
Feasible constructions of Bi@CNT materials with extremely high rate and Na+ storage performance†
Jianke
Li
 a,
Xincheng
Miao
*c,
Beibei
Han
d,
Kun
Wang
*a,
Baigang
An
a,
Chengguo
Sun
a,
Guiying
Xu
*a,
Dongying
Ju
b,
Lin
Tao
a and
Weimin
Zhou
*a
a,
Xincheng
Miao
*c,
Beibei
Han
d,
Kun
Wang
*a,
Baigang
An
a,
Chengguo
Sun
a,
Guiying
Xu
*a,
Dongying
Ju
b,
Lin
Tao
a and
Weimin
Zhou
*a
aKey Laboratory of Energy Materials and Electrochemistry Research Liaoning Province, University of Science and Technology Liaoning, No. 189, Qianshan Middle Road, Lishan District, Anshan City, Liaoning Province, Anshan 114051, China. E-mail: aszhou15242870697@163.com; ustl15542731203@163.com; xuguiying751107@163.com
bAdvanced Science Research Laboratory, Saitama Institute of Technology, 1690 Fusaiji, Fukaya, Japan
cSchool of Materials and Metallurgy, University of Science and Technology Liaoning, No. 189, Qianshan Middle Road, Lishan District, Anshan City, Liaoning Province, Anshan 114051, China. E-mail: asust_msn@163.com
dKey Laboratory of Advanced Fuel Cells and Electrolyzers Technology of Zhejiang Province, Ningbo Institute of Materials Technology and Engineering, Chinese Academy of Sciences, No. 1219 Zhongguan West Road, Ningbo, Zhejiang 315201, China
First published on 11th April 2025
Abstract
Micro-sized Bi is well-known for its high capacity and optimal operating potential, which makes it a compelling option for the negative electrodes of SIBs. To maximize the sodium storage capacity of Bi, developing its advantages and mitigating its disadvantages during storage processes are pivotal considerations. Our studies innovatively synthesized Bi@CNT materials constructed from micro-sized Bi and carbon nanotubes (CNTs). Moreover, it is intriguingly found that some nano-sized Bi particles are embedded on the surface of the CNTs and inserted into the CNTs. These unique structures enable Bi@CNT to possess excellent Na+ energy storage capacity and rate performance. At a current density of 0.2 A g−1, the maximal reversible capacity of Bi@CNT-2.61 is 462 mA h g−1, which surpasses the theoretical specific capacity of pure Bi. Surprisingly, the discharge-specific capacity of Bi@CNT-2.61, with a mass loading of 0.89 mg cm−2, is maintained at 248.1 mA h g−1 after 5450 cycles at an enormously high current density of 50 A g−1. When full cells (Bi@CNT-2.61//NVP) are assembled with Na3V2(PO4)3 (NVP) as the cathode and Bi@CNT-2.61 as the anode, it is found that the Bi@CNT-2.61//NVP demonstrates a high energy density of 192.1 W h kg−1. The high capacity retentions of the Bi@CNT-2.61//NVP are 96.67% and 98.64% after 445 and 400 cycles at current densities of 1.0 and 5.0 A g−1, respectively. The exceptional stability and charge transport of Bi@CNTs were analyzed in detail by ex situ XRD measurements and DFT computations.
1. Introduction
Due to the abundance and low cost of sodium resources, sodium-ion batteries (SIBs) have received extensive attention as a potential solution to address the lithium resource bottleneck in the development of electrochemical energy storage systems (EESS). Although SIBs have been developed, their low energy density, poor rate performance, and poor cycle stability restrict their practical applications. The fundamental problem lies in the design and fabrication of anode materials, which directly impact the energy density, rate performance, and cycle performance of SIBs. Therefore, designing and fabricating anode materials with excellent energy density, rate performance and cycle stability has become a more pivotal research subject than ever before.1–3 In terms of specific design, energy density is closely related to the elemental compositions of electrodes, while rate performance and cycle stability mainly rely on the electronic conductivity and structural stability of materials.4A variety of anode materials have been explored in this field, including carbon-based materials, metallic compounds, transition metal oxides/sulfides, titanium-based compounds, and fluorides.5 Specifically, hard carbons, which possess low potential plateaus, have been extensively studied and utilized in the fabrication of SIBs.6–8 However, the significantly poor rate performance of hard carbons restricts their practical applications.9–11 To overcome the shortcomings of hard carbons, research on finding other Na+ anode materials with low voltage plateaus, high storage capacity and excellent rate performance has become a hot topic.5,12
Among them, Bi, belonging to the alloy storage type, stands out as a promising candidate for the anode material of SIBs due to its high theoretical specific capacity (386 mA h g−1) and moderate operating potentials (0.5–0.75 V). However, Bi undergoes a significant volume expansion of up to 250% during the Na+ de-intercalation processes while employed as a negative electrode in SIBs. The severe fragmentations and depletions of active materials by the intense volume expansion of Bi particles result in the interruption of electrical contact and rapid capacity decay. Furthermore, this volume alteration also plays a role in the continuous degradation and regeneration of the solid-electrolyte interphase (SEI) layer, resulting in the irreversible depletion of the electrolyte and an increase in the charge transfer resistance, which leads to the continuing decline in the capacity of Bi electrodes.
Prior research has demonstrated that combining conductive carbon substances with nanoscale Bi to form composites is a feasible approach to improving the electrochemical characteristics of Bi. For instance, Li et al. fabricated Bi nanosheet microspheres with an open pore structure, which exhibit a notable capacity of 361 mA h g−1 at a current density of 5 A g−1 over 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles.13 The composites of Bi nanoparticles covered by the nitrogen-doped carbons display excellent cyclic stability, showing a capacity of 326.9 mA h g−1 after 5000 cycles at 2 A g−1.14 Yu's group successfully fabricated composite materials constructed by Bi nanorods enveloped with N-doped carbon nanotubes. It manifests a high capacity of 302 mA h g−1 after 1000 cycles under a current density of 1 A g−1.15 Ma et al. fabricated Bi nanoflowers coated with N-doped carbons. The obtained composite material sustains the significantly reversible specific capacity of 363 mA h g−1 over 4000 cycles at a current density of 2 A g−1.16 These approaches primarily improve the electrochemical performance of Bi-based materials from two aspects, such as ensuring a sufficient number of active sites and adjusting the size of Bi particles at the nanoscale.13–16
000 cycles.13 The composites of Bi nanoparticles covered by the nitrogen-doped carbons display excellent cyclic stability, showing a capacity of 326.9 mA h g−1 after 5000 cycles at 2 A g−1.14 Yu's group successfully fabricated composite materials constructed by Bi nanorods enveloped with N-doped carbon nanotubes. It manifests a high capacity of 302 mA h g−1 after 1000 cycles under a current density of 1 A g−1.15 Ma et al. fabricated Bi nanoflowers coated with N-doped carbons. The obtained composite material sustains the significantly reversible specific capacity of 363 mA h g−1 over 4000 cycles at a current density of 2 A g−1.16 These approaches primarily improve the electrochemical performance of Bi-based materials from two aspects, such as ensuring a sufficient number of active sites and adjusting the size of Bi particles at the nanoscale.13–16
From a perspective of cost-effectiveness, controlling the sizes of Bi particles at the micrometer level rather than at the nanometer level is appealing.17 The micro-sized Bi particles in composites can offer advantages, such as increasing the mass loading, volume capacity, energy density, coulomb efficiency, and practicality of SIBs.18 Nevertheless, it is crucial to address the noticeable volume changes of micro-sized Bi particles during the discharge–charge cycles, which can induce the instability of the SEI layers and decline in capacity. Furthermore, bigger micro-sized Bi particles also lead to sluggish Na+ storage kinetics, causing uneven Na alloying processes. It is thought that carbon coating is one of the effective methods for tackling the aforementioned issues.18 On the other hand, distributing the micro-sized Bi onto the carbon matrix to establish the robust interface connection is another efficient way to enhance the electrochemical performance of the Bi-based anode electrodes.16
Based on the comprehensive analyses, the CNTs as a carbon matrix and Bi(NO3)3·5H2O as the Bi source are utilized to synthesize Bi@CNT materials. After detailed structural analyses, it was found that the micro-sized Bi balls are tightly packaged by the CNTs. What is even more impressive is that the nano-sized Bi particles are embedded on the walls of tubes and inserted inside the tubes. These characteristic distributions of nano-sized Bi particles are significantly beneficial to constructing more complex conductive networks on the surface of micro-sized Bi balls, which ideally overcome the conductive anisotropy of CNTs. Meanwhile, the nano-sized Bi particles also possess significant contributions for energy storage because the nano-sized Bi particles facilely exhibit the pseudocapacitance behavior.19 The distinctive structures of Bi@CNT materials contribute to their exceptional performance. As an example, the Bi@CNT-2.61 exhibits the maximum reversible capacity of 462 mA h g−1 at a current density of 0.2 A g−1, surpassing the theoretical specific capacity of pure Bi. The discharge-specific capacities of the Bi@CNT-2.61 electrode reached 400.1, 394.0, 401.2, 403.6, 402.1, 395.7, 390.4, 382, 366.5, 340.8, and 293.6 mA h g−1, respectively, at current densities of 0.1, 0.2, 0.5, 1, 2, 5, 10, 20, 30, 40, and 50 A g−1. Moreover, when the mass loading was 0.89 mg cm−2, the discharge specific capacity at a high current density of 50 A g−1 is at 248.1 mA h g−1 after 5450 cycles.
The full battery (Bi@CNT-2.61//NVP) was assembled using Na3V2(PO4)3 as a cathode and Bi@CNT-2.61 as an anode and exhibited excellent rate performance. For instance, when the current densities are set at 1 A g−1, 2 A g−1, 3 A g−1, 4 A g−1, 5 A g−1, 6 A g−1, and 7 A g−1, the specific discharge capacities are 195.4, 184.8, 176, 157.2, 132.4, 87.7, and 53.5 mA h g−1, respectively. The high energy density of Bi@CNT-2.61//NVP is 192 W h kg−1, which is higher than that of most fabricated Na+ full cells. These electrochemical evaluations strongly supported the fact that the fabricated Bi@CNT materials possess significant potential in the fields of EESS.
2. Experimental
2.1. Chemicals
Bi(NO3)3·5H2O (AR 99%) was purchased from RON Technology Co., LTD. Carbon nanotubes (CNT, length 0.5–2 μm, diameter 5–15 nm, purity 99%) were procured from Chengdu Organic Chemistry Co., LTD, Chinese Academy of Sciences. Ethylene glycol (AR) was purchased from Sinopharm Group Chemical Reagent Co., LTD. No additional purification was conducted on the aforementioned materials and reagents.2.2. Preparations of Bi, CNT, and Bi@CNT materials
First, 1.74 g, 2.61 g, and 4.43 g of Bi(NO3)3·5H2O were individually placed in three beakers, each containing 50 mL glycol solution. These mixtures were stirred for 1 hour to dissolve the solid substance completely. The carbon nanotubes (CNTs) (0.1 g) were added to the mixture solutions, and these solutions were stirred for 3 hours to form uniform solutions. These mixture solutions were transferred into a polytetrafluoroethylene container (100 ml), and the syntheses proceeded for 12 h at 180 °C. After cooling to room temperature, the reacted liquid was filtered, and the obtained solids were washed with deionized water and ethanol. The obtained black solids were dried in a drying oven at 80 °C for 12 h to remove excess water. Finally, the dried samples were placed in a tube furnace filled with nitrogen atmosphere and heated to 800 °C at a heating rate of 5 °C min−1. The temperature of the tube furnace was kept at 800 °C for 2 hours. The obtained materials were named Bi@CNT-1.74, Bi@CNT-2.61, and Bi@CNT-4.43, according to the different amounts of Bi(NO3)3·5H2O.Based on the same preparation procedure of Bi@CNT materials, pure Bi and CNTs were prepared without adding CNTs and Bi(NO3)3·5H2O, respectively.
2.3. Characterization
The crystal structure of the sample was analyzed using an X-ray diffractometer (Bruker D8 ADVANCE, Germany) equipped with a Cu Kα radiation source (wavelength 0.15406 nm). The morphologies and microstructures of the samples were examined using scanning electron microscopy (SEM) (ZEISS Gemini SEM 300, Germany) and transmission electron microscopy (TEM) (JEOL JEM-F200, Japan). The elemental distribution was identified using energy-dispersive X-ray spectroscopy (EDS). Raman spectra were obtained using a Raman spectrometer equipped with an excitation wavelength of 325 nm (Horiba LabRAM HR Evolution, Japan). X-ray photoelectron spectroscopy (XPS) analyses were conducted using the Thermo Scientific K-Alpha instrument (USA). The N2 adsorption/desorption isotherms were obtained using the Quadrasorbautosorb-iQ2 (USA) at 77 K. The specific surface area of the material was determined using the Brunauer–Emmett–Teller (BET) method, while the pore size distribution was assessed using density functional theory (DFT). The thermogravimetric analysis was conducted using the Rigaku TG/DTA8122 instrument (Japan). The carbon contents of the composite materials were measured using the Elementar Unicube instrument (Germany).2.4. Electrochemical measurements
:20:10. N-Methyl-2-pyrrolidone (NMP) was employed to blend the active materials, Super-P carbon black and PVDF. Subsequently, the obtained slurries were evenly placed on the copper foil. The pre-dried copper foils with slurries were dried at 80 °C for 1 h to eliminate the NMP solvent. Finally, they were dried in a vacuum oven at 120 °C for 12 hours. After cooling down to room temperature, the copper foils with active materials were cut into discs (Φ = 11 mm).
The counter electrodes are sodium chips, the separators are the glass fiber disk (GF/D), and the electrolyte is 1 M NaPF6 dissolved in 1, 2-dimethoxyethane (DME) (Suzhou Duoduo Reagent Co., LTD). The electrochemical performance of the half-cells was evaluated using the CT3002A LANHE battery test system manufactured by the Wuhan Landian Company. The cut-off voltage for charge and discharge processes ranged from 0.01 to 1.2 V. Cyclic voltammetry (CV) and electrochemical impedance analyses were conducted using the Shanghai Chenhua CHI660E electrochemical workstation. The scanning voltage range of CV is 0.01–1.2 V. The electrochemical impedance test was performed in a frequency range from 100 kHz to 0.01 Hz, with an applied amplitude of 5 mV.
3 Results and discussion
The specific synthesis procedures of pure Bi and CNT materials are described in the ESI (Fig. S1†). Fig. 1 illustrates the one-step synthesis procedure of the Bi@CNT materials. Fig. 2a shows the XRD patterns of the materials. It can be seen that the diffraction peaks at 22.47°, 27.17°, 37.95°, 39.62°, 44.55°, 45.86°, 48.70°, 56.02°, 62.18°, 64.51°, 70.78°, 71.88°, and 85.34° correspond to the (003), (012), (104), (110), (015), (006), (202), (024), (116), (122), (214), (300), and (220) crystal planes, respectively, of hexagonal Bi (ICDD No. 01-85-1329, space group R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m), suggesting that the Bi and Bi@CNT composites were prepared successfully. In addition, the sharp peak at 26.3° corresponding to the diffraction pattern of CNTs suggests that they possess a high degree of graphitization after the hydrothermal synthesis process. To perform a detailed analysis of the crystal structures of Bi and Bi@CNT materials, the diffraction data were refined using the Rietveld refinement method (Fig. S2†). Table S1† exhibits the lattice parameters and micro-strain data. It can be seen that the Bi@CNT-2.61 exhibits the largest unit cell volume while maintaining the smallest micro-strain among all samples. This finding provides complementary evidence that Bi@CNT-2.61 exhibits enhanced sodium storage performance. In order to explore the mass proportions of Bi in the Bi@CNT materials, thermogravimetric analyses were conducted, and the results are presented in Fig. 2b. The Bi contents of Bi@CNT-1.74, Bi@CNT-2.61, and Bi@CNT-4.43 composites were calculated to be 87.00%, 91.98%, and 95.08%, respectively (eqn S1†). Furthermore, the carbon contents in the Bi@CNT materials were also determined by elemental analyses (Table S2†). Table S2† illustrates the carbon contents in Bi@CNT-1.74, Bi@CNT-2.61, and Bi@CNT-4.43 composite materials, which are 11.08%, 8.82%, and 5.27%, respectively. The corresponding Bi contents in Bi@CNT-1.74, Bi@CNT-2.61, and Bi@CNT-4.43 composite materials were calculated to be 88.92%, 91.18% and 94.73%, respectively, which are close to the thermogravimetric analysis (TGA) results (Fig. 2b). X-ray photoelectron spectroscopy (XPS) analyses were performed to reveal the surface chemical state of the Bi@CNT materials, and the results are shown in Fig. 2c–f, S3 and Table S3.†Fig. 2c–f exhibit the XPS spectra of the Bi@CNT-2.61 material. As displayed in Fig. 2c, the C (284.6 eV), O (531.3 eV) and Bi (5d (26.08 eV), 5p (102.08 eV), 4f (159–165 eV), 4d (442.08 eV) and 4p (532.08 eV)) were distinctly observed, indicating that the Bi@CNT-2.61 material is comprised of C, O and Bi elements. In the HRXPS spectrum of Bi 4f, two strong peaks of Bi 4f located at 160.7 and 164.9 eV correspond to the Bi 4f7/2 and Bi 4f5/2 of Bi, respectively (Fig. 2d).4,16 The other peaks at 159.6 and 166.1 eV can be assigned to the Bi–O bonds. Associating with the fact that no Bi2O3 signals were detected in the XRD patterns, we conjecture that oxidations only occurred on the surface of the Bi.26
m), suggesting that the Bi and Bi@CNT composites were prepared successfully. In addition, the sharp peak at 26.3° corresponding to the diffraction pattern of CNTs suggests that they possess a high degree of graphitization after the hydrothermal synthesis process. To perform a detailed analysis of the crystal structures of Bi and Bi@CNT materials, the diffraction data were refined using the Rietveld refinement method (Fig. S2†). Table S1† exhibits the lattice parameters and micro-strain data. It can be seen that the Bi@CNT-2.61 exhibits the largest unit cell volume while maintaining the smallest micro-strain among all samples. This finding provides complementary evidence that Bi@CNT-2.61 exhibits enhanced sodium storage performance. In order to explore the mass proportions of Bi in the Bi@CNT materials, thermogravimetric analyses were conducted, and the results are presented in Fig. 2b. The Bi contents of Bi@CNT-1.74, Bi@CNT-2.61, and Bi@CNT-4.43 composites were calculated to be 87.00%, 91.98%, and 95.08%, respectively (eqn S1†). Furthermore, the carbon contents in the Bi@CNT materials were also determined by elemental analyses (Table S2†). Table S2† illustrates the carbon contents in Bi@CNT-1.74, Bi@CNT-2.61, and Bi@CNT-4.43 composite materials, which are 11.08%, 8.82%, and 5.27%, respectively. The corresponding Bi contents in Bi@CNT-1.74, Bi@CNT-2.61, and Bi@CNT-4.43 composite materials were calculated to be 88.92%, 91.18% and 94.73%, respectively, which are close to the thermogravimetric analysis (TGA) results (Fig. 2b). X-ray photoelectron spectroscopy (XPS) analyses were performed to reveal the surface chemical state of the Bi@CNT materials, and the results are shown in Fig. 2c–f, S3 and Table S3.†Fig. 2c–f exhibit the XPS spectra of the Bi@CNT-2.61 material. As displayed in Fig. 2c, the C (284.6 eV), O (531.3 eV) and Bi (5d (26.08 eV), 5p (102.08 eV), 4f (159–165 eV), 4d (442.08 eV) and 4p (532.08 eV)) were distinctly observed, indicating that the Bi@CNT-2.61 material is comprised of C, O and Bi elements. In the HRXPS spectrum of Bi 4f, two strong peaks of Bi 4f located at 160.7 and 164.9 eV correspond to the Bi 4f7/2 and Bi 4f5/2 of Bi, respectively (Fig. 2d).4,16 The other peaks at 159.6 and 166.1 eV can be assigned to the Bi–O bonds. Associating with the fact that no Bi2O3 signals were detected in the XRD patterns, we conjecture that oxidations only occurred on the surface of the Bi.26
 | ||
| Fig. 1 Synthetic schematic diagram of the Bi@CNT materials. (A) Dissolution process of Bi(NO3)3·5H2O. (B) Hydrothermal reaction process of Bi(NO3)3·5H2O and CNT. (C) High-temperature carbonization process. | ||
 | ||
| Fig. 2 (a) XRD patterns of Bi, CNTs, and Bi@CNT materials, (b) TGA curves of Bi@CNT materials, and (c–f) XPS spectra of the Bi@CNT-2.61 material. | ||
As shown in the O 1s spectrum in Fig. 2e, the peaks at 530.1, 531.8, and 533.3 eV corresponding to Bi–O–Bi, C–O–Bi, and C–O/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds are observed clearly.3 The presence of Bi–O–Bi indicates that a small amount of bismuth oxide is present in Bi@CNT-2.61, and the C–O–Bi bond reveals discernible connections between Bi and the CNTs. The C–O–Bi bonds facilitate the swift ion and electron transfer between Bi and the CNTs matrix during electrochemical processes, consequently augmenting the rapid charge–discharge ability.3,4 In the HRXPS spectrum of C 1s, three distinct peaks at 284.6, 285.6, and 290.5 eV correspond to the C–C, C–O, and CO bonds, respectively (Fig. 2f). Moreover, the XPS spectra of Bi@CNT-1.74 and Bi@CNT-4.43 materials are presented in Fig. S3.† The contents of chemical bonds in the O 1s spectra of Bi@CNT materials are illustrated in Table S3.† The content of C–O–Bi bonds in Bi@CNT-2.61 is more than that in Bi@CNT-1.74 and Bi@CNT-4.43 materials (Table S3†). A large number of C–O–Bi bonds are beneficial for constructing a conductive network on the surface of Bi particles, which can prominently enhance the electrochemical performance of Bi@CNT-2.61.3,4,16,18
O bonds are observed clearly.3 The presence of Bi–O–Bi indicates that a small amount of bismuth oxide is present in Bi@CNT-2.61, and the C–O–Bi bond reveals discernible connections between Bi and the CNTs. The C–O–Bi bonds facilitate the swift ion and electron transfer between Bi and the CNTs matrix during electrochemical processes, consequently augmenting the rapid charge–discharge ability.3,4 In the HRXPS spectrum of C 1s, three distinct peaks at 284.6, 285.6, and 290.5 eV correspond to the C–C, C–O, and CO bonds, respectively (Fig. 2f). Moreover, the XPS spectra of Bi@CNT-1.74 and Bi@CNT-4.43 materials are presented in Fig. S3.† The contents of chemical bonds in the O 1s spectra of Bi@CNT materials are illustrated in Table S3.† The content of C–O–Bi bonds in Bi@CNT-2.61 is more than that in Bi@CNT-1.74 and Bi@CNT-4.43 materials (Table S3†). A large number of C–O–Bi bonds are beneficial for constructing a conductive network on the surface of Bi particles, which can prominently enhance the electrochemical performance of Bi@CNT-2.61.3,4,16,18
The Raman spectra of pure Bi, CNTs, and Bi@CNT materials are shown in Fig. S4.† The characteristic peak at 305 cm−1 of pure Bi and Bi@CNT materials is assigned to the Bi nanocrystals, indicating the presence of Bi in the Bi@CNT materials, which is consistent with the XRD and XPS results.1 Two peaks at 1343 cm−1 and 1596 cm−1 are ascribed to the D and G bands of the carbon matrix, corresponding to the bond stretching of sp3-C atoms of disordered carbon and sp2-C atoms of ordered carbon, respectively.13 The ID/IG values of CNTs, Bi@CNT-1.74, Bi@CNT-2.61, and Bi@CNT-4.43 are 1.05, 1.03, 0.92, and 0.99, respectively. The lowest ID/IG value of the Bi@CNT-2.61 material implies that Bi@CNT-2.61 has more excellent electrical conductivity than the others, which provides the basic conditions for rapid Na+ charging and discharging.1,3,4,13
Fig. S5† illustrates the adsorption–desorption isotherms and pore size distribution curves of Bi, CNTs, and Bi@CNT materials. As shown in Fig. S5a,† the N2 absorption–desorption curves of the materials are assigned to the type IV features with H4 hysteresis loops, suggesting the presence of mesopores in the materials. The pore sizes of mesopores in the materials are demonstrated in Fig. S5b.† In general, it is considered that the presence of mesoporous structures promotes electrolyte infiltration into the materials, allowing the primary Bi@CNT active materials to enhance the storage capacity of SIBs. From the DFT method, the specific surface areas of Bi, CNT, Bi@CNT-1.74, Bi@CNT-2.61, and Bi@CNT-4.43 were calculated as 26.69, 218.98, 26.38, 26.42 and 24.73 m2 g−1, respectively (Table S4†). In addition, the limited number of micropore structures in Bi@CNT materials can contribute to the appropriate specific surface area, thereby increasing the electrochemical reaction site (Fig. S5b†).26–28 It is found that the specific surface area of Bi@CNT materials is nearly identical to that of pure Bi but different from that of CNTs materials (Table S4†). One of the reasons is that some Bi nanoparticles blocked the pore structures of CNTs materials, resulting in a sharp decrease in the specific surface area and pore volume size of Bi@CNT materials (Fig. S6†). On account of the fact that the Bi@CNT-2.61 possesses the smallest total pore volume, it is reasonable to infer that more Bi particles are present inside the CNTs of Bi@CNT-2.61 (Table S4†). BET results give hard evidence that Bi nanoparticles are partially stuffed into the tubes of CNTs in Bi@CNT materials.
To gain a deeper understanding of the structural conversions of Bi@CNT materials, the precursor materials such as Bi@CNT-1.74-uncarbonized, Bi@CNT-2.61-uncarbonized, and Bi@CNT-4.43-uncarbonized were fabricated using the same hydrothermal method before carbonizations. XRD and SEM analyses were utilized to analyze the structural conversions of precursor materials in detail (Fig. S7† and 3). It is observed that the precursor materials mainly comprise Bi components and a small amount of Bi2O3, indicating that most of Bi(NO3)3·5H2O is mainly reduced to the metallic Bi in the hydrothermal process (Fig. S7†). Moreover, micro-sized Bi balls with three-dimensional nanoflower-like structures are distinctly observed (Fig. 3a–c). After carbonizations in the N2 atmosphere, the cellular structures of Bi balls disappeared, and CNTs with the bird's nest-like structures enveloped the Bi balls (Fig. 3d–l). The average particle sizes of Bi balls in the carbonized Bi@CNT-1.74, Bi@CNT-2.61 and Bi@CNT-4.43 materials are 4.70, 4.85 and 7.35 μm, respectively (Fig. S8†). The low melting point (271.3 °C) of Bi leads us to consider that Bi undergoes a process similar to annealing during the carbonization process. To validate this hypothesis, the Bi@CNT materials were subjected to a 20 Mpa pressure for 5 minutes to crush the Bi@CNT materials, and it was found that they could withstand it. Fig. S9† suggests our conjecture that phase separations among the Bi particles and CNTs occurred when the carbonization temperature was reduced to room temperature. Ultimately, the Bi and C elements are homogeneously distributed in Bi@CNT-2.61, as shown in the SEM-EDS images (Fig. S10†).
 | ||
| Fig. 3 SEM morphologies of Bi@CNT before carbonizations (a–c), Bi@CNT-1.74 after carbonizations (d–f), Bi@CNT-2.61 after carbonizations (g–i) and Bi@CNT-4.43 after carbonizations (j–l). | ||
For more detailed microscopic observations, TEM images of Bi@CNT-2.61 and CNTs were analyzed (Fig. 4 and S11†). It is obvious that the carbonized CNTs show a strong cohesion and form a tight enclosure around the Bi particles (Fig. 4). Additionally, it can be inferred that the nearly parallel carbon lattice stripes exist on the tube wall, indicating that the CNTs possess a high degree of graphitization (Fig. S11c†), which is perfectly aligned with the XRD results. The higher graphitization degree of the pure CNTs can provide excellent electrical conductivity for Bi@CNT composites, thereby improving their rate performance significantly. As shown in Fig. 4b, the lattice spacings of 0.395 nm, 0.328 nm, and 0.1555 nm are assigned to the (003), (012), and (107) crystal planes of Bi (ICDD No. 01-85-1329, space group Rm), which further confirms the existence of Bi. Moreover, the clear interfaces between Bi and CNTs indicate that the Bi particles are effectively packaged by the CNTs, bringing the perfect inhibition for the volume expansions of Bi particles during the discharge–charge processes (Fig. 4b). Intriguingly, it is clearly observed that the nano-sized Bi particles exist inside and outside CNTs (Fig. 4c–d). These structures can not only increase the isotropy of electrical conductivity of CNTs, but the nano Bi particles can also play a role in Na+ storage. From the XPS (more C–O–Bi groups exist in the Bi@CNT-2.61) and BET (Bi@CNT-2.61, with the smallest total pore volume) results, the fact that more nano Bi particles are incorporated into the nanotubes in Bi@CNT-2.61 can be considered natural.
 | ||
| Fig. 4 (a and c) TEM images, (b and d) HRTEM images of the Bi@CNT-2.61 material. | ||
The sodium storage behaviors of Bi, CNTs, and Bi@CNT negative electrodes of SIBs were studied using half batteries in a voltage range of 0.01–1.20 V (Fig. 5). As shown in Fig. 5a, Bi@CNT-1.74, Bi@CNT-2.61, and Bi@CNT-4.43 electrodes exhibit the discharge specific capacities of 312.9, 462.8, and 357.3 mA h g−1 after 100 cycles at a current density of 0.2 A g−1, respectively. On the contrary, the reversible discharge specific capacity of pure Bi and CNTs are only about 127 mA h g−1 and 60 mA h g−1, respectively. These results illustrate that neither Bi nor CNT has a substantial capacity for storing Na+. The capacity of the Bi@CNT-2.61 electrode, surpassing the theoretical specific capacity, should be attributed to additional contributions from the characteristic structures of the electrode materials (see descriptions below). Fig. 5b demonstrates the rate performance of Bi, CNTs, and Bi@CNT electrodes, while the current densities are set as 0.1, 0.2, 0.5, 1, 2, 5, 10, 20, 30, 40 and 50 A g−1, respectively. Among them, the Bi@CNT-2.61 exhibits excellent rate performance. The discharge-specific capacities of Bi@CNT-2.61 are 400.1, 394.0, 401.2, 403.6, 402.1, 395.7, 390.4, 382, 366.5, 340.8 and 293.6 mA h g−1 at 0.1, 0.2, 0.5, 1, 2, 5, 10, 20, 30, 40 and 50 A g−1, respectively. Bi@CNT-2.61 still manifests the specific discharge capacity of 377.6 mA h g−1, with 2200 discharge–charge cycles at a current density of 5 A g−1 (Fig. S12†). Following 2600, 2000 and 1490 cycles at a current density of 10 A g−1, the Bi@CNT-2.61 with the mass loads of 0.99, 1.22 and 1.36 mg cm−2 display the specific discharge capacity of 329.3, 335.5 and 338.9 mA h g−1, respectively (Fig. S13†).
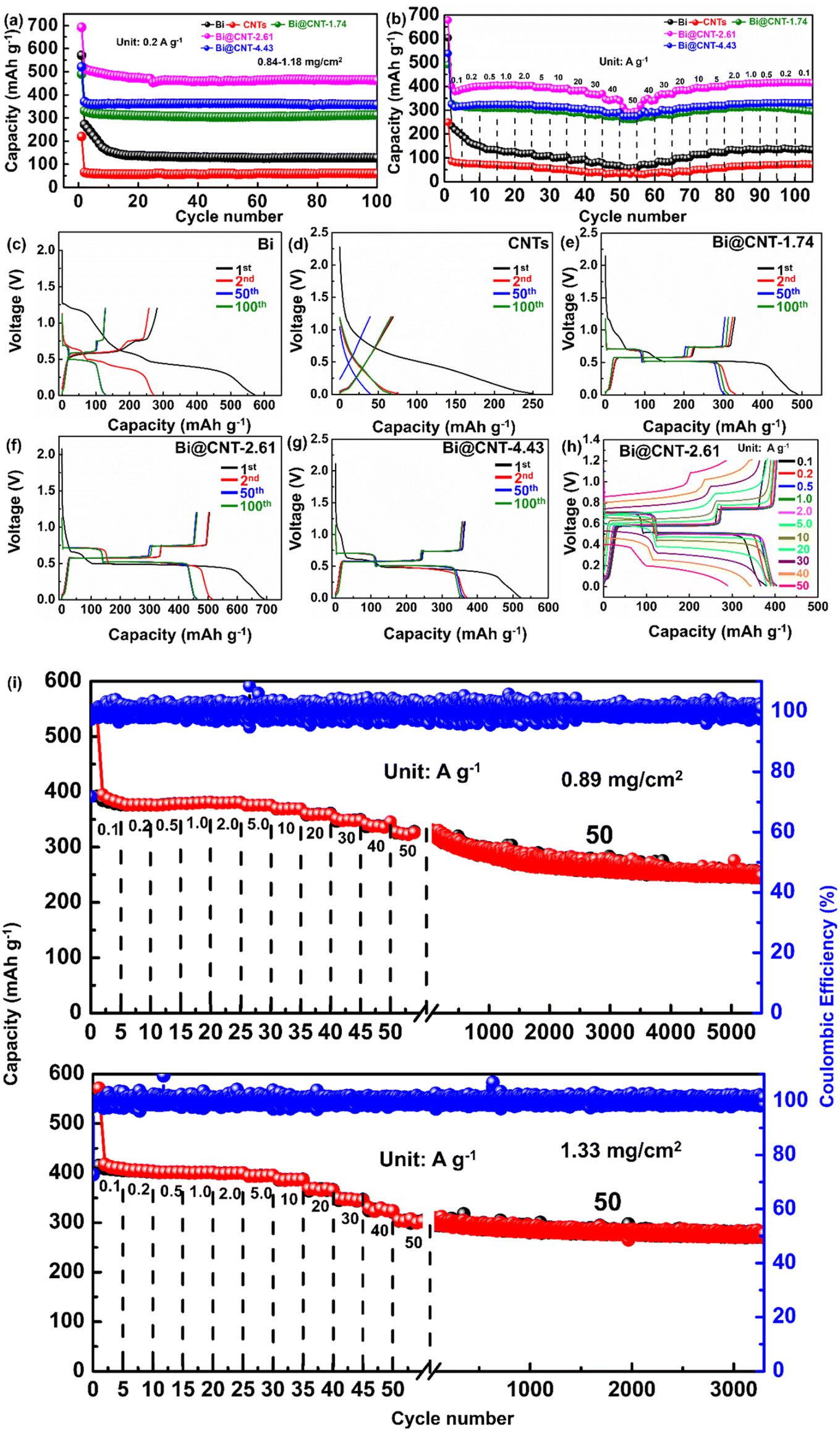 | ||
| Fig. 5 (a) Cycling performance of Bi, CNTs, and Bi@CNT electrodes at 0.2 A g−1. (b) Rate performance of Bi, CNTs, and Bi@CNT electrodes. (c–g) Charge/discharge profiles at 0.2 A g−1 of Bi, CNTs, and Bi@CNT electrodes, respectively. (h) Charge/discharge profiles at various current densities of the Bi@CNT-2.61 electrode. (i) Long-term cycle curve of the Bi@CNT-2.61 electrode at 50 A g−1. | ||
Surprisingly, when the mass loadings of the Bi@CNT-2.61 electrode are 0.89 and 1.33 mg cm−2, the specific discharge capacities of Bi@CNT-2.61 are 248.1 and 277.3 mA h g−1, respectively, after 5450 and 3250 cycles at a current density of 50 A g−1. The stability under a supercurrent (50 A g−1) is far superior to the Bi-based anodes reported in recent years (Fig. 5i and Table S5†).4,17,29–33 In addition, even increasing the mass load of Bi@CNT-2.61 to 3.20 mg cm−2, a reversible discharge specific capacity of 373.5 mA h g−1 is achieved after 1690 cycles at a current density of 1 A g−1 (Fig. S14†). The morphological evolutions of Bi@CNT-2.61 electrodes after 100 cycles at 0.2 A g−1 and 5450 cycles at 50 A g−1 were investigated through SEM characterizations (Fig. S15†). After 100 cycles at 0.2 A g−1, the Bi particles in the Bi@CNT-2.61 electrode transform into nanorods, which are firmly embedded within the conductive carbon matrix (Fig. S15b†). The root image was used to describe the process of Fig. S15b in S15g.† On the contrary, the Bi particles of the Bi@CNT-2.61 electrode after 5450 cycles at a current density of 50 A g−1 were also broken due to huge stress (Fig. S15c†). The difference from the SEM image after a cycle at a low current is that the initial Bi becomes a sea urchin-like structure with burrs of different sizes on the surface. Also, the smaller urchin-like structures cluster together to form a three-dimensional network (Fig. S15c–f and S15h†). This three-dimensional network structure facilitates the rapid diffusion of sodium ions in the Bi@CNT-2.61 electrode material.
Fig. 5c–g show the charge and discharge curves of Bi, CNT, and Bi@CNT electrodes at a current density of 0.2 A g−1. As can be seen from Fig. 5c–g, the initial charge/discharge specific capacities of Bi, CNTs, Bi@CNT-1.74, Bi@CNT-2.61, and Bi@CNT-4.43 electrodes were 281.31/570.96 mA h g−1, 69.19/250.25 mA h g−1, 329.59/489.21 mA h g−1, 504.42/691.69 mA h g−1 and 365.14/521.16 mA h g−1, corresponding to the initial coulombic efficiencies (ICE) of 49.27%, 27.65%, 67.37%, 72.93% and 70.06%, respectively. The irreversibility of the initial capacity loss can be attributed to the decomposition of the electrolyte in the SEI films and the appearance of side reactions between the active groups on the carbon matrix surface and sodium ions.34–40 In subsequent cycles, the coulombic efficiency rapidly increased to nearly 100%, indicating that the irreversible reactions mainly occurred during the initial charge–discharge step. It is considered that the Bi@CNT-2.61 electrode owns the minimal polarization, as the charge and discharge curves nearly overlapped (Fig. 5h). The small polarization can be ascribed to the fact that the Bi@CNT-2.61 electrode possesses excellent conductivities, which can be further explained by the analyses of electrochemical impedance spectroscopy (EIS) as follows.
Fig. 6a–e show the cyclic voltammetry (CV) curves of Bi, CNTs, and Bi@CNT electrodes in a voltage range of 0.01–1.20 V at a sweep speed of 0.1 mV s−1. In Fig. 6c–e, the first reduction peak at 0.61–0.67 V corresponds to the alloying reaction of Na with Bi to form NaBi.41–46 The subsequent peak at 0.48 V is attributed to NaBi reacting with Na+ further to form a Na3Bi alloy.47 During the first desodiation process, the two oxidation peaks of 0.62 V and 0.77 V correspond to the dealloying of Na3Bi to NaBi and Bi (Fig. 6e–g).3,48–50 It is worth pointing out that the redox peaks of the Bi electrode material differ from that of the Bi@CNT electrodes, which may be caused by the presence of a small amount of Bi2O3 in the pure Bi (Fig. 6a).1–4,21 Moreover, the reduction peak at 0.50 V is observed during the initial discharge of the CNT electrode, indicating the formation of the SEI layer (Fig. 6b).9 Additionally, the apparent reduction peak at 0.01 V is associated with the insertion of sodium ions (Na+) into the CNTs electrode (Fig. 6b).9 The oxidation peak at 0.08 V during the initial charge process is attributed to the extraction of Na+ from the carbon layer of the CNTs (Fig. 6b). In the subsequent cycle, the CV curves overlapped very well, revealing that CNTs have stable characteristics of Na+ storage (Fig. 6b).
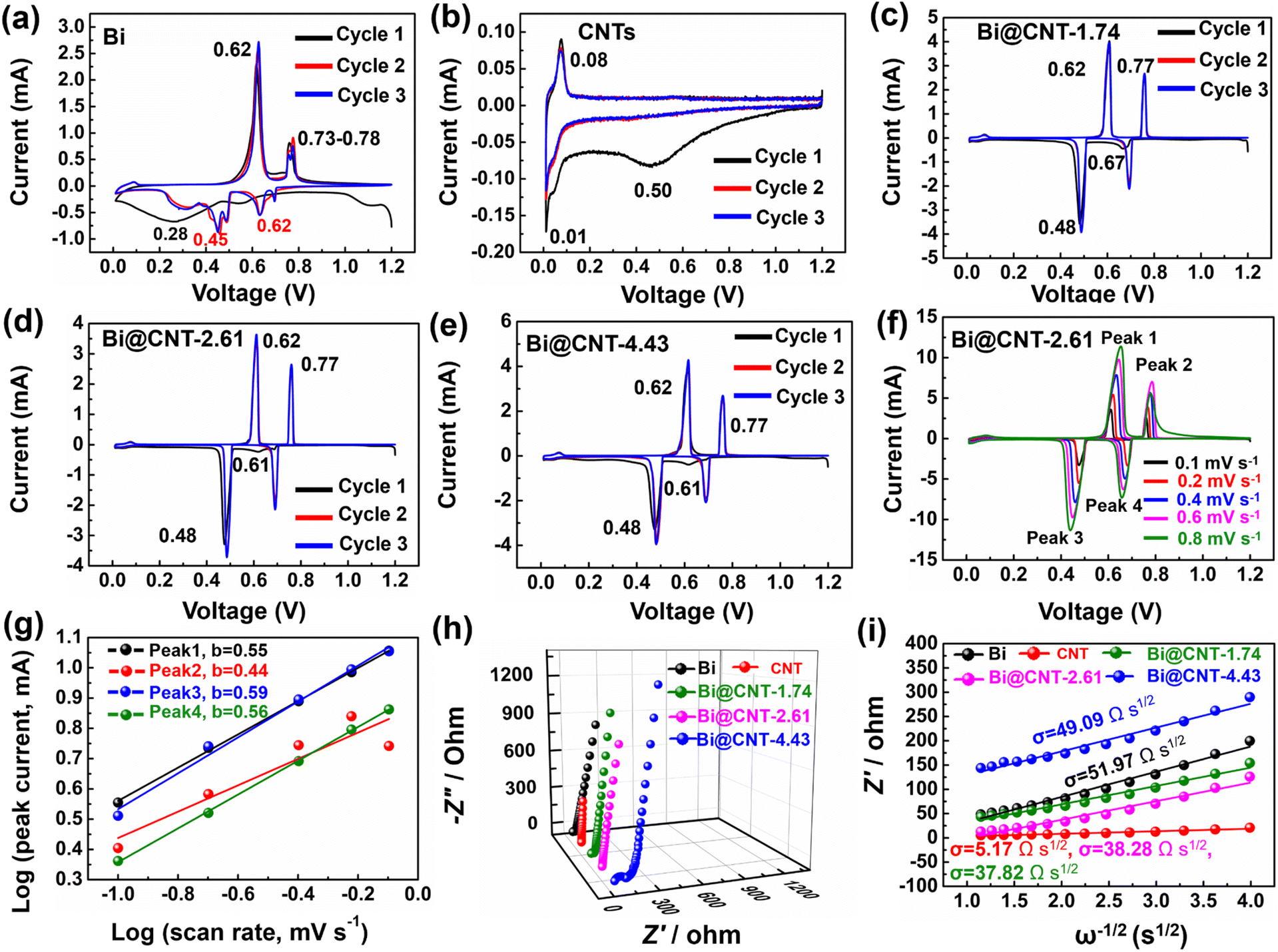 | ||
| Fig. 6 CV curves of Bi, CNTs, and Bi@CNT electrodes (a–e). (f and g) CV curves at various scan rates and the corresponding log(peak current) vs. log(scan rate) of the Bi@CNT-2.61 electrode. (h and i) Nyquist plots and illustrations of relationships between Z′ and ω−1/2 in the low-frequency region of Bi, CNTs, and Bi@CNT electrodes. | ||
To investigate the energy storage mechanism of the Bi@CNT-2.61 material, CV tests at various sweep speeds were conducted (Fig. 6f). Those two pairs of redox peaks, such as peak 3/peak 1 and peak 4/peak 2, are attributed to the alloying/dealloying reactions between Bi and sodium. The alterations of redox potentials of Bi@CNT materials with the changes in the sweeping speeds were minimal, and the peak behaviors barely changed, suggesting that the Bi@CNT-2.61 materials have rapid electrode kinetic rates and high reversibility.48
The relationships between the peak current (i, mA) and scan rate (ν, mV s−1) are expressed as shown in eqn S2 and S3.† Among them, a and b are the empirical parameters, and the values of a and b can be calculated using the log(ν)–log(i) diagrams. Generally, a and b values of 0.5 or 1, reflecting the energy storage types of the materials, are attributed to the diffusion control or surface control processes, respectively. The b values can be calculated from the linear curves of the redox peaks in Fig. 6g.49 After calculations, it is confirmed that the b values of peak 1 (0.55), peak 2 (0.44), peak 3 (0.59) and peak 4 (0.56) are close to 0.5, indicating that the Na+ storage type is mainly cataloged to the diffusion type (Fig. 6g and S16†). Due to the capacitance effect of CNTs in the Bi@CNT composite, the specific capacity of the Bi@CNT composite exceeds the theoretical capacity of Bi (386 mA h g−1) (Fig. S17†). It is also observed that the b values of reductive peak 3 and peak 4 are larger than those of oxidative peak 1 and peak 2, which suggests that the dealloying process proceeds faster than alloying in the storage processes of Bi@CNT materials. The primary factor is ascribed to the relatively high carbon contents in Bi@CNT-1.74 and Bi@CNT-2.61, which are conducive to improving the Na+ transport kinetics.
To gain deeper insights into the higher sodium storage capacity of Bi@CNT-2.61 compared to other materials, the EIS tests were conducted on Bi, CNTs, and Bi@CNT electrodes (Fig. 6h and S18†). Based on the fitting of equivalent circuit diagrams (Fig. S18†), the charge transfer impedances of various electrode materials were calculated to be 12.25 Ω, 0.74 Ω, 16.41 Ω, 0.72 Ω, and 66.34 Ω, respectively (Table S6†). Especially, the Bi@CNT-2.61 electrode exhibits the lowest charge transfer impedance (<1 Ω), which is a key factor for its excellent rate capability and long-cycle stability. The exceedingly excellent conductivity of Bi@CNT-2.61 is ascribed to its specific structure, in which more nano Bi particles were incorporated inside the CNTs, decreasing the anisotropy of conduction and thereby significantly reducing the polarization phenomenon.39,40
The correlations between Z′ and ω−1/2 in the low-frequency region of the Bi, CNTs, and Bi@CNT electrodes are illustrated in Fig. 6i. In general, the σ value (Warburg factor) relating to the slope of the line Z′–ω−1/2 is utilized to estimate the Na+ transfer of the fabricated materials.14,27 After calculations, the σ values of the Bi, CNT, Bi@CNT-1.74, Bi@CNT-2.61, and Bi@CNT-4.43 electrodes were calculated to be 51.97, 5.17, 37.82, 38.28, and 49.09 Ω s1/2, respectively.51
In addition, the diffusion coefficient of sodium ions (DNa+) of Bi@CNT-1.74, Bi@CNT-2.61, and Bi@CNT-4.43 electrodes can be calculated using eqn S4†.27,51 By substituting the aforementioned σ values into eqn S4,† the DNa+ values of Bi@CNT-1.74, Bi@CNT-2.61, and Bi@CNT-4.43 are calculated as 3.54 × 10−13, 3.46 × 10−13 and 2.10 × 10−13 cm2 s−1, indicating that Bi@CNT-1.74 and Bi@CNT-2.61 anodes own the fast Na+ transport kinetics. The aforementioned EIS analyses indicate that the Bi@CNT-2.61 exhibits a comprehensively fabulous electronic and ionic conductivity, which is attributed to the distinctive structure of Bi@CNT-2.61, as confirmed by SEM and TEM analyses (Fig. 3 and 4).
The more detailed analyses of the electrochemical processes of Bi@CNT-2.61 were further investigated by the in situ galvanostatic electrochemical impedance spectra (GEIS) (Fig. 7). Fig. 7a–d show the Nyquist plots of the Bi@CNT-2.61 half-cell in the discharge–charge processes on the first cycle and third cycle. The specific electrochemical processes can be distinguished by the distribution of relaxation time (DRT).17 The time domain-based DRT spectra are obtained by the mathematical transformations from the frequency domain-based Nyquist diagrams (Fig. 7a1–d1). As shown in Fig. 7a1–d1, there are two main peaks in the DRT curves in the discharge and charge processes belonging to the first and third cycles, which are labeled as P1 and P2, respectively.
 | ||
| Fig. 7 (a and b) The in situ GEIS plots and (a1 and b1) the DRT curves of the Bi@CNT-2.61 electrode during the first discharge/charge process. (c and d) The in situ GEIS plots and (c1 and d1) the DRT curves of the Bi@CNT-2.61 electrode during the third discharge/charge process. | ||
The time constants of P1 peaks at a range of around 10−5 s in the DRT curves generally correspond to the contact resistances (between the active material and the collector), which are demonstrated in the Nyquist diagrams.17,52–54 It is found that the relaxation time at the voltage range of 0.7–0.8 V in the discharge–charge processes on the first and third cycle is shorter than that in the other voltages, revealing that the Na+ de-intercalation reaction (the reversible reaction of NaBi dealloyed to the Na and Bi) mainly occurs near 0.7–0.8 V, which has the fastest rate in the entire electrochemical case. By contrast, the relaxation time of Na+ de-intercalation occurring near 0.4–0.5 V is attributed to the reversible reactions in which Na3Bi is dealloyed to NaBi and NaBi is alloyed to the Na3Bi (Fig. 7a1–d1).17,52–54 The larger relaxation time at a voltage range of 0.4–0.5 V indicates that the reversible reactions between Na3Bi and NaBi are the rate-determining step in the entire dealloying reactions, which has not been reported in the literature (Fig. 7a1–d1).
The relaxation times of the P2 peak in the range of 10−4–10−2 s represent the impedance of the SEI layer.17 In addition, the fact that peak intensities have minor alternation with time conversions implies that the SEI layer has a high level of stability (Fig. 7a1 and b1). It provides the basis for excellent cyclic stability in the subsequent cycles.17 Similar to changes in the P2 peak intensities of the first cycle, the same alternations also occur in the charging and discharging processes of the third cycle (Fig. 7c1 and d1), supporting the stability of the SEI layer. This strongly supports the fact that the Bi@CNT-2.61 material possesses a stable structure, and no obvious volume expansion occurs in the alloying/dealloying processes.
The ex situ XRD technique was utilized to further clarify the phase evaluation of the Bi@CNT-2.61 electrode material during the Na+ de-intercalation processes (Fig. 8a). Fig. 8b and c are the enlarged images of the two regions at 2θ = 24°–28° and 36°–40° in Fig. 8a. During the Na+ intercalation process, the peak intensity of the metallic phase of Bi diminishes until it disappears entirely. It is attributed to the fact that the Bi reacted with the Na+ to form NaBi (2θ = 26.3° and 37.2°) first, and NaBi continuously reacted with the Na+ to form the Na3Bi (2θ = 38.8°) within a potential range of 0.70–0.50 V.47 Upon discharging to 0.01 V, the peaks corresponding to the NaBi phase converted to those of the Na3Bi phase (Fig. 8b and c).47 In contrast, when increasing the voltage from 0.74 to 1.20 V, the peak intensities of Na3Bi (2θ = 38.8°) and NaBi phases at around 2θ (26.3° and 37.2°) will gradually diminish and disappear, and the Bi-phase appears again. It is ascribed to the fact that Na3Bi (2θ = 38.8°) is reduced to NaBi, and NaBi can be reduced to Bi further in the Na+ extraction process.3,21,48–50 The phase transitions during the de-intercalation processes of Na+ in Bi@CNT-2.61 are consistent with the redox peaks in the CV curve.
 | ||
| Fig. 8 (a) Contour plot of ex situ XRD patterns at different voltage states of Bi@CNT-2.61. (b and c) Enlarged images of (a). (d) The diagrams of sodium migration paths from the top and side views on the Bi (012) surface. (e) The diagrams of sodium migration paths from top and side views on the Bi@CNT surface. (f) The migration energy barriers on the Bi and Bi@CNT surface. (g) Comparison of the calculated density of states (DOS) of Bi and Bi@CNT. The Fermi level is denoted by the green line. | ||
To examine the energy transfer barrier of sodium ions in Bi@CNT and Bi electrode materials, detailed DFT (Density Functional Theory) calculations were conducted. Considering the complexity of CNT structures, the monolayer graphene serves as a simplified calculation model, representing the carbon layer of the Bi@CNT material. To demonstrate the existence of interactions within the established mixed-layer model, the Na+ adsorption energies were calculated for the individual Bi layer, individual carbon layer, and mixed layer, combining both layers, respectively (Fig. S19†). As a result, the adsorption energies of Na+ on the graphene surface, pristine Bi surface, and Bi@CNT materials were systematically calculated as −4.29 eV, −6.53 eV and −8.53 eV, respectively. The enhanced Na+ adsorption energy of Bi@CNT proves the existence of interactions between the Bi and the carbons in our calculation model. Fig. 8d and e illustrate the migration pathways of sodium ions in Bi and Bi@CNT materials, respectively, while Fig. 8f presents the migration energy barrier for sodium ions in both Bi and Bi@CNT materials. Fig. 8f reveals that the transport energy barrier for sodium ions in Bi@CNT (0.69 eV) is significantly lower than that of Bi (1.95 eV), indicating that the Bi@CNT composite structure effectively facilitates sodium ion diffusion on its surface. This led us to consider that the covering of CNT on the surface of Bi can not only restrict volume expansion but also remarkably improve sodium ion diffusion.21 Furthermore, as illustrated by the total state density in Fig. 8g, the distribution area of the density of states (DOS) for the Bi@CNT composite is markedly elevated compared to that of Bi alone. It is conducive to improving the charge transport dynamics, thereby augmenting the rate performance of the Bi@CNT electrode.21,55,56 The high density of states at the Fermi level indicates that pure Bi possesses a large number of free charge carriers. Associating with Fig. S19,† the decrease in the Fermi level of Bi@CNT is probably attributed to the electronic coupling between the Bi and carbons, which leads to the redistribution of electron density toward the carbons in Bi@CNT. Consequently, the enhanced delocalization of electrons contributes to improved electrical conductivity and adsorption energy of Bi@CNT.
To further probe the feasibility of Bi@CNT-2.61 as an anode electrode for SIBs, we assembled a full battery using the Bi@CNT-2.61 anode and commercial Na3V2(PO4)3 (NVP) cathode. The NVP is selected as a cathode due to its excellent cycling stability, rapid Na+ diffusion, and superior thermal stability. Before assembling the full cells, the physical and chemical properties of the NVP were analyzed in detail. It is found that the XRD patterns of NVP are coincident with the standard sample (COD No. 96-222-5133), and the sharp patterns indicate that NVP has a highly crystalline structure (Fig. S20†). The cycle stability, GCD profiles, CV, and rate performance of NVP are illustrated in Fig. S21.† The reversible capacity of the NVP cathode is 102.3 mA h g−1 when cycling 180 times at a current density of 1 A g−1. It is indicative of the NVP cathode having superior structural stability (Fig. S21a†). A pair of redox peaks/platforms between the CV and GCD curves at 3.44/3.26 V represents a reversible reaction process between V3+/V4+ (Fig. S21b and S21d†).55 Additionally, the NVP also has excellent rate properties (Fig. S21c†). The specific reversible charging capacities of the NVP cathode are 106.5, 104.5, 103.3, 102.0, 96.5 and 77.6 mA h g−1 when the current densities are set as 0.1 A g−1, 0.2 A g−1, 0.5 A g−1, 1 A g−1, 2 A g−1 and 5 A g−1, respectively. The capacity degradation exhibits minimal attenuation with increasing current densities.
The electrochemical reversibility of the Bi@CNT-2.61//NVP full battery is also validated by two distinct redox couples of 2.55/2.68 V and 2.72/2.97 V in the CV curves (Fig. 9b).33 In accordance with the charge/discharge platform (3.44 V/3.26 V) of NVP and the discharge/charge plateaus of Bi@CNT-2.61 (0.61 V, 0.48 V/0.77 V and 0.62 V), the potentials in a range of (2.49 V ∼2.96 V) of a full battery (Bi@CNT-2.61//NVP) are obtained. At different current densities, good overlaps of the GCD curves of the obtained full battery support the conclusion that there is no obvious polarization phenomenon at a high current density (Fig. 9c).57 After performing the electrochemical evaluations, this full battery shows a competitive electrochemical performance. For instance, the specific reversible charging capacities are 195.4, 184.8, 176, 157.2, 132.4, 87.7, and 53.5 mA h g−1 at the current densities of 1 A g−1, 2 A g−1, 3 A g−1, 4 A g−1, 5 A g−1, 6 A g−1, and 7 A g−1, respectively (Fig. 9d). Furthermore, it exhibits impressive capacity retentions of 96.64% and 98.64% after 445 and 400 cycles at the current densities of 1 A g−1 and 5 A g−1, respectively (Fig. 9d and e). On comparing with additional reports, this Bi@CNT-2.61//NVP full battery also manifests the exceedingly excellent storage performance, indicating its great application potential (Fig. S22 and Table S7†).13–16,19 As shown in Fig. S22 and Table S7,† the energy density of the Bi@CNT-2.61//NVP full battery is 192.1total W h kg−1 at 979 W kg−1. Even at a high-power density of 4982.4 W kg−1, the energy density of the full battery could still be maintained at 130.1total W h kg−1.
 | ||
| Fig. 9 Electrochemical performance of Bi@CNT-2.61//NVP Na-ion full cell. (a) Schematic working mechanism. (b) CV curves at 0.2 mV s−1. (c) Charge/discharge curves at various current densities. (d) Rate performance at various current densities. (e) Cycle performance at 5 A g−1. | ||
4. Conclusions
Using a one-step hydrothermal method, the Bi@CNT materials were successfully synthesized from Bi(NO3)3·5H2O and CNTs. After performing detailed structural analyses, the unique structures of Bi@CNT-2.61 are distinctly confirmed. Namely, the micro-sized Bi balls are completely covered by the CNTs. Furthermore, it is intriguingly found that nano-sized Bi particles are embedded into the walls of tubes and packed inside the tubes. These original structures can bring the following favorable characteristics in energy storage. (i) The optimal carbon content in Bi@CNT effectively preserves the crystal structure of Bi, ensuring the continuous progression of alloying/de-alloying reactions between Na+ and Bi. (ii) The exceptional electrical conductivity and effective interface connections between Bi and CNTs are crucial in promoting the ion/electron transport in the Bi@CNT composites. (iii) A suitable specific surface area and pore size structure can not only enhance the infiltration of the electrolyte in Bi@CNT but also increase the electrochemical reaction site, thereby increasing the sodium storage capacity of Bi@CNT materials. The ex situ XRD and DFT calculations show the remarkable stability and improvement in charge transport dynamics of Bi@CNT. On account of its unique structure, Bi@CNT-2.61 exhibits a significantly higher Na+ storage capacity and excellent rate performance. For instance, Bi@CNT-2.61 maintains a discharge-specific capacity of 248.1 mA h g−1 after 5450 cycles at a current density of 50 A g−1. The Bi@CNT-2.61//NVP full battery also exhibits a high energy density of 192.1 W h kg−1, with capacity retentions of 96.64% and 98.64% after 445 and 400 cycles at current densities of 1 A g−1 and 5 A g−1, respectively. The exceptionally high Na+ storage capacity and remarkable full battery performance strongly support the great potential of Bi@CNT-2.61 in the application of SIBs.Data availability
Because of privacy concerns, the raw data underpinning this research is not publicly available. However, the data supporting the plots in this paper will be made accessible upon reasonable request from the corresponding author, Dr Sandya Rani Mangishetti (https://orcid.org/0000-0003-3112-0016). The data and figures supporting this article are included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the support of the University of Science and Technology Liaoning (601009816-39), LJKQZ2021126 and 2017RC03. This work was supported by the Liaoning Province Education Department of China (Grant No. 601009887-16). Additionally, this work is partly funded by the National Natural Science Foundation of China (Grant No. 51672117 and 51672118).References
- S. Wei, W. Li, Z. Ma, X. Deng, Y. Li and X. Wang, Small, 2023, 19, 2304265 CrossRef CAS PubMed.
- J. Gao, M. Zhang, X. Zhang, R. Bai, G. Xin and G. Wang, Chem. Eng. J., 2022, 433, 133521 Search PubMed.
- X. Zhang, X. Qiu, J. Lin, Z. Lin, S. Sun, J. Yin, H. N. Alshareef and W. Zhang, Small, 2023, 19, 2302071 CrossRef CAS PubMed.
- T. Li, Q. Gao, S. Liu, X. Zhang, Y. Zhang, Y. Liu, L. Hou and C. Yuan, ACS Appl. Energy Mater., 2023, 6, 3071 CrossRef CAS.
- X. Yang and A. L. Rogach, Adv. Energy Mater., 2020, 10, 2000288 Search PubMed.
- S. Alvin, D. Yoon, C. Chandra, H. S. Cahyadi, J.-H. Park, W. Chang, K. Y. Chung and J. Kim, Carbon, 2019, 145, 67 CrossRef CAS.
- D. Wu, F. Sun, Z. Qu, H. Wang, Z. Lou, B. Wu and G. Zhao, J. Mater. Chem. A, 2022, 10, 17225 Search PubMed.
- T. Xu, X. Qiu, X. Zhang and Y. Xia, Chem. Eng. J., 2023, 452, 139514 CrossRef CAS.
- N. Qin, Y. Sun, C. Hu, S. Liu, Z. Luo, X. Cao, S. Liang and G. Fang, J. Energy Chem., 2023, 77, 310 Search PubMed.
- X. Chen, C. Liu, Y. Fang, X. Ai, F. Zhong, H. Yang and Y. Cao, Carbon Energy, 2022, 4, 1133 CrossRef CAS.
- N. Jiang, L. Chen, H. Jiang, Y. Hu and C. Li, Small, 2022, 18, 2108092 Search PubMed.
- T. Perveen, M. Siddiq, N. Shahzad, R. Ihsan, A. Ahmad and M. I. Shahzad, Renew. Sust. Energy Rev., 2020, 119, 109549 CrossRef CAS.
- Y. Li, X. Zhong, X. Wu, M. Li, W. Zhang and D. Wang, J. Mater. Chem. A, 2021, 9, 22364 Search PubMed.
- L. Chen, X. He, H. Chen, S. Huang and M. Wei, J. Mater. Chem. A, 2021, 9, 22048 RSC.
- P. Xue, N. Wang, Z. Fang, Z. Lu, X. Xu, L. Wang, Y. Du, X. Ren, Z. Bai, S. Dou and G. Yu, Nano Lett., 2019, 19, 1998 CrossRef CAS PubMed.
- H. Ma, T. Wang, J. Li, J. Yang, Z. Liu, N. Wang, D. Su and C. Wang, Chem.–Asian J., 2021, 16, 2314 CrossRef CAS PubMed.
- Z. Chen, X. Wu, Z. Sun, J. Pan, J. Han, Y. Wang, H. Liu, Y. Shen, J. Li, D. Peng and Q. Zhang, Adv. Energy Mater., 2024, 14, 2400132 Search PubMed.
- W. Hong, A. Wang, L. Li, T. Qiu, J. Li, Y. Jiang, G. Zou, H. Peng, H. Hou and X. Ji, Adv. Funct. Mater., 2021, 31, 2000756 CrossRef CAS.
- J. Chen, G. Zhang, J. Xiao, J. Li, Y. Xiao, D. Zhang, H. Gao, X. Guo, G. Wang and H. Liu, Adv. Funct. Mater., 2024, 34, 2307959 CrossRef CAS.
- B. Delley, J. Chem. Phys., 2000, 113, 7756–7764 CrossRef CAS.
- Q. Yao, C. Zheng, K. Liu, M. Wang, J. Song, L. Cui, D. Huang, N. Wang, S. X. Dou, Z. Bai and J. Yang, Adv. Sci., 2024, 11, 2401730 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B:Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- Y. Qiu, J. Zhang, J. Jin, J. Sun, H. Tang, Q. Chen, Z. Zhang, W. Sun, G. Meng, Q. Xu, Y. Zhu, A. Han, L. Gu, D. Wang and Y. Li, Nat. Commun., 2021, 12, 5273 Search PubMed.
- T. A. Halgren and W. N. Lipscomb, Chem. Phys. Lett., 1977, 49, 225–232 CrossRef CAS.
- J. Li, X. Miao, B. Han, K. Wang, B. An, G. Xu, D. Ju, Y. Li, H. Yu and W. Zhou, Catal. Today, 2024, 439, 114822 Search PubMed.
- Y. Mu, D. Zhang, J. Li, B. Han, G. Xu, K. Wang, B. An, D. Ju, L. Li and W. Zhou, Electrochim. Acta, 2023, 437, 141532 Search PubMed.
- J. Pan, K. Yu, H. Mao, L. Li, Y. Zhang, Y. Li, P. J. Ferreira and J. Yang, Chem. Eng. J., 2020, 380, 122624 Search PubMed.
- H. Yang, R. Xu, Y. Yao, S. Ye, X. Zhou and Y. Yu, Adv. Funct. Mater., 2019, 29, 1809195 CrossRef.
- H. Long, X. Yin, X. Wang, Y. Zhao and L. Yan, J. Energy Chem., 2022, 67, 787 CrossRef CAS.
- X. Wu, Z. Li, W. Feng, W. Luo, L. Liao, H. Cai, X. Chen, Z. Deng, J. Wu, B. Xing, J. Ren, Z. Lou and L. Mai, Energy Storage Mater., 2024, 66, 103219 CrossRef.
- B. Pu, Y. Liu, J. Bai, X. Chu, X. Zhou, Y. Qing, Y. Wang, M. Zhang, Q. Ma, Z. Xu, B. Zhou and W. Yang, ACS Nano, 2022, 16, 18746 CrossRef CAS PubMed.
- Y. Liu, Y. Wang, H. Wang, S. Dou, H. Tian, W. Gan and Q. Yuan, J. Mater. Chem. A, 2023, 11, 5945 RSC.
- C. Wang, D. Du, M. Song, Y. Wang and F. Li, Adv. Energy Mater., 2019, 9, 1900022 CrossRef.
- S. Wei, Y. Yang, J. Chen, X. Shi, Y. Sun, P. Li, X. Tian, H. Chen, Z. Luo and Z. Chen, Adv. Energy Mater., 2024, 14, 2401825 CrossRef CAS.
- B. Long, Z. Qiao, J. Zhang, S. Zhang, M.-S. Balogun, J. Lu, S. Song and Y. Tong, J. Mater. Chem. A, 2019, 7, 11370 RSC.
- J. Huang, X. Lin, H. Tan and B. Zhang, Adv. Energy Mater., 2018, 8, 1703496 CrossRef.
- J. Chen, X. Fan, X. Ji, T. Gao, S. Hou, X. Zhou, L. Wang, F. Wang, C. Yang, L. Chen and C. Wang, Energy Environ. Sci., 2018, 11, 1218 RSC.
- X. Zou, S. Ye, C. Ou, X. Zheng, F. Tian, D. Lei and C. Wang, Energy Storage Mater., 2024, 71, 103573 CrossRef.
- J. Yang, J. Xian, Q. Liu, Y. Sun and G. Li, J. Energy Chem., 2022, 69, 524 CrossRef CAS.
- X. Wang, Y. Wu, P. Huang, P. Chen, Z. Wang, X. Xu, J. Xie, J. Yan, S. Li, J. Tu and Y.-L. Ding, Nanoscale, 2020, 12, 23682 RSC.
- C. Hu, Y. Zhu, G. Ma, F. Tian, Y. Zhou, J. Yang and Y. Qian, Electrochim. Acta, 2021, 365, 137379 CrossRef CAS.
- J. Zhou, J. Chen, M. Chen, J. Wang, X. Liu, B. Wei, Z. Wang, J. Li, L. Gu, Q. Zhang, H. Wang and L. Guo, Adv. Mater., 2019, 31, 1807874 CrossRef PubMed.
- C. Wang, L. Wang, F. Li, F. Cheng and J. Chen, Adv. Mater., 2017, 29, 1702212 CrossRef PubMed.
- L. Xie, Z. Yang, J. Sun, H. Zhou, X. Chi, H. Chen, A. X. Li, Y. Yao and S. Chen, Nano–Micro Lett., 2018, 10, 50 CrossRef PubMed.
- W. Sun, X. Rui, D. Zhang, Y. Jiang, Z. Sun, H. Liu and S. Dou, J. Power Sources, 2016, 309, 135 CrossRef CAS.
- X. Li, J. Ni, S. V. Savilov and L. Li, Chem.–Eur. J., 2018, 24, 13719 CrossRef CAS PubMed.
- Y. Liang, N. Song, Z. Zhang, W. Chen, J. Feng, B. Xi and S. Xiong, Adv. Mater., 2022, 34, 2202673 CrossRef CAS PubMed.
- Z. Li, W. Zhong, D. Cheng and H. Zhang, J. Mater. Sci., 2021, 56, 11000 CrossRef CAS.
- P. Xiong, P. Bai, A. Li, B. Li, M. Cheng, Y. Chen, S. Huang, Q. Jiang, X. Bu and Y. Xu, Adv. Mater., 2019, 31, 1904771 CrossRef CAS PubMed.
- S. Zhang, G. Wang, B. Wang, J. Wang, J. Bai and H. Wang, Adv. Funct. Mater., 2020, 30, 2001592 CrossRef CAS.
- Y. Lu, C.-Z. Zhao, J.-Q. Huang and Q. Zhang, Joule, 2022, 6, 1172 CrossRef CAS.
- J. Liu and F. Ciucci, J. Electrochem. Soc., 2020, 167, 026506 CrossRef CAS.
- B. Manikandan, V. Ramar, C. Yap and P. Balaya, J. Power Sources, 2017, 361, 300 CrossRef CAS.
- J. Zhu, L. Cai, X. Yin, Z. Wang, L. Zhang, H. Ma, Y. Ke, Y. Du, S. Xi, A. T. S. Wee, Y. Chai and W. Zhang, ACS Nano, 2020, 14, 5600 CrossRef CAS PubMed.
- H. Zhu, F. Wang, L. Peng, T. Qin, F. Kang and C. Yang, Angew. Chem., Int. Ed., 2023, 62, e202212439 CrossRef CAS PubMed.
- M. Liu, Z. Yang, Y. Shen, S. Guo, J. Zhang, X. Ai, H. Yang and J. Qian, J. Mater. Chem. A, 2021, 9, 5639–5647 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ta01206f |
| This journal is © The Royal Society of Chemistry 2025 |