
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceToughening self-healable and recyclable PDMS supramolecular elastomers through an end-capping agent and a metallic crosslinker†
Rou-Han
Lai‡
 a,
Yi-An
Chen‡
a,
Chung-Ying
Chou
b,
Hung-Yi
Huang
a,
Wassana
Mongkonkan
c,
Chia-An
Chiu
a,
Yan-Heng
Chen
a,
Min-Han
Yu
a,
Chi-Chang
Hu
a,
Siriporn
Jungsuttiwong
c and
Ho-Hsiu
Chou
*ade
a,
Yi-An
Chen‡
a,
Chung-Ying
Chou
b,
Hung-Yi
Huang
a,
Wassana
Mongkonkan
c,
Chia-An
Chiu
a,
Yan-Heng
Chen
a,
Min-Han
Yu
a,
Chi-Chang
Hu
a,
Siriporn
Jungsuttiwong
c and
Ho-Hsiu
Chou
*ade
aDepartment of Chemical Engineering, National Tsing Hua University, Hsinchu 300044, Taiwan. E-mail: hhchou@mx.nthu.edu.tw
bDepartment of Electrical Engineering and Institute of Electronics Engineering, National Tsing Hua University, Hsinchu 30013, Taiwan
cDepartment of Chemistry, Faculty of Science, Ubon Ratchathani University, Ubon Ratchathani, 34190, Thailand
dPhotonics Research Center, National Tsing Hua University, Hsinchu 300044, Taiwan
eCollege of Semiconductor Research, National Tsing Hua University, Hsinchu 300044, Taiwan
First published on 10th March 2025
Abstract
Elastomers are indispensable in wearable electronics due to their elasticity and flexibility. Among them, poly(dimethylsiloxane) (PDMS) is particularly valued for its nontoxicity and chemical stability. However, conventional PDMS materials lack recyclability and self-healing properties, while most self-healable PDMS materials reported in the literature suffer from insufficient mechanical performance, making it challenging to simultaneously achieve high toughness and efficient self-healing in a single material. To address this, we developed a high-toughness, self-healing, and recyclable PDMS elastomer by introducing 2,4-pentanedione (Hacac) as a capping agent and coordination site, simplifying the synthesis process and enhancing coordination tunability. By incorporating coordination bonds between aluminum metal ions and molecular chain segments, along with the synergistic effect of introducing counter ions, the resulting PUIP-Hac-AlOTf elastomer achieved remarkable mechanical properties (toughness: 48.73 MJ m−3) and self-healing efficiency (>95% in 12 hours). Beyond its outstanding mechanical performance, this material demonstrates versatility in wearable applications such as electrocardiogram (ECG) monitoring, hand motion detection, and voice signal sensing. Compared to commercial hydrogel-based electrodes, PUIP-Hac-AlOTf-based patches offer enhanced durability, reusability, and resistance to drying, ensuring stable signal quality over extended use. Its self-healing and recyclable properties, coupled with biocompatibility, make it a groundbreaking solution for intelligent and sustainable healthcare systems.
1. Introduction
The excellent elasticity, toughness, and high controllability of elastomers have led to their widespread use in daily life as well as in fields such as military1,2 and medical3–5 applications. However, this extensive use has resulted in significant annual waste generation and pollution. For instance, in 2019, global rubber consumption reached 29 million tons.6 The inability to recycle commercial elastomers exacerbates the environmental impact of discarded materials, posing a significant challenge. Consequently, self-healing and recyclable elastomers have garnered significant attention in recent years, including applications in wearable electronic devices,7,8 electronic skins9 and soft robotics.10,11 These elastomers can efficiently repair themselves, restoring their original mechanical properties after damage. Additionally, their recyclability significantly enhances reusability, extending product lifespan, reducing costs, and minimizing environmental pollution.12–14Among all common commercial materials, poly(dimethylsiloxane) (PDMS) is an ideal choice for use in flexible electronics due to its nontoxicity, thermal stability, outstanding stretchability, and chemical stability.15–17 Nevertheless, commonly used commercial PDMS materials such as Sylgard 184 cannot be effectively recycled or remanufactured due to their highly cross-linked thermoset nature.18,19 Therefore, incorporating self-healing and recyclable properties into PDMS materials would greatly enhance their application potential and be highly attractive.
Self-healable PDMS elastomers typically introduce dynamic covalent bonds, such as Diels–Alder reactions,20,21 disulfide bonds,21–24 imine bonds,25–27 B–O bonds,28 or reversible non-covalent interactions, including hydrogen bonds,29–32 metal coordination bonds,33–36 and π–π stacking interactions,37,38 to endow the material with self-healing ability. Yet, a significant challenge faced by self-healable materials is the inevitable trade-off between mechanical properties and self-healing ability. In applications like sensors and actuators, elastomers frequently endure large loads.39 Achieving a balance between high stress, toughness, and flexibility is essential for ensuring functionality and durability. And high-toughness self-healing materials have also been widely recognized as ideal candidates for sensors, sensing electrodes, and triboelectric nanogenerators (TENGs) due to their superior mechanical robustness and long-term reliability. Unfortunately, most reported self-healable PDMS elastomers suffer from poor mechanical properties, which limits their extended application in various scenarios.40–43 Consequently, the fabrication of high-toughness, self-healable, and recyclable PDMS materials has recently garnered extensive attention and become a focal point in the field of intelligent materials.44–48
Over the past few years, significant efforts have been made to fabricate robust, recyclable, and self-healable PDMS elastomers. Metal–ligand coordination has emerged as a popular strategy within polymer networks, offering materials with high stiffness and self-healing capabilities due to its thermodynamic stability, kinetic lability, and adjustability.36 For example, Liu et al. incorporated metal–ligand coordination bonds into a self-healing PDMS-based elastomer by introducing modified bipyridine and cross-linking with Zn2+ metal ions, endowing the elastomer with improved mechanical properties (0.9 MPa, 600%).49 Similarly, Jiang et al. reported a high-adhesion and healable PDMS elastomer with a Young's modulus of 17.9 MPa (1.2 MPa, 230%) by incorporating 3,5-diethynylpyridine into azido-terminated PDMS via click polymerization, followed by Zn2+ coordination with a tridentate ligand.50 These abovementioned methods improve the mechanical properties of self-healing PDMS materials by incorporating metal coordination while their additional chemical modification processes may pose challenges for practical applications. On the other hand, in a recent study published in 2022 by Bai et al., aluminum acetylacetonate was introduced directly as a crosslinking agent without additional modifications.51 This strategy enabled dynamic bonding within the polyurethane network, allowing the elastomer to achieve high recyclability through hot pressing and solvent redissolution. While this method simplified the fabrication process by introducing a dynamic coordination network with a metal linker, its reliance on a specific crosslinking agent resulted in a fixed and singular coordination bonding mode. This limitation restricts adaptability in integrating various metals and counter anions, reducing the versatility of the system. Thus, synthesizing coordinated self-healing materials through a simple yet versatile method that retains tunability and multiplicity of coordination remains a significant challenge.
