
Ligand-protected and lowered-temperature hydrothermal synthesis of platinum encapsulated in TON zeolite for shape-selective hydrogenation of furfural to furfuryl alcohol†
Xuelin
Wang
ab,
Congxin
Wang
*a,
Wentao
Bi
ab,
Wei
Qu
a and
Zhijian
Tian
 *a
*a
aDalian National Laboratory for Clean Energy, State Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, 457 Zhongshan Road, Dalian 116023, China. E-mail: wangcx@dicp.ac.cn; tianz@dicp.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
First published on 13th November 2024
Abstract
Encapsulating metal in zeolite is an effective tactic to upregulate the catalytic selectivity of metal/zeolite catalysts in hydrogenation reactions by the spatial confinement of the zeolite microchannels. Herein, we present the synthesis of Pt encapsulated in zeolite with TON topology (Pt@ZSM-22) by adopting a ligand-protected and lowered-temperature hydrothermal crystallization. XRD, SEM, TEM, TG-DSC-MS, and 13C CP/MAS NMR are used to track the hydrothermal process. The experimental results indicate that the decomposition and reduction of the metal precursor ([Pt(en)2]2+), which usually occurs at harsh hydrothermal conditions, are effectively restrained at a lowered hydrothermal temperature (140 °C) and with ligand protection (ethylenediamine). The intact [Pt(en)2]2+ is electrostatically adsorbed onto the amorphous silicate nanoparticles (the zeolite precursor) and is encapsulated inside the ZSM-22 crystals as these nanoparticles are crystallized. The highly dispersed and uniform Pt particles embedded inside the ZSM-22 zeolite are successfully obtained by adopting a direct H2 reduction to remove the template and reduce [Pt(en)2]2+. The hydrogenation of furfural to furfuryl alcohol was conducted to evaluate the selective hydrogenation performance of the encapsulated Pt@ZSM-22. The reaction results reveal that the selectivity of furfuryl alcohol reaches as high as 97.6% at a conversion of 99.5% over the encapsulated Pt@ZSM-22, which is superior to the supported Pt/ZSM-22. The excellent selectivity of furfuryl alcohol reflects the shape selectivity conferred by the spatial confinement of the one-dimensional microchannels of ZSM-22. The CO-FT-IR, XPS, XAFS and FT-IR of adsorbed furfural are used to disclose the structure–activity relationship of Pt@ZSM-22. Our work not only successfully realizes the direct hydrothermal synthesis of metal encapsulation in zeolite with 1D straight channels but also demonstrates the great application potentials of such catalysts in selective catalysis.
Introduction
Metal/zeolite catalysts, exhibiting outstanding catalytic activity and selectivity, have been widely used in many important chemical processes, such as hydrogenation/dehydrogenation,1,2 hydroisomerization,3 hydrocracking4,5 and aromatization.6 The cooperation between the active metal sites and the zeolite micropores offers excellent catalytic performance for the catalysts. Lv et al.7 reported that Pt distribution played a key role in the hydroisomerization performance of the Pt/SAPO-11 catalyst. They found that the distribution of Pt sites near the pore mouth of SAPO-11 can promote the pore mouth shape-selectivity catalysis and improve the isomerization selectivity of Pt/SAPO-11. Xu et al.6 investigated the effect of the Pt location on the aromatization performances of Pt/KL catalysts. It was discovered that the catalyst with Pt inside the zeolite micropores, integrating the dehydrocyclization of Pt sites with the shape selectivity of the micropores, exhibited superior aromatization selectivity.Despite the good performance of metal/zeolite in selective catalysis, the controllable distribution of the metal sites in or on the zeolite remains challenging. The metals randomly distributed on the surface always cause poor catalytic selectivity. For instance, Gu et al.8 reported the Pt/ZSM-5 with the nonuniform distribution of Pt on the zeolite surface and revealed the poor selectivity toward aminostyrene in nitrostyrene hydrogenation. Wang et al.9 established Pd randomly supported on the SOD zeolite surface, and with this metal distribution, the Pd/SOD exhibited lower ethylene selectivity in acetylene hydrogenation.
Many efforts have been made to control the distribution of metal in zeolite, such as ion exchange with the exchangeable sites inside the zeolite micropores,10 selectively removing the metals from the zeolite surface by extraction.11 Among them, encapsulating metal inside zeolite to obtain unique metal catalysts named metal@zeolite catalysts is a versatile strategy to control the metal distribution and improve the catalytic selectivity. Weisz et al.12 reported the reactant shape selectivity in the hydrogenation of olefins over Pt@LTA. They found that only 1-butene was hydrogenated into butane with a mixture of small 1-butene and bulky isobutene as the reactant. The reactant shape selectivity was also discovered in the hydrogenation of olefins and the oxidation of alcohols over metals@LTA.13 It was discovered that the reaction rate of small molecules was remarkably higher than that of the bulky molecules. Cho et al.14 studied the tandem aldol condensation and hydrogenation of furfural and acetone and found improved selectivity toward the tandem reaction over the encapsulated Pt@HZSM-5 compared with the supported Pt/HZSM-5. For Pt/HZSM-5, the access of furfural to the supported Pt sites was unimpeded; thus, the undesired hydrogenation and decarbonylation of furfural prevailed. In the case of Pt@HZSM-5, the furfural and acetone molecules first encountered the Brønsted acid sites, and the aldol condensation occurred. The generated aldol adducts passed through the microchannels to contact the encapsulated Pt sites and were hydrogenated into the target C8 products. The above-encapsulated catalysts combined the shape selectivity of zeolite microchannels with the encapsulated active sites and exhibited shape-selective catalysis to improve catalytic selectivity. The change in the distribution of the active sites via encapsulation also benefited the improved reaction selectivity.
Hence, great interest has been aroused in the synthesis of metal@zeolite due to its advanced nature in selective catalysis. Geol et al.15 reported a series of metals (Pt, Pd, Ru, and Rh) encapsulated inside SOD and GIS zeolites. Encapsulation was achieved when the metal precursors stabilized by the ammonia or organic amine ligands were introduced into the hydrothermal system of zeolite. Similarly, Wang et al.16 synthesized Pd@MFI under hydrothermal conditions by adding the mixture of Pd precursor [Pd(NH2CH2CH2NH2)]Cl2 and ethylenediamine ligand into the synthesis gel. They found that the ultra Pd clusters were encaged within the intersectional channels of MFI zeolite. Choi and collaborators17 developed the mercaptosilane-assisted hydrothermal method for the encapsulation of metals (Pt, Pd, Ir, Rh and Ag) in NaA zeolite. The bifunctional (3-mercaptopropyl)trimethoxysilane ligands were used to coordinate with cationic metal centers via the mercapto groups and to form covalent linkages with the zeolite crystal nucleus via the alkoxysilane moieties, both of which synergistically led to the encapsulation of metals. By adopting this strategy, Deng et al.18 successfully synthesized the Pt@Y with the single-atom Pt encapsulated within the six-membered rings connecting the sodalite cages and supercages. The encapsulation of Pt and Ru in beta zeolite was also achieved with the bifunction ligands to stabilize metal precursors.19,20 The above studies show that direct hydrothermal synthesis is a general and straightforward strategy that contains synthetic steps similar to that of the pure zeolites besides the addition of metal precursors and ligands into the synthesis gel. Until now, the metals have been successfully encapsulated in zeolites with intersectional channels, cages or supercages using this method.
The metal/zeolite catalysts with one-dimensional straight channels, especially those with ten-membered pore windows (e.g., the ZSM-22 with TON topology), have been successfully applied in hydroisomerization.21,22 The unique pore structures introduce the pore mouth and key–lock shape-selective catalysis, resulting in the excellent isomerization selectivity of the catalysts. It has been reported that the metal encapsulated in the ZSM-22 zeolite was achieved by adopting the dry-gel conversion method.23,24 Metal species grabbed with the ligand group were first grafted onto amorphous aluminosilicate solid gel and then encapsulated into ZSM-22 zeolite with the crystallization of the solid gel. This method involved multiple synthetic steps and inevitably resulted in time-consuming processes for practical application compared with direct hydrothermal synthesis. However, the direct hydrothermal synthesis of the metal@zeolite with one-dimensional straight channels has rarely been reported. One of the main reasons is that the metal precursors always undergo premature precipitation and are transformed into bulky metal hydroxides or particles even in the presence of ligands (e.g., ammonia, organic amine, and mercaptosilane) due to the harsh hydrothermal environment (high temperature and pH), causing the failure of encapsulation inside the zeolite crystals with straight channels.15 Moreover, the subsequent oxidative calcination is always adopted to remove the ligands and organic template.15–18 Mobile metal oxides, such as PtO2 species, are generated and migrate out of the zeolite microchannels to fail encapsulation.25,26 The ability of straight channels to capture and immobilize metal oxide species is inferior to that of the zeolite cages and intersectional channels. Consequently, the metal oxide species might migrate easily out of the zeolites along the straight channels. This is also detrimental to the metal encapsulation inside the zeolites with one-dimensional straight channels.
Herein, we report the strategy of ligand-protected and lowered-temperature hydrothermal crystallization, followed by the direct H2 reduction for the synthesis of ZSM-22 encapsulating Pt (Pt@ZSM-22). ZSM-22 is added as the seed crystals to accelerate the zeolite crystallization. The lowered temperature (140 °C) and the added ligand (ethylenediamine) prevent the decomposition and reduction of Pt precursor [Pt(en)2]2+ into large Pt particles. This strategy leads to the intact [Pt(en)2]2+ encapsulated into ZSM-22 crystals. The encapsulated [Pt(en)2]2+ is directly reduced into the metallic Pt whose growth is restricted by the rigid zeolite framework. Hence, the highly dispersed and uniform Pt particles embedded inside the ZSM-22 zeolite are successfully obtained. In the furfural hydrogenation reaction, the Pt@ZSM-22 catalyst exhibits a selectivity of furfuryl alcohol as high as 97.6% at a conversion of 99.5%.
Experimental
Chemicals and materials
Colloidal silica (LUDOX HS-40, 40%, Sigma Aldrich), potassium hydroxide (KOH, AR, Sinopharm Chemical Reagent Co., Ltd), 1,6-hexanediamine (HDA, AR, Sinopharm Chemical Reagent Co., Ltd), aluminum sulfate (Al2(SO4)3·18H2O, AR, Tianjin Kemiou Chemical Reagent Co., Ltd), [Pt(en)2]Cl2 aqueous solution (0.02 g Pt per mL, Lab homemade), ethylenediamine (NH2CH2CH2NH2, ≥99.0%, Sinopharm Chemical Reagent Co., Ltd), tetraammineplatinum(II) chloride aqueous solution (Pt(NH3)4Cl2, 0.03 g Pt per mL, Lab homemade), ammonium hydroxide (NH3·H2O, 25–28%, Tianjin Damao Chemical Reagent Factory).Catalyst preparation
The Pt@ZSM-22 and ZSM-22 supports in this study are quasi-pure-silicon. It is difficult to synthesize high-purity pure-silicon ZSM-22 by applying the hydrothermal method. To solve this problem, few ZSM-22 seeds were added to the synthesis gel to promote the zeolite crystallization. The ZSM-22 seeds were synthesized by the conventional hydrothermal synthesis under rotation (50 rpm) at 160 °C for 48 h according to the method of Jamil et al.27 from the starting gel with the molar composition of 1.67 Al2O3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100 SiO2:14.1 K2O:30.2 HDA:4031H2O.
:100 SiO2:14.1 K2O:30.2 HDA:4031H2O.
Pt@ZSM-22 was synthesized using the one-pot hydrothermal method at a lowered temperature (140 °C) in the presence of ZSM-22 seeds and ethylenediamine chelating ligand, followed by direct H2 reduction. The quasi-pure-silicon Pt@ZSM-22 was obtained because the synthesis gel was pure-silicon, and no other heteroatoms were introduced except for the few aluminum atoms contained in the added ZSM-22 seeds. As a typical run, KOH, HDA and deionized water were mixed under stirring to obtain a clear solution. After 30 min, a mixture of ethylenediamine and [Pt(en)2]Cl2 aqueous solution was added and stirred for 10 min. LUDOX HS-40 was added and stirred for 1 hour to obtain the initial gel with the molar composition of 100 SiO2:37.5 HDA:5 K2O:3800 H2O:8.86 NH2CH2CH2NH2:y [Pt(en)2]Cl2 (y = 0.061, 0.123, 0.185, 0.247, 0.308). Then, ZSM-22 seeds were added and stirred for 10 min. The quantity of the seeds added was 1 wt% based on the weight of SiO2. The resultant mixture was transferred into a 100 mL Teflon-lined autoclave and crystalized for 24 h under rotation (50 rpm). The solid product was washed, filtered and then dried at 120 °C for 3 h to obtain the as-synthesized samples (Ptx@ZSM-22-as). Ptx@ZSM-22-as were directly reduced with a temperature ramping to 400 °C for 2 h and holding for 2 h under a H2 atmosphere (100 mL min−1). The obtained samples were denoted as Ptx@ZSM-22-H, where x represents the Pt loading (wt%) determined by ICP-OES.
In contrast to the direct-reduction method, the conventional calcination–reduction method was also adopted to treat the Pt0.6@ZSM-22-as. The calcination was conducted at 550 °C for 12 h under an air atmosphere to obtain the Pt0.6@ZSM-22-C. The subsequent reduction was performed at 400 °C for 4 h under a H2 atmosphere to obtain the Pt0.6@ZSM-22-C-H.
The ZSM-22 support (ZSM-22-H) synthesized followed procedures similar to those of Ptx@ZSM-22-H except for the addition of the mixture of ethylenediamine and [Pt(en)2]Cl2 aqueous solution.
The supported Pt0.6/ZSM-22-H catalyst was prepared using a strong electrostatic adsorption method. Typically, the support (ZSM-22-H) was dispersed in diluted ammonium hydroxide solution with pH = 12.0 and stirred for 30 min. Then, the Pt(NH3)4Cl2 solution was added and stirred for another 1 h. The solid products were obtained after filtration, washed with deionized water and dried at 120 °C for 3 h. The ratio of the support relative to the solution was 1 g/50 mL. The theoretical Pt loading was 0.6 wt%, and the volume of Pt(NH3)4Cl2 was calculated based on the mass of the support. After direct H2 reduction, the Pt0.6/ZSM-22-H catalyst was obtained. The practical Pt loading is 0.33 wt% (ICP-OES, Table S1†) for Pt0.6/ZSM-22-H, which is lower than the theoretical loading because of the insufficient hydroxyl on the external surface of ZSM-22-H to load sufficient [Pt (NH3)4]2+.28
Characterization
Powder X-ray diffraction (XRD) patterns were collected using a PANalytical Empyrean-100 X-ray diffractometer with a Cu Kα radiation source (λ = 1.5418 Å, 40 kV, 40 mA) in the 2θ range of 5–60°.Nitrogen physical adsorption was performed at −196 °C using a Micromeritics ASAP 2420 analyzer to obtain the specific surface area and pore volume of the sample. Before the measurement, the sample was degassed by evacuating at 350 °C for 8 h.
The Pt loading of the catalysts was determined by Inductively Coupled Plasma-Optical Emission Spectroscopy (ICP-OES) measurement performed using a PerkinElmer Optima 7300DV instrument.
The H2 chemisorption was conducted on a Micromeritics AutoChemII 2920 instrument using a pulse injection of 10 vol% H2/Ar at 40 °C to determine the Pt surface site concentration, which was defined as the number of Pt sites per unit mass of the sample and calculated under the assumption H/Pt = 1. Before adsorption, the sample was pretreated at 400 °C for 1 h under 10 vol% H2/Ar and then cooled down to 40 °C under Ar.
Scanning electron microscopy (SEM) images were obtained using a JSM-7800F filed-emission scanning electron microscope with an accelerating voltage of 3 kV.
Transmission microscope microscopy (TEM) images were recorded using a JEM-2100 electron microscope at 200 kV. Metal particle size distribution was obtained by counting at least 300 particles. The average size was calculated using dTEM = ∑nidi4/∑nidi3 based on the volume-weighted particle size statistics from the TEM results.
Ultramicrotomy combined with high-resolution scanning transmission electron microscopy (STEM) imaging was conducted to image the Pt species in Pt@ZSM-22-H. The sample was dispersed in epoxy resin and then dried at 80 °C for 24 h. The resin-embedded sample was sliced into sections of 30 nm in thickness. The resultant microtomed sections were imaged by applying a JEOL JEM F200.
Thermogravimetric analyses (TGs) were conducted using a TA TGA55 thermal analyzer in 20% O2/N2 with a heating rate of 10 °C min−1.
Thermogravimetric and mass spectrometric analyses (TG-MS) were recorded using a simultaneous thermal analyzer (NETZSCH, STA449F5) equipped with a mass spectrometer (Pfeiffer Vacuum, GSD350 ThermoStar) in 20% O2/N2 with a heating rate of 10 °C min−1. In this characterization, the TG curve was collected as the weight loss of the sample occurred by heating or oxidation burning. Simultaneously, the resulting gas products were monitored by the MS.
13C CP/MAS NMR spectra were collected using an AVANCE III HD 600 WB spectrometer at a resonance of 150.9 MHz with a spinning rate of 10 kHz and a MAS probe of 4 mm.
The chemical composition of the samples was determined by X-ray fluorescence (XRF) spectroscopy using a PANalytical Zetium instrument.
The acid properties of the catalysts were determined by Fourier transform infrared spectroscopy of adsorbed pyridine (Py-FT-IR) using a ThermoFisher Scientific Nicolet iS50 spectrometer equipped with a DTGS KBr detector. Before the test, the sample was prepared into a self-supported wafer with a diameter of 13 mm, and the wafer was treated at 350 °C for 1 h under a high vacuum (<10−3 Pa) to remove physiosorbed water. After being cooled to 25 °C, the IR spectrum of the dehydrated sample was collected; then, the wafer was exposed to the pyridine vapor. The IR spectrum of the pyridine-adsorbed sample was collected after evacuating the pyridine-adsorbed samples for 30 min at 150 °C. The difference spectrum was obtained by subtracting the spectrum of the dehydrated sample from the spectrum of the pyridine-adsorbed one.
In situ CO-Fourier transform infrared spectra (CO-FT-IR) were carried out using a Brucker TENSOR27 spectrometer equipped with an MCT detector (resolution: 4 cm−1): 20 mg of sample was prepared into a self-supported wafer with a diameter of 13 mm, followed by pre-treatment at 350 °C for 1 h under a H2 atmosphere in a quartz sample cell. Then, the sample cell was purged by N2 flow and cooled to about 25 °C before a background IR spectrum was collected. CO was introduced at ambient temperature for 30 min; then, the sample cell was purged by N2 flow for 1 hour to remove gaseous CO before the IR spectrum of adsorbed CO was recorded. The CO-FT-IR spectrum was obtained by subtracting the background spectrum from the CO-adsorbed spectrum.
The X-ray photoelectrons spectroscopy (XPS) spectra were collected at a ThermoFisher Escalab 250 Xi+ with Al Kα radiation as the excitation source. All binding energies were calibrated with the C 1s peak at 284.8 eV.
The X-ray absorption fine structure spectra (XAFS) (Pt L3-edge) were collected at the BL14W beamline in the Shanghai Synchrotron Radiation Facility (SSRF). The storage rings of SSRF were operated at 3.5 GeV with a stable current of 200 mA. Using a Si (111) double-crystal monochromator, the data collection was carried out in fluorescence mode using an SDD detector. Before the test, all the samples were reduced under 10 vol% H2/Ar at 350 °C for 1 h and then cooled to room temperature. The reduced samples were sealed in the plastic centrifuge tubes using the sealing film in a glove box under the N2 atmosphere to avoid exposure to air. All spectra were collected under ambient conditions. The X-ray adsorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) data were processed and analyzed according to standard procedures using the Athena module implemented in the IFEFFIT software packages. The EXAFS spectra were obtained by subtracting the post-edge background from the overall absorption and then normalizing it with respect to the edge-jump step. Subsequently, the χ(k) data were Fourier transformed into real (R) space using Hanning windows (dk = 1.0 Å−1) to separate the EXAFS contributions from different coordination shells. To obtain the quantitative structural parameters around the central atoms, least-squares curve parameter fitting was performed using the ARTEMIS module of the IFEFFIT software packages. The amplitude reduction factor (S02) was fixed to 0.874 according to the experimental EXAFS fit of Pt foil by fixing CN as the known crystallographic value.
The adsorption behavior of furfural was studied by Fourier transform infrared spectroscopy of adsorbed furfural (FFL-FT-IR) using a ThermoFisher Scientific Nicolet iS50 spectrometer equipped with a DTGS KBr detector. The sample wafer with a diameter of 13 mm was treated at 350 °C for 1 h under 10 vol% H2/Ar and then cooled to room temperature (about 30 °C). The spectrum of the pretreated sample was collected after evacuating below 10−3 Pa. In the glove box (O2 and H2O content below 0.5 ppm), furfural was added dropwise onto the pretreated wafer using a pipette. The spectrum of the furfural-adsorbed sample was recorded after evacuation (<10−3 Pa) at different temperatures (30 °C, 80 °C, 130 °C and 180 °C). The difference spectrum was obtained by subtracting the spectrum of the pretreated sample from the spectrum of the furfural-adsorbed one.
Catalytic test
The hydrogenation of furfural was performed at a 25 mL batch-type stainless-steel autoclave equipped using a magnetic stirrer. Typically, the reactor was charged with a 0.10 g catalyst and a mixture of 10 mL isopropanol and 0.10 g furfural. After repeatedly purging with 1 MPa N2 and H2 three times under magnetic stirring at 1000 rpm, the reactor was pressurized with hydrogen to the desired pressure (P) at room temperature. Then, the reactor was heated to a certain reaction temperature (T), and the reaction time (t) was recorded. The reactor was naturally cooled to room temperature and depressurized carefully after a certain reaction time. The solid catalyst and the liquid were separated by filtration, and the liquid was analyzed using an Agilent 8890-7250 GC-MS and an Agilent 7890A GC equipped with a flame ionization detector (FID) and a DB-WAX capillary column (30 m × 0.200 mm × 0.5 μm). The quantification was conducted using the inner standard method with n-hexanol as the inner standard. The conversion of furfural and product selectivity were calculated by applying the following equations:
where nfeeding, nresidue and nproduct(i) represent the mole number of furfural feeding, unconverted furfural and product i, respectively; Si represents the selectivity of product i.
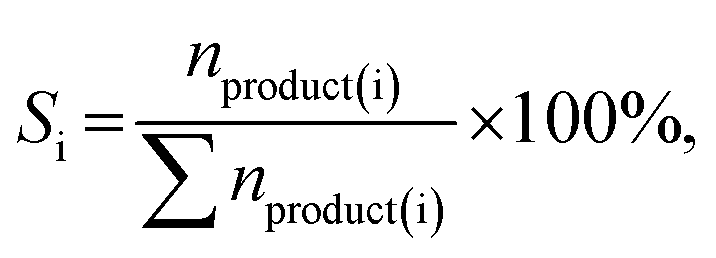
Results and discussion
Synthesis of Pt encapsulated in ZSM-22
ZSM-22 encapsulating Pt (Pt@ZSM-22) is obtained using ligand-protected and lowered-temperature one-pot hydrothermal synthesis and subsequent direct H2 reduction. By tuning the amount of [Pt(en)2]Cl2 in the initial gel, the Ptx@ZSM-22-H with the Pt loading of 0.2–1.0 wt% is obtained, as listed in Table 1. XRD patterns (Fig. 1a) show that all the samples possess the intense characteristic diffraction peaks as those of the ZSM-22 support (ZSM-22-H) and no peaks of impurity are detected. This demonstrates the good ZSM-22 crystallinity and high Pt dispersion. SEM images show that all the samples possess a similar morphology of interpenetrating shuttles as ZSM-22-H (Fig. S1†). The texture properties of all the Ptx@ZSM-22-H are almost identical to those of ZSM-22-H (Table 1), indicating that the introduction of Pt barely causes the plug of zeolite channels.| Sample | S BET (m2 g−1) | S micropore (m2 g−1) | V micropore (cm3 g−1) | Pt loadinga (wt%) | C H2 (μmol g−1) | d TEM (nm) |
|---|---|---|---|---|---|---|
| a By ICP-OES. b Concentration of Pt surface sites determined by H2 chemisorption. c Average particle size of Pt determined by TEM. | ||||||
| Pt0.2@ZSM-22-H | 203.1 | 183.7 | 0.090 | 0.20 | 2.3 | 2.8 |
| Pt0.4@ZSM-22-H | 208.3 | 185.2 | 0.091 | 0.41 | 5.5 | 2.8 |
| Pt0.6@ZSM-22-H | 205.7 | 179.0 | 0.088 | 0.62 | 9.8 | 3.0 |
| Pt0.8@ZSM-22-H | 216.1 | 186.7 | 0.092 | 0.84 | 11.3 | 3.4 |
| Pt1.0@ZSM-22-H | 223.3 | 184.2 | 0.091 | 1.05 | 14.4 | 3.6 |
| ZSM-22-H | 203.3 | 183.1 | 0.090 | — | — | — |
 | ||
| Fig. 1 XRD patterns of samples. Direct-reduction samples of (a) Ptx@ZSM-22-H and ZSM-22-H. The as-synthesized samples obtained (b) at 130–150 °C with ethylenediamine and (c) at 140 °C without ethylenediamine. | ||
The TEM images and particle size statistic results (Fig. 2a–e) reveal that Pt exists as small particles that are intensively distributed between 1 and 3 nm for all the Ptx@ZSM-22-H. With the increase in the Pt loading, the number of the Pt particles above 3 nm slightly increases. The average particle size, calculated based on the statistical results, shows a slight increase from 2.8 nm to 3.6 nm (Table 1). Notably, TEM provides a two-dimensional projection of the sample. If the Pt particles were supported on the zeolite external surface, they would be observed at the edge of the ZSM-22 crystals and overlapping with the crystals in the projected image. The supported Pt/ZSM-22 (i.e. the Pt0.6/ZSM-22-H) is prepared with Pt(NH3)4Cl2 as a precursor using the electrostatic adsorption method. Because the kinetic diameter of Pt(NH3)42+ (0.48 nm) is close to the pore size of ZSM-22 (0.46 nm × 0.57 nm),29 it is difficult for Pt(NH3)42+ to enter into the zeolite micropores. As shown in Fig. 2g, the Pt particles with an average size of 2.5 nm have an intensive distribution of 1–3 nm. Apart from those overlapping with the zeolite crystals, a considerable number of Pt particles are at the crystal edge (indicated by the yellow arrows), which is consistent with the above description of the TEM image of the Pt particles located on the zeolite external surface. As compared with the supported Pt0.6/ZSM-22-H, the Ptx@ZSM-22-H exhibits a different Pt distribution in the TEM images (Fig. 2a–e). The Pt particles almost exist in the manner of overlapping with the ZSM-22 crystals, except for very few Pt particles located at the crystal edge (indicated by the yellow arrows) for the samples with high loading. It is deduced that the Pt particles of Ptx@ZSM-22-H are located at the inner part of the zeolite crystals, except for a few particles of high-loading samples that are on the external surface.
 | ||
| Fig. 2 TEM images and Pt particle size distribution of (a) Pt0.2@ZSM-22-H, (b) Pt0.4@ZSM-22-H, (c) Pt0.6@ZSM-22-H, (d) Pt0.8@ZSM-22-H, (e) Pt1.0@ZSM-22-H, (f) Pt0.6@ZSM-22-H-C, (g) Pt0.6/ZSM-22-H and (h) Pt0.6/ZSM-22-H-C; (i) high-resolution STEM image of sliced Pt0.6@ZSM-22-H with a slice thickness of 30 nm. All the samples were reduced at 400 °C before the images were collected. | ||
To further examine the Pt location of Pt@ZSM-22, the cross section of Pt0.6@ZSM-22-H is analyzed. The Pt0.6@ZSM-22-H was embedded into the resins and then cut into a slice of 30 nm thickness by ultramicrotomy to expose the cross section. The obtained slice was imaged using high-resolution STEM. Fig. 2i shows a high-resolution STEM image of the sliced sample. It is observed that all the Pt particles are dispersed inside the zeolite crystals, while almost no Pt particles are on the zeolite surface. Combined with the above TEM results, it is concluded that the Pt particles with uniform particle sizes of 1–3 nm are encapsulated inside ZSM-22.
The encapsulated Pt particles of the Pt@ZSM-22 exhibit remarkable resistance to sintering. The Pt0.6@ZSM-22-H was calcinated at 500 °C for 6 h in air to obtain the Pt0.6@ZSM-22-H-C. The results of the TEM images and particle size statistics show a similar Pt particle size distribution and average size for both samples (Fig. 2c and f, Tables 1, and S1†), which means that the Pt particles are significantly anti-sintering. For comparison, the supported Pt/ZSM-22 (Pt0.6/ZSM-22-H) catalyst was also calcinated at 500 °C for 6 h. The Pt particles on the obtained Pt0.6/ZSM-22-H-C present a considerable increase in average size (from 2.5 nm to 9.9 nm) and a significantly wider size distribution compared with those of the Pt0.6/ZSM-22-H (Fig. 2g, h, Table S1†), indicating the severe sintering. It was proposed that the sintering of the Pt under an oxidative atmosphere obeyed the molecular migration model with PtO2 molecules as the mobile species,26 which was further sustained by the EXAFS study30 and the sintering simulation.31 The outer-layer Pt atoms of one Pt particle were oxidized into PtO2, then migrated and attached to another Pt particle to grow this particle. The oxidization of the Pt atoms, the migration of the generated PtO2 and the growth of the Pt particle on the zeolite external surface are inclined to occur for the supported Pt0.6/ZSM-22-H because of the weak Pt–zeolite interaction. The sintering resistance of the encapsulated Pt particles reflects the enhanced Pt–zeolite interaction, which restricts the oxidization–migration–growth process.
The experimental parameters for the synthesis of the encapsulated Pt@ZSM-22 are explored. It is found that the key aspects of the successful synthesis are lowered hydrothermal temperature (140 °C), the addition of ZSM-22 seeds, the addition of ethylenediamine chelating ligand and the direct hydrogen reduction after the crystallization.
The hydrothermal synthesis of ZSM-22 zeolite always requires a high temperature (160–200 °C) to obtain good crystallinity.27 Given that the Pt precursor tends to precipitate and grow into big Pt particles under harsh hydrothermal conditions (high temperature and high pH), the hydrothermal temperature is reduced to make the hydrothermal environment milder. However, the well-crystallized zeolite is usually not available at a lowered temperature, and the ZSM-22 seeds are added to accelerate the lowered-temperature zeolite crystallization. The cristobalite peak and the Pt (111) peak are also observed in addition to the ZSM-22 peaks in the XRD patterns of the samples crystallized at 150 °C (Fig. 1b). The appearance of Pt diffraction peak means that the [Pt(en)2]Cl2 is decomposed and reduced into big Pt particles. Despite the protection of the ethylenediamine ligand, the high temperature (150 °C) and high pH along with the reductive organic amine (HDA) still cause the occurrence of the undesired decomposition and reduction reaction. This undesired reaction is suppressed by further reducing the temperature to 130 °C and 140 °C. In the presence of ZSM-22 seeds, 140 °C is enough to obtain ZSM-22 with good crystallinity, while 130 °C is too low to obtain it (Fig. 1b). Hence, the lowered temperature of 140 °C is suitable for obtaining the sample with good crystallinity and highly dispersed Pt.
The ethylenediamine added to the synthesis gel acts as the chelating ligand to prevent the [Pt(en)2]Cl2 from decomposition and reduction. In the absence of ethylenediamine, a broad Pt (111) diffraction peak is observed besides the intense ZSM-22 peaks in the XRD patterns (Fig. 1c), indicating the generation of big Pt particles. This means that the decomposition and reduction of [Pt(en)2]Cl2 occur in the lack of the protection of ethylenediamine. In the presence of ethylenediamine, the Pt (111) diffraction peak disappears (Fig. 1b, 140 °C), indicating the well-dispersed Pt species. The decomposition–reduction process is suppressed under the protection of ethylenediamine.
The template inside the as-synthesized metal@zeolite should be removed in a post-synthesis treatment to expose the zeolite microchannels. Heating under H2 atmosphere (direct H2 reduction) was proved to be an efficient and mild approach to remove the template.32,33 Herein, the as-synthesized Pt@ZSM-22 was treated using direct H2 reduction. This approach can remove the template and reduce the encapsulated Pt species. The TG curve of Pt0.6@ZSM-22-H (Fig. S2†) presents a slight weight loss of about 0.7% between 200 °C and 550 °C, which is attributed to the few residues of hydrocracked template, indicating the highly efficient removal of the template by adopting the direct H2 reduction. TEM result reveals the Pt particles with an averaged particle size of 3.0 nm (Table 1) and a relatively narrow size distribution for the direct-reduction Pt0.6@ZSM-22-H (Fig. 2c). In the direct reduction, the Pt species in the as-synthesized Pt0.6@ZSM-22 (Pt0.6@ZSM-22-as) are directly reduced into metallic Pt, which tends to coalesce into Pt particles due to the high surface energy. Owing to the hindrance of the rigid zeolite framework, this coalescence occurs in a limited manner, which contributes to the formation of small particles.
In contrast, the conventional calcination and subsequent H2 reduction were also conducted for the post-synthesis treatment of the Pt0.6@ZSM-22-as. The calcination-reduction Pt0.6@ZSM-22-C-H shows larger Pt particles with a mean size of 8.9 nm (Table S1†) and a wider size distribution, accompanied by partially large particles on the external surface of ZSM-22 (Fig. S3,† indicating the yellow arrow). In the calcination, the encapsulated Pt species is transformed into the mobile PtO2 species, similar to that produced in the oxidative sintering of Pt particles, which would have a chance to migrate out of the ZSM-22 crystals along the straight channels. Once out of the channel, it is difficult to migrate back because the channel is too small. They agglomerate into large particles on the zeolite external surface due to the weak interaction. After the subsequent reduction, the large Pt particles are obtained on the surface. Consequently, avoiding the calcinating process and adopting the direct H2 reduction is effective in inhibiting the migration of the encapsulated Pt species out of the ZSM-22 microchannels.
In brief, the highly dispersed Pt with a uniform particle size is encapsulated inside the ZSM-22 crystals by employing the above strategy. The lowered hydrothermal temperature and the added ligand offer a guarantee of the high dispersion of Pt species inside ZSM-22. The post-synthesis treatment by the direct H2 reduction leads to smaller Pt particles and avoids the aggregation of Pt particles on the zeolite surface compared with the conventional calcination–reduction process.
Investigation of the encapsulation mechanism of Pt@ZSM-22
The Pt-encapsulated crystallization process of Pt@ZSM-22 is investigated. Fig. S4† shows the XRD patterns of the samples obtained at different times and the corresponding crystallization curves. A 100% relative crystallinity is achieved with a crystallization time of 24 h, which consists of an induction period of 12 h and a growth period of 12 h. Fig. 3a–f shows the corresponding SEM images of the samples. In the induction period (6 h and 12 h), the obtained samples (Fig. 3a and b) are mainly composed of amorphous silicate nanoparticles. In the growth period (15–24 h), these nanoparticles gradually fade away, and the mutually interpenetrating shuttles are fully formed at 24 h (Fig. 3c–f). | ||
| Fig. 3 SEM images of the samples obtained with the molar composition of 100 SiO2:37.5 HDA:5 K2O:3800 H2O:8.86 NH2CH2CH2NH2:0.185 [Pt(en)2]Cl2 at different crystallization times: (a) 6 h, (b) 12 h, (c) 15 h, (d) 18 h, (e) 21 h, and (f) 24 h. TEM images of the direct-reduction samples obtained at different crystallization times: (g) 12 h, (h) 15 h, and (i) 24 h. | ||
The morphology of the samples crystallized for 12 h, 15 h and 24 h is imaged via TEM to further investigate the evolution of the silicate nanoparticles. The filtration, washing and drying are conducted to separate the solid products from the suspension mixture at different crystallization times. The separated solid products are treated by direct H2 reduction to obtain the samples for TEM imaging. This direct reduction can remove the template contained in the solid products and reduce the Pt species, which is beneficial in obtaining the TEM images with better contrast. The sample at 12 h is composed of a mass of amorphous silicate nanoparticles adhered to the surface of the zeolite shuttles (Fig. 3g). The Pt particles are observed on these nanoparticles and shuttles. Even though the samples are filtered and washed when they are collected, the Pt cation species ([Pt(en)2]2+ or its derivatives) are still located on the samples, indicating that they are strongly adsorbed to avoid being washed off. Given that the samples are synthesized at a pH of about 13 above the point of zero charge (PZC) of silica (about 4),34 these silica nanoparticles are negatively charged. The strong adsorption between the Pt cation species and the negatively charged silica nanoparticles is most likely due to electrostatic attraction. Prolonging the crystallization to 15 h, the attached silicate nanoparticles diminish dramatically; correspondingly, many new nanorods parallel to the long axis of shuttles are generated (Fig. 3h). This means that these disappeared silicate nanoparticles are transformed into the nanorods, which are assembled into the zeolite shuttles through the sideway fusion. Finally, the interpenetrating shuttle-like zeolite composed of nanorods is obtained at 24 h (Fig. 3i). This is accompanied by the disappearance of the amorphous nanoparticles and the incorporation of Pt particles inside the zeolite shuttles. According to the above results, it could be considered that the Pt species are adsorbed on the amorphous silicate nanoparticles by electrostatic attraction in the crystallization process of Pt@ZSM-22. These silicate nanoparticles crystallize to form the nanorods assembled into the zeolite. The adsorbed Pt species are encapsulated inside zeolite during the crystallization and assembly.
The TG-MS characterization was conducted under 20% O2/N2 to probe the organic components contained in the as-synthesized Pt@ZSM-22 (Ptx@ZSM-22-as) by analyzing the oxidation burning products, with the ZSM-22 support (ZSM-22-as) as the reference sample. The TG curves and the corresponding MS curves are shown in Fig. 4a–c. Three stages of mass loss caused by heating or oxidation burning are observed in the TG curves of the ZSM-22-as (Fig. 4a). The loss below 200 °C is accompanied by the appearance of one peak (150 °C) in the MS curve of H2O (Fig. 4b), which is attributed to the desorption of physically adsorbed water (1.3%). The loss of 200–310 °C is accompanied by the appearance of two peaks (265 °C) in the two MS curves of H2O and CO2, and the loss of 360–550 °C by the appearance of three peaks (430 °C) in the three MS curves of H2O and CO2 and NO2, both of which are attributed the burning of organic compounds. Because the HDA template is the only organic compound added to the initial gel of ZSM-22-as, it is concluded that the last two losses are caused by the two-stage burning of HDA. In the case of Pt0.6@ZSM-22-as, another weight loss of 310–360 °C (0.8%) in the TG curve (Fig. 4a) together with another three peaks (340 °C) in the three MS curves (Fig. 4c) are observed apart from those similar to the ZSM-22-as, corresponding to the burning of another compound. Because ethylenediamine is also incorporated in the initial gel besides HDA, it is deduced that the extra weight loss peak and exothermic peak are attributed to the burning of ethylenediamine. Thus, the TG-DSC-MS results confirm that ethylenediamine is incorporated in the as-synthesized Pt@ZSM-22.
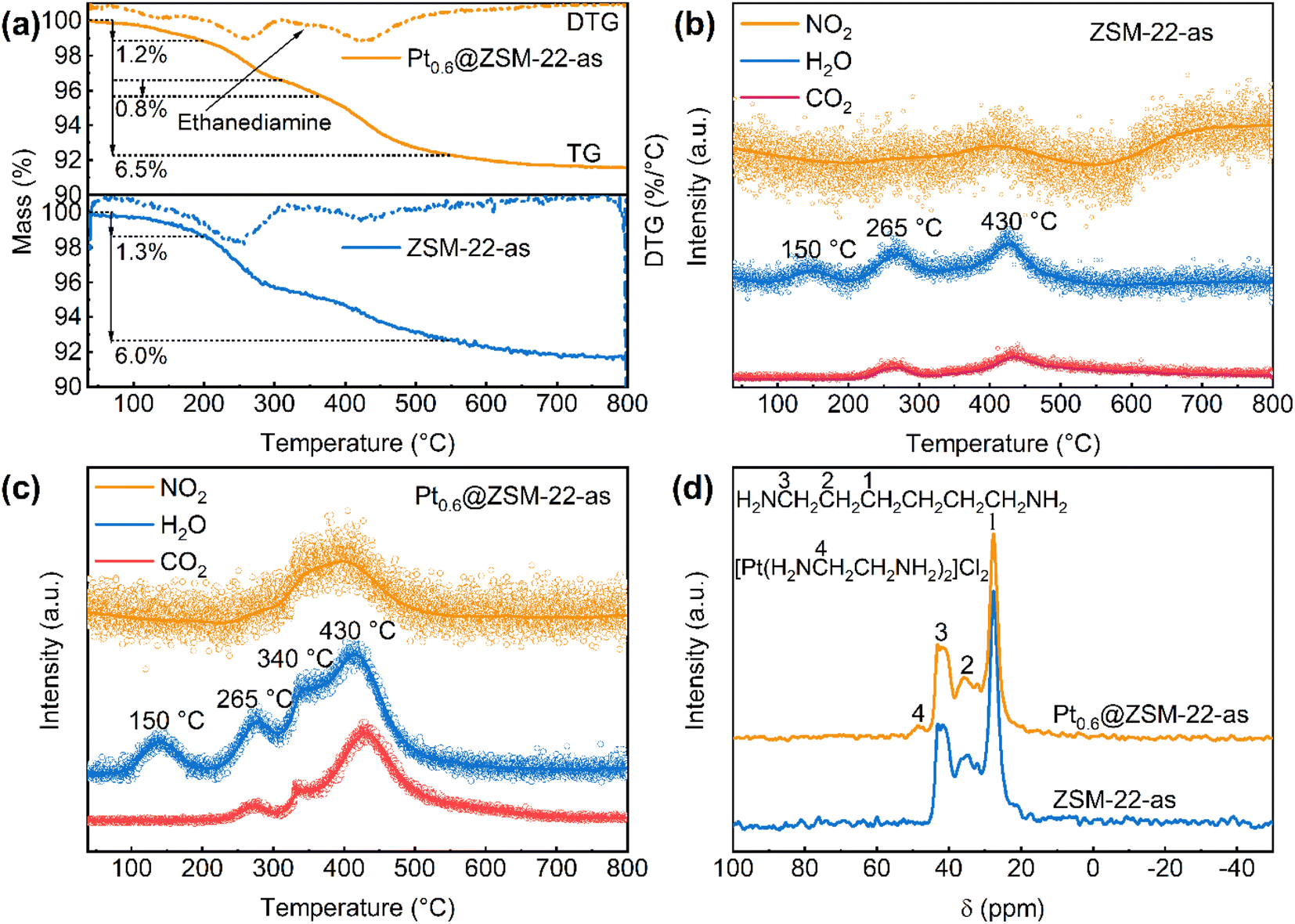 | ||
| Fig. 4 (a) TG-DTG curves of Pt0.6@ZSM-22-as and ZSM-22-as. TG-MS ion curves of (b) ZSM-22-as and (c) Pt0.6@ZSM-22-as monitored for H2O (m/z = 18), CO2 (m/z = 44) and NO2 (m/z = 30). (d) 13C CP/MS NMR spectra of Pt0.6@ZSM-22-as and ZSM-22-as. | ||
The solid-state 13C CP/MAS NMR was measured for Pt0.6@ZSM-22-as and ZSM-22-as, which can provide information about the local structure and coordination of organic compounds. As shown in Fig. 4d, three peaks (peaks 1, 2 and 3) located between 20 ppm and 45 ppm for both samples are associated with HDA. Wang et al.35 reported similar resonance peaks of HDA inside MFI and FER. The unique resonance peak at about 48 ppm (peak 4) in the spectra of Pt0.6@ZSM-22-as is ascribed to –CH2– of ethylenediamine in [Pt(en)2]2+. The [Pt(en)2]2+ in MFI exhibited a similar resonance peak.25 This indicates that the ethylenediamine in Pt0.6@ZSM-22-as acts as the chelating ligand of [Pt(en)2]2+. Pt existing as the cationic complex of [Pt(en)2]2+ confirms its high dispersion in the as-synthesized Pt@ZSM-22. Meanwhile, the above results demonstrate that [Pt(en)2]2+ cations are not decomposed under the protection of ethylenediamine at the lowered hydrothermal temperature (140 °C) and they are the adsorbed Pt species on the silicate nanoparticles in hydrothermal crystallization.
Hence the Pt encapsulated within ZSM-22 is successfully synthesized by adopting the ligand-protected and lowered-temperature one-pot hydrothermal synthesis, followed by subsequent direct H2 reduction. The graphical encapsulation mechanism is illustrated in Scheme 1. Owing to the synergy of the lowered hydrothermal temperature (140 °C) and the added ethylenediamine ligand, the Pt precursor [Pt(en)2]2+ remains intact during hydrothermal crystallization. ZSM-22 is added as the crystal seeds to expedite the lowered-temperature crystallization of zeolite. Under the alkaline hydrothermal condition (pH of about 13), the amorphous silicate nanoparticles are negatively charged because the pH is higher than the PZC of silica (about 4). The intact [Pt(en)2]2+ cations are adsorbed on the negatively charged amorphous silicate nanoparticles by electrostatic attraction and then are encapsulated as these nanoparticles are crystallized into zeolite. After crystallization, the as-synthesized Pt@ZSM-22 is treated by direct H2 reduction to reduce [Pt(en)2]2+ into metallic Pt. The coalescence and growth of the metallic Pt are restricted by applying the rigid zeolite framework. Therefore, the well-dispersed and uniform Pt particles are encapsulated inside ZSM-22 crystals.
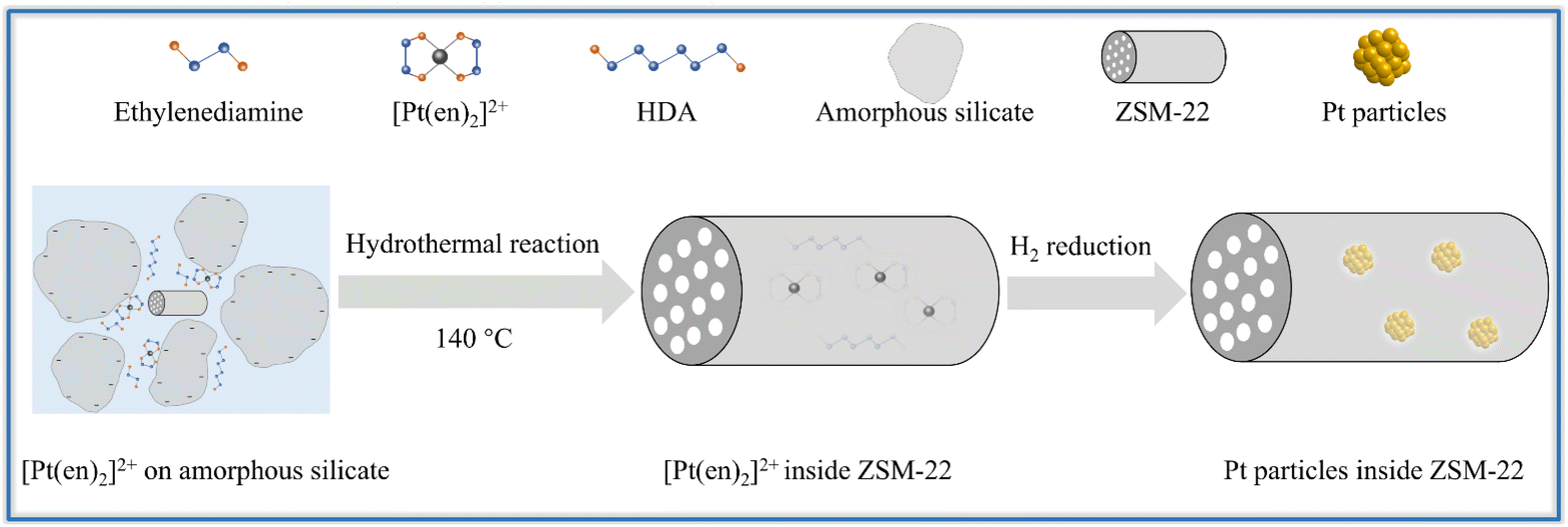 | ||
| Scheme 1 Schematic of the encapsulation mechanism of Pt in ZSM-22 by direct hydrothermal synthesis. | ||
Characterization of acid properties and Pt structure features of Pt@ZSM-22
The encapsulated Pt@ZSM-22 is quasi-pure-silicon (SiO2 of 98.54 wt% and Al2O3 of 0.15 wt%, Table S2†) because no other heteroatoms are introduced into the pure-silicon synthesis gel except for few aluminum atoms contained in the added ZSM-22 seeds. The acid properties of the Pt@ZSM-22 are analyzed by the FT-IR spectra of adsorbed pyridine. As shown in Fig. 5a, the peak at 1445 cm−1 ascribed to Lewis acid sites (LASs) is observed for the encapsulated Pt0.6@ZSM-22-H, along with the supported Pt0.6/ZSM-22-H and the ZSM-22-H support, while no peak at 1540 cm−1 ascribed to Brønsted acid sites (BASs) is detected. The smaller LAS peak of Pt0.6/ZSM-22-H than that of Pt0.6@ZSM-22-H and ZSM-22-H reflects the diminished LAS. This is perhaps due to the partial occupation of LAS by the Pt particles on the ZSM-22 external surface.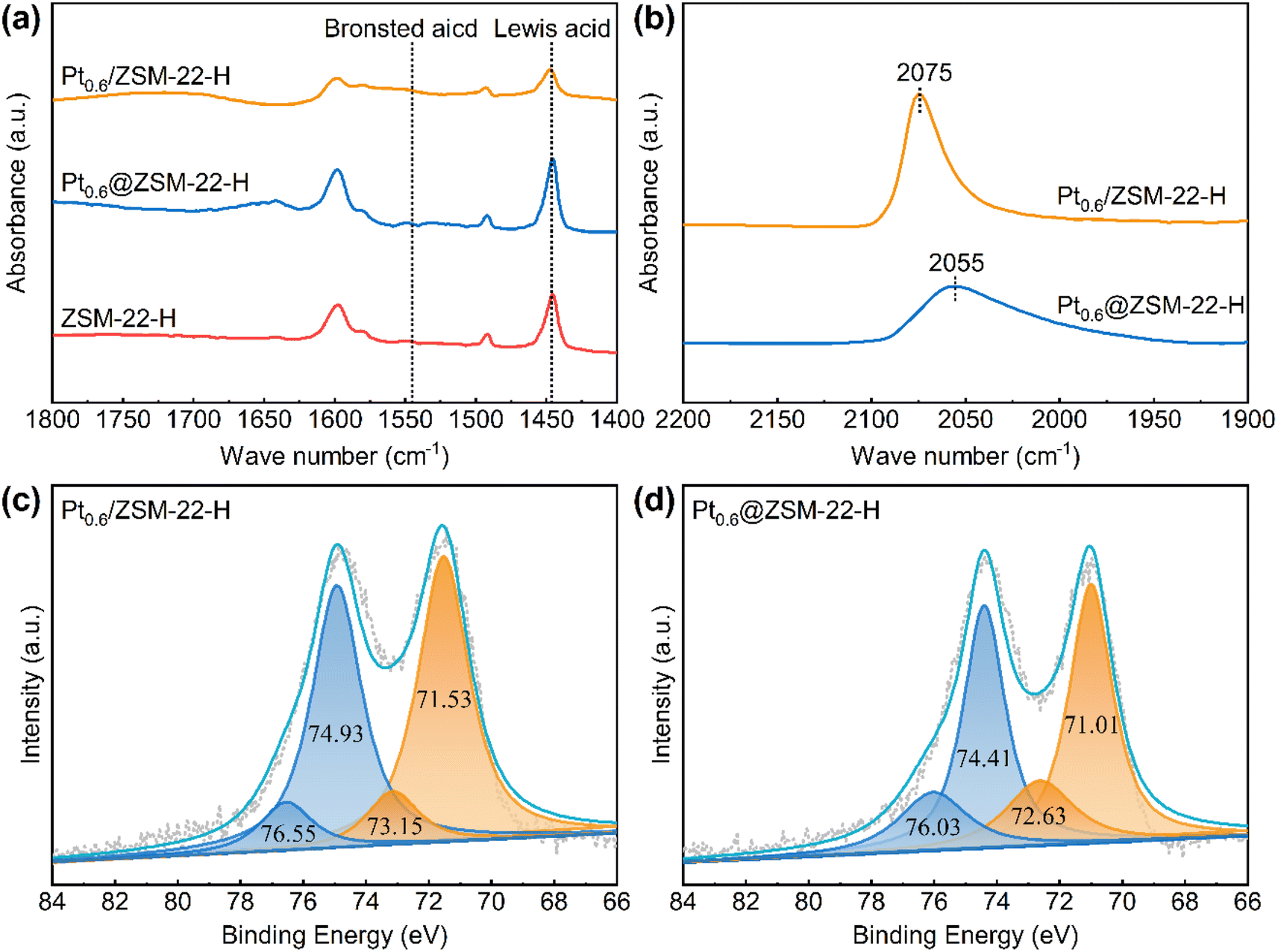 | ||
| Fig. 5 (a) Py-FT-IR spectra of Pt0.6@ZSM-22-H, Pt0.6/ZSM-22-H and ZSM-22-H after pyridine desorption by evacuating at 150 °C; (b) in situ CO-FT-IR spectra of Pt0.6@ZSM-22-H and Pt0.6/ZSM-22-H; XPS spectrum of (c) Pt0.6/ZSM-22-H and (d) Pt0.6@ZSM-22-H. | ||
The electronic structure of the encapsulated Pt particles in ZSM-22 is investigated using CO-FT-IR and XPS. The CO-FT-IR spectra are shown in Fig. 5b. Only one adsorption peak centered at 2075 cm−1 assigned to the linear-adsorbed CO on the Pt particles is observed for the supported Pt0.6/ZSM-22-H, while the peak shows a redshift for the encapsulated Pt0.6@ZSM-22-H (2055 cm−1). It was believed that the redshift of the vibration frequency of adsorbed CO was caused by the stronger π-back-donation from the Pt surface site to CO when the particle size decreased and the surface electron density increased.36,37 This redshift is unlikely to be caused by the decrease in Pt particle size because Pt0.6@ZSM-22-H (3.0 nm, Table 1) possesses a Pt particle size similar to Pt0.6/ZSM-22-H (2.5 nm, Table S1†). Hence, the significant decrease in CO vibration frequency adsorbed on Pt0.6@ZSM-22-H reflects the increased Pt surface electron density. Furthermore, the CO adsorption peak intensity of the Pt0.6@ZSM-22-H is remarkably lower than that of the Pt0.6/ZSM-22-H. This could be related to the encapsulation of Pt particles in the ZSM-22 crystals. Partial Pt sites of the encapsulated Pt0.6@ZSM-22-H could be hidden and not accessible to the CO molecules due to the obstruction of the ZSM-22 framework. This causes a decreased quantity of adsorbed CO on the encapsulated Pt and thus the lowered intensity of the CO adsorption peak. The XPS spectra of Pt 4f are plotted in Fig. 5c and d. The XPS peaks of Pt0.6@ZSM-22-H exhibit a negative shift of 0.52 eV relative to the peaks assigned to metallic Pt0 (71.53 eV and 74.93 eV) and the peaks assigned to Ptδ+ (73.15 eV and 76.55 eV) of Pt0.6/ZSM-22-H, which further supports the fact that the electron density of Pt in Pt0.6@ZSM-22-H increases. The encapsulated Pt inside KL zeolite38 and MCM-22 zeolite39 also exhibited similar results with increased electron density. Thus, the above results suggest that the encapsulated Pt of Pt@ZSM-22 is electron-rich.
The Pt loading of the encapsulated Pt0.6@ZSM-22-H (0.62 wt%, Table 1) is about twice that of the supported Pt0.6/ZSM-22-H (0.33 wt%, Table S1†). The similarity in Pt average particle size for both samples means that the Pt surface site concentration of the Pt0.6@ZSM-22-H should be twice that of the Pt0.6/ZSM-22-H. However, the results of H2 chemisorption reveal a similar Pt surface site concentration (9.8 μmol g−1vs. 10.5 μmol g−1, Tables 1 and S1†). This might correlate with the electron-rich feature of the encapsulated Pt. The Pt serves as the electron acceptor in the chemisorption of H2. The electron rich of the encapsulated Pt is adverse to the chemisorption of H2 and thus leads to the lowered amount of Pt surface sites determined by H2 chemisorption.
The X-ray adsorption fine structure (XAFS) spectra were collected to obtain the structure characteristics of the encapsulated Pt of Pt0.6@ZSM-22-H with the supported Pt of Pt0.6/ZSM-22-H as a reference. The Pt-L3 edge X-ray adsorption near structure (XANES) spectrum (Fig. 6a) shows that the white-line intensity of both samples is slightly higher than that of Pt foil and significantly lower than that of PtO2, indicating that the Pt is not a totally metallic state and part of the Pt atoms carry positive charges. The Fourier transforms of k3-weighted extended X-ray adsorption fine structure (EXAFS) spectrum (Fig. 6b) presents a major peak ascribed to the Pt–Pt scattering path (∼2.6 Å) and a minor peak ascribed to Pt–O scattering path (∼1.6 Å) for both catalysts, revealing that the metallic Pt is the dominant species and the positively charged Pt atoms are bonded with O atoms. The Pt–O path is ascribed to the irreducible interfacial Pt atoms bonding with the O atoms of the zeolite framework because the reduced samples are used for the characterization. The EXAFS fitting curves are shown in Fig. 6c and d with the fitting results, as listed in Table S3.† The coordination number (CN) of Pt–O for Pt0.6@ZSM-22-H (CNPt–O = 1.3) is smaller than that for Pt0.6/ZSM-22-H (CNPt–O = 2.5). The CN of Pt–Pt for Pt0.6@ZSM-22-H (CNPt–Pt = 4.0) is larger than that for Pt0.6/ZSM-22-H (CNPt–Pt = 3.0). The differences in the CNPt–O and CNPt–Pt for both samples are related to the difference in the shape and the size of Pt particles. A more flattened shape (like a raft) and smaller size could cause the larger CNPt–O and smaller CNPt–Pt.40 Because the Pt average particle size of Pt0.6@ZSM-22-H (3.0 nm, Table 1) is similar to that of Pt0.6/ZSM-22-H (2.5 nm, Table S1†), it is deduced that the smaller CNPt–O and larger CNPt–Pt for Pt0.6@ZSM-22-H is caused by the rounded shape of Pt particles, while the larger CNPt–O and smaller CNPt–Pt for the supported Pt0.6/ZSM-22-H reflects the flat shape of Pt particles. The CNPt–Pt for Pt0.6@ZSM-22-H is significantly lower than that of Pt foil (CN = 12), meaning that the Pt particles are highly dispersed.
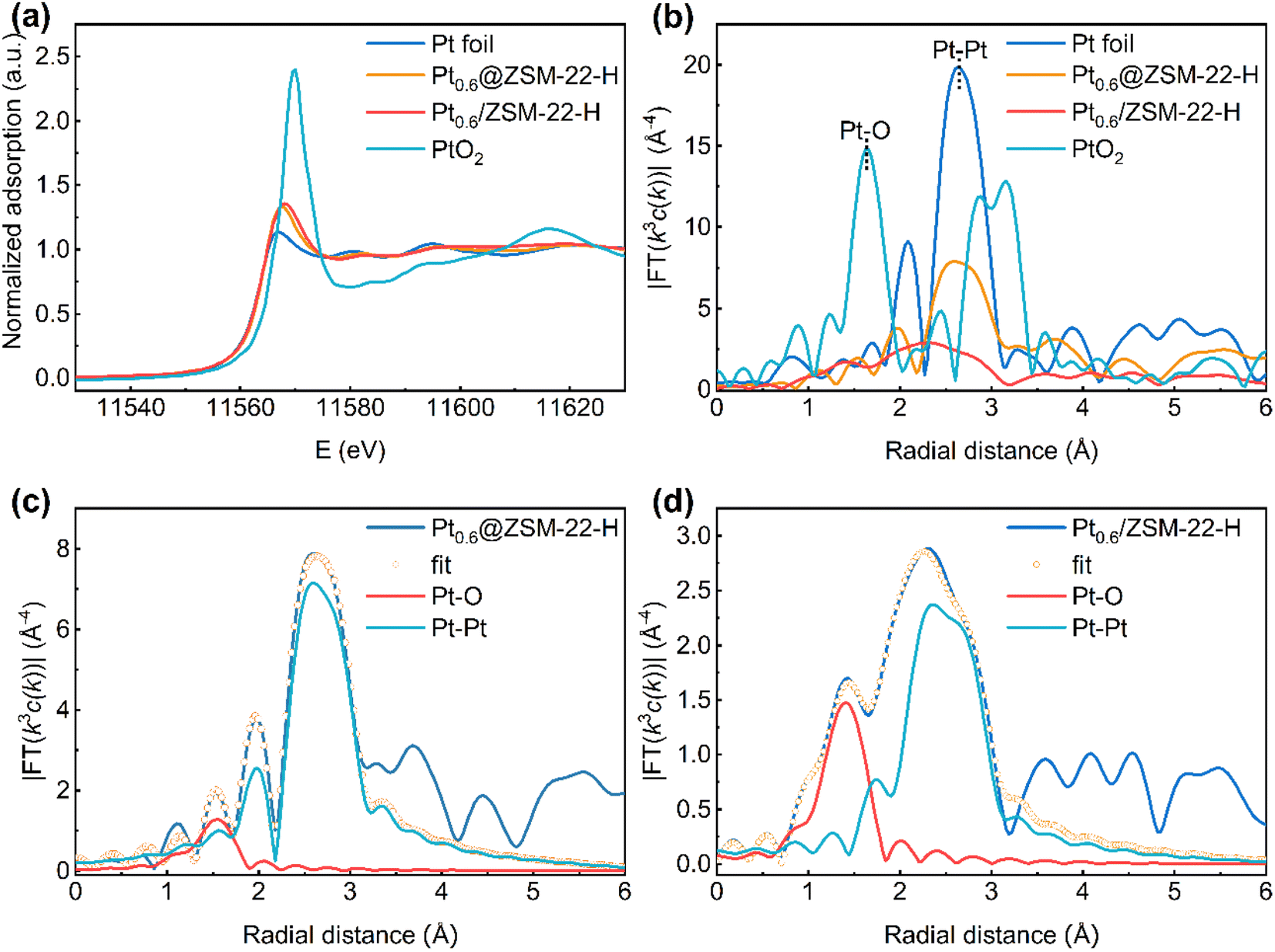 | ||
| Fig. 6 (a) XANES of the Pt L3-edge for Pt0.6@ZSM-22-H and Pt0.6/ZSM-22-H with the reference Pt foil and PtO2. (b) Fourier transforms of k3-weighted Pt L3-edge EXAFS data for Pt foil, Pt0.6@ZSM-22-H, Pt0.6/ZSM-22-H and PtO2. Fitting of the magnitude of the Fourier transforms of the k3-weighted EXAFS for (c) Pt0.6@ZSM-22-H and (d) Pt0.6/ZSM-22-H. | ||
A combination of the above results (CO-FT-IR, XPS and XAFS) indicates that the Pt particles of the encapsulated Pt0.6@ZSM-22-H are round and are bonded with fewer O atoms of the zeolite matrix, while the Pt particles of the supported Pt0.6/ZSM-22-H are flat in shape and are bonded with more O atoms. The zeolite matrix withdraws the electrons from the Pt particles through the Pt–O bonding. Fewer Pt–O bonds lead to fewer electrons being withdrawn, which could account for the electron-rich features of the Pt particles of Pt0.6@ZSM-22-H.
Catalytic performance in furfural hydrogenation
The hydrogenation of furfural (FFL) to furfuryl alcohol (FOL) was carried out to evaluate the selective hydrogenation performance of the catalysts with Pt encapsulated in ZSM-22 (Pt@ZSM-22). FFL is a major biomass-derived platform molecule that produces valuable chemicals and biofuels. The high-value conversion of renewable FFL is crucial to deal with the environmental pollution and energy shortage caused by the excessive exploitation of fossil reserves.41–43 The FOL is the highly desired product because of its various applications in manufacturing resins, dispersants, lysine and vitamin C.44 About 65% of the FFL produced worldwide yearly is converted to FOL owing to its high value-added utilization.45However, over the supported catalysts, the FFL molecule, containing multiple active functional groups (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and CO) is always converted through the multistep routes, leading to low FOL selectivity. The main reaction pathways are illustrated in Scheme 2. The FFL conversion could occur at the CO to obtain unsaturated products, such as hydrogenation into FOL, hydrodeoxygenation into 2-methylfuran (2-MFA) and decarboxylation into furan. The CC of these unsaturated products could be further hydrogenated to the corresponding saturated products, such as tetrahydrofurfuryl alcohol (THFOL), tetrahydrofuran (THF), and 2-methyl tetrahydrofuran (2-THMFA). The isopropanol is a common solvent because its high hydrogen donating ability and polarity lead to excellent FFL conversion and FOL selectivity.46,47 However, the isopropanol solvent can react with the FOL to generate furfuryl isopropyl ether (FIE) by etherification, which lowers the FOL selectivity. Bhogeswararao and Srinivas48 investigated the FFL hydrogenation over Pt(5 wt%)/Al2O3 at 180 °C and obtained a FOL selectivity of 89.1%, 2-MFA selectivity of 2.6% and furan selectivity of 5.5% at a conversion of 91.0%. Byun and Lee49 reported the complex production distribution, including FOL, THFOL, THF, 2-MFA, and 2-THMFA, in the FFL hydrogenation over the Pt-supported hierarchical porous carbon with the FOL selectivity as low as 1% at the conversion of 100%. Agote-Arán50 reported the FIE selectivity of 5.2% and FOL selectivity of 91.9% at the FFL conversion of 58% over the P-modified Pt/Al2O3 (Al2O3-Pt-2P) with isopropanol as solvent. The conversion of FFL over the above-supported catalysts underwent complex pathways and showed low selectivity toward FOL.
C and CO) is always converted through the multistep routes, leading to low FOL selectivity. The main reaction pathways are illustrated in Scheme 2. The FFL conversion could occur at the CO to obtain unsaturated products, such as hydrogenation into FOL, hydrodeoxygenation into 2-methylfuran (2-MFA) and decarboxylation into furan. The CC of these unsaturated products could be further hydrogenated to the corresponding saturated products, such as tetrahydrofurfuryl alcohol (THFOL), tetrahydrofuran (THF), and 2-methyl tetrahydrofuran (2-THMFA). The isopropanol is a common solvent because its high hydrogen donating ability and polarity lead to excellent FFL conversion and FOL selectivity.46,47 However, the isopropanol solvent can react with the FOL to generate furfuryl isopropyl ether (FIE) by etherification, which lowers the FOL selectivity. Bhogeswararao and Srinivas48 investigated the FFL hydrogenation over Pt(5 wt%)/Al2O3 at 180 °C and obtained a FOL selectivity of 89.1%, 2-MFA selectivity of 2.6% and furan selectivity of 5.5% at a conversion of 91.0%. Byun and Lee49 reported the complex production distribution, including FOL, THFOL, THF, 2-MFA, and 2-THMFA, in the FFL hydrogenation over the Pt-supported hierarchical porous carbon with the FOL selectivity as low as 1% at the conversion of 100%. Agote-Arán50 reported the FIE selectivity of 5.2% and FOL selectivity of 91.9% at the FFL conversion of 58% over the P-modified Pt/Al2O3 (Al2O3-Pt-2P) with isopropanol as solvent. The conversion of FFL over the above-supported catalysts underwent complex pathways and showed low selectivity toward FOL.
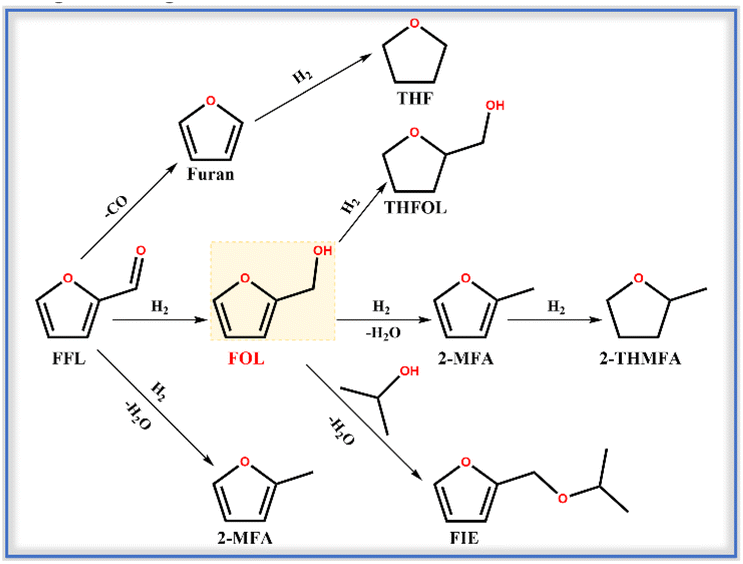 | ||
| Scheme 2 Main reaction pathways for FFL hydrogenation. | ||
The time-dependent catalytic performance of the encapsulated Pt0.6@ZSM-22-H is shown in Fig. 7a. The FFL conversion increases as the reaction time increases, and the selectivity of the FOL almost remains unchanged. The FOL selectivity is 97.6% at the FFL conversion of 99.5% after 6 h (Table 2). This suggests the hydrogenation of CO of FFL to yield that FOL is dominant and the other side reactions are effectively suppressed over Pt0.6@ZSM-22-H. The FFL hydrogenation over Ptx@ZSM-22-H with different Pt loading (x = 0.2, 0.4, 0.8, 1.0) was also carried out. The FOL selectivity is about 98% over Pt0.2@ZSM-22-H and Pt0.4@ZSM-22-H, while it decreases over Pt0.8@ZSM-22-H and Pt1.0@ZSM-22-H but still reaches 96.9% and 96.1%, respectively (Table 2). Due to the difference in the reaction conditions (solvent, pressure, temperature, etc.), the direct comparison of the catalytic performances of different catalysts is difficult. A reasonable comparison can be made by a comprehensive consideration of the catalytic activity (conversion, selectivity, etc.). Compared with these reported Pt-based or zeolite-based catalysts, the Pt0.6@ZSM-22-H in this work presents a high or at least comparable furfural conversion and furfuryl alcohol selectivity (Table S4†). This means that Pt0.6@ZSM-22-H can serve as an effective catalyst for the hydrogenation of furfural into furfuryl alcohol. The above results prove the excellent FOL selectivity in FFL hydrogenation over the encapsulated Pt@ZSM-22. The encapsulation leads to the combination of the hydrogenation activity of the Pt particles and the shape selectivity of the ZSM-22 microchannels. The high selectivity toward FOL might be related to the shape selectivity of the microchannels.
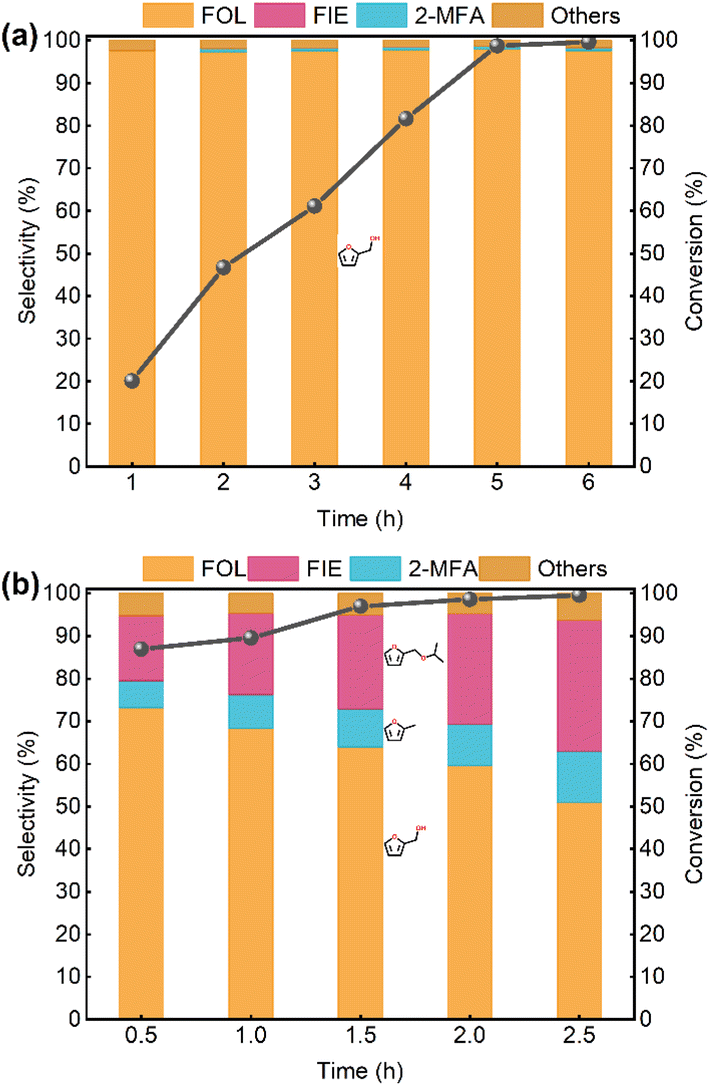 | ||
| Fig. 7 Time-dependent catalytic performance of (a) Pt0.6@ZSM-22-H and (b) Pt0.6/ZSM-22-H in FFL hydrogenation. Reaction conditions: 0.10 g furfural, 10 mL isopropanol, T = 180 °C and P = 4 MPa. FOL, 2-MFA and FIE indicate furfuryl alcohol, 2-methyl furan and furfuryl isopropyl ether, respectively. | ||
| Catalysts | Conversion (%) | Selectivity (%) | |||
|---|---|---|---|---|---|
| FOL | FIE | 2-MFA | Others | ||
| a Reaction conditions: 0.10 g catalysts, 0.10 g furfural, 10 mL isopropanol, reaction temperature 180 °C, initial H2 pressure 4.0 MPa, reaction time 6 h. FOL, FIE and 2-MFA indicate furfuryl alcohol, furfuryl isopropyl ether and 2-methyl furan, respectively. | |||||
| Pt0.2@ZSM-22-H | 32.2 | 97.8 | 0.0 | 0.4 | 1.8 |
| Pt0.4@ZSM-22-H | 87.6 | 98.3 | 0.0 | 0.5 | 1.2 |
| Pt0.6@ZSM-22-H | 99.5 | 97.6 | 0.0 | 0.7 | 1.7 |
| Pt0.8@ZSM-22-H | 91.0 | 96.9 | 0.0 | 0.7 | 2.4 |
| Pt1.0@ZSM-22-H | 95.7 | 96.1 | 0.0 | 0.8 | 3.1 |
| Pt0.6/ZSM-22-H | 100 | 43.3 | 34.5 | 15.4 | 6.8 |
| ZSM-22-H | 3.5 | 93.7 | 0 | 0.5 | 5.8 |
For comparison, the FFL hydrogenation over the supported Pt0.6/ZSM-22-H catalyst was conducted. As shown in Fig. 7b, a complex product distribution is obtained. FIE and 2-MFA are significantly detected besides FOL. As the reaction time is prolonged, the FFL conversion increases from 86.9% to 99.6%. The selectivity of FOL decreases from 73.2% to 50.9%, and the selectivity of 2-MFA increases from 6.4% to 12.0%. When the reaction time is prolonged to 6 h, the selectivity of FOL further decreases to 43.3% and the selectivity of 2-MFA increases to 15.4% (Table 2). The increase in 2-MFA is accompanied by the consumption of FOL, indicating that the generated FOL is further converted to 2-MFA by hydrodeoxygenation.
Similarly, over Pt0.6/ZSM-22-H, the selectivity of FIE increases at the expense of the selectivity of FOL as the reaction time increases, i.e. FOL is converted to FIE. The generation of FIE followed the intermolecular etherification between FOL and isopropyl alcohol.50 The intermolecular etherification requires acid sites (BASs or LASs) as catalytic active sites.51,52 Given the fact that both Pt0.6@ZSM-22-H and Pt0.6/ZSM-22-H possess LASs but not BASs, the LASs seem to be the active sites of etherification. However, the FIE is undetected over Pt0.6@ZSM-22-H, which indicates that the LASs act as the active sites. Hence, BASs are deduced to be the active sites. Ebitani et al.53 reported the H2 spillover from the Pt sites to the SO42+–ZrO2 support to transform the LAS on the support to BAS, which catalyzed the skeletal isomerization of alkanes. The H2 spillover from metal sites to LASs in zeolite was also found on Pt/NaA.54 Hence, it is speculated that the activated H species from the dissociation of H2 on the Pt particles would transfer to the adjacent LAS to form BAS, which could act as the catalytic active sites for the formation of FIE.
Insights into the structure–activity relationship of Pt@ZSM-22
Based on the above catalytic test results, it can be concluded that the encapsulated Pt@ZSM-22 exhibits superior FOL selectivity to the supported Pt/ZSM-22. Pt@ZSM-22 couples the hydrogenation activity of the encapsulated Pt particles with the shape selectivity of ZSM-22 microchannels. This coupling might result in the shape-selective catalysis and thus the excellent FOL selectivity of Pt@ZSM-22. The classical shape selectivity was mainly classified into three types, i.e., reactant, transition state and product selectivity.55 The reactant shape selectivity occurred in the case that only the partial reactant molecules were small enough to pass through the zeolite microchannels. The transition state shape selectivity occurred when the transition state required a larger space than was available in the zeolite microchannels. The product shape selectivity indicated that the product molecules were too large to diffuse out of the zeolite channels, which would either block the channels to deactivate the catalyst or be converted to small molecules and diffuse out.ZSM-22 possesses one-dimension straight microchannels with a pore size of 0.46 nm × 0.57 nm. The kinetic diameters of FFL and FOL molecules are 0.55 nm and 0.57 nm, respectively,56 both of which match the pore size of ZSM-22. This means that both molecules can pass through the ZSM-22 microchannels, and the hydrogenation of FFL into FOL over the encapsulated Pt can successfully proceed.
The bulky FIE is not detected over the encapsulated Pt@ZSM-22. It is considered that the transition state shape selectivity leads to the absence of FIE over the Pt@ZSM-22. The transference of the dissociated H species from the Pt particles to the neighboring LAS results in the transformation of the LAS to the BAS. The transformational BAS could catalyze the etherification between FOL and isopropanol to generate FIE. However, the ZSM-22 microchannels around the encapsulated Pt particles and the neighboring LAS could not provide sufficient space for the transition state involved in the etherification reaction, i.e. the transition state to the bulky FIE is too large to be formed due to the spatial confinement of the ZSM-22 microchannels.
To gain more insight into the catalytic reaction mechanism of FFL hydrogenation into FOL over the encapsulated Pt@ZSM-22, the FT-IR spectra of adsorbed FFL (FFL-FT-IR) were collected because the substrate adsorption configuration played a key role in the reaction pathway and the product selectivity.57,58 The spectra were collected at room temperature after adding FFL dropwise to the catalyst and evacuating at different temperatures.
Fig. 8a shows the FFL-FT-IR spectra of the support (ZSM-22-H). The absorption band at 1698 cm−1 is assigned to the ν(CO) of physiosorbed FFL. The band centered at 1670 cm−1 is ascribed to ν(CO) of chemisorbed FFL through aldehydic O atom (η1(O), Fig. 8d).59 The absorption bands assigned to furan ring breath (1570 cm−1, 1474 cm−1, 1466 cm−1, and 1396 cm−1) and ν(CC) (1570 cm−1, 1474 cm−1, and 1466 cm−1) are intense and similar to those of the unadsorbed FFL (Table S5†), suggesting no significant interaction between the furan ring and the adsorption sites.60–63 These results suggest that the ZSM-22-H support adsorbs FFL through η1(O) configuration. The Lewis acid sites (LASs) on the ZSM-22-H, which are capable of accepting the unbonded electron pairs of aldehydic O in FFL, are considered the adsorption sites of FFL. The very low FFL conversion over ZSM-22-H (3.5%, Table 2) verifies that the LAS contributed barely to FFL hydrogenation though they can adsorb FFL. The catalysts containing Pt (Pt@ZSM-22 and Pt/ZSM-22) exhibited significant FFL hydrogenation conversion (Fig. 7 and Table 2). The above results indicate that Pt is the active sites of the FFL hydrogenation.
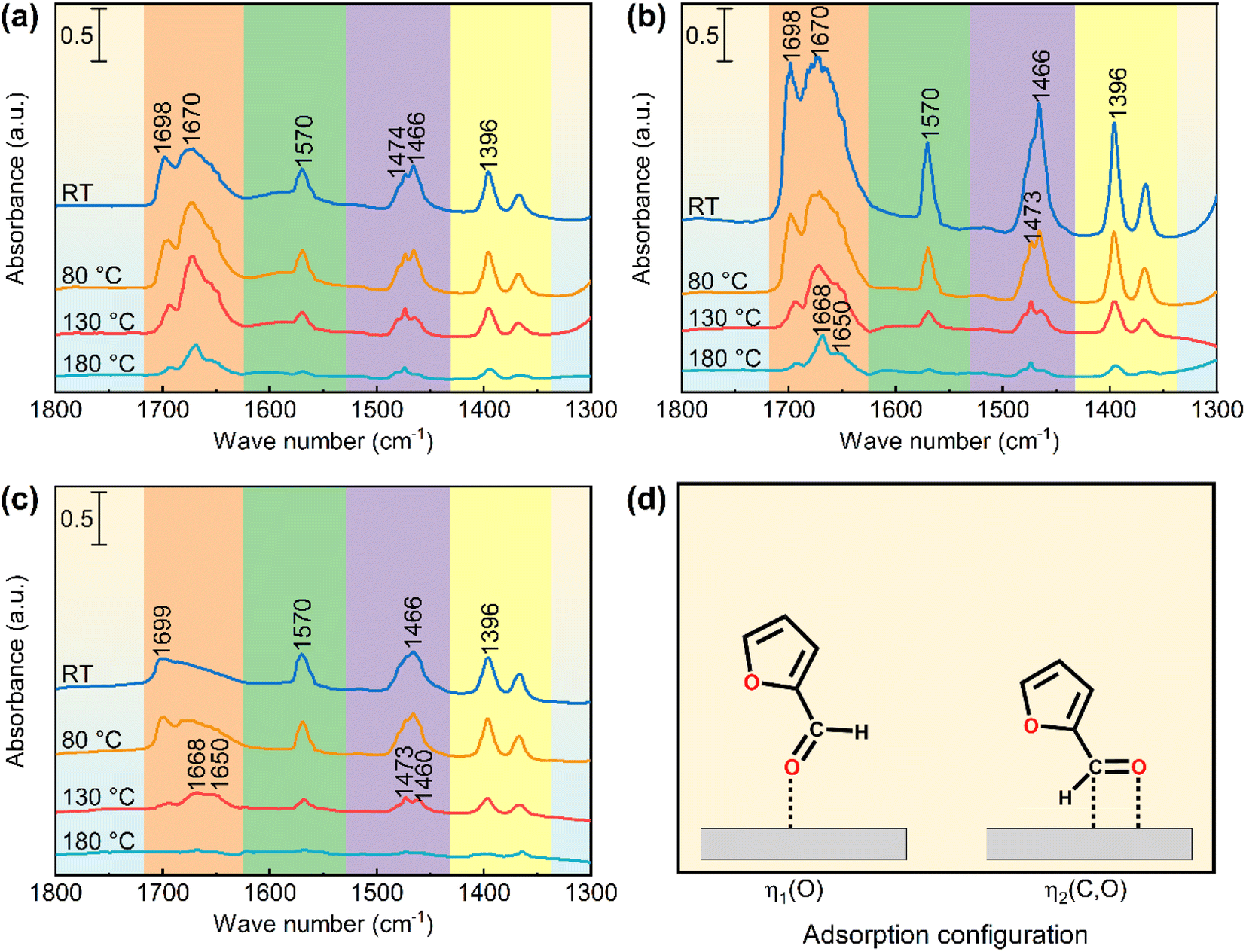 | ||
| Fig. 8 FT-IR spectra of adsorbed furfural (FFL-FT-IR) on (a) ZSM-22-H, (b) Pt0.6@ZSM-22-H and (c) Pt0.6/ZSM-22-H after successive evacuation at room temperature (RT), 80 °C, 130 °C and 180 °C. (d) Adsorption configuration of FFL over catalyst surface through aldehyde group. | ||
Fig. 8b shows the FFL-FT-IR spectra of the encapsulated catalyst Pt0.6@ZSM-22-H. Compared with the ZSM-22-H, the spectra exhibit a similar adsorption band position, suggesting the FFL is chemisorbed on Pt0.6@ZSM-22-H by η1(O) configuration. The higher the band intensity, the more FFL molecules are chemisorbed on Pt0.6@ZSM-22-H. Besides the LAS for the chemisorption of FFL, the encapsulated Pt particles in Pt0.6@ZSM-22-H can chemisorb FFL. Hence, the increased amount of the chemisorbed FFL on Pt0.6@ZSM-22-H results from the chemisorption of FFL on the Pt particles. For the spectrum obtained after evacuating at room temperature (RT), the band at 1670 cm−1 is wide. Further evacuating at 80 °C, 130 °C and 180 °C, the intensity of all absorption bands in the corresponding spectrum gradually decreases. The original wide ν(CO) band is divided into two bands at 1668 cm−1 and 1650 cm−1: the former band assigned to the adsorbed FFL on LAS and the latter band to the adsorbed FFL on the Pt particle. The above results suggest that the chemisorbed FFL molecules interact with the encapsulated Pt particles of Pt0.6@ZSM-22-H by η1(O) adsorption configuration.
Fig. 8c shows the FFL-FT-IR spectra of the supported catalyst Pt0.6/ZSM-22-H. The adsorption band intensity in the spectra of Pt0.6/ZSM-22-H is remarkably lower than that of Pt0.6@ZSM-22-H. The fewer adsorbed sites lead to the fewer adsorbed FFL molecules, which lead to the less intense adsorption band. The lower band intensity indicates that fewer FFL molecules are adsorbed over the Pt0.6/ZSM-22-H, which possesses fewer adsorbed sites. Given that both catalysts possess almost equivalent concentrations of Pt sites (Tables 1 and S1†) to adsorb FFL, the fewer LASs on Pt0.6/ZSM-22-H, which is proved by the lower intensity of the band ascribed to LAS in the Py-FT-IR spectra (Fig. 5a), afford to the fewer adsorbed FFL molecules. Apart from the η1(O)-FFL chemisorbed on the Pt particles (1650 cm−1), the unique band at 1460 cm−1 assigned to the ν(CO) of FFL chemisorbed on the supported Pt particles through η2(C, O) configuration (η2(C, O)-FFL, Fig. 8d) emerges in the spectrum obtained upon evacuating at 130 °C. The simultaneous adsorption of C and O might contribute to the large redshift of the ν(CO) of η2(C, O)-FFL.64 Hence, the FFL is adsorbed on the supported Pt particles of Pt0.6/ZSM-22-H by η1(O) and η2(C, O) adsorption configuration.
Based on the above results of FFL-FT-IR, the selectivity toward the adsorption configuration of FFL occurs on the encapsulated Pt0.6@ZSM-22-H. FFL molecules have to penetrate the unidimensional straight microchannels of ZSM-22 to reach and adsorb on the encapsulated Pt particles. The ZSM-22 microchannels select the FFL adsorption configuration based on the shape and size. Obviously, the η1(O) configuration is chosen, and the η2(C, O) configuration is excluded by the ZSM-22 microchannels. Sitthisa et al.58 reported the preferred η1(O)-FFL on the Cu/SiO2. They used the DFT calculations to study the hydrogenation reaction pathway and found that the η1(O)-FFL can be converted to FOL via either an alkoxide or hydroxyalkyl intermediate. Therefore, it is inferred that the selectivity toward η1(O)-FFL on Pt@ZSM-22 results in the preferred generation of FOL.
The electronic property of Pt also affects the catalytic selectivity. Zhu et al. reported that the electron-deficient atomic Pt sites were in favor of the O-terminal (hydroxyl oxygen) adsorption of FOL to activate and break the C–OH, resulting in the generation of 2-MFA.65 For the encapsulated Pt@ZSM-22 in this study, the Pt sites are electron-rich as confirmed by the results of CO-FT-IR and XPS. Because the O-terminal adsorption of FOL is formed by donating the unbonded electrons of the O atoms to the Pt sites, the increased surface electron density of Pt restricts this adsorption of FOL. Hence, the electron-rich nature facilitates the desorption of the generated FOL from the encapsulated Pt sites. The timely desorption of FOL might suppress the further conversion of FOL, which could contribute to the FOL selectivity to some extent.
In view of all the above results, the remarkable catalytic selectivity to FOL in FFL hydrogenation over the encapsulated Pt@ZSM-22 originates from the spatial confinement of the one-dimensional microchannels of ZSM-22. The proposed mechanism of FFL hydrogenation over Pt@ZSM-22 is illustrated in Scheme 3. Because FFL molecules and FOL molecules possess a kinetic diameter equivalent to the zeolite pore size, they can pass through the ZSM-22 microchannels and interact with the encapsulated Pt particles. With the spatial-constraint effect, the FFL molecules are adsorbed on the encapsulated Pt sites selectively through η1(O) configuration, which then leads to the generation of FOL by hydrogenation. The electron-rich feature of the encapsulated Pt particles benefits the easier desorption of the generated FOL. The desorbed FOL molecules then diffuse out of the ZSM-22 microchannels. The generation of products other than FOL is refrained because the shape selectivity conferred by the ZSM-22 microchannels inhibits the formation of the corresponding transition states and intermediates. Therefore, the excellent selectivity of FOL on Pt@ZSM-22-H in FFL hydrogenation is achieved.
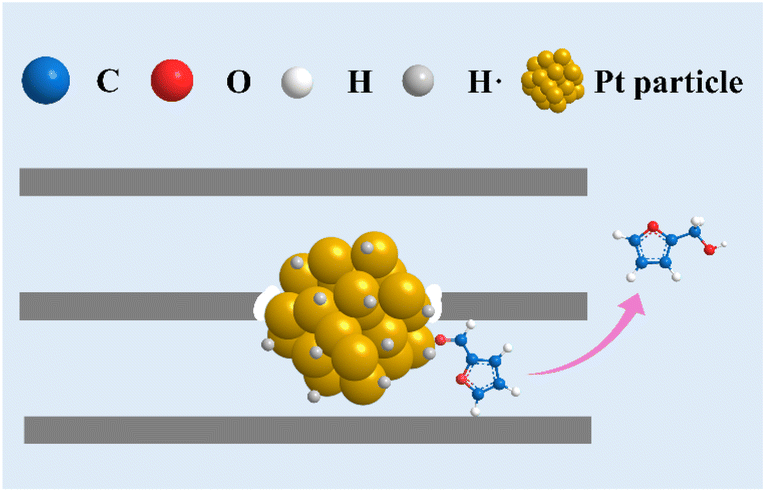 | ||
| Scheme 3 Illustration of the proposed mechanism of FFL hydrogenation over Pt@ZSM-22. | ||
Conclusions
In summary, we report a facile one-pot hydrothermal synthesis approach to prepare Pt@ZSM-22 catalyst. The ZSM-22 seeds are added to expedite the lowered-temperature crystallization of the zeolite. We disclose that the ligand-protected and lowered-temperature crystallization strategy is effective in preventing the decomposition and reduction of the metal precursor ([Pt(en)2]2+), which usually occurs in harsh hydrothermal environments. The intact [Pt(en)2]2+ is adsorbed on the silicate nanoparticles by electrostatic adsorption and wrapped within the ZSM-22 crystals with the crystallization of these nanoparticles. The post-synthesis treatment by direct reduction is adopted for the template removal and the reduction of the encapsulated [Pt(en)2]2+. This approach leads to the highly dispersed and uniform Pt particles encapsulated in ZSM-22 crystals. The encapsulated Pt particles are round and form fewer Pt–O bonds with the zeolite matrix, leading to the electron-rich Pt.The resultant Pt@ZSM-22 catalyst exhibits excellent catalytic performance in furfural hydrogenation with a furfural alcohol selectivity of 97.6% at a furfural conversion of 99.5%. The investigation of the structure–activity relationship reveals that the spatial confinement of the one-dimensional microchannels ZSM-22 boosts the outstanding furfuryl alcohol selectivity of Pt@ZSM-22. The η1(O) adsorption of furfural on the Pt sites is selected by the ZSM-22 microchannels, benefiting the generation of furfuryl alcohol. The electron-rich property of the encapsulated Pt facilitates the desorption of furfuryl alcohol from the Pt sites. Additionally, the shape selectivity conferred by the ZSM-22 microchannels inhibits the formation of the transition states and intermediates, leading to the by-products.
Our work extends the direct hydrothermal synthesis protocol to the encapsulation of noble metal in zeolites with one-dimensional straight channels and raises the prospects of the encapsulation of other metals inside such zeolites, which have great application potentials in selective hydrogenation reactions.
Data availability
The data supporting this article have been included within the article and the ESI.†Author contributions
C. W. and Z. T.: conceptualization, funding acquisition, resources, supervision, writing – review & editing; X. W.: investigation, data curation, formal analysis, writing – original draft; W. B. and W. Q.: investigation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R&D Program of China (2020YFB0606405), and the Strategic Priority Research Program of the Chinese Academy of Sciences (XDA21021201).Notes and references
- S. Ma and Z.-P. Liu, Nat. Commun., 2022, 13, 2716 CrossRef CAS PubMed.
- K. Xu, Y. Chen, H. Yang, Y. Gan, L. Wu, L. Tan, Y. Dai and Y. Tang, Appl. Catal., B, 2024, 341, 123244 CrossRef CAS.
- Y. Peng, X. Wang, C. Wang, W. Bi, Q. Jiang and Z. Tian, Chem Catal., 2023, 3, 100557 CrossRef.
- B. C. Vance, Z. Yuliu, S. Najmi, E. Selvam, J. E. Granite, K. Yu, M. G. Ierapetritou and D. G. Vlachos, Chem. Eng. J., 2024, 487, 150468 CrossRef CAS.
- C. Mu, J. Sun, C. Xie, J. Bao, X. Guo, H. Zhang, Y. Zhao, S. Wang and X. Ma, ACS Catal., 2024, 14, 1394–1404 CrossRef CAS.
- D. Xu, S. Wang, B. Wu, C. Huo, Y. Qin, B. Zhang, J. Yin, L. Huang, X. Wen, Y. Yang and Y. Li, J. Catal., 2018, 365, 163–173 CrossRef CAS.
- G. Lv, C. Wang, K. Chi, H. Liu, P. Wang, H. Ma, W. Qu and Z. Tian, Catal. Today, 2018, 316, 43–50 CrossRef CAS.
- J. Gu, Z. Zhang, P. Hu, L. Ding, N. Xue, L. Peng, X. Guo, M. Lin and W. Ding, ACS Catal., 2015, 5, 6893–6901 CrossRef CAS.
- S. Wang, Z. J. Zhao, X. Chang, J. Zhao, H. Tian, C. Yang, M. Li, Q. Fu, R. Mu and J. Gong, Angew. Chem., Int. Ed., 2019, 58, 7668–7672 CrossRef CAS.
- D. J. Ostgard, L. Kustov, K. R. Poeppelmeier and W. M. H. Sachtler, J. Catal., 1992, 133, 342–357 CrossRef CAS.
- D. V. Peron, V. L. Zholobenko, M. R. de la Rocha, M. O. de Souza, L. A. Feris, N. R. Marcilio, V. V. Ordomsky and A. Y. Khodakov, J. Mater. Sci., 2019, 54, 5399–5411 CrossRef CAS.
- P. B. Weisz, V. J. Frilette, R. W. Maatman and E. B. Mower, J. Catal., 1962, 1, 307–312 CrossRef CAS.
- Z. Wu, S. Goel, M. Choi and E. Iglesia, J. Catal., 2014, 311, 458–468 CrossRef CAS.
- H. J. Cho, D. Kim, J. Li, D. Su and B. Xu, J. Am. Chem. Soc., 2018, 140, 13514–13520 CrossRef CAS PubMed.
- S. Goel, Z. Wu, S. I. Zones and E. Iglesia, J. Am. Chem. Soc., 2012, 134, 17688–17695 CrossRef CAS.
- N. Wang, Q. Sun, R. Bai, X. Li, G. Guo and J. Yu, J. Am. Chem. Soc., 2016, 138, 7484–7487 CrossRef CAS PubMed.
- Z. W. Minkee Choi and E. Iglesia, J. Am. Chem. Soc., 2010, 132, 9129–9137 CrossRef.
- X. Deng, B. Qin, R. Liu, X. Qin, W. Dai, G. Wu, N. Guan, D. Ma and L. Li, J. Am. Chem. Soc., 2021, 143, 20898–20906 CrossRef CAS PubMed.
- X. Liu, Y. Liu, Y. Wu, S. Dong, G. Qi, C. Chen, S. Xi, P. Luo, Y. Dai, Y. Han, Y. Zhou, Y. Guo and J. Wang, J. Hazard. Mater., 2023, 458, 131848 CrossRef CAS.
- Y. Wu, C. Chen, Z. Xiong, M. Xu, Y. Zhao, Y. Dai, Y. Han, Y. Zhou, X. Liu and J. Wang, Chem. Eng. J., 2024, 493, 152457 CrossRef CAS.
- M. C. Claude and J. A. Martens, J. Catal., 2000, 190, 39–48 CrossRef CAS.
- M. Zhang, Y. Chen, L. Wang, Q. Zhang, C.-W. Tsang and C. Liang, Ind. Eng. Chem. Res., 2016, 55, 6069–6078 CrossRef CAS.
- J. Tang, P. Liu, X. Liu, L. Chen, H. Wen, Y. Zhou and J. Wang, ACS Appl. Mater. Interfaces, 2020, 12, 11522–11532 CrossRef CAS.
- J. Lan, P. Liu, P. Fu, X. Liu, M. Xie, S. Jiang, H. Wen, Y. Zhou and J. Wang, Microporous Mesoporous Mater., 2021, 322, 111161 CrossRef CAS.
- Q. Sun, N. Wang, Q. Fan, L. Zeng, A. Mayoral, S. Miao, R. Yang, Z. Jiang, W. Zhou, J. Zhang, T. Zhang, J. Xu, P. Zhang, J. Cheng, D. Yang, R. Jia, L. Li, Q. Zhang, Y. Wang, O. Terasaki and J. Yu, Angew. Chem., Int. Ed., 2020, 59, 19450–19459 CrossRef CAS PubMed.
- R. M. J. Fiedorow and S. E. Wanke, J. Catal., 1976, 43, 34–42 CrossRef CAS.
- A. K. Jamil and O. Muraza, Microporous Mesoporous Mater., 2016, 227, 16–22 CrossRef CAS.
- L. Jiao and J. R. Regalbuto, J. Catal., 2008, 260, 329–341 CrossRef CAS.
- M. Schreier, S. Teren, L. Belcher, J. R. Regalbuto and J. T. Miller, Nanotechnology, 2005, 16, S582–S591 CrossRef PubMed.
- A. Borgna, F. Lenormand, T. Garetto, C. R. Apesteguia and B. Moraweck, Catal. Lett., 1992, 13, 175–188 CrossRef CAS.
- P. N. Plessow and F. Abild-Pedersen, ACS Catal., 2016, 6, 7098–7108 CrossRef CAS.
- X. Liu, L. Xu, B. Zhang and X. Liu, Microporous Mesoporous Mater., 2014, 193, 127–133 CrossRef CAS.
- S. Zhao, L. Lang, X. Yin, W. Yang and C. Wu, Acta Phys.-Chim. Sin., 2015, 31, 793–799 CAS.
- E. A. Kyriakidou, O. S. Alexeev, A. P. Wong, C. Papadimitriou, M. D. Amiridis and J. R. Regalbuto, J. Catal., 2016, 344, 749–756 CrossRef CAS.
- Y. Wang, T. Lv, H. L. Wang, Y. Zhao, C. Meng and H. Liu, Microporous Mesoporous Mater., 2015, 208, 66–71 CrossRef CAS.
- J. Zhu, T. Wang, X. Xu, P. Xiao and J. Li, Appl. Catal., B, 2013, 130–131, 197–217 CrossRef CAS.
- P. Bazin, O. Saur, J. C. Lavalley, M. Daturi and G. Blanchard, Phys. Chem. Chem. Phys., 2005, 7, 187–194 RSC.
- A. Y. Stakheev, E. S. Shpiro, N. I. Jaeger and G. Schulzekloff, Catal. Lett., 1995, 32, 147–158 CrossRef CAS.
- L. Liu, U. Diaz, R. Arenal, G. Agostini, P. Concepcion and A. Corma, Nat. Mater., 2017, 16, 132–138 CrossRef CAS PubMed.
- G. Jacobs, F. Ghadiali, A. Pisanu, A. Borgna, W. E. Alvarez and D. E. Resasco, Appl. Catal., A, 1999, 188, 79–98 CrossRef CAS.
- C. Wang, L. Wang, J. Zhang, H. Wang, J. P. Lewis and F. Xiao, J. Am. Chem. Soc., 2016, 138, 7880–7883 CrossRef CAS PubMed.
- Z. An and J. Li, Green Chem., 2022, 24, 1780–1808 RSC.
- Z. An, P. Yang, D. Duan, J. Li, T. Wan, Y. Kong, S. Caratzoulas, S. Xiang, J. Liu, L. Huang, A. I. Frenkel, Y.-Y. Jiang, R. Long, Z. Li and D. G. Vlachos, Nat. Commun., 2023, 14, 6666 CrossRef CAS PubMed.
- B. M. Nagaraja, A. H. Padmasri, B. D. Raju and K. S. R. Rao, J. Mol. Catal. A:Chem., 2007, 265, 90–97 CrossRef CAS.
- A. Jaswal, P. P. Singh and T. Mondal, Green Chem., 2022, 24, 510–551 RSC.
- A. B. Merlo, V. Vetere, J. F. Ruggera and M. L. Casella, Catal. Commun., 2009, 10, 1665–1669 CrossRef CAS.
- S. Thongratkaew, S. Kiatphuengporn, A. Junkaew, S. Kuboon, N. Chanlek, A. Seubsai, B. Rungtaweevoranit and K. Faungnawakij, Catal. Commun., 2022, 169, 106468 CrossRef CAS.
- S. Bhogeswararao and D. Srinivas, J. Catal., 2015, 327, 65–77 CrossRef CAS.
- M. Y. Byun and M. S. Lee, J. Ind. Eng. Chem., 2021, 104, 406–415 CrossRef CAS.
- M. Agote-Arán, S. Alijani, C. Coffano, A. Villa and D. Ferri, Catal. Lett., 2022, 152, 980–990 CrossRef.
- P. Neves, M. M. Antunes, P. A. Russo, J. P. Abrantes, S. Lima, A. Fernandes, M. Pillinger, S. M. Rocha, M. F. Ribeiro and A. A. Valente, Green Chem., 2013, 15, 3367–3376 RSC.
- J. Luo, J. Yu, R. J. Gorte, E. Mahmoud, D. G. Vlachos and M. A. Smith, Catal. Sci. Technol., 2014, 4, 3074–3081 RSC.
- K. Ebitani, J. Konishi and H. Hattori, J. Catal., 1991, 130, 257–267 CrossRef CAS.
- J. Im, H. Shin, H. Jang, H. Kim and M. Choi, Nat. Commun., 2014, 5, 3370 CrossRef.
- S. M. Csicsery, Zeolites, 1984, 4, 202–213 CrossRef CAS.
- J. Jae, G. A. Tompsett, A. J. Foster, K. D. Hammond, S. M. Auerbach, R. F. Lobo and G. W. Huber, J. Catal., 2011, 279, 257–268 CrossRef CAS.
- P. Claus, Top. Catal., 1998, 5, 51–62 CrossRef CAS.
- S. Sitthisa, T. Sooknoi, Y. Ma, P. B. Balbuena and D. E. Resasco, J. Catal., 2011, 277, 1–13 CrossRef CAS.
- Q. Wang, J. Feng, L. Zheng, B. Wang, R. Bi, Y. He, H. Liu and D. Li, ACS Catal., 2020, 10, 1353–1365 CrossRef CAS.
- G. Allen and H. J. Bernstein, Can. J. Chem., 1955, 33, 1055–1061 CrossRef CAS.
- H. Ashish and P. Ramasami, Mol. Phys., 2008, 106, 175–185 CrossRef CAS.
- X. Meng, Y. Yang, L. Chen, M. Xu, X. Zhang and M. Wei, ACS Catal., 2019, 9, 4226–4235 CrossRef CAS.
- W. Liu, Y. Yang, L. Chen, E. Xu, J. Xu, S. Hong, X. Zhang and M. Wei, Appl. Catal., B, 2021, 282, 119569 CrossRef CAS.
- J. L. Davis and M. A. Barteau, Surf. Sci., 1990, 235, 235–248 CrossRef CAS.
- Y. Zhu, W. Zhao, J. Zhang, Z. An, X. Ma, Z. Zhang, Y. Jiang, L. Zheng, X. Shu, H. Song, X. Xiang and J. He, ACS Catal., 2020, 10, 8032–8041 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta07243j |
| This journal is © The Royal Society of Chemistry 2025 |