
High-performance, ambient-processable organic solar cells achieved by single terpene-based entirely eco-friendly process†
Hyerin
Jeon
a,
Jin-Woo
Lee
a,
Kihyun
Bae
a,
Tan
Ngoc-Lan Phan
a,
Chulhee
Lim
 a,
Jaeyoung
Choi
a,
Cheng
Wang
b,
Seungjin
Lee
*c and
Bumjoon J.
Kim
*a
a,
Jaeyoung
Choi
a,
Cheng
Wang
b,
Seungjin
Lee
*c and
Bumjoon J.
Kim
*a
aDepartment of Chemical and Biomolecular Engineering, Korea Advanced Institute of Science and Technology (KAIST), Daejeon, 34141, Republic of Korea. E-mail: bumjoonkim@kaist.ac.kr
bAdvanced Light Source, Lawrence Berkeley National Laboratory, 1 Cyclotron Road, Berkeley, CA 94720, USA
cPhotoenergy Research Center, Korea Research Institute of Chemical Technology (KRICT), Daejeon, 34114, Republic of Korea. E-mail: 332sjin@krict.re.kr
First published on 13th November 2024
Abstract
Conventional processing solvents for organic electronics pose significant health/environmental risks, prompting the search for greener/safer alternatives. Herein, we develop organic solar cells (OSCs), processed from a single terpene solvent, eucalyptol (Eu), with almost no environmental hazards and toxicity. Notably, a record-high power conversion efficiency (PCE) of 15.1% is achieved without any additive, which is particularly significant given the low PCEs (0.1–3.0%) of previous OSCs using a single terpene. First, we design a small-molecule acceptor (MYBO) with optimized side-chains, offering sufficient solubility while maintaining excellent optoelectronic properties. Second, we develop a processing technique which controls the film-formation kinetics to independently tune the aggregation of polymer donor and MYBO. This enables the formation of well-developed MYBO crystallites embedded within interconnected polymer fibrillar structures. And, all the solution processing can be performed in air without using a glove box, thanks to the eco-friendly Eu process. The devices also exhibit excellent air-stability, retaining more than 92% of the initial PCE after 2300 h in air. This work provides important guidelines for material designs and processing methods to achieve eco-friendly processed, high-performance OSCs.
Introduction
Organic solar cells (OSCs) have attracted considerable attention as promising renewable energy sources due to their advantageous properties of being lightweight, semi-transparent, flexible/stretchable, and solution-processable.1–3 Over the past few years, significant advancements in power conversion efficiencies (PCEs) of OSCs have been proceeded, with PCEs reaching 19–20% through the development of non-fullerene small molecule acceptors (SMAs).4–13 Typically, the active layer of high-performance SMA-based OSCs is processed using halogenated solvents (e.g., chloroform (CF) and chlorobenzene), which are effective in achieving an optimal blend film morphology with sufficient solubility. However, the use of halogenated solvents poses major reproductive/carcinogenic hazards to humans and eco-toxicological risks, restricting their use in commercial applications.14–16 Thus, non-halogenated aromatic solvents (e.g., o-xylene and toluene) have been widely employed as alternatives to toxic halogenated solvents.17–25 While these solvents offer more sustainable production conditions compared to the halogenated ones, their aromatic structures still present environmental challenges, which limit their application in industry. For instance, the toxics release inventory (TRI) program tracks the industrial management of aromatic solvents including o-xylene and toluene, with a 1.0% de minimis threshold.26 Therefore, eliminating harmful halogenated or aromatic solvents in industrial processes is essential and requires immediate attention.In this context, terpenes, a group of natural organic compounds produced by various plants and fruit peels, have recently garnered great attention for their potential applications as a new class of processing solvents.27 It is noteworthy that this solvent category, including limonene (LM) and eucalyptol (Eu), offers almost no environmental hazards and toxicity. In addition, these solvents have a lower carbon footprint than typical organic solvents. For instance, LM, derived from lemon and orange peels, is commonly used to produce a citrus scent in food, air fresheners, and cosmetics.28 Also, Eu, the primary compound of eucalyptus essential oil, is used in mouthwash and medicines.29,30 Given these environmental benefits, a few research groups are actively exploring the application of terpene solvents in the fabrication of OSCs. For example, the Ade group employed a sequential deposition method to fabricate bilayer OSCs using LM and 2-methyltetrahydrofuran (2-MeTHF) as a pair of solvents for FTAZ donor and IT-M SMA, respectively. In this study, LM enhanced the molecular ordering of FTAZ, leading to an impressive PCE of 12.5%.31 More recently, the Baran group successfully demonstrated that incorporating tetralin, indan, or ethyl phenyl sulfide into terpene solvents can effectively improve the solubility and OSC performance; as a result, the optimal Eu![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :tetralin ratio (52:48) led to a high PCE of 15.7% for PM6:BTP-eC9-based OSCs.27
:tetralin ratio (52:48) led to a high PCE of 15.7% for PM6:BTP-eC9-based OSCs.27
Despite the significant efforts and advancements made by the researchers, there have been no reports, to date, of high-performance OSC systems employing a single terpene solvent. Indeed, OSCs fabricated thus far with a single terpene solvent exhibited extremely low performance (PCE = 0.1–3.0%).32,33 Thus, the current terpene-based OSCs heavily rely on secondary solvents (e.g., tetralin and 2-MeTHF) as exemplified above, which still present significant safety/environmental concerns for industrial applications. For example, tetralin is a detrimental aromatic solvent with a low median lethal dose (LD50) of 1620 μL kg−1,34 which threatens human health (here, the LD50 value indicates the oral dose of tetralin per unit body weight required to cause the death of half of the tested rat population). In addition, although 2-MeTHF is classified as a representative halogen-free and non-aromatic green solvent, it is a highly flammable liquid with a low flash point (ca. −10 °C) and must be handled with safety protocols in industry.35,36 Thus, the development of OSCs fabricated by a single terpene solvent is in immediate demand. One of the main reasons for the poor performance of single terpene-based OSCs is the low processability of typical SMAs in terpenes due to their highly fused ladder-type backbones and short side chains. This induces strong aggregation and precipitation of SMAs, creating charge trap sites and hindering the reproducibility/scalability of the devices. Furthermore, the exceptionally high boiling point of terpenes (>150 °C) causes prolonged crystallization and severe phase separation during the solvent evaporation process, adversely affecting the charge generation. Therefore, it is imperative to establish SMA design strategies and new processing methods tailored for eco-friendly OSCs based on a single terpene solvent process.
Herein, we develop high-performance, air-stable OSCs using a fully eco-/human-friendly process with a single terpene solvent (Eu). The combination of new photoactive materials and advanced processing techniques enables the creation of eco-friendly OSCs with a record-high PCE of 15.1%, the highest value among all single terpene-based OSCs reported to date. First, we design a new SMA (MYBO: 2,2′-((2Z,2′Z)-((3,9-bis(2-butyloctyl)-12,13-bis(2-octyldodecyl)-12,13-dihydro-[1,2,5]thiadiazolo[3,4-e]thieno[2′′,3′′:4′,5′]thieno[2′,3′:4,5]pyrrolo[3,2-g]thieno[2′,3′:4,5]thieno[3,2-b]indole-2,10-diyl)bis(methaneylylidene))bis(5,6-dichloro-3-oxo-2,3-dihydro-1H-indene-2,1-diylidene))dimalononitrile), featuring optimized alkyl chain structures that promote high solubility in Eu, while maintaining strong SMA intermolecular assembly during Eu processing owing to its strong backbone crystallinity. Poly[(thiophene)-alt-(6,7-difluoro-2-(2-hexyldecyloxy)quinoxaline)] (PTQ10) is selected as the polymer donor for its superior optoelectronic properties, despite its simple structure, providing sufficient processability in Eu. Notably, we meticulously tuned the Eu processing conditions to independently optimize the aggregation structures of MYBO and PTQ10, as well as their quenching times during film formation. This fine-tuned optimization is the key to achieving strong crystalline structures of both MYBO and PTQ10 in the blend with appropriate domain purity and length-scale, leading to high PCEs in the OSCs. We note that the solution processing can be performed in air without using a glove box due to the eco-friendly Eu process without any additive. Moreover, these OSCs exhibit remarkable air stability, maintaining more than 92% of the initial PCE after 2300 h in ambient conditions without encapsulation. In contrast, reference OSCs based on PTQ10:2,2′-((2Z,2′Z)-((12,13-bis(2-ethylhexyl)-3,9-(2-butyloctyl)-12,13-dihydro-[1,2,5]thiadiazolo[3,4-e]thieno[2′′,3′′:4′,5′]thieno[2′,3′:4,5]pyrrolo[3,2-g]thieno[2′,3′:4,5]thieno[3,2-b]indole-2,10-diyl)bis(methanylylidene))bis(5,6-difluoro-3-oxo-2,3-dihydro-1H-indene-2,1-diylidene))dimalononitrile (L8-BO) display very poor performance (PCE = 0.13%) due to the limited solubility of L8-BO in Eu. This research offers valuable insights into material selection and processing strategies essential for boosting the performance of single terpene-based devices, which are pivotal to advancing sustainable OSC technology.
Results and discussion
Basic material characteristics
Terpenes have great potential as environmentally friendly solvents for organic electronics due to their minimal toxicity to living creatures and their production from sustainable, carbon-recycling resources such as plants and fruits (Fig. 1a). Notably, the distinctively high boiling points (BP, >150 °C) of terpene solvents are advantageous for fabricating large-area modules using printing techniques (e.g., bar coating, blade-coating, and slot-die coating), which require much longer processing time for uniform film formation compared to lab-scale spin-coating processes. While several groups have reported high-performance OSCs (PCE >15%) using a co-solvent strategy that combines terpenes with other types of aromatic solvents with high toxicity, OSCs utilizing terpene solvents alone have shown significantly lower PCEs of only 0.1–3.0%.32,33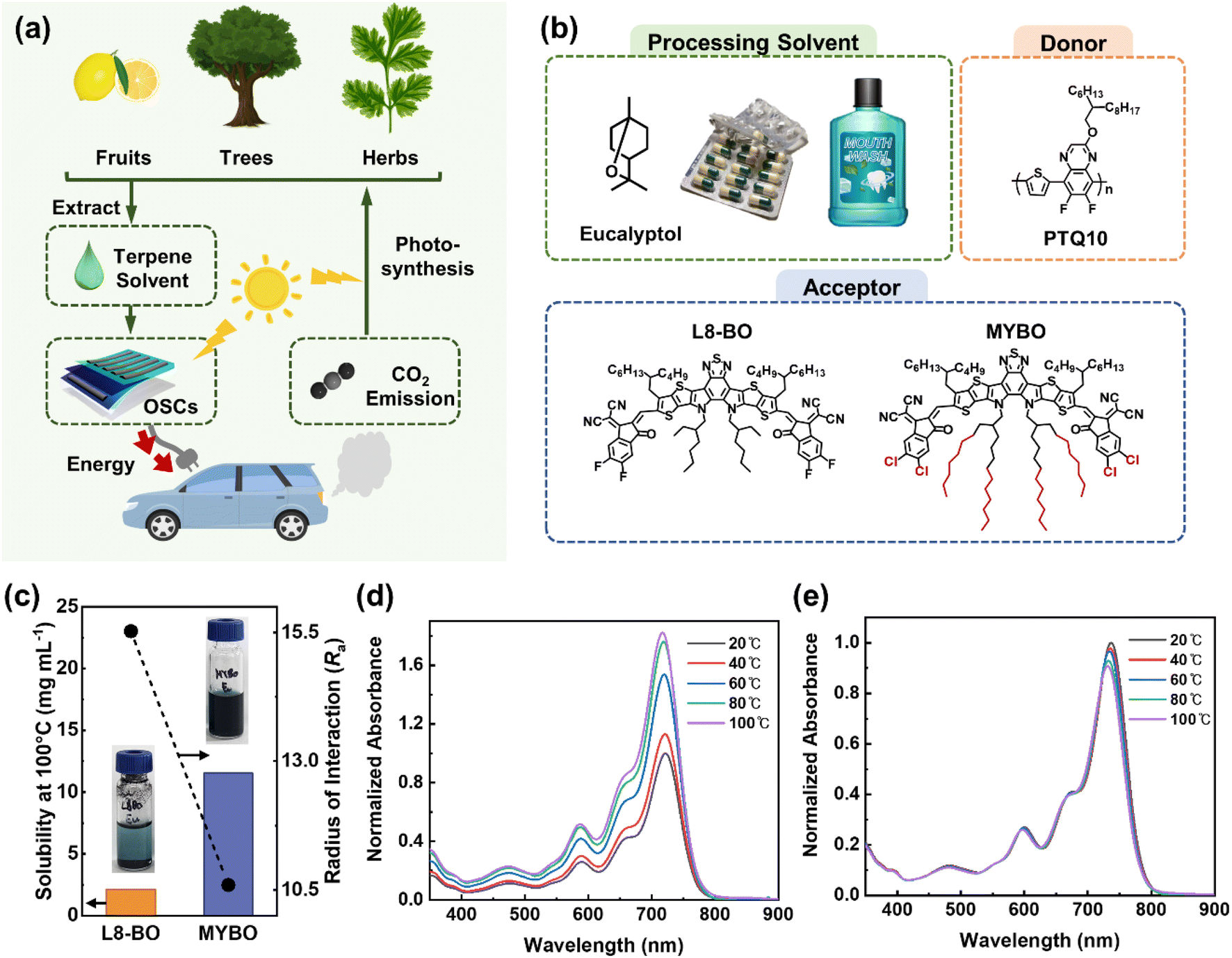 | ||
| Fig. 1 (a) Schematic diagram of renewable terpene-based solvent use in OSCs and carbon resource circulation. (b) Processing solvent and photoactive materials used in this study. (c) Solubility and Ra of L8-BO and MYBO with respect to Eu. Temperature-dependent absorption spectra of (d) L8-BO and (e) MYBO in Eu solution (0.01 mg mL−1) normalized by their maximum absorbance at 20 °C. | ||
The poor PCE of the previous single terpene-based OSCs is primarily due to two factors: (1) the limited processability of conventional active materials in terpene solvents, and (2) the excessive crystallization of SMA molecules and the resulting unoptimized blend morphology, caused by the prolonged film formation kinetics during terpene processing. Most high-performance active materials in the OSC field have been designed for halogenated solvent processes (e.g., CF), where the solubility has been a lower priority in material design. Instead, the focus has been on promoting proper pre-aggregation of active components in solution to achieve strongly developed crystalline microstructures in film.37–39 However, these designs have resulted in most existing high-performance active materials being unsuitable for processing with terpene solvents due to very limited solubility in them.27,40 In addition, the slow evaporation rate of terpene solvents leads to excessive crystallization of SMA molecules or over-purification of domains during the film formation, which results in the formation of large agglomerates and excessive phase separation in blend films. This process is particularly accelerated by the molecular structures of conventional SMAs designed to enhance pre-aggregation in conventional halogenated and non-halogenated solvents. Therefore, to address the challenges in single terpene-based OSCs, two key strategies are imperative: (1) design of SMAs that offer sufficient solubility and reduced pre-aggregation in terpene solvents, and (2) meticulous control of film formation kinetics during terpene processing to prevent excessive crystallization of SMAs and over-purified blend domains.
To investigate the effects of photoactive molecular structure and film-formation kinetics on the performance of terpene-based OSCs, we established a model system as shown in Fig. 1b. We selected L8-BO as a reference high-performance SMA, which is currently one of the leading SMAs for OSCs using halogenated solvents.41,42 Furthermore, to develop an SMA more suitable for terpene processing while maintaining the excellent optoelectronic properties of L8-BO, we designed a MYBO SMA. Specifically, we retained the main backbone structure of L8-BO while increasing the length of the inner side chains from 2-ethylhexyl to 2-octyldodecyl. The lengthening of the inner side chains serves three main purposes. First, the increased side chain length enhances the volume fraction that interacts with the solvent, thereby improving solubility.43,44 Simultaneously, the increased volume fraction weakens intermolecular backbone-to-backbone interactions in solution, reducing pre-aggregation of the SMA molecules.45,46 Furthermore, tuning the structure of the alkyl side chains has minimal impact on the optoelectronic properties, allowing the preservation of the excellent optoelectronic characteristics inherent in the L8-BO backbones.47,48 The MYBO SMAs were synthesized by two-step reaction as shown in Scheme S1.†49,50 The detailed synthetic procedures are provided in the ESI.† The successful synthesis was confirmed by nuclear magnetic resonance (NMR) and matrix-assisted laser desorption/ionization time of flight (MALDI-ToF) measurements, as shown in Fig. S1–S3.† For polymer donor materials to pair with these SMAs, we selected PTQ10 due to its good solubility in non-halogenated solvents as well as its excellent optoelectronic properties.51–53 To further investigate the impact of donor molecular weights and optimize the photovoltaic performance of the terpene-processed OSCs, we synthesized PTQ10 with different number-average molecular weights (Mn) ranging from 21 to 33 kg mol−1 (Table S1†). In addition, using these photoactive materials, we fabricated OSCs with two different solvents of halogenated solvent (CF) and terpene solvent (Eu) to compare the photovoltaic performances based on the solvent types and SMA structures. Electrochemical properties of the photoactive materials were measured by cyclic voltammetry (CV) (Fig. S4† and Table 1). The lowest unoccupied molecular orbital energy level (ELUMO) and highest occupied molecular orbital energy level (EHOMO) of MYBO were measured to be −4.36 and −5.77 eV, respectively, while the ELUMO and EHOMO of L8-BO were −4.24 and −5.67 eV, respectively. Thus, both L8-BO and MYBO exhibited well-matched energy levels with PTQ10 for efficient exciton dissociation.
| Material | Solubility in Eua (mg mL−1) | Radius of interaction (Ra) | λ solmax (nm) |
λ
filmmaxb (nm) |
E
optgc (eV) |
E HOMO (eV) | E LUMO (eV) |
|---|---|---|---|---|---|---|---|
| a Measured at 100 °C. b Obtained from the UV-vis spectra of the thin film state spin-coated from the Eu solution. c Obtained from the absorption onsets in thin films spin-coated from the Eu solution using Eoptg = 1240/λedgefilm. d Measured by CV. e E LUMO = EHOMO + Eoptg. | |||||||
| PTQ10 | 13.6 | 9.9 | 566 | 606 | 1.89 | −5.58 | −3.69 |
| L8-BO | 2.1 | 15.5 | 722 | 797 | 1.43 | −5.67 | −4.24 |
| MYBO | 16.7 | 10.6 | 736 | 815 | 1.41 | −5.77 | −4.36 |
To estimate the processability of the active materials in terpene solvent, the radius of interaction (Ra) between the active materials and Eu was calculated based on the Hansen solubility parameters (HSPs) (Fig. 1c).54 The detailed procedures of Ra calculation are provided in the ESI and Tables S2–S4.† Between the SMAs, MYBO was calculated to have a significantly smaller Ra of 10.6 compared to L8-BO (Ra = 15.5). This result suggests that MYBO has significantly improved interactions with Eu than L8-BO, as a result of the lengthening of the inner side chains. In addition, the PTQ10 has similarly low Ra (9.9) with Eu, suggesting high processability of both PTQ10 donor and MYBO SMA in Eu. Indeed, experimental measurements of the solubility of the materials in Eu at 100 °C supported the predictions based on HSPs. The measured solubility values of MYBO, L8-BO, and PTQ10 were 16.7, 2.1, and 13.6 mg mL−1, respectively. To note, similar to L8-BO, other representative high-performance SMAs (e.g., Y6, Y6-BO, and BTP-eC9) designed for halogenated solvents processing exhibited extremely low solubility in Eu (Fig. S5†).
Optical properties of the materials were characterized by measuring ultraviolet-visible (UV-vis) light absorption spectra. In film state, MYBO showed a distinct peak at λ = 815 nm, regardless of processing solvent, while the intensity of L8-BO absorption peak showed significant variation between CF and Eu (Fig. S6†). To examine the aggregation behavior of the SMAs in solution state, we obtained their temperature-dependent UV-vis absorption spectra in toluene (Fig. S7†) and Eu (Fig. 1d and e) in the temperature ranges of 20–100 °C. Note that, instead of CF, we selected toluene with a higher BP to investigate the temperature-dependent pre-aggregation property of SMAs over a broader temperature range. In toluene, the absorption spectra of both L8-BO and MYBO solutions were slightly blue-shifted and their maximum absorbance decreased as the temperature increased (Fig. S7†). Specifically, L8-BO and MYBO exhibited the I100°Cmax/I20°Cmax value of 0.87 and 0.84, respectively, indicating slightly reduced pre-aggregation property of MYBO compared to L8-BO (Table S5†). Notably, in Eu solution, L8-BO and MYBO showed significantly different aggregation properties depending on temperature (Fig. 1d and e). L8-BO exhibited a substantial decrease in the absorbance at reduced temperatures, whereas MYBO experienced a slight increase in absorbance under the same conditions. This result can be attributed to the severe aggregation properties of L8-BO in Eu, which are in stark contrast to MYBO.
To investigate the thin-film crystallinity of the pristine SMAs processed by CF and Eu, differential scanning calorimetry (DSC) measurements were conducted (Fig. S8 and Table S6†). For the sample preparation, the SMA solutions were spin-coated onto glass substrates with same fabrication procedures and the resulting films were subsequently scrubbed to transfer into DSC pans. The DSC thermograms were obtained during the first heating cycle to retain the thermal history of the spin-coated films to directly examine crystalline features of SMAs in thin film.55 For the Eu-processed SMAs, a reduction in melting temperature (Tm) and an increase in melting enthalpy (ΔHm) were observed when compared to the pristine SMAs processed with CF. Specifically, the Tm of the pristine L8-BO significantly decreased from 309 °C to 299 °C, while the ΔHm value increased from 35.1 to 51.2 J g−1 when the processing solvent was changed from CF to Eu. The reduction in Tm is likely due to diminished interaction between SMA and Eu, relative to CF, resulting in the formation of imperfect crystals that reach the melting state more easily. The increase in ΔHm value can be attributed to the formation of larger aggregate domains, induced by partially soluble L8-BO in Eu. Moreover, this phenomenon is likely influenced by the significantly higher BP of Eu (177 °C) compared to CF (61 °C), which affords a longer crystallization time during spin-coating. In comparison, the change in the Tm of the pristine MYBO was smaller (from 255 to 249 °C), indicating that the crystalline properties of MYBO thin film were less affected depending on the solvent type compared to the L8-BO film.
To understand charge transport capabilities of the pristine SMAs, the space-charge limited current (SCLC) values were measured (Table S6†).56,57 For the neat SMA films, the electron mobility (μe) value of L8-BO in CF (1.7 × 10−4 cm2 V−1 s−1) was higher than the μe value of MYBO in CF (1.2 × 10−4 cm2 V−1 s−1), due to higher crystallinity of L8-BO film. In contrast, for the Eu, the trend in the μe of SMA was very different. Whereas the μe value of MYBO (1.1 × 10−4 cm2 V−1 s−1) in Eu remained comparable to the value in CF, the μe value of L8-BO (8.8 × 10−7 cm2 V−1 s−1) dropped dramatically.
Photovoltaic properties
We then investigated the photovoltaic properties of the PTQ10:L8-BO and PTQ10:MYBO blend systems by fabricating OSCs with a conventional device architecture (Fig. 2a and Table 2). To analyze the effects of SMA and solvent types on the performance of OSCs, we fabricated OSCs using both CF and Eu solvents. Detailed OSC fabrication procedures are described in the ESI and Tables S7–S13† (solution temperature, active layer thickness, molecular weight of PTQ10 and etc. were optimized). For the OSCs processed using CF, the PTQ10:L8-BO (CF) and PTQ10:MYBO (CF) systems exhibited similar PCEs of ∼15%. To note, the PCE of PTQ10:L8-BO (CF) device is comparable to the previously reported result.58 Among them, the PTQ10:L8-BO (CF)-based OSCs showed a slightly higher PCE of 15.04% compared to the PTQ10:MYBO (CF)-based OSCs (PCE = 14.84%), owing to higher VOC (0.93 V vs. 0.91 V) value. In contrast to the CF-processed devices, the OSCs based on PTQ10:L8-BO (Eu) and PTQ10:MYBO (Eu) blend films exhibited completely different photovoltaic performances. Specifically, the PTQ10:L8-BO (Eu) OSCs showed an extremely low PCE value of 0.13%, with a significant reduction in all photovoltaic parameters (VOC = 0.42 V, JSC = 0.69 mA cm−2, and FF = 0.43) compared to the CF-processed OSCs (VOC = 0.93 V, JSC = 24.06 mA cm−2, and FF = 0.67). In stark contrast, the PTQ10:MYBO (Eu)-based OSCs achieved a high PCE of 15.14%, which is comparable to that of the CF-processed devices (PCE = 14.84%). The PCE values of both PTQ10:MYBO (CF) and PTQ10:MYBO (Eu)-based OSCs were optimized with the same donor:acceptor ratio (1:1.2) and similar film thickness of ∼100–110 nm. Between the solvent types, PTQ10:MYBO (Eu) showed a slightly higher FF (0.69) compared to the PTQ10:MYBO (CF). To the best of our knowledge, 15.14% of PCE is the highest value among all reported OSCs fabricated using a single terpene solvent (Fig. 2b, S9 and Table S14†). This achievement is particularly noteworthy given the extremely low PCEs (∼0.1–3.0%) observed in previous OSCs processed with a single terpene solvent. Furthermore, compared to the terpene mixture systems including a harmful solvent, our system offers a distinct advantage of fully eco-friendly processing. The Gaussian PCE distributions of the OSCs are shown in Fig. 2c, indicating minimal deviation in the PCE values and high reproducibility. External quantum efficiency (EQE) spectra of the OSCs are shown in Fig. 2d. The calculated JSC values from the EQE profiles were well-matched with the device JSC within an error margin of 5%.
 | ||
| Fig. 2 (a) Current density (J)–voltage (V) curves. (b) Summary of PCEs of terpene-based OSCs depending on the toxicity from the literature and this work. (c) PCE distribution with Gaussian fitting. (d) EQE spectra of PTQ10:L8-BO and PTQ10:MYBO devices fabricated with different processing solvents. (e) Jph–Veff curves of the PTQ10-based OSCs. (f) Light intensity-dependent VOC plots of PTQ10-based OSCs. | ||
| SMA | Solvent | V OC (V) | J SC (mA cm−2) | Calc. JSCa (mA cm−2) | FF | PCEmax (PCEavg)b (%) |
|---|---|---|---|---|---|---|
| a Calculated from EQE spectra. b Average values obtained from at least 10 independent devices. | ||||||
| L8-BO | CF | 0.93 (0.92 ± 0.00) | 24.06 (24.03 ± 0.40) | 23.05 | 0.67 (0.67 ± 0.01) | 15.04 (14.74 ± 0.24) |
| Eu | 0.42 (0.40 ± 0.05) | 0.69 (0.56 ± 0.19) | 0.70 | 0.43 (0.41 ± 0.05) | 0.13 (0.09 ± 0.03) | |
| MYBO | CF | 0.91 (0.91 ± 0.00) | 24.91 (24.63 ± 0.43) | 24.46 | 0.65 (0.66 ± 0.01) | 14.84 (14.68 ± 0.23) |
| Eu | 0.88 (0.88 ± 0.01) | 25.23 (24.84 ± 0.29) | 25.24 | 0.69 (0.68 ± 0.01) | 15.14 (14.71 ± 0.22) | |
To better understand the different photovoltaic properties of the OSCs, we investigated charge generation, transport, and recombination properties of the OSCs. The charge generation properties were examined by measuring photocurrent density (Jph) of the OSCs under effective voltage (Veff). From the Jphvs. Veff profiles, exciton dissociation probability (P(E,T)) of the OSCs was estimated by dividing JSC by saturated current density (Jsat, at Veff = 3 V) (Fig. 2e).59 While the PTQ10:L8-BO (CF), PTQ10:MYBO (CF), and PTQ10:MYBO (Eu) OSCs exhibited excellent charge generation properties with high P(E,T) values exceeding 93%, the P(E,T) of PTQ10:L8-BO (Eu) was not able to be calculated due to incomplete collection of photo-generated charge carriers at the electrodes. Charge transport properties of the blend films were estimated by measuring SCLC charge mobility (Table S15†). For blend films, the hole mobility (μh) values of PTQ10:L8-BO (CF), PTQ10:MYBO (CF), and PTQ10:MYBO (Eu) were similar in the range of 3.5 × 10−4 to 3.7 × 10−4 cm2 V−1 s−1. In comparison, despite the use of the same polymer donor, the charge transport property of PTQ10 in the PTQ10:L8-BO (Eu) system was inferior, exhibiting a lower μh value of 1.5 × 10−4 cm2 V−1 s−1. Meanwhile, PTQ10:L8-BO (Eu) featured a significantly decreased μe value of 4.0 × 10−6 cm2 V−1 s−1, leading to highly deviated μe/μh value of 0.03 from unity compared to those of the other blend systems (μe/μh = 1.64–1.97). This result suggests that continuous carrier transport pathway was not formed in PTQ10:L8-BO (Eu) since L8-BO in Eu induced excessive phase separation, explaining lower FF value (0.43) in the OSCs. This will be discussed in the later section associated with the morphological behavior.
Next, the dependence of JSC and VOC on illuminating light intensity (P) was measured (Fig. S10† and 2f).60 The parameter α representing the slope of the Jscvs. P plot indicates the degree of bimolecular recombination. Additionally, the slope (S) in a Vocvs. P plot (kB = Boltzmann constant, T = temperature in Kelvin, and q = elementary charge) corresponds to the monomolecular/trap-assisted recombination, with S approaching unity indicating rarer occurrence of this recombination mechanism. PTQ10:MYBO (Eu) showed the most suppressed bimolecular recombination (α = 0.98) and monomolecular/trap-assisted recombination (S = 1.01 kBT q−1) compared to the other systems, aligning with the highest JSC and FF values. PTQ10:L8-BO (CF) and PTQ10:MYBO (CF) exhibited similar α (0.97) and S values (1.04 kBT q−1), corresponding to their similar JSC and FF values. However, both α (0.85) and S (1.81 kBT q−1) values of PTQ10:L8-BO (Eu) were considerably deviated from unity, indicating a larger degree of recombination via both bimolecular and monomolecular/trap-assisted recombination pathways compared to the previous three systems. The higher occurrence of charge recombination in PTQ10:L8-BO (Eu)-OSCs accounts for their poor photovoltaic performances.
Morphological properties
To elucidate the origin of the differences in the photovoltaic performance, the morphological characteristics of the blend films were investigated. In atomic force microscopy (AFM) height images (Fig. 3a), PTQ10:MYBO (Eu) showed a smooth surface with a root-mean-square roughness (Rq) value of only 0.8 nm. In stark contrast, PTQ10:L8-BO (Eu) displayed extremely large aggregates with a significantly higher Rq of 44.8 nm. These findings reflect the high processability of MYBO, which leads to smooth active layer surface conducive to conformal contact to the adjacent layers and high device performance.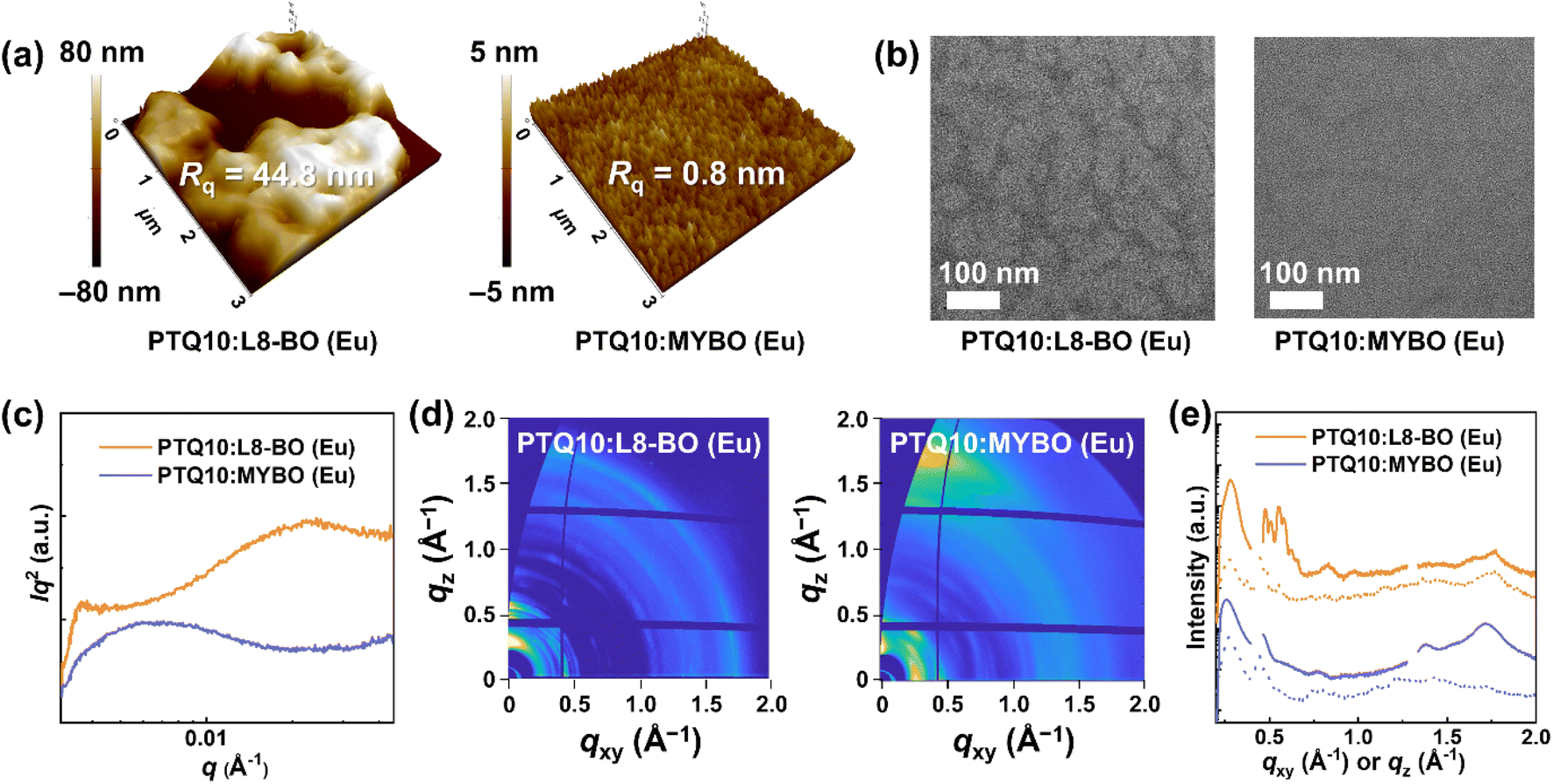 | ||
| Fig. 3 (a) AFM height images of the blend films processed with Eu. (b) TEM images of the PTQ10-based blend film processed with Eu (scale bars are 100 nm). (c) RSoXS profiles of blend films. (d) GIWAXS 2D images of PTQ10-based film in Eu. (e) GIWAXS line–cut profiles in the OOP (solid lines) and IP (dotted lines) directions. | ||
The internal morphology of the blend films was examined by transmission electron microscopy (TEM) and resonant soft X-ray scattering (RSoXS). In the TEM images (Fig. 3b), PTQ10:MYBO (Eu) displayed more miscible features and suppressed phase separation with less distinct domain features compared to the L8-BO counterpart. For more quantitative investigation, the RSoXS technique was employed (Fig. 3c and Table 3),61 revealing the same trend as that observed in TEM. In the RSoXS profiles, PTQ10:MYBO (Eu) showed a single scattering peak at q ∼ 0.007 Å−1, corresponding to a domain size of 44 nm. On the other hand, PTQ10:L8-BO (Eu) exhibited two distinct peaks in the low- and high-q regions, respectively. The low-q peak (q ∼ 0.004 Å−1, domain size = 85 nm) is likely due to excessive liquid–liquid phase separation, while the high-q peak (q ∼ 0.024 Å−1, domain size = 13 nm) may result from liquid–solid phase separation.62 Moreover, the L8-BO blend showed significantly higher relative domain purity (r-DP = 1.00) compared to the MYBO blend (r-DP = 0.67), corroborating the largely phase-separated morphology of PTQ10:L8-BO (Eu). The smaller domains and enhanced miscibility of PTQ10:MYBO (Eu) account for the suppressed charge recombination characteristics and superior device performance.63
Grazing incidence wide-angle X-ray scattering (GIWAXS) measurements were conducted to examine the crystalline structures of the blend films (Fig. 3d and e). All the samples were prepared using the same conditions as those used for OSC fabrication. The most pronounced difference in the GIWAXS patterns was the molecular packing orientation. PTQ10:MYBO (Eu) showed dominantly face-on orientation, evidenced by a distinct (010) π–π stacking peak in the out-of-plane (OOP) direction and a (100) lamellar scattering peak in the in-plane (IP) direction. In contrast, PTQ10:L8-BO (Eu) displayed edge-on/face-on mixed orientation, confirmed by the emergence of (010) and (100) peaks in the IP and OOP directions, respectively. For a more quantitative comparison, the ratio of the face-on fraction relative to the edge-on fraction (Af/Ae) was estimated from pole figures (Fig. S11, Table 3, and S16†).50,64 The detailed procedures to obtain the pole figures and Af/Ae values are described in the ESI.† As a result, the MYBO blend showed an 11-fold higher Af/Ae value (9.1) than the L8-BO blend (Af/Ae = 0.8). This result is in line with the well-known tendency that highly aggregated materials in solution favor edge-on orientation in film.65 The face-on-oriented MYBO blend film, favorable for the vertical charge transport in OSCs,66 is likely to contribute to the high device performance.
To investigate the crystallinity of the blend films processed by CF and Eu, DSC measurements were conducted (Fig. S12 and Table S17†). For the sample preparation, all the samples were prepared using the same conditions as those used for OSC fabrication. The resulting films were subsequently scrubbed to transfer into DSC pans. The DSC thermograms were obtained during the first heating cycle to retain the thermal history of the spin-coated films to directly examine crystalline features of polymers and SMAs in thin film.55 While the Tm value significantly decreased from 306 °C for the PTQ10:L8-BO (CF) to 290 °C for the PTQ10:L8-BO (Eu), the ΔHm value increased from 21.3 to 48.3 J g−1. This indicates that the excessive aggregation of L8-BO in Eu led to large-sized, but less-ordered defective crystals in film. Additionally, PTQ10:L8-BO (Eu) system exhibited a broader pre-melting transition from 262 °C to 290 °C, which corresponds to the early stage of the melting process where less stable or disordered crystals begin to partially melt before the main melting process. These impure crystal forms are likely to act as traps within the density of states (DOS), thereby leading to inferior photovoltaic parameters. In contrast, the difference in the Tm and ΔHm of the PTQ10:MYBO (CF) (Tm = 251 °C and ΔHm = 7.4 J g−1) and PTQ10:MYBO (Eu) (Tm = 242 °C and ΔHm = 9.8 J g−1) was relatively marginal, indicating that the well-ordered and small-sized crystalline structures of MYBO in CF-processed films were well-preserved in Eu-processed ones.
Morphology optimization by Eu processing control
Although the use of high-BP solvent is advantageous to large-area printing process for uniform film coating, it is susceptible to excessive phase separation due to prolonged morphology evolution time. In particular, since Eu has an even higher BP (177 °C) compared to typical high-BP solvents such as o-xylene (139 °C), chlorobenzene (132 °C), and toluene (111 °C), controlling its evaporation rate for tuning the crystalline structures of donor and SMA as well as their phase-separated domains is especially important. With careful modulation of the processing time, the crystalline structures of donor and SMA should form sufficiently, while suppressing excessive phase separation in the blend. In this context, we devised a new processing technique that varies the film-formation kinetics while keeping the film thickness constant and, thus maintaining similarly high light harvesting capability. In detail, the concentration of active solution (14.0 → 20.0 → 30.0 mg mL−1) and spin-coating speed (1800 → 2500 → 4000 rpm) were simultaneously controlled to yield three different morphology quenching conditions, labeled as Eu (slow), Eu (medium), and Eu (fast), respectively. It is noted that all the films have almost identical thickness of 105–108 nm (Table S18†).To examine the photovoltaic performance under different processing conditions, OSCs were fabricated (Fig. 4a and Table 4). Notably, although all the active layers had the same film thickness and PTQ10:SMA ratio, the PCE remarkably increased from 6.59 and 10.52 to 15.14% as the morphology quenching time was reduced (slow → medium → fast). For example, PTQ10:MYBO (Eu, fast) showed significantly higher performance (PCE = 15.14%, JSC = 25.23 mA cm−2, and FF 0.69) compared to PTQ10:MYBO (Eu, slow) (PCE = 6.59%, JSC = 17.66 mA cm−2, and FF = 0.49), indicating that the charge generation and transport behaviors are markedly different in the two systems. To gain a deeper understanding of the origin of those differences, we investigated the blend morphology of the active layers. It is noteworthy that the degree of phase separation was successfully reduced by using a shorter processing time. For example, compared to the Eu (slow) system, Eu (fast) had much smoother film as indicated by the substantial decrease in surface roughness (Rq = 3.5 → 0.8 nm) (Fig. 4b). TEM images in Fig. 4c reveal that the PTQ10:MYBO (Eu, fast) exhibited a blend morphology with smaller domain sizes and larger fraction of intermixed domains, in contrast to the distinctly phase-segregated blend with larger domain sizes (∼100 nm) observed for PTQ10:MYBO (Eu, slow) (Fig. 4c). Considering limited exciton diffusion length scales (<30 nm) in photoactive layers, the better intermixed blend morphology in PTQ10:MYBO (Eu, fast) films led to suppressed charge recombination and significant improvement in JSC and FF values.
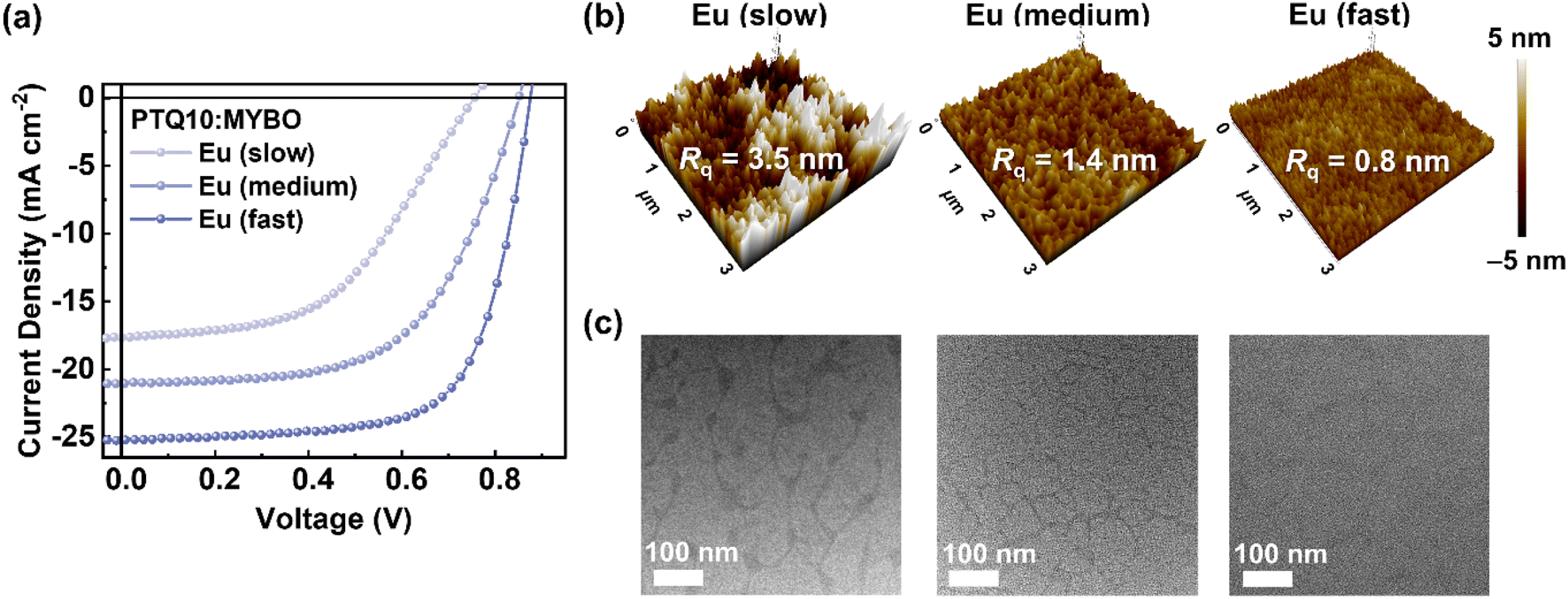 | ||
| Fig. 4 (a) J–V curves of the Eu-processed PTQ10:MYBO devices depending on the processing condition. (b) AFM height images and (c) TEM images of the active layers (scale bars are 100 nm). | ||
| System | Solvent | V OC (V) | J SC (mA cm−2) | FF | PCEmax (PCEavg)b (%) |
|---|---|---|---|---|---|
| a The total concentration and spin-coating speed for the Eu (slow), Eu (medium), and Eu (fast) conditions are (14.0 mg mL−1 & 1800 rpm), (20.0 mg mL−1 & 2500 rpm), and (30.0 mg mL−1 & 4000 rpm), respectively. All the films have almost identical thickness of 105–108 nm. b Average values obtained from at least 10 independent devices. | |||||
| PTQ10:MYBO | Eu (slow)a | 0.76 (0.75 ± 0.02) | 17.66 (17.37 ± 0.76) | 0.49 (0.47 ± 0.03) | 6.59 (6.04 ± 0.53) |
| Eu (medium)a | 0.85 (0.86 ± 0.01) | 21.07 (20.38 ± 0.80) | 0.59 (0.56 ± 0.03) | 10.52 (9.81 ± 0.49) | |
| Eu (fast)a | 0.88 (0.88 ± 0.01) | 25.23 (24.84 ± 0.29) | 0.69 (0.68 ± 0.01) | 15.14 (14.71 ± 0.22) | |
To investigate the film-formation kinetics of the PTQ10:MYBO blend systems in Eu and explore their correlation with the device performance/blend morphology, in situ UV-vis absorption spectroscopy was employed (Fig. 5a).67 Each blend was spin-coated under the same conditions applied for device fabrication and their absorbance was tracked as a function of spin-coating time at λmax of PTQ10 and MYBO (Fig. 5b). Intriguingly, the blend solution showed significantly different absorbance saturation time (τsat) depending on the processing time (Fig. 5c). The τsats of both PTQ10 (τPTQ10sat) and MYBO (τMYBOsat) decreased with shorter quenching time. For example, the τPTQ10sat and τMYBOsat significantly decreased from 33.20 and 33.85 s in the Eu (slow) system to 23.70 and 26.30 s in the Eu (fast) system, respectively. Importantly, PTQ10 showed earlier crystallization than MYBO (τPTQ10sat < τMYBOsat) in all three blends, but the gap became larger with shorter processing time (τMYBOsat− τPTQ10sat = 0.65 → 1.40 → 2.60 s) (Table S18†). This suggests that in the Eu (fast) process, the MYBO SMAs have sufficient time for developing their crystalline structures within the pre-formed PTQ10 network scaffold.
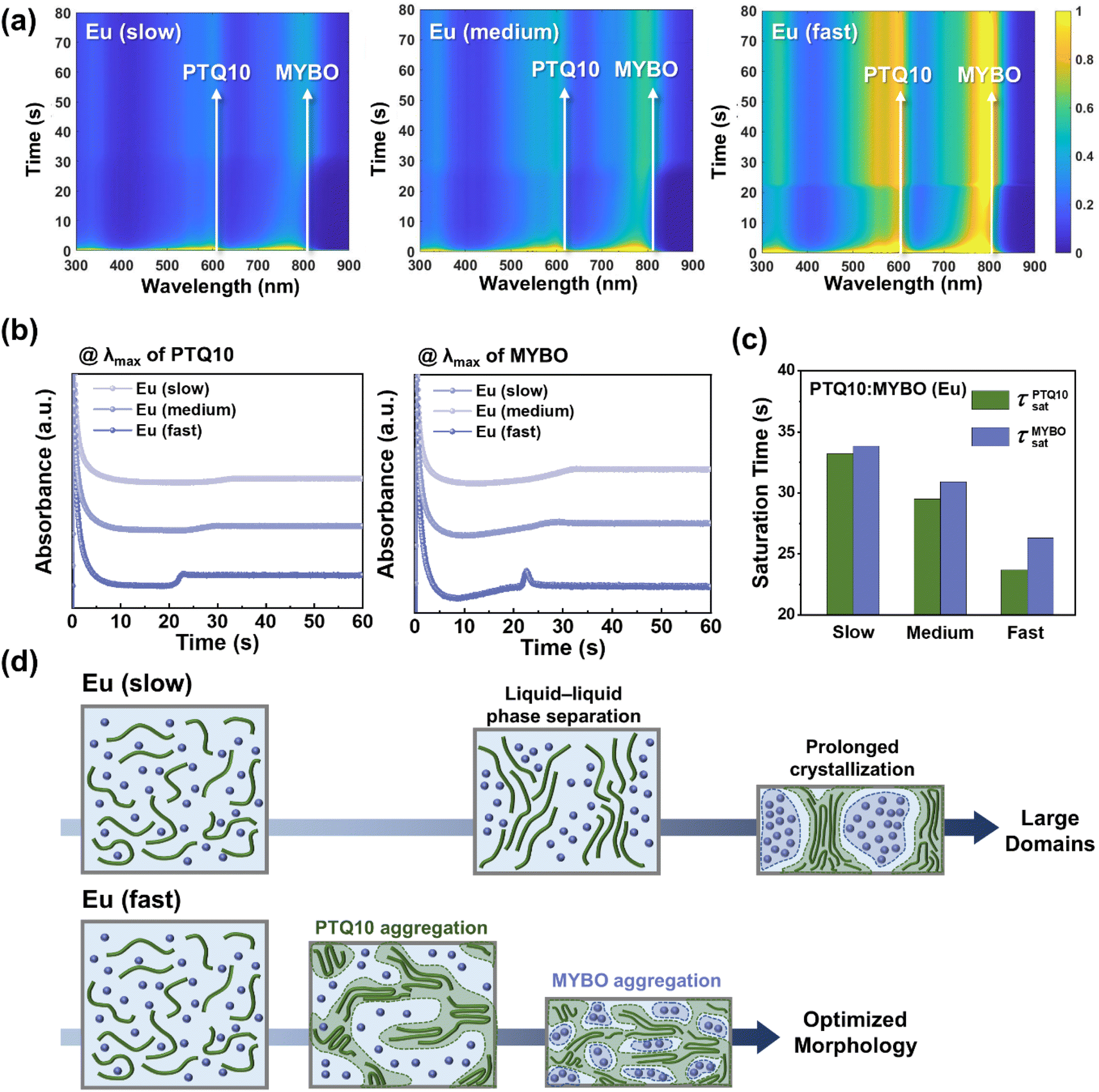 | ||
| Fig. 5 (a) 2D contour maps of in situ UV-vis absorption spectra of PTQ10:MYBO systems processed with Eu depending on the processing condition and their (b) changes in absorbance at the λmax of PTQ10 and MYBO. (c) Saturation time of PTQ10 and MYBO depending on the processing condition. (d) Schematic illustration displaying the blend morphology evolution during the film drying process of PTQ10:MYBO (Eu). | ||
Based on the observations above, a schematic illustration showing the morphology evolution during the film drying processes is presented in Fig. 5d. In Eu (slow), the extended evaporation allows more time for spontaneous separation of a homogeneous active solution into the donor and acceptor phases (liquid–liquid phase separation) before solidifying into a thin film.68 This prolonged drying time can lead to the excessively phase-separated blend morphology with large domain scale and domain purity, which align with the results in Fig. 4b and c. In contrast, under the Eu (fast) condition, PTQ10 aggregates first, forming fibrillary interconnected networks before excessive liquid–liquid demixing occurs. Due to this earlier aggregation and precipitation of PTQ10, we propose that MYBO molecules become embedded and dispersed within the PTQ10 matrix. Then, the large difference in τMYBOsat and τPTQ10sat allows more time for the MYBO to develop their crystalline structures and enhance the optoelectronic properties.69,70 The individually formed donor and acceptor domains can finally form bi-continuous interpenetrating networks for efficient charge generation and transport in OSC devices. In addition, it is noted that the smaller τPTQ10sat and τMYBOsat themselves in Eu (fast) compared to Eu (slow) help prevent the formation of large-scale and over-purified domains. This kinetics control, achieved by tuning the evaporation rate of Eu, enables the remarkable increase in PCE of the Eu-processed OSCs from 6.59 to 15.14%, synergistically driven by improvements in both JSC and FF values. Overall, these results signify the importance of controlling the film-formation kinetics in the eco-friendly processing based on high-BP Eu.
Air stability of OSCs fabricated with eco-friendly solvent
Ensuring air stability is crucial for the potential commercialization of OSCs fabricated with environmentally benign solvents.71 In industrial setting, exposure of OSCs to ambient conditions during roll-to-roll processing is unavoidable, given the challenges and costs of maintaining an inert atmosphere in large-scale production.72 However, ambient air, which contains moisture and oxygen, affects morphology and film-formation kinetics, often resulting in lower PCE for air-processed OSCs compared to those fabricated in a glove box (Fig. 6a).73,74 In this context, we compared the performance of OSCs fabricated in a glove box (inert N2 atmosphere) to those fabricated in ambient conditions. Interestingly, the eco-friendly OSCs based on PTQ10:MYBO (Eu) processed in ambient conditions showed comparable performance to those fabricated in inert environments with only marginal changes in JSC and FF (Fig. 6b and Table S19†). This result underscores the potential of Eu processing for large-area printing applications.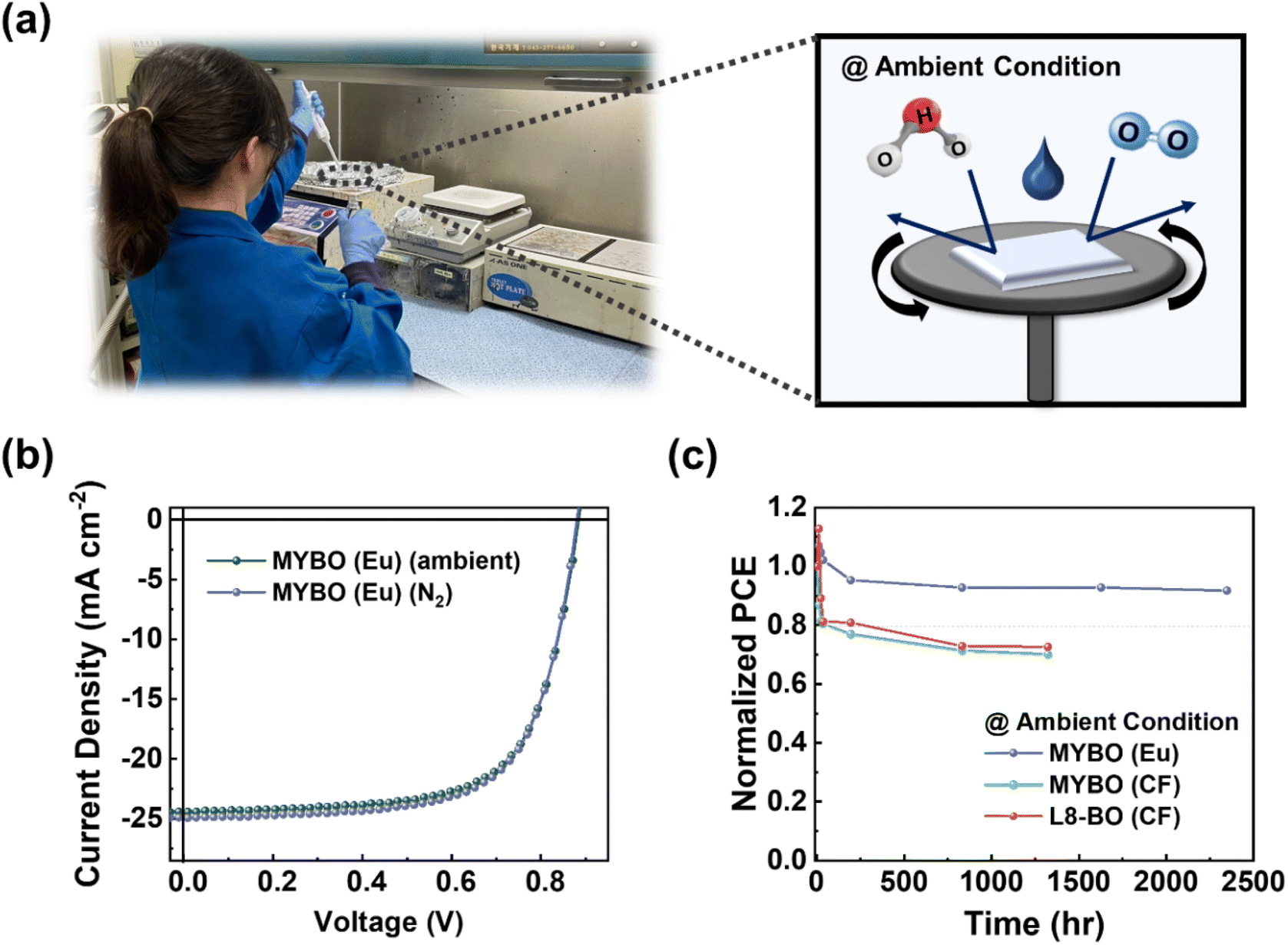 | ||
| Fig. 6 (a) Fabrication of OSCs under ambient conditions. (b) J–V curves of PTQ10:MYBO OSCs fabricated in a glove box (N2) and ambient conditions. (c) Air stability of PTQ10:MYBO (Eu), PTQ10:MYBO (CF), and PTQ10:L8-BO (CF). All the devices were fabricated in ambient conditions. The resulting devices were stored in ambient and dark conditions without encapsulation. | ||
To further assess the air stability, we fabricated PTQ10:L8-BO (CF), PTQ10:MYBO (CF), and PTQ10:MYBO (Eu)-based OSCs under ambient conditions and stored them in air without encapsulation (relative humidity: 70–80%). To mitigate the adverse effects of moisture on device stability, an inverted configuration was employed, enabling the exclusion of hygroscopic poly(3,4-ethylenedioxythiophene):polystyrene sulfonate (PEDOT:PSS) layer in the conventional type. Notably, the PTQ10:MYBO (Eu) system exhibited superior air stability, retaining 92% of its initial PCE even after 2300 h of exposure to ambient conditions (Fig. 6c). In contrast, the other systems processed with CF showed significantly inferior air stability, reaching 80% of their initial PCEs in less than 200 h. We speculate that this result may be attributed to the slower evaporation rate of high-BP Eu than low-BP CF. Since most blend films are kinetically trapped after film-casting processes, the solvent evaporation time plays a crucial role in determining the initial blend morphology.75,76 Given that the τsat of Eu-processed blend films (>20 s) is around 20 times longer than CF-processed blend films (∼1 s) (Fig. S13 and Table S20†), it is likely that photoactive materials in the former case are closer to the thermodynamic equilibrium,77 leading to superior stability. In the stability test, the burn-in-loss of PTQ10:MYBO (Eu) was significantly improved compared to the other CF-processed devices, highlighting the importance of eco-friendly and high-BP Eu processing for enhancing the air stability of OSCs.
Conclusions
In this study, we developed high-performance, air-stable, and truly human-/eco-friendly OSCs processed using a single terpene solvent (Eu). To achieve both high solubility and electrical properties with Eu processing, we designed a MYBO SMA, featuring an optimized alkyl-chain structure and strong backbone crystallinity. When paired with PTQ10 polymer donor, a remarkable PCE of 15.1% was achieved, which is the highest value for all single terpene-based OSCs. We highlight that the breakthrough in PCE was enabled by meticulous control of the evaporation rate of Eu, leading to independent aggregation of PTQ10 and MYBO and formation of well-developed MYBO crystalline structures embedded in interconnected polymer networks. Combined results from morphological analyses, in situ UV-vis spectroscopy, and electrical property measurements manifest the importance of this blend morphology control in optimizing the charge generation/transport and device performance. Additionally, the OSCs exhibited excellent air-stability, retaining 92% of the initial PCE after 2300 h of air exposure. This work provides valuable insights into the potential of using eco-friendly, single terpene solvents in the development of high-performance and air-stable OSCs.Experimental
Full details of ESI† (e.g., 1H NMR, MALDI-ToF, CV, UV-vis spectra, DSC, and additional device fabrication data) and experimental procedures can be found in the ESI.†Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
B. J. Kim supervised the study and revised the manuscript. H. Jeon fabricated the solar cells and wrote the original draft. J.-W. Lee and S. Lee reviewed and revised the manuscript. K. Bae assisted with the fabrication of solar cells. T. N.-L. Phan and C. Lim synthesized the photoactive materials. J. Choi assisted with the TEM measurement. C. Wang assisted with the RSoXS measurement. All authors commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation (NRF) of Korea (RS-2024-00432362 and RS-2024-00441837). This work was also supported by the Korea Research Institute of Chemical Technology (KRICT) (No. KS2422-10) of the Republic of Korea. This research used resources of the Advanced Light Source, which is a DOE Office of Science User Facility under contract no. DE-AC02-05CH11231.References
- Y. Li, G. Xu, C. Cui and Y. Li, Adv. Energy Mater., 2018, 8, 1701791 CrossRef.
- J. S. Park, G.-U. Kim, S. Lee, J.-W. Lee, S. Li, J.-Y. Lee and B. J. Kim, Adv. Mater., 2022, 34, 2201623 CrossRef CAS.
- Q. Wan, S. Seo, S.-W. Lee, J. Lee, H. Jeon, T.-S. Kim, B. J. Kim and B. C. Thompson, J. Am. Chem. Soc., 2023, 145, 11914–11920 CrossRef CAS PubMed.
- L. Zhan, S. Li, T.-K. Lau, Y. Cui, X. Lu, M. Shi, C.-Z. Li, H. Li, J. Hou and H. Chen, Energy Environ. Sci., 2020, 13, 635–645 RSC.
- C. Dou, X. Long, Z. Ding, Z. Xie, J. Liu and L. Wang, Angew. Chem., Int. Ed., 2016, 55, 1436–1440 CrossRef CAS.
- W. Gao, F. Qi, Z. Peng, F. R. Lin, K. Jiang, C. Zhong, W. Kaminsky, Z. Guan, C.-S. Lee, T. J. Marks, H. Ade and A. K.-Y. Jen, Adv. Mater., 2022, 34, 2202089 CrossRef CAS.
- Y. Cui, Y. Xu, H. Yao, P. Bi, L. Hong, J. Zhang, Y. Zu, T. Zhang, J. Qin, J. Ren, Z. Chen, C. He, X. Hao, Z. Wei and J. Hou, Adv. Mater., 2021, 33, 2102420 CrossRef CAS.
- R. Sun, Y. Wu, X. Yang, Y. Gao, Z. Chen, K. Li, J. Qiao, T. Wang, J. Guo, C. Liu, X. Hao, H. Zhu and J. Min, Adv. Mater., 2022, 34, 2110147 CrossRef CAS.
- L. Zhu, M. Zhang, J. Xu, C. Li, J. Yan, G. Zhou, W. Zhong, T. Hao, J. Song, X. Xue, Z. Zhou, R. Zeng, H. Zhu, C.-C. Chen, R. C. I. MacKenzie, Y. Zou, J. Nelson, Y. Zhang, Y. Sun and F. Liu, Nat. Mater., 2022, 21, 656–663 CrossRef CAS.
- B. Liu, W. Xu, R. Ma, J.-W. Lee, T. A. Dela Peña, W. Yang, B. Li, M. Li, J. Wu, Y. Wang, C. Zhang, J. Yang, J. Wang, S. Ning, Z. Wang, J. Li, H. Wang, G. Li, B. J. Kim, L. Niu, X. Guo and H. Sun, Adv. Mater., 2023, 35, 2308334 CrossRef CAS.
- S. Luo, C. Li, J. Zhang, X. Zou, H. Zhao, K. Ding, H. Huang, J. Song, J. Yi, H. Yu, K. S. Wong, G. Zhang, H. Ade, W. Ma, H. Hu, Y. Sun and H. Yan, Nat. Commun., 2023, 14, 6964 CrossRef CAS.
- Y.-J. Xue, Z.-Y. Lai, H.-C. Lu, J.-C. Hong, C.-L. Tsai, C.-L. Huang, K.-H. Huang, C.-F. Lu, Y.-Y. Lai, C.-S. Hsu, J.-M. Lin, J.-W. Chang, S.-Y. Chien, G.-H. Lee, U. S. Jeng and Y.-J. Cheng, J. Am. Chem. Soc., 2024, 146, 833–848 CrossRef CAS.
- J.-W. Lee, C. Sun, J. Lee, D. J. Kim, W. J. Kang, S. Lee, D. Kim, J. Park, T. N.-L. Phan, Z. Tan, F. S. Kim, J.-Y. Lee, X. Bao, T.-S. Kim, Y.-H. Kim and B. J. Kim, Adv. Energy Mater., 2024, 14, 2303872 CrossRef CAS.
- Z. Ma, B. Zhao, Y. Gong, J. Deng and Z. a. Tan, J. Mater. Chem. A, 2019, 7, 22826–22847 RSC.
- C. Kim, S. Chen, J. S. Park, G.-U. Kim, H. Kang, S. Lee, T. N.-L. Phan, S.-K. Kwon, Y.-H. Kim and B. J. Kim, J. Mater. Chem. A, 2021, 9, 24622–24630 RSC.
- S. Lee, D. Jeong, C. Kim, C. Lee, H. Kang, H. Y. Woo and B. J. Kim, ACS Nano, 2020, 14, 14493–14527 CrossRef CAS.
- C.-C. Chueh, K. Yao, H.-L. Yip, C.-Y. Chang, Y.-X. Xu, K.-S. Chen, C.-Z. Li, P. Liu, F. Huang, Y. Chen, W.-C. Chen and A. K. Y. Jen, Energy Environ. Sci., 2013, 6, 3241–3248 RSC.
- J. Zhao, Y. Li, G. Yang, K. Jiang, H. Lin, H. Ade, W. Ma and H. Yan, Nat. Energy, 2016, 1, 15027 CrossRef CAS.
- H. Chen, H. Lai, Z. Chen, Y. Zhu, H. Wang, L. Han, Y. Zhang and F. He, Angew. Chem., Int. Ed., 2021, 60, 3238–3246 CrossRef CAS.
- C.-Y. Liao, Y. Chen, C.-C. Lee, G. Wang, N.-W. Teng, C.-H. Lee, W.-L. Li, Y.-K. Chen, C.-H. Li, H.-L. Ho, P. H.-S. Tan, B. Wang, Y.-C. Huang, R. M. Young, M. R. Wasielewski, T. J. Marks, Y.-M. Chang and A. Facchetti, Joule, 2020, 4, 189–206 CrossRef CAS.
- Y. Tang, H. Sun, Z. Wu, Y. Zhang, G. Zhang, M. Su, X. Zhou, X. Wu, W. Sun, X. Zhang, B. Liu, W. Chen, Q. Liao, H. Y. Woo and X. Guo, Adv. Sci., 2019, 6, 1901773 CrossRef CAS PubMed.
- Y. Wang, H. Tatsumi, R. Otsuka, T. Mori and T. Michinobu, J. Mater. Chem. C, 2018, 6, 5865–5876 RSC.
- T. Kumari, S. Jung, Y. Cho, H.-P. Kim, J. W. Lee, J. Oh, J. Lee, S. M. Lee, M. Jeong, J. M. Baik, W. Jo and C. Yang, Nano Energy, 2020, 68, 104327 CrossRef CAS.
- B. Liu, H. Sun, J.-W. Lee, J. Yang, J. Wang, Y. Li, B. Li, M. Xu, Q. Liao, W. Zhang, D. Han, L. Niu, H. Meng, B. J. Kim and X. Guo, Energy Environ. Sci., 2021, 14, 4499–4507 RSC.
- J. Zhang, Q. Huang, K. Zhang, T. Jia, J. Jing, Y. Chen, Y. Li, Y. Chen, X. Lu, H. Wu, F. Huang and Y. Cao, Energy Environ. Sci., 2022, 15, 4561–4571 RSC.
- U. S. E. P. Agency, TRI Chemical List, http://www.epa.gov/toxics-release-inventory-tri-program/tri-listed-chemicals, accessed June 10, 2024 Search PubMed.
- D. Corzo, D. Rosas-Villalva, A. C, G. Tostado-Blázquez, E. B. Alexandre, L. H. Hernandez, J. Han, H. Xu, M. Babics, S. De Wolf and D. Baran, Nat. Energy, 2023, 8, 62–73 CrossRef.
- C. Ravichandran, P. C. Badgujar, P. Gundev and A. Upadhyay, Food Chem. Toxicol., 2018, 120, 668–680 CrossRef CAS PubMed.
- M. M. Masadeh, S. F. Gharaibeh, K. H. Alzoubi, S. I. Al-Azzam and W. M. Obeidat, J. Clin. Med. Res., 2013, 5, 389–394 Search PubMed.
- M. Bhowal and M. Gopal, J. Pharm. Sci., 2015, 5, 125–131 CAS.
- L. Ye, Y. Xiong, Z. Chen, Q. Zhang, Z. Fei, R. Henry, M. Heeney, B. T. O'Connor, W. You and H. Ade, Adv. Mater., 2019, 31, 1808153 CrossRef.
- C. Sprau, A. M. Cruz, L. Bautista, L. Molina, M. Wagner, C. L. Chochos, M. Della Pirriera and A. Colsmann, Adv. Energy Sustainability Res., 2021, 2, 2100043 CrossRef CAS.
- M.-J. Li, B.-B. Fan, W.-K. Zhong, Z.-M.-Y. Zeng, J.-K. Xu and L. Ying, Chin. J. Polym. Sci., 2020, 38, 791–796 CrossRef CAS.
- T. C. Industry, Material Safety Data Sheet, http://www.tcichemicals.com/KR/ko/, accessed June 10, 2024 Search PubMed.
- I. Deneme, T. A. Yıldız, N. Kayaci and H. Usta, J. Mater. Chem. C, 2024, 12, 3854–3864 RSC.
- NFPA30, Flammable and Combustible Liquids Code 2021 Edition, NFPA, 2021 Search PubMed.
- B. Fan, F. Lin, J. Oh, H. Fu, W. Gao, Q. Fan, Z. Zhu, W. J. Li, N. Li, L. Ying, F. Huang, C. Yang and A. K.-Y. Jen, Adv. Energy Mater., 2021, 11, 2101768 CrossRef CAS.
- H. Hu, K. Zhao, N. Fernandes, P. Boufflet, J. H. Bannock, L. Yu, J. C. de Mello, N. Stingelin, M. Heeney, E. P. Giannelis and A. Amassian, J. Mater. Chem. C, 2015, 3, 7394–7404 RSC.
- N.-K. Kim, S.-Y. Jang, G. Pace, M. Caironi, W.-T. Park, D. Khim, J. Kim, D.-Y. Kim and Y.-Y. Noh, Chem. Mater., 2015, 27, 8345–8353 CrossRef CAS.
- S. Pang, Z. Chen, J. Li, Y. Chen, Z. Liu, H. Wu, C. Duan, F. Huang and Y. Cao, Mater. Horiz., 2023, 10, 473–482 RSC.
- C. Chen, L. Wang, W. Xia, K. Qiu, C. Guo, Z. Gan, J. Zhou, Y. Sun, D. Liu, W. Li and T. Wang, Nat. Commun., 2024, 15, 6865 CrossRef CAS.
- L. Wang, C. Chen, Y. Fu, C. Guo, D. Li, J. Cheng, W. Sun, Z. Gan, Y. Sun, B. Zhou, C. Liu, D. Liu, W. Li and T. Wang, Nat. Energy, 2024, 9, 208–218 CrossRef CAS.
- Z. Abbas, S. U. Ryu, M. Haris, C. E. Song, H. K. Lee, S. K. Lee, W. S. Shin, T. Park and J.-C. Lee, Nano Energy, 2022, 101, 107574 CrossRef CAS.
- K. Jiang, Q. Wei, J. Y. L. Lai, Z. Peng, H. K. Kim, J. Yuan, L. Ye, H. Ade, Y. Zou and H. Yan, Joule, 2019, 3, 3020–3033 CrossRef CAS.
- S. Dong, T. Jia, K. Zhang, J. Jing and F. Huang, Joule, 2020, 4, 2004–2016 CrossRef CAS.
- H. Bai, R. Ma, W. Su, T. A. D. Peña, T. Li, L. Tang, J. Yang, B. Hu, Y. Wang, Z. Bi, Y. Su, Q. Wei, Q. Wu, Y. Duan, Y. Li, J. Wu, Z. Ding, X. Liao, Y. Huang, C. Gao, G. Lu, M. Li, W. Zhu, G. Li, Q. Fan and W. Ma, Nanomicro Lett., 2023, 15, 241 CAS.
- C. Li, J. Zhou, J. Song, J. Xu, H. Zhang, X. Zhang, J. Guo, L. Zhu, D. Wei, G. Han, J. Min, Y. Zhang, Z. Xie, Y. Yi, H. Yan, F. Gao, F. Liu and Y. Sun, Nat. Energy, 2021, 6, 605–613 CrossRef CAS.
- Q. He, P. Ufimkin, F. Aniés, X. Hu, P. Kafourou, M. Rimmele, C. L. Rapley and B. Ding, SusMat, 2022, 2, 591–606 CrossRef CAS.
- C. Sun, J.-W. Lee, C. Lee, D. Lee, S. Cho, S.-K. Kwon, B. J. Kim and Y.-H. Kim, Joule, 2023, 7, 416–430 CrossRef CAS.
- J.-W. Lee, C. Sun, T. N.-L. Phan, D. C. Lee, Z. Tan, H. Jeon, S. Cho, S.-K. Kwon, Y.-H. Kim and B. J. Kim, Energy Environ. Sci., 2023, 16, 3339–3349 RSC.
- S. Qin, Z. Jia, L. Meng, C. Zhu, W. Lai, J. Zhang, W. Huang, C. Sun, B. Qiu and Y. Li, Adv. Funct. Mater., 2021, 31, 2102361 CrossRef CAS.
- H. Huang, X. Li, C. Sun, I. Angunawela, B. Qiu, J. Du, S. Qin, L. Meng, Z. Zhang, H. Ade and Y. Li, J. Mater. Chem. C, 2020, 8, 7718–7724 RSC.
- C. Sun, F. Pan, H. Bin, J. Zhang, L. Xue, B. Qiu, Z. Wei, Z.-G. Zhang and Y. Li, Nat. Commun., 2018, 9, 743 CrossRef.
- C. M. Hansen, Hansen Solubility Parameters: A User's Handbook, CRC Press, Boca Raton, 2007 Search PubMed.
- J. Kim, G.-U. Kim, D. J. Kim, S. Lee, D. Jeong, S. Seo, S.-J. Ko, S. C. Yoon, T.-S. Kim and B. J. Kim, J. Mater. Chem. A, 2023, 11, 4808–4817 RSC.
- Z. Chiguvare and V. Dyakonov, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 235207 CrossRef.
- H. Kang, K.-H. Kim, T. E. Kang, C.-H. Cho, S. Park, S. C. Yoon and B. J. Kim, ACS Appl. Mater. Interfaces, 2013, 5, 4401–4408 CrossRef CAS PubMed.
- S. Zhang, Z. Xue, Z. He, Q. Wei, N. Yang, X. Wu, C. C. Chen, C. Sun and H. Zhong, Chem. Eng. J., 2024, 481, 148728 CrossRef CAS.
- J. Jeon, K. Doi, H. D. Kim, H. Ogawa, M. Takenaka and H. Ohkita, Polym. J., 2023, 55, 477–487 CrossRef CAS.
- S. R. Cowan, A. Roy and A. J. Heeger, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 245207 CrossRef.
- B. A. Collins, Z. Li, J. R. Tumbleston, E. Gann, C. R. McNeill and H. Ade, Adv. Energy Mater., 2013, 3, 65–74 CrossRef CAS.
- L. Nian, Y. Kan, K. Gao, M. Zhang, N. Li, G. Zhou, S. B. Jo, X. Shi, F. Lin, Q. Rong, F. Liu, G. Zhou and A. K. Y. Jen, Joule, 2020, 4, 2223–2236 CrossRef CAS.
- H. Benten, D. Mori, H. Ohkita and S. Ito, J. Mater. Chem. A, 2016, 4, 5340–5365 RSC.
- D. Jeong, I.-Y. Jo, S. Lee, J. H. Kim, Y. Kim, D. Kim, J. R. Reynolds, M.-H. Yoon and B. J. Kim, Adv. Funct. Mater., 2022, 32, 2111950 CrossRef CAS.
- G. G. Jeon, M. Lee, J. Nam, W. Park, M. Yang, J.-H. Choi, D. K. Yoon, E. Lee, B. Kim and J. H. Kim, ACS Appl. Mater. Interfaces, 2018, 10, 29824–29830 CrossRef CAS.
- T. J. Aubry, A. S. Ferreira, P. Y. Yee, J. C. Aguirre, S. A. Hawks, M. T. Fontana, B. J. Schwartz and S. H. Tolbert, J. Phys. Chem. C, 2018, 122, 15078–15089 CrossRef CAS.
- Y.-F. Shen, H. Zhang, J. Zhang, C. Tian, Y. Shi, D. Qiu, Z. Zhang, K. Lu and Z. Wei, Adv. Mater., 2023, 35, 2209030 CrossRef CAS PubMed.
- D. Yang, S. Grott, X. Jiang, K. S. Wienhold, M. Schwartzkopf, S. V. Roth and P. Müller-Buschbaum, Small Methods, 2020, 4, 2000418 CrossRef CAS.
- J. J. van Franeker, M. Turbiez, W. Li, M. M. Wienk and R. A. J. Janssen, Nat. Commun., 2015, 6, 6229 CrossRef CAS.
- M. Deng, X. Xu, Y. Duan, L. Yu, R. Li and Q. Peng, Adv. Mater., 2023, 35, 2210760 CrossRef CAS PubMed.
- B. Li, X. Yang, S. Li and J. Yuan, Energy Environ. Sci., 2023, 16, 723–744 RSC.
- T. Kumari, J. Oh, S. M. Lee, M. Jeong, J. Lee, B. Lee, S.-H. Kang and C. Yang, Nano Energy, 2021, 85, 105982 CrossRef CAS.
- S. Rasool, J. W. Kim, H. W. Cho, Y.-J. Kim, D. C. Lee, C. B. Park, W. Lee, O.-H. Kwon, S. Cho and J. Y. Kim, Adv. Energy Mater., 2023, 13, 2203452 CrossRef CAS.
- Y. Xu, J. Yuan, S. Zhou, M. Seifrid, L. Ying, B. Li, F. Huang, G. C. Bazan and W. Ma, Adv. Funct. Mater., 2019, 29, 1806747 CrossRef.
- Z. Wang, K. Gao, Y. Kan, M. Zhang, C. Qiu, L. Zhu, Z. Zhao, X. Peng, W. Feng, Z. Qian, X. Gu, A. K. Y. Jen, B. Z. Tang, Y. Cao, Y. Zhang and F. Liu, Nat. Commun., 2021, 12, 332 CrossRef CAS PubMed.
- B. Li, Y. Kong, T. Li, H. Li, H. Zhao, P. Cheng and J. Yuan, Adv. Mater., 2024, 2408988 CrossRef CAS.
- L. Ye, S. Li, X. Liu, S. Zhang, M. Ghasemi, Y. Xiong, J. Hou and H. Ade, Joule, 2019, 3, 443–458 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta07223e |
| This journal is © The Royal Society of Chemistry 2025 |