Herein, a simple synthesis method is needed to simultaneously combine high toughness, self-healing ability, and recyclability in a single PDMS elastomer. In our work, we devised a strategy to fabricate high-toughness, self-healing and recyclable PDMS elastomers by introducing an end-capping agent – 2,4-pentanedione (Hacac) – with coordination ability, greatly simplifying the synthesis process. Notably, Hacac is a unique ligand with multiple coordination modes with metal ions,34,52 which endows the polymer with a more dynamic network, enhancing mechanical toughness and self-healing efficiency. Three different aluminum salts (aluminum acetylacetonate (Al(acac)3), aluminum trifluoromethanesulfonate (Al(OTf)3), and aluminum perchlorate (Al(ClO4)3)) were introduced into the polymer system to develop metal–ligand coordination with the Hacac group.51 Leveraging the synergistic effect of hydrogen bonding between the urea groups and the metal–ligand coordination of Al3+ ions with Hacac and various counter anions, we successfully fabricated three series of self-healing PDMS supramolecular elastomers: PUIP-Hac-Alac, PUIP-Hac-AlOTf, and PUIP-Hac-AlClO4, each exhibiting distinct mechanical properties. Instead of directly introducing a metal acetylacetonate complex as a crosslinking agent, we first employed Hacac as a chain-end capping agent, followed by the incorporation of metal salts to build coordination linkages. This approach not only overcomes the coordination limitations of traditional metal crosslinkers, increasing the range of metals that can be introduced, but also offers greater potential and flexibility in the development and tunability of elastomers by pairing with different counter anions. Upon further characterization and analysis, PUIP-Hac-AlOTf was found to have stronger binding energy between aluminum ions and oxygen atoms compared to the other series, exhibiting exceptional properties, including high transparency (99.5% at 500 nm), outstanding mechanical properties (yielding a toughness of 48.73 MJ m−3), and highly efficient self-healing ability (>95% healing efficiency within 12 hours). These results underscore the high tunability and versatility of our approach. Furthermore, PUIP-Hac-AlOTf shows great potential for application in intelligent fields, such as signal monitoring for heart rate detection and human motion.
2. Experimental
The characterization instruments and detailed experimental methods are provided in the ESI.† Additionally, informed consent was obtained for experiments involving human participants.2.1 Materials
Bis(3-aminopropyl)-terminated poly(dimethylsiloxane) (NH2-PDMS-NH2, Mn = 2500), aluminum acetylacetonate (Al(acac)3) and 2,4-pentaneedione (Hacac) were purchased from Nova-Matls. Bis(3-aminopropyl)-terminated poly(dimethylsiloxane) (NH2-PDMS-NH2, Mn = 900–1000) was sourced from Gelest. Aluminum trifluoromethanesulfonate (Al(OTf)3) was obtained from Acros Organics. Aluminum perchlorate nonahydrate (Al(ClO4)3) was purchased from Strem Chemicals, Inc. Other reagents were commercial chemicals and used without further purification.2.2 Synthesis of the PUIP-Hac elastomer
NH2-PDMS-NH2 (Mn = 900∼1000) (0.05 mmol, 50 mg) and NH2-PDMS-NH2 (Mn = 2500) (0.2 mmol, 500 mg) were mixed in a 20 mL vial. First, the mixed soft segments were heated to 80 °C under vacuum for 1 h to remove moisture and then cooled to 60 °C. Then, 3 mL of anhydrous THF was added into the vial to dissolve the mixed soft segments under a N2 atmosphere. IPDI (0.3 mmol, 66.69 mg) dissolved in anhydrous THF (3 mL) was dropwise added and stirred. This solution was continuously stirred at 60 °C for 2 h to obtain the prepolymer solution. Afterward, for further synthesizing PUIP-Hac, Hacac (0.1 mmol, 10.013 mg) dissolved in anhydrous THF (2 mL) was also dropwise added in the prepolymer solution and stirred for 4 h at 60 °C.2.3 Synthesis of PUIP-Hac-Alx (x represents the introduced aluminum salt)
For the synthesis of PU-Hac-Alx, the aluminum salt (Al(acac)3, Al(OTf)3 and Al(ClO4)3) (0.0625 mmol) dissolved in THF (2 mL) was added to the PUIP-Hac solution, and the solution was stirred at room-temperature overnight.2.4 Preparation of free-standing films
The reacted solutions were respectively poured into Teflon molds and the solvent was evaporated at 50 °C for 12 hours to obtain free-standing films.2.5 Fabrication of the AgFK-PUIP-Hac-AlOTf electrodes
250 mg of PUIP-Hac-AlOTf polymer was dissolved in 5 mL of THF solvent. Subsequently, 250 mg of silver nanoflakes (AgFKs) was added to the solution, which was then subjected to ultrasonic agitation for 30 minutes. Following this, the solution was stirred magnetically for approximately two hours. The AgFK-PUIP-Hac-AlOTf solution was then poured into a PTFE mold, and the THF solvent was evaporated in an environment maintained at 50 °C. Once the solvent started to evaporate, a metal snap button was placed at the center of the membrane to ensure firm adhesion of the snap button to the patches during the film formation process. Finally, after 24 hours, the AgFK-PUIP-Hac-AlOTf electrodes were carefully peeled off from the mold.3. Results and discussion
3.1 Synthesis and characterization of PUIP-Hac and PUIP-Hac-Alac, OTf and ClO4
We developed these elastomers, denoted as PUIP-Hac and PUIP-Hac-Alx (where x represents different counter anions: “ac” for acetylacetonate, “OTf” for trifluoromethanesulfonate, and “ClO4” for perchlorate), through a one-pot polycondensation reaction. The design and synthesis process of the PUIP-Hac series polymers are shown in Fig. 1 and S1.† PUIP-Hac was synthesized via a condensation reaction between bis(3-aminopropyl)-terminated poly(dimethylsiloxane) (NH2-PDMS-NH2) with different molecular weights (Mn = 900–1000 and 2500, molar ratio = 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8) and isophorone diisocyanate (IPDI), yielding a linear poly-urea prepolymer which could establish hydrogen bonds between the urea groups. Additionally, the combination of different lengths of NH2-PDMS-NH2 and IPDI results in the random formation of urea groups within the polymer network, leading to varying densities of hydrogen bonding.53 In low-density hydrogen bond regions, chain segments exhibit higher mobility, enhancing self-healing capability and facilitating efficient energy dissipation upon external impact. Conversely, high-density hydrogen bond regions contain chain segments with higher energy barriers, providing rigidity to the network and contributing to the material's excellent mechanical properties. And then the prepolymer was end-capped with 2,4-pentanedione (Hacac) to terminate the polymerization reaction while also serving as the coordination site.
:8) and isophorone diisocyanate (IPDI), yielding a linear poly-urea prepolymer which could establish hydrogen bonds between the urea groups. Additionally, the combination of different lengths of NH2-PDMS-NH2 and IPDI results in the random formation of urea groups within the polymer network, leading to varying densities of hydrogen bonding.53 In low-density hydrogen bond regions, chain segments exhibit higher mobility, enhancing self-healing capability and facilitating efficient energy dissipation upon external impact. Conversely, high-density hydrogen bond regions contain chain segments with higher energy barriers, providing rigidity to the network and contributing to the material's excellent mechanical properties. And then the prepolymer was end-capped with 2,4-pentanedione (Hacac) to terminate the polymerization reaction while also serving as the coordination site.
 | ||
| Fig. 1 Designed structure and schematic illustrating the dynamic intermolecular interaction and properties of PUIP-Hac, PUIP-Hac-Alac, AlOTf and AlClO4 polymers. | ||
In our strategy, NH2-PDMS-NH2 was utilized as the soft segment due to its high polymer chain mobility, which allows it to absorb external impact forces. Meanwhile, the hard urea segment, capable of forming hydrogen bonds between polymer chains, imparted structural rigidity and facilitated dynamic deformation. The successful synthesis of PUIP-Hac was first confirmed by Fourier Transform Infrared (FTIR) spectroscopy, which showed a gradual disappearance of the isocyanate peaks (around 2250 cm−1),54 as illustrated in Fig. 2a. As the reaction time increased, the isocyanate peak progressively weakened. After the addition of the end-capping agent (Hacac) and a subsequent four-hour reaction period, the peak nearly vanished. Furthermore, Gel Permeation Chromatography (GPC) analysis provided the number-average molecular weights (Mn), corroborating the elongation of the polymer chains and the polymerization of the elastomers (Table S1†). Variable-temperature 1H NMR spectroscopy (Fig. S2†) revealed the presence of hydrogen bonding in PUIP-Hac networks. With the increasing temperature, hydrogen bonds within the system tend to dissociate, weakening the shielding effect and causing signals to shift to high field.21,40 Additionally, temperature-dependent FTIR analysis was performed to further confirm the formation of hydrogen bonds (Fig. 2b). As the temperature increased from 25 °C to 100 °C, the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration peak at 1630 cm−1 and the N–H stretching vibration peak at 3360 cm−1 exhibited a blue shift, while the N–H bending vibration peak at 1570 cm−1 exhibited a red shift. These shifts are attributed to the dissociation of hydrogen bonds, resulting in the transition of the CO and N–H groups from bonded to free states, thereby shortening the bond lengths of the CO and N–H groups.55,56
O stretching vibration peak at 1630 cm−1 and the N–H stretching vibration peak at 3360 cm−1 exhibited a blue shift, while the N–H bending vibration peak at 1570 cm−1 exhibited a red shift. These shifts are attributed to the dissociation of hydrogen bonds, resulting in the transition of the CO and N–H groups from bonded to free states, thereby shortening the bond lengths of the CO and N–H groups.55,56
 | ||
| Fig. 2 (a) FT-IR spectra for tracing the polymerization process of PUIP-Hac. (b) FT-IR spectra of PUIP-Hac recorded at an increasing temperature from 25 °C to 100 °C. (c) X-ray photoelectron spectra of PUIP-Hac-Alac, AlOTf and AlClO4. (d) Thermogravimetric analysis (TGA) curves of PUIP-Hac and PUIP-Hac-Alac, AlOTf, AlClO4 (heating rate of 10 °C per minute). (e) UV-vis spectra (spanning the wavelength range from 300 nm to 700 nm) and digital photograph of PUIP-Hac and PUIP-Hac-Alac, AlOTf and AlClO4 (thickness ≈ 0.5 mm and size of 3 cm × 3 cm). | ||
After the end-capping reaction, we introduced aluminum acetylacetonate (Al(acac)3), aluminum trifluoromethanesulfonate (Al(OTf)3) and aluminum perchlorate (Al(ClO4)3) respectively to fabricate PUIP-Hac-Alac, PUIP-Hac-AlOTf and PUIP-Hac-AlClO4, forming dynamic metal–ligand coordination bonds between the Hacac group and Al3+ ions. The formation of these metal–ligand coordination bonds was confirmed by FTIR spectroscopy, as indicated by the peak at 606 cm−1 (Al–O)57 observed in the PUIP-Hac-Alac, PUIP-Hac-AlOTf, and PUIP-Hac-AlClO4 polymers, as shown in Fig. S3.† X-ray photoelectron spectroscopy (XPS) further confirmed the dynamic Al–O coordination bonds between aluminum and the Hacac group58,59 (Fig. 2c). Detailed data supporting these findings are presented in Table S2.† All three elastomers exhibited a predominant Al–O bond ratio within the polymer network, and it was noted that some aluminum ions also coordinated with the urea groups in the main chain (Al–N). The Thermogravimetric Analysis (TGA) results indicated that the thermal decomposition temperatures of all series materials exceed 250 °C, as shown in Fig. 2d and Table S3† suggesting excellent thermal stability for practical applications in everyday scenarios. In terms of optical properties, except for the PUIP-Hac-Alac series, all other elastomers demonstrated over 98% transparency at a wavelength of 500 nm. The PUIP-Hac-Alac series, due to the incorporation of Al(acac)3, appeared milky white with a transparency of only 80.6%, as detailed in Fig. 2e. Through dynamic mechanical analysis (DMA), it was confirmed that the glass transition temperature (Tg) of the series materials was approximately −100 °C, significantly lower than room temperature (PDMS itself has a Tg point of around −120 °C).60 This indicates that the materials possess good polymer chain mobility at room temperatures, suggesting potential for self-repair capabilities (Fig. S4 and Table S4†). Through water contact angle (WCA) measurement experiments (Fig. S5 and Table S5†), we employed untreated glass slides as control substrates and observed that after coating with the PUIP-Hac elastomers, all coated glass substrates exhibited high hydrophobicity (>105°), further highlighting the potential of our materials for use in wearable devices for daily use. Energy-dispersive X-ray (EDS) analysis confirmed the uniform distribution of added aluminum ions, counter anions (acac−, OTf−, and ClO4−), and other elements (C, N, Si, and O) throughout the polymer matrix, as shown in Fig. S6–S9.† This also signifies the homogeneous presence of dynamic bonds, such as hydrogen bonds and metal coordination bonds, within the polymer network.
3.2 Mechanical properties
Mechanical properties were evaluated on films (measuring 3 cm × 1 cm × 0.5 mm) using a uniaxial tensile tester at a rate of 100 mm min−1. The stress–strain curves of different elastomers were obtained, as shown in Fig. 3a. It can be observed that the PUIP-Hac elastomer without the introduction of aluminum metal salts has inferior mechanical performance, with a lower elongation at break (507%) and tensile stress (1.03 ± 0.18 MPa). With the introduction of aluminum salts, Al3+ ions coordinate with the Hacac group, forming metal coordination bonds, which enhance the molecular network's dynamicity and robustness, resulting in a significant improvement in mechanical properties overall (Table S6†). These outstanding mechanical properties are attributed to the synergistic effects of the partitioning of high and low-density hydrogen bonds within the polymer system, as well as the highly dynamic coordination bonds between Al3+ ions and the Hacac group of the polymer chain. Additionally, Hacac, with its ability to form multiple coordination modes, can create diverse and dynamic coordination modes with Al3+ ions, greatly enhancing the toughness and stretchability of the molecular network. Specifically, among these three groups of metal-added elastomers, the PUIP-Hac-Alac series exhibits only a slight increase in tensile stress (1.62 ± 0.27 MPa) compared to PUIP-Hac, accompanied by a decrease in elongation at break (300%). This phenomenon is attributed to the introduction of Al(acac)3, where the counter anion involved (acac−) and the coordination sites provided by the polymer matrix (Hacac group) are compatible. However, Al3+ ions tend to favor the formation of metal coordination bonds with the free anionic state of acac− instead of the polymer chains capped by the Hacac group due to its larger steric hindrance (Fig. 3g(I)). Therefore, there is only a partial enhancement in stress and a slight decrease in elongation for the PUIP-Hac-Alac elastomer, due to the limited dynamic bonding introduced. In contrast, both PUIP-Hac-AlOTf and PUIP-Hac-AlClO4 exhibit significant improvements in tensile stress and elongation at break compared to the pristine PUIP-Hac elastomer (Fig. 3a and b). The PUIP-Hac-AlOTf group demonstrates excellent tensile strength (4.36 ± 0.03 MPa) and elongation (1352 ± 110%), while PUIP-Hac-AlClO4 also displays outstanding tensile stress performance (4.44 ± 0.32 MPa) and elongation (712 ± 111%). The difference in tensile properties between PUIP-Hac-AlOTf and PUIP-Hac-AlClO4 is attributed to the different coordination abilities of their involved anions (OTf− and ClO4−) within the molecular network. OTf− anions exhibit stronger coordination ability compared to ClO4− anions.35 During material stretching, the coordination bonds between aluminum ions and Hacac groups in the molecular network may break under external stress. At this point, OTf− anions can temporarily occupy vacant coordination sites to stabilize the polymer network while stretching, resulting in outstanding ductility and higher toughness in PUIP-Hac-AlOTf (Fig. 3g(II)). Conversely, ClO4− anions have lower coordination ability; thus during molecular chain elongation, they are less capable of stabilizing the polymer network, leading to lower elongation at break in PUIP-Hac-AlClO4 compared to PUIP-Hac-AlOTf (Fig. 3g(III)). To elucidate the aforementioned mechanical properties, shear stress relaxation experiments were performed at a shear strain of 3% (Fig. 3c). We observed that the elastomers in each group followed the trend consistent with the tensile test results. PUIP-Hac exhibited the fastest stress dissipation due to the absence of metal coordination bond breaking and reformation. Similarly, PUIP-Hac-Alac showed a relatively fast stress dissipation rate because the coordination bonds formed between Al3+ ions and Hacac groups were limited. In contrast, PUIP-Hac-AlOTf and PUIP-Hac-AlClO4 displayed slower stress dissipation rates, as energy dissipation involved the reformation of Al-Hacac bonds and needed longer time for polymer chain rearrangement. In addition, the residual strain after the cyclic mechanical test with a strain of 50% was smaller due to the formation of metal coordination bonds in the polymer networks (Fig. 3d), suggesting that the polymer networks become more elastic with the dynamic bonds formed between the Al3+ ions and Hacac groups. In the cyclic loading/unloading test (Fig. S10†), the energy dissipation of each elastomer series increased with the maximum tensile strain (ranging from 50% to 250%), as higher strain led to the breaking of more dynamic bonds (such as hydrogen bonds and metal coordination bonds), resulting in greater energy dissipation. We further used density functional theory (DFT) calculations to evaluate the bonding energy (Eb) differences between the aluminum metal ions and the oxygen atoms of the Hacac group in PUIP-Hac-Alac, PUIP-Hac-AlOTf, and PUIP-Hac-AlClO4 elastomers (Fig. 3h). The Eb is calculated by eqn (1).56| Eb = E(PUIP-Hac-Alx) − E(PUIP-Hac) − E(metal salt) | (1) |
 | ||
| Fig. 3 (a) Stress–strain curves of PUIP-Hac, PUIP-Hac-Alac, PUIP-Hac-AlOTf, and PUIP-Hac-AlClO4. (b) Young's modulus and toughness of four series of samples. (c) Stress relaxation curves of the four series of samples at room temperature under a shear strain of 3%. (d) Cyclic curves of the polymer films with a sample width of 5 mm and a thickness of 0.4–0.5 mm under cyclic loading (loading rate: 100 mm min−1). (e) PUIP-Hac-AlOTf sample could bear over 22000 times its weight. (f) Comparison of toughness with previously reported self-healable PDMS elastomers.33,44,47,48,61–66 (g) The schematic illustration of the proposed mechanical mechanism of PUIP-Hac-Alac (I), PUIP-Hac-AlOTf (II) and PUIP-Hac-AlClO4 (III) elastomers. (h) DFT calculations and molecular structure of the PUIP-Hac, PUIP-Hac-Alac, PUIP-Hac-AlOTf, and PUIP-Hac-AlClO4. (i) Bonding energy (Eb) of the Al–O bonds in PUIP-Hac-Alac, PUIP-Hac-AlOTf, and PUIP-Hac-AlClO4. | ||
The results revealed significant differences among the systems studied. The calculated binding energies for PUIP-Hac-Alac, PUIP-Hac-AlOTf, and PUIP-Hac-AlClO4 are 69.41, −14.53, and 21.68 kcal mol−1, respectively (Fig. 3i). Interestingly, these values indicate that the formation of the Al–O coordination bond in PUIP-Hac-Alac is the least thermodynamically favorable, requiring the highest energy input. Conversely, the negative binding energy of PUIP-Hac-AlOTf suggests a spontaneous and thermodynamically favorable coordination process, indicating that the formed complex possesses high stability. PUIP-Hac-AlClO4, with its intermediate binding energy, represents a moderately stable coordination bond. These findings highlight the significant impact of different ligands on the stability and formation energetics of Al–O coordination bonds in these polymer systems and further demonstrate the tunable dynamic coordination network in our strategy. The results indicate that the Al–O bond in PUIP-Hac-AlOTf is more stable and robust, requiring more energy to break, suggesting that during the stretching process, the dynamic metal coordination bonds in PUIP-Hac-AlOTf can withstand greater tensile forces. In contrast, the Al–O binding energy in PUIP-Hac-Alac suggests that the bond in the system is unstable and tends to break, corresponding to its inferior mechanical performance. Given the excellent mechanical properties of PUIP-Hac-AlOTf, we demonstrate its superior mechanical performance. It can withstand a load exceeding 20000 times its own weight without breaking or forming cracks (Fig. 3e). Also, compared to many previously published self-healing PDMS materials, PUIP-Hac-AlOTf exhibits remarkable toughness (48.7 MJm−3), indicating its high potential for application in a broader range of scenarios, such as sports and medical monitoring (Fig. 3f and Table S7†).
3.3 Self-healing properties and recyclability of the PUIP-Hac-Alx elastomer
To investigate the self-healing ability of the samples, we first conducted a scratch test by using a surgical blade to create artificial wounds on the surface of the films. As shown in Fig. S11,† all series of the elastomers have the self-healing capability and demonstrate varying degrees of repair abilities in a room temperature environment. PUIP-Hac and PUIP-Hac-Alac with higher polymer chain mobility displayed great self-healing ability after 1 day of recovery. In contract, PUIP-Hac-AlOTf and PUIP-Hac-AlClO4, possessing robust polymer networks, exhibited a certain degree of self-healing performance, with PUIP-Hac-AlClO4 demonstrating poorer healing effects due to the weak coordination ability of its anion, ClO4−, making its molecular network less dynamic. To further quantify the self-healing capability of each elastomer, we conducted fracture tests on all samples (Fig. S12†). After 12 hours of healing at 50 °C, PUIP-Hac, PUIP-Hac-Alac, and PUIP-Hac-OTf exhibited nearly 100% mechanical recovery. In contrast, PUIP-Hac-ClO4, which is relatively rigid and lacks dynamic coordination, demonstrated inferior healing performance (healing efficiency of toughness: 24%). To further investigate the differences in the self-healing abilities of these elastomers, we conducted temperature-dependent stress relaxation tests55 (Fig. S13†). By analyzing the spectra, we calculated the activation energy (Ea) of each polymer system. Among the four polymers, PUIP-Hac-ClO4 exhibited the highest activation energy (41.66 kJ mol−1), indicating the greatest energy barrier for molecular mobility. This result correlates well with its relatively poor self-healing performance. In contrast, PUIP-Hac, PUIP-Hac-Alac, and PUIP-Hac-OTf displayed activation energies of 14.99 kJ mol−1, 28.86 kJ mol−1, and 36.02 kJ mol−1, respectively, suggesting better molecular chain mobility and consequently enhanced self-healing capabilities.73–76PUIP-Hac-AlOTf, with the best overall mechanical properties, was selected to conduct further detailed tests to study the self-healing ability. As shown in Fig. 4b, we allowed the PUIP-Hac-AlOTf elastomer to heal in a 50 °C environment, and the scratch exhibited significant recovery within only ten minutes. Subsequently, we also conducted fracture tests to quantify its healing efficiency (η). The healing efficiency was calculated based on toughness. The elastomer was completely cut into half with a blade, and then the two separated pieces were brought into contact and placed in a 50 °C environment for different durations to heal the damage (Fig. 4c). After different healing times, tensile tests were performed on the healing samples. As shown in Fig. 4a and d, the repairing efficiency increased with the extended healing time. The stress–strain curve of the 12 h-healed sample closely resembled the pristine curve. The healing efficiency of the PUIP-Hac-AlOTf elastomer reached about 95% (toughness) and the stress of the healed sample also recovered to 4.4 MPa (∼100%), showing its outstanding self-healing ability. The remarkable self-healing performance of the PUIP-Hac-AlOTf elastomer is entirely attributed to the synergistic effects of the high mobility of the siloxane polymer chain segment, rapid reformation of hydrogen bonds, and the dynamic nature of Al-Hacac coordination bonds in the polymer network (Fig. 4e). When the fractured surfaces are brought into contact, the polymer chains on the fractured surface diffuse toward each other and the broken hydrogen bonds gradually reform; meanwhile the Al-Hacac coordination bonds also rearrange as the healing time increases. Also, Hacac, as a bidentate functional group, can adopt various coordination modes, thereby bringing different polymer chains closer together more quickly. It is noteworthy that the PUIP-Hac-AlOTf elastomer greatly balances the trade-off between the mechanical properties and self-healing ability, standing out among many robust and self-healing PDMS-based materials (Fig. 4f and Table S8†). It has a robust network, outstanding mechanical performance, and excellent self-healing capability. Meanwhile, the PUIP-Hac-AlOTf elastomer can also be recycled using a solvent method. The films are cut into pieces and re-dissolved in THF solvent, followed by drop-casting onto a PTFE mold to re-form elastomer films (Fig. 4h). As shown in Fig. 4i, the recycled elastomers exhibit almost the same mechanical performance as the pristine ones after three recycling processes, which indicate the excellent recyclability of the PUIP-Hac-AlOTf elastomers. We further performed FT-IR tests on the recycled elastomer film to verify whether there were any changes in the chemical structure of the material before and after the recycling process. The FT-IR spectra before and after recycling are almost identical, indicating the stability and consistency of the chemical composition of the material after recycling (Fig. S14†). This also significantly demonstrates the promising potential of the material as a substrate for devices. The self-healing capability of the material not only extends the product's lifespan but also greatly reduces usage costs due to its excellent recyclability, making it more environmentally friendly.
 | ||
| Fig. 4 (a) Stress–strain curves of pristine and healed PUIP-Hac-AlOTf elastomers at different healing times at 50 °C. (b) Optical microscope images of scratch tests on PUIP-Hac-AlOTf, conducted at 50 °C. (c) Digital photograph of the fracture testing process. PUIP-Hac-AlOTf sample healed at 50 °C for 12 h. (d) Self-healing efficiency of the fractured PUIP-Hac-AlOTf elastomers healed for different times (2 h, 4 h, 6 h, 8 h and 12 h). (e) Schematic illustration of the self-healing mechanism of the PUIP-Hac-AlOTf. (f) Comprehensive comparison of self-healing ability and mechanical properties with previously reported self-healing PDMS elastomers.33,40,41,44,45,47,48,62,66–72 (g) The solvent recycling process of the PUIP-Hac-AlOTf elastomer. (h) Stress–strain curves of the PUIP-Hac-AlOTf elastomer for the first, second and third cycles of the recycling test. | ||
3.4 Application of the PUIP-Hac-AlOTf elastomer
Electrocardiogram (ECG) is an essential tool in the medical field for observing patients' cardiac function. Especially in long-term care, it is essential for monitoring human health and diagnosing diseases. However, the commonly used commercial ECG patches are composed of Ag/AgCl electrodes and conductive hydrogels, forming wet electrodes. The drawbacks of these wet electrodes stem from the hydrogel properties, as repeated use or prolonged storage can lead to the drying out of the conductive gel, which in turn weakens the signal and reduces clarity. Additionally, hydrogel usage would potentially cause skin irritation or allergies. Consequently, wet electrode patches are disposable and not suitable for everyone. In contrast, flexible and soft dry electrodes present a highly feasible alternative.77 These soft and elastic electrode patches can conform well to the skin surface without causing allergic reactions.We incorporated silver nanoflakes (AgFKs) into PUIP-Hac-AlOTf to prepare conductive composites named AgFK-PUIP-Hac-AlOTf, which were subsequently developed into flexible and soft dry electrode patches. The detailed fabrication and testing processes are shown in Fig. 5a. To further investigate the electrochemical properties of AgFK-PUIP-Hac-AlOTf, we conducted cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) measurements, as presented in Fig. S15.† The CV curves in Fig. S15a† demonstrate that despite being embedded in the PUIP-Hac-AlOTf self-healing polymer, Ag flakes retain excellent electrochemical reversibility. The nearly overlapping curves in the 1st and 2nd cycles indicate high stability and reproducibility, suggesting that the incorporation of silver nanoflakes does not hinder charge transfer but instead preserves Ag's intrinsic redox activity. The smooth and well-defined profile, without noticeable distortion, further confirms the electrode's electrochemical robustness, which is essential for ensuring stable and reliable performance in bioelectronic applications, particularly in ECG monitoring, where consistent electrode behavior is critical for reducing signal artifacts. Meanwhile, Fig. S15b† illustrates the influence of scan rate on electrochemical response. With increasing scan rates, the current response scales proportionally while maintaining a symmetrical shape, indicating efficient and reversible charge transfer kinetics. This behavior suggests a hybrid charge storage mechanism, involving both capacitive (non-faradaic) and faradaic contributions. The incorporation of silver nanoflakes enhances electrical conductivity, while the polymer matrix provides mechanical flexibility, creating a well-balanced charge transfer system that retains the redox properties of Ag while ensuring the electrode's durability. The EIS spectra in Fig. S15c† further confirm the electrode's favorable impedance behavior following the incorporation of Ag flakes. The presence of a well-defined semicircle in the high-frequency region suggests efficient charge transfer at the electrode–electrolyte interface, while the linear region in the low-frequency domain exhibits a 45° slope, indicative of Warburg-type diffusion impedance. This feature suggests that the integration of Ag nanoflakes enables the polymer matrix to retain the faradaic characteristics of Ag, while simultaneously leveraging the polymer's flexibility and mechanical resilience. This balance between electrical conductivity and mechanical adaptability demonstrates that the AgFK-PUIP-Hac-AlOTf composite holds significant potential as a high-performance conductive material, making it a promising candidate for flexible electrode applications.78
 | ||
| Fig. 5 (a) Process of fabricating the AgFK-PUIP-Hac-AlOTf electrodes. (b) Real-time ECG voltage signals recorded by Ag/AgCl electrodes (orange), pristine AgFK-PUIP-Hac-AlOTf electrodes (dark green), and self-healed electrodes (light green), alongside optical images of the corresponding electrodes attached to the skin. (c) Signal-to-noise ratio (SNR) of the ECG signals obtained with commercial Ag/AgCl electrodes, pristine AgFK-PUIP-Hac-AlOTf electrodes, and self-healed electrodes. (d) Comparison of ECG signals recorded using commercial electrodes and AgFK-PUIP-Hac-AlOTf electrodes. (e) Preparation of the self-healed AgFK-PUIP-Hac-AlOTf electrodes. (f) ECG signals and heart rate detected by AgFK-PUIP-Hac-AlOTf electrodes at rest, after walking, and after running. (g) The recycling process of the AgFK-PUIP-Hac-AlOTf electrodes: (i) soaking the electrodes in THF solvent, (ii) separating the polymer solution and electronic components, (iii) re-molding the conductive polymer solution and installing the metal snap button, and (iv) peeling off the electrodes and ECG signals collected from the pristine AgFK-PUIP-Hac-AlOTf electrodes and the recycled ones. (h) The optical image and corresponding signals for the flexing movements of the ring, middle and index fingers. (i) The corresponding signals for speaking different words (“OK”, “ni hao” and “hello”) and the optical image of the electrode patches attached to the volunteer's neck. | ||
Three ECG electrodes are applied to the right wrist, left wrist, and right arm of a 25-year-old man to record the 3-electrode ECG (ESI Video 1†). Paper surgical tape is used to secure the electrode patches to the skin. For comparative analysis, real-time ECG signals are captured using commercial Ag/AgCl electrodes with conductive hydrogels, pristine AgFK-PUIP-Hac-AlOTf electrodes, and self-healed AgFK-PUIP-Hac-AlOTf electrodes, respectively, as depicted in Fig. 5b. The self-healed AgFK-PUIP-Hac-AlOTf electrodes were prepared by cutting the electrode patches into four segments using a surgical knife, followed by a 12-hour healing process at 50 °C (Fig. 5e). An ECG waveform typically includes peaks known as P, Q, R, S, and T waves. The P wave denotes atrial activation, the QRS complex signifies ventricular activation and depolarization, and the T wave indicates ventricular repolarization.79 From Fig. 5d, it is evident that both AgFK-PUIP-Hac-AlOTf electrodes and Ag/AgCl electrodes can clearly measure ECG waveform including P, Q, R, S, and T peaks. Meanwhile, the peaks recorded by AgFK-PUIP-Hac-AlOTf electrodes exhibit clearer signals with less noise. We proceed to calculate the signal-to-noise ratio (SNR) for each electrode to further assess the quality of the measured signals. The SNR is computed using eqn (2), where Vrms-signal and Vrms-noise represent the root mean square of the QRS peaks and the remaining noise,80 respectively.
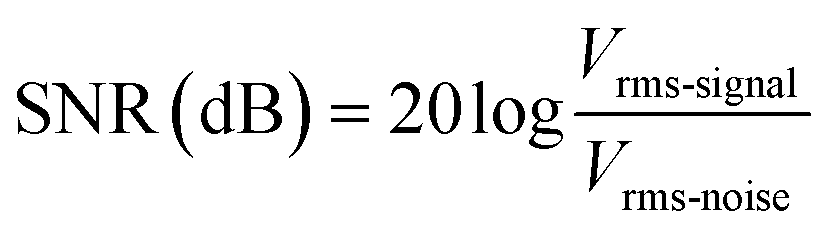 | (2) |
The SNR values for the Ag/AgCl electrode, AgFK-PUIP-Hac-AlOTf electrode, and self-healed AgFK-PUIP-Hac-AlOTf electrode are estimated to be 37.9, 61.5, and 44.5 dB, respectively (Fig. 5c). This indicates that our self-healable ECG electrode exhibits a performance comparable to that of the conventional electrode. Even the self-healed electrodes exhibited signal quality comparable to that of commercial electrodes, indicating the promising application potential of our material.
To confirm the stable operation of the self-healing patches, ECG signals were recorded while the test volunteer exercised (rest, walking and running) with the patches attached to his wrist. As shown in Fig. 5f, the ECG signals collected over the same period show a reduction in the interval between two R peaks: from 0.75 seconds at rest, to 0.6 seconds after walking, and 0.48 seconds after running. This demonstrates an accelerated heart rate in response to increased physical activity. This reveals that our electrode patches can reliably monitor the heart rate of volunteers under varying motion conditions. Additionally, the self-healable electrode patches can be recycled and reused. The patches can be soaked in THF solvent and sonicated for 30 minutes to separate the electronic components from the patches. After separation, the polymer solution containing AgFKs and metal snap buttons can be used to fabricate the recycled patches following the same process. As shown in Fig. 5g, ECG measurements taken with the recycled patches show that they can still produce stable and clear electrocardiogram signals. We further utilized the patches to monitor volunteer's movements such as hand and speaking activities. As illustrated in Fig. 5h, the patches were attached to the forearm of a volunteer to detect finger movements. The results demonstrate distinct and corresponding signals for each finger movement. Notably, strong signals were recorded during finger flexion, while the signals stabilized during relaxation. Moreover, electrode patches were applied to the volunteer's neck. As depicted in Fig. 5i, clear signal variations were observed when the volunteer articulated test words (“OK”, “ni hao”, and “Hello”). Characteristic signal peaks corresponded to vocal cord vibrations during speech, with the signals returning to a stable state during periods of silence. Importantly, the waveforms generated by repeated utterances of the same word were nearly identical, enabling the identification of spoken content through waveform analysis. These applications demonstrate significant potential for use in medical diagnostics and motion monitoring.
4. Conclusion
In summary, we introduced a strategy utilizing 2,4-pentanedione (Hacac) as a capping agent for polyurethane-urea, further incorporating aluminum metal salts to form metal coordination bonds, thereby significantly simplifying the synthesis process and maintaining the diversity of metal coordination. This approach enabled the development of a dynamic network featuring both hydrogen bonds and multimodal metal coordination bonds. Among the synthesized polymers, PUIP-Hac-AlOTf demonstrated exceptional mechanical properties, with toughness reaching up to 48.73 MJ m−3, which is remarkable compared to reported self-healing PDMS materials. Additionally, it exhibited excellent self-healing ability, with a recovery efficiency of nearly 95%. The synergy between hydrogen bonds and the multiple dynamic metal coordination bonds formed by Hacac within the system is responsible for its superior mechanical properties and self-healing performance. Furthermore, the material's recyclability underscores its potential for reducing production costs and promoting environmental sustainability. Moreover, the flexible electrode patch fabricated from PUIP-Hac-AlOTf and silver flakes conforms well to human skin and can be reused for applications such as ECG monitoring and daily activity detection. This work highlights the potential of integrating multi-dynamic metal coordination capping agents into PDMS materials, offering a significant advancement in the development of robust elastomers with enhanced self-healing capabilities.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Rou-Han Lai: conceived the original idea, designed the project, conducted the experiments, and primarily wrote the manuscript. Yi-An Chen: co-first author, provided helpful suggestions and polished the manuscript. Chung-Ying Chou, Hung-Yi Huang, and Wassana Mongkonkan: assisted with experiments and measurements. Chia-An Chiu: contributed to experimental work and manuscript refinement. Yan-Heng Chen and Min-Han Yu: provided valuable suggestions for the experiments. Chi-Chang Hu and Siriporn Jungsuttiwong: supervised the students involved in the experiments. Ho-Hsiu Chou: corresponding author. All authors commented on the results and approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to express their sincere gratitude for the financial support provided by the National Science and Technology Council (NSTC), under the grant numbers NSTC 113-2923-E-007-007-MY3, NSTC 113-2823-8-007-003, NSTC 112-2622-E-007-032, NSTC 112-2223-E-007-006-MY3, and NSTC 112-2221-E-007-081. Additionally, the authors appreciate thankful for the analytical and measurement support extended by the Precision Instrument Support Center at National Tsing Hua University. The authors appreciate the NSRF via the Program Management Unit for Human Resource & Institutional Development, Research and Innovation [B16f640099].References
- N. Mandlekar, M. Joshi and B. S. Butola, Adv. Ind. Eng. Polym. Res., 2022, 5, 33–45 Search PubMed.
- A. Kanti Sikder and S. Reddy, Propellants, Explos., Pyrotech., 2013, 38, 14–28 CrossRef CAS.
