Cu0.06Si2PO12 |
| LAGP | Li1.5Al0.5Ge1.5(PO4)3 |
| LATP | Li1+xAlxTi2−x(PO4)3 |
| LCO | LiCoO2 |
| LFP | LiFePO4 |
| LGLZO | Li6.25Ga0.25La3Zr2O12 |
| LGPS | Li10GeP2S12 |
| LIC | Li3InCl6 |
| LLZC | Li2.5Lu0.5Zr0.5Cl6 |
| LLZO | Li7La3Zr2O12 |
| LLZCO | Li7.5La3Zr1.5Co0.5O12 |
| LLZTO | Li6.5La3Zr1.5Ta0.5O12 |
| LLZWO | Li6.3La3Zr1.65W0.35O12 |
| LNMO | LiNi0.5Mn1.5O4 |
| LSZC | Li2.375Sc0.375Zr0.625Cl6 |
| LSiPSCl | Li10.02Si1.47P1.56S11.7Cl0.3 |
| LTO | Li4Ti5O12 |
| LYB | Li3YBr6 |
| LYC | Li3YCl6 |
| Mg-doped NZSP-0.128 | Na3.456Zr1.872Mg0.128Si2.2P0.8O12 |
| NCM622 | LiNi0.6Co0.2Mn0.2O2 |
| NCM811 | LiNi0.8Co0.1Mn0.1O2 |
| NCM83 | LiNi0.83Co0.11Mn0.06O2 |
| NCM85 | LiNi0.85Co0.1Mn0.05O2 |
| NCM88 | LiNi0.88Co0.09Mn0.03O2 |
| NCM90 | LiNi0.9Co0.05Mn0.05O2 |
| NLZSP | Na3.3La0.3Zr1.7Si2PO12 |
| NMNFO | Na0.85Mn0.5Ni0.4Fe0.1O2 |
| NZSP | Na3Zr2Si2PO12 |
| NZSP-ATO | Na3Zr2Si2PO12 with 5 wt% antimony tin oxide |
| NLZSP-NBO3 | Na3.3La0.3Zr1.7Si2PO12 with 3 wt% Na2B4O7 |
| NVPF | Na3V2(PO4)2F3 |
| SbCl3-doped LGPS | Li9.88GeP1.96Sb0.04S11.88Cl0.12 |
| Te-doped LAGP | Li1.5Al0.5Te0.06Ge1.44(PO4)3 |
List of abbreviations
| AB | acetylene black |
| ACN | acetonitrile |
| AGG | abnormal grain growth |
| AIMD | Ab initio molecular dynamics |
| ALD | atomic layer deposition |
| ASR | area-specific resistance |
| ASSBs | all-solid-state batteries |
| ASSLIBs | all-solid-state lithium-ion batteries |
| ASSSIBs | all-solid-state sodium-ion batteries |
| BVEL | bond valence energy landscape |
| BVSE | bond-valence site energy |
| CAD | computer-aided design |
| CAM | cathode active materials |
| CB | carbon black |
| CCP | cubic close-packed |
| CEI | cathode interphase layer |
| CI-NEB | climbing-image nudged elastic band |
| CNT | carbon nanotube |
| CSP | cold sintering process |
| CVD | chemical vapor deposition |
| DDSE | dynamic database of solid-state electrolyte |
| DFT | density functional theory |
| DME | 1,2-dimethoxyethane |
| Ea | activation energy |
| EDS | energy-dispersive X-ray spectroscopy |
| EIS | electrochemical impedance spectroscopy |
| GITT | galvanostatic intermittent titration technique |
| HCF | high-coordination frameworks |
| HCF-Sm | high coordination frameworks-Na0.75Sm1.75Cl6 |
| HCF-La | high coordination frameworks-Na0.75La1.75Cl6 |
| HCP | hexagonal close-packed |
| HPLT | high-pressure low-temperature |
| HSAB | hard and soft acids and bases |
| ISEs | inorganic solid electrolytes |
| LCF | low-coordination frameworks |
| LCF-Ta | low coordination frameworks-NaTaCl6 |
| LCF-Zr | low coordination frameworks-Na2ZrCl6 |
| LCF-Hf | low coordination frameworks-Na2HfCl6 |
| LIBs | lithium-ion batteries |
| LiRAP | Li-rich antiperovskite |
| LISICON | lithium superionic conductor |
| LPS | lithium thiophosphates |
| MAS NMR | magic angle spinning nuclear magnetic resonance |
| MEM | maximum entropy method |
| MSD | mean squared displacement |
| MWCNT | multi-walled carbon nanotube |
| NASICON | natrium superionic conductor |
| NMP | N-methylpyrrolidone |
| NPD | neutron powder diffraction |
| pair distribution function | |
| p(MMA/nBA) | poly(methyl methacrylate)/n-butyl acrylate |
| R gb | grain boundary resistance |
| RT | room temperature |
| RTP | rapid thermal processing |
| SEI | solid interphase layer |
| SEM | scanning electron microscopy |
| SIBs | sodium-ion batteries |
| SSBs | solid-state batteries |
| SPS | spark plasma sintering |
| THF | tetrahydrofuran |
| UHS | ultrafast high-temperature sintering |
| XRD | X-ray diffraction |
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was upheld by the National Research Council of Thailand (NRCT) and Chulalongkorn University (N42A660383) and the National Science, Research and Innovation Fund (NSRF) via the Program Management Unit for Human Resources & Institutional Development, Research and Innovation (B41G670026). Muhammad Muzakir & Karnan Manickavasakam would like to thank Chulalongkorn University in Thailand for their C2F scholarship sponsorship. Eric Jianfeng Cheng thanks the financial support of the Basic Research grant from TEPCO Memorial Foundation.References
- X. Chang, G. Liu, M. Wu, M. Chang, X. Zhao, G. Z. Chen, K. L. Fow and X. Yao, Battery Energy, 2023, 2, 20220051 CrossRef CAS.
- J. Fu, S. Wang, J. Liang, S. H. Alahakoon, D. Wu, J. Luo, H. Duan, S. Zhang, F. Zhao, W. Li, M. Li, X. Hao, X. Li, J. Chen, N. Chen, G. King, L.-Y. Chang, R. Li, Y. Huang, M. Gu, T.-K. Sham, Y. Mo and X. Sun, J. Am. Chem. Soc., 2023, 145, 2183–2194 CrossRef CAS.
- X. Lu, A. Windmüller, D. Schmidt, S. Schöner, C.-L. Tsai, H. Kungl, X. Liao, Y. Chen, S. Yu, H. Tempel and R.-A. Eichel, ACS Appl. Mater. Interfaces, 2023, 15, 34973–34982 CrossRef CAS.
- P. Kokmat, P. Matsayamat, K. Wongrach, P. Surinlert and A. Ruammaitree, J. Met., Mater. Miner., 2023, 33, 1779 CrossRef CAS.
- W. Zhang, Y. Wu, Z. Xu, H. Li, M. Xu, J. Li, Y. Dai, W. Zong, R. Chen, L. He, Z. Zhang, D. J. L. Brett, G. He, Y. Lai and I. P. Parkin, Adv. Energy Mater., 2022, 12, 2201065 CrossRef CAS.
- M. Jin, Z. Yang, S. Cheng and Y. Guo, ACS Appl. Energy Mater., 2022, 5, 15578–15585 CrossRef CAS.
- T. Ito, S. Hori, M. Hirayama and R. Kanno, J. Mater. Chem. A, 2022, 10, 14392–14398 RSC.
- K. Lolupiman, P. Wangyao and J. Qin, J. Met., Mater. Miner., 2019, 29, 120–126 CAS.
- H. Ouyang, S. Min, J. Yi, X. Liu, F. Ning, J. Qin, Y. Jiang, B. Zhao and J. Zhang, Green Energy Environ., 2023, 8, 1195–1204 CrossRef CAS.
- J. Cao, D. Zhang, X. Zhang, M. Sawangphruk, J. Qin and R. Liu, J. Mater. Chem. A, 2020, 8, 9331–9344 RSC.
- C. Li, D. Zhang, J. Cao, P. Yu, M. Okhawilai, J. Yi, J. Qin and X. Zhang, ACS Appl. Energy Mater., 2021, 4, 7821–7828 CrossRef CAS.
- D. Yan, S. Xiao, X. Li, J. Jiang, Q. He, H. Li, J. Qin, R. Wu, X. Niu and J. S. Chen, Energy Mater. Adv., 2024, 4, 12 CrossRef.
- H. Zhong, D. Liu, X. Yuan, X. Xiong and K. Han, Energy Fuels, 2024, 38, 7693–7732 CrossRef CAS.
- W. Tong, Y. Chen, S. Gong, S. Zhu, J. Tian, J. Qin, W. Chen and S. Chen, J. Power Sources, 2024, 591, 233887 CrossRef CAS.
- S. Min, X. Liu, A. Wang, F. Ning, Y. Liu, J. Qin, J. Zhang, S. Lu and J. Yi, Chin. Chem. Lett., 2023, 34, 108586 CrossRef CAS.
- W. Arnold, V. Shreyas, Y. Li, M. K. Koralalage, J. B. Jasinski, A. Thapa, G. Sumanasekera, A. T. Ngo, B. Narayanan and H. Wang, ACS Appl. Mater. Interfaces, 2022, 14, 11483–11492 CrossRef CAS.
- B. Helm, N. Minafra, B. Wankmiller, M. T. Agne, C. Li, A. Senyshyn, M. R. Hansen and W. G. Zeier, Chem. Mater., 2022, 34, 5558–5570 CrossRef CAS.
- G. Huang, Y. Zhong, X. Xia, X. Wang, C. Gu and J. Tu, J. Colloid Interface Sci., 2023, 632, 11–18 CrossRef CAS PubMed.
- M. Shimoda, M. Maegawa, S. Yoshida, H. Akamatsu, K. Hayashi, P. Gorai and S. Ohno, Chem. Mater., 2022, 34, 5634–5643 CrossRef CAS.
- T.-T. Zuo, F. Walther, J. H. Teo, R. Rueß, Y. Wang, M. Rohnke, D. Schröder, L. F. Nazar and J. Janek, Angew. Chem., Int. Ed., 2023, 62, e202213228 CrossRef CAS PubMed.
- S. Shiba, A. Miura, Y. Fujii, K. Tadanaga, K. Terai, F. Utsuno and H. Higuchi, RSC Adv., 2023, 13, 22895–22900 RSC.
- R. Li, P. Lu, X. Liang, L. Liu, M. Avdeev, Z. Deng, S. Li, K. Xu, J. Feng, R. Si, F. Wu, Z. Zhang and Y.-S. Hu, ACS Energy Lett., 2024, 9, 1043–1052 CrossRef CAS.
- J. Li, Y. Li, S. Zhang, D. Li and L. Ci, eTransportation, 2023, 16, 100236 CrossRef.
- W. Feng, L. Zhu, X. Dong, Y. Wang, Y. Xia and F. Wang, Adv. Mater., 2023, 35, 2210365 CrossRef CAS.
- J. Roh, J. Lyoo and S.-T. Hong, ACS Appl. Energy Mater., 2023, 6, 5446–5455 CrossRef CAS.
- P.-C. Tsai, S. Mair, J. Smith, D. M. Halat, P.-H. Chien, K. Kim, D. Zhang, Y. Li, L. Yin, J. Liu, S. H. Lapidus, J. A. Reimer, N. P. Balsara, D. J. Siegel and Y. Chiang, Adv. Energy Mater., 2023, 13, 2203284 CrossRef CAS.
- A. Jodlbauer, J. Spychala, K. Hogrefe, B. Gadermaier and H. M. R. Wilkening, Chem. Mater., 2024, 36, 1648–1664 CrossRef CAS.
- I. Yang, S.-G. Kim, S. H. Kwon, M.-S. Kim and J. C. Jung, Electrochim. Acta, 2017, 223, 21–30 CrossRef CAS.
- S. Xiong, Y. Liu, P. Jankowski, Q. Liu, F. Nitze, K. Xie, J. Song and A. Matic, Adv. Funct. Mater., 2020, 30, 2001444 CrossRef CAS.
- J. Fu, S. Wang, D. Wu, J. Luo, C. Wang, J. Liang, X. Lin, Y. Hu, S. Zhang, F. Zhao, W. Li, M. Li, H. Duan, Y. Zhao, M. Gu, T. Sham, Y. Mo and X. Sun, Adv. Mater., 2024, 36, e2308012 CrossRef.
- H. J. Lee, B. Darminto, S. Narayanan, M. Diaz-Lopez, A. W. Xiao, Y. Chart, J. H. Lee, J. A. Dawson and M. Pasta, J. Mater. Chem. A, 2022, 10, 11574–11586 RSC.
- R. Rajagopal, M.-H. Park, Y. Subramanian, Y. J. Jung and K.-S. Ryu, J. Electroanal. Chem., 2021, 895, 115477 CrossRef CAS.
- J. C. Bachman, S. Muy, A. Grimaud, H.-H. Chang, N. Pour, S. F. Lux, O. Paschos, F. Maglia, S. Lupart, P. Lamp, L. Giordano and Y. Shao-Horn, Chem. Rev., 2016, 116, 140–162 CrossRef CAS.
- M. Nasir, J. Seo, J. S. Park and H. J. Park, J. Eur. Ceram. Soc., 2024, 44, 4606–4611 CrossRef CAS.
- X. Chen, T. Wang, W. Lu, T. Cao, M. Xue, B. Li and C. Zhang, J. Alloys Compd., 2018, 744, 386–394 CrossRef CAS.
- X. Li, R. Li, S. Chu, K. Liao, R. Cai, W. Zhou and Z. Shao, J. Alloys Compd., 2019, 794, 347–357 CrossRef CAS.
- J. Awaka, A. Takashima, K. Kataoka, N. Kijima, Y. Idemoto and J. Akimoto, Chem. Lett., 2011, 40, 60–62 CrossRef.
- E. J. Cheng, A. Sharafi and J. Sakamoto, Electrochim. Acta, 2017, 223, 85–91 CrossRef CAS.
- P. Jiang, G. Du, Y. Shi, F. She, P. Guo, G. Qian, X. Lu, F. Xie and X. Lu, Chem. Eng. J., 2023, 451, 138771 CrossRef CAS.
- J. Wang, T. He, X. Yang, Z. Cai, Y. Wang, V. Lacivita, H. Kim, B. Ouyang and G. Ceder, Nat. Commun., 2023, 14, 5210 CrossRef CAS.
- Z. Wang, Z. Wang, M. Li, C. Tang, K. Yu, P. Lv and W. Wei, Solid State Ionics, 2023, 396, 116229 CrossRef CAS.
- S. Deng, M. Jiang, A. Rao, X. Lin, K. Doyle-Davis, J. Liang, C. Yu, R. Li, S. Zhao, L. Zhang, H. Huang, J. Wang, C. V. Singh and X. Sun, Adv. Funct. Mater., 2022, 32, 2200767 CrossRef CAS.
- Z. Huang, S. Yoshida, H. Akamatsu, K. Hayashi and S. Ohno, ACS Mater. Lett., 2024, 6, 1732–1738 CrossRef CAS.
- X. Li, J. T. Kim, J. Luo, C. Zhao, Y. Xu, T. Mei, R. Li, J. Liang and X. Sun, Nat. Commun., 2024, 15, 53 CrossRef CAS.
- X. Li, J. Liang, X. Yang, K. R. Adair, C. Wang, F. Zhao and X. Sun, Energy Environ. Sci., 2020, 13, 1429–1461 RSC.
- X. Liu, Y. Zhou, F. Mi, X. Ma and C. Sun, Energy Storage Mater., 2024, 72, 103737 CrossRef.
- T. Kimura, R. Izawa, C. Hotehama, K. Fujii, A. Sakuda, M. Yashima, M. Tatsumisago and A. Hayashi, Solid State Ionics, 2023, 399, 116287 CrossRef CAS.
- R. Wang, Z. Wu, C. Yu, C. Wei, L. Peng, L. Wang, S. Cheng and J. Xie, Front. Energy Res., 2023, 10, 1108789 CrossRef.
- X. Zhao, P. Xiang, J. Wu, Z. Liu, L. Shen, G. Liu, Z. Tian, L. Chen and X. Yao, Nano Lett., 2023, 23, 227–234 CrossRef CAS PubMed.
- P. Qiu, X. Chen, W. Zhang, G. Zhang, Y. Zhang, Z. Lu, Y. Wu and X. Chen, Angew. Chem., Int. Ed., 2024, 63, e202401480 CrossRef CAS PubMed.
- Z. Yang, M. Jin, S. Cheng, X. Ma, Z. Qin, J. Zhang, Y. Yang and Y. Guo, J. Mater. Chem. A, 2022, 10, 7186–7194 RSC.
- C. Zhou, Y. Yan and T. R. Jensen, ACS Appl. Energy Mater., 2023, 6, 7346–7352 CrossRef CAS.
- S. J. Turrell, H. J. Lee, M. Siniscalchi, S. Narayanan, M. Pasta, S. C. Speller and C. R. M. Grovenor, Mater. Adv., 2022, 3, 8995–9008 RSC.
- X. Li, Y. Xu, C. Zhao, D. Wu, L. Wang, M. Zheng, X. Han, S. Zhang, J. Yue, B. Xiao, W. Xiao, L. Wang, T. Mei, M. Gu, J. Liang and X. Sun, Angew. Chem., Int. Ed., 2023, 62, e202306433 CrossRef CAS PubMed.
- Y.-Y. Sun, Q. Zhang, L. Yan, T.-B. Wang and P.-Y. Hou, Chem. Eng. J., 2022, 437, 135179 CrossRef CAS.
- J. Liang, E. van der Maas, J. Luo, X. Li, N. Chen, K. R. Adair, W. Li, J. Li, Y. Hu, J. Liu, L. Zhang, S. Zhao, S. Lu, J. Wang, H. Huang, W. Zhao, S. Parnell, R. I. Smith, S. Ganapathy, M. Wagemaker and X. Sun, Adv. Energy Mater., 2022, 12, 2103921 CrossRef CAS.
- S. Zhang, F. Zhao, L.-Y. Chang, Y.-C. Chuang, Z. Zhang, Y. Zhu, X. Hao, J. Fu, J. Chen, J. Luo, M. Li, Y. Gao, Y. Huang, T.-K. Sham, M. D. Gu, Y. Zhang, G. King and X. Sun, J. Am. Chem. Soc., 2024, 146, 2977–2985 CrossRef CAS.
- C. Sun, J. Liu, Y. Gong, D. P. Wilkinson and J. Zhang, Nano Energy, 2017, 33, 363–386 CrossRef CAS.
- H. Braun, R. Asakura, A. Remhof and C. Battaglia, ACS Energy Lett., 2024, 9, 707–714 CrossRef CAS.
- X. G. Cao, X. H. Zhang, T. Tao and H. Y. Zhang, Ceram. Int., 2020, 46, 8405–8412 CrossRef CAS.
- E. J. Cheng, T. Kimura, M. Shoji, H. Ueda, H. Munakata and K. Kanamura, ACS Appl. Mater. Interfaces, 2020, 12, 10382–10388 CrossRef CAS PubMed.
- E. J. Cheng, T. Yang, Y. Liu, L. Chai, R. Garcia-Mendez, E. Kazyak, Z. Fu, G. Luo, F. Chen, R. Inada, V. Badilita, H. Duan, Z. Wang, J. Qin, H. Li, S. Orimo and H. Kato, Mater. Today Energy, 2024, 44, 101644 CrossRef CAS.
- X. Feng, P.-H. Chien, Z. Zhu, I.-H. Chu, P. Wang, M. Immediato-Scuotto, H. Arabzadeh, S. P. Ong and Y.-Y. Hu, Adv. Funct. Mater., 2019, 29, 1807951 CrossRef.
- N. Hamao, K. Kataoka, N. Kijima and J. Akimoto, J. Ceram. Soc. Jpn., 2016, 124, 678–683 CrossRef CAS.
- L. Lv, N. Ahmad, C. Zeng, P. Yu, T. Song, Q. Dong and W. Yang, ACS Appl. Mater. Interfaces, 2022, 14, 39985–39995 CrossRef CAS.
- Q. Luo, C. Yu, C. Wei, S. Chen, S. Chen, Z. Jiang, L. Peng, S. Cheng and J. Xie, Ceram. Int., 2023, 49, 11485–11493 CrossRef CAS.
- J. Liang, X. Li, J. T. Kim, X. Hao, H. Duan, R. Li and X. Sun, Angew. Chem., Int. Ed., 2023, 62, e202217081 CrossRef CAS PubMed.
- J. M. Lee, Y. S. Park, J.-W. Moon and H. Hwang, Front. Chem., 2021, 9, 778057 CrossRef CAS PubMed.
- M. Jin, D. Xu, Z. Su, Z. He, X. Chen, R. Wu and Y. Guo, ACS Energy Lett., 2024, 9, 1176–1183 CrossRef CAS.
- M. Jin, S. Cheng, Z. Yang, Y. Luo and Y. Guo, Chem. Eng. J., 2023, 455, 140904 CrossRef CAS.
- Y. Jang, H. Seo, Y. Lee, S. Kang, W. Cho, Y. W. Cho and J. Kim, Adv. Sci., 2023, 10, 2204942 CrossRef PubMed.
- L. Gao, X. Zhang, J. Zhu, S. Han, H. Zhang, L. Wang, R. Zhao, S. Gao, S. Li, Y. Wang, D. Huang, Y. Zhao and R. Zou, Nat. Commun., 2023, 14, 6807 CrossRef CAS PubMed.
- E. J. Cheng, Y. Kushida, T. Abe and K. Kanamura, ACS Appl. Mater. Interfaces, 2022, 14, 40881–40889 CrossRef CAS.
- K. Yoshikawa, T. Yamamoto, M. K. Sugumar, M. Motoyama and Y. Iriyama, Energy Fuels, 2021, 35, 12581–12587 CrossRef CAS.
- L. Zhou, T. Zuo, C. Li, Q. Zhang, J. Janek and L. F. Nazar, ACS Energy Lett., 2023, 8, 3102–3111 Search PubMed.
- B. Shen, L. Li, X. Yao and B. Huang, Ceram. Int., 2024, 50, 7150–7155 CrossRef CAS.
- W. S. Scheld, J. N. Ebert, M. Scherer, L. Fulanovic, L. Porz, C. Dellen, M. Ihrig, S. Uhlenbruck, M. Finsterbusch, O. Guillon, D. Fattakhova-Rohlfing and W. Rheinheimer, J. Eur. Ceram. Soc., 2024, 44, 3039–3048 CrossRef CAS.
- B. Pandit, M. Johansen, B. P. Andersen, C. S. Martínez-Cisneros, B. Levenfeld, D. B. Ravnsbæk and A. Varez, Chem. Eng. J., 2023, 472, 144509 CrossRef CAS.
- O. Maus, M. T. Agne, T. Fuchs, P. S. Till, B. Wankmiller, J. M. Gerdes, R. Sharma, M. Heere, N. Jalarvo, O. Yaffe, M. R. Hansen and W. G. Zeier, J. Am. Chem. Soc., 2023, 145, 7147–7158 CrossRef CAS PubMed.
- T. Yabuzaki, M. Sato, H. Kim, K. Watanabe, N. Matsui, K. Suzuki, S. Hori, K. Hikima, S. Obokata, H. Muto, A. Matsuda, R. Kanno and M. Hirayama, J. Ceram. Soc. Jpn., 2023, 131, 23070 CrossRef.
- T. Ma, Z. Wang, D. Wu, P. Lu, X. Zhu, M. Yang, J. Peng, L. Chen, H. Li and F. Wu, Energy Environ. Sci., 2023, 16, 2142–2152 RSC.
- J. Zhou, Y. Chen, Z. Yu, M. Bowden, Q. R. S. Miller, P. Chen, H. T. Schaef, K. T. Mueller, D. Lu, J. Xiao, J. Liu, W. Wang and X. Zhang, Chem. Eng. J., 2022, 429, 132334 CrossRef CAS.
- H. Cai, T. Yu, D. Xie, B. Sun, J. Cheng, L. Li, X. Bao and H. Zhang, J. Alloys Compd., 2023, 939, 168702 Search PubMed.
- M. Brighi, F. Murgia and R. Černý, Adv. Mater. Interfaces, 2022, 9, 2101254 CrossRef CAS.
- J. Peng, D. Wu, P. Lu, Z. Wang, Y. Du, Y. Wu, Y. Wu, W. Yan, J. Wang, H. Li, L. Chen and F. Wu, Energy Storage Mater., 2023, 54, 430–439 CrossRef.
- F. Lu, J. Xu, C. Wang, Z. Liu, X. Yue, X. Duan, J. Lin, X. Li, X. Zhang, P. Lin, T. Lin and P. He, Energy Storage Mater., 2024, 65, 103101 Search PubMed.
- S. Wang, X. Xu, C. Cui, C. Zeng, J. Liang, J. Fu, R. Zhang, T. Zhai and H. Li, Adv. Funct. Mater., 2022, 32, 2108805 CrossRef CAS.
- A. Morscher, B. B. Duff, G. Han, L. M. Daniels, Y. Dang, M. Zanella, M. Sonni, A. Malik, M. S. Dyer, R. Chen, F. Blanc, J. B. Claridge and M. J. Rosseinsky, J. Am. Chem. Soc., 2022, 144, 22178–22192 CrossRef CAS.
- S. Valiyaveettil-SobhanRaj, P. Głuchowski, P. López-Aranguren, F. Aguesse, R. Sampathkumar, T. Thompson, C. Rojviriya, W. Manalastas Jr, M. Srinivasan and M. Casas-Cabanas, Materialia, 2024, 33, 101999 CrossRef CAS.
- B. Hitesh and A. Sil, J. Am. Ceram. Soc., 2023, 106, 6743–6754 CrossRef CAS.
- L. Liu, X. Cui, Z. Jie, Y. Lin, C. Zhang, J. Song, L. Wang, J. Ma and L. Ma, J. Power Sources, 2023, 575, 233137 CrossRef CAS.
- W. Arnold, V. Shreyas, S. Akter, Y. Li, S. Halacoglu, M. B. Kalutara Koralalage, X. Guo, D. Vithanage, W. Wei, G. Sumanasekera, J. B. Jasinski, B. Narayanan and H. Wang, J. Phys. Chem. C, 2023, 127, 11801–11809 CrossRef CAS.
- E. van der Maas, T. Famprikis, S. Pieters, J. P. Dijkstra, Z. Li, S. R. Parnell, R. I. Smith, E. R. H. van Eck, S. Ganapathy and M. Wagemaker, J. Mater. Chem. A, 2023, 11, 4559–4571 RSC.
- Y. Yang, J. Han, M. DeVita, S. S. Lee and J. C. Kim, Front. Chem., 2020, 8, 562549 CrossRef CAS.
- X. Luo, A. Rawal, M. S. Salman and K.-F. Aguey-Zinsou, ACS Appl. Nano Mater., 2022, 5, 373–379 CrossRef CAS.
- A. Manthiram, X. Yu and S. Wang, Nat. Rev. Mater., 2017, 2, 16103 CrossRef CAS.
- M. Wu, M. Li, Y. Jin, X. Chang, X. Zhao, Z. Gu, G. Liu and X. Yao, J. Energy Chem., 2023, 79, 272–278 CrossRef CAS.
- Z. Bi, R. Shi, X. Liu, K. Liu, M. Jia and X. Guo, Adv. Funct. Mater., 2023, 33, 2307701 CrossRef CAS.
- W. Li, M. Li, P.-H. Chien, S. Wang, C. Yu, G. King, Y. Hu, Q. Xiao, M. Shakouri, R. Feng, B. Fu, H. Abdolvand, A. Fraser, R. Li, Y. Huang, J. Liu, Y. Mo, T.-K. Sham and X. Sun, Sci. Adv., 2023, 9, 1–14 Search PubMed.
- K. Yoshikawa, Y. Suzuki, A. Shiota and Y. Iriyama, ACS Appl. Energy Mater., 2023, 6, 4191–4197 CrossRef CAS.
- C. K. Moon, H.-J. Lee, K. H. Park, H. Kwak, J. W. Heo, K. Choi, H. Yang, M.-S. Kim, S.-T. Hong, J. H. Lee and Y. S. Jung, ACS Energy Lett., 2018, 3, 2504–2512 CrossRef CAS.
- C. Zhao, M. Lyu, C. Bi, S. Huo, S. Li and W. Xue, Results Chem., 2022, 4, 100468 CrossRef CAS.
- N. Masuda, K. Kobayashi, F. Utsuno and N. Kuwata, J. Solid State Electrochem., 2024, 28, 4409–4417 CrossRef CAS.
- E. A. Wu, C. S. Kompella, Z. Zhu, J. Z. Lee, S. C. Lee, I.-H. Chu, H. Nguyen, S. P. Ong, A. Banerjee and Y. S. Meng, ACS Appl. Mater. Interfaces, 2018, 10, 10076–10086 CrossRef CAS PubMed.
- S. P. Culver, R. Koerver, W. G. Zeier and J. Janek, Adv. Energy Mater., 2019, 9, 1900626 CrossRef.
- J. Liang, Y. Zhu, X. Li, J. Luo, S. Deng, Y. Zhao, Y. Sun, D. Wu, Y. Hu, W. Li, T.-K. Sham, R. Li, M. Gu and X. Sun, Nat. Commun., 2023, 14, 146 CrossRef CAS PubMed.
- J. Song, C. Zhang, Z. Zheng, S. Huo, Y. Lin, F. Yang and L. Liu, J. Colloid Interface Sci., 2024, 663, 132–142 CrossRef CAS.
- D. Kitsche, Y. Tang, H. Hemmelmann, F. Walther, M. Bianchini, A. Kondrakov, J. Janek and T. Brezesinski, Small Sci., 2023, 3, 2200073 CrossRef CAS.
- W. Xie, M. Chang, W. Fan, M. Yang, F. Tian, X. Xue, X. Zhao, H. He and X. Yao, Mater. Chem. Front., 2023, 7, 2844–2850 RSC.
- M. Jiang, W. Fan, G. Liu, W. Weng, L. Cai and X. Yao, Electrochim. Acta, 2021, 398, 139280 CrossRef CAS.
- Y. Subramanian, R. Rajagopal and K.-S. Ryu, J. Energy Storage, 2024, 78, 109943 CrossRef.
- T. A. Hendriks, M. A. Lange, E. M. Kiens, C. Baeumer and W. G. Zeier, Batteries Supercaps, 2023, 6, e202200544 CrossRef CAS.
- Q. Liu, L. Jiang, P. Zheng, J. Sun, C. Liu, J. Chai, X. Li, Y. Zheng and Z. Liu, Chem. Rec., 2022, 22, e202200116 CrossRef CAS.
- C. Wang, J. T. Kim, C. Wang and X. Sun, Adv. Mater., 2023, 35, 2209074 CrossRef CAS.
- J. Wu, R. Zhang, Q. Fu, J. Zhang, X. Zhou, P. Gao, C. Xu, J. Liu and X. Guo, Adv. Funct. Mater., 2021, 31, 2008165 CrossRef CAS.
- M. York, K. Larson, K. C. Harris, E. Carmona, P. Albertus, R. Sharma, M. Noked, E. Strauss, H. Ragones and D. Golodnitsky, J. Solid State Electrochem., 2022, 26, 1851–1869 CrossRef CAS.
- M. Shoji, E. Cheng, T. Kimura and K. Kanamura, J. Phys. D Appl. Phys., 2019, 52, 103001 CrossRef.
- Y. Ren, K. Chen, R. Chen, T. Liu, Y. Zhang and C.-W. Nan, J. Am. Ceram. Soc., 2015, 98, 3603–3623 CrossRef CAS.
- J. Gai, E. Zhao, F. Ma, D. Sun, X. Ma, Y. Jin, Q. Wu and Y. Cui, J. Eur. Ceram. Soc., 2018, 38, 1673–1678 CrossRef CAS.
- F. Han, Y. Zhu, X. He, Y. Mo and C. Wang, Adv. Energy Mater., 2016, 6, 1501590 CrossRef.
- Y. Li, Z. Wang, Y. Cao, F. Du, C. Chen, Z. Cui and X. Guo, Electrochim. Acta, 2015, 180, 37–42 CrossRef CAS.
- S. Li, Z. Huang, Y. Xiao and C. Sun, Mater. Chem. Front., 2021, 5, 5336–5343 RSC.
- S. Saffirio, M. Falco, G. B. Appetecchi, F. Smeacetto and C. Gerbaldi, J. Eur. Ceram. Soc., 2022, 42, 1023–1032 CrossRef CAS.
- F. Stainer, B. Gadermaier, A. Kügerl, L. Ladenstein, K. Hogrefe and H. M. R. Wilkening, Solid State Ionics, 2023, 395, 116209 CrossRef CAS.
- W. Wang, W. Yuan, Z. Zhao, D. Zou, P. Zhang, Z. Shi, J. Weng and P. Zhou, J. Electroanal. Chem., 2023, 937, 117405 CrossRef CAS.
- X. Shen, Q. Zhang, T. Ning, T. Liu, Y. Luo, X. He, Z. Luo and A. Lu, Mater. Today Chem., 2020, 18, 100368 CrossRef CAS.
- V. Thangadurai, H. Kaack and W. J. F. Weppner, J. Am. Ceram. Soc., 2003, 86, 437–440 CrossRef CAS.
- R. Murugan, V. Thangadurai and W. Weppner, Angew. Chem., Int. Ed., 2007, 46, 7778–7781 Search PubMed.
- C. Wang, W. Ping, Q. Bai, H. Cui, R. Hensleigh, R. Wang, A. H. Brozena, Z. Xu, J. Dai, Y. Pei, C. Zheng, G. Pastel, J. Gao, X. Wang, H. Wang, J.-C. Zhao, B. Yang, X. (Rayne) Zheng, J. Luo, Y. Mo, B. Dunn and L. Hu, Science, 2020, 368, 521–526 CrossRef CAS PubMed.
- Y. Liu, Q. Sun, Y. Zhao, B. Wang, P. Kaghazchi, K. R. Adair, R. Li, C. Zhang, J. Liu, L.-Y. Kuo, Y. Hu, T.-K. Sham, L. Zhang, R. Yang, S. Lu, X. Song and X. Sun, ACS Appl. Mater. Interfaces, 2018, 10, 31240–31248 CrossRef CAS PubMed.
- H. Aono, E. Sugimoto, Y. Sadaoka, N. Imanaka and G. Adachi, J. Electrochem. Soc., 1990, 137, 1023 CrossRef CAS.
- J.-F. Wu, W. K. Pang, V. K. Peterson, L. Wei and X. Guo, ACS Appl. Mater. Interfaces, 2017, 9, 12461–12468 CrossRef CAS.
- X. Huang, Y. Lu, Z. Song, K. Rui, Q. Wang, T. Xiu, M. E. Badding and Z. Wen, Energy Storage Mater., 2019, 22, 207–217 CrossRef.
- J. Košir, S. Mousavihashemi, B. P. Wilson, E.-L. Rautama and T. Kallio, Solid State Ionics, 2022, 380, 115943 CrossRef.
- D. Rettenwander, P. Blaha, R. Laskowski, K. Schwarz, P. Bottke, M. Wilkening, C. A. Geiger and G. Amthauer, Chem. Mater., 2014, 26, 2617–2623 CrossRef CAS.
- P. Zhao, Y. Xiang, Y. Wen, M. Li, X. Zhu, S. Zhao, Z. Jin, H. Ming and G. Cao, J. Eur. Ceram. Soc., 2018, 38, 5454–5462 CrossRef CAS.
- J. Liu, F. Yin, Y. Mao and C. Sun, ACS Appl. Mater. Interfaces, 2024, 16, 31191–31200 CrossRef CAS PubMed.
- A. J. Samson, K. Hofstetter, S. Bag and V. Thangadurai, Energy Environ. Sci., 2019, 12, 2957–2975 RSC.
- J. Košir, S. Mousavihashemi, M. Suominen, A. Kobets, B. P. Wilson, E.-L. Rautama and T. Kallio, Mater. Adv., 2024, 5, 5260–5274 RSC.
- R. H. Brugge, J. A. Kilner and A. Aguadero, Solid State Ionics, 2019, 337, 154–160 CrossRef CAS.
- Y. Zhang, J. Deng, D. Hu, F. Chen, Q. Shen, L. Zhang and S. Dong, Electrochim. Acta, 2019, 296, 823–829 CrossRef CAS.
- Y. Jiang, X. Zhu, S. Qin, M. Ling and J. Zhu, Solid State Ionics, 2017, 300, 73–77 CrossRef CAS.
- W. Zhang and C. Sun, J. Phys. Chem. Solids, 2019, 135, 109080 CrossRef CAS.
- X. Zhou, L. Huang, O. Elkedim, Y. Xie, Y. Luo, Q. Chen, Y. Zhang and Y. Chen, J. Alloys Compd., 2022, 891, 161906 CrossRef CAS.
- J. Wang, X. Li, X. Wang, G. Liu, W. Yu, X. Dong and J. Wang, Ceram. Int., 2024, 50, 6472–6480 CrossRef CAS.
- R. Yan, X. Cheng, D. Zhang, R. Yang, Q. Yan, Y. Jiang, X. Pu and X. Zhu, J. Energy Storage, 2024, 87, 111481 CrossRef.
- Y. Lu, X. Meng, J. A. Alonso, M. T. Fernández-Díaz and C. Sun, ACS Appl. Mater. Interfaces, 2019, 11, 2042–2049 CrossRef CAS.
- T. Zhang, T. D. Christopher, S. Huang, Y. Liu, W. Gao, T. Söhnel and P. Cao, Ceram. Int., 2019, 45, 20954–20960 CrossRef CAS.
- Z. Zhang, Z. Zou, K. Kaup, R. Xiao, S. Shi, M. Avdeev, Y. S. Hu, D. Wang, B. He, H. Li, X. Huang, L. F. Nazar and L. Chen, Adv. Energy Mater., 2019, 9, 1902373 CrossRef CAS.
- F. Yin, Z. Zhang, Y. Fang and C. Sun, J. Energy Storage, 2023, 73, 108950 CrossRef.
- F. Lalère, J. B. Leriche, M. Courty, S. Boulineau, V. Viallet, C. Masquelier and V. Seznec, J. Power Sources, 2014, 247, 975–980 CrossRef.
- J. P. Boilot, G. Collin and P. Colomban, Mater. Res. Bull., 1987, 22, 669–676 CrossRef CAS.
- S. A. Ahmed, T. Pareek, S. Dwivedi, M. Badole and S. Kumar, J. Solid State Electrochem., 2020, 24, 2407–2417 CrossRef CAS.
- M. Illbeigi, A. Fazlali, M. Kazazi and A. H. Mohammadi, Solid State Ionics, 2016, 289, 180–187 CrossRef CAS.
- A. Chakraborty, R. Thirupathi, S. Bhattacharyya, K. Singh and S. Omar, J. Power Sources, 2023, 572, 233092 CrossRef CAS.
- Y. Lu, J. A. Alonso, Q. Yi, L. Lu, Z. L. Wang and C. Sun, Adv. Energy Mater., 2019, 9, 1901205 CrossRef.
- Z. Khakpour, Electrochim. Acta, 2016, 196, 337–347 CrossRef CAS.
- L. Ran, A. Baktash, M. Li, Y. Yin, B. Demir, T. Lin, M. Li, M. Rana, I. Gentle, L. Wang, D. J. Searles and R. Knibbe, Energy Storage Mater., 2021, 40, 282–291 CrossRef.
- G. Taveri, A. Güneren, M. Barlog, M. Hnatko, I. Zhukova, Z. Netriova, E. Šimon, M. Mičušik, M. Mikolášek and H. Kaňková, J. Power Sources, 2024, 592, 233917 CrossRef CAS.
- L. Zhang, Y. Liu, J. Han, C. Yang, X. Zhou, Y. Yuan and Y. You, ACS Appl. Mater. Interfaces, 2023, 15, 44867–44875 CrossRef CAS.
- F. Sun, Y. Xiang, Q. Sun, G. Zhong, M. N. Banis, Y. Liu, R. Li, R. Fu, M. Zheng, T. Sham, Y. Yang, X. Sun and X. Sun, Adv. Funct. Mater., 2021, 31, 2102129 CrossRef CAS.
- J. Wang, C.-W. Sun, Y.-D. Gong, H.-R. Zhang, J. A. Alonso, M. T. Fernández-Díaz, Z.-L. Wang and J. B. Goodenough, Chin. Phys. B, 2018, 27, 128201 CrossRef CAS.
- H. Gan, W. Zhu, L. Zhang and Y. Jia, Electrochim. Acta, 2022, 423, 140567 CrossRef CAS.
- E. A. Kurzina, I. A. Stenina, A. Dalvi and A. B. Yaroslavtsev, Inorg. Mater., 2021, 57, 1035–1042 CrossRef CAS.
- J. Li, Z. Li, S. Tang, J. Hao, T. Wang, C. Wang and L. Pan, Inorg. Chem. Front., 2022, 9, 4962–4973 RSC.
- Q. Wu, Y. Ma, S. Zhang, X. Chen, J. Bai, H. Wang and X. Liu, Small, 2024, 2404660 CrossRef CAS.
- C. Gao, J. Zhou, Q. Zhang, P. Cui, Q. Zhang and W. Wei, Funct. Mater. Lett., 2023, 16, 2350001 CrossRef CAS.
- G. Kaur, M. D. Singh, S. C. Sivasubramanian and A. Dalvi, Mater. Res. Bull., 2022, 145, 111555 CrossRef CAS.
- Y. Kee, N. Dimov, K. Minami and S. Okada, Electrochim. Acta, 2015, 174, 516–520 CrossRef CAS.
- A. G. Sabato, M. Nuñez Eroles, S. Anelli, C. D. Sierra, J. C. Gonzalez-Rosillo, M. Torrell, A. Pesce, G. Accardo, M. Casas-Cabanas, P. López-Aranguren, A. Morata and A. Tarancón, J. Mater. Chem. A, 2023, 11, 13677–13686 RSC.
- K. Hikima, I. Kusaba, H. Gamo, N. H. H. Phuc, H. Muto and A. Matsuda, ACS Omega, 2022, 7, 16561–16567 CrossRef CAS.
- D. H. S. Tan, E. A. Wu, H. Nguyen, Z. Chen, M. A. T. Marple, J. M. Doux, X. Wang, H. Yang, A. Banerjee and Y. S. Meng, ACS Energy Lett., 2019, 4, 2418–2427 CrossRef CAS.
- Z. Yu, S. L. Shang, Y. Gao, D. Wang, X. Li, Z. K. Liu and D. Wang, Nano Energy, 2018, 47, 325–330 CrossRef CAS.
- H. Guo, Q. Wang, A. Urban and N. Artrith, Chem. Mater., 2022, 34, 6702–6712 CrossRef CAS.
- Y. Morino, H. Sano, A. Shiota, K. Kawamoto, T. Takahashi, N. Miyashita, A. Sakuda and A. Hayashi, Electrochemistry, 2023, 91, 23–00003 CrossRef.
- T. Krauskopf, S. P. Culver and W. G. Zeier, Inorg. Chem., 2018, 57, 4739–4744 CrossRef CAS PubMed.
- C. Mi and S. R. Hall, RSC Adv., 2023, 13, 27066–27076 RSC.
- L. Gigli, D. Tisi, F. Grasselli and M. Ceriotti, Chem. Mater., 2024, 36, 1482–1496 CrossRef CAS.
- D. Park, H. Park, Y. Lee, S.-O. Kim, H.-G. Jung, K. Y. Chung, J. H. Shim and S. Yu, ACS Appl. Mater. Interfaces, 2020, 12, 34806–34814 CrossRef CAS.
- L. Zheng, J. Shi, G. Ren, T. Tang, Y. Yang, S. Shen and Z. Yao, Energy Fuels, 2024, 38, 3470–3476 CrossRef CAS.
- T. A. Tu, T. V. Toan, L. T. Anh, L. Van Thang and N. H. H. Phuc, RSC Adv., 2024, 14, 5764–5770 RSC.
- B. Tao, C. Ren, H. Li, B. Liu, X. Jia, X. Dong, S. Zhang and H. Chang, Adv. Funct. Mater., 2022, 32, 2203551 CrossRef CAS.
- J. A. Dawson and M. S. Islam, ACS Mater. Lett., 2022, 4, 424–431 CrossRef CAS.
- H. Wang, Y. Chen, Z. D. Hood, G. Sahu, A. S. Pandian, J. K. Keum, K. An and C. Liang, Angew. Chem., Int. Ed., 2016, 55, 8551–8555 CrossRef CAS.
- B. T. Leube, C. M. Collins, L. M. Daniels, B. B. Duff, Y. Dang, R. Chen, M. W. Gaultois, T. D. Manning, F. Blanc, M. S. Dyer, J. B. Claridge and M. J. Rosseinsky, Chem. Mater., 2022, 34, 4073–4087 CrossRef CAS PubMed.
- L. Zhou, N. Minafra, W. G. Zeier and L. F. Nazar, Acc. Chem. Res., 2021, 54, 2717–2728 CrossRef CAS PubMed.
- L. Peng, C. Yu, C. Wei, C. Liao, S. Chen, L. Zhang, S. Cheng and J. Xie, Acta Phys.-Chim. Sin., 2022, 39, 2211034 Search PubMed.
- Z. Zhang, L. Zhang, X. Yan, H. Wang, Y. Liu, C. Yu, X. Cao, L. van Eijck and B. Wen, J. Power Sources, 2019, 410–411, 162–170 CrossRef CAS.
- Z. Deng, D. Ni, D. Chen, Y. Bian, S. Li, Z. Wang and Y. Zhao, InfoMat, 2022, 4, e12252 CrossRef CAS.
- M. Dixit, N. Muralidharan, A. Bisht, C. J. Jafta, C. T. Nelson, R. Amin, R. Essehli, M. Balasubramanian and I. Belharouak, ACS Energy Lett., 2023, 8, 2356–2364 CrossRef CAS.
- W. Feng, Z. Lai, X. Dong, P. Li, Y. Wang and Y. Xia, iScience, 2020, 23, 101071 CrossRef CAS.
- B. H. Sjølin, P. B. Jørgensen, A. Fedrigucci, T. Vegge, A. Bhowmik and I. E. Castelli, Batteries Supercaps, 2023, 6, e202300041 CrossRef.
- E. Ahiavi, J. A. Dawson, U. Kudu, M. Courty, M. S. Islam, O. Clemens, C. Masquelier and T. Famprikis, J. Power Sources, 2020, 471, 228489 CrossRef CAS.
- Y. Sun, Y. Wang, X. Liang, Y. Xia, L. Peng, H. Jia, H. Li, L. Bai, J. Feng, H. Jiang and J. Xie, J. Am. Chem. Soc., 2019, 141, 5640–5644 CrossRef CAS.
- Z. Zhang and L. F. Nazar, Nat. Rev. Mater., 2022, 7, 389–405 CrossRef.
- S. Wang, S. Liu, W. Chen, Y. Hu, D. Chen, M. He, M. Zhou, T. Lei, Y. Zhang and J. Xiong, Adv. Sci., 2024, 11, 2401889 CrossRef CAS.
- X. Nie, J. Hu and C. Li, Interdiscip. Mater., 2023, 2, 365–389 Search PubMed.
- K. Tuo, F. Yin, F. Mi and C. Sun, J. Energy Chem., 2023, 87, 12–23 CrossRef CAS.
- K. Tuo, F. Yin and C. Sun, ACS Sustain. Chem. Eng., 2024, 12, 7012–7025 CrossRef CAS.
- K. Tuo, C. Sun, C. A. López, M. T. Fernández-Díaz and J. A. Alonso, J. Mater. Chem. A, 2023, 11, 15651–15662 RSC.
- L. Lu, Y. Lu, J. A. Alonso, C. A. López, M. T. Fernández-Díaz, B. Zou and C. Sun, ACS Appl. Mater. Interfaces, 2021, 13, 42927–42934 CrossRef CAS.
- L. Lu, C. Sun, J. Hao, Z. Wang, S. F. Mayer, M. T. Fernández-Díaz, J. A. Alonso and B. Zou, Energy Environ. Mater., 2023, 6, e12364 CrossRef CAS.
- S. Balijapelly, P. Sandineni, A. Adhikary, N. N. Gerasimchuk, A. V. Chernatynskiy and A. Choudhury, Dalton Trans., 2021, 50, 7372–7379 RSC.
- F. Strauss, J. Lin, L. Karger, D. Weber and T. Brezesinski, Inorg. Chem., 2022, 61, 5885–5890 CrossRef CAS PubMed.
- E. P. Ramos, N. Kim, A. Assoud, I. Kochetkov, L. Wan and L. F. Nazar, ACS Mater. Lett., 2023, 5, 144–154 CrossRef CAS.
- K. Hikima, K. Ogawa, H. Gamo and A. Matsuda, Chem. Commun., 2023, 59, 6564–6567 RSC.
- F. Strauss, T. Zinkevich, S. Indris and T. Brezesinski, Inorg. Chem., 2020, 59, 12954–12959 CrossRef CAS PubMed.
- J. Lin, G. Cherkashinin, M. Schäfer, G. Melinte, S. Indris, A. Kondrakov, J. Janek, T. Brezesinski and F. Strauss, ACS Mater. Lett., 2022, 4, 2187–2194 CrossRef CAS.
- Y. Liu, H. Peng, H. Su, Y. Zhong, X. Wang, X. Xia, C. Gu and J. Tu, Adv. Mater., 2022, 34, 2107346 CrossRef CAS.
- D.-X. Ni, Y.-D. Liu, Z. Deng, D.-C. Chen, X.-X. Zhang, T. Wang, S. Li and Y.-S. Zhao, Chin. Phys. Lett., 2022, 39, 028201 CrossRef CAS.
- H. Zhang, Z. Zeng, X. Shi, C. Wang and Y. Du, EcoMat, 2023, 5, e12315 CrossRef CAS.
- K. Wang, Q. Ren, Z. Gu, C. Duan, J. Wang, F. Zhu, Y. Fu, J. Hao, J. Zhu, L. He, C.-W. Wang, Y. Lu, J. Ma and C. Ma, Nat. Commun., 2021, 12, 4410 CrossRef CAS.
- W. S. Scheld, S. Lobe, C. Dellen, M. Ihrig, G. Häuschen, L. C. Hoff, M. Finsterbusch, S. Uhlenbruck, O. Guillon and D. Fattakhova-Rohlfing, J. Power Sources, 2022, 545, 231872 CrossRef CAS.
- X. Xu, Z. Wen, X. Yang, J. Zhang and Z. Gu, Solid State Ionics, 2006, 177, 2611–2615 CrossRef CAS.
- D. Rettenwander, C. A. Geiger, M. Tribus, P. Tropper and G. Amthauer, Inorg. Chem., 2014, 53, 6264–6269 CrossRef CAS PubMed.
- K. Tuo, C. Sun and S. Liu, Electrochem. Energy Rev., 2023, 6, 17 CrossRef CAS.
- M. He, Z. Cui, C. Chen, Y. Li and X. Guo, J. Mater. Chem. A, 2018, 6, 11463–11470 RSC.
- S. Ohta, T. Kobayashi and T. Asaoka, J. Power Sources, 2011, 196, 3342–3345 CrossRef CAS.
- T. Koç, F. Marchini, G. Rousse, R. Dugas and J.-M. Tarascon, ACS Appl. Energy Mater., 2021, 4, 13575–13585 CrossRef.
- G. T. Hitz, D. W. McOwen, L. Zhang, Z. Ma, Z. Fu, Y. Wen, Y. Gong, J. Dai, T. R. Hamann, L. Hu and E. D. Wachsman, Mater. Today, 2019, 22, 50–57 CrossRef CAS.
- H. Gamo, A. Nagai and A. Matsuda, Sci. Rep., 2021, 11, 21097 CrossRef PubMed.
- Z. Liu, W. Fu, E. A. Payzant, X. Yu, Z. Wu, N. J. Dudney, J. Kiggans, K. Hong, A. J. Rondinone and C. Liang, J. Am. Chem. Soc., 2013, 135, 975–978 CrossRef CAS PubMed.
- K. Yamamoto, S. Yang, M. Takahashi, K. Ohara, T. Uchiyama, T. Watanabe, A. Sakuda, A. Hayashi, M. Tatsumisago, H. Muto, A. Matsuda and Y. Uchimoto, ACS Appl. Energy Mater., 2021, 4, 2275–2281 CrossRef CAS.
- R. C. Xu, X. H. Xia, Z. J. Yao, X. L. Wang, C. D. Gu and J. P. Tu, Electrochim. Acta, 2016, 219, 235–240 CrossRef CAS.
- P. Paetzold, H. F. Bettinger and O. Volkov, Z. Anorg. Allg. Chem., 2007, 633, 846–850 CrossRef CAS.
- H. Shen, E. Yi, M. Amores, L. Cheng, N. Tamura, D. Y. Parkinson, G. Chen, K. Chen and M. Doeff, J. Mater. Chem. A, 2019, 7, 20861–20870 RSC.
- Q. Chen, C. Ouyang, Y. Liang, H. Liu and H. Duan, Energy Storage Mater., 2024, 69, 103418 CrossRef.
- K. H. Park, Q. Bai, D. H. Kim, D. Y. Oh, Y. Zhu, Y. Mo and Y. S. Jung, Adv. Energy Mater., 2018, 8, 1800035 CrossRef.
- Z. Gao, J. Yang, H. Yuan, H. Fu, Y. Li, Y. Li, T. Ferber, C. Guhl, H. Sun, W. Jaegermann, R. Hausbrand and Y. Huang, Chem. Mater., 2020, 32, 3970–3979 CrossRef CAS.
- Y. Zhu, X. He and Y. Mo, Adv. Sci., 2017, 4, 1600517 CrossRef PubMed.
- Y. Zhu, X. He and Y. Mo, ACS Appl. Mater. Interfaces, 2015, 7, 23685–23693 CrossRef CAS.
- J. Liang, X. Li, S. Wang, K. R. Adair, W. Li, Y. Zhao, C. Wang, Y. Hu, L. Zhang, S. Zhao, S. Lu, H. Huang, R. Li, Y. Mo and X. Sun, J. Am. Chem. Soc., 2020, 142, 7012–7022 CrossRef CAS.
- P. Hu, Y. Zhang, X. Chi, K. Kumar Rao, F. Hao, H. Dong, F. Guo, Y. Ren, L. C. Grabow and Y. Yao, ACS Appl. Mater. Interfaces, 2019, 11, 9672–9678 CrossRef CAS PubMed.
- Y. Tian, T. Shi, W. D. Richards, J. Li, J. C. Kim, S.-H. Bo and G. Ceder, Energy Environ. Sci., 2017, 10, 1150–1166 RSC.
- S. Wenzel, T. Leichtweiss, D. A. Weber, J. Sann, W. G. Zeier and J. Janek, ACS Appl. Mater. Interfaces, 2016, 8, 28216–28224 CrossRef CAS.
- S. J. Baek, E. Cha, D. G. Kim, J. H. Yun and D. K. Kim, Ceram. Int., 2022, 48, 34828–34836 CrossRef CAS.
- S. Zhang, Q. Sun, G. Hou, J. Cheng, L. Dai, J. Li and L. Ci, Nano Res., 2023, 16, 6825–6832 CrossRef CAS.
- J. A. S. Oh, L. He, B. Chua, K. Zeng and L. Lu, Energy Storage Mater., 2021, 34, 28–44 CrossRef.
- H. Zheng, G. Li, R. Ouyang, Y. Han, H. Zhu, Y. Wu, X. Huang, H. Liu and H. Duan, Adv. Funct. Mater., 2022, 32, 2205778 CrossRef CAS.
- H. Huo, J. Luo, V. Thangadurai, X. Guo, C.-W. Nan and X. Sun, ACS Energy Lett., 2020, 5, 252–262 CrossRef CAS.
- S. G. Kang and D. S. Sholl, J. Phys. Chem. C, 2014, 118, 17402–17406 CrossRef CAS.
- C.-L. Tsai, V. Roddatis, C. V. Chandran, Q. Ma, S. Uhlenbruck, M. Bram, P. Heitjans and O. Guillon, ACS Appl. Mater. Interfaces, 2016, 8, 10617–10626 CrossRef CAS.
- C. Wang, H. Xie, L. Zhang, Y. Gong, G. Pastel, J. Dai, B. Liu, E. D. Wachsman and L. Hu, Adv. Energy Mater., 2018, 8, 1701963 CrossRef.
- T. Wang, J. Duan, B. Zhang, W. Luo, X. Ji, H. Xu, Y. Huang, L. Huang, Z. Song, J. Wen, C. Wang, Y. Huang and J. B. Goodenough, Energy Environ. Sci., 2022, 15, 1325–1333 RSC.
- L. Chen, G. Chen, W. Tang, H. Wang, F. Chen, X. Liu and R. Ma, Mater. Today Energy, 2020, 18, 100520 CrossRef CAS.
- X. Ma and Y. Xu, Electrochim. Acta, 2023, 441, 141789 CrossRef CAS.
- L. Porz, T. Swamy, B. W. Sheldon, D. Rettenwander, T. Frömling, H. L. Thaman, S. Berendts, R. Uecker, W. C. Carter and Y.-M. Chiang, Adv. Energy Mater., 2017, 7, 1701003 CrossRef.
- Y. Wang, Z. Chen, K. Jiang, Z. Shen, S. Passerini and M. Chen, Small, 2024, 20, 2402035 CrossRef CAS PubMed.
- C. Yuan, X. Gao, Y. Jia, W. Zhang, Q. Wu and J. Xu, Nano Energy, 2021, 86, 106057 CrossRef CAS.
- E. Kazyak, R. Garcia-Mendez, W. S. LePage, A. Sharafi, A. L. Davis, A. J. Sanchez, K.-H. Chen, C. Haslam, J. Sakamoto and N. P. Dasgupta, Matter, 2020, 2, 1025–1048 CrossRef.
- T. Swamy, R. Park, B. W. Sheldon, D. Rettenwander, L. Porz, S. Berendts, R. Uecker, W. C. Carter and Y.-M. Chiang, J. Electrochem. Soc., 2018, 165, A3648 CrossRef CAS.
- Y. Qi, C. Ban and S. J. Harris, Joule, 2020, 4, 2599–2608 CrossRef CAS.
- M. B. Dixit, B. S. Vishugopi, W. Zaman, P. Kenesei, J.-S. Park, J. Almer, P. P. Mukherjee and K. B. Hatzell, Nat. Mater., 2022, 21, 1298–1305 CrossRef CAS.
- F. Han, A. S. Westover, J. Yue, X. Fan, F. Wang, M. Chi, D. N. Leonard, N. J. Dudney, H. Wang and C. Wang, Nat. Energy, 2019, 4, 187–196 CrossRef CAS.
- H.-K. Tian, Z. Liu, Y. Ji, L.-Q. Chen and Y. Qi, Chem. Mater., 2019, 31, 7351–7359 CrossRef CAS.
- M. Feng, J. Pan and Y. Qi, J. Phys. Chem. C, 2021, 125, 15821–15829 CrossRef CAS.
- Y. Zou, H. Zheng, S. Wu, ZehuanHei, H. Liu and H. Duan, Mater. Lett., 2021, 297, 129959 CrossRef CAS.
- Y. Zhang, Y. Mei, X. Gao, Y. Xiao, Z. Tang, X. Xiang and J. Deng, J. Alloys Compd., 2024, 971, 172746 CrossRef CAS.
- Y. Zhang, X. Gao, Y. Mei, Z. Tang, D. Luo and J. Deng, J. Alloys Compd., 2023, 933, 167736 CrossRef CAS.
- H. Fu, Q. Yin, Y. Huang, H. Sun, Y. Chen, R. Zhang, Q. Yu, L. Gu, J. Duan and W. Luo, ACS Mater. Lett., 2020, 2, 127–132 CrossRef CAS.
- Z. Wang, Y. Mao, L. Sheng and C. Sun, ACS Appl. Mater. Interfaces, 2024, 16, 12706–12716 CrossRef CAS.
- Y. Wang, Q. Zhang, Z.-C. Xue, L. Yang, J. Wang, F. Meng, Q. Li, H. Pan, J.-N. Zhang, Z. Jiang, W. Yang, X. Yu, L. Gu and H. Li, Adv. Energy Mater., 2020, 10, 2001413 CrossRef CAS.
- E. J. Cheng, N. J. Taylor, J. Wolfenstine and J. Sakamoto, J. Asian Ceram. Soc., 2017, 5, 113–117 CrossRef.
- E. J. Cheng, Interface Engineering of Solid-State Li Metal Batteries with Garnet Electrolytes, Kyoto University, 2023 Search PubMed.
- Y. Gao, S. Sun, X. Zhang, Y. Liu, J. Hu, Z. Huang, M. Gao and H. Pan, Adv. Funct. Mater., 2021, 31, 2009692 CrossRef CAS.
- X. Miao, H. Di, X. Ge, D. Zhao, P. Wang, R. Wang, C. Wang and L. Yin, Energy Storage Mater., 2020, 30, 170–178 CrossRef.
- Z. Xie, Y. Zhang, A. Yuan and J. Xu, J. Alloys Compd., 2019, 787, 429–439 CrossRef CAS.
- Y. S. Byeon, D. Kim, S. A. Han, J. H. Kim and M.-S. Park, Adv. Energy Sustainability Res., 2024, 2400135 CrossRef CAS.
- E. Wang, Y. Zhao, D. Xiao, X. Zhang, T. Wu, B. Wang, M. Zubair, Y. Li, X. Sun and H. Yu, Adv. Mater., 2020, 32, 1906070 CrossRef CAS.
- C. Wang, X. Li, Y. Zhao, M. N. Banis, J. Liang, X. Li, Y. Sun, K. R. Adair, Q. Sun, Y. Liu, F. Zhao, S. Deng, X. Lin, R. Li, Y. Hu, T.-K. Sham, H. Huang, L. Zhang, R. Yang, S. Lu and X. Sun, Small Methods, 2019, 3, 1900261 CrossRef CAS.
- A. Banerjee, K. H. Park, J. W. Heo, Y. J. Nam, C. K. Moon, S. M. Oh, S.-T. Hong and Y. S. Jung, Angew. Chem., Int. Ed., 2016, 55, 9634–9638 CrossRef CAS PubMed.
- T. Koç, M. Hallot, E. Quemin, B. Hennequart, R. Dugas, A. M. Abakumov, C. Lethien and J.-M. Tarascon, ACS Energy Lett., 2022, 7, 2979–2987 CrossRef.
- G. Liu, Y. Lu, H. Wan, W. Weng, L. Cai, Z. Li, X. Que, H. Liu and X. Yao, ACS Appl. Mater. Interfaces, 2020, 12, 28083–28090 CrossRef CAS.
- X. Xu, G. Du, C. Cui, J. Liang, C. Zeng, S. Wang, Y. Ma and H. Li, ACS Appl. Mater. Interfaces, 2022, 14, 39951–39958 CrossRef CAS.
- L. Wu, G. Liu, H. Wan, W. Weng and X. Yao, J. Power Sources, 2021, 491, 229565 CrossRef CAS.
- Y. Kato, S. Hori, T. Saito, K. Suzuki, M. Hirayama, A. Mitsui, M. Yonemura, H. Iba and R. Kanno, Nat. Energy, 2016, 1, 16030 CrossRef CAS.
- N. Kamaya, K. Homma, Y. Yamakawa, M. Hirayama, R. Kanno, M. Yonemura, T. Kamiyama, Y. Kato, S. Hama, K. Kawamoto and A. Mitsui, Nat. Mater., 2011, 10, 682–686 CrossRef CAS PubMed.
- R. Chen, Q. Li, X. Yu, L. Chen and H. Li, Chem. Rev., 2020, 120, 6820–6877 CrossRef CAS PubMed.
- L. Chen, S. Huang, D. Ma, Y. Xiong, J. Qiu, G. Cao and H. Zhang, Energy Fuels, 2021, 35, 13411–13418 CrossRef CAS.
- F. Yang, E. Campos dos Santos, X. Jia, R. Sato, K. Kisu, Y. Hashimoto, S. Orimo and H. Li, Nano Mater. Sci., 2024, 6, 256–262 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |