
Linkage engineering regulated π-conjugated covalent organic framework (COF)-based anodes for high-performance LIBs†
Changyu
Weng
a,
Hongmei
Yuan
a,
Jie
Wang
a,
Longlong
Ma
 ab and
Jianguo
Liu
*a
ab and
Jianguo
Liu
*a
aKey Laboratory of Energy Thermal Conversion and Control of Ministry of Education, School of Energy and Environment, Southeast University, Nanjing 210096, P. R. China. E-mail: liujg@seu.edu.cn
bDepartment of Thermal Science and Energy Engineering, University of Science and Technology of China, Hefei 230026, P. R. China
First published on 19th November 2024
Abstract
COFs have garnered widespread attention and demonstrated significant potential in energy storage and conversion, attributed to their predesigned structures, high surface areas, and excellent stability. However, designing and preparing COFs with high-performance Li-ion storage capabilities among different types of linkages remains challenging. In this work, we synthesized two types of COFs with distinct linkages through linkage engineering. COFs@BOA, incorporating oxazole linkages, and COFs@IM, incorporating imine linkages, both exhibited fine crystallinity and stability. Due to the semiconductor properties of the COFs, we employed an in situ growth approach to deposit the COFs onto the surface of carbon nanotubes. The resulting COFs@BOA-30 and COFs@IM-30 composites demonstrated highly reversible capacity, with the latter exhibiting a superior capacity of up to 1100.3 mA h g−1 at 100 mA g−1. The capacity contribution of COFs@IM was calculated to be 1696.0 mA h g−1 in the COFs@IM-30 composite, while COFs@BOA contributed only 925.8 mA h g−1. DFT calculations suggest that the discrepancy in capacities may be attributed to the lower LUMO–HOMO gap of COFs@IM. Additionally, electrical conductivity measurements indicate that COFs@IM has better conductivity than COFs@BOA, highlighting the superior performance of COFs@IM. This study underscores the significance of linkage engineering in designing COFs to improve the performance of organic electrodes in LIBs.
1 Introduction
Rechargeable lithium-ion batteries (LIBs) serve as essential energy storage devices and converters, playing a significant role in various portable electronic equipment and other fields.1–3 However, with the ever-increasing demand for LIBs, there is an urgent need to develop novel electrode materials with high-performance Li-ion storage and high capacity.4 Over the past few decades, research and development efforts have primarily focused on inorganic materials (such as transition metal oxides and layer-structured inorganic materials) as electrode materials in both the scientific community and industry.5 However, these materials suffer from several drawbacks, including resource scarcity, high toxicity, complex preparation procedures, high cost, and rapid capacity decay during the discharge process.6,7 Consequently, traditional inorganic material-based electrodes have encountered a bottleneck.8 Against this backdrop, researchers are gradually shifting their focus toward exploring novel electrode materials. Among the plethora of materials, organic compounds are emerging as potential alternatives to current inorganic electrode materials due to their abundant sources, comprising a large number of light elements (C, H, O, N, and S), relatively low cost, and environmental friendliness.9,10 Organic small molecules, a subset of organic compounds, have also been investigated for electrode manufacturing. However, they often exhibit unsatisfactory performance due to their intrinsic poor electrical conductivity and susceptibility to dissolution in liquid organic electrolytes.11 One of the most effective approaches to address these challenges is to anchor organic small molecules with redox-active groups onto organic polymer frameworks.12COFs have garnered considerable research interest among polymers due to their highly ordered porous organic structures, characterized by pre-designable architectures, large surface areas, open channels, and high chemical and thermal stabilities.13,14 Consequently, COFs can be synthesized using pre-selected monomers containing active sites such as C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and forming CN,15 which are conducive to Li-ion storage.16 As a result, these unique polymers not only address the issue of dissolution commonly encountered with organic small molecules in electrolytes but also serve as sophisticated electrode materials rich in active sites.17 Moreover, the high surface area of COFs allows for better exposure of active sites to the electrolyte than traditional electrodes, improving utilization of active sites.17,18 Furthermore, the open channels and layered structures of COFs facilitate rapid insertion/extraction of lithium ions, thereby improving the utilization of the active sites.17 Consequently, the diffusion kinetics of lithium ions during the electrochemical process are significantly enhanced within COF structures.19 However, despite the application of COFs as electrode materials in LIBs, several challenges persist. These include unsatisfactory electrical conductivity and limited utilization of active sites, which hinder further development of COF-based electrodes.20 In light of these challenges, approaches such as linkage engineering may be employed to manipulate the intrinsic conductivity of materials and optimize the availability of active sites.
O and forming CN,15 which are conducive to Li-ion storage.16 As a result, these unique polymers not only address the issue of dissolution commonly encountered with organic small molecules in electrolytes but also serve as sophisticated electrode materials rich in active sites.17 Moreover, the high surface area of COFs allows for better exposure of active sites to the electrolyte than traditional electrodes, improving utilization of active sites.17,18 Furthermore, the open channels and layered structures of COFs facilitate rapid insertion/extraction of lithium ions, thereby improving the utilization of the active sites.17 Consequently, the diffusion kinetics of lithium ions during the electrochemical process are significantly enhanced within COF structures.19 However, despite the application of COFs as electrode materials in LIBs, several challenges persist. These include unsatisfactory electrical conductivity and limited utilization of active sites, which hinder further development of COF-based electrodes.20 In light of these challenges, approaches such as linkage engineering may be employed to manipulate the intrinsic conductivity of materials and optimize the availability of active sites.
In this study, two similar COFs with different linkages, oxazole linkage and imine linkage, were synthesized using two groups of slightly different monomers via solvothermal synthesis as anode materials for LIBs. Our design aimed to generate nearly identical frameworks using these two groups of monomers, differing only in the direction of the linkage. However, the two COFs exhibited significant differences in the linkage of their frameworks. As depicted in Fig. 1a, the COFs linked by oxazole (referred to as COFs@BOA) were prepared by condensing 1,3,5-triformylbenzene (TFB) and 2,5-diaminohydroquinone dihydrochloride (DABD) in mixed solvents of N-methylpyrrolidone and 1,3,5-trimethylbenzene. Correspondingly, the COFs linked by imine (referred to as COFs@IM) were synthesized by condensing benzene-1,3,5-triamine trihydrochloride (TAB) and 2,5-dihydroxyterephthalaldehyde (DHA) in mixed solvents of mesitylene and 1,4-dioxane. Fourier-transform infrared (FT-IR) and solid-state 13C nuclear magnetic resonance techniques were applied to investigate the linkage of both COFs, and the differences in linkage were verified. To examine the influence of linkage engineering on lithium storage performance, pure COFs@BOA and COFs@IM were synthesized as electrodes, respectively. However, due to their low intrinsic conductivity, both pure COFs were found to have a relatively low specific capacity of 352 mA h g−1 and 474 mA h g−1. To enhance their electrochemical performance in LIBs, both COF composites with different ratios of CNTs were prepared via in situ polycondensation on carbon nanotubes (CNTs). The composites with a 30% ratio of CNTs displayed satisfactory Li-ion storage performance, with the specific capacity of COFs@BOA-30 (853.1 mA h g−1) being approximately 2.5 times higher than that of pure COFs@BOA and about 1.8 times higher than that of COFs@BOA-15. A similar phenomenon was observed for COFs@IM-30; the specific capacity of COFs@IM-30 (1100.3 mA h g−1) is 2.3 times higher than that of pure COFs@IM and 2.2 times higher that of COFs@IM-15. After analysis, it was determined that the main contribution to the capacity of the CNT-composited electrodes came from the incorporated COFs. Notably, an enormous reversible capacity contribution of 925.8 mA h g−1 was achieved for the COFs in COFs@BOA-30, while for COFs@IM-30, the contribution of COFs@IM reached 1696.0 mA h g−1. The higher contribution of COFs@IM can be attributed to its low yield and favorable chemical structure. Moreover, the differences in the internal properties of COFs@IM and COFs@BOA are likely the primary factors driving the observed variation in capacity, with electrical conduction measurements and DFT calculations providing further support for this hypothesis. Moreover, density functional theory (DFT) calculations revealed that COFs@IM had a relatively lower LUMO–HOMO gap level than COFs@BOA, indicating superior electron conductivity. In conclusion, we believe that this study may guide the development of high-performance COF-based electrode materials for LIBs through linkage engineering.
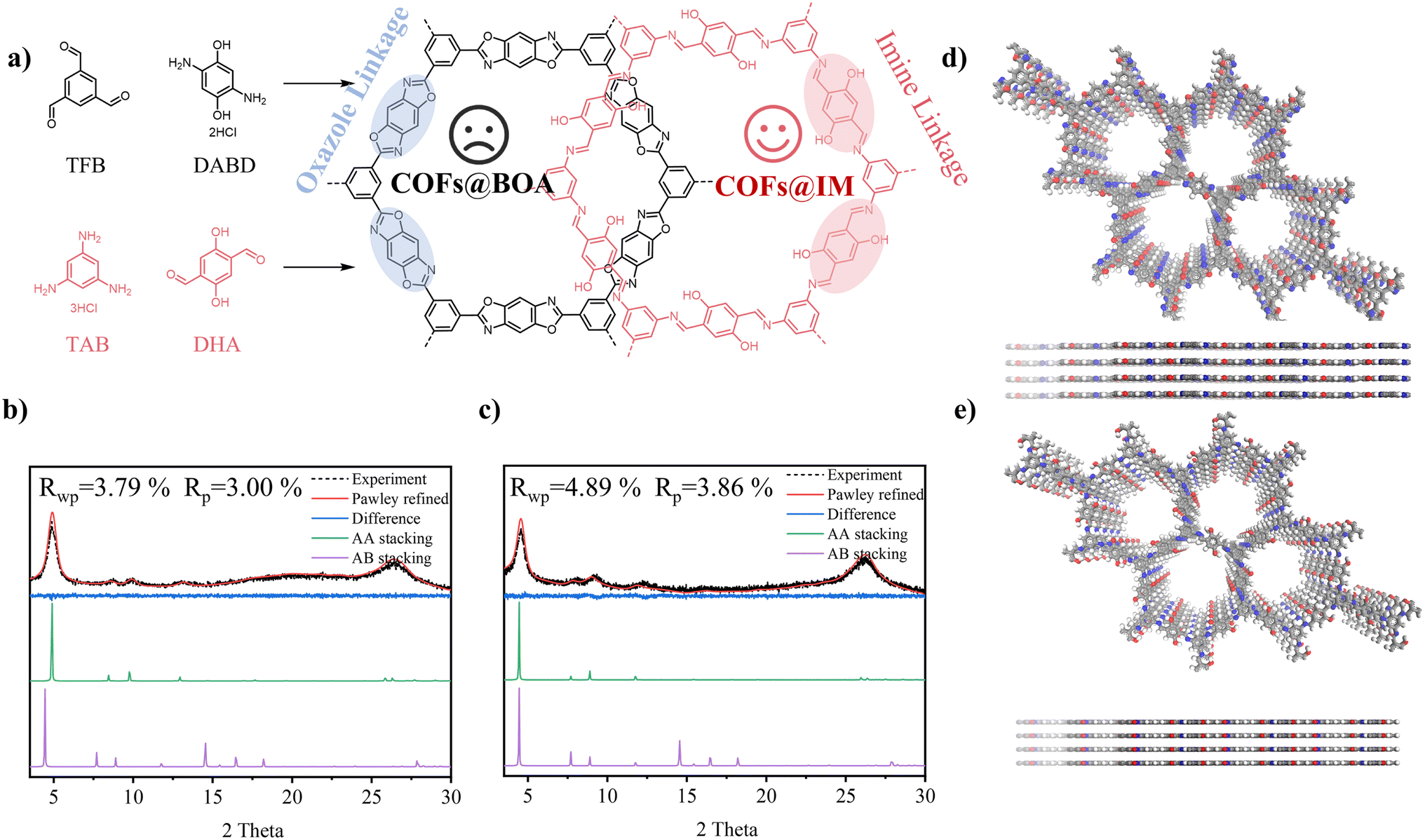 | ||
| Fig. 1 (a) Synthetic scheme of COFs@BOA and COFs@IM. (b and c) Experimental XRD pattern and Pawley refinement of COFs@BOA (b) and COFs@IM (c): experiment (black), Pawley refined (red), the difference between experimental and calculated data (blue), calculated for AA-stacking (green), and calculated for AB-stacking (violet). (d and e) AA stacking models for COFs@BOA (d) and COFs@IM (e) with C: gray, N: blue, O: red, and H: white. | ||
2 Experimental
2.1 Materials
All reagents were obtained from commercial suppliers and used as received. N-Methylpyrrolidone (99.0%), 1,3,5-trimethylbenzene (99.0%), benzimidazole (99.0%), 1,3,5-triformylbenzene (TFB, 99.0%), 2,5-diaminohydroquinone dihydrochloride (DABD, 98.0%), benzene-1,3,5-triamine trihydrochloride (TAB, 98.0%), 2,5-dihydroxyterephthalaldehyde (DHA, 98.0%), 1,4-dioxane (99.5%), acetic acid (99.0%), tetrahydrofuran (THF, 99.5%), methanol (99.9%), and carbon nanotubes (CNTs, purity: >95 wt%, OD: <2 nm, length: 5–30 μm) were purchased from Tansoole. Acetone (99.0%) was purchased from Sinopharm Chemical Reagent Limited Corporation. Acetylene black and polyvinylidene fluoride (PVDF) were purchased from Canrd.2.2 Synthesis of COFs
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 V/V, 5 mL). After the mixture was sonicated for 20 min, benzimidazole (159.5 mg, 1.35 mmol) was added. Subsequently, the Pyrex tube was frozen at 77 K using a liquid N2 bath and degassed by three freeze–pump–thaw cycles, then sealed under vacuum and heated at 120 °C for 72 hours. The resulting COFs@BOA precipitate was isolated by vacuum filtration and washed with THF, methanol, and acetone and then dried at 100 °C under vacuum for 24 hours to give the desired COFs@BOA.
:1 V/V, 2 mL). After the mixture was sonicated for 20 minutes, acetic acid (6 M, 0.2 mL) was added. Subsequently, the Pyrex tube was frozen at 77 K using a liquid N2 bath and degassed by three freeze–pump–thaw cycles, then sealed under vacuum and heated at 120 °C for 72 hours. The resulting COFs@IM precipitate was isolated by vacuum filtration and washed with THF, methanol, and acetone and then dried at 100 °C under vacuum for 24 hours to give the desired COFs@IM.
:1 V/V, 5 mL). After the mixture was sonicated for 20 min, benzimidazole (159.5 mg, 1.35 mmol) was added. Subsequently, the Pyrex tube was frozen at 77 K using a liquid N2 bath and degassed by three freeze–pump–thaw cycles, then sealed under vacuum and heated at 120 °C for 72 hours. The resulting COFs@BOA precipitate was isolated by vacuum filtration and washed with THF, methanol, and acetone and then dried at 100 °C under vacuum for 24 hours to give the desired COFs@BOA.
:1 V/V, 2 mL). After the mixture was sonicated for 20 minutes, acetic acid (6 M, 0.2 mL) was added. Subsequently, the Pyrex tube was frozen at 77 K using a liquid N2 bath and degassed by three freeze–pump–thaw cycles, then sealed under vacuum and heated at 120 °C for 72 hours. The resulting COFs@IM precipitate was isolated by vacuum filtration and washed with THF, methanol, and acetone and then dried at 100 °C under vacuum for 24 hours to give the desired COFs@IM.
3 Results and discussion
3.1 Synthesis and characterization of COF-based electrodes
As shown in the synthetic routes in Fig. 1a, two COFs with different linkages were synthesized using similar monomers. The details of the synthesis process are provided in the Experimental section. Fourier transform infrared (FT-IR) spectra were used to analyze the linkage of COFs. From Fig. 2a, two apparent peaks at wavenumbers of 1118 cm−1 and 1620 cm−1 were found. The former peak was ascribed to the stretching vibration of the C–O bond, while the latter can be attributed to NC–O. These two peaks manifested the formation of the benzoxazole ring.21,22 Additionally, the disappearance of characteristic peaks of C–N (1197 cm−1) and CO (1694 cm−1) also proved the formation of COFs@BOA and the corresponding nanotube complex. As for COFs@IM (Fig. 2b), the newly formed peak at 1617 cm−1 was due to the stretching vibration of the CN linkage.23 At the same time, the intensity of N–H (at around 3220 cm−1, 3225 cm−1 and 3408 cm−1) and CO (1667 cm−1) experienced a reduction in strength. Meanwhile, the peak intensity at 3424 cm−1 (O–H) still existed, reflecting the presence of oxhydryl in COFs@IM. To further validate the formation of the different linkages, 13C cross-polarization solid-state nuclear magnetic resonance (13C CP/MAS NMR) spectroscopy was performed. As shown in Fig. 2c, the spectra of COFs@BOA exhibited signals at 163, 148, and 140 ppm, originating from the N-heterocyclic rings' carbon atoms. Specifically, the signal at 163 ppm indicated the formation of benzoxazole rings.24 Regarding COFs@IM, the signal at 161 ppm could be ascribed to the imine linkage25 and the signal at 148 ppm might be the peak of the phenolic hydroxyl group.26 The other signals of both COFs are attributed to the carbon atoms attached to benzene rings and are specifically labeled in the figure. Based on elemental analysis, the weight contents of nitrogen, carbon and hydrogen elements in both COFs and their composites with CNTs are summarized in Table S1.† Hence, we can infer that COFs@BOA-30 consists of 70% COFs@BOA and 30% CNTs, while COFs@IM-30 consists of 46% COFs@IM and 54% CNTs. This discrepancy may arise from differences in the yield of the COFs, which could be influenced by variations in the active monomers used and the different synthesis conditions (including mixed solvents and catalysts).
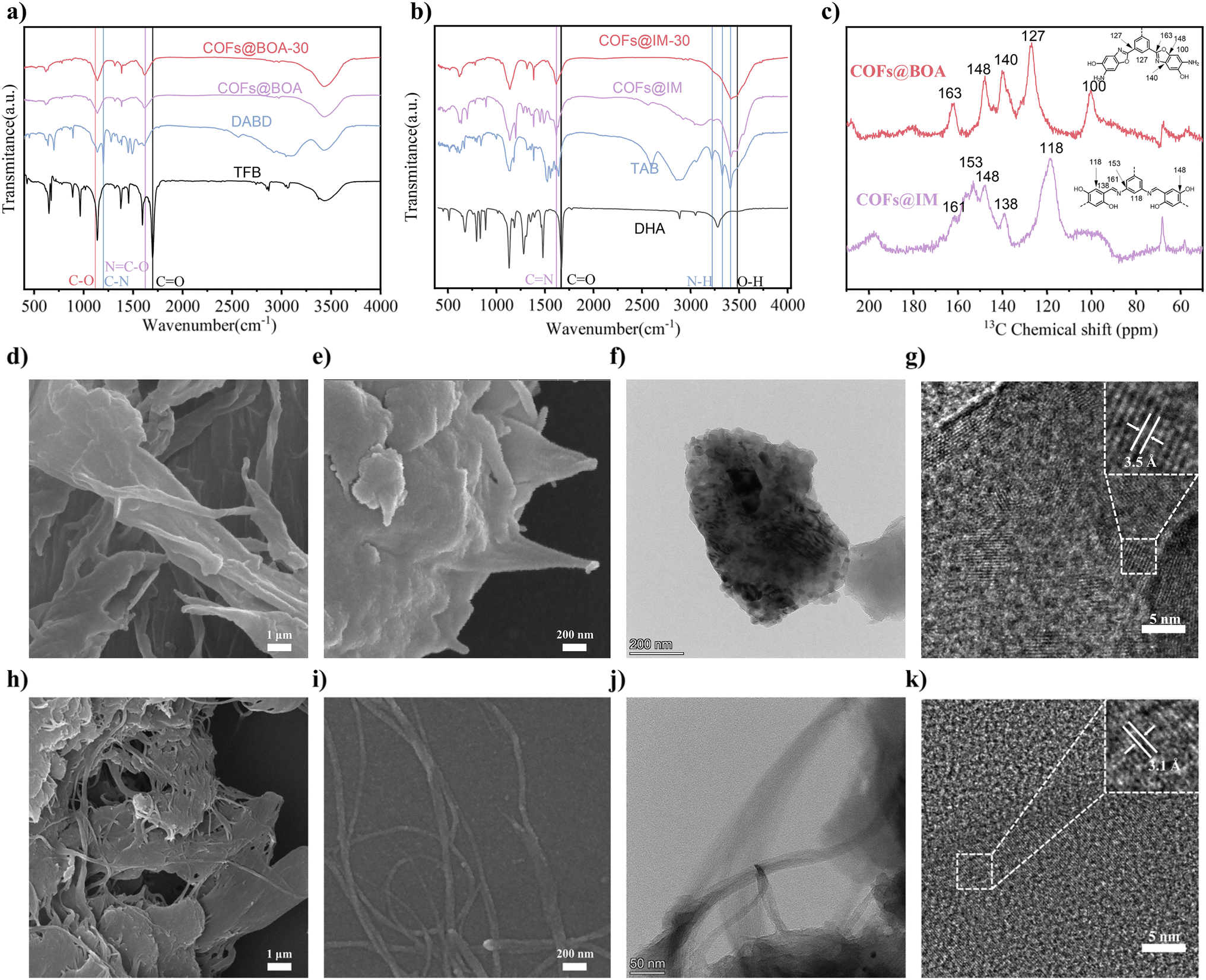 | ||
| Fig. 2 (a) FT-IR spectra of TFB, DABD, COFs@BOA and COFs@BOA-30. (b) FT-IR spectra of DHA, TAB, COFs@IM and COFs@IM-30. (c) 13C CP/MAS NMR spectra of COFs@BOA and COFs@IM. (d and e) SEM images of COFs@BOA-30. (f and g) TEM image of COFs@BOA-30. (h and i) SEM images of COFs@IM-30. (j and k) TEM image of COFs@IM-30. | ||
X-ray diffraction (XRD) patterns were obtained to analyze the crystallinities of COFs@BOA, COFs@IM, and their CNT composites. As shown in Fig. 1b and S1,† the observed XRD pattern of COFs@BOA exhibited characteristic peaks at 4.8°, 8.5°, 9.8°, 12.9° and 26.3°, corresponding to the 100, 110, 200, 210 and 001 reflections, respectively, as determined through computational simulation and Pawley refinement. Similarly, the experimental XRD pattern of COFs@IM (Fig. 1c and S2†) showed five peaks at 4.5°, 8.1°, 9.1°, 12.3° and 26.2°, which exposed planes with 100, 110, 200, 210 and 001 reflections, respectively. Computational simulation and Pawley refinement were used to reconstruct the optimized theoretical structures. The Pawley-refined XRD patterns were in good agreement with the experimental data, as confirmed by the small residual values (Fig. 1b, COFs@BOA: Rp = 3.00% and Rwp = 3.79%; Fig. 1c, COFs@IM: Rp = 3.86% and Rwp = 4.89%). Through refinement, it was verified that both structures of the two COFs satisfied AA stacking (Fig. 1d and e). All of the results revealed the optimized unit cell parameters of COFs@BOA (a = b = 20.9018 Å, c = 3.4460 Å, α = β = 90°, and γ = 120°) and COFs@IM (a = b = 22.9668 Å, c = 3.4304 Å, α = β = 90°, and γ = 120°), both belonging to the P6/M space group. In addition, both COF composites exhibited some characteristic peaks of corresponding pure COFs in the XRD of COFs@BOA composites (Fig. S1†) and COFs@IM composites (Fig. S2†). Hence, the two COFs with different ratios of CNT composites were successfully fabricated. Subsequently, the specific surface area of the pure COFs, their composites, and CNTs was characterized by the N2 adsorption/desorption analysis. Fig. S4–S8† show the N2 sorption/desorption isotherms of both COFs, CNTs, and their composites, and the Brunauer–Emmett–Teller (BET) surface area (Table S2†) can be calculated to be 809 m2 g−1 (COFs@BOA), 57 m2 g−1 (COFs@IM), 1300 m2 g−1 (CNTs), 445 m2 g−1 (COFs@BOA-30) and 155 m2 g−1 (COFs@IM-30), respectively. As for COFs@BOA-30, it exhibited a smaller surface area than COFs@BOA and CNTs, possibly because COFs@BOA may block the pores of CNTs, leading to the obstruction of some pores. However, COFs@IM-30 displayed a larger surface area than pure COFs@IM, which may be caused by the large surface area of CNTs, indicating that COFs@IM was dispersed on the surface of the CNTs. Both pure COFs displayed fine thermal stability performance in the thermogravimetric analysis (TGA) test (Fig. S9†). However, their CNT composites exhibited better thermal stability performance than the corresponding COFs, which can be attributed to the excellent thermal stability of CNTs. In addition, the electrical conduction measurements showed that the electrical conductivity of COFs@IM (9.85 × 10−7 S m−1) was about 8.5 times much higher than that of COFs@BOA (1.16 × 10−7 S m−1), which was consistent with theoretical calculations.
The microstructures of the materials were extensively examined using scanning electron microscopy (SEM) and high-resolution transmission electron microscopy (HRTEM). As depicted in Fig. S10 and S11†, both COFs@BOA and COFs@IM consisted of nanoparticles with sizes of approximately 1 μm. As observed in Fig. 2d, e, h and i, both COFs exhibited better growth on the exterior of the CNT fiber, as evidenced by SEM images. COFs@IM-30 and COFs@BOA-30 displayed rough tubular structures similar to pure CNTs (Fig. S12†). Furthermore, HRTEM images of COFs@BOA (Fig. S13†) and COFs@IM (Fig. S15†) revealed distinct lattice fringes with lattice distances of 0.36 nm and 0.31 nm, respectively, which approximately matched the interplanar distances of both COFs as determined by computational simulation and the XRD results. The corresponding elemental mapping results showed an even distribution of the primary constituent elements, C, N, and O, in both pure COF samples (Fig. S14 and S16†). In the case of COF composites, the interplanar distances of COFs@BOA-30 (Fig. 2g) and COFs@IM-30 (Fig. 2k) were measured to be 0.35 nm and 0.31 nm, respectively. Both interplanar distances were approximately consistent with those of the corresponding pure COFs. Correspondingly, elemental mapping confirmed the even distribution of C, N, and O in the composites (Fig. S17 and S18†). Therefore, the above analyses indicate successful synthesis of COFs@BOA, COFs@IM, and their composites with fine crystallinity, which may be beneficial for the diffusion and migration of Li ions.
3.2 Electrochemical properties
After confirming the structure of COFs and their composites, their electrochemical performances were evaluated within a voltage range of 0.005–3.0 V using CR2032 coin cells. Cyclic voltammetry (CV) with a scan rate of 0.1 mV s−1 was initially employed to analyze the lithiation/delithiation behavior of the as-prepared electrode materials. From Fig. S20,† it can be observed that the first scan of COFs@BOA-30 exhibited a strong cathodic peak at 0.28 V during the negative scan, attributed to the irreversible reduction formed by the solid electrochemical interphase (SEI).27 The cathodic peak/anodic peak pair at 0.01/0.13 V could be attributed to the reversible lithiation/delithiation behavior derived from the benzene ring.2 In the subsequent three cycles, overlapped CV curves were observed, indicating excellent reversibility and cyclability of COFs@BOA-30.28 Similarly, for COFs@IM-30 (Fig. S24†), similar phenomena, including the first scan and the subsequent three scans, were observed, indicating that COFs@IM-30 also formed the SEI and exhibited sophisticated reversibility and cyclability. In addition, COFs@IM, COFs@IM-15, COFs@IM/30, COFs@BOA, COFs@BOA-15, and COFs@BOA/30 (Fig. S21–S27†) also had the process of forming the SEI and the overlapping CVs in the second and fourth laps, but COFs@IM-30 and COFs@BOA-30 showed higher current intensity than relevant pure COFs and their other composites, which revealed the high performance of COFs@IM-30 and COFs@BOA-30 to some extent.To investigate the specific capacities of both COFs and their CNT composites, galvanostatic charge–discharge (GCD) tests were implemented at a current density of 100 mA g−1. COFs@BOA-30 displayed initial discharge/charge capacities of 1510.6/777.3 mA h g−1 with a corresponding coulombic efficiency (CE) of 51.5% (Fig. S28†). The irreversible capacity in the first cycle was attributed to the formation of the SEI along with electrolyte decomposition, a common phenomenon in organic anodes.29 For COFs@IM-30, the initial discharge/charge capacities of 1687.3/821.3 mA h g−1 with a corresponding CE of 48.7% were obtained (Fig. 3a). The discharge–charge profiles of COFs@BOA-30 (Fig. S28†) showed a capacity decrease to 703.2 mA h g−1, followed by an increase to 834.8 mA h g−1 and finally stabilizing at a current density of 100 mA g−1. And the CE displayed a different trend, initially increasing and then stabilizing at about 100%. COFs@BOA-30 exhibited fine performance, whereas COFs@BOA-15 (Fig. S29†) (436 mA h g−1), pure COFs@BOA (Fig. S30†) (352 mA h g−1), and COFs@BOA/30 (Fig. S31†) (516 mA h g−1) displayed unsatisfactory performance at the same current density. Similar phenomena were observed in COFs@IM-30. Although the capacity of COFs@IM-30 followed a similar trend to COFs@BOA-30, it had a higher capacity of 1100.3 mA h g−1 (Fig. 3a). Similarly, COFs@IM-30 also showed better performance than COFs@IM (Fig. S32†) (474 mA h g−1), COFs@IM-15 (Fig. S33†) (494 mA h g−1), and COFs@IM/30 (Fig. S34†) (554 mA h g−1). In addition, COFs@IM-30 displayed the fine performance of Li-ion storage, comparable to other reported COF-based electrodes (Table S3†). Pure CNTs exhibited low capacities of 616.7 mA h g−1 (Fig. S35†). Thus, we infer that CNTs played a significant role in the COF-based electrode, likely due to their excellent conductivity. Subsequently, the rate performances of the prepared materials were examined at various current densities. As presented in Fig. 3b and S36–S39,† COFs@IM-30 showed the most satisfactory rate performance among the pure COFs, CNT, COFs@IM-15, and their composites. COFs@IM-30 delivered high reversible capacities of 823.5, 737.9, 615.9, 504.6 and 411.2 mA h g−1 at 100, 200, 500, 1000, and 2000 mA g−1, respectively. When the current density decreased to 100 mA g−1, the specific capacities of COFs@IM-30 increased to 916.0 mA h g−1, demonstrating satisfactory stability and fine electrochemical properties.30 COFs@BOA-30 (Fig. S40†) showed slightly lower capacities of 809.3, 651.8, 531.4, 418 and 329.1 mA h g−1 at 100, 200, 500, 1,000, and 2000 mA g−1, respectively, compared with COFs@IM-30. But COFs@BOA-30 was better than CNTs, COFs@BOA-15, COFs@BOA, and COFs@BOA/30 (Fig. S41–S43†). Meanwhile, the pure COFs and CNT electrodes exhibited relatively low capacity and cycling stability at the corresponding current densities, indicating that the fine rate performance of the CNT-composited electrode may be related to the excellent conductivity of CNTs. The long-term cycle performance of COFs@IM-30, COFs@BOA-30, COFs@IM-15, COFs@BOA-15, pure COFs@IM, pure COFs@BOA, COFs@IM/30, COFs@BOA/30, and CNTs was also evaluated at 100 mA g−1. COFs@IM-30 exhibited a high initial capacity of 1687.3 mA h g−1, followed by a capacity decay to 932.5 mA h g−1 after 7 cycles, and then increasing to about 1100 mA h g−1 by the 120th cycle. A similar phenomenon occurred with COFs@BOA-30. This phenomenon was ascribed to the electrode-activated procedure mainly caused by the low electronic conductivity of these materials. During repeated Li-ion insertion/extraction, the SEI film stabilized, and the inner action sites of COFs were gradually exposed and activated, resulting in a capacity increase.31 The increasing capacity usually results from the improved Li-ion diffusion kinetics during repetitive cycling and can be observed in other COF anodes.32 However, COFs@IM-30 had the highest capacity of 1100.3 mA h g−1 among the prepared electrodes in long-cycle testing, approximately 130% of the specific capacity of COFs@BOA-30. This difference may be related to the different linkages of the related COFs. In particular, the CE of COFs@BOA-30 and COFs@IM-30 anodes were notably close to 100% throughout the cycle process except for the first few cycles, further highlighting the highly reversible lithiation and delithiation process. However, the initial capacity of pure COFs@IM and COFs@BOA was only about 100 mA h g−1, attributed to the low conductivity of pure COFs. Therefore, we investigated the composite capacity of COFs@CNT, which can be regarded as the contribution from the two components of COFs and CNTs.27,29 CNTs exhibited a reversible capacity of about 600 mA h g−1 without substantial capacity fading during long-term cycling. Thus, the capacity contribution from COFs in the composite can be calculated based on the weight ratio of COFs and CNTs, attainable from the elemental analysis. A large reversible capacity contribution of 1696.0 mA h g−1 was achieved for COFs@IM in COFs@IM-30 (Fig. 3c), even higher than the theoretical capacities of traditional inorganic anodes (700–1000 mA h g−1) such as transitional metal oxides (CuO, Fe2O3, Co3O4, NiO, etc.).33 However, COFs@BOA only had a reversible capacity contribution of 925.8 mA h g−1 in COFs@BOA-30 (Fig. S44†). These differences may be attributed to the varying yields and the intrinsic properties of the COFs, with the latter being the primary factor.
 | ||
| Fig. 3 (a) Cycling performances of the COFs@IM-30 electrode at a current density of 100 mA g−1. (b) The rate capability of the COFs@IM-30 electrode at different current densities. (c) Capacity contribution of COFs@IM (based on the mass of COFs) in COFs@IM-30 at 100 mA g−1. (d) The EIS of COFs@BOA-30, COFs@IM-30, COFs@BOA-15, COFs@IM-15, COFs@IM, COFs@BOA, and CNTs. (e) Capacitive and diffusion contribution of the COFs@IM-30 anode at multiple scan rates of 0.2–1.0 mV s−1. (f) GITT curve and diffusion coefficient of the COFs@IM-30 electrode. | ||
The kinetics evolutions of COFs@BOA-30 and COFs@IM-30 were investigated by electrochemical impedance spectroscopy (EIS). Fig. 3d presents the EIS curves of the initial CNT composite electrode, CNTs, and pure COFs. The solution resistance (Rs) and charge transfer resistance (Rct) of both COFs@BOA-30 and COFs@IM-30 were lower than those of the pure COFs and COFs with 15% CNTs,34 suggesting the rapid and stable reaction kinetics.35 Between the four COF composites, COFs@IM-30 exhibited smaller resistance, indicating better conductivity. Conversely, both pure COFs displayed larger Rct, likely due to their lower conductivity. Subsequently, the reaction kinetics of COF composites were further analyzed using the CVs at various scan rates (Fig. S45–S52†). The relation between peak current (i) and the scan rate (v) followed the equation: i = avb (a and b were adjustable parameters) while b = 1 and 0.5 indicate capacitive- and diffusion-controlled processes, respectively.36 For the reduction/oxidation peak of COFs@IM-30 (Fig. S53†), the b values were counted to be 0.55 and 0.96, respectively, while for COFs@BOA-30 (Fig. S54†), the b values of reduction/oxidation peaks were 0.62 and 0.94, respectively. This evidence suggests that the charge storage of both COF composites involves a combination of Li-ion diffusion and capacitance-controlled processes. Fig. 3e, S55 and S56† display the capacitance and diffusion-controlled contributions of COFs@IM-30, COFs@BOA-30, and other prepared anodes, respectively. With an increasing scanning rate, the capacitive contribution of both COF composites gradually increased, attributed to the fast dynamics mainly occurring during capacitor storage, explaining the electrode's higher rate performance and longer cycle life.37 When the scan rate increased to 1 mV s−1, the capacitive contribution was approximately 83% for COFs@IM-30 and 91% for COFs@BOA-30. In addition, Li-ion diffusion coefficients were investigated for the COFs@BOA-30 and COFs@IM-30 electrodes using the galvanostatic intermittent titration technique (GITT).38 The Li-ion diffusion coefficient of COFs@IM-30 (Fig. 3f) was slightly higher than that of COFs@BOA-30 (Fig. S57†), possibly due to differences in COFs' linkage and varied yield of COFs. Overall, COFs@IM-30 exhibited faster kinetics than COFs@BOA-30 and other prepared anodes.
3.3 The mechanism of charge storage in COF composites
Ex situ X-ray photoelectron spectroscopy (XPS) analysis was conducted to investigate the reaction mechanism of the COF composite-based electrode. For COFs@IM-30 (Fig. S58†), the high-resolution C 1s XPS peak of the initial electrode could be resolved into several major components: CC (284.2 eV), C–C (284.8 eV), C–O/C–N (285.9 eV), CN (287.6 eV), CO (288.8 eV), and π–π* (290.0 eV).28 The N 1s spectra of the initial COFs@IM-30 showed peaks for C–N and CN, while the O 1s spectra exhibited peaks for CO and C–O, with no sign of Li in the Li 1s spectra. Upon discharge to 0.005 V, changes were observed in the XPS spectra: the intensity of the CC peak decreased while that of C–C increased, along with enhancement in the C–O/C–N peak. This suggests the involvement of CC and CN in Li-ion storage, as supported by changes in the N 1s and C 1s spectra. In the O 1s spectra, the appearance of the O–Li peak and the absence of the CO peak, alongside the enhanced C–O peak, indicate the insertion of Li ions by CO. The involved CO may be formed during the electrochemical oxidation of oxhydryl.39 The Li 1s spectra provide evidence of reversible redox reactions involving Li–C, Li–O, Li–N, Li–Nx, and Li–Cx, corresponding to CN, CO, and CC bonds.40 Upon charging to 3.0 V, the XPS spectra resembled those of the initial anode material, with significant enhancement in CC, CN, and CO peaks and reduction in C–C, C–O, and C–N peaks, confirming the involvement of CC, CN, and CO bonds in Li-ion insertion/de-insertion. Based on this XPS information and the contribution of COFs@IM, the lithiation/delithiation mechanism of COFs@IM in the composite is proposed (Fig. 4a). For COFs@BOA-30 (Fig. S59†), the C 1s spectra showed a similar trend to those of COFs@IM-30. Upon discharge to 0.005 V, the peaks corresponding to CC and CN decreased while the corresponding peaks increased. Upon charging to 3.0 V, the CC and CN peaks returned to their initial state, indicating reversible Li-ion restoration. However, the C–O–C peak did not appear, possibly due to its instability, while the Li–O peak persisted, likely related to the SEI. The possible redox mechanism of COFs@BOA in the composite is illustrated in Fig. S60.†
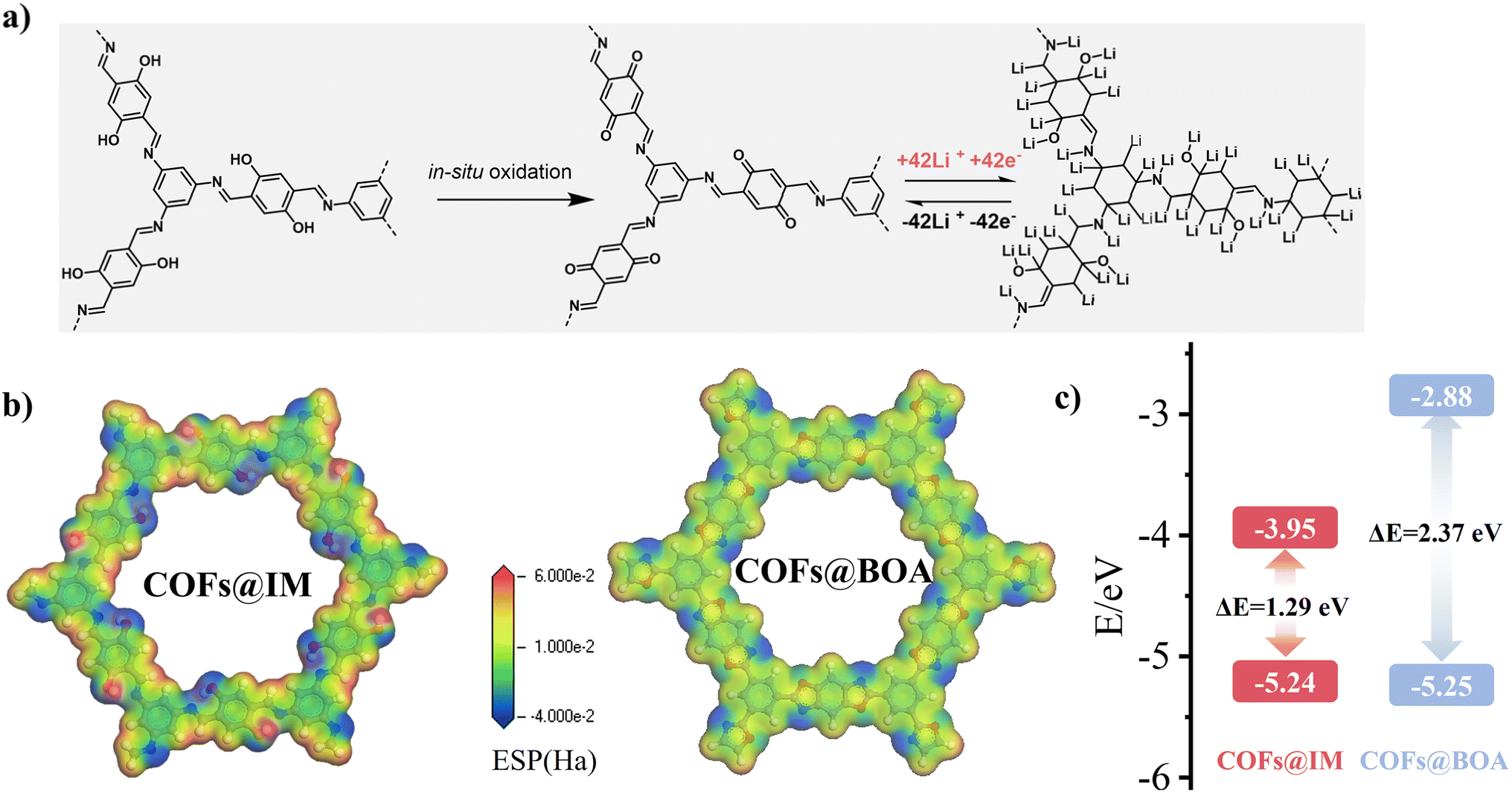 | ||
| Fig. 4 (a) Structural evolution of COFs@IM during the lithiation/delithiation procedure. (b) The MESP of COFs@IM and COFs@BOA. (c) The simulated HOMO and LUMO energy levels of COFs@IM and COFs@BOA. | ||
Density functional theory (DFT) was employed to analyze the intrinsic characteristics of COFs with varied linkages. Initially, the molecular electrostatic potential (MESP) of both COFs (Fig. 4b) was depicted to reveal potential Li-ion storage sites. The equivalent minima of MESP areas were located around the oxygen and nitrogen atoms of both frameworks, suggesting their roles as redox-active centers for Li-ion storage in COFs@BOA and COFs@IM.41 However, a comparison of the MESP indicated that the electrostatic potential of COFs@IM was more negative, theoretically making it easier to store Li ions compared to COFs@BOA.39 Moreover, the LUMO–HOMO gaps of COFs@IM (1.29 eV) were smaller than those of COFs@BOA (2.37 eV) (Fig. 4c), suggesting better electronic conductivity of COFs@IM.42 Additionally, the LUMO of COFs@IM was lower than that of COFs@BOA, indicating that COFs@IM facilitates electro-activity as an anode and has high working potentials.43,44 Furthermore, DFT calculations for the energy gap (Eg) revealed that the energy level of COFs@BOA was 2.397 eV (Fig. S61†), while COFs@IM obtained a relatively lower level of 1.783 eV (Fig. S62†), highlighting the high electron conductivity and enhanced redox process of COFs@IM.45
4 Conclusion
In this study, we employed linkage engineering to utilize two groups of similar monomers to prepare two COFs with different linkages (oxazole and imine), namely COFs@BOA and COFs@IM. Due to the inherent semiconductor properties of COFs, composites were formed by incorporating CNTs through in situ growth. Electrochemical measurements demonstrated that COFs@BOA-30 and COFs@IM-30 exhibited excellent Li-ion storage performance, attributed to their enhanced conductivity compared to other prepared anodes. The COFs@BOA-30 anode for lithium batteries achieved a reversible capacity of 853.1 mA h g−1 at 100 mA g−1, while COFs@IM-30 reached 1100.3 mA h g−1 under the same conditions. The capacity contribution of COFs@BOA-30 was 925.8 mA h g−1, whereas COFs@IM-30 demonstrated a higher capacity contribution of 1696.0 mA h g−1 in the COF composite. This difference may be attributed to the different yields and internal properties of the COFs. DFT calculations revealed that COFs@IM exhibited superior conductivity compared to COFs@BOA. Given that both COFs only differ in their linkage, it can be inferred that the imine linkage is more suitable for constructing COF-based anodes than the oxazole linkage. Therefore, this work may contribute to advancing COF-based materials for energy storage and conversion applications.Data availability
Data supporting the findings of this study are available from the corresponding authors upon reasonable request.Author contributions
J. G. L. supervised and designed the research. C. Y. W. performed the experiments and data analysis. L. L. M. performed useful discussion. C. Y. W. wrote the original paper. J. G. L. revised and corrected the manuscript. All authors discussed the results and assisted during manuscript preparation.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (51976225 and 52236010) and the Fundamental Research Funds for the Central Universities (2242022R10058).References
- G. Harper, R. Sommerville, E. Kendrick, L. Driscoll, P. Slater, R. Stolkin, A. Walton, P. Christensen, O. Heidrich, S. Lambert, A. Abbott, K. Ryder, L. Gaines and P. Anderson, Nature, 2019, 575, 75–86 CrossRef CAS PubMed
.
- J. Wu, X. Rui, G. Long, W. Chen, Q. Yan and Q. Zhang, Angew. Chem., Int. Ed., 2015, 54, 7354–7358 CrossRef CAS PubMed
- X. Shen, R. Zhang, X. Chen, X. B. Cheng, X. Li and Q. Zhang, Adv. Energy Mater., 2020, 10, 1903645 CrossRef CAS
- Z. Luo, L. Liu, J. Ning, K. Lei, Y. Lu, F. Li and J. Chen, Angew. Chem., Int. Ed., 2018, 57, 9443–9446 CrossRef CAS PubMed
- Y. Tong, Z. Sun, J. Wang, W. Huang and Q. Zhang, SmartMat, 2022, 3, 685–694 CrossRef CAS
- L. Sieuw, A. Jouhara, É. Quarez, C. Auger, J.-F. Gohy, P. Poizot and A. Vlad, Chem. Sci., 2019, 10, 418–426 RSC
- P. Li, S. Luo, L. Zhang, Q. Liu, Y. Wang, Y. Lin, C. Xu, J. Guo, P. Cheali and X. Xia, J. Energy Chem., 2024, 89, 144–171 CrossRef CAS
- T. Sun, J. Xie, W. Guo, D. S. Li and Q. Zhang, Adv. Energy Mater., 2020, 10, 1904199 CrossRef CAS
- Y. Lu and J. Chen, Nat. Rev. Chem, 2020, 4, 127–142 CrossRef CAS
- Q. Zhang, Y. Dou, Q. He, S. Deng, Q. Huang, S. Huang and Y. Yang, Energy Environ. Mater., 2022, 5, 1037–1059 CrossRef CAS
- X. Cui, J. Chen, Z. Sun, L. Wang, Q. Peng, B. Xiao, L. Zhao, H. Zheng, Y. Wang, J. Wang, X. Chen, Q. Zhang and S. Chen, Adv. Funct. Mater., 2023, 33, 2212100 CrossRef CAS
- A. Wang, R. Tan, C. Breakwell, X. Wei, Z. Fan, C. Ye, R. Malpass-Evans, T. Liu, M. A. Zwijnenburg, K. E. Jelfs, N. B. McKeown, J. Chen and Q. Song, J. Am. Chem. Soc., 2022, 144, 17198–17208 CrossRef CAS PubMed
- R. Liu, K. T. Tan, Y. Gong, Y. Chen, Z. Li, S. Xie, T. He, Z. Lu, H. Yang and D. Jiang, Chem. Soc. Rev., 2021, 50, 120–242 RSC
- K. Geng, T. He, R. Liu, S. Dalapati, K. T. Tan, Z. Li, S. Tao, Y. Gong, Q. Jiang and D. Jiang, Chem. Rev., 2020, 120, 8814–8933 CrossRef CAS
- J. L. Segura, M. J. Mancheno and F. Zamora, Chem. Soc. Rev., 2016, 45, 5635–5671 RSC
- S. Haldar, A. Schneemann and S. Kaskel, J. Am. Chem. Soc., 2023, 145, 13494–13513 CrossRef CAS PubMed
- L. Zhou, S. Jo, M. Park, L. Fang, K. Zhang, Y. Fan, Z. Hao and Y. M. Kang, Adv. Energy Mater., 2021, 11, 2003054 CrossRef CAS
- K. Roy, A. Banerjee and S. Ogale, ACS Appl. Mater. Interfaces, 2022, 14, 20326–20348 CrossRef CAS
- B. Zhang, W. Wang, L. Liang, Z. Xu, X. Li and S. Qiao, Coord. Chem. Rev., 2021, 436, 213782 CrossRef CAS
- J. Sun, Y. Xu, Y. Lv, Q. Zhang and X. Zhou, CCS Chem., 2023, 5, 1259–1276 CrossRef CAS
- D. A. Pyles, J. W. Crowe, L. A. Baldwin and P. L. McGrier, ACS Macro Lett., 2016, 5, 1055–1058 CrossRef CAS
- X. Li, S. Yang, M. Liu, X. Yang, Q. Xu, G. Zeng and Z. Jiang, Angew. Chem., Int. Ed., 2023, 62, e202304356 CrossRef CAS
- J. Yang, A. Acharjya, M. Y. Ye, J. Rabeah, S. Li, Z. Kochovski, S. Youk, J. Roeser, J. Grüneberg, C. Penschke, M. Schwarze, T. Wang, Y. Lu, R. van de Krol, M. Oschatz, R. Schomäcker, P. Saalfrank and A. Thomas, Angew. Chem., Int. Ed., 2021, 60, 19797–19803 CrossRef CAS
- P.-F. Wei, M.-Z. Qi, Z.-P. Wang, S.-Y. Ding, W. Yu, Q. Liu, L.-K. Wang, H.-Z. Wang, W.-K. An and W. Wang, J. Am. Chem. Soc., 2018, 140, 4623–4631 CrossRef CAS
- S. An, X. Li, S. Shang, T. Xu, S. Yang, C. X. Cui, C. Peng, H. Liu, Q. Xu, Z. Jiang and J. Hu, Angew. Chem., Int. Ed., 2023, 62, e202218742 CrossRef CAS
- C. Gao, J. Li, S. Yin, J. Sun and C. Wang, Nat. Commun., 2020, 11, 4919 CrossRef CAS
- J. Zhao, M. Zhou, J. Chen, L. Wang, Q. Zhang, S. Zhong, H. Xie and Y. Li, Small, 2023, 19, e2303353 CrossRef
- Y. Zhang, Y. Wu, Y. An, C. Wei, L. Tan, B. Xi, S. Xiong and J. Feng, Small Methods, 2022, 6, e2200306 CrossRef
- H. Kang, H. Liu, C. Li, L. Sun, C. Zhang, H. Gao, J. Yin, B. Yang, Y. You, K. C. Jiang, H. Long and S. Xin, ACS Appl. Mater. Interfaces, 2018, 10, 37023–37030 CrossRef CAS PubMed
- L. Bai, Q. Gao and Y. Zhao, J. Mater. Chem. A, 2016, 4, 14106–14110 RSC
- Q. Geng, Z. Xu, J. Wang, C. Song, Y. Wu and Y. Wang, Chem. Eng. J., 2023, 469, 143941 CrossRef CAS
- X. Li, W. Liu, Y. Wang, L. Lv, H. Feng, W. Huang, Y. Sun, W. Xiong and H. Zheng, Chem. Eng. J., 2023, 473, 145310 CrossRef CAS
- Z. Lei, Q. Yang, Y. Xu, S. Guo, W. Sun, H. Liu, L. P. Lv, Y. Zhang and Y. Wang, Nat. Commun., 2018, 9, 576 CrossRef PubMed
- P. Wang, J. Bai, K. Li, H. Ma, W. Li, X. Zhu, Y. Sun and B. Zhao, Chem. Eng. J., 2021, 425, 130607 CrossRef CAS
- H. Zhang, W. Sun, X. Chen and Y. Wang, ACS Nano, 2019, 13, 14252–14261 CrossRef CAS PubMed
- T. Brezesinski, J. Wang, S. H. Tolbert and B. Dunn, Nat. Mater., 2010, 9, 146–151 CrossRef CAS
- J. Wu, J. Liu, Z. Wang, X. Gong, M. Qi and Y. Wang, J. Mater. Chem. A, 2019, 7, 11347–11354 RSC
- Y.-C. Chien, H. Liu, A. S. Menon, W. R. Brant, D. Brandell and M. J. Lacey, Nat. Commun., 2023, 14, 2289 CrossRef CAS PubMed
- Z. Xiong, L. Gu, Y. Liu, H. Wang, L. Shi, X. Wu, L. Liu and Z. Chen, CCS Chem., 2024, 1–10, DOI:10.31635/ccschem.024.202403899
- S. Haldar, K. Roy, S. Nandi, D. Chakraborty, D. Puthusseri, Y. Gawli, S. Ogale and R. Vaidhyanathan, Adv. Energy Mater., 2018, 8, 1702170 CrossRef
- X. Xu, S. Zhang, K. Xu, H. Chen, X. Fan and N. Huang, J. Am. Chem. Soc., 2023, 145, 1022–1030 CrossRef CAS
- Y. Cao, H. Fang, C. Guo, W. Sun, Y. Xu, Y. Wu and Y. Wang, Angew. Chem., Int. Ed., 2023, 62, e202302143 CrossRef CAS
- Z. Song, H. Zhan and Y. Zhou, Angew. Chem., Int. Ed., 2010, 49, 8444–8448 CrossRef CAS
- Y. Zhang, J. Wang and S. N. Riduan, J. Mater. Chem. A, 2016, 4, 14902–14914 RSC
- Z. Yang, Y. Song, X. Ren, C. Zhang, X. Hu, X. Li, K. Wang, J. Li and C. Huang, Carbon, 2021, 182, 413–421 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta06789d |
| This journal is © The Royal Society of Chemistry 2025 |