- R. Yoda, J. Biomater. Sci., Polym. Ed., 1998, 9, 561–626 CrossRef CAS PubMed.
- G. Demirci, M. J. Niedźwiedź, N. Kantor-Malujdy and M. El Fray, Polymers, 2022, 14, 1822 CrossRef CAS PubMed.
- M. C. Serrano, E. J. Chung and G. A. Ameer, Adv. Funct. Mater., 2010, 20, 192–208 CrossRef CAS.
- M. H. Zainol, R. Khimi Shuib, I. Ibrahim, F. S. Abd Razak, N. F. Mohd Sani and T. D. Lam, Pure Appl. Chem., 2024, 96, 1227–1244 CrossRef CAS.
- J. Chen, Q. Peng, T. Thundat and H. Zeng, Chem. Mater., 2019, 31, 4553–4563 CrossRef CAS.
- J. Kang, J. B.-H. Tok and Z. Bao, Nat. Electron., 2019, 2, 144–150 CrossRef.
- Y. Gai, H. Li and Z. Li, Small, 2021, 17, 2101383 CrossRef CAS PubMed.
- E. Roels, S. Terryn, F. Iida, A. W. Bosman, S. Norvez, F. Clemens, G. Van Assche, B. Vanderborght and J. Brancart, Adv. Mater., 2022, 34, 2104798 CrossRef CAS PubMed.
- S. Terryn, J. Langenbach, E. Roels, J. Brancart, C. Bakkali-Hassani, Q.-A. Poutrel, A. Georgopoulou, T. G. Thuruthel, A. Safaei and P. Ferrentino, Mater. Today, 2021, 47, 187–205 CrossRef CAS.
- Y. Zhang, A. A. Broekhuis and F. Picchioni, Macromolecules, 2009, 42, 1906–1912 CrossRef CAS.
- N. Zheng, Y. Xu, Q. Zhao and T. Xie, Chem. Rev., 2021, 121, 1716–1745 CrossRef CAS PubMed.
- K. Cerdan, M. Thys, A. C. Cornellà, F. Demir, S. Norvez, R. Vendamme, P. Van Puyvelde and J. Brancart, Prog. Polym. Sci., 2024, 101816 CrossRef.
- J. Chen, J. Zheng, Q. Gao, J. Zhang, J. Zhang, O. M. Omisore, L. Wang and H. Li, Appl. Sci., 2018, 8, 345 CrossRef.
- A. Larmagnac, S. Eggenberger, H. Janossy and J. Vörös, Sci. Rep., 2014, 4, 7254 CrossRef CAS PubMed.
- D. Qi, K. Zhang, G. Tian, B. Jiang and Y. Huang, Adv. Mater., 2021, 33, 2003155 CrossRef CAS PubMed.
- I. D. Johnston, D. K. McCluskey, C. K. Tan and M. C. Tracey, J. Manuf. Syst., 2014, 24, 035017 CAS.
- S. Park, K. Mondal, R. M. Treadway III, V. Kumar, S. Ma, J. D. Holbery and M. D. Dickey, ACS Appl. Mater. Interfaces, 2018, 10, 11261–11268 CrossRef CAS PubMed.
- Y.-L. Liu and T.-W. Chuo, Polym. Chem., 2013, 4, 2194–2205 RSC.
- C.-M. Yeh, C.-H. Lin, T.-Y. Han, Y.-T. Xiao, Y.-A. Chen and H.-H. Chou, J. Mater. Chem. A, 2021, 9, 6109–6116 RSC.
- J. Canadell, H. Goossens and B. Klumperman, Macromolecules, 2011, 44, 2536–2541 CrossRef CAS.
- K. Chang, H. Jia and S.-Y. Gu, Eur. Polym. J., 2019, 112, 822–831 CrossRef CAS.
- Y. Xu and D. Chen, Macromol. Chem. Phys., 2016, 217, 1191–1196 CrossRef CAS.
- A. Chao, I. Negulescu and D. Zhang, Macromolecules, 2016, 49, 6277–6284 CrossRef CAS.
- J. Hu, R. Mo, X. Sheng and X. Zhang, Polym. Chem., 2020, 11, 2585–2594 RSC.
- F. Song, Z. Li, P. Jia, M. Zhang, C. Bo, G. Feng, L. Hu and Y. Zhou, J. Mater. Chem. A, 2019, 7, 13400–13410 RSC.
- A. P. Bapat, B. S. Sumerlin and A. Sutti, Mater. Horiz., 2020, 7, 694–714 RSC.
- J. Cao, C. Lu, J. Zhuang, M. Liu, X. Zhang, Y. Yu and Q. Tao, Angew. Chem., 2017, 129, 8921–8926 CrossRef.
- L. Chen, J. Xu, M. Zhu, Z. Zeng, Y. Song, Y. Zhang, X. Zhang, Y. Deng, R. Xiong and C. Huang, Mater. Horiz., 2023, 10, 4000–4032 RSC.
- A. Faghihnejad, K. E. Feldman, J. Yu, M. V. Tirrell, J. N. Israelachvili, C. J. Hawker, E. J. Kramer and H. Zeng, Adv. Funct. Mater., 2014, 24, 2322–2333 CrossRef CAS.
- Z. Xie, B.-L. Hu, R.-W. Li and Q. Zhang, ACS Omega, 2021, 6, 9319–9333 CrossRef CAS PubMed.
- J.-C. Lai, X.-Y. Jia, D.-P. Wang, Y.-B. Deng, P. Zheng, C.-H. Li, J.-L. Zuo and Z. Bao, Nat. Commun., 2019, 10, 1164 CrossRef PubMed.
- H. Park, T. Kang, H. Kim, J.-C. Kim, Z. Bao and J. Kang, Nat. Commun., 2023, 14, 5026 CrossRef CAS PubMed.
- Y.-L. Rao, A. Chortos, R. Pfattner, F. Lissel, Y.-C. Chiu, V. Feig, J. Xu, T. Kurosawa, X. Gu and C. Wang, J. Am. Chem. Soc., 2016, 138, 6020–6027 CrossRef CAS PubMed.
- C. H. Li and J. L. Zuo, Adv. Mater., 2020, 32, 1903762 CrossRef CAS PubMed.
- Y.-A. Chen, S.-J. Chen, L.-Y. Lee, R.-H. Lai, C.-M. Yeh, C.-A. Chiu, J.-Y. Lai, Y.-C. Lai and H.-H. Chou, Nano Energy, 2023, 117, 108882 CrossRef CAS.
- S. Burattini, H. M. Colquhoun, B. W. Greenland and W. Hayes, Faraday Discuss., 2009, 143, 251–264 RSC.
- W. A. D. M. Jayathilaka, K. Qi, Y. Qin, A. Chinnappan, W. Serrano-García, C. Baskar, H. Wang, J. He, S. Cui and S. W. Thomas, Adv. Mater., 2019, 31, 1805921 CrossRef PubMed.
- C.-H. Li, C. Wang, C. Keplinger, J.-L. Zuo, L. Jin, Y. Sun, P. Zheng, Y. Cao, F. Lissel and C. Linder, Nat. Chem., 2016, 8, 618–624 CrossRef CAS PubMed.
- H. Guo, Y. Han, W. Zhao, J. Yang and L. Zhang, Nat. Commun., 2020, 11, 2037 CrossRef CAS PubMed.
- P. F. Cao, B. Li, T. Hong, J. Townsend, Z. Qiang, K. Xing, K. D. Vogiatzis, Y. Wang, J. W. Mays and A. P. Sokolov, Adv. Funct. Mater., 2018, 28, 1800741 CrossRef.
- B. Zhang, P. Zhang, H. Zhang, C. Yan, Z. Zheng, B. Wu and Y. Yu, Macromol. Rapid Commun., 2017, 38, 1700110 CrossRef PubMed.
- J.-H. Gao, B. Wan, M.-S. Zheng, L. Luo, H. Zhang, Q.-L. Zhao, G. Chen and J.-W. Zha, Mater. Horiz., 2024, 11, 1305–1314 RSC.
- J. Kang, D. Son, G. J. N. Wang, Y. Liu, J. Lopez, Y. Kim, J. Y. Oh, T. Katsumata, J. Mun, Y. Lee, L. Jin, J. Tok and Z. Bao, Adv. Mater., 2018, 30, 1706846 CrossRef PubMed.
- C. L. He, F. C. Liang, L. Veeramuthu, C. J. Cho, J. S. Benas, Y. R. Tzeng, Y. L. Tseng, W. C. Chen, A. Rwei and C. C. Kuo, Adv. Sci., 2021, 8, 2102275 CrossRef CAS PubMed.
- J. Fan, J. Huang, Z. Gong, L. Cao and Y. Chen, ACS Appl. Mater. Interfaces, 2020, 13, 1135–1144 CrossRef PubMed.
- X. Wu, J. Wang, J. Huang and S. Yang, ACS Appl. Mater. Interfaces, 2019, 11, 7387–7396 CrossRef CAS PubMed.
- G. Ye, Z. Song, T. Yu, Q. Tan, Y. Zhang, T. Chen, C. He, L. Jin and N. Liu, ACS Appl. Mater. Interfaces, 2019, 12, 1486–1494 CrossRef PubMed.
- Y. Zhang, J. Chen, G. Zhang, J. Xv, J. Xv, Y. Hu, H. Guo, J. Fu and W. Jiang, Chem. Eng. J., 2021, 425, 130665 CrossRef CAS.
- J. Bai, L. Zhang, Z. Shi and X. Jiang, Chem. Mater., 2022, 34, 10172–10180 CrossRef CAS.
- A. Hübner, D. Stroybusch, H.-W. Lerner and M. Bolte, J. Chem. Crystallogr., 2008, 38, 953–957 CrossRef.
- T. Jing, X. Heng, T. Jingqing, L. Haozhe, L. Li, L. Pingyun and G. Xiaode, Chem. Eng. J., 2023, 465, 142887 CrossRef CAS.
- Y. Xue, J. Lin, T. Wan, Y. Luo, Z. Ma, Y. Zhou, B. T. Tuten, M. Zhang, X. Tao and P. Song, Adv. Sci., 2023, 10, 2207268 CrossRef CAS PubMed.
- Y. Li, W. Li, A. Sun, M. Jing, X. Liu, L. Wei, K. Wu and Q. Fu, Mater. Horiz., 2021, 8, 267–275 RSC.
- Y. Wang, X. Huang and X. Zhang, Nat. Commun., 2021, 12, 1291 CrossRef CAS PubMed.
- M. Okuno, N. Zotov, M. Schmücker and H. Schneider, J. Non-Cryst. Solids, 2005, 351, 1032–1038 CrossRef CAS.
- Y.-A. Chen, R.-H. Lai, W.-C. Lin, H.-Y. Huang, S.-J. Chen, C.-M. Yeh, H.-L. Huang, M. M. Elsenety, C.-C. Hu, C.-H. Yu and H.-H. Chou, ACS Appl. Polym. Mater., 2024, 6(12), 6976–6987 CrossRef CAS.
- W.-C. Liao, C.-T. Chiang, J.-I. Chyi and Y.-M. Hsin, J. Electrochem. Soc., 2015, 162, E160 CrossRef CAS.
- D. Fragiadakis, P. Pissis and L. Bokobza, Polymer, 2005, 46, 6001–6008 CrossRef CAS.
- Y. Zhang, L. Yuan, G. Liang and A. Gu, J. Mater. Chem. A, 2018, 6, 23425–23434 RSC.
- W. Wang, W. Wang, F. Wang, X. Xie, G. Yi and Z. Li, J. Mater. Chem. A, 2022, 10, 23375–23383 RSC.
- Z. Wang, Y. Liu, D. Zhang, K. Zhang, C. Gao and Y. Wu, Compos. Sci. Technol., 2021, 216, 109042 CrossRef CAS.
- C. Lv, J. Wang, Z. Li, K. Zhao and J. Zheng, Composites, Part B, 2019, 177, 107270 CrossRef CAS.
- H. Sun, X. Liu, S. Liu, B. Yu, N. Ning, M. Tian and L. Zhang, Chem. Eng. J., 2020, 384, 123242 CrossRef CAS.
- H. Li, F. Xu, J. Wang, J. Zhang, H. Wang, Y. Li and J. Sun, Nano Energy, 2023, 108, 108243 CrossRef CAS.
- S. Liu, S. Chen, W. Shi, Z. Peng, K. Luo, S. Xing, J. Li, Z. Liu and L. Liu, Adv. Funct. Mater., 2021, 31, 2102225 CrossRef CAS.
- K. Zhang, X. Shi, J. Chen, T. Xiong, B. Jiang and Y. Huang, Chem. Eng. J., 2021, 412, 128734 CrossRef CAS.
- Z. Yang, H. Li, C. Li, X. Lai and X. Zeng, Chem. Eng. J., 2022, 430, 133103 CrossRef CAS.
- K. Zhao, C. Lv and J. Zheng, Sci. China: Technol. Sci., 2020, 63, 740–747 CrossRef CAS.
- B. Li, S. Ge, S. Zhao, K. Xing, A. P. Sokolov, P.-F. Cao and T. Saito, Mater. Horiz., 2023, 10, 2868–2875 RSC.
- C. Liu, J. T. Kim, D. S. Yang, D. Cho, S. Yoo, S. R. Madhvapathy, H. Jeong, T. Yang, H. Luan and R. Avila, Adv. Funct. Mater., 2023, 33, 2302256 CrossRef CAS.
- J. Chen, Z. Wang, B. Yao, Y. Geng, C. Wang, J. Xu, T. Chen, J. Jing and J. Fu, Adv. Mater., 2024, 2401178 CrossRef CAS PubMed.
- F. Sun, L. Liu, T. Liu, X. Wang, Q. Qi, Z. Hang, K. Chen, J. Xu and J. Fu, Nat. Commun., 2023, 14, 130 CrossRef CAS PubMed.
- F. Sun, J. Zhang, T. Liu, H. Yao, L. Wang, H. Meng, Y. Gao, Y. Cao, B. Yao and J. Xu, Adv. Mater., 2024, 36, 2410650 CrossRef CAS PubMed.
- J. Xu, J. Chen, Y. Zhang, T. Liu and J. Fu, Angew. Chem., 2021, 60, 7947–7955 CrossRef CAS PubMed.
- A. C. Myers, H. Huang and Y. Zhu, RSC Adv., 2015, 5, 11627–11632 RSC.
- R. Hu, B. Yao, Y. Geng, S. Zhou, M. Li, W. Zhong, F. Sun, H. Zhao, J. Wang and J. Ge, Adv. Mater., 2024, 36, 2403111 CrossRef CAS PubMed.
- T. Hlaing, T. DiMino, P. R. Kowey and G. X. Yan, Ann. Noninvasive Electrocardiol., 2005, 10, 211–223 CrossRef PubMed.
- M. Park, W. Yang, J. W. Kim, Y. Choi, S. Kim, Y. Lee, D. S. Kim, J. Kim, D. K. Lim and J. S. Ha, Adv. Funct. Mater., 2024, 2402508 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta08955c |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |