
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh-pressure synthesis of PbN2, the missing group 14 AN2-type compound†
Ken Niwa *ab,
Hirokazu Ogasawarac,
Takuya Sasakib,
Shunsuke Nomurab,
Gendai Azumab,
Sylvain Pitiéd,
Gilles Frapperd and
Masashi Hasegawaab
*ab,
Hirokazu Ogasawarac,
Takuya Sasakib,
Shunsuke Nomurab,
Gendai Azumab,
Sylvain Pitiéd,
Gilles Frapperd and
Masashi Hasegawaab
aResearch Center for Crystalline Materials Engineering, Nagoya University, Furo-cho, Chikusa-ku, Nagoya, Aichi 464-8603, Japan. E-mail: niwa@mp.pse.nagoya-u.ac.jp; Fax: +81-52-789-3252; Tel: +81-52-789-5515
bDepartment of Materials Physics, Nagoya University, Furo-cho, Chikusa-ku, Nagoya, Aichi 464-8603, Japan
cDepartment of Crystalline Materials Science, Nagoya University, Furo-cho, Chikusa-ku, Nagoya, Aichi 464-8603, Japan
dApplied Quantum Chemistry Group, E4 team, IC2MP UMR 7285, Université de Poitiers – CNRS, 4, rue Michel Brunet TSA 51106 – 86073, Poitiers, France
First published on 23rd May 2025
Abstract
The direct reaction of Pb with molecular nitrogen was investigated using a laser-heated diamond anvil cell combined with high-pressure in situ synchrotron X-ray powder diffraction measurements and Raman spectroscopy. From the results of the experiments and DFT calculations, it was revealed that a novel PbN2 was successfully synthesized above 50 GPa, and it was observed to crystallize in a tetragonal system (indexation in the CuAl2-type structure, I4/mcm), comprising PbN8 units connected via N–N dimers. Room-temperature compression and decompression experiments revealed that PbN2 retained its structural integrity up to a pressure of approximately 90 GPa, while it decomposed into lead and nitrogen at around 15 GPa. The experimental evidence indicates that the encapsulated nitrogen dumbbells act as anionic units, (N2)2− interaction with the lead lattice. The newly synthesized PbN2 exhibits a distinct crystal structure in comparison to the pyrite form of the other group-14 pernitrides (SiN2, GeN2, and SnN2) that have been previously reported. This finding provides novel insights into the high-pressure crystal chemistry of nitrogen-based compounds.
Introduction
The development of ultrahigh-pressure generation techniques has facilitated new insights into material science, including the study of superconductivity and superhard materials.1 In particular, the laser-heated diamond-anvil cell (LHDAC), when combined with advanced characterization methods (such as synchrotron radiation, infrared spectroscopy, electron diffraction spectroscopy, and so forth) represents a promising tool for the synthesis of novel and attractive materials.2,3 Furthermore, the LHDAC enables the convenient loading of gaseous samples, transportation to the synchrotron facility, and setup at the latter location.4,5 A quarter of a century ago, these noteworthy features led to synthesizing novel nitrogen-based compounds, including γ-Si3N4 (ref. 6) and PtN2 (ref. 7 and 8) among others.9–11 Subsequently, numerous novel nitrides [N3−], dumbbells [N2q−], and polynitrogen-based units [Nxq−] have been discovered in almost all binary nitrogen-based systems to date [e.g. some examples in the transition metal–nitrogen system].12–19 The recent synthesis of new nitrogen-based compounds identified in the pnictogen (Pn)–nitrogen systems provides an illustrative example of this high-pressure chemistry.20–23 The synthesis of AsN and BiN was successfully achieved at 30–40 GPa and 50 GPa, respectively.20,22 Furthermore, nitrogen-rich phases (Sb2N3, SbCN3) were synthesized through the nitridation experiments of Sb at approximately 32 GPa.21,24 What about the nitrides of group 14 elements? They are of significant importance in the field of functional ceramics.25 Two structural polymorphs of Si3N4 have been identified at ambient pressure,26–29 and a third polymorph, γ-Si3N4,6 was discovered through direct nitridation of silicon at 15 GPa. Subsequently, the γ-phase was also found in Ge3N4 under high pressure.30 The synthesis of γ-Sn3N4 has also been achieved without the use of high-pressure conditions,31 while the high-pressure technique was employed to prevent the decomposition of nitride under high temperatures.32 Subsequent high-pressure nitridation experiments on Si, Ge, and Sn were successful in synthesizing the pyrite-type AN2 (A = Si, Ge, and Sn) above approximately 60 GPa.33,34The pyrite type is one of the high-density structures observed in MX2-type compounds. Therefore, the exploration of higher-density phases than pyrite-type represents a promising avenue of research, offering insights not only into high-pressure crystal chemistry but also in the development of novel functional nitrogen-based ceramics. In accordance with the phase transition sequence upon pressure in AO2 (A = Si, Ge, Sn, and Pb), the crystal structure of the high-pressure phase that manifests in low-Z oxides is analogous to that of high-Z oxides at low pressure.35–40 It is therefore important to investigate the Pb–N system under conditions of elevated pressure and at atmospheric conditions, where the only observed binary phase is Pb(N3)2 azide.41–43 Lead typically assumes the +2 and +4 oxidation states. The Pb2+-containing compounds exhibit remarkable dielectricity due to the distortion of the unit cell induced by the lone-pair electrons,44–46 although the use of lead in material development is discouraged. The effect of lone-pair electrons on structural stability was also highlighted in the high-pressure synthesis of the novel PnN compounds.22 As evidenced by the synthesis of PnxNy compounds,20–22 the high-pressure technique represents a more effective and advantageous approach for accessing the synthesis of the remaining group-14 pernitride, PbN2 (Pb2+N22− or Pb4+N24−). However, to the best of our knowledge, only the explosive material, lead azide Pb(N3)2, has been identified as a thermodynamically metastable compound within the Pb–N binary system.41–43 In contrast to these experimental findings, a recent theoretical study has been conducted to investigate the Pb–N phase diagram under pressure using evolutionary algorithms combined with density functional theory (DFT) calculations.47 This study revealed the phase stability and crystal chemistry of novel PbNz phases (z = 2–4, 6, 8, and 10) when pressure is increased up to 100 GPa, including the hitherto unidentified pernitride PbN2 of group 14 elements.47 The Pb–N2 system offers a diverse range of opportunities for the exploration of pernitrides and polynitrides, which could result in the development of high-energy materials. The objective of the present study is to conduct high-pressure synthesis experiments (LHDAC) on the Pb–N2 system up to approximately 90 GPa, with a particular focus on the PbN2.
Experimental
We performed five independent experiments (sample number: OH#45, OH#52, NS#1, AG#1, AG#2) and the details of the experimental setup are given below. Further additional information for each experiment is given in ESI (please refer to Experimental details of the ESI†). The high-pressure synthesis experiments were conducted using a laser-heated diamond anvil cell (LHDAC) equipped with a 350 μm culet size anvil. Stainless steel (OH#45, OH#52, AG#1, AG#2) or rhenium (NS#1) plate with an initial thickness of approximately 280 μm was used as a gasket material. The gasket material was indented, and the intended area was drilled with a pulsed infrared laser to make a sample chamber with a diameter of 1/2 or 1/3 of the culet size. Lead foil (99.99%, The Nilaco Corporation) was stretched, thinned, and then shaped with less than approximately 80 × 80 μm2. After that, the lead thin film was placed in the sample hole with ruby chips as a pressure marker and then the sample chamber was filled with liquid nitrogen. The pressure was increased up to the desired pressure under room temperature, and then, the sample was heated by irradiating the infrared laser (SPI, Ltd λ = 1090 nm) from both sides. The sample was monitored during laser heating. The temperature was not measured in this study however, intense thermal radiation was observed during laser irradiation, which might correspond to over 2000 K. The unheated regions were probably due to the insufficient thickness of the thermal insulating layer. The heated sample was evaluated by optical microscope observation, Raman scattering, and X-ray diffraction measurements under high pressures. Argon ion (488 nm, <100 mW) or diode-pumped (473 nm, <100 mW) laser was used as an excitation source for Raman scattering measurements. The incident laser was focused with a size of less than approximately 10 μm through the diamond. The Raman signal was detected with the liquid nitrogen-cooled CCD detector. High-pressure in situ X-ray diffraction experiments were conducted at BL2S1, the Nagoya University beamline of Aichi Synchrotron Radiation Center, Aichi Science & Technology Foundation,48 and BL10XU, SPring-8.49 The distance between the sample and detector, the wavelength of the X-ray, the tilting of the detector, etc. were calibrated based on the diffraction profile of standard material CeO2 before the sample measurements. At BL2S1, the monochromatic X-ray with the size of 75–100 μm was introduced to the sample chamber at 15 degrees tilting from the compression axis, which allows measuring the wide 2-theta range, while for the experiments at BL10XU, the X-rays with the size of approximately 10 μm were introduced parallel to the compression axis. The diffracted X-rays were recorded with a typical exposure time of 3–5 minutes and 2 seconds for the experiments at BL2S1 and BL10XU, respectively. Details of high-pressure in situ XRD measurements are described elsewhere.48,49 The 2-dimensional (2-D) diffraction pattern was converted to the conventional 1-D profile by using IPAnalyzer.50 The phase identification and indexing analysis were conducted using dedicated software of PDIndexer50 and DICVOL06, respectively.51 Le Bail refinement was carried out using RIETAN-FP.52 The pressure was evaluated based on the pressure dependence of the ruby fluorescence peak53 and the equation of the state of rhenium (gasket material),54 solid nitrogen,55 and residual lead.56,57 The recovered sample was characterized by a scanning electron microscope equipped with energy-dispersive spectroscopy (FlexSEM 1000 II, Hitachi High-Tech Corporation, accelerating voltage: 15 kV).The energy and crystal structure calculations of PbN2 phases with different nitrogen dimer arrangement in 2 × 2 × 2 supercell was carried out by density functional theory (DFT) calculations performed using the CASTEP ab initio program58 on the Materials Studio package. The generalized gradient approximation (GGA-PBE)59,60 was applied, and wave functions were characterized by a plane wave basis set, whereas ultrasoft pseudopotentials were employed with a cutoff energy of 570 eV. k-Point grids of 4 × 4 × 4 were used for geometry optimizations. The energy, atomic force, and displacement convergence criteria were set to 5 × 10−6 eV per atom, 1 × 10−2 eV Å−1, and 5 × 10−4 Å, respectively. To obtain the Raman spectra, we employed the VASP code61,62 or the computation of phonons. These calculations were performed at the PBE level of theory using the finite displacement method implemented in the PHONOPY code (for additional details regarding the phonon calculation, please refer to Section S1.1–S1.3 of the ESI†).63,64 Based on these phonon calculations, we extracted and computed the intensity of the active Raman bands using the Phonopy-Spectroscopy package.65,66
Frequency calculations were performed at Γ, thanks to the Phonopy package with a finite difference approach (0.01 Å displacements), employing VASP 5.4.4 as the force calculator. IR intensities and Raman activities were evaluated, thanks to the Phonopy-spectroscopy framework. A 2 × 2 × 1 and 2 × 1 × 1 supercells were needed for the I4/mcm PbN2 and R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c Pb(N2)3 crystals to ensure convergence.
c Pb(N2)3 crystals to ensure convergence.
Results and discussion
Fig. 1(a) and (b) show the high-pressure in situ Raman spectra (AG#1) obtained prior to and following laser heating of lead in nitrogen, respectively. The application of laser heating at approximately 50 GPa resulted in a notable alteration of the sample's colour, transitioning from a metallic to a black colour. The Raman scattering measurements conducted following the heating process revealed the emergence of several additional sharp peaks and a notable reduction in the intensity of the nitrogen pressure medium. These findings provide compelling evidence that the synthesis of new PbxNy compounds can be achieved by reacting between lead and nitrogen under high-pressure conditions. Then, the pressure was decreased from approximately 55 GPa to 40 GPa, while Raman spectra were recorded. The newly emerged Raman peaks underwent a shift, yet no additional peaks or those that had disappeared were observed (see Fig. S1†). These findings suggest that the novel PbxNy compound was stable at pressures down to approximately 40 GPa. Notably, the Raman peak situated at approximately 2100 cm−1 shifted to a higher wavenumber with decreasing pressure, which represents an inverse pressure response to that observed in conventional solid-state materials and will be discussed subsequently. | ||
| Fig. 1 Raman spectra and XRD profiles under high pressures. (a) and (b) shows the Raman spectra corresponding to the Pb–N2 sample (AG#1) before and after laser heating at approximately 50 GPa. Two wavenumber regions were separately displayed. Intense peaks after laser-heating are marked with arrows. The horizontal arrows show the wavenumber region derived from solid molecular nitrogen. (c) High-pressure in situ X-ray diffraction profiles acquired from the heated and unheated areas. The diffraction peak positions of Pb, and N2 are represented with tics at the bottom of the profiles. The intense peak positions derived from the new phase that appeared after laser heating are also indicated with tics. The peak marked with an asterisk might be derived from the device. | ||
The newly synthesized compounds were characterized by high-pressure in situ powder X-ray diffraction measurements at synchrotron facilities. Recently, a technique using high-intensity synchrotron radiation has been widely used to characterize the crystal structure by extracting single-domain-derived diffraction spots from a multi-grain sample.67 Unfortunately, we have used conventional powder diffraction technique in this study, while have attempted to access the details of newly synthesized compound by combining the results of Raman scattering measurements and first-principles calculations in addition to an information of the powder diffraction pattern. A finely focused X-ray beam at BL10XU, SPring-8, allows the diffracted X-rays from heated and unheated areas to be measured separately, as illustrated in Fig. 1(c). The diffraction peaks from the unheated area were assigned using lead56,57 and solid molecular nitrogen.55 Conversely, several sharp diffraction peaks distinct from those observed in solid nitrogen and lead were identified in the heated region. The crystal symmetry was investigated through the assignment of over fifteen intense diffraction peaks using dedicated indexing software, leading to the identification of a tetragonal cell with lattice parameters a = 3.297(1) Å and c = 3.089(2) Å as the most plausible candidate (see Table S1†). Additionally, the orthorhombic cell was identified as a potential candidate, but some XRD peak positions could not be accurately fitted. The Miller indices of the tetragonal unit cell follow the extinction rule of the P lattice. Further analysis by the charge-flipping method68 utilizing the extracted intensity data revealed the Pb position at the vertex of the tetragonal cell. Two Pb–Pb distances were identified within the tetragonal Pb-lattice. One (d ≈ 3.1 Å) exhibited a comparable value to that observed in the high-pressure hexagonal phase of lead (d ≈ 3.1 Å),56,57 while the other (d ≈ 3.3 Å) demonstrated a slightly longer distance. The longer Pb–Pb distance may be attributed to the incorporation of nitrogen atoms or dimers into the Pb lattice.
As illustrated in Fig. 2(a), modifying the charge density map contrast results in a diminished charge density at the intermediate sites among Pb, which signifies the incorporation and alignment of the nitrogen dimer within the Pb lattice. The state of nitrogen was additionally evaluated through a volumetric comparison with Pb + N2. It can be postulated that the nitrogen dimer existed in the tetragonal Pb lattice, forming the Pb(N2)x. The unit cell is assumed to be Z = 1 and the volume is compared with that of Pb + N2 using the equation of state for lead and solid molecular nitrogen. It was determined that the formation of PbN2 via the reaction between lead and nitrogen molecules resulted in a reasonable volume reduction of approximately 18%. To gain insight into the details of the newly synthesized PbN2, several model structures were proposed. In consideration of the alignments of the nitrogen dimer, the 2 × 2 × 2 size of the initial Pb-lattice was postulated, and the total electronic energies of these structural models were calculated, with the experimentally determined lattice parameters held constant (constant volume). It should be noted that N2 units can align in different directions within each Pb8 tetragonal cage, a condition that is considered in our models. As illustrated in Fig. 2(b)–(d) and S2,† the DFT computational analysis indicated that the most energetically favorable structure involved the alternate alignment of the nitrogen dimers within the Pb lattice. Considering the data yielded by the experiments, the CuAl2-type structure (I4/mcm, no. 140) was put forth as a potential hypothesis. The unit cell of the I lattice is equivalent to the √2a × √2a × 2c of the initially obtained P lattice unit cell (Fig. 2(b)–(d)). This equivalence can be confirmed by the existence of 213 diffraction in the I lattice setting, as no peak corresponding to this index exists in the P lattice. Upon examination of the unrolled Debye-ring recorded on a two-dimensional detector, weak diffraction spots corresponding to 213 of the I lattice were identified (Fig. S3†). Fig. 2(e) represents the Le Bail refinement of the XRD profile based on the space group of I4/mcm with the lattice parameters of a = 4.6266(1) Å and c = 6.1278(2) Å. In this CuAl2-type phase, each Pb atom coordinates to eight nitrogen atoms, in which a coordination number is higher than that observed in the pyrite form of other group-14 pernitrides. Furthermore, structural diversity is permitted for CuAl2-type compounds, exemplifying the considerable variation in axial length ratio contingent on the constituent elements.69 Lead exhibits compressive behavior,56,57 a known property of Pb, whereby the nitrogen dimer may align flexibly within the structure and the structure diversity may be yielded. The present result, which is based on modeling that has been guided by experimental insights and DFT calculations, is in perfect agreement with the ab initio evolutionary (EA-DFT) study that has been conducted to investigate the potential emergence of PbxNy compounds in the Pb–N system up to a pressure of 100 GPa.47 Firstly, the crystal structure prediction (CSP EA-DFT) searches identified the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 composition as lying on the convex hull, i.e. PbN2 is a dynamically and thermodynamically stable compound above 32 GPa and at least up to 100 GPa. Secondly, PbN2 crystallizes in the CuAl2-type structure within the aforementioned pressure range. At 50 GPa, the a- and c-lattices were calculated to be 4.588 Å and 6.007 Å, respectively (meta-GGA r2SCAN level of theory), and these calculated values are in perfect agreement with the experimental data.
:2 composition as lying on the convex hull, i.e. PbN2 is a dynamically and thermodynamically stable compound above 32 GPa and at least up to 100 GPa. Secondly, PbN2 crystallizes in the CuAl2-type structure within the aforementioned pressure range. At 50 GPa, the a- and c-lattices were calculated to be 4.588 Å and 6.007 Å, respectively (meta-GGA r2SCAN level of theory), and these calculated values are in perfect agreement with the experimental data.
 | ||
| Fig. 2 Charge density map, atomic arrangement, and refined XRD profile of newly synthesized PbN2. (a) Charge density map and corresponding atomic assignment of the tetragonal cell. Large and small half-filled balls correspond to Pb and N, respectively. (b) Atomic arrangements in the 2 × 2 × 2 extended cell of the initially obtained one. The nitrogen dimers alternatively align in the neighbor cell which was obtained by the energy calculation results. The initial obtained P lattice cell and extended cell (I lattice) are represented with the dashed and dot-dashed lines, respectively. (c) and (d) Projected atomic arrangement along the c-axis and [110] direction, respectively. (e) LeBail refinement based on the tetragonal symmetry having the I lattice. The 2θ regions where nitrogen diffraction peaks appeared are excluded from the refinements. The crystal structures are drawn with 3D visualization program VESTA.70 | ||
The phase stability of PbN2 was investigated based on the additional high-pressure in situ XRD measurements and Raman spectroscopy along with the compression and decompression process. As illustrated in Fig. 3, the XRD peaks derived from the PbN2 remained intact up to 86.9 GPa under room temperature compression. In that experiment, it is possible that the pressure was overestimated because the pressure was determined by the equation of state of the rhenium gasket which was uniaxially compressed. Upon decompression process at room temperature, PbN2 underwent decomposition at approximately 15 GPa. Our experiments did not yield substantial evidence for the other metastable low-pressure phase prior to decomposition. Furthermore, the room temperature re-compression of the decomposed sample (AG#1) was conducted, yet no PbN2 was observed to reappear (for details, please refer to Fig. S4†). The pressure dependence of the cell volume per formula unit and Pb–Pb distances are displayed in Fig. 4. Additionally, the pressure-volume relation for Pb + N2 is also shown there. The zero-pressure bulk modulus (K0) of 65(3) GPa and unit cell volume (V0) of 44.7(3) Å3 were preliminarily obtained by fitting the pressure–volume data (OH#45) to the 2nd-order Birch–Murnaghan equation of state. The zero-pressure bulk modulus is markedly lower than that of other group-14 pernitrides33 SiN2, GeN2, and SnN2, reflecting the weaker Pb–N interactions. It is approximately 1.5 times that of lead.56,57 This phenomenon is likely attributable to the incorporation of the nitrogen dimer into the Pb lattice, resulting in a charge transfer from Pb to N2. The experimentally observed volume reduction is reasonably consistent with the result of the first principles calculation (41% from 0 GPa to 100 GPa) if the uncertainty of the pressure determination was considered. In addition to bulk compressibility, axial compressibility and ratio were evaluated (see Fig. S5†). It was established that PbN2 displays enhanced compressibility along the c-axis relative to the a-axis, which leads to the decreasing of c/a with increasing the pressure. This finding aligns with the incorporation of nitrogen dimers that are alternatively aligned in the a–b plane.
 | ||
| Fig. 3 Pressure evolution of X-ray diffraction profiles (OH#45 and NS#1) (a) the XRD profile of the sample (OH#45) acquired during the decompression from 47 GPa. The top profile corresponds to the sample after heating at 47 GPa. Newly appeared peaks are marked with arrows. XRD patterns were acquired along the decompression to ambient pressure at room temperature. (b) The XRD profile of the sample (NS#1) was acquired during the compression from 54.2 GPa. The bottom profile corresponds to the laser-heated sample. The arrows correspond to the peaks of the newly synthesized compound as well as (a). XRD patterns were acquired along the compression to approximately 90 GPa under room temperature. Non-marked peaks might be derived from nitrogen and weak peaks from the rhenium gasket are detected with the labeling of “Re” in (b). There are still a few unidentified peaks that might represent other new compounds, which should be investigated further. | ||
 | ||
| Fig. 4 Pressure dependence of volume per formula unit and Pb–Pb distances. The bulk modulus volume at ambient pressure (half-filled square) was determined by fitting pressure-volume data for OH#45 to B-M EOS. The equimolar volume of Pb + N2 is represented as a dashed line calculated from the reported P–V data for Pb and N2. The Pb–Pb distances parallel to the a- and c-axes correspond to the lattice parameters of the tetragonal Pb-lattice, respectively. | ||
Raman scattering measurements provide useful information to assess the newly synthesized material's phase stability and bonding nature. Fig. 5(a) and (b) shows the Raman spectra of the samples (AG#2) acquired during the decompression process. As mentioned earlier, the peaks around 200 and 2100 cm−1 became less intense, shifting to lower and higher wavenumbers respectively with decreasing pressure. A lower wavenumber peak could not be detected below 23 GPa due to the cutting-edge wavenumber of the long pass filter for this sample. The high-resolution Raman spectra were also obtained using an argon ion laser (488 nm), which allows the detection of low wavenumber peaks down to ambient pressure by using a suitable long-pass filter (see Fig. S6†). The nature of remaining weak peaks is unclear but may be related to the slow decompression rate, high crystallinity, high detector sensitivity, etc. On the other hand, the higher wavenumber peak eventually disappeared or merged with the N2 pressure medium (see Fig. 5(b)), although it could be detected down to around 10 GPa when the Raman spectra were measured from different positions (see Fig. S7†).
 | ||
| Fig. 5 Pressure dependence of the measured (AG#2) and calculated Raman spectra. Two wavenumber regions are separately shown in (a) and (b). Newly appeared peaks are represented with νi (i = 1–4). Wavenumber regions corresponding to the N2 pressure medium are represented with horizontal arrows. Spiky peaks at low wavenumber regions at low pressures might be derived from the instruments or atmospheric species. Calculated Raman spectra of I4/mcm PbN2 at 0 K (c) 50 GPa and (d) 0 GPa. | ||
The nitrogen dimer [NN]q− gives a vibrational wavenumber depending on the q negative charge, which is reflected in the bond order in N2 dumbbells. As the bond order increases, i.e. one in [N–N]4−, two in [N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N]2− and three in neutral [N
N]2− and three in neutral [N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N], the vibrational frequency increases, i.e. approximatively 900, 1300, and 2400 cm−1, respectively.7,71–75 The vibrational mode wavenumbers associated with the [N–N]4− stretching mode in group-14 element pernitrides of SiN2, GeN2, and SnN2 are observed around 1000–1100 cm−1.33 However, the Raman wavenumber of PbN2 is largely different from the other group-14 element pernitrides. What are the Raman active modes and spectra of the CuAl2-type PbN2? To give a response, we have conducted a study of the Raman spectra of the ground state I4/mcm PbN2 phase at 50 GPa using DFT calculations. The simulated Raman spectra are shown in Fig. 5(c) and (d), and the corresponding wavenumbers are provided in Table S2.† The pressure dependences of measured and calculated wavenumbers are plotted in Fig. 6. A factor group analysis predicts the presence of five Raman-active modes: Eg, Eg, B1g, B2g, and A1g. In the wavenumber range below 800 cm−1, the Raman spectrum of I4/mcm PbN2 at 50 GPa is distinguished by an intense and sharp mode at 130 cm−1 (Eg), accompanied by lower intensity satellite modes at 543 cm−1 (Eg) and 609 cm−1 (B1g).
N], the vibrational frequency increases, i.e. approximatively 900, 1300, and 2400 cm−1, respectively.7,71–75 The vibrational mode wavenumbers associated with the [N–N]4− stretching mode in group-14 element pernitrides of SiN2, GeN2, and SnN2 are observed around 1000–1100 cm−1.33 However, the Raman wavenumber of PbN2 is largely different from the other group-14 element pernitrides. What are the Raman active modes and spectra of the CuAl2-type PbN2? To give a response, we have conducted a study of the Raman spectra of the ground state I4/mcm PbN2 phase at 50 GPa using DFT calculations. The simulated Raman spectra are shown in Fig. 5(c) and (d), and the corresponding wavenumbers are provided in Table S2.† The pressure dependences of measured and calculated wavenumbers are plotted in Fig. 6. A factor group analysis predicts the presence of five Raman-active modes: Eg, Eg, B1g, B2g, and A1g. In the wavenumber range below 800 cm−1, the Raman spectrum of I4/mcm PbN2 at 50 GPa is distinguished by an intense and sharp mode at 130 cm−1 (Eg), accompanied by lower intensity satellite modes at 543 cm−1 (Eg) and 609 cm−1 (B1g).
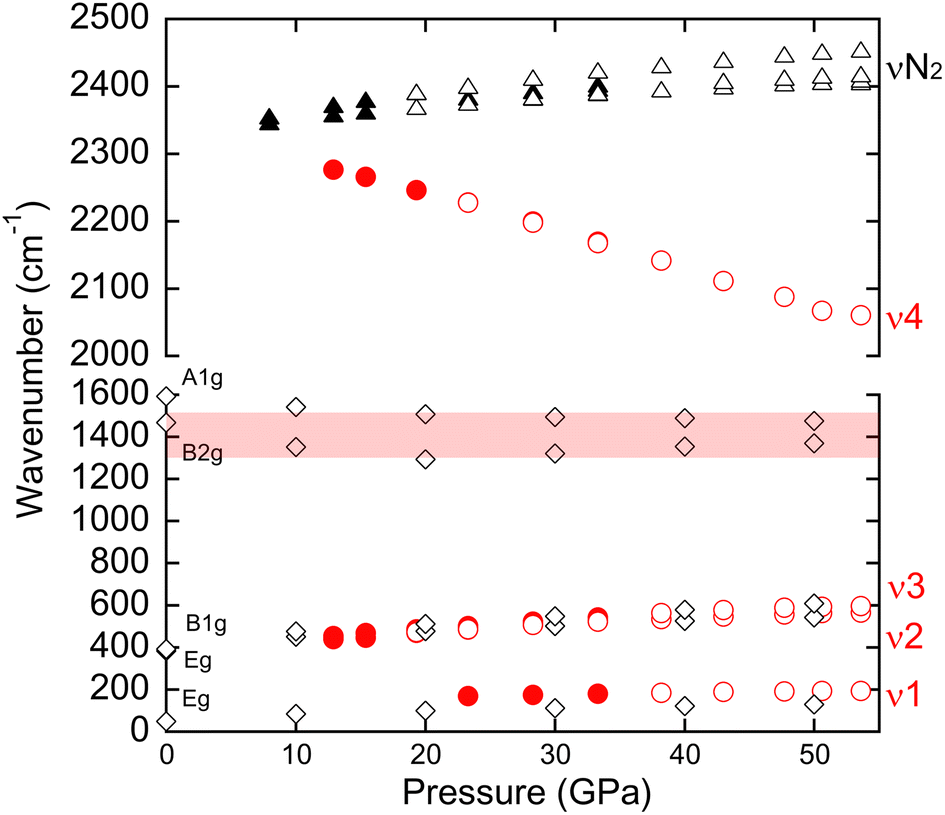 | ||
| Fig. 6 Pressure dependence of experimentally detected wavenumber (AG#2) together with wavenumbers of PbN2 obtained by DFT calculation. Open and solid circles correspond to the different positions (position 1 and position 2) on the same sample, respectively. The spectra of position 2 are shown in Fig. S7.† Open and solid triangles correspond to the wavenumbers of nitrogen from the same positions. The open diamond represents the wavenumbers of PbN2 obtained by DFT calculation. The data are listed in Table S2.† The Diamond-Raman band with the range of 1300–1500 cm−1 is hutched. | ||
As the pressure is reduced from 50 GPa to atmospheric pressure, the intensity of the Eg mode diminishes, and its wavenumber decreases from 130 cm−1 to 49 cm−1. This behavior is in complete accordance with the experimental Raman spectra, as illustrated in Fig. 5, 6, S6, and S7.† Conversely, the relative intensity of the B2g mode is observed to increase with a decrease in pressure, becoming the most intense and sharp mode at atmospheric pressure. Moreover, a slight shift of this B2g mode is observed when the pressure increases, from 1468 cm−1 to 1369 cm−1 at 0 and 50 GPa, respectively. It would be reasonable to posit that an increase in pressure would increase wavenumbers (and a concomitant decrease in lattice volume and bond lengths). However, the data indicate that this is not the case for both B2g and A1g modes, where the opposite effect is observed. In I4/mcm PbN2, the N–N bond length exhibits a slight but sensitive increase from 1.21 Å to 1.27 Å as the pressure rises from 0 to 100 GPa. This apparent anomaly in high-pressure chemistry can be attributed to the relative positioning of the 6s (Pb) level in relation to the antibonding NN π*orbitals.47 A 6s (Pb) band contribution is observed below the π* (N2) levels at atmospheric pressure. In contrast, at 50 GPa, the 6s (Pb) band is above the vacant π* (N2) levels. This alteration in electronic density is an unambiguous indication of the oxidation of Pb in the I4/mcm PbN2 as pressure increases (reduction when the applied external pressure tends to zero). Furthermore, this behavior can also account for the instability of PbN2 at atmospheric pressure, given that the Pb(2−δ)+ oxidation state is not thermodynamically favorable. However, the calculated B2g and A1g Raman-active bands located in the 1300–1600 cm−1 region are not visible in the experimental Raman spectra. This could be due to the essential technical problems of the DAC and the Raman band of approximately 1300–1500 cm−1 region is masked by the Diamond-Raman band (see Fig. S8†).
Moreover, the experimental band observed at around 2050 cm−1 is absent in the theoretical Raman spectra of I4/mcm PbN2 at 50 GPa. This band value is typically observed in activated N2 species with a bond order of 3 or slightly less.75 This DFT study demonstrates unequivocally that the measured high wavenumber of 2000–2300 cm−1 is not associated with the CuAl2-type I4/mcm PbN2. To modelize the high wavenumbers (2000–2300 cm−1) obtained from the samples, the need for a second model (or more) of the Pb–N system should be considered in which the N2 dimers undergo a minor activation process when mixing with electropositive metals. We propose two hypotheses in the following section.
The first one is that an increase in the N2/Pb ratio will result in a reduction in the negative charge of each anionic N2q− dimer, thus the +2 |e| charge given by Pb would be shared by several N2 dimers, decreasing the negative charge of N2q− dimer. Fortunately, some of us recently showed that stable Pb(N2)z phases comprising several N2 dumbbells per unit cell emerge on the convex hull of the Pb–N system when the pressure increases.47 We selected the nitrogen-rich R3![[c with combining macron]](https://www.rsc.org/images/entities/i_char_0063_0304.gif) Pb(N2)3 phase as a model of N2-rich compounds for the investigation of its Raman spectra. Hexagonal Rc has nine Raman-active modes with A1g calculated at 2175 cm−1 and Eg at 2041 cm−1 at 50 GPa (2154 and 2023 cm−1 at 10 GPa, respectively), as illustrated in Fig. S9.† Nevertheless, the wavenumbers slightly increased up to 50 GPa. It can thus be concluded that the N2-rich model in question, Rc Pb(N2)3, does not provide an adequate explanation of the experimental Raman spectra in the 2000–2300 cm−1 region. To account for the observed increase in Raman bands at lower pressures, it is necessary, at least, to consider a decrease in the bond order of N2, i.e. an increase in electron occupation of the antibonding N2 π* levels.47 This phenomenon is not observed in Rc Pb(N2)3 from 100 to 0 GPa, as in other high-pressure stable Pb(N2)z in Fig. S10,† which depicts the evolution of N–N interatomic distances upon pressure.
Pb(N2)3 phase as a model of N2-rich compounds for the investigation of its Raman spectra. Hexagonal Rc has nine Raman-active modes with A1g calculated at 2175 cm−1 and Eg at 2041 cm−1 at 50 GPa (2154 and 2023 cm−1 at 10 GPa, respectively), as illustrated in Fig. S9.† Nevertheless, the wavenumbers slightly increased up to 50 GPa. It can thus be concluded that the N2-rich model in question, Rc Pb(N2)3, does not provide an adequate explanation of the experimental Raman spectra in the 2000–2300 cm−1 region. To account for the observed increase in Raman bands at lower pressures, it is necessary, at least, to consider a decrease in the bond order of N2, i.e. an increase in electron occupation of the antibonding N2 π* levels.47 This phenomenon is not observed in Rc Pb(N2)3 from 100 to 0 GPa, as in other high-pressure stable Pb(N2)z in Fig. S10,† which depicts the evolution of N–N interatomic distances upon pressure.
The other hypothesis, among others, is in which N2 dimers are adsorbed on the crystallite surfaces of the samples. However, the DFT modeling of such PbN2 surfaces and N2 adsorption is beyond the scope of the present study. Further advanced study would find reasonable solutions for this issue, including ones other than mentioned above.
The present XRD, Raman measurements, and bonding analysis indicate a weak interaction between the Pb lattice and the nitrogen dimers at low pressure as evidenced by the decomposition of PbN2 into Pb and nitrogen molecules upon decompression. The recovered sample was characterized by scanning electron microscopy equipped with energy-dispersive spectroscopy (see Fig. S11†). The surface texture of the heated area was different from that of the unheated one and no nitrogen was detected anywhere. It is noteworthy that the CSP EA-DFT study indicated the ambient pressure recoverability of PbN2, Pb(N2)4, and Pb(cyclo-N5)2 based on the phonon mode analysis and ab initio molecular dynamic simulations at atmospheric conditions compounds.47 It is important to note that experimental metastability is dependent on several factors, including decompression speed, crystallinity, and other variables. Therefore, further rigorous experimentation with comprehensive analysis is essential to gain a deeper understanding of the Pb–N binary phase diagram under pressure.
To date, the CuAl2-type structure was found for TiN2 and VN2 compounds, which can be synthesized at pressure exceeding 70 GPa.13,19 The coordination number of cations in the CuAl2-type structure is eight, which is greater than that of the six in the pyrite-type structure, as shown in Fig. S12.† Considering the high-pressure phase transition analogy,35–40 this result suggests that a phase transition from pyrite-type to CuAl2-one may occur with increasing pressure. However, FeS2, a prototype of the pyrite structure, was revealed to be stable up to 80 GPa and 2400 K.76 For the high-pressure experiments on the intermetallic compound CrSb2, the CuAl2-type CrSb2 was synthesized above 5.5 GPa and 600 °C, and the boundary between the low pressure phase of marcasite-type, which have a similar crystallographic aspect to pyrite-type, and high pressure phase with CuAl2-type, was established.77 These experimental results indicate that pyrite-type structure is stable under wide pressure and temperature, and it seems difficult to find the compounds that transform directly from pyrite- to CuAl2-type structure, probably due to large rearrangement of atomic coordination including the increscent of coordination number of cations through the transition. Even in the case of pernitrides, the recent advanced high-pressure study revealed that pyrite-type SiN2 does not undergo a phase transition to a denser phase up to 140 GPa.34 As previously outlined, lead is a compressive element,56,57 and the influence of pressure on the chemical bonding may be amplified, resulting in the synthesis of PbN2, distinct from the pyrite-type group-14 pernitrides. Theoretical calculations have also indicated the potential for forming several new Pb(N2)z phases, contingent on the pressure.47 The recent findings from both experiments and calculations have significantly advanced our understanding of the high-pressure crystal chemistry of group-14 pernitrides. However, there are still some unclear issues, indicating a need for further advanced experimental and theoretical approaches to gain a deeper understanding of pernitrides material science.
Conclusions
The chemistry of the lead-nitrogen binary system was investigated under high pressure using a laser-heated diamond-anvil cell combined with synchrotron XRD measurements, Raman spectroscopy, and DFT modeling. A novel compound, PbN2, was successfully synthesized above 50 GPa, and crystallized in a tetragonal system having Pb atom bonded to 8N atoms and encapsulated anionic N2 dimers in Pb8 cages. Both experimental and DFT calculations analysis led to the CuAl2-type crystal of PbN2 at 50 GPa. This differs from the pyrite-form other group-14 pernitrides (SiN2, GeN2, and SnN2) that have been synthesized to date. This compound was found to remain stable up to 90 GPa at room temperature, however, it was unable to be recovered into ambient conditions. The presence of a Raman band at around 2000–2200 cm−1 remains unexplained and requires further experimental and theoretical investigation. The newly synthesized PbN2 offers new insight into the high-pressure crystal chemistry of nitrogen rich compounds in terms of the interaction between nitrogen dimer and host-lattice.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Author contributions
The manuscript was written through the contributions of all authors. All authors have approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was partially supported by JSPS KAKENHI (Grant Number 15H05542, 20K21080, 23K23039), and JST FOREST Program (JPMJFR213T). Synchrotron X-ray diffraction measurements were conducted at the BL2S1 of Aichi Synchrotron Radiation Center, Aichi Science & Technology Foundation, Aichi, Japan (Proposal Number 2016N1001, 2016N3001, 2019N4001, 2019N5011, 2023N3002, 2023N5002) with the financial and technical support of the Synchrotron Radiation Research Center, Nagoya University, and at the BL10XU SPring-8 (Proposal Number 2023B1301). The authors would like to thank Venture Business Laboratory, Nagoya University, for supporting the use of the Materials Studio package for DFT calculations. This research was made possible by two grants from the French National Research Agency (ANR) for postdoctoral fellowships S. P. (PRC TcPredictor, PRC Hydroptere). This work was partially supported by the European Union (ERDF), and Région Nouvelle Aquitaine (France). We thank the PHC Sakura 2019 (MEAE/MESR France and Japon) for supporting this France-Japon collaborative research. The authors acknowledge the High-Performance Computing Centers (HPC) of Irène/TGCC, and Jean Zay/IDRIS of GENCI (France) under projects no. A0140807539 and A0160815101 and “Grand Challenge 2023 TGCC” Session A14 spe00040 for generous allocations of computing time.References
- L. Zhang, Y. Wang, J. Lv and Y. Ma, Nat. Rev. Mater., 2017, 2, 17005 CrossRef CAS.
- H. B. Elisabeta, R. Riedel, A. Zerr, P. F. Mc Millan, G. Auffermann, Y. Prots, W. Bronger, R. Kniep and P. Kroll, Chem. Soc. Rev., 2006, 35, 987–1014 RSC.
- A. Zerr, R. Riedel, T. Sekine, J. E. Lowther, W. Y. Ching and I. Tanaka, Adv. Mater., 2006, 18, 2933–2948 CrossRef CAS.
- M. Rivers, V. Prakapenka, A. Kubo, C. Pullins, C. Holl and S. Jacobsen, High Pressure Res., 2008, 28, 273–292 Search PubMed.
- G. Shen and H. K. Mao, Rep. Prog. Phys., 2017, 80, 016101 CrossRef PubMed.
- A. Zerr, G. Miehe, G. Serghiou, M. Schwarz, E. Kroke, R. Riedel, H. Fueß, P. Kroll and R. Boehler, Nature, 1999, 400, 340–342 CrossRef CAS.
- J. C. Crowhurst, A. F. Goncharov, B. Sadigh, C. L. Evans, P. G. Morrall, J. L. Ferreira and A. J. Nelson, Science, 2006, 311, 1275–1278 CrossRef CAS PubMed.
- E. Gregoryanz, C. Sanloup, M. Somayazulu, J. Badro, G. Fiquet, H. K. Mao and R. J. Hemley, Nat. Mater., 2004, 3, 294–297 CrossRef CAS PubMed.
- A. F. Young, C. Sanloup, E. Gregoryanz, S. Scandolo, R. J. Hemley and H. Mao, Phys. Rev. Lett., 2006, 96, 155501 CrossRef PubMed.
- J. A. Montoya, A. D. Hernandez, C. Sanloup, E. Gregoryanz and S. Scandolo, Appl. Phys. Lett., 2007, 90, 011909 CrossRef.
- J. C. Crowhurst, A. F. Goncharov, B. Sadigh, J. M. Zaug, D. Aberg, Y. Meng and V. B. Prakapenka, J. Mater. Res., 2008, 23, 1–5 CrossRef CAS.
- W. P. Clark, S. Steinberg, R. Dronskowski, C. McCammon, I. Kupenko, M. Bykov, L. Dubrovinsky, L. G. Akselrud, U. Schwarz and R. Niewa, Angew. Chem., Int. Ed., 2017, 56, 7302–7306 CrossRef CAS PubMed.
- S. Asano, K. Niwa, T. Sasaki, N. A. Gaida and M. Hasegawa, Dalton Trans., 2022, 51, 2656–2659 RSC.
- M. Bykov, E. Bykova, E. Koemets, T. Fedotenko, G. Aprilis, K. Glazyrin, H. Liermann, A. V Ponomareva, J. Tidholm, F. Tasn, I. A. Abrikosov, N. Dubrovinskaia and L. Dubrovinsky, Angew. Chem., Int. Ed., 2018, 57, 9048–9053 CrossRef CAS PubMed.
- K. Niwa, T. Terabe, D. Kato, S. Takayama, M. Kato, K. Soda and M. Hasegawa, Inorg. Chem., 2017, 56, 6410–6418 CrossRef CAS PubMed.
- D. Laniel, A. Dewaele, S. Anzellini and N. Guignot, J. Alloys Compd., 2018, 733, 53–58 CrossRef CAS.
- M. Bykov, E. Bykova, G. Aprilis, K. Glazyrin, E. Koemets, I. Chuvashova, I. Kupenko, C. McCammon, M. Mezouar, V. Prakapenka, H.-P. Liermann, F. Tasnádi, A. V. Ponomareva, I. A. Abrikosov, N. Dubrovinskaia and L. Dubrovinsky, Nat. Commun., 2018, 9, 2756 CrossRef CAS PubMed.
- M. Bykov, S. Chariton, E. Bykova, S. Khandarkhaeva, T. Fedotenko, A. V. Ponomareva, J. Tidholm, F. Tasnádi, I. A. Abrikosov, P. Sedmak, V. Prakapenka, M. Hanfland, H. P. Liermann, M. Mahmood, A. F. Goncharov, N. Dubrovinskaia and L. Dubrovinsky, Angew. Chem., Int. Ed., 2020, 59, 10321–10326 CrossRef CAS PubMed.
- V. S. Bhadram, D. Y. Kim and T. A. Strobel, Chem. Mater., 2016, 28, 1616–1620 CrossRef CAS.
- K. Glazyrin, A. Aslandukov, A. Aslandukova, T. Fedotenko, S. Khandarkhaeva, D. Laniel, M. Bykov and L. Dubrovinsky, Front. Chem., 2023, 11, 1257942 CrossRef CAS PubMed.
- M. Ceppatelli, M. Serrano-Ruiz, M. Morana, K. Dziubek, D. Scelta, G. Garbarino, T. Poręba, M. Mezouar, R. Bini and M. Peruzzini, Angew. Chem., Int. Ed., 2024, 63, e202319278 CrossRef CAS PubMed.
- M. Ceppatelli, D. Scelta, M. Serrano-Ruiz, K. Dziubek, M. Morana, V. Svitlyk, G. Garbarino, T. Poręba, M. Mezouar, M. Peruzzini and R. Bini, Angew. Chem., Int. Ed., 2022, 61, e202114191 CrossRef CAS PubMed.
- K. Landskron, H. Huppertz, J. Senker and W. Schnick, Angew. Chem., Int. Ed., 2001, 40, 2643–2645 CrossRef CAS PubMed.
- L. Brüning, N. Jena, E. Bykova, P. L. Jurzick, N. T. Flosbach, M. Mezouar, M. Hanfland, N. Giordano, T. Fedotenko, B. Winkler, I. A. Abrikosov and M. Bykov, Angew. Chem., Int. Ed., 2023, 62, e202311519 CrossRef PubMed.
- F. L. Riley, J. Am. Ceram. Soc., 2000, 83, 245–265 CrossRef CAS.
- P. Yang, H. K. Fun, I. A. Rahman and M. I. Saleh, Ceram. Int., 1995, 21, 137–142 CrossRef CAS.
- H. L. Kornberg and H. A. Krebs, Nature, 1957, 180, 756–757 CrossRef PubMed.
- P. Popper and S. N. Ruddlesden, Nature, 1957, 179, 1129 CrossRef CAS.
- S. N. Ruddlesden and P. Popper, Acta Crystallogr., 1958, 11, 465–468 CrossRef CAS.
- G. Serghiou, G. Miehe, R. Boehler, O. Tschauner and A. Zerr, J. Chem. Phys., 2002, 111, 4659–4662 CrossRef.
- N. Scotti, W. Kockelmann and J. Senker, Z. Anorg. Allg. Chem., 1999, 624, 1435–1439 CrossRef.
- M. P. Shemkunas, G. H. Wolf, K. Leinenweber and W. T. Petuskey, J. Am. Ceram. Soc., 2002, 85, 101–104 CrossRef CAS.
- K. Niwa, H. Ogasawara and M. Hasegawa, Dalton Trans., 2017, 46, 9750–9754 RSC.
- P. L. Jurzick, G. Krach, L. Brüning, W. Schnick and M. Bykov, Acta Crystallogr., Sect. E:Crystallogr. Commun., 2023, 79, 923–925 CrossRef CAS PubMed.
- E. C. T. Chao, J. J. Fahey and J. Littler, J. Geophys. Res., 1962, 67, 419–421 CrossRef CAS.
- L. C. Ming and M. H. Manghnani, Phys. Earth Planet. Inter., 1983, 33, 26–30 CrossRef CAS.
- J. Haines and J. M. Léger, Phys. Rev. B, 1997, 55, 11144–11154 CrossRef CAS.
- J. Haines, J. M. Leger and O. Schulte, J. Phys.:Condens. Matter, 1996, 8, 1631–1646 CrossRef CAS.
- J. Haines, J. M. Léger and O. Schulte, Science, 1996, 271, 629–631 CrossRef CAS.
- Y. Kuwayama, K. Hirose, N. Sata and Y. Ohishi, Science, 2005, 309, 923–925 CrossRef CAS PubMed.
- Th. Curtius, Ber. Dtsch. Chem. Ges., 1891, 24, 3341–3349 CrossRef.
- M. Frank Douglas, J. Chem. Soc., 1931, 2532–2542 Search PubMed.
- L. V. AzÁroff, Z. Kristallogr., 1956, 107, 362–369 CrossRef.
- Y. Kuroiwa, S. Aoyagi, A. Sawada, J. Harada, E. Nishibori, M. Takata and M. Sakata, Phys. Rev. Lett., 2001, 87, 217601 CrossRef CAS PubMed.
- R. V. Shpanchenko, V. V. Chernaya, A. A. Tsirlin, P. S. Chizhov, D. E. Sklovsky, E. V. Antipov, E. P. Khlybov, V. Pomjakushin, A. M. Balagurov, J. E. Medvedeva, E. E. Kaul and C. Geibel, Chem. Mater., 2004, 16, 3267–3273 CrossRef CAS.
- A. A. Belik, M. Azuma, T. Saito, Y. Shimakawa and M. Takano, Chem. Mater., 2005, 17, 269–273 CrossRef CAS.
- S. Pitié, K. Niwa and G. Frapper, J. Phys. Chem. C, 2024, 128, 19410–19422 CrossRef.
- N. Watanabe, T. Nagae, Y. Yamada, A. Tomita, N. Matsugaki and M. Tabuchi, J. Synchrotron Radiat., 2017, 24, 338–343 CrossRef CAS PubMed.
- S. Kawaguchi-Imada, R. Sinmyo, K. Ohta, S. Kawaguchi and T. Kobayashi, J. Synchrotron Radiat., 2024, 31, 343–354 CrossRef CAS PubMed.
- Y. Seto, D. Nishio-Hamane, T. Nagai and N. Sata, Rev. High Pressure Sci. Technol., 2010, 20, 269–276 CrossRef.
- A. Boultif and D. Loue, J. Appl. Crystallogr., 2004, 37, 724–731 CrossRef CAS.
- F. Izumi and K. Momma, Solid State Phenom., 2007, 130, 15–20 CAS.
- H. K. Mao, J. Xu and P. M. Bell, J. Geophys. Res., 1986, 91, 4673 CrossRef CAS.
- C. S. Zha, W. A. Bassett and S. H. Shim, Rev. Sci. Instrum., 2004, 75, 2409–2418 CrossRef CAS.
- H. Olijnyk, J. Chem. Phys., 1990, 93, 8968–8972 CrossRef CAS.
- T. Takahashi, H. K. Mao and W. A. Bassett, Science, 1969, 165, 1352–1353 CrossRef CAS PubMed.
- H. K. Mao, Y. Wu, J. F. Shu, J. Z. Hu, R. J. Hemley and D. E. Cox, Solid State Commun., 1990, 74, 1027–1029 CrossRef CAS.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. I. J. Probert, K. Refson and M. C. Payne, Z. Kristallogr. - Cryst. Mater., 2005, 220, 567–570 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B, 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- A. Togo, J. Phys. Soc. Jpn., 2023, 92, 012001 CrossRef.
- A. Togo, L. Chaput, T. Tadano and I. Tanaka, J. Phys.: Condens. Matter, 2023, 35, 353001 Search PubMed.
- J. M. Skelton, L. A. Burton, A. J. Jackson, F. Oba, S. C. Parker and A. Walsh, Phys. Chem. Chem. Phys., 2017, 19, 12452–12465 RSC.
- https://github.com/skelton-group/Phonopy-Spectroscopy.
- E. Bykova, PhD thesis, University of Bayreuth, 2015, 1.
- L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786–790 CrossRef CAS.
- R. W. G. Wyckoff, Crystal Structures, Second., 1963, vol. 1 Search PubMed.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- M. I. Eremets, A. G. Gavriliuk, I. A. Trojan, D. A. Dzivenko and R. Boehler, Nat. Mater., 2004, 3, 558–563 CrossRef CAS PubMed.
- G. V Vajenine, G. Auffermann, Y. Prots, W. Schnelle, R. K. Kremer, A. Simon and R. Kniep, Inorg. Chem., 2001, 40, 4866–4870 CrossRef PubMed.
- S. B. Schneider, R. Frankovsky and W. Schnick, Inorg. Chem., 2012, 51, 2366–2373 CrossRef CAS PubMed.
- M. E. Fieser, D. H. Woen, J. F. Corbey, T. J. Mueller, J. W. Ziller and W. J. Evans, Dalton Trans., 2016, 45, 14634–14644 RSC.
- H. Schneider, W. Hiifner, A. Wokaun and H. Olijnyk, J. Chem. Phys., 1992, 96, 8046–8053 CrossRef CAS.
- E. C. Thompson, B. A. Chidester, R. A. Fischer, G. I. Myers, D. L. Heinz, V. B. Prakapenka and A. J. Campbell, Am. Mineral., 2016, 101, 1046–1051 CrossRef.
- H. Takizawa, K. Uheda and T. Endo, J. Alloys Compd., 1999, 287, 145–149 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental details; measured Raman spectra; XRD pattern and analysis results; scanning electron microscope image, additional calculations details with computational methods; calculated Raman spectra. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5ta01488c |
| This journal is © The Royal Society of Chemistry 2025 |