
In vivo transplantation of intrahepatic cholangiocyte organoids with decellularized liver-derived hydrogels supports hepatic cellular proliferation and differentiation in chronic liver injury†
Impreet
Kaur‡
a,
Ashwini
Vasudevan‡
ab,
Natalia
Sanchez-Romero
cde,
Arka
Sanyal
f,
Aarushi
Sharma
f,
Hamed
Hemati
g,
Pinky
Juneja
a,
Aarti
Sharma
a,
Iris
Pla Palacin
c,
Archana
Rastogi
h,
Pooja
Vijayaragavan
b,
Sourabh
Ghosh
f,
Seeram
Ramakrishna
 i,
Shiv K.
Sarin
a,
Pedro M.
Baptista
cjkl,
Dinesh M.
Tripathi
*a and
Savneet
Kaur
*a
i,
Shiv K.
Sarin
a,
Pedro M.
Baptista
cjkl,
Dinesh M.
Tripathi
*a and
Savneet
Kaur
*a
aDepartment of Molecular and Cellular Medicine, Institute of Liver and Biliary Sciences, New Delhi, India. E-mail: dineshmanitripathi@gmail.com; savykaur@gmail.com
bAmity Institute of Biotechnology, Amity University Uttar Pradesh, Sector-125, Noida 201301, Uttar Pradesh, India
cInstituto de Investigación Sanitária de Aragón (IIS Aragón), Zaragoza, Spain
dBe Cytes Biotechnologies, Barcelona, Spain
eFacultad de Ciencias de la Salud, Universidad San Jorge, Campus Universitario, Autov A23 km 299, 50830, Villanueva de Gallego, Zaragoza, Spain
fDepartment of Textile and Fibre Engineering, Indian Institute of Technology, Delhi, India
gDepartment of Toxicology and Cancer Biology, University of Kentucky, KY, USA
hDepartment of Pathology, ILBS, New Delhi, India
iNational University of Singapore, Singapore
jCentro de Investigación Biomédica en Red en el Área Temática de Enfermedades Hepáticas (CIBERehd), Madrid, Spain
kFundación ARAID, Zaragoza, Spain
lDepartment of Biomedical and Aerospace Engineering, Universidad Carlos III de Madrid, Madrid, Spain
First published on 27th November 2024
Abstract
The limited replicative potential of primary hepatocytes (Hep) is a major hurdle for obtaining sufficient quantity and quality hepatocytes during cell therapy in patients with liver failure. Intrahepatic cholangiocyte organoids (ICOs) derived from intrahepatic bile ducts differentiate into both hepatocytes and cholangiocytes in vitro. Here, we studied in vivo effects of transplanting ICOs and Hep in chronic liver injury mice models. Well characterized primary mouse ICOs and Hep were mixed in decellularized liver (DCL) matrix hydrogels and injected into the subcapsular left lateral liver lobe of CCl4-induced liver injury models whereas mice given DCL alone were in the sham group. Two weeks post-transplantation, transplanted liver lobes were collected and studied by histology and RNA sequencing. Transplanted animals did not exhibit any tumors, mortality or morbidity. Mice livers transplanted with ICOs had increased cellular proliferation and vascularization as compared to Hep transplanted mice or sham. Collagen deposition in the liver was significantly reduced and serum albumin levels were significantly increased in transplanted groups compared to the sham group. Expression of genes associated with hepatocyte differentiation was highest in Hep transplanted livers among the three groups, but they were also upregulated in ICO transplanted livers compared to sham. Our study demonstrates that ICOs encapsulated in DCL hydrogels when transplanted in chronically injured mice livers engraft well and show hepatocyte differentiation and reduction of fibrosis, indicating that hydrogel transplanted cholangiocyte organoids may serve as an efficient cell source and therapy for renewal of hepatocytes, restoration of hepatocyte functions and resolution of liver injury.
1. Introduction
Liver transplantation is a well-established strategy for end-stage liver disease and liver failure, but it is largely restricted by the acute shortage of suitable donor organs. The imbalance between the qualified recipients for transplants and the available donors has always been outnumbered at a ratio of approximately 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100.1,2 Alternative strategies for liver transplantation and effective bridging therapies for patients with end-stage liver diseases are consistently being experimented. In the past few decades, hepatocyte transplantation has been widely explored in patients with acute liver failure, to rebuild the regenerative capacity of the liver, thereby surpassing the need for whole organ replacement.3
:100.1,2 Alternative strategies for liver transplantation and effective bridging therapies for patients with end-stage liver diseases are consistently being experimented. In the past few decades, hepatocyte transplantation has been widely explored in patients with acute liver failure, to rebuild the regenerative capacity of the liver, thereby surpassing the need for whole organ replacement.3
Limitations of hepatocyte transplantation include insufficient in vivo engraftment, immunological reactions towards the transplanted hepatocytes, and less availability due to the limited proliferative capacity of the mature hepatocytes ex vivo. Previous studies have reported that at least 5% of successful engraftment of the transplanted hepatocytes with respect to the whole liver mass is required to aid liver regeneration and restore normal liver functions.4 However, the most effective route of hepatocyte transplantation, i.e., via the portal vein has been reported to achieve only 0.5% of successful graft efficiency.5 Moreover, portal vein injection has been reported to cause portal vein thrombosis and portal hypertension along with an instant blood-mediated inflammatory reaction (IBMIR).6,7 Also, as soon as hepatocytes are isolated, their functions and viability decline with time, and hence the availability of fully mature hepatocytes for transplantation is limited.8,9
Mesenchymal stem cells and other stem cell therapies have been used as an alternative for primary hepatocyte cultures, yet these cells cannot replace the primary hepatocytes for in vivo transplantations which remains the gold standard therapy for hepatic reconstitution.10,11 The use of embryonic stem cells or induced pluripotent stem (iPS) cell-derived human hepatocytes represents a lucrative therapeutic alternative but the lack of complete differentiation of these cells into mature adult liver cells ex vivo is one of the major limitations of these cells.12 These hurdles have led to the search for other sources of resident hepatic cells that have optimal hepatocyte functions, can be expanded in vitro and could be transplanted in vivo.13 Organoids present a venture of new possibilities as an autologous cell source for tissue engineering or cell therapy and can be easily obtained from liver tissue or biopsy and expanded in culture for years, remaining genetically stable.14,15 Organoid generation strategies have evolved over the years with advancements such as scaffold-based and microfluidic chip-based development for prolonged cultures and to facilitate vasculature inside the developing organoids.16,17 Cholangiocyte organoids derived from both intrahepatic and extrahepatic cholangiocytes (ECOs) are being widely used as a transplant strategy to repair damaged bile ducts.18 Intrahepatic cholangiocyte organoids (ICOs) have been reported to differentiate into both hepatocytes and cholangiocytes in vitro,19 which is beneficial in repopulating the damaged bile ducts as well as the hepatocytes, on the other hand, ECOs lack the bipotent nature and could differentiate only into cholangiocyte phenotype with appropriate growth factors.20 Extracellular matrix (ECM) derived hydrogels have been widely used since the 2000s for the culture of organoids as well as for transplantation purposes owing to their bio-compatibility and low immunogenicity.21–24 However, there are challenges in using ECM-based hydrogels for transplantation purposes such as incomplete decellularization, and fragmented, cleaved, or partially denatured macromolecules as a result of enzymatic digestion and solubilization. The remaining cellular components in the ECM can cause unfavorable host immune responses. Thus, an effective decellularization process is essential to improve the quality and clinical translation of decellularized liver (DCL) extracellular matrix hydrogels.
In view of the promising benefits of ICOs for transplantation and the dearth of data available for post-transplantation effects of these organoids in the liver, in the present study, we investigated the effects of in vivo transplantation of ICOs and compared them with the transplantation of primary hepatocytes (Hep) in CCl4-induced chronic liver injury mice models. For effective engraftment of organoids and cells in the liver, we used DCL as a delivery vehicle for transplanting the cells directly beneath the liver capsule.
2. Methods
2.1 Animal experiment and ethics
All animals used in this study were housed and fed according to guidelines of the Institutional Animal Ethics Committee (IAEC) at the Centre for Comparative Medicine, Institute of Liver and Biliary Sciences (ILBS), New Delhi duly approved by the Animal Ethics Committee of ILBS via two protocols (IAEC/ILBS/22/11 and IAEC/ILBS/23/13). The study utilized Sprague Dawley rats (male, 8 weeks) for the preparation of the decellularized liver ECM, and C57BL/6 mice (male, 8 weeks) for cell isolation and preparation of chemically induced [carbon tetrachloride (CCl4)] liver injury animal models and transplantation.2.2 Liver decellularization and preparation of the DCL
Decellularization was done as previously reported.25 Briefly, preparation of the DCL was performed by solubilizing the powdered decell tissues and mixing with 0.1 N HCl and pepsin (Himedia Labs) keeping a 5:1:2 (mg:mL:mg) proportion, under constant stirring at 4 °C until no traces of powder were visible and a homogenous solution was formed. After pepsin solubilization, pH neutralization (to pH 7.4) was performed at 4 °C with 0.5 N NaOH. The solution was then filtered using a sterile 100 μm sieve, centrifuged at 3000g for 10 minutes, aliquoted, and stored at −20 °C until further use. For gelation, the solution (final concentration: 10 mg mL−1) was incubated at 37 °C for one hour.
2.3 Culture and characterization of ICOs
ICOs were isolated and cultured from mice livers as described earlier.26 A freshly isolated liver was immersed in fresh medium in a Petri dish and after mincing, it was transferred to a 50 mL falcon tube with 1% DMEM. After vigorous pipetting, the fragments were allowed to settle under gravity. After observing a clear supernatant, the liver pieces were subjected to enzymatic digestion with collagenase I (Gibco). For this, the liver tissue with the medium was kept in a shaking water bath at 37 °C and at 200 rpm for 1 hour. After discarding the supernatant, the tissue was incubated with a fresh enzymatic solution for 30 minutes at 37 °C with agitation. After every 30 minutes of digestion, the supernatant was transferred to a new falcon tube and stored at 4 °C, and the fresh enzymatic solution was added to the cells. The procedure was repeated for 30 minutes, three to four times, and observed under a microscope for the presence of any cells. Supernatants obtained after each digestion were centrifuged at 210g for 5 minutes at 4 °C. After discarding the supernatant, the pellet was washed once with 8 mL of cold 1% DMEM and centrifuged. After aspirating the supernatant completely and making the pellet dry it was mixed with matrigel and cultured in an array of growth factors according to the earlier studies (Table S1, ESI†). ICOs were passaged after two weeks and characterized by hematoxylin and eosin staining (H&E) and immunostaining with a biliary marker, CK19 (Cytokeratin 19) and GGT (Gamma-glutamyl transpeptidase). For characterization of the organoids, they were recovered with cell recovery solution, fixed with paraformaldehyde, embedded in paraffin, and cut as thin sections of approximately 2–5 μm, later the sections were deparaffinized and dehydrated in a series of alcohol solutions. The sections were then stained with H&E. For immunostaining, the organoids were fixed with 4% paraformaldehyde for 30 minutes at 4 °C, and then they were permeabilized with 0.1% Triton-X 100 for 15 minutes; later, the organoids were blocked with 2% BSA for 1 hour. The CK19 primary antibody (1:100) was then added to them and incubated overnight at 4 °C. The next day, the organoids were washed with PBS thrice and incubated with a secondary antibody (1:500) for 1 hour and imaged under a confocal microscope.
2.4 Transplantation of Hep and ICOs in animal models of chronic liver injury
For transplantation, specific pathogen-free C57BL/6 mice, with weights (∼25 g to 30 g) were used. The surgical procedure was carried out in a sterile environment and the animal was anesthetized using a gaseous form of anesthesia, isoflurane. The concentration of isoflurane used was 1.5% to 3% with a sustained oxygen flow of 0.5 L min−1. We have used ∼3 × 106 primary hepatocytes/50 μL of hydrogel and ICOs between passages 2–3 (106 organoids per 50 μL of hydrogels) for transplantation onto the exposed subcapsular region of the left lateral lobe (∼50 μL of the transplant material) using a 27G syringe. To track the cells in vivo, they were labeled with GFP (green fluorescent protein). The transplant groups were divided and designated as follows, n = 6 per group, (i) Hep w/o the ECM (primary hepatocytes transplanted without any carrier), (ii) Hep-cECM (primary hepatocytes transplanted with the commercial ECM), (iii) Hep-DCL (primary hepatocytes transplanted with DCL), (iv) ICOs w/o the ECM (ICOs transplanted without any carrier), (v) ICO-cECM (ICOs transplanted with the commercial ECM), and (vi) ICO-DCL (ICOs transplanted with DCL). Matrigel (Corning) was the commercial ECM used in this study. The injected site was then sealed with the Tisseel-fibrin sealant (Baxter). The abdominal wall was then sutured after confirming hemostasis. After the completion of the surgical procedure, inhalation anesthesia was withdrawn and the animal was allowed to recover. The animals were given antibiotics for 3 days to prevent any infection post-surgery.2.5 RNA sequencing and analysis
Transplanted liver lobes were harvested and proceeded for RNA extraction. The RNA quality was assessed with a NanoDrop spectrometer. Three independent samples for each treatment were collected and their RNA expression profiles were analyzed using the Illumina NextSeq Platform. For transcriptomics analysis, all processing of RNA samples (cDNA synthesis and labelling hybridization) was performed by DNA Xperts Pvt Ltd. The Illumina transcriptome data were mapped to the reference mice genome GRCm39 (accession no. GCF_000001635.27). edgeR software was used for calculating transcript abundance which is represented as CPM (counts per minute). Heatmaps were constructed using the log-transformed and normalized value of genes based on the Pearson uncentered correlation distance and average linkage method. Significant differentially expressed genes (DEGs) were identified and enriched using the gene set enrichment analysis (GSEA)27 website for pathways in Kyoto Encyclopedia of Genes and Genomes (KEGG,28 and gene ontology-biological processes (false discovery rate (FDR) ≤ 0.05). Molecular Signatures Database v7.5.1 was used to retrieve the cell markers and the list of genes involved in signaling pathways.29 Hierarchical clustering analysis was performed using Perseus software.30P < 0.05 was considered significant.2.6 Statistical analysis
A power analysis software program (G*Power 3 software, Germany) was used to calculate the number of animals used for transplantation which came out to be n = 6. Data were reported as mean ± standard deviation (SD). Two groups have been compared using Student's t-test and P < 0.05 has been considered significant.3. Results
3.1 Characterization of the decellularized liver
Decellularization of the liver through portal vein cannulation was performed according to established protocols and the perfusion of detergents resulted in a shiny white appearance of the liver indicating complete removal of the liver cells (Fig. 1b). Characterization of the decellularized liver was performed to assess the efficiency of decellularization in terms of acellularity and the ECM protein content. H&E staining and DAPI staining revealed a complete absence of cells in the decellularized liver (Fig. 1c and d). We performed DNA quantification in control and decell tissues which revealed a significant DNA decrease in decell liver tissue compared to control livers (P ≤ 0.001, Fig. 1e). The presence of intact ECM matrix proteins in decell tissue was clearly seen by MT (Masson Trichome) staining specific for collagen proteins, Alcian blue for the glycosaminoglycans (GAGs), and orcein staining for elastin proteins (Fig. 1f–h).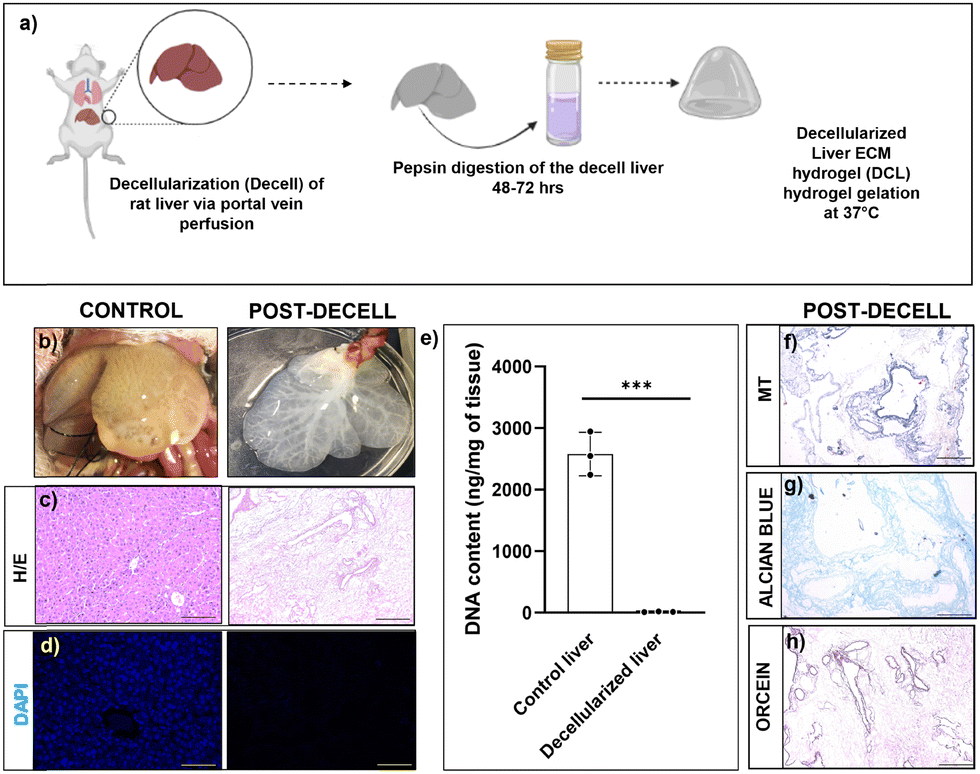 | ||
| Fig. 1 Characterization of decell liver tissue. (a) Schematic representation of the decellularization process (b) appearance of the liver after the decellularization process. (c) H&E staining showing the presence and absence of cells before and after the decellularization process; scale bar 100 μm. (d) DAPI staining showing the presence and absence of cell nucleus before and after the decellularization process; scale bar 50 μm. (e) DNA contents of control and decellularized livers. Statistical analysis performed with GraphPad Prism and Student's t-test between the control and decellularized livers (***P < 0.001). (f) MT staining showing the presence of collagen matrix protein after the decellularization process. (g) Alcian blue staining showing the presence of GAGs after the decellularization process. (h) Orcein staining showing the presence of the elastin matrix protein after the decellularization process; scale bar 100 μm. All the estimations were carried out in triplicate (n = 3). | ||
Biophysically, DCL hydrogels behaved as moderately low-viscosity fluids. Both, 4 mg mL−1 and 10 mg mL−1 DCL hydrogels showed a relatively constant viscosity and flow behavior of Newtonian fluid within the shear rate range of 10−2 to 10 s−1. However, beyond the shear rate of 100 s−1, the DCL hydrogels exhibited a shear thinning behavior with a significant decrease in viscosity, which is a property desirable for injectable hydrogels (Fig. 2a). The injectability of 10 mg mL−1 DCL was possible as there was shear thinning behaviour observed upon the application of strain. In the amplitude sweep experiments, the storage modulus (G′) of DCL increased from 134.68 Pa to 905.06 Pa upon increasing the DCL concentration from 4 mg mL−1 to 10 mg mL−1. The storage modulus (G′) was greater than the loss modulus (G′′) at both concentrations of DCL as shown in (Fig. 2b). To prevent sample disintegration throughout the experiment, 1% strain was employed for subsequent frequency sweep analysis. Results from angular frequency sweep showed that the G′ values for both the DCL groups (4 mg mL−1 and 10 mg mL−1) were dependent on the frequency values, which is a characteristic of physically crosslinked hydrogels (Fig. 2c). At low frequency values, the G′′ is seen to be higher than the G′, indicative of weak gel formation. With an increase in the angular frequency, both G′ and G′′ increased, and a crossover point is observed for both groups. As expected, increasing the DCL concentration from 4 mg mL−1 to 10 mg mL−1 was associated with a broader range of frequency values for which the G′ became higher than the G′′ underscoring the improved stability of the formed hydrogels. The irregularity in the modulus curve was observed as hydrogels were a suspension mix of the DCL.
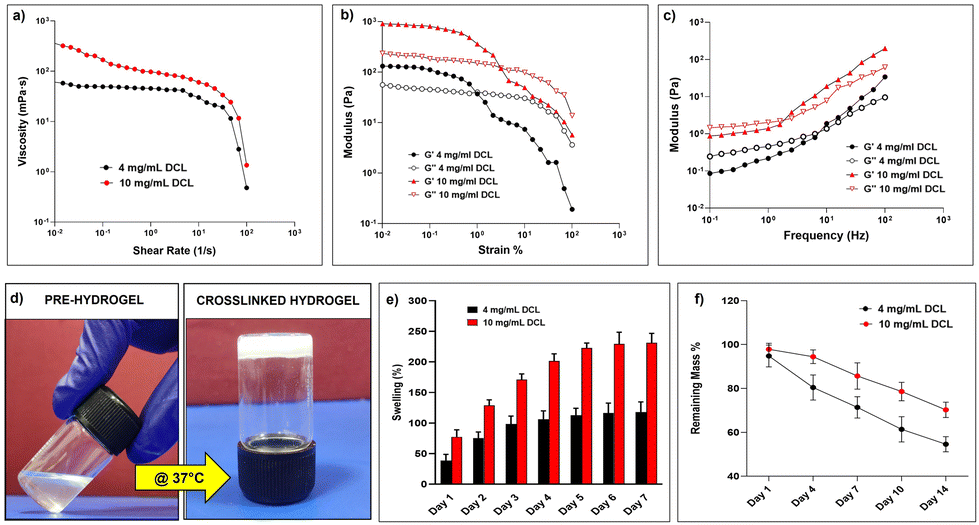 | ||
| Fig. 2 Physical characterization of the DCL. (a) Flow curves of the DCL at two different concentrations (b) Amplitude sweep of the DCL at two different concentrations. (c) Frequency sweep of the DCL at two different concentrations. (d) Tube inversion test demonstrating temperature-dependent sol–gel transition of 10 mg mL−1 DCL. (e) Comparative representation of the water uptake activity of 4 mg mL−1 and 10 mg mL−1 DCL expressed as a swelling percentage (***P < 0.001). (f) Comparative representation of in vitro degradation of 4 mg mL−1 and 10 mg mL−1 DCL expressed as the remaining mass percentage (**P < 0.005). All the estimations were carried out in triplicate (n = 3); statistical analysis was performed with GraphPad Prism and Student's t-test between the two concentrations of DCL. | ||
The tube inversion method was used to investigate the thermo-responsive behaviours of the DCL, and both samples demonstrated temperature-dependent sol–gel transitions. The 10 mg mL−1 DCL group molecules were in solution at room temperature but formed opaque hydrogels in the physiological temperature range following 60 minutes of incubation at 37 °C (Fig. 2d). Although the 4 mg mL−1 DCL group demonstrated a similar sol–gel behaviour, the gelation duration was unfavourably greater (data not shown). The gelation of decellularized tissue hydrogels is essentially a collagen-based self-assembly process and the native biochemical profile of the source tissue and the concentration of proteins remaining after the decellularization process have a direct correlation with the polymerization kinetics in the resulting hydrogel.31 Moreover, in the context of minimally invasive injectable hydrogels an optimal time period is required for the delivery of the pre-gel solution to selected anatomic sites before gelation is complete; the gelation kinetics of the 10 mg mL−1 hydrogel warrants its applicability as an injectable hydrogel. In general, the liver ECM is primarily composed of collagen and proteoglycans, both of which are inherently good at absorbing water. The relative proportions of these components in the decellularized matrix obtained vary considerably depending upon the decellularization protocol implemented and thus can be correlated with the water absorption potential of hydrogels prepared thereafter. Water absorption analysis of the DCL showed a concentration-dependent behaviour wherein the 10 mg mL−1 DCL group exhibited a comparatively higher water absorption profile (231.17 ± 15.48%) than the 4 mg mL−1 group (118.07 ± 16.41%) after 7 days of incubation (Fig. 2e). The excellent swelling properties of the DCL suggest that they can effectively mimic the hydrated environment of the native liver ECM and can support cell adhesion and protein sequestration of the encapsulated cells.32 The in vitro degradation kinetics revealed that the DCL exhibited a consistent decrease in their mass percent starting on day 1; by the end of day 14, the remaining mass percent of 4 mg mL−1 and 10 mg mL−1 DCL were observed to be 54.61 ± 3.47% and 70.27 ± 3.51% respectively (Fig. 2f). It could be inferred that the DCL owing to their increased water uptake and rapid diffusibility undergoes bulk erosion due to easy access to water and collagenase throughout the hydrogel lattice enabling hydrolytic or enzymatic cleavage of bonds.
To assess the immunocompatibility of the fabricated DCL hydrogels, we performed a flow-cytometry based analysis to determine any possible increase in infiltrating immune cells, post-transplantation of the rat derived DCL in control C57BL/6 mice. Results showed the presence of 17.90 ± 02.12% vs. 12.3 ± 01.59% of CD4+ T-cells; 22.80 ± 09.97% vs. 23.40 ± 05.48% of CD8+ T-cells; 89.70 ± 13.85% vs. 95.90 ± 04.44% of neutrophils; 16.20 ± 10.11% vs. 26.30 ± 08.87% of monocytes in control vs. transplant groups Fig. S1 (ESI†) with which we could infer that there was no heightened systemic immune response with the rat ECM derived DCL transplantation in mice.
3.2 Characterization of Hep and ICOs
Hepatocyte isolation from rats was performed by in situ perfusion of the liver via the portal vein with collagenase digestion mixture followed by in vitro digestion as depicted in Fig. S2a (ESI†). The viability of the isolated primary mouse hepatocytes was >95% as evidenced by trypan blue dye exclusion. About 90% of the cells were adherent within 24 h after plating (Fig. S2b, ESI†). The hepatocytes stained positive for asialoglycoprotein receptor 1 (ASGR1) (Fig. S2c, ESI†), and albumin (Fig. S2d, ESI†) day 1 post-isolation.The intrahepatic cholangiocytes were isolated and cultured according to established protocols as depicted in Fig. 3a. During the first week of culture, small organoids emerged from matrigel-embedded cells (Fig. 3b and c). The organoids expanded within 10–15 days (Fig. 3d). The viability of the ICOs at day14 was verified with calcein/PI staining, and the images depict the viable cells (calcein stained) within the core of ICOs along with very few PI-stained dead cells (marked with arrows) (Fig. 3e), H&E staining of the cryosectioned organoids showed viable cells inside the core of organoids (Fig. 3f). Characteristic markers of cholangiocytes such as CK19 and GGT were stained positive in the cultured ICOs at day 14 (Fig. 3g and h).
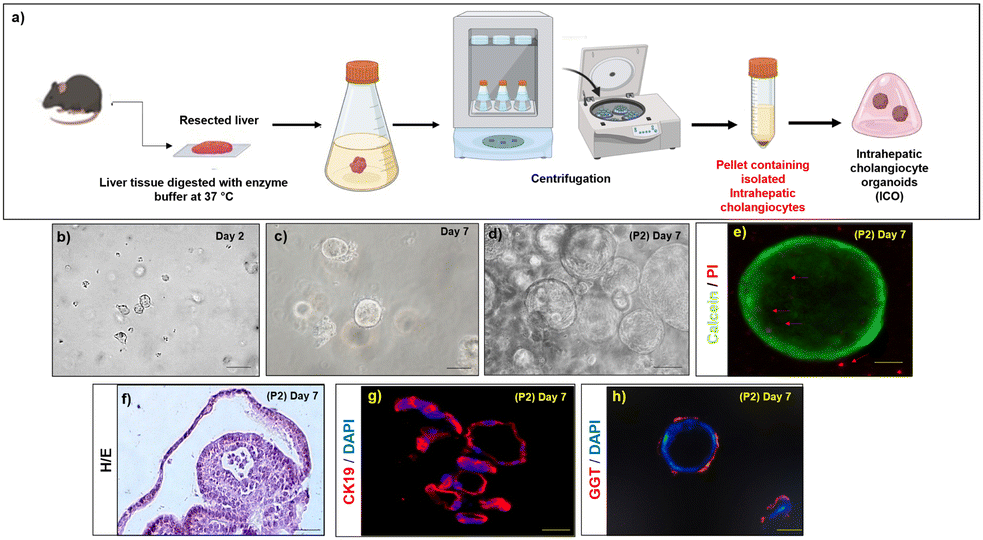 | ||
| Fig. 3 Isolation and characterization of ICOs (a) schematic representation of the isolation method used to obtain ICOs. Bright-field images of ICOs in vitro (b) at day 2; scale bar 500 μm (c) at day 7; scale bar 500 μm and (d) passage 2; at day 14; scale bar 500 μm. (e) Whole mount calcein/PI staining of the ICOs at day 14; scale bar 10 μm (arrows indicate dead cells) (f) H&E staining of the cryosectioned ICOs at day 14; scale bar 100 μm. (g) CK19 immunostaining of the cryosectioned ICOs at day 14; scale bar 100 μm. (h) Whole mount GGT immunostaining of the ICOs at day 14; scale bar 50 μm. | ||
3.3 Transplantation of Hep and ICOs in chronic liver injury models
We next performed intrahepatic transplantation of Hep and ICOs in chronic liver injury mice models. The developed chronic liver injury models showed damaged tissue architecture (F2-stage, classified according to a Metavir grading system) with portal-to-portal fibrous septa (Fig. S3b, ESI†). For transplantation, primary hepatocytes (3 × 106 cells/50 μL of hydrogel) and ICOs passage 2 (106 organoids/50 μL of hydrogel) were mixed and injected beneath the liver capsule in the prepared CCl4 mice models. There was no mortality, tumor formation or necrosis observed at the transplanted site two weeks post-transplantation in any of the transplanted groups (Fig. S4, ESI†). To assess the engraftment efficiency of the transplanted cells, GFP-tagged cells in the recipient mice liver tissues were detected at day 14 post-transplantation using a confocal microscope (Fig. 4a–f). Quantification results revealed that in vivo engraftment efficiency (GFP+ area/field) was higher when both Hep and ICOs were transplanted with DCL compared to those transplanted with the cECM (∼2 ± 1.8 in Hep-cECM vs. ∼4 ± 1.13 in Hep-DCL; P = 0.016); (∼3 ± 0.26 in ICO-cECM vs. ∼6 ± 0.54 in ICO-DCL; P = 0.006). Also, engraftment was the lowest when the cells were transplanted without any carrier (∼0.8 ± 1.2 in ICOs w/o ECM vs. ∼6 ± 0.54 in ICO-DCL; P ≤ 0.0001 and ∼0.5 ± 0.25 in Hep w/o ECM vs. ∼4 ± 1.13 in Hep-DCL; P ≤ 0.0001) (Fig. 4g). Since the highest engraftment efficiency was observed only in the Hep-DCL and ICO-DCL groups, these two groups were only considered for evaluation further throughout the study. The liver lobes of Hep-DCL and ICO-DCL revealed increased expression of nuclear (proliferating cell nuclear antigen) PCNA (Fig. 4h–j), proliferation marker after two weeks of transplantation in comparison to that of sham, with maximum PCNA positive cells observed in the ICO-DCL group (about 8-fold increase, P = 0.0004, Fig. 4k).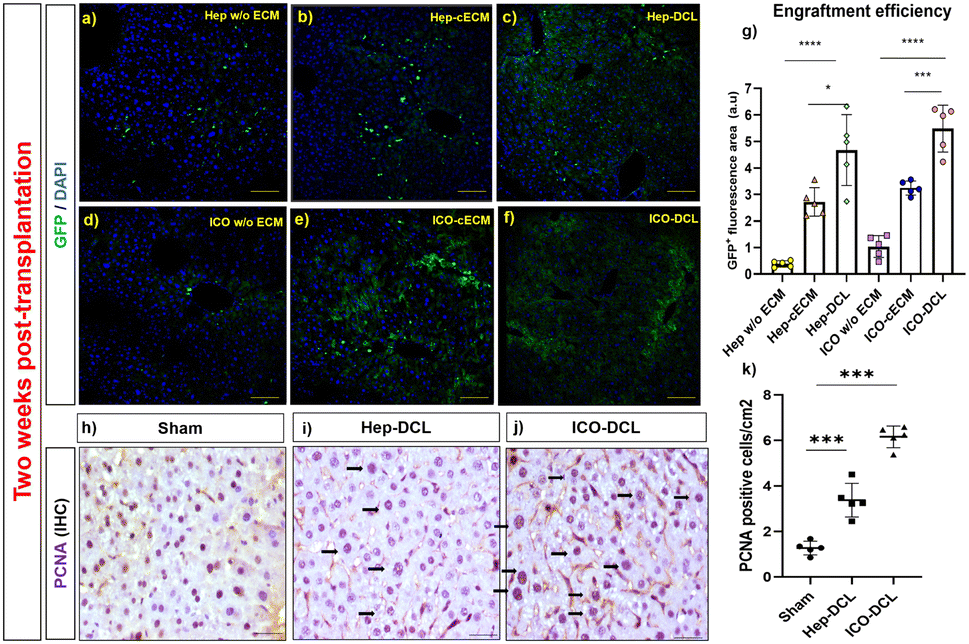 | ||
| Fig. 4 Transplantation of Hep and ICOs in chronic liver injury models. (a)–(f) Representative images of in vivo engrafted GFP labelled Hep and ICOs in mouse liver lobe two weeks post-transplantation; scale bars 100 μm. (g) Quantification of engraftment was determined using ImageJ software, n = 5 fields per group. Statistical analysis was performed with GraphPad Prism and t-test. P value represented as *P ≤ 0.05, ***P ≤ 0.001, and ****P ≤ 0.0001. (h)–(j) PCNA staining in the transplanted lobes in comparison to the sham liver (arrows indicating proliferating cell nucleus); scale bar 50 μm. (k) Graph showing a number of PCNA positive cells quantified in sham and transplanted liver lobes with ImageJ software, n = 5. Statistical analysis was performed with GraphPad Prism and Student's t-test. The P value represented as sham vs. the transplanted lobes (***P ≤ 0.001). | ||
To determine the migration of the transplanted cells in the host liver, we verified the presence of GFP+ cells on the non-transplanted liver lobes two weeks post transplantation with confocal microscopy (Fig. S5, ESI†). We observed GFP positive cells in both Hep-DCL and ICO-DCL groups indicating cell migration of the transplanted cells within the liver, outside the transplanted lobe.
3.4 Expression of hepatocyte and cholangiocyte-specific markers in transplanted mice
To further assess the differentiation of engrafted cells in transplanted mice, we analysed the transplanted liver lobe for expression of hepatic and cholangiocyte markers. To determine the cells differentiating towards hepatic phenotype, immunofluorescence imaging specific for hepatocyte markers albumin and ASGR1 was performed. The results clearly showed that albumin expression was increased in Hep-DCL vs. sham (2-fold; P = 0.005; Fig. 5a and b) and also in ICO-DCL vs. sham (2-fold; P = 0.001; Fig. 5a and b). ASGR1 expression was almost similar in sham vs. Hep-DCL; Fig. 5c and d) with significant increase in ICO-DCL vs. sham (1-fold; P = 0.05; Fig. 5c and d). Further, the cells differentiating towards cholangiocyte phenotype were studied by the immunofluorescence staining specific for biliary markers, GGT and CK19. GGT expression was significantly increased in Hep-DCL vs. sham (1-fold; P = 0.056), as well as ICO-DCL vs. sham (3-fold; P = 0.001) (Fig. 5e and f). CK19 expression was significantly higher in both the transplant groups than in the sham group (Hep-DCL vs. sham; 3-fold; P = 0.001) (ICO-DCL vs. sham; 4-fold; and P = 0.001) (Fig. 5g and h).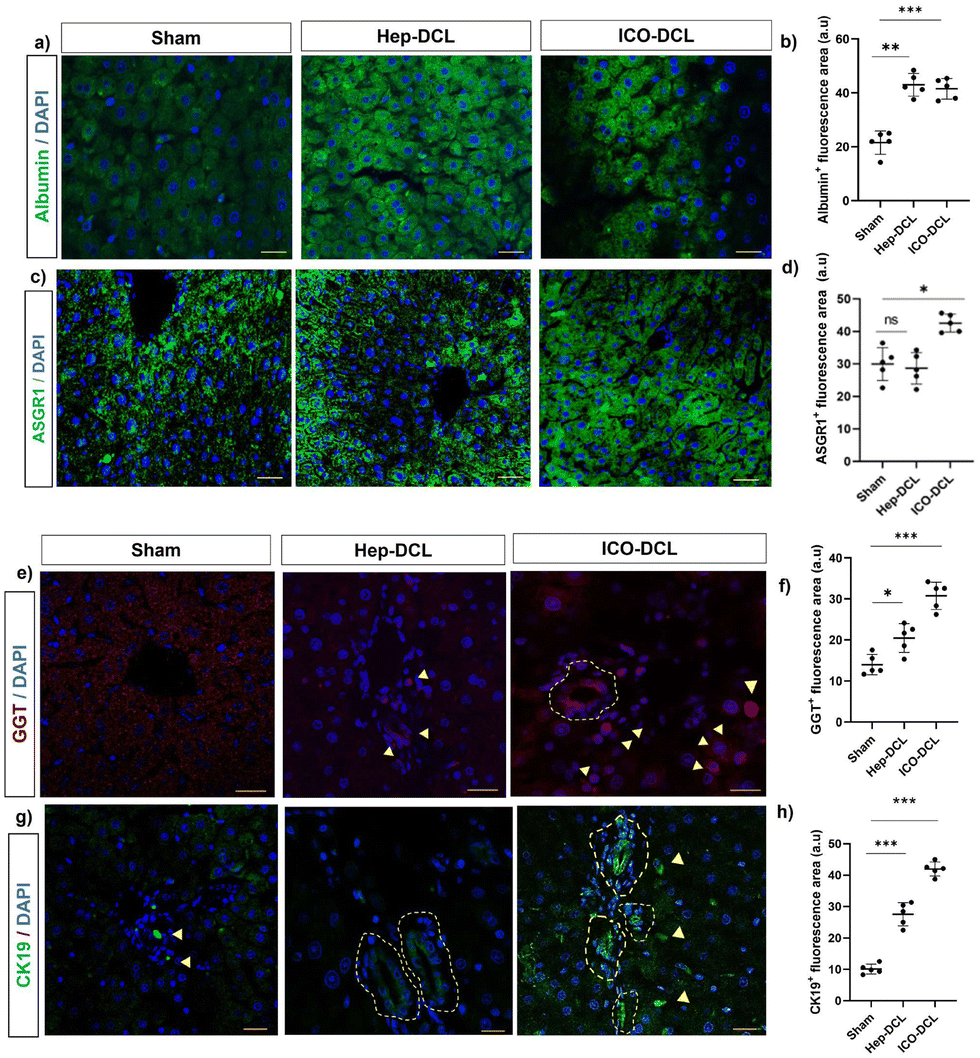 | ||
| Fig. 5 Immunofluorescence staining for hepatic and biliary markers two-weeks post-transplantation. (a) Albumin expression in sham and transplanted animals, scale bar- 50 μm. (b) Graph showing area positive for albumin quantified with ImageJ. (c) ASGR1 expression in sham and transplanted animals, scale bars 50 μm. (d) Graph showing area positive for ASGR1 quantified with ImageJ. (e) GGT expression in sham and transplanted animals, scale bars 50 μm. (f) Graph showing area positive for GGT quantified with ImageJ software (g) CK19 expression in sham and transplanted animals, scale bars 50 μm. (h) Graph showing area positive for CK19 quantified with ImageJ software. Statistical analysis was performed with GraphPad Prism and Student's t-test. The P value represented as sham vs. the transplanted lobes (*P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, and ****P ≤ 0.0001); (n = 5). | ||
CD31 staining was performed on the sham and transplanted liver lobes to determine the presence of new blood vessel formation post-transplantation. It was observed that ICO-DCL liver lobes showed increased sinusoidal capillarization and angiogenesis compared to sham (Fig. 6a–c). Quantification showed increased (2-fold; ****P ≤ 0.0001) capillarization in the ICO-DCL group compared to that in the sham group (Fig. 6d).
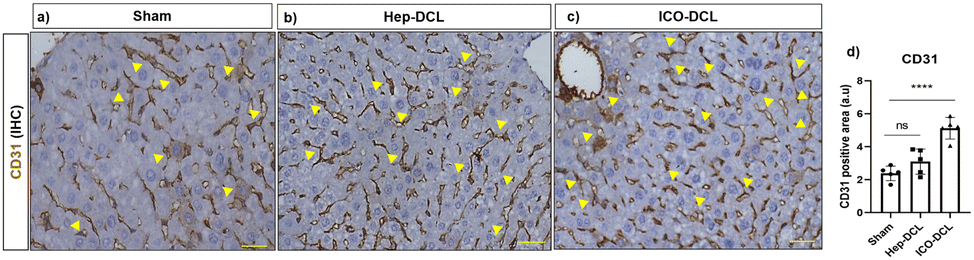 | ||
| Fig. 6 Characterization of neo-vascularization two weeks post-transplantation. CD31 expression in (a) Sham, (b) Hep-DCL and (c) ICO-DCL groups, scale bar 50 μm. (d) Graph showing the quantification of the CD31 positive area quantified with ImageJ software n = 5 fields per group. Statistical analysis was performed with GraphPad Prism and Student's t-test. The P value represented as sham vs. the transplanted lobes (****P ≤ 0.0001). | ||
3.5 Transcriptomic analysis of the transplanted liver lobes
We next compared the gene expression markers specific to hepatocytes, cholangiocytes, and hepatic progenitor cells in the sham, Hep-DCL, and ICO-DCL transplanted livers using RNA sequencing. Both biliary and hepatocyte-specific markers such as Sox9 (SRY-Box transcription factor 9), Tspan8 (tetraspanin-8), genes, including Alb (albumin), ApoA1 (apolipoprotein A1), and Apoa2 (apolipoprotein A2), were upregulated in the hep transplanted livers (Fig. 7a and c). Expression of genes such as Fgfr2 (fibroblast growth factor receptor 2) and krt19 (cytokeratin 19), which are present in hepatic progenitor cells or are known to increase in the liver after injury, and were seen to be upregulated in Hep-DCL as well as ICO-DCL livers (Fig. 7b). ICO-DCL livers also showed upregulation of hepatocyte markers however lower than the Hep-DCL livers (Fig. 7c). Hepatocyte differentiation genes, including Hnf4a (hepatocyte nuclear factor 4 alpha), Bmp4 (bone morphogenetic protein 4), Foxa3 (forkhead box a3) were highly upregulated and robust in Hep-DCL livers but also higher in ICO-DCL than in the sham group (Fig. 7d). Genes associated with liver regeneration, such as a Gli1 (GLI family zinc finger 1) were upregulated in ICO, however, downregulated in Hep-DCL livers. Ezh1 (enhancer of zeste 1 polycomb repressive complex 2), an epigenetic regulator of liver regeneration, and IL-6 (interleukin 6), a major driver of liver regeneration was highly expressed in Hep transplanted livers (Fig. 7e). Among the genes involved in hepatocyte proliferation and regeneration, critical genes involved in cell proliferation, including E2f 1, 3 and 4 (eukaryotic transcription factor), and Ki67 were upregulated in both Hep and ICO-DCL livers compared to sham (Fig. 7f). Among the major signaling pathways involved in modulating hepatic injury and repair, the expression of gene members of the Notch signaling pathway was significantly higher in both Hep and ICO-DCL liver lobes compared to that in the sham group (Fig. 7g and h).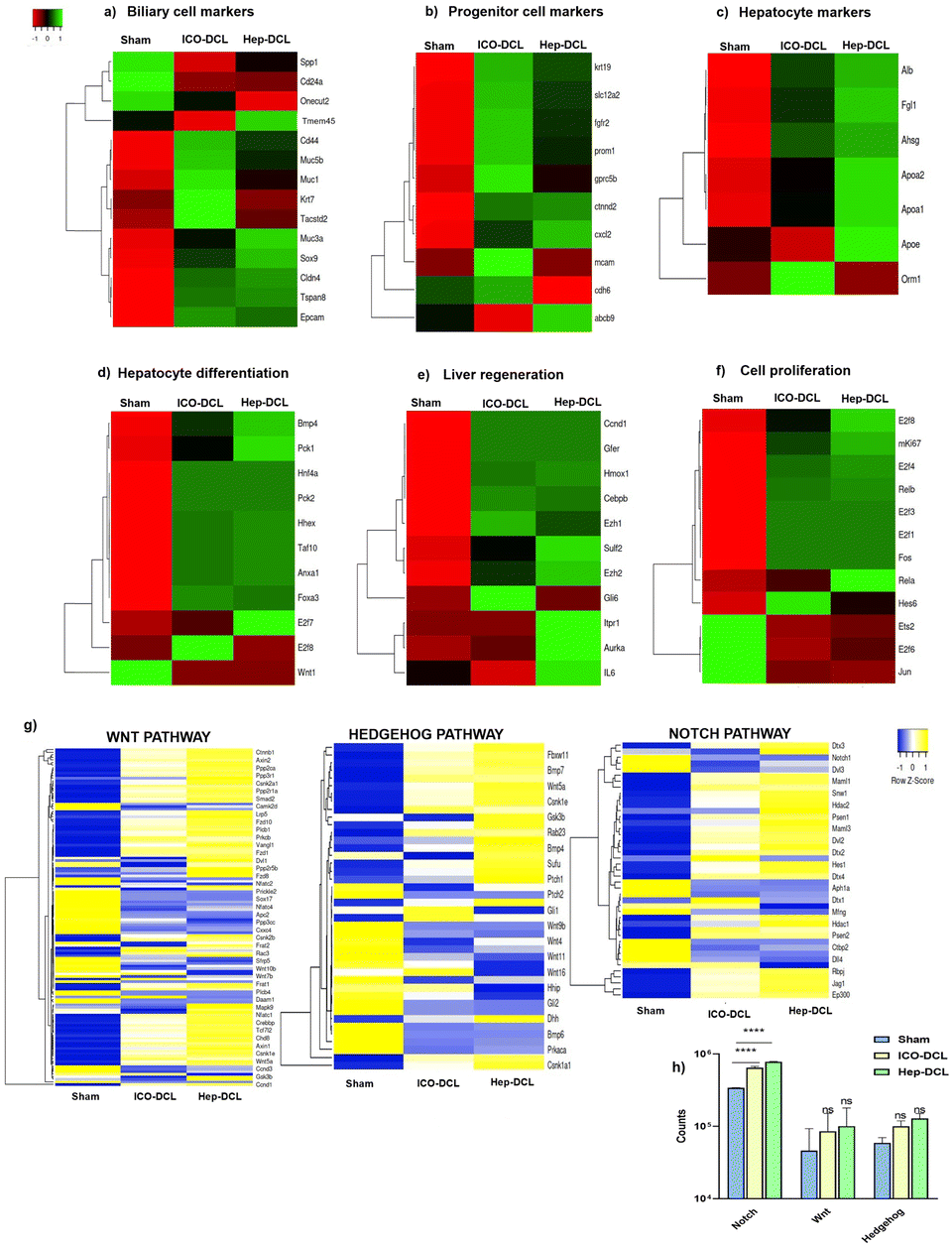 | ||
| Fig. 7 Gene expression profiling of the transplanted liver lobes two weeks post-transplantation by RNA sequencing. (a) Biliary epithelial markers observed in sham, Hep-DCL and ICO-DCL livers. (b) Progenitor cell markers observed in sham, Hep-DCL and ICO-DCL livers. (c) Hepatocyte markers observed in sham, Hep-DCL and ICO-DCL livers. (d) Hepatocyte differentiation genes observed in sham, Hep-DCL and ICO-DCL livers. (e) Genes involved in cell proliferation observed in sham, Hep-DCL and ICO-DCL livers. (f) Liver regeneration genes observed in sham, Hep-DCL and ICO-DCL livers. (g) Comparison of major pathways between the sham and DCL groups based on the expression of the respective member genes. (h) Graph showing NOTCH, WNT and Hedgehog pathway in sham and transplanted lobes. Statistical analysis was performed with GraphPad Prism. One-way ANOVA performed between sham and transplanted lobes (*P ≤ 0.001 and **P ≤ 0.05). All the estimations were carried out in triplicate (n = 3). | ||
The specific cluster for each group was identified using the hierarchical clustering analysis. Further gene ontology analysis of the Hep and ICO clusters showed that they were enriched for the biological processes associated with cell cycle and cellular proliferation (Fig. S6, ESI†). In addition, cluster-specific for ICO transplanted livers specifically was enriched for the immune system and biological adhesion-related processes, while biosynthetic and tissue homeostatic processes were highly enriched in the cluster-specific for Hep transplanted livers (Fig. S6, ESI†). To validate the RNAseq results we performed RT-PCR analysis, and the results showed that the biliary cell marker namely SOX9 (SRY-box transcription factor 9), was upregulated in the Hep-DCL (4-fold change; P = 0.01) and in the ICO-DCL (3-fold change; P = 0.0004) vs. the sham group, progenitor cell marker namely CK19 was upregulated in the Hep-DCL (3.5-fold change; P = 0.0006), and non-significantly upregulated in the ICO-DCL group (1-fold change; P = ns) vs. the sham group. Hepatocyte differentiation markers such as HNF4a were upregulated within the Hep-DCL (5-fold change; P < 0.0001) and in the ICO-DCL (6-fold change; P < 0.0001) vs. the sham group. Liver regeneration marker namely Ki67 showed in the Hep-DCL (4-fold change; P = 0.001), in the ICO-DCL (5-fold change; P = 0.001) vs. the sham group, and proliferation marker e2f6 (eukaryotic transcription factor 6) showed in the Hep-DCL (2-fold change; P = 0.001), and in the ICO-DCL (4-fold change; P < 0.0001) vs. the sham group (Fig. S7a–e, ESI†).
3.6 Assessment of liver functions and fibrosis in the transplanted liver
We next studied the effects of Hep and organoid transplantation on liver injury and functions. MT staining of the transplanted liver lobe indicated that collagen deposition was significantly reduced in mice treated either with Hep (P ≤ 0.001) or ICOs (P ≤ 0.001) compared to sham animals (Fig. 8). We also verified the collagen deposition in the non-transplanted liver lobes to determine if there had been a global effect of cell transplantation contributing to a reduction in fibrosis as well as an improvement in overall liver function (Fig. S8, ESI†). We observed that two weeks post transplantation there was negligible collagen positive area in the non-transplanted liver lobes of the Hep-DCL and ICO-DCL animals vs. the non-transplanted liver lobes of sham animals. We did not however observe any significant difference in the serum aspartate aminotransferase (AST) and alanine transaminase (ALT) levels in all three groups (Fig. S9a and b, ESI†). In terms of liver functions, levels of serum albumin were significantly increased in animals treated with hepatocytes compared to sham (P = 0.035, Fig. S9c, ESI†). Albumin levels were also increased in ICO-DCL animals in comparison to sham albeit less than the Hep-DCL (P = 0.02, Fig. S9c, ESI†).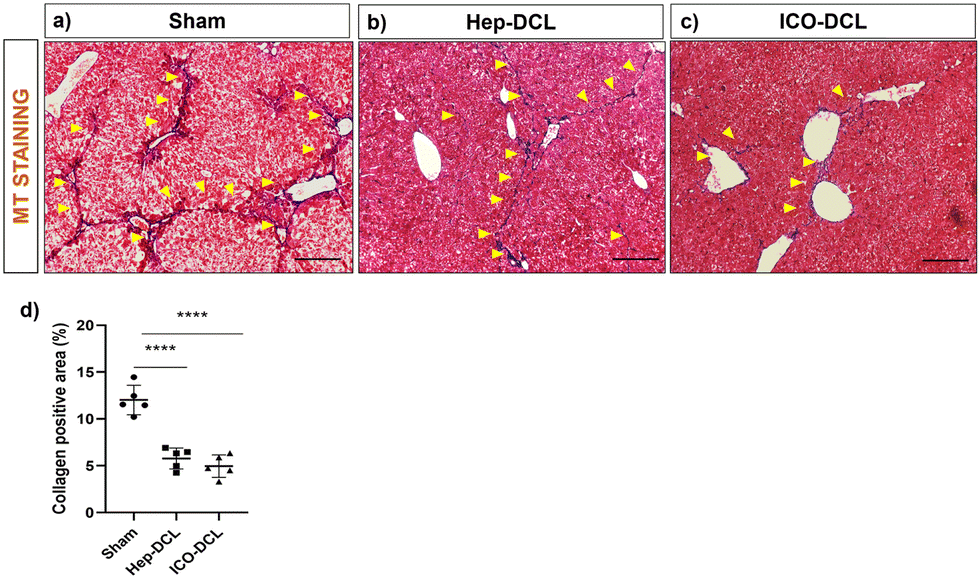 | ||
| Fig. 8 Assessment of reduction in fibrosis of the transplanted lobes two-weeks post transplantation. (a)–(c) Representative images of MT staining of sham and transplanted liver lobes, collagen deposition marked with arrows; scale bars 200 μm. (d) Quantification of the collagen positive area in each group with respect to sham with ImageJ software, n = 5 fields per group. Statistical analysis was performed with Student's t-test; the P value represented as sham vs. transplanted groups (****P ≤ 0.0001). | ||
4. Discussion
ECM-based polymer hydrogels have emanated as the most appropriate scaffolds for supporting cell growth and differentiation both in vitro and in vivo.23,33–37 The biocompatibility of DCL hydrogels with liver cells has been reported earlier in the in vitro and in vivo studies but only little work has been done to explore its efficacy to deliver cells directly into the liver. In the current study, we developed and characterized a decellularized rat liver ECM-based hydrogel and used it as a vehicle to transplant mouse primary hepatocytes and ICOs directly into the mouse liver to achieve enhanced cell engraftment. We used DCL to transplant primary mice Hep and organoids as we can get the liver ECM from a single rat in abundance when compared to the amount of ECM obtained from mice livers. Interspecies utilization of ECM derived scaffolds and hydrogels has been demonstrated to support the growth, differentiation, and functions of hepatic cells in previous studies.23,34,37 In our study, a negligible increase in the circulating blood immune cells in mice clearly demonstrated the immunocompatibility of rat derived DCL. We used DCL at a concentration of 10 mg mL−1 for cell transplantation because this concentration was found to exhibit better mechanical properties and rapid gelation. The shear thinning behaviour of the DCL with higher storage modulus than loss modulus supported post-injection stability of the hydrogel and remained localized at the point of injection. An earlier study used 20 mg mL−1 liver extracellular matrix for in vivo hepatocyte transplantation. The study has shown that hepatocyte transplantation using DCL is efficient in immunodeficient mice via the subcutaneous route.33 The biodegradability and minimal systemic immune response clearly revealed the safety of fabricated DCL when transplanted via the sub-capsular region of the liver in healthy control mice models. Previous studies have illustrated that freshly isolated or cryopreserved hepatocytes given through the splenic route or via the portal vein repopulate the damaged liver parenchyma.9–11 However, there might be a cell loss during the transfer of hepatocytes from the spleen to the liver, causing reduced engraftment of cells in the liver and transplantation of hepatocytes via the portal vein can result in portal vein occlusion as stated earlier.5–7 We attempted to deliver DCL encapsulated cells and organoids in the liver beneath the liver capsule to ensure that a greater number of cells are engrafted in the liver and at the same time the chances of portal vein occlusion are mitigated. Our observations revealed that as compared to cells without DCL or with the commercial ECM, cells encapsulated within the DCL had better engraftment in the liver (Fig. 4g). Also, the observation that proliferation of cells (PCNA staining), hepatocyte-specific gene expression and albumin levels were significantly higher in the Hep/ICO-DCL animals compared to sham, clearly indicated that the transplanted cells significantly contributed towards repopulating the damaged parenchyma. As compared to Hep-DCL, ICO-DCL liver lobes exhibited a significant increase in the number of proliferating cells. We also observed an increased neo-vascularization in the ICO-DCL as compared to the Hep-DCL liver lobes, suggesting a combined role of ECM proteins and growth factors of both DCL and ICO respectively in inducing greater cellular proliferation and angiogenesis when compared to sham and Hep-DCL livers.Studies have shown that injured hepatocytes activate hepatic stellate cells and increase collagen deposition.38–40 In terms of transplantation few studies have shown that transplantation of mesenchymal stem cells and induced pluripotent stem cells improves liver function post-injury by ameliorating fibrosis and this is achieved by differentiation of the transplanted cells into hepatocytes thereby replenishing the damaged parenchymal population of the liver.41–44 Similarly in our study, Hep and ICO transplantation has resulted in global reduction in fibrosis of the damaged liver (Fig. S8, ESI†), and an overall improvement in liver function, certainly due to the migration and differentiation of the transplanted cells (Fig. S5, ESI†).
ICOs have been successfully isolated from adult bile ducts and also cultured from Lgr5+ liver stem cells under specific in vitro conditions. They are bipotential cells and have the ability of long-term propagation in vitro.45 The ability of human cholangiocyte organoids to repair biliary epithelium has been recently shown.46 In another study, the authors have reported that human embryonic stem cell-derived expandable hepatic organoids can efficiently engraft into and repopulate the liver parenchyma of FRG mice (mice used as a robust model to characterize in vivo liver engraftment during transplantation assays) and at three months following transplantation show increased human albumin levels in the serum. It has been earlier reported that cell differentiation and functions in organoid grafts are maximally attained at about one month.47 Yang et al.48 reported transplantation of bioprinted hepatocyte organoids derived from HepaRG cells in animal models of liver failure. They observed that post-organoid transplantation, there was a decrease in liver injury enzymes and an improvement in liver function after four weeks. In accordance with these results, cell proliferation (Fig. 4h–k) and neovascularization (Fig. 6) were clearly observed in organoid transplanted animals on day 14 in our study as well, along with an increased expression of albumin and CK19 in the transplanted liver lobes. Transcriptomics analysis of the transplanted liver lobes also revealed the upregulation of genes involved in cell proliferation and regeneration (Ki67, e2f6) in ICO and Hep transplanted animals w.r.t sham (Fig. 7). Therefore, we believe that our ICO and Hep transplanted animals would have shown further improvement in liver functions after three or four weeks if the study had been evaluated at later time points.
A recent study described the establishment of a long-term 3D organoid culture system from mouse and human primary hepatocytes.49 The adult hepatocyte-derived organoids exhibited transcriptional profiles resembling those of proliferating hepatocytes after partial hepatectomy (PHx). We also observed a significant upregulation of many genes associated with hepatocyte proliferation and regeneration in both hepatocyte and ICO transplanted liver lobes in comparison to that seen in the transplanted liver lobes of sham animals. Genes such as Ezh1, which is an epigenetic regulator of liver regeneration and IL-6, have been shown to be upregulated during PHx.50,51 In our study, a higher expression of biliary markers was also observed in the hep-DCL liver lobes as compared to sham liver lobes. It has been earlier published using genetic tracing techniques that hepatocytes do contribute towards the formation of primitive ductules in response to chronic liver damage.52 Some of the progenitor cell marker genes such as Prom1 (prominin 1), Cdh6 (cadherin 6), Mcam, and Slc2a2 (solute carrier family 2 member 2) were increased in CCl4 sham groups without any cell transplantation, possibly because of the progenitor cell response to injury in these animals. It is possible that adult healthy mature hepatocytes might have also undergone in vivo reprogramming into proliferative bipotent progenitor cells in response to chronic liver injury.53–55 In our study, we observed an increase in liver function related proteins such as albumin which may have occurred due to the improvement in overall liver function on account of the cell/organoid transplantation followed by a series of cellular events such as replacement of the damaged parenchymal cells, along with maturation and differentiation of the organoids into hepatocytes. Reduction in liver injury enzymes such as AST and ALT levels in the serum is a physiological event that takes a longer time period to reverse, hence, we did not observe an improvement in liver enzymes during our study duration. A study by Yang et al.48 also showed that only after four weeks of bioprinted organoid transplantation, the levels of liver injury enzymes such as AST, ALT and bilirubin have significantly reduced w.r.t sham.
Tracking of the labelled transplanted cells would provide clear insights into the contribution and differentiation of the transplanted cells vis-à-vis native resident cells of the liver.
5. Conclusion
In this study, we report the successful delivery of mouse primary Hep and ICOs encapsulated in DCL hydrogels directly into an injured mouse liver. The transplanted ICOs show enhanced engraftment, proliferation and angiogenesis in vivo restoring some of the liver functions and reducing liver fibrosis without any adverse effects. Using ICOs can serve as an effective regenerative therapy of the liver in patients with chronic liver injury.Author contributions
Conceptualization: Savneet Kaur and Pedro M. Baptista; methodology: Dinesh Mani Tripathi, Natalia Sanchez-Romero, Pedro M. Baptista, and Sourabh Ghosh; experimentation: Impreet Kaur, Ashwini Vasudevan, Aarushi Sharma, Arka Sanyal, Iris Pla Palacin, Natalia Sanchez-Romero, and Aarti Sharma; formal analysis and investigation: Hamed Hemati, Archana Rastogi, Savneet Kaur, and Dinesh Mani Tripathi; writing – original draft preparation: Impreet Kaur and Ashwini Vasudevan; writing – review and editing: Ashwini Vasudevan, Savneet Kaur, and Seeram Ramakrishna; funding acquisition: Savneet Kaur and Dinesh Mani Tripathi; and supervision: Savneet Kaur, Dinesh Mani Tripathi, Pooja Vijayaraghavan, Sourabh Ghosh, and Shiv K Sarin.Data availability
All data supporting this article are provided within the main text as well as in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The study was partly funded by the Department of Science and Technology through an Indo-ASEAN project (CRD/2019/000120) and from an ILBS-IITD collaborative project. The visit of Prof. Pedro Baptista to ILBS and a part of the study was funded by the TATA fellowship. We also acknowledge funding from the Science & Engineering Research Board (SERB), Government of India (SRG/2019/002128). The authors I. Kaur and A. Vasudevan were supported by the ICMR-SRF fellowship ICMR-45/5/2020-PHY/BMS and ICMR-5/3/8/27/ITR-F/2022-ITR.References
- J. J. Nostedt, A. M. James Shapiro, D. H. Freed and D. L. Bigam, Can. J. Surg., 2020, 63, E135 CrossRef PubMed.
- M. Lauerer, K. Kaiser and E. Nagel, Visc. Med., 2016, 32, 278–285 CrossRef PubMed.
- K. A. Soltys, A. Soto-Gutiérrez, M. Nagaya, K. M. Baskin, M. Deutsch, R. Ito, B. L. Shneider, R. Squires, J. Vockley, C. Guha, J. Roy-Chowdhury, S. C. Strom, J. L. Platt and I. J. Fox, J. Hepatol., 2010, 53, 769–774 CrossRef.
- H. Yamana, A. Inagaki, T. Imura, Y. Nakamura, H. Nishimaki, T. Katano, K. Ohashi, S. Miyagi, T. Kamei, M. Unno and M. Goto, Transplantation, 2022, 106, 1963–1973 CrossRef CAS.
- H. Ogasawara, A. Inagaki, I. Fathi, T. Imura, H. Yamana, Y. Saitoh, M. Matsumura, K. Fukuoka, S. Miyagi, Y. Nakamura, K. Ohashi, M. Unno, T. Kamei and M. Goto, Cell Transplant., 2021, 30, 9636897211040012 Search PubMed.
- M. Goto, C. G. Groth, B. Nilsson and O. Korsgren, Xenotransplantation, 2004, 11, 195–202 CrossRef PubMed.
- M. Goto, J. Tjernberg, D. Dufrane, G. Elgue, D. Brandhorst, K. N. Ekdahl, H. Brandhorst, L. Wennberg, Y. Kurokawa, S. Satomi, J. D. Lambris, P. Gianello, O. Korsgren and B. Nilsson, Xenotransplantation, 2008, 15, 225–234 CrossRef PubMed.
- R. R. Mitry, R. D. Hughes, M. M. Aw, C. Terry, G. Mieli-Vergani, R. Girlanda, P. Muiesan, M. Rela, N. D. Heaton and A. Dhawan, Cell Transplant., 2003, 12, 69–74 Search PubMed.
- H. Nagata, R. Nishitai, C. Shirota, J. L. Zhang, C. A. Koch, J. Cai, M. Awwad, H. J. Schuurman, U. Christians, M. Abe, J. Baranowska-Kortylewicz, J. L. Platt and I. J. Fox, Gastroenterology, 2007, 132, 321–329 CrossRef CAS PubMed.
- T. Tricot, J. De Boeck and C. Verfaillie, Cells, 2020, 9, 566 CrossRef CAS.
- H. El Baz, Z. Demerdash, M. Kamel, S. Atta, F. Salah, S. Hassan, O. Hammam, H. Khalil, S. Meshaal and I. Raafat, Exp. Clin. Transplant., 2018, 16, 81–89 Search PubMed.
- G. Brolén, L. Sivertsson, P. Björquist, G. Eriksson, M. Ek, H. Semb, I. Johansson, T. B. Andersson, M. Ingelman-Sundberg and N. Heins, J. Biotechnol., 2010, 145, 284–294 CrossRef.
- Y. Duan, A. Catana, Y. Meng, N. Yamamoto, S. He, S. Gupta, S. S. Gambhir and M. A. Zern, Stem Cells, 2007, 25, 3058–3068 CrossRef CAS.
- S. Nantasanti, A. de Bruin, J. Rothuizen, L. C. Penning and B. A. Schotanus, Stem Cells Transl. Med., 2016, 5, 325 CrossRef CAS PubMed.
- G. Calà, B. Sina, P. De Coppi, G. G. Giobbe and M. F. M. Gerli, Front. Bioeng. Biotechnol., 2023, 11, 1058970 CrossRef.
- D. T. U. H. Lam, Y. Y. Dan, Y. S. Chan and H. H. Ng, Cell Regener., 2021, 10, 1–19 CrossRef.
- C. Sang, J. Lin, S. Ji and Q. Gao, Clin. Cancer Bull., 2024, 3, 1–18 CrossRef.
- C. A. Rimland, S. G. Tilson, C. M. Morell, R. A. Tomaz, W. Y. Lu, S. E. Adams, N. Georgakopoulos, F. Otaizo-Carrasquero, T. G. Myers, J. R. Ferdinand, R. L. Gieseck, F. Sampaziotis, O. C. Tysoe, A. Ross, J. M. Kraiczy, B. Wesley, D. Muraro, M. Zilbauer, G. C. Oniscu, N. R. F. Hannan, S. J. Forbes, K. Saeb-Parsy, T. A. Wynn and L. Vallier, Hepatology, 2021, 73, 247–267 CrossRef CAS PubMed.
- F. J. M. Roos, H. Wu, J. Willemse, R. Lieshout, L. A. M. Albarinos, Y. Kan, J. Poley, M. J. Bruno, J. de Jonge, R. Bártfai, H. Marks, J. N. M. IJzermans, M. M. A. Verstegen and L. J. W. van der Laan, Clin. Transl. Med., 2021, 11, e566 CrossRef CAS.
- M. M. A. Verstegen, F. J. M. Roos, K. Burka, H. Gehart, M. Jager, M. de Wolf, M. J. C. Bijvelds, H. R. de Jonge, A. I. Ardisasmita, N. A. van Huizen, H. P. Roest, J. de Jonge, M. Koch, F. Pampaloni, S. A. Fuchs, I. F. Schene, T. M. Luider, H. P. J. van der Doef, F. A. J. A. Bodewes, R. H. J. de Kleine, B. Spee, G. J. Kremers, H. Clevers, J. N. M. IJzermans, E. Cuppen and L. J. W. van der Laan, Sci. Rep., 2020, 10, 1–16 CrossRef.
- H. Ijima, S. Nakamura, R. P. Bual and K. Yoshida, J. Biosci. Bioeng., 2019, 128, 365–372 CrossRef CAS.
- A. Ravichandran, B. Murekatete, D. Moedder, C. Meinert and L. J. Bray, Sci. Rep., 2021, 11, 1–12 CrossRef PubMed.
- A. E. Loneker, D. M. Faulk, G. S. Hussey, A. D