
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An atomically precise Pt17 nanocluster: its electronic structure and high activity for the hydrogen evolution reaction†
Kazutaka
Oiwa‡
a,
Kaoru
Ikeda‡
a,
Ryuki
Kurosaki
a,
Kotaro
Sato
a,
Naoki
Nishi
a,
Haruna
Tachibana
a,
Md. Ahsanul
Haque
 a,
Tokuhisa
Kawawaki
*acd,
Kenji
Iida
*b and
Yuichi
Negishi
*acd
a,
Tokuhisa
Kawawaki
*acd,
Kenji
Iida
*b and
Yuichi
Negishi
*acd
aDepartment of Applied Chemistry, Faculty of Science, Tokyo University of Science, 1-3 Kagurazaka, Shinjuku-ku, Tokyo 162-8601, Japan
bInstitute for Catalysis, Hokkaido University, Sapporo, Hokkaido 001-0021, Japan. E-mail: k-iida@cat.hokudai.ac.jp
cCarbon Value Research Center, Research Institute for Science and Technology, Tokyo University of Science, 2641 Yamazaki, Noda, Chiba, Japan
dInstitute of Multidisciplinary Research for Advanced Materials, Tohoku University, Katahira 2-1-1, Aoba-ku, Sendai 980-8577, Japan. E-mail: yuichi.negishi.a8@tohoku.ac.jp; tokuhisa.kawawaki.d8@tohoku.ac.jp
First published on 3rd March 2025
Abstract
Pt nanoclusters (Pt NCs) approximately 1 nm in size show potential as catalysts owing to their large specific surface areas and unique electronic structures, which are influenced by quantum size effects. However, synthesizing Pt NCs with atomic precision under ambient conditions remains challenging, with [Pt17(CO)12(PPh3)8]z (z = 1+ or 2+; CO = carbon monoxide; PPh3 = triphenylphosphine) being the only current example of such a NC. It exhibits extraordinary stability, and its electronic structure and catalytic utility in a range of reactions are topics of widespread interest. In this study, we reveal its electronic structure and explore its catalytic activity in the hydrogen evolution reaction (HER). Our findings revealed that [Pt17(CO)12(PPh3)8]z possesses a discrete electronic structure, with the HOMO and LUMO primarily constituted by the s, p, and d orbitals of Pt; that a Pt17 NC-supported carbon-black catalyst (Pt17/CB) achieves 3.59 times the HER mass activity of a commercially available Pt/CB catalyst; and that the optimal electronic structure of the surface Pt atoms in Pt17/CB significantly enhances its HER activity. These insights underscore the potential of leveraging atomically precise Pt NCs in the design and development of highly active electrocatalysts for water splitting.
Introduction
The synthesis of ∼1 nm-sized Pt nanoclusters (Pt NCs) and their alloy NCs has garnered significant interest owing to their unique properties and potential applications in catalysis. Using carbon monoxide (CO) ligands,1,2 Pt NCs (Ptn(CO)m NCs; where n and m are the numbers of Pt atoms and CO ligands, respectively) can be synthesized with atomic precision.3–6 Most such NCs are not stable under atmospheric conditions, and thus few studies have been conducted on their application. However, a recently developed method for the synthesis of Pt NCs under atmospheric conditions using CO and phosphines (PR3) or thiolates (SR) as ligands (Ptn(CO)m(PR3)l or Ptn(CO)m(SR)o NCs, respectively) has emerged as a simple and convenient method to generate such NCs in large quantities.7–12In particular, [Pt17(CO)12(PPh3)8]z (z = 1+ or 2+; PPh3 = triphenylphosphine; Fig. 1a) represents a unique form of Pt NC that can be synthesized under atmospheric conditions and exhibits remarkable stability. These NCs not only are structurally distinct but also serve as highly effective precursors of catalysts for reactions, including CO oxidation, propylene oxidation, and oxygen reduction.8,11,13 Their robust catalytic activity highlights their potential, making them invaluable in the development of high-performance catalysts for various fields. However, the origins of the unique electronic properties of [Pt17(CO)12(PPh3)8]z remain unclear. In particular, its excellent catalytic activity is thought to be based on the electronic structure of [Pt17(CO)12(PPh3)8]z, and it is desirable to clarify this. [Pt17(CO)12(PPh3)8]z is composed of (i) CO, (ii) μ2-CO, (iii) PPh3, and (iv) capping Pt2(μ2-CO)(PPh3)2 units around an icosahedral Pt13 core,11 but there are few methods that can clarify how its electronic structure is composed of each of these parts at each energy level. Here, density functional theory (DFT) calculations may provide valuable insights into the underlying reasons for the high stability and catalytic activity of [Pt17(CO)12(PPh3)8]z by clarifying its electronic structure. Such a study could also help predict the geometric and electronic structures of other potentially stable Ptn(CO)m(PR3)l NCs that could be synthesized in the future.14–19
 | ||
| Fig. 1 Schematic of the work performed in this study. (a) Geometric and (b) calculated electronic structure of [Pt17(CO)12(PPh3)8]z and its catalytic application to the (c) HER for (d) water electrolysis. | ||
With ongoing depletion of fossil resources and global warming, the transition to a hydrogen (H2)-based energy economy is increasingly necessary. Currently, H2 is predominantly produced from methane derived from natural gas and coal, which are fossil fuels. Thus, to reduce its environmental impact, water electrolysis using electricity supplied by renewable energy sources has gained significant attention as a method for H2 production (Fig. S1†).20,21 Water electrolysis involves both the hydrogen evolution reaction (HER) and the oxygen evolution reaction (OER), where H2 is generated through the reduction of water in the HER. Among the various water electrolysis systems, proton exchange membrane water electrolyzers (PEMWEs) are particularly attractive owing to their compact size, high energy efficiency, high H2-production rate, and quick response to voltage fluctuations. However, the widespread adoption of PEMWEs faces a major challenge: the need of stable and active electrocatalysts that can operate effectively under acidic conditions.
Currently, Pt metals, which are precious and expensive, are the only materials that meet these requirements.22,23 Consequently, reducing the amount of Pt used in PEMWE electrocatalysts is critical for making this technology more economically viable and environmentally sustainable. Using [Pt17(CO)12(PPh3)8]z (Fig. 1a) as a precursor to deposit Pt17 NCs on carbon black (CB; Pt17/CB) is a promising strategy to enhance the activity of HER catalysts while decreasing overall Pt usage. Using ∼1 nm-Pt NCs increases the number of active sites for the reaction, potentially leading to catalysts that are more efficient than typical CB catalysts loaded with larger (∼2–3 nm) Pt nanoparticles (Pt NPs) commonly used in PEMWEs. Additionally, DFT calculations can provide insight into the electronic structure, high stability, and catalytic activity of these small Pt NCs, guiding the future design of highly active and stable electrocatalysts for the HER.13
Accordingly, in the present study, we aimed to elucidate the electronic structure of [Pt17(CO)12(PPh3)8]z using DFT calculations (Fig. 1b); develop an HER catalyst with enhanced activity compared with commercial Pt NPs/CB by using [Pt17(CO)12(PPh3)8]z as a precursor (Fig. 1c); and determine the origin of the high HER activity exhibited by Pt17/CB (Fig. 1d). Our results revealed that the transition from the highest occupied molecular orbital (HOMO) to the lowest unoccupied molecular orbital (LUMO) in [Pt17(CO)12(PPh3)8]2+ is forbidden and that the orbitals neighbouring them primarily comprise the s, p, and d orbitals of Pt. Additionally, we successfully created a Pt17/CB catalyst with an HER mass activity 3.59 times that of commercially available Pt NPs/CB. The DFT calculations also strongly suggested that the high HER activity of Pt17/CB arises from the presence of Pt atoms with an optimal electronic structure for the HER.
Results and discussion
Elucidation of the electronic structure of atomically precise [Pt17(CO)12(PPh3)8]z
[Pt17(CO)12(PPh3)8]z was synthesized through a polyol reduction and ligand-exchange method,24–29 similar to that in previous reports.11,13,30 Briefly, the Pt precursor was heated to 120 °C in ethylene glycol with sodium hydroxide under atmospheric conditions. The solution was then cooled to room temperature, and PPh3 was added to form Ptn(CO)m(PPh3)l NCs. From the obtained products, [Pt17(CO)12(PPh3)8]z was selectively isolated using solvent extraction. The resulting product was confirmed to be high-purity [Pt17(CO)12(PPh3)8]zvia electrospray ionization mass spectrometry (ESI-MS; Fig. S2†). Fig. 2a shows the optical absorption spectrum of [Pt17(CO)12(PPh3)8]z, which presents prominent peaks at approximately 1.10, 1.20, 1.85, and 2.15 eV, indicating a discrete electronic structure similar to that of Au25(PET)18 (PET = 2-phenylethanethiolate) and its alloy NCs.31–34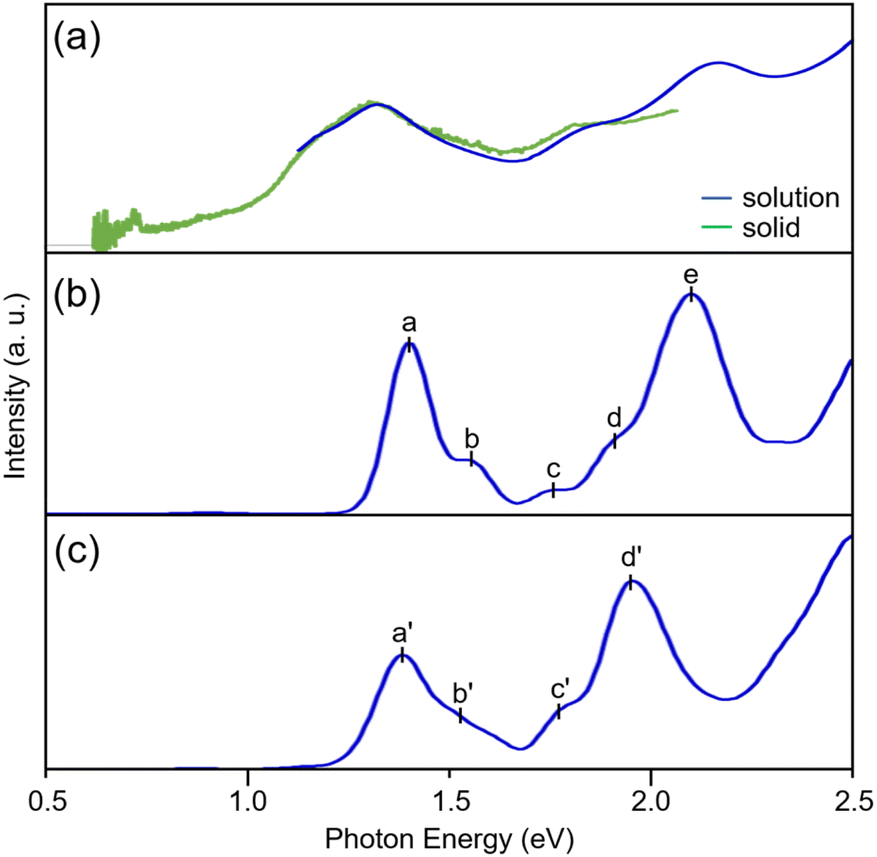 | ||
| Fig. 2 (a) Optical absorbance (blue) and diffused reflectance (green) spectra of [Pt17(CO)12(PPh3)8]z (z = 1+, 2+), and the calculated absorption spectra of (b) [Pt17(CO)12(P(CH3)3)8]2+ and (c) [Pt17(CO)12(PPh3)8]2+. | ||
DFT calculations were conducted to elucidate the origin of peaks observed in the optical absorption spectrum of [Pt17(CO)12(PPh3)8]z using the Gaussian 16 program,35 and the B3LYP functional36–38 was adopted (basis set; the Stuttgard/Dresden basis set with effective core potentials39 for Pt, 6-311+G* for P, C, O, and H). This study focused on the closed-shell electronic systems of [Pt17(CO)12(PPh3)8]2+ owing to the computational complexity of the open-shell systems in [Pt17(CO)12(PPh3)8]1+. To reduce the computational burden, the phenyl groups (Ph3) were replaced with methyl groups (CH3)3.40 Geometry optimization was performed based on the structure of [Pt17(CO)12(PPh3)8]2+ obtained by single crystal X-ray diffraction (Fig. 3a), followed by time-dependent (TD)-DFT calculations for the optical absorption spectrum using the optimized geometric structure of [Pt17(CO)12(PPh3)8]2+.
 | ||
| Fig. 3 Geometric structure of (a) [Pt17(CO)12(PPh3)8]2+ determined by SC-XRD from ref. 11 and the calculated most stable structures of (b) [Pt17(CO)12(P(CH3)3)8]2+ and (c) [Pt17(CO)12(PPh3)8]2+. Hydrogen and solvent atoms are omitted for clarity. Pt: grey, P: yellow, O: red, and carbon: light grey. | ||
The optimized structure [Pt17(CO)12(P(CH3)3)8]2+ (Fig. 3b) features an icosahedral Pt13 core surrounded by CO, μ2-CO, P(CH3)3, and capping Pt2(μ2-CO)2(P(CH3)3)2 ligands. This structure closely matches that obtained experimentally for [Pt17(CO)12(PPh3)8]2+ (Fig. 3a and S3†).11 The calculated spectrum of [Pt17(CO)12(P(CH3)3)8]2+ (Fig. 2b) shows peaks at approximately 1.40, 1.55, 1.75, and 2.10 eV, which correspond well with the experimental spectrum of [Pt17(CO)12(PPh3)8]z (Fig. 2a).
We also conducted calculations using the Los Alamos (LANL2DZ) basis set with effective core potentials and the CAM-B3LYP functional41–43 to examine how the choice of basis set affects the spectral shape. The results revealed that both the geometric structure (Fig. S4†) and optical absorption spectrum (Fig. S5†) remain largely consistent with those obtained using other basis functions and DFT functionals. To explore the impact on the spectral characteristics of substituting Ph3 with CH3, we performed calculations for [Pt17(CO)12(PPh3)8]2+ using the LANL2DZ basis function, and obtained a geometric structure (Fig. 3c) and optical absorption spectrum (Fig. 2c) similar to those of [Pt17(CO)12(P(CH3)3)8]2+. The overall similarity in both the geometric structure and the spectral shape indicates that (1) the calculated optical absorption spectra are relatively insensitive to the choice of basis set and functional group substitution and (2) the calculated results are in good agreement with experimental observations.
Fig. 4A shows the density of states (DOSs) for the orbitals ranging from HOMO−20 to LUMO+10, calculated for [Pt17(CO)12(P(CH3)3)8]2+ (basis set; SDD for Pt, 6-311+G* for P, C, O, and H). It was revealed that the orbitals near the HOMO and LUMO primarily comprise the s, p, and d orbitals of Pt (Fig. 4A and B). Owing to the high symmetry of [Pt17(CO)12(P(CH3)3)8]2+, the transition from the HOMO to the LUMO is forbidden (Fig. 3b), and thus it is not observed (at 0.74 eV) in the experimental optical absorption spectrum (Fig. 2a). The peak around 1.40 eV is attributed to transitions from HOMO−1,2,3,4 to the LUMO or LUMO+1, whereas the peak near 1.55 eV is due to transitions from HOMO−5 to LUMO+1. The peak around 2.10 eV involves multiple transitions, including those from orbitals below HOMO−10 to the LUMO, as well as transitions from orbitals near the HOMO to LUMO+1 and the orbitals above. Further analysis using the LANL2DZ basis set confirmed that the DOSs of [Pt17(CO)12(PPh3)8]2+ are not significantly influenced by the choice of basis set or by the functional group substitutions (Fig. S6†).
 | ||
| Fig. 4 (A) Calculated energy diagram and (B) related molecular orbitals of [Pt17(CO)12(P(CH3)3)8]2+. (a) LUMO, (b) LUMO+1, (c) LUMO+2, (d) HOMO, (e) HOMO−1, and (f) HOMO−2 states. | ||
Thus, this study successfully elucidated the electronic structure and the origin of the optical absorption spectrum of [Pt17(CO)12(PPh3)8]z using both experimental and theoretical approaches. These findings align with previous research44–49 on ligand-protected Au-, Ag-, and Cu NCs. Specifically, several key findings regarding the electronic structure have been observed: (1) a distinct peak structure appears in the optical absorption spectrum when the metal core is refined to ∼1 nm;50–53 (2) the peak structure in the visible region is mainly caused by the absorption of the metal core;31 (3) differences in the structure of the functional groups at the end of the ligands have a negligible effect on optical absorption.54–56
Electrocatalytic activity of Pt17/CB for the HER
Having established the geometrical and electronic structures of the Pt NCs, we next aimed to create highly active catalysts by increasing the number of active sites through miniaturization. Although many highly active Pt NCs have been reported,57–67 most require special equipment and reagents. Accordingly, those methods are not suitable for the preparation of a practical Pt NC catalyst. Although simple methods have also been reported for the preparation of Pt NCs, Pt NCs synthesized by those methods typically include the variation in the number of constituent atoms. Accordingly, they do not necessarily show high activity specialized in the NC-size region. Therefore, in this work, we attempted to prepare Pt NC catalysts using [Pt17(CO)12(PPh3)8]z, which can be obtained with atomic precision by simple synthesis in air, as a precursor to establish a method for preparing a practical NC catalyst with high HER activity. It can be expected that using such an atomically precise Pt NC as catalyst also helps to understand the mechanisms of the HER which occurs over NC catalysts.67In the experiment, following a previously established method with a slight modification,13 Pt17 NCs were supported on CB (Fig. 1c) with a Pt loading of 1.0 wt% through chemisorption, and the ligands were removed by calcination at 200 °C. Transmission electron microscopy (TEM) images of the calcined Pt17 NC-supported CB catalyst (Pt17/CB(1.0 wt% Pt)) showed that the particle size of Pt17 NC remains largely unchanged during the adsorption and calcination processes (Fig. 5a, b and S7†). X-ray photoelectron spectroscopy (XPS) indicated that the Pt in Pt17/CB(1.0 wt% Pt) is in a largely metallic electronic state with a slight positive charge, similar to the previously reported one (Fig. 5c and S8†).13
 | ||
| Fig. 5 TEM images and resulting histograms of particle-size distribution for (a) [Pt17(CO)12(PPh3)8]z and (b) Pt17/CB(1.0 wt% Pt). (c) Pt 4f7/2 XPS spectra (blue line) and their fitting results (red, green, and magenta lines) for [Pt17(CO)12(PPh3)8]z and Pt17/CB(1.0 wt% Pt). In (c), grey vertical lines indicate the position of Pt(0) and Pt(II). | ||
To evaluate their HER activities, a catalyst slurry was prepared using Pt17/CB(1.0 wt% Pt) and then deposited onto a glassy carbon electrode (GCE) to create the working electrode. After electrochemical cleaning68 to remove residual organic substances, the HER activity was measured and compared with that of a catalyst made by diluting commercial Pt NPs/CB(46.9 wt% Pt; TEC10E50E) with CB until 1.0 wt% Pt to prepare Pt NPs/CB(1.0 wt% Pt).
Fig. 6a and S9† show the linear sweep voltammetry (LSV) curves for Pt17/CB(1.0 wt% Pt) and Pt NPs/CB(1.0 wt% Pt). The results show that Pt17/CB(1.0 wt% Pt) generates a larger reduction current than Pt NPs/CB(1.0 wt% Pt) starting around 0.0 V vs. the reversible hydrogen electrode (RHE), indicating that Pt17/CB(1.0 wt% Pt) has superior HER activity to Pt NPs/CB(1.0 wt% Pt). Comparison of current values at −0.055 V vs. RHE with the appropriate Tafel slope in the HER (Fig. 6b) revealed that Pt17/CB(1.0 wt% Pt) has 1.26 times the HER activity (7.54 vs. 5.98 mA cm−2; Fig. 6c) of Pt NPs/CB(1.0 wt% Pt). Additionally, Pt17/CB(1.0 wt% Pt) provided Tafel slopes similar to the theoretical one for the Volmer step of 120 mV dec−1 assuming that H+ adsorption on the catalyst surface serves as the rate determining step for the two-step HER. The reduction potential at which a current of −10 mA cm−2 is obtained, which is commonly used for HER activity comparisons, is −0.075 V vs. RHE for Pt17/CB(1.0 wt% Pt), indicating a significantly decreased overvoltage for the HER compared with −0.103 V vs. RHE for Pt NPs/CB(1.0 wt% Pt). Furthermore, the obtained mass activity of Pt17/CB(1.0 wt% Pt) at −0.05 V vs. RHE is significantly higher than the HER mass activities reported in previous experimental studies using Pt NCs or single-atom Pt catalysts (Table S1†).
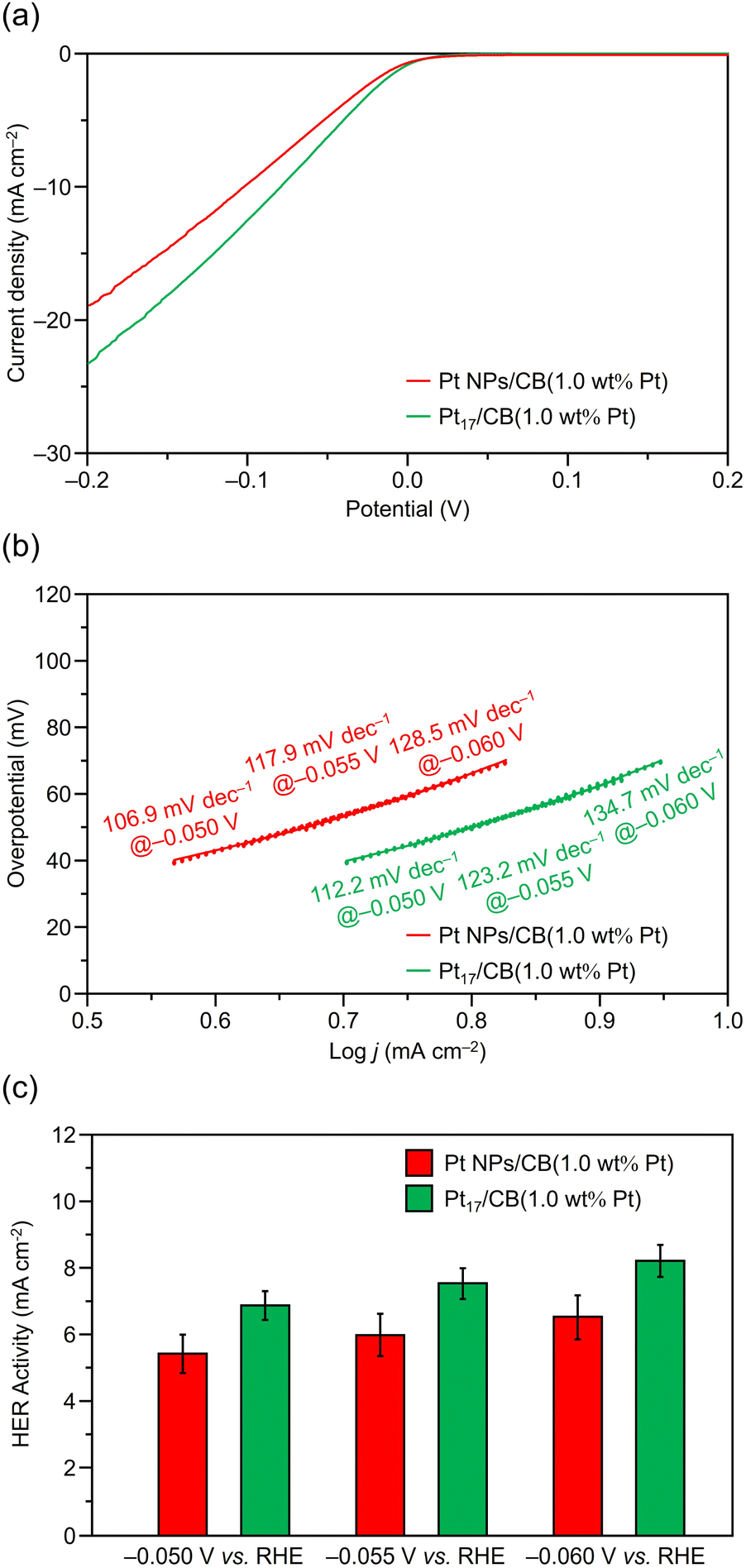 | ||
| Fig. 6 (a) Representative LSV curves. (b) Tafel slopes and (c) HER activity of Pt17/CB(1.0 wt% Pt) and Pt NPs/CB(1.0 wt% Pt). For (b) and (c), data obtained at −0.050, −0.055, and −0.060 V vs. RHE are also shown for comparison. | ||
To evaluate the ease of proton (H+) adsorption, we attempted to calculate the electrochemical surface area (ECSA) based on H+ adsorption from cyclic voltammetry (CV) curves. Owing to the low Pt loading (1.0 wt% Pt), the current value for H+ adsorption was insufficient for accurate ECSA calculation (Fig. S10†). Therefore, we prepared a catalyst with a higher Pt loading (20.0 wt% Pt) for further investigation. Pt17/CB(20.0 wt% Pt) was prepared by loading [Pt17(CO)12(PPh3)8]z with 20.0 wt% Pt followed by calcination using a similar method to that used to prepare Pt17/CB(1.0 wt% Pt). The calcination conditions were more severe (250 °C, 120 min) than for Pt17/CB(1.0 wt% Pt), considering that the number of ligands present on the support also increased. For the obtained Pt17/CB(20.0 wt% Pt), a slight increase in average particle size based on the aggregation of Pt17 NCs was observed (Fig. S11†). This is thought to be because Pt17 NCs were mainly loaded on CB by physisorption, different from the case of Pt17/CB(1.0 wt% Pt). Because of such a weak interaction between Pt17 NCs and CB, a part of Pt17 NCs aggregated during the calcination process. Electrochemical cleaning was performed on the obtained Pt17/CB(20.0 wt% Pt), followed by electrochemical measurements. A commercial catalyst (TEC10E50E; Pt NPs/CB(46.9 wt% Pt)) was used as a comparison (Fig. S12†). The HER mass activity at different Pt loadings was also calculated and compared. As a result, Pt17/CB(20.0 wt% Pt) showed 3.59 times higher HER mass activity (1.76 vs. 0.49 A mgPt−1 at −0.055 V vs. RHE; Fig. S13† and 7) than Pt NPs/CB(46.9 wt% Pt). The long-term stability of the catalyst during the HER was also evaluated. As no significant decrease in current density was observed in chronoamperometry measurements (Fig. S14†), it was suggested that Pt17/CB(20.0 wt% Pt) exhibits almost the same stability as Pt NPs/CB(46.9 wt% Pt).
 | ||
| Fig. 7 HER mass activity of Pt17/CB(20.0 wt% Pt) and Pt NPs/CB(46.9 wt% Pt) obtained from the LSVs in Fig. S11.† Data obtained at −0.050, −0.055, and −0.060 V vs. RHE are also shown for comparison. | ||
In terms of the ECSAs estimated by H+ adsorption, Pt17/CB(20.0 wt% Pt) has a surface area 1.07 times (101.3 vs. 94.6 m2 gPt−1; Fig. S15 and S16a†) that of Pt NPs/CB(46.9 wt% Pt). Thus, the activity enhancement of Pt17/CB(20.0 wt% Pt) compared with Pt NPs/CB(46.9 wt% Pt) is higher than that of ECSA. Consequently, when specific activity (SA), the activity per surface active site, is calculated by dividing ECSA by HER mass activity, Pt17/CB(20.0 wt% Pt) shows 3.39 times higher SA than that of Pt NPs/CB(46.9 wt% Pt) (Fig. S16b†). Although it is difficult to accurately calculate the active sites from the ECSA because, unlike Pt NPs, no specific crystalline planes are exposed in Pt17 NCs, our estimation implies that the Pt atoms of Pt17 NCs might have better HER properties than those of Pt NPs. Therefore, it is reasonable to expect that further improvement in activity can be achieved if it becomes possible to prepare catalysts with a higher Pt loading ratio while suppressing the aggregation of Pt17 NCs.
Mechanism of HER activity for Pt17/CB
We examined the origins of the high HER activity of Pt17/CB. Recent studies have suggested that miniaturization creates surface Pt atoms with diverse electronic states, and that high activity occurs when these electronic states are favourable for the progression of reactions.69,70 In our previous study,13 DFT calculations revealed that in the optimized structure of Pt17/graphite (Fig. 8A), Pt atoms with various charges and local DOSs are mixed (Fig. S17†). Specifically, it was revealed that (1) two Pt atoms (I = 6 and 12) contact the graphite, (2) Pt atoms on terraces (I = 1, 4, 7, and 17) are positively charged, and (3) Pt atoms at steps and corners (I = 2, 3, 5, 8–11, and 13–16) are negatively charged. These calculations used graphite as a support instead of CB to reduce computational costs and employed the TEM-observed structure of Pt17NCs as the initial configuration, which was then optimized.13 | ||
| Fig. 8 Results of DFT calculations. (A) Intermediate structure optimized for (a) (H + H)/Pt17(i)/graphite, (b) (H + H)/Pt17(ii)/graphite, (c) (H + H)/Pt17(iii)/graphite (d) (H + H)/Pt17(iv)/graphite and (e) (H + H)/Pt17(v)/graphite. (B) Free-energy diagram for the Volmer–Tafel mechanism in the HER on Pt17(X)/graphite (X = i, ii, iii or iv) or Pt(111) under an applied potential of 0.0 V vs. SHE. In this figure, Pt17(X)/graphite and Pt(111) are abbreviated as Cat. In (A), the red arrows indicate adsorbed H. | ||
To obtain insights into the HER mechanisms on Pt17/graphite, we optimized the structures of the reaction intermediates for the HER. In the calculation, four pairs of Pt atoms (Fig. S18a†) were selected as potential adsorption sites for hydrogen atoms (Hads) on the Pt17/graphite surface, referred to as Pt17(X)/graphite (X = i, ii, iii, or iv). The reaction intermediates (Hads/Pt17(X)/graphite) were then obtained by adsorbing Hads onto Pt17(X)/graphite (X = i, ii, iii, or iv). In the calculation of the intermediates, we also found another structure in which the pair of Hads is on the identical Pt atom (I = 11), referred to as Pt17(v)/graphite.
The HER generally proceeds through two main pathways. Initially, H+ is adsorbed and reduced as Hads on the electrode surface (the Volmer step). Subsequently, the reaction can proceed through either: (1) the combination of two Hads to form H2, which then desorbs (the Volmer–Tafel mechanism), or (2) the incorporation of another H+ and electron onto Hads to form H2, which then desorbs (the Volmer–Heyrovsky mechanism). Here, the mechanism by which the reaction proceeds depends largely on the electrode material. In the case of the Pt(111) surface, which is considered to be present in Pt NPs/CB, the HER is likely to proceed by the Volmer–Tafel mechanism.71–74 Thus, we have calculated the reaction when it proceeds by the Volmer–Tafel mechanism (Fig. 8B). The free-energy change in the Tafel process in Pt17(iii)/graphite and Pt17(v)/graphite was smaller than that of Pt(111). This HER energy diagram implies that the smaller H adsorption energy on Pt17(X)/graphite compared to that on a large Pt nanoparticle causes the high HER activity of Pt17/CB(20.0 wt% Pt).
Finally, we discuss the reason why H adsorption energy on Pt17(X)/graphite is smaller than that on a large Pt NP. The variation in the H adsorption energy is attributed to three factors. The first factor is the H adsorption structure. The bridge positions in sites i and iv result in the large stabilization of Hads compared to the on-top positions. The second is the low coordination-number site in Pt17(X)/graphite. The pair of Hads on the identical Pt atom (I = 11) at site v results in the small adsorption energy. The last is the variety in the electronic properties among the Pt atoms of Pt17(X)/graphite. In the case of site iii (I = 9, 11), both Hads are bound to Pt in the on-top manner. In addition, in this structure, both Pt atoms are negatively charged (Fig. S17b†). Accordingly, both Hads on site iii are negatively charged (Table S2†). Although the higher electronegativity of Pt compared with H (2.28 for Pt cf. 2.20 for H, respectively) could promote the electron transfer from H to Pt, a negatively charged Pt atom is less likely to facilitate electron transfer from H to Pt, causing less stabilization. Accordingly, the adsorption of Hads is weak at site iii, and thereby HER progression occurs more easily than at other reaction sites. This is in contrast to site ii where both Hads are on the on-top position in a similar manner to the case of site iii. In this case, the pair of Pt atoms at site ii are positively charged, resulting in the strong adsorption of H via the electron transfer from H to Pt.
Conclusion
In this study, we used DFT calculations to elucidate the origin of the electronic structure of [Pt17(CO)12(PPh3)8]z with atomic precision. Furthermore, the HER activity of Pt17/CB catalysts using [Pt17(CO)12(PPh3)8]z as a precursor was evaluated. The origin of the high HER activity of the Pt17/CB catalyst was also investigated using DFT calculations. As a result, the following conclusions were obtained:(1) The HOMO-to-LUMO transition in [Pt17(CO)12(PPh3)8]2+ is forbidden, with nearby orbitals primarily comprising the s, p, and d orbitals of Pt.
(2) Pt17/CB(20.0 wt% Pt) exhibits 3.59 times the HER mass activity of the commercial Pt NPs/CB(46.9 wt% Pt) catalyst.
(3) Pt17/graphite generates surface Pt atoms more favourable for the HER than the Pt(111) surface in Pt NPs/CB, leading to higher HER activity.
These findings offer clear design guidelines for understanding the electronic structure of atomically precise Pt NCs, developing highly active HER catalysts using Pt NCs, and reducing Pt usage in PEMWE applications. It is also expected that alloying these NCs will lead to the creation of even more active catalysts.75–77
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
T. Kawawaki and Y. Negishi designed the experiments and conducted the measurements with K. Oiwa, K. Ikeda, R. Kurosaki, K. Sato, N. Nishi, H. Tachibana, and A. Haque. K. Iida performed the DFT calculations. T. Kawawaki, K. Iida, and Y. Negishi wrote the paper. All authors approved the final version of the manuscript.Conflicts of interest
The authors declare that there are no conflicts of interest.Acknowledgements
This work was based on results obtained from a project commissioned by the New Energy and Industrial Technology Development Organization (NEDO). It was also supported by the Japan Society for the Promotion of Science (JSPS) KAKENHI (grant numbers 22K19012, 23KK0098, 23H00289, and 24K01459). Additional funding was provided by the Ogasawara Foundation for the Promotion of Science and Engineering, the Carbon Recycling Fund Institute, and the Japan Gas Association.References
- I. Ciabatti, C. Femoni, M. C. Iapalucci, G. Longoni and S. Zacchini, J. Cluster Sci., 2014, 25, 115–146 CrossRef
.
- M. Paolieri, I. Ciabatti and M. Fontani, J. Cluster Sci., 2019, 30, 1623–1631 CrossRef
- S. S. Kurasov, N. K. Eremenko, Y. L. Slovokhotov and Y. T. Struchkov, J. Organomet. Chem., 1989, 361, 405–408 Search PubMed
- N. d. Silva and L. F. Dahl, Inorg. Chem., 2005, 44, 9604–9606 CrossRef PubMed
- I. Ciabatti, C. Femoni, M. C. Iapalucci, G. Longoni, T. Lovato and S. Zacchini, Inorg. Chem., 2013, 52, 4384–4395 CrossRef PubMed
- B. Berti, C. Femoni, M. C. Iapalucci, S. Ruggieri and S. Zacchini, Eur. J. Inorg. Chem., 2018, 2018, 3285–3296 CrossRef CAS
- T. Kawawaki, N. Shimizu, K. Funai, Y. Mitomi, S. Hossain, S. Kikkawa, D. J. Osborn, S. Yamazoe, G. F. Metha and Y. Negishi, Nanoscale, 2021, 13, 14679–14687 RSC
- T. Kawawaki, N. Shimizu, Y. Mitomi, D. Yazaki, S. Hossain and Y. Negishi, Bull. Chem. Soc. Jpn., 2021, 94, 2853–2870 CrossRef CAS
- D. Yazaki, T. Kawawaki, D. Hirayama, M. Kawachi, K. Kato, S. Oguchi, Y. Yamaguchi, S. Kikkawa, Y. Ueki, S. Hossain, D. J. Osborn, F. Ozaki, S. Tanaka, J. Yoshinobu, G. F. Metha, S. Yamazoe, A. Kudo, A. Yamakata and Y. Negishi, Small, 2023, 19, 2208287 CrossRef CAS
- D. Yazaki, T. Kawawaki, T. Tanaka, D. Hirayama, Y. Shingyouchi and Y. Negishi, Energy Adv., 2023, 2, 1148–1154 RSC
- L. V. Nair, S. Hossain, S. Wakayama, S. Takagi, M. Yoshioka, J. Maekawa, A. Harasawa, B. Kumar, Y. Niihori, W. Kurashige and Y. Negishi, J. Phys. Chem. C, 2017, 121, 11002–11009 CrossRef
- C. Schmitt, N. D. Roit, M. Neumaier, C. B. Maliakkal, D. Wang, T. Henrich, C. Kübel, M. Kappes and S. Behrens, Nanoscale Adv., 2024, 6, 2459–2468 RSC
- T. Kawawaki, Y. Mitomi, N. Nishi, R. Kurosaki, K. Oiwa, T. Tanaka, H. Hirase, S. Miyajima, Y. Niihori, D. J. Osborn, T. Koitaya, G. F. Metha, T. Yokoyama, K. Iida and Y. Negishi, Nanoscale, 2023, 15, 7272–7279 RSC
- S. Reid and H. Hernández, J. Phys. Chem. A, 2023, 127, 4237–4244 CrossRef
- A. K. Satheesan, L. V. Nair, J. S. Gopinath, P. Parameswaran and C. Keloth, J. Phys. Chem. C, 2023, 127, 568–576 CrossRef
- J. Wei, R. Marchal, D. Astruc, S. Kahlal, J.-F. Halet and J.-Y. Saillard, Nanoscale, 2022, 14, 3946–3957 RSC
- A. K. Satheesan, N. P. Purayil, J. Singh and C. Keloth, ACS Appl. Nano Mater., 2024, 7, 7486–7495 CrossRef
- J. M. Guevara-Vela, T. Rocha-Rinza, P. L. Rodríguez-Kessler and A. Muñoz-Castro, Phys. Chem. Chem. Phys., 2023, 25, 28835–28840 RSC
- H. Hirase, K. Iida and J.-y. Hasegawa, Phys. Chem. Chem. Phys., 2024, 26, 18530–18537 RSC
- J. Zhu, L. Hu, P. Zhao, L. Y. S. Lee and K.-Y. Wong, Chem. Rev., 2020, 120, 851–918 CrossRef PubMed
- M. Chatenet, B. G. Pollet, D. R. Dekel, F. Dionigi, J. Deseure, P. Millet, R. D. Braatz, M. Z. Bazant, M. Eikerling, I. Staffell, P. Balcombe, Y. Shao-Horn and H. Schäfer, Chem. Soc. Rev., 2022, 51, 4583–4762 RSC
- J. N. Hansen, H. Prats, K. K. Toudahl, N. M. Secher, K. Chan, J. Kibsgaard and I. Chorkendorff, ACS Energy Lett., 2021, 6, 1175–1180 Search PubMed
- C. C. L. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2015, 137, 4347–4357 Search PubMed
- F. Fievet, J. P. Lagier and M. Figlarz, MRS Bull., 2013, 14, 29–34 CrossRef
- C. Bock, C. Paquet, M. Couillard, G. A. Botton and B. R. MacDougall, J. Am. Chem. Soc., 2004, 126, 8028–8037 CrossRef CAS PubMed
- H. Dong, Y.-C. Chen and C. Feldmann, Green Chem., 2015, 17, 4107–4132 RSC
- C. M. Pelicano, M. Saruyama, R. Takahata, R. Sato, Y. Kitahama, H. Matsuzaki, T. Yamada, T. Hisatomi, K. Domen and T. Teranishi, Adv. Funct. Mater., 2022, 32, 2202987 CrossRef CAS
- B.-J. Hwang, L. S. Sarma, C.-H. Chen, C. Bock, F.-J. Lai, S.-H. Chang, S.-C. Yen, D.-G. Liu, H.-S. Sheu and J.-F. Lee, J. Phys. Chem. C, 2008, 112, 19922–19929 CrossRef CAS
- I. Schrader, J. Warneke, S. Neumann, S. Grotheer, A. A. Swane, J. J. K. Kirkensgaard, M. Arenz and S. Kunz, J. Phys. Chem. C, 2015, 119, 17655–17661 CrossRef CAS
- Y. Negishi, N. Shimizu, K. Funai, R. Kaneko, K. Wakamatsu, A. Harasawa, S. Hossain, M. E. Schuster, D. Ozkaya, W. Kurashige, T. Kawawaki, S. Yamazoe and S. Nagaoka, Nanoscale Adv., 2020, 2, 669–678 RSC
- M. Zhu, C. M. Aikens, F. J. Hollander, G. C. Schatz and R. Jin, J. Am. Chem. Soc., 2008, 130, 5883–5885 Search PubMed
- M. W. Heaven, A. Dass, P. S. White, K. M. Holt and R. W. Murray, J. Am. Chem. Soc., 2008, 130, 3754–3755 CrossRef CAS
- C. M. Aikens, J. Phys. Chem. A, 2009, 113, 10811–10817 CrossRef CAS PubMed
- M. J. Hartmann, H. Häkkinen, J. E. Millstone and D. S. Lambrecht, J. Phys.
Chem. C, 2015, 119, 8290–8298 CrossRef CAS
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 Search PubMed
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS
- M. Dolg, U. Wedig, H. Stoll and H. Preuss, J. Chem. Phys., 1987, 86, 866–872 CrossRef CAS
- K. A. Kacprzak, L. Lehtovaara, J. Akola, O. Lopez-Acevedo and H. Häkkinen, Phys. Chem. Chem. Phys., 2009, 11, 7123–7129 Search PubMed
- P. J. Hay and W. R. J. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef
-
T. H. Dunning Jr and P. J. Hay, in Modern Theoretical Chemistry, ed. H. F. Schaefer III, Plenum, New York, 1976, vol. 3, pp. 1–28 Search PubMed
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef
- B. Yin and Z. Luo, Coord. Chem. Rev., 2021, 429, 213643 CrossRef
- T. Kawawaki, Y. Imai, D. Suzuki, S. Kato, I. Kobayashi, T. Suzuki, R. Kaneko, S. Hossain and Y. Negishi, Chem.–Eur. J., 2020, 26, 16150–16193 CrossRef PubMed
- X. Kang, Y. Li, M. Zhu and R. Jin, Chem. Soc. Rev., 2020, 49, 6443–6514 RSC
- J. Fang, B. Zhang, Q. Yao, Y. Yang, J. Xie and N. Yan, Coord. Chem. Rev., 2016, 322, 1–29 CrossRef
- I. Chakraborty and T. Pradeep, Chem. Rev., 2017, 117, 8208–8271 CrossRef
- Y. Du, H. Sheng, D. Astruc and M. Zhu, Chem. Rev., 2020, 120, 526–622 CrossRef PubMed
- T. Kawawaki and Y. Negishi, Dalton Trans., 2023, 52, 15152–15167 RSC
- K. Kwak, V. D. Thanthirige, K. Pyo, D. Lee and G. Ramakrishna, J. Phys. Chem. Lett., 2017, 8, 4898–4905 Search PubMed
- M. Zhou, C. Zeng, Y. Song, J. W. Padelford, G. Wang, M. Y. Sfeir, T. Higaki and R. Jin, Angew. Chem., Int. Ed., 2017, 56, 16257–16261 Search PubMed
- M. Zhou, X. Du, H. Wang and R. Jin, ACS Nano, 2021, 15, 13980–13992 Search PubMed
- Y. Song, J. Zhong, S. Yang, S. Wang, T. Cao, J. Zhang, P. Li, D. Hu, Y. Pei and M. Zhu, Nanoscale, 2014, 6, 13977–13985 RSC
- S. Takano, S. Ito and T. Tsukuda, J. Am. Chem. Soc., 2019, 141, 15994–16002 CrossRef PubMed
- T. Omoda, S. Takano and T. Tsukuda, Small, 2021, 17, 2001439 CrossRef
- W. Liu, Z. Xiang, A. Tan, K. Wan, Z. Fu and Z. Liang, Adv. Funct. Mater., 2023, 33, 2212752 CrossRef
- J. Klein, A. K. Engstfeld, S. Brimaud and R. J. Behm, Phys. Chem. Chem. Phys., 2020, 22, 19059–19068 RSC
- J. Han, C. Gong, C. He, P. He, J. Zhang and Z. Zhang, J. Mater. Chem. A, 2022, 10, 16403–16408 RSC
- Y. Lai, Z. Zhang, Z. Zhang, Y. Tan, L. Yu, W. Wu, Z. Wang, T. Jiang, S. Gao and N. Cheng, Chem. Eng. J., 2022, 435, 135102 CrossRef
- N. Cheng, S. Stambula, D. Wang, M. N. Banis, J. Liu, A. Riese, B. Xiao, R. Li, T.-K. Sham, L.-M. Liu, G. A. Botton and X. Sun, Nat. Commun., 2016, 7, 13638 CrossRef PubMed
- Y. Zhao, P. V. Kumar, X. Tan, X. Lu, X. Zhu, J. Jiang, J. Pan, S. Xi, H. Y. Yang, Z. Ma, T. Wan, D. Chu, W. Jiang, S. C. Smith, R. Amal, Z. Han and X. Lu, Nat. Commun., 2022, 13, 2430 CrossRef PubMed
- S. Kumari, T. Masubuchi, H. S. White, A. Alexandrova, S. L. Anderson and P. Sautet, J. Am. Chem. Soc., 2023, 145, 5834–5845 CrossRef CAS PubMed
- M. Zhou, S. Bao and A. J. Bard, J. Am. Chem. Soc., 2019, 141, 7327–7332 CrossRef CAS PubMed
- Z. Zeng, S. Küspert, S. E. Balaghi, H. E. M. Hussein, N. Ortlieb, M. Knäbbeler-Buß, P. Hügenell, S. Pollitt, N. Hug, J. Melke and A. Fischer, Small, 2023, 19, 2205885 CrossRef CAS
- C. Dong, Y. Li, D. Cheng, M. Zhang, J. Liu, Y.-G. Wang, D. Xiao and D. Ma, ACS Catal., 2020, 10, 11011–11045 CrossRef CAS
- R. Wang, D. Chen, L. Fang, W. Fan, Q. You, G. Bian, Y. Zhou, W. Gu, C. Wang, L. Bai, J. Li, H. Deng, L. Liao, J. Yang and Z. Wu, Angew. Chem., Int. Ed., 2024, 63, e202402565 CrossRef CAS PubMed
- Y. Lu, Y. Jiang, X. Gao and W. Chen, Chem. Commun., 2014, 50, 8464–8467 Search PubMed
- F. Calle-Vallejo, J. Tymoczko, V. Colic, Q. H. Vu, M. D. Pohl, K. Morgenstern, D. Loffreda, P. Sautet, W. Schuhmann and A. S. Bandarenka, Science, 2015, 350, 185–189 CrossRef CAS
- B. Garlyyev, J. Fichtner, O. Piqué, O. Schneider, A. S. Bandarenka and F. Calle-Vallejo, Chem. Sci., 2019, 10, 8060–8075 RSC
- G. Ramos-Sanchez and P. B. Balbuena, Phys. Chem. Chem. Phys., 2013, 15, 11950–11959 RSC
- E. Skúlason, V. Tripkovic, M. E. Björketun, S. Gudmundsdóttir, G. Karlberg, J. Rossmeisl, T. Bligaard, H. Jónsson and J. K. Nørskov, J. Phys. Chem. C, 2010, 114, 18182–18197 CrossRef
- S. Kumari, T. Masubuchi, H. S. White, A. Alexandrova, S. L. Anderson and P. Sautet, J. Am. Chem. Soc., 2023, 145, 5834–5845 CrossRef
- M. Zhou, S. Bao and A. J. Bard, J. Am. Chem. Soc., 2019, 141, 7327–7332 Search PubMed
- L. Zhang, Y. Lei, Y. Yang, D. Wang, Y. Zhao, X. Xiang, H. Shang and B. Zhang, Adv. Sci., 2024, 2407475 Search PubMed
- L. Zhang, Y. Lei, W. Xu, D. Wang, Y. Zhao, W. Chen, X. Xiang, X. Pang, B. Zhang and H. Shang, Chem. Eng. J., 2023, 460, 141119 CrossRef
- Y. Lei, L. Zhang, W. Xu, C. Xiong, W. Chen, X. Xiang, B. Zhang and H. Shang, Nano Res., 2022, 15, 6054–6061 CrossRef
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta08004a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |