
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStoichiometric anion exchange by a low-dielectric-constant solvent for highly-doped conjugated polymers with enhanced environmental stability†
Daegun Kim‡
a,
Jiwoo Min‡b,
Kyeong-Jun Jeong‡c,
Eunsol Okb,
Jaemin Imd,
Hyun Ho Choid,
Giwon Lee e,
Chang Yun Son*c and
Kilwon Cho*b
e,
Chang Yun Son*c and
Kilwon Cho*b
aSchool of Chemical, Biological and Battery Engineering, Gachon University, Seongnam 13120, Korea
bDepartment of Chemical Engineering, Pohang University of Science and Technology, Pohang 37673, Korea. E-mail: kwcho@postech.ac.kr
cDepartment of Chemistry, Seoul National University, Seoul 08826, Korea. E-mail: changyunson@snu.ac.kr
dDepartment of Materials Engineering and Convergence Technology, Gyeongsang National University, Jinju 52828, Korea
eDepartment of Chemical Engineering, Kwangwoon University, Seoul 01899, Korea
First published on 23rd May 2025
Abstract
High-degree doping of conjugated polymers often employs a strong redox agent, which facilitates polymer ionization but results in poor environmental stability for the counter-ion. Here, we demonstrate an anion-exchange doping using a model study that systematically investigates the effect of the solvent dielectric constant on both doping and anion exchange. The dielectric constant significantly affects the initial doping of poly(2,5-bis(3-hexadecylthiophene-2-yl)thieno[3,2-b]thiophene) (PBTTT) films using FeCl3, as well as in the subsequent anion exchange of [FeCl4]− to dodecylbenzenesulfonate ([DBS]−). A solvent with a higher dielectric constant improves the FeCl3 doping efficiency but hinders the subsequent anion exchange. Such conflicting effects can be resolved by stepwise immersion in separate solutions of FeCl3 and dodecylbenzenesulfonic acid (DBSA). Stepwise anion-exchange doping achieves high electrical conductivity with improved environmental stability, while also allowing for the application of desired anions that require extended time for the direct doping method, such as in Brønsted acid doping.
1. Introduction
Conducting polymers (CPs) are promising candidates as active materials or electrodes for flexible electronics, offering advantages such as low processing costs and good mechanical flexibility.1,2 However, as-synthesized CPs often exhibit low carrier densities (n) and low carrier mobilities (μ), both of which cause very low electrical conductivities (σ).3 Doping CPs can increase n, thereby enhancing σ.4–7 Doping of CPs typically exploits a redox reaction to generate charge carriers in CPs via charge transfer with small dopant molecules.5,8,9 For efficient doping of CPs, the dopant should have a high redox potential for oxidation (p-type) or reduction (n-type) of CPs4,10,11 and a sufficiently small size for facile incorporation in CPs.12–14 For example, polyacetylene exhibited an excellent σ ∼ 105 S cm−1 by iodine doping, for which the Nobel prize in chemistry was granted in 2000.15 However, such a small size of atomic dopant resulted in poor environmental stability of the counter-ion which is readily degraded in the CP film by various environmental factors: heat or reaction with oxygen or water molecules.16 Hence, molecular dopants, such as FeCl3,17,18 2,3,5,6-tetrafluoro-tetracyanoquinodimethane (F4TCNQ),14,19,20 and tetramethylhydroquinone (TMQH),21 have been developed to induce a stable and high-degree doping of CPs. Nonetheless, the sizes of such molecular dopants are made to be sufficiently small, representing the trade-off between doping efficiency (diffusion into CPs) and environmental stability (degradation in CPs).12Recently, H. Sirringhaus and coworkers reported a high degree of doping in CPs using ion-exchange doping, in which the counter-ions formed by the redox reaction of the dopant molecules were subsequently replaced by anions from an added ionic liquid.22,23 It was claimed that the redox potential of the dopant molecule serves as the driving force for anion exchange in a manner analogous to the applied electrical potential in electrochemical doping. Doping of CPs with simultaneous counter-ion exchange offers insight into how the trade-off between doping efficiency and environmental stability when using a single dopant molecule can be resolved. However, the reported ion-exchange doping methods homogeneously added molecular dopants and ionic liquids using the same solvent, necessitating the use of a highly polar solvent that dissolves the molecular dopants and stabilizes their redox reactions.5,17,24–26 The polar solvent also limits the choice of replaceable ions, as the solubility in the polar solvent requires an ionic salt to have high polarity. An effective counter-ion should have a wide electrochemical window to prevent charge transfer from the counter-ion back to CPs, good miscibility with CPs,22 and a sufficiently large size for environmental stability.27 To meet all these requirements in a single counter-ion, the counter-ion should possess not only an electrochemically stable ionic functional group but also a considerably large hydrocarbon, which results in poor solubility in polar solvents.28 However, using a solvent with a low dielectric constant to dissolve such ions provides insufficient stabilization for the charged species after the redox reaction, thus suppressing the carrier density increment upon doping of CPs.22,29 The mismatch of the required solvent dielectric constant for the dopant and the counter-ion cannot be resolved in homogeneous doping.
In this work, ion-exchange doping was conducted through a two-step process using two separate solutions for the redox reaction and the ion exchange, respectively. By separating the redox reaction and the ion exchange, different solvents can be adopted for each process. To systematically study the optimal solvent conditions for these processes, solvents with varied dielectric constants were employed to investigate the effect of solvent on the environment around the charge carriers and anions during the anion-replacement step in a model system. Dodecylbenzenesulfonic acid (DBSA) was selected for the model study, as the ionized dodecylbenzenesulfonate ([DBS]−) is a decent counter-ion exhibiting good miscibility in solvents with a wide range of dielectric constants. DBSA can also directly oxidize CPs via Brønsted-acid doping, allowing for an evident comparison between stepwise ion-exchange doping and direct doping. Poly(2,5-bis(3-hexadecylthiophen-2-yl)thieno[3,2-b]thiophene) (PBTTT) films were initially doped using FeCl3 and subsequently immersed in DBSA diluted in solvents with varying dielectric constants. Quantitative evaluation of carrier densities and computational calculations on the binding state between the CP and counter-ion revealed that a partially polar solvent was favored for the redox reaction, whereas a non-polar solvent was favored for Brønsted-acid doping and anion exchange. With suitable solvents, the stepwise ion-exchange doping not only facilitated the incorporation of bulky and non-polar counter-ions into CPs but also achieved electrical conductivity comparable to that obtained by conventional direct doping.
2. Results and discussion
The solvent effect during anion exchange was systematically investigated in a model system, where PBTTT film was first doped by FeCl3, and then the [FeCl4]− anion was sequentially exchanged for the [DBS]− anion under various solvent conditions. PBTTT is a solution-processable conjugated polymer widely used in semiconducting devices, such as field-effect transistors, chemical sensors and thermoelectric generators, because of its high carrier mobility30–33 (Fig. 1a). The electrical conductivity of PBTTT can be readily enhanced when the polymer backbone is charged through a redox reaction with an added dopant. PBTTT is typically oxidized to achieve p-type character by either solution-mixing or sequential doping with strong oxidants. FeCl3 is a commonly used oxidant for p-type doping as it forms a relatively stable CP-anion pair. DBSA is a strong Brønsted acid that protonates polymer or organic solvent, leaving the [DBS]− anion. The benzene ring in the [DBS]− anion donates electrons, so the [DBS]− anion can experience significant stabilization when paired with a positive polaron. Therefore, competition between [DBS]− and [FeCl4]− anions as counter-ions is expected when DBSA is sequentially applied to FeCl3-doped PBTTT film. DBSA is also soluble in both polar and non-polar solvents due to the coexistence of a polar sulfonic acid group and a long alkyl chain.34 Hence, DBSA is suitable for use in the model study to evaluate the effect of various solvents across a wide range of dielectric constants (Fig. 1b).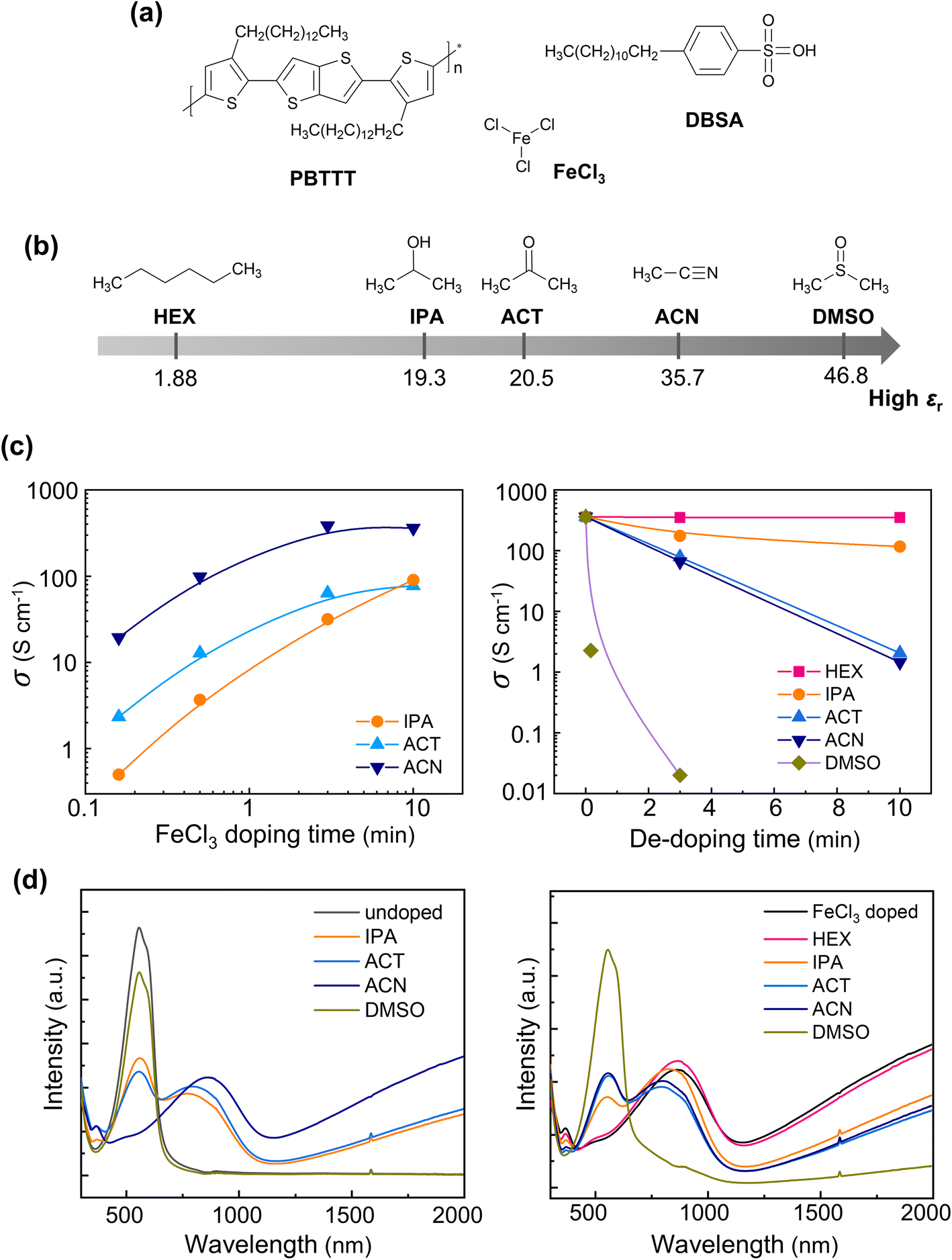 | ||
| Fig. 1 (a) Chemical structures of PBTTT, FeCl3, and DBSA. (b) Chemical structures of solvents with different dielectric constants εr. (c) The electrical conductivity σ and (d) the UV-vis-NIR spectra of PBTTT films after FeCl3 doping with various solvents (left) and FeCl3-doped PBTTT films rinsed (dedoped) with various solvents (right). | ||
To evaluate the ion-exchange behavior, PBTTT films were first doped by immersing spin-coated PBTTT films into an FeCl3 solution for the desired time. The polar FeCl3 can only be dissolved in polar solvents. FeCl3 doping induced higher electrical conductivity in PBTTT films with acetonitrile (ACN) compared to isopropanol (IPA) or acetone (ACT) (Fig. 1c). The electrical conductivity of PBTTT films barely exceeded 0.1 S cm−1 when the FeCl3 solution was prepared with dimethyl sulfoxide (DMSO), which has the highest dielectric constant among the solvents used in this study. PBTTT films doped by FeCl3 in ACN were then immersed in pure solvents to assess the stability of the [FeCl4]− anion. The electrical conductivity of the PBTTT films dramatically decreased with increasing dielectric constant of the solvent. The UV-vis-NIR absorption spectra of the PBTTT films supported the electrical measurements (Fig. 1d). The pristine PBTTT film exhibited a notable absorption at 560 nm. Upon doping, this absorption decreased while new absorptions appeared at 800 nm and broadly at wavelengths over 1200 nm. These absorptions correspond to the neutral PBTTT chain, charged PBTTT chain, and generated polaron, respectively.35 Doping with FeCl3 in ACN, which produced the highest electrical conductivity among the solvents used, almost completely suppressed the absorption of the neutral PBTTT chain and resulted in the strongest absorptions for the charged PBTTT chain and polarons, compared to the other solvents. This result was consistent with the electrical conductivity results. Rinsing the doped PBTTT films with solvents led to the opposite effect in the UV-vis-NIR spectra. Rinsing with polar solvents, such as IPA, ACT, ACN, and DMSO caused decreases in the absorptions associated with the charged PBTTT chain and polarons, while increasing the absorption of the neutral PBTTT chain. On the other hand, rinsing with the non-polar solvent hexane (HEX) did not cause significant changes in either the electrical conductivity or the UV-vis-NIR absorption spectrum of the FeCl3-doped PBTTT film.
The effect of the dielectric constant on doping efficiency is primarily related to the stabilization of the oxidized PBTTT-anion pair and the swelling of the PBTTT film during sequential doping.29 A solvent with a higher dielectric constant can enhance doping by stabilizing ion species, including oxidized PBTTT and ionized dopant molecules, while a solvent with a lower dielectric constant can cause significant swelling of the PBTTT film, promoting the diffusion of dopant molecules. Hence, the dielectric constant should be carefully selected for a specific dopant-CP system. PBTTT films were not effectively doped by FeCl3 in DMSO, which has a higher dielectric constant than the other solvents. The poor doping efficiency in DMSO was attributed to the poor solubilization of PBTTT in DMSO, which exhibited a higher contact angle on the PBTTT surface compared to the other solvents (Fig. S1†). The strong coordinating ability of DMSO may also contribute to the low electrical conductivity, as it can reduce the oxidizing power of FeCl3. On the other hand, dedoping of the doped PBTTT films through solvent rinsing became progressively stronger when a solvent with a higher dielectric constant was used. This result indicates that the [FeCl4]− anion is unstable and readily loses coulombic interaction with the [PBTTT]+ chain in polar environments. Such poor stability of the counter-ion in polar environments is common for various molecular dopants due to their strong polarity indices and small sizes.36 Therefore, the counter-ion should be replaced with an ion with low polarity and large size to improve the stability of the doped state in the PBTTT film.
The PBTTT film was first doped with FeCl3, which triggered the redox reaction with PBTTT. The FeCl3-doped PBTTT film was then immersed in a DBSA solution for a desired duration to induce anion exchange between the unstable [FeCl4]− anion and the [DBS]− anion (Fig. 2a). The electrical conductivity of the PBTTT film was around 100 S cm−1 after doping with FeCl3 in ACN for 30 s, during which only a small portion of the PBTTT repeat units was oxidized, resulting in a relatively low charge carrier density. The resulting electrical conductivity after doping would be primarily determined by the doping efficiency, as the PBTTT morphology did not vary significantly depending on the solvent (Fig. S2†). The sequential ion exchange following 30 s of FeCl3 doping was significantly affected by the solvent choice (Fig. 2b). When immersed in a DBSA solution with polar solvents, the FeCl3-doped PBTTT film exhibited a substantial decrease in electrical conductivity, with the decrement being larger for solvents with a higher dielectric constant. Conversely, the electrical conductivity of the FeCl3-doped PBTTT film increased following DBSA treatment in HEX. An increase in electrical conductivity upon DBSA treatment indicated additional doping by DBSA, which presumably generates charge carriers through protonation of the PBTTT chain.37,38 The results confirmed that the dielectric constant was a primary property affecting both doping and the anion exchange process, though the electrical conductivity after DBSA treatment was not completely proportional to the dielectric constants of the solvents used. This minor inconsistency was possibly attributed to solvent properties such as pKa, catalytic effect, and stabilization of protonation, all of which are related to direct doping by DBSA. The direct doping by DBSA will be examined in the final section. Similar results were obtained for DBSA treatment on highly doped PBTTT films, which had undergone FeCl3 doping for 3 min (Fig. 2c). The electrical conductivity of the highly doped PBTTT film decreased from 350 S cm−1 to below 10 S cm−1 after DBSA treatment in polar solvents, whereas only a slight decrease in the electrical conductivity was observed for DBSA treatment in HEX. The results of the DBSA treatment were analogous to those of solvent rinsing, where polar solvents significantly removed polarons and ionized dopants from the PBTTT film, while the non-polar solvent did not.
 | ||
| Fig. 2 (a) Schematic process of sequential ion-exchange doping. The σ values of PBTTT films after doping of FeCl3 (ACN) for (b) 30 s or (c) 3 min followed by the DBSA treatment in various solvents. (d) XPS Cl 2p and S 2p spectra, (e) UV-vis-NIR absorption spectra, and (f) GIWAXS patterns along the qz direction of FeCl3-doped (dotted) and FeCl3/DBSA-doped (solid, color) PBTTT films. FeCl3 was doped with CAN for 3 min. The DBSA solution for the anion exchange was prepared with the labelled solvent. The DBSA treatment time was 1 min, unless it is specified (10 s or 60 s). | ||
The anion exchange between the [FeCl4]− and [DBS]− anions was further investigated using X-ray photoelectron spectroscopy (XPS) (Fig. 2d). The as-doped PBTTT film exhibited notable Cl 2p peaks originating from [FeCl4]− anions. Two distinguishable peaks were attributed to spin–orbit splitting. When the FeCl3-doped PBTTT film underwent anion exchange by DBSA treatment, the Cl 2p peaks disappeared regardless of the solvent used, indicating that DBSA treatment completely removed the [FeCl4]− anions from the PBTTT film by inducing anion exchange or by annihilating polarons (Fig. S3†). On the other hand, DBSA treatment generated new peaks in the S 2p spectra. The as-doped PBTTT film exhibited two distinct S 2p peaks at binding energies between 162 and 166 eV, which arose from the S atoms in the thiophene and thienothiophene rings in the PBTTT chain.22 The chemical environments around S atoms in the thiophene and thienothiophene are very similar, so their S 2p peaks are hardly distinguishable. The existence of two peaks were again attributed to spin–orbit splitting.39 Anion exchange by DBSA yielded new S 2p peaks at binding energies between 167 and 170 eV, which originated from the sulfonate group in the [DBS]− anions.40 These peaks were more intense when a non-polar solvent was used compared to a polar solvent. Considering the electrical conductivities after DBSA treatment, the XPS results provided direct evidence that the [DBS]− anions replaced the [FeCl4]− anions to stabilize oxidized PBTTT chains. The XPS results also suggested that polar solvents washed away the [FeCl4]− anions and reduced the oxidation level of the PBTTT chain, rather than inducing a stoichiometric replacement of the [FeCl4]− anions with the [DBS]− anions.
The UV-vis-NIR absorption spectra also confirmed that DBSA treatment in a polar solvent decreased the oxidation level of the PBTTT chain (Fig. 2e). As observed during pure solvent rinsing, the use of a polar solvent for DBSA treatment caused a significant increase in absorption by the neutral PBTTT chain at 560 nm, along with decreases in absorption by the oxidized PBTTT chain at 800 nm and broad absorption by polarons at wavelengths over 1200 nm. The reduction in the oxidation level of the PBTTT chain by anion exchange was greater when a solvent with a higher dielectric constant was used. On the other hand, the content of the [DBS]− anions in PBTTT films showed a relatively weak correlation with the dielectric constant of the solvent used for DBSA treatment. For example, according to the XPS spectra (Fig. 2d, right), a higher [DBS]− content was expected for ACN compared to IPA or ACT, both of which have lower dielectric constants than ACN. The mismatch between XPS and UV-vis-NIR spectra suggested that physically adsorbed DBSA in polar circumstances did not fully dissociate to form [DBS]− anions that could replace [FeCl4]− anions.
The incorporation of the [DBS]− anion in the crystalline PBTTT domain was investigated using grazing-incidence wide-angle X-ray scattering (GIWAXS) (Fig. 2f and S4†). The GIWAXS patterns of undoped and doped PBTTT films showed several (h00) peaks that represent the lamellar spacing (d spacing) between PBTTT lamellae along the out-of-plane direction. FeCl3 doping shifted the (100) peak to a lower q value, indicating an increase in the d spacing as a result of incorporating [FeCl4]− anions between the PBTTT lamellae. FeCl3 doping increased the d spacing from 20.9 to 24.1 Å. Anion exchange in all solvents, except for DMSO, shifted the (100) peak further to lower qz values along the out-of-plane direction, indicating that DBSA or [DBS]− anions were incorporated between PBTTT lamellae, thus expanding the d spacing. When DMSO was used as the solvent for DBSA treatment, it effectively washed away the incorporated [FeCl4]− anions instead of replacing them with [DBS]− anions, causing the expanded PBTTT lamellar structure to shrink back, resulting in a shift of the (100) peak to higher qz values. The degree of lamellar expansion in FeCl3-doped PBTTT films by anion exchange depended on the solvent and the immersion duration. DBSA treatment with HEX as the solvent (DBSA/HEX treatment) increased the d spacing from 24.1 to 27.6 Å after 10 s of immersion. The d spacing was further increased with a longer immersion time of 60 s. In contrast, DBSA/IPA, DBSA/ACT, and DBSA/ACN treatments increased the d spacing after 10 s of immersion, but the d spacing decreased with longer immersion of 60 s.
Note that the lamellar expansion after 10 s of DBSA treatment monotonically decreased with increasing dielectric constant. The lamellar expansion upon DBSA treatment indicated the incorporation of DBSA or [DBS]− ions between the PBTTT lamellae. Considering the short immersion time, the diffusion of DBSA into the PBTTT film would predominantly affect the lamellar expansion, rather than the anion exchange rate. Therefore, the lamellar expansion by the DBSA treatment for 10 s would increase with greater miscibility with PBTTT, whose long alkyl chain interacts strongly with solvents of low dielectric constants. With extended DBSA treatment, the lamellar expansion by DBSA incorporation increased in non-polar environments, whereas the use of a polar solvent led to lamellar shrinkage.
The observed lamellar shrinkage in polar environments indicated that a significant portion of the DBSA intercalated with the PBTTT film diffused out without undergoing anion exchange. The diffusion of DBSA in and out of the PBTTT film may have originated from the rapid escape of the [FeCl4]− anion, whose redox potential acted as a driving force to promote DBSA diffusion into the PBTTT film.22 When the DBSA solution first contacts the PBTTT surface, DBSA diffuses into the PBTTT film due to the strong electrochemical potential exerted by the embedded [FeCl4]− anions. Non-polar solvents did not dissolve the [FeCl4]− anion, which led to continued diffusion of DBSA and an increased probability of anion exchange with increasing treatment time. In contrast, polar solvents caused the [FeCl4]− anions to escape quickly from the PBTTT film, reducing both the driving force for DBSA diffusion and the probability of anion exchange over treatment time. The DBSA treatment using ACT as the solvent exhibited relatively small lamellar shrinkage after 60 s of treatment, possibly due to the stabilization of DBSA within the PBTTT film via hydrogen bonding. Though a considerable amount of DBSA was expected to remain between the crystalline PBTTT lamellae, the decrease in electrical conductivity after the DBSA/ACT treatment suggested that most DBSA molecules were unionized.
The solvent effect on the anion exchange was further investigated through quantitative characterization of the carrier mobility and carrier density of PBTTT films. The carrier mobility of doped PBTTT films was measured using a four-point probe field-effect transistor (FET) configuration, according to our previous work41 (Fig. 3a). Briefly, four electrodes were deposited in a line with equal spacing onto a SiO2/Si wafer, followed by spin-coating of the PBTTT film and subsequent doping and anion exchange processes (Fig. 3b). Among the four electrodes, the outer two served as the source and drain, where the drain voltage VSD was applied, while the inner two acted as probes to measure the voltage drop V4p, thereby excluding the voltage drops caused by Schottky-barrier contact resistance from the calculation of carrier mobility. The FET mobility μ2p in the conventional source-drain (two-point probe) configuration is given by the following equation:
 | (1) |
 | (2) |
 where t is the thickness of the PBTTT film. This equation is identical to that used in a conventional four-point probe measurement to determine conductivity. The doped PBTTT film had a very high carrier density, which showed a noticeable linear correlation with the applied VG in the range between −100 and +100 V, despite a small change induced by the field effect. The electrical conductivity of the PBTTT film was measured using both two- and four-point probe methods with respect to VG. The conductivity σ2p in the two-probe configuration is given by
where t is the thickness of the PBTTT film. This equation is identical to that used in a conventional four-point probe measurement to determine conductivity. The doped PBTTT film had a very high carrier density, which showed a noticeable linear correlation with the applied VG in the range between −100 and +100 V, despite a small change induced by the field effect. The electrical conductivity of the PBTTT film was measured using both two- and four-point probe methods with respect to VG. The conductivity σ2p in the two-probe configuration is given by  The value of σ2p was far smaller than that of σ4p, implying a considerable voltage loss due to contact resistance (Fig. 3c). Carrier mobilities were obtained from the slope of σ2p(σ4p) vs. VG in the two-point probe (four-point probe) configuration as follows:
The value of σ2p was far smaller than that of σ4p, implying a considerable voltage loss due to contact resistance (Fig. 3c). Carrier mobilities were obtained from the slope of σ2p(σ4p) vs. VG in the two-point probe (four-point probe) configuration as follows:
 | (3) |
 | ||
| Fig. 3 (a) and (b) The device structure for four-point probe FET measurement where L2p = 1000 μm, L4p = 500 μm and W = 500 μm. (c) Obtained σ values of FeCl3-doped (left) and FeCl3/DBSA-doped (right) PBTTT films by performing two-point and four-point probe FET measurement. (d) The carrier mobility μ4p obtained in four-point probe FET (column) and the carrier density n (line) of FeCl3-doped and FeCl3/DBSA-doped PBTTT films. (e) Changes in σ of FeCl3-doped and FeCl3/DBSA-doped PBTTT films at room temperature with relative humidity (RH) of 20%. (f) (100) peaks along the out of plane direction of FeCl3-doped and FeCl3/DBSA-doped PBTTT films with and without exposure to humid conditions (RH = 50%) for 1 h. FeCl3 was doped with ACN for 3 min. The DBSA was applied with HEX as the solvent unless it is specified. | ||
The FeCl3-doped PBTTT film exhibited carrier mobilities of μ2p = 10.1 ± 0.01 cm2 V−1 s−1 and μ4p = 11.9 ± 0.06 cm2 V−1 s−1. The FeCl3-doped PBTTT film after the DBSA/HEX treatment (FeCl3/DBSA-doped PBTTT film) had mobilities of μ2p = 8.4 ± 0.03 cm2 V−1 s−1 and μ4p = 10.8 ± 0.02 cm2 V−1 s−1. The difference in carrier mobilities of these PBTTT films was 17% and 9% by the two-point and four-point probe measurements, respectively. The enlarged difference in carrier mobilities in the two-point probe measurement originated from the underestimation of the carrier mobility at an increased contact resistance.
The carrier mobilities of PBTTT films were therefore obtained using the four-point probe FET measurement for explicit comparison. The carrier density n of a PBTTT film was calculated from the conductivity σ and μ4p; i.e., n = σ/eμ4p, where e is the charge of an electron. PBTTT film was highly-doped, so using FET mobility (μ4p) would not make a significant error in estimating n. Both n and μ4p of the FeCl3-doped PBTTT film were largely maintained after the DBSA/HEX treatment (Fig. 3d). In contrast, the DBSA treatment with a polar solvent critically decreased both n and μ4p. The decrease in μ4p associated with dedoping was attributed to a reduction in the probability of activation, in which a charge carrier has sufficient energy to reach an extended number of accessible states. This occurred because the density of states (DOS) near the Fermi level decreased dramatically as the Fermi level shifted toward the forbidden region as a result of dedoping.42 The decrease in both n and μ4p upon DBSA treatment in polar environments underscored the importance of solvent selection in anion exchange processes.
With the proper choice of solvent for each doping and anion exchange step, the FeCl3/DBSA-doped film showed improved environmental stability compared to the FeCl3-doped PBTTT film (Fig. 3e). The electrical conductivities of FeCl3- and FeCl3/DBSA-doped PBTTT films decreased upon exposure to ambient conditions at room temperature with 20% humidity, because of polaron annihilation and counter-ion solvation by chemicals in the ambient air. However, anion exchange from the [FeCl4]− to the [DBS]− anion suppressed the decrease in electrical conductivity, reducing the change by almost half after 24 h of exposure to ambient conditions. It is noteworthy that almost 80% of electrical conductivity was preserved after 24 h exposure to ambient conditions with anion replacement, considering doped conjugated polymer films often lose significant portions of their electrical conductivity upon exposure to ambient conditions for a few hours.43 Exposure to humid conditions with 50% humidity for 1 h shifted the (100) peaks of FeCl3- and FeCl3/DBSA-doped PBTTT films to higher values, indicating lamellar shrinkage caused by the escape of counter-ions from the PBTTT film due to solvation by water molecules (Fig. 3f). The anion exchange from the [FeCl4]− to the [DBS]− anion also mitigated the shift of the (100) peak, suggesting that the [DBS]− anion improved the water resistance of the doped PBTTT film, possibly due to the bulky and non-polar alkyl chain of the [DBS]− anion.
To elucidate the mechanism of the anion exchange process, the effect of the solvent dielectric constant on the binding of anions to polarons and on the doping level was evaluated using DFT calculations. Benzenesulfonate [BS]− and C0-BTTT were used as surrogates for the [DBS]− ion and PBTTT, respectively, to maintain computational tractability without significantly deviating from the real chemical environment (see Fig. S5 and S6 in ESI Section 2† for details). The energy level of the [BTTT]+–[FeCl4]− pair or the [BTTT]+–[BS]− pair decreased considerably when the intermolecular distance was reduced from an infinitely large distance (separated) to an optimized distance (Fig. 4a). The energy decrement upon ion-pair formation corresponds to the binding energy, which represents the stabilization of charged species via coulombic interaction. A larger binding energy of the [BTTT]+–[BS]− pair than that of the [BTTT]+–[FeCl4]− pair indicated that the difference in binding energy acted as the driving force of anion exchange. Notice that the binding energies of the [BTTT]+–[BS]− and [BTTT]+–[FeCl4]− pairs and their difference ΔEex both diminished in DMSO compared to the values in HEX. The reduced binding energies in a polar environment can be attributed to the stabilization of charged species by the solvent. The weakening of [BTTT]+–[FeCl4]− binding in a polar environment would promote the escape of the [FeCl4]− anion from the polaron. The dissociation of the [FeCl4]− anion can initiate the anion exchange process, but the reduction in ΔEex in a polar solvent weakened the driving force for anion exchange, thereby requiring a longer time in a polar solvent compared to a non-polar solvent. As a result, in a polar solvent, the [FeCl4]− anion would quickly dissociate from the polaron prior to the completion of anion exchange.
 | ||
| Fig. 4 Structures and energetics of polaron-dopant anion interactions calculated by density functional theory (DFT). (a) Energy diagram for the dopant ion exchange between [BTTT]+–[FeCl4]− and [BTTT]+–[BS]− in hexane and DMSO. (b) Polaron-anion binding energies (ΔE([BTTT]+–[BS]−) − ΔE([BTTT]+–[FeCl4]−)) for the stepwise doping processes with respect to the solvent's dielectric constant. (c) Optimized geometries and binding energies of π-stacked [BTTT]3+ paired with [FeCl4]− (left) and [BS]− in HEX (right) in a front view. In panel (c), the dotted lines and numbers illustrate the interatomic distance in units of Å. | ||
The binding energies of the [BTTT]+–[FeCl4]− and [BTTT]+–[BS]− pairs, along with their difference ΔEex, decreased monotonically with increasing dielectric constant for all investigated solvents (Fig. 4b). The binding energy of the [BTTT]+–[FeCl4]− pair had the strongest correlation with the dielectric constant, even between ACN and DMSO, both of which exhibited similar binding energies for the [BTTT]+–[BS]− pair and ΔEex values. In conjunction with a dramatic decrease in the electrical conductivity of the FeCl3-doped PBTTT film after DBSA/DMSO treatment compared to DBSA/ACN treatment, the DFT calculations suggest that the rapid dissociation of the [FeCl4]− anion from the polaron in a polar solvent played a dominant role in causing dedoping instead of anion exchange during the DBSA treatment.
The DFT-optimized geometries of the polaron-anion dimers revealed that, in all investigated solvents, both [FeCl4]− and [BS]− anions preferentially bound to the side of the thienothiophene ring in [BTTT]+ (Fig. 4c). Consequently, the two anions were expected to compete for binding to the same site on the [BTTT]+ during the anion exchange, resulting in the replacement of the more weakly bound [FeCl4]− anion. This dopant-ion binding motif predicted by our DFT model aligns with the results of an experimental study on FeCl3-doped C12-PBTTT, where the dopant ionic species were observed to intercalate into the space between the lamellae of a C12-PBTTT crystal. We note that the predicted orientation of anion binding did not interfere with the π–π stacking of PBTTT chains, and the relative binding strengths of the anions to polarons were consistent in DFT calculations considering either a single chain or three stacked chains of BTTT (Fig. S7†), as well as in auxiliary DFT calculations with extended BTTT trimer systems to eliminate the artifact caused by confining the polaron charge in a monomer of BTTT (Fig. S8–S10†). The anion binding orientations and the exothermicity of ion exchange in the trimeric polaron systems were consistent with the results for the monomeric polaron systems, supporting the validity of the molecular physical arguments for the monomeric systems.
Finally, the anion exchange process was compared to direct doping. DBSA itself can generate charge carriers when added to PBTTT film. Although the mechanism of charge generation by DBSA has not been clearly elucidated, a recent report on the doping mechanism of poly[2,6-(4,4-bis(2-hexadecyl)-4H-cyclopenta[2,1-b;3,4-b′]dithiophene)-alt-4,7(2,1,3-benzothiadiazole)] (PCPDTBT) by the tris(pentafluoro-phenyl)borane–H2O (BCF–H2O) complex suggests that charge generation may occur via Brønsted-acid doping,37 in which protonation oxidizes the conjugated polymer backbones. When a PBTTT film was doped by immersion in a DBSA solution with HEX as the solvent, the electrical conductivity of the PBTTT film gradually increased, reaching a value comparable to that achieved by FeCl3 doping after an immersion time of 5 h (Fig. 5a). Such doping of conjugated polymers via direct protonation was unlikely to occur in doping via anion exchange, because the [FeCl4]− anion would quickly react with the protonated conjugated polymer, forming FeCl3 and HCl.44 The very short time (<1 min) required for DBSA doping via anion exchange suggests that PBTTT underwent negligible protonation, which would otherwise require a much longer time (>1 h) to induce a noticeable enhancement in electrical conductivity.
 | ||
| Fig. 5 (a) σ of FeCl3(ACN)/DBSA(HEX)-doped and DBSA-doped PBTTT films with respect to the doping time. The inset is the magnification for σ of FeCl3(ACN)/DBSA(HEX)-doped PBTTT films with a doping time < 5 min. Chemical processes of (b) anion-exchange doping and (c) direct Brønsted acid doping. | ||
The choice of solvent significantly affected both direct doping and anion exchange. The use of a non-polar solvent improved the electrical conductivity of PBTTT films doped by both anion exchange and direct doping. However, doping via anion exchange required an immersion time of less than 10 min for each dipping in FeCl3 and DBSA solutions, whereas the direct doping by DBSA did not exhibit a plateau in the electrical conductivity after immersion in the DBSA solution for 5 h. Stepwise doping via anion exchange effectively shortened the overall processing time by using a strong redox agent (FeCl3) to oxidize the conjugated polymer and form an intermediate ion-pair, where the anion was vulnerable to environmental factors. The subsequent exchange of this anion with a bulky ion formed a stable ion-pair with a strong binding state (Fig. 5b). In contrast, doping directly such a bulky ion via Brønsted-acid doping required a much longer time to oxidize the conjugated polymer due to its low redox potential (Fig. 5c). Interestingly, the best solvent to achieve a high electrical conductivity varied for each step: FeCl3 doping was most effective with a partially polar solvent (ACN), while anion exchange with DBSA was favored with a non-polar solvent (HEX). The model study using DBSA emphasizes the importance of choosing the proper solvent, potentially opening new possibilities of achieving both high doping levels and a stable doped state in organic semiconductors.
3. Conclusion
We demonstrated sequential ion-exchange doping of PBTTT film using FeCl3 and a Brønsted acid, DBSA, in solvents with various dielectric constants. The molecular mechanism involved several stages, including the diffusion of the second anion solution into the crystal interlayer space of the PBTTT film, the replacement of [FeCl4]− with [DBS]− at the polaron sites, and the long-term stabilization of the polaron-dopant pair after completion of anion exchange. The final doping level achieved by ion exchange depended on the ability of the solvent to preserve the polaron-dopant pairs without causing polaron annihilation and on the thermodynamic spontaneity of the ion replacement process at polaron sites. Using a solvent with a low dielectric constant for the second anion solution resulted in a high electrical conductivity due to the following factors throughout the process: (i) enhanced access of the second anion to polaron sites, originating from good anion solubility and CP miscibility, and (ii) improved rate of anion exchange under non-polar conditions. Replacing [FeCl4]− with [DBS]− in a non-polar solvent improved the environmental stability of the doped PBTTT film under ambient conditions, preserving the high carrier density achieved by FeCl3 doping. Additionally, DBSA required a long time to achieve high electrical conductivity through direct doping as a Brønsted acid, whereas rapid ion exchange between [DBS]− and [FeCl4]− achieved comparably high electrical conductivity in a very short time. We believe that this work deepens the understanding of the interactions among counter-ions, CPs, and solvent environments during ion-exchange doping of CP and contributes to the advancement of highly conductive organic semiconductors.4. Experimental
FeCl3/DBSA doped PBTTT film preparation
PBTTT (Mw > 50 kDa), FeCl3·6H2O, DBSA, and all used solvents were purchased from Sigma-Aldrich. 0.5 wt% PBTTT solution was prepared by dissolving PBTTT powder in a mixed solvent (dichlorobenzene![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :cholorobenzene = 1:1). The solution was stirred for more than 3 h at 120 °C, and then was spun coated onto glass substrates at 1000 rpm for 30 s at room temperature. After that, PBTTT films were thermally annealed at 150 °C for 25 min to increase the crystallinity. 0.1 mM FeCl3·6H2O solution was prepared by dissolving FeCl3 powder in acetonitrile. PBTTT films were immersed in FeCl3 solution with the desired doping time, then blown out with N2. Next, highly FeCl3-doped PBTTT films were dipped in 0.1 mM DBSA solution for a desired period of time. The film was washed with solvent to remove the remaining DBSA. For Brønsted acid doping, undoped PBTTT film was directly dipped into 0.1 mM DBSA solution.
:cholorobenzene = 1:1). The solution was stirred for more than 3 h at 120 °C, and then was spun coated onto glass substrates at 1000 rpm for 30 s at room temperature. After that, PBTTT films were thermally annealed at 150 °C for 25 min to increase the crystallinity. 0.1 mM FeCl3·6H2O solution was prepared by dissolving FeCl3 powder in acetonitrile. PBTTT films were immersed in FeCl3 solution with the desired doping time, then blown out with N2. Next, highly FeCl3-doped PBTTT films were dipped in 0.1 mM DBSA solution for a desired period of time. The film was washed with solvent to remove the remaining DBSA. For Brønsted acid doping, undoped PBTTT film was directly dipped into 0.1 mM DBSA solution.
Contact angle measurement
The contact angle was measured by using a SmartDrop_Plus (Femtobiomed Inc.).45 20 μL of solvent droplet was controlled, then dropped onto the undoped PBTTT film.Measurement of electrical properties
σ was obtained by using a four-point probe measurement system (MSTECH) and AFM.46 The thickness of the undoped PBTTT film was about 25 nm, and it increased to 40 nm after doping.Four-point probe FET methods
An FET device with bottom-gate, bottom-contact structure was prepared. 5/50 nm of Ti/Au electrodes were thermal evaporated on SiO2 1000 nm/Si substrates. After that, the substrates were ODTS-treated in 0.5 vol% ODTS diluted in toluene. The active layer was prepared as described in the film preparation part. The electrical properties of OFETs were measured by using Keithley 2400 and 6514 source/measure units in the probe station under vacuum conditions.Computational details
Computational details are provided in ESI Section 2.†Data availability
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean Government (MSIT) (RS-2023-00234757, RS-2025-00516030). GIWAXS was carried out on the 9A U-SAXS and 3C SAXS-I beamlines at the Pohang Accelerator Laboratory (PLS-II), Republic of Korea.References
- B. Russ, A. Glaudell, J. J. Urban, M. L. Chabinyc and R. A. Segalman, Nat. Rev. Mater., 2016, 1, 1–14 CrossRef.
- O. Bubnova and X. Crispin, Energy Environ. Sci., 2012, 5, 9345–9362 RSC.
- C. G. Shuttle, R. Hamilton, J. Nelson, B. C. O'Regan and J. R. Durrant, Adv. Funct. Mater., 2010, 20, 698–702 CrossRef CAS.
- I. Salzmann and G. Heimel, J. Electron Spectrosc. Relat. Phenom., 2015, 204, 208–222 CrossRef CAS.
- I. E. Jacobs and A. J. Moulé, Adv. Mater., 2017, 29, 1703063 CrossRef PubMed.
- S. H. Kim, H. Yook, W. Sung, J. Choi, H. Lim, S. Chung, J. W. Han and K. Cho, Adv. Mater., 2023, 35, 2207320 CrossRef CAS PubMed.
- A. M. Glaudell, J. E. Cochran, S. N. Patel and M. L. Chabinyc, Adv. Energy Mater., 2015, 5, 1401072 CrossRef.
- W. Zhao, J. Ding, Y. Zou, C.-a. Di and D. Zhu, Chem. Soc. Rev., 2020, 49, 7210–7228 RSC.
- I. Salzmann, G. Heimel, M. Oehzelt, S. Winkler and N. Koch, Acc. Chem. Res., 2016, 49, 370–378 CrossRef CAS PubMed.
- Y. Liu, B. Nell, K. Ortstein, Z. Wu, Y. Karpov, T. Beryozkina, S. Lenk, A. Kiriy, K. Leo and S. Reineke, ACS Appl. Mater. Interfaces, 2019, 11, 11660–11666 CrossRef CAS.
- Y. Karpov, T. Erdmann, I. Raguzin, M. Al-Hussein, M. Binner, U. Lappan, M. Stamm, K. L. Gerasimov, T. Beryozkina and V. Bakulev, Adv. Mater., 2016, 28, 6003–6010 CrossRef CAS PubMed.
- K. Kang, S. Watanabe, K. Broch, A. Sepe, A. Brown, I. Nasrallah, M. Nikolka, Z. Fei, M. Heeney and D. Matsumoto, Nat. Mater., 2016, 15, 896–902 CrossRef CAS.
- J. Min, J. Im, S. H. Kim, H. H. Choi and K. Cho, Adv. Funct. Mater., 2023, 33, 2212825 CrossRef CAS.
- J. Min, D. Kim, S. G. Han, C. Park, H. Lim, W. Sung and K. Cho, Adv. Electron. Mater., 2022, 8, 2101142 CrossRef CAS.
- Y. W. Park, A. J. Heeger, M. A. Druy and A. G. MacDiarmid, J. Chem. Phys., 1980, 73, 946–957 CrossRef CAS.
- K. Walzer, B. Maennig, M. Pfeiffer and K. Leo, Chem. Rev., 2007, 107, 1233–1271 CrossRef CAS PubMed.
- V. Vijayakumar, Y. Zhong, V. Untilova, M. Bahri, L. Herrmann, L. Biniek, N. Leclerc and M. Brinkmann, Adv. Energy Mater., 2019, 9, 1900266 CrossRef.
- N.-H. Park, E. S. Shin, G.-S. Ryu, J. Kwon, D. Ji, H. Park, Y. H. Kim and Y.-Y. Noh, J. Inf. Disp., 2022, 24, 109–118 CrossRef.
- I. E. Jacobs, E. W. Aasen, J. L. Oliveira, T. N. Fonseca, J. D. Roehling, J. Li, G. Zhang, M. P. Augustine, M. Mascal and A. J. Moulé, J. Mater. Chem. C, 2016, 4, 3454–3466 RSC.
- E. Lim, K. A. Peterson, G. M. Su and M. L. Chabinyc, Chem. Mater., 2018, 30, 998–1010 CrossRef CAS.
- H. Tang, Y. Liang, C. Liu, Z. Hu, Y. Deng, H. Guo, Z. Yu, A. Song, H. Zhao, D. Zhao, Y. Zhang, X. Guo, J. Pei, Y. Ma, Y. Cao and F. Huang, Nature, 2022, 611, 271–277 CrossRef CAS PubMed.
- I. E. Jacobs, Y. Lin, Y. Huang, X. Ren, D. Simatos, C. Chen, D. Tjhe, M. Statz, L. Lai, P. A. Finn, W. G. Neal, G. D'Avino, V. Lemaur, S. Fratini, D. Beljonne, J. Strzalka, C. B. Nielsen, S. Barlow, S. R. Marder, I. McCulloch and H. Sirringhaus, Adv. Mater., 2022, 34, 2102988 CrossRef CAS PubMed.
- Y. Yamashita, J. Tsurumi, M. Ohno, R. Fujimoto, S. Kumagai, T. Kurosawa, T. Okamoto, J. Takeya and S. Watanabe, Nature, 2019, 572, 634–638 CrossRef CAS PubMed.
- C. Zhong, Y. Deng, W. Hu, J. Qiao, L. Zhang and J. Zhang, Chem. Soc. Rev., 2015, 44, 7484–7539 RSC.
- C. Moreau and G. Douhéret, J. Chem. Thermodyn., 1976, 8, 403–410 CrossRef CAS.
- S. N. Patel, A. M. Glaudell, K. A. Peterson, E. M. Thomas, K. A. O'Hara, E. Lim and M. L. Chabinyc, Sci. Adv., 2017, 3, e1700434 CrossRef PubMed.
- O. Zapata-Arteaga, A. Perevedentsev, M. Prete, S. Busato, P. S. Floris, J. Asatryan, R. Rurali, J. Martin and M. Campoy-Quiles, ACS Energy Lett., 2024, 9, 3567–3577 CrossRef CAS PubMed.
- C. V. Krishnan and H. L. Friedman, J. Phys. Chem., 1971, 75, 3598–3606 CrossRef CAS.
- S. E. Yoon, J. M. Han, B. E. Seo, S.-W. Kim, O.-P. Kwon, B.-G. Kim and J. H. Kim, Org. Electron., 2021, 90, 106061 CrossRef CAS.
- I. McCulloch, M. Heeney, C. Bailey, K. Genevicius, I. MacDonald, M. Shkunov, D. Sparrowe, S. Tierney, R. Wagner and W. Zhang, Nat. Mater., 2006, 5, 328–333 CrossRef CAS PubMed.
- T. Ma, B. X. Dong, G. L. Grocke, J. Strzalka and S. N. Patel, Macromolecules, 2020, 53, 2882–2892 CrossRef CAS.
- H. Tanaka, K. Kanahashi, N. Takekoshi, H. Mada, H. Ito, Y. Shimoi, H. Ohta and T. Takenobu, Sci. Adv., 2020, 6, eaay8065 CrossRef CAS PubMed.
- Y. Kim, S. Chung, K. Cho, D. Harkin, W. T. Hwang, D. Yoo, J. K. Kim, W. Lee, Y. Song and H. Ahn, Adv. Mater., 2019, 31, 1806697 CrossRef PubMed.
- N. A. Mustaffa, Q. Ahsan, M. A. Azam and L. C. Abdullah, Malays. J. Anal. Sci., 2017, 21, 950–957 Search PubMed.
- J. E. Cochran, M. J. Junk, A. M. Glaudell, P. L. Miller, J. S. Cowart, M. F. Toney, C. J. Hawker, B. F. Chmelka and M. L. Chabinyc, Macromolecules, 2014, 47, 6836–6846 CrossRef CAS.
- Y. Yamashita, J. Tsurumi, T. Kurosawa, K. Ueji, Y. Tsuneda, S. Kohno, H. Kempe, S. Kumagai, T. Okamoto, J. Takeya and S. Watanabe, Commun. Mater., 2021, 2, 45 CrossRef CAS.
- B. Yurash, D. X. Cao, V. V. Brus, D. Leifert, M. Wang, A. Dixon, M. Seifrid, A. E. Mansour, D. Lungwitz and T. Liu, Nat. Mater., 2019, 18, 1327–1334 CrossRef CAS PubMed.
- E. Alveroglu, J. Mol. Struct., 2015, 1086, 86–92 CrossRef CAS.
- J. Fraxedas, A. Vollmer, N. Koch, D. de Caro, K. Jacob, C. Faulmann and L. Valade, Materials, 2021, 14, 2058 CrossRef CAS PubMed.
- P. Kappen, N. Brack, P. S. Hale, W. Prissanaroon, E. Welter and P. J. Pigram, Appl. Surf. Sci., 2005, 243, 287–295 CrossRef CAS.
- H. H. Choi, Y. I. Rodionov, A. F. Paterson, J. Panidi, D. Saranin, N. Kharlamov, S. I. Didenko, T. D. Anthopoulos, K. Cho and V. Podzorov, Adv. Funct. Mater., 2018, 28, 1707105 CrossRef.
- K. Kang, S. Schott, D. Venkateshvaran, K. Broch, G. Schweicher, D. Harkin, C. Jellett, C. B. Nielsen, I. McCulloch and H. Sirringhaus, Mater. Today Phys., 2019, 8, 112–122 CrossRef.
- E. M. Thomas, K. A. Peterson, A. H. Balzer, D. Rawlings, N. Stingelin, R. A. Segalman and M. L. Chabinyc, Adv. Electron. Mater., 2020, 6, 2000595 CrossRef CAS.
- P. Taylor, Electrophilic Aromatic Substitution Reactions, The Open University, UK, 2002 Search PubMed.
- J. Son, G. Y. Bae, S. Lee, G. Lee, S. W. Kim, D. Kim, S. Chung and K. Cho, Adv. Mater., 2021, 33, 2102740 CrossRef CAS PubMed.
- D. Kim, D. Ju and K. Cho, Adv. Mater. Technol., 2018, 3, 1700335 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ta01703c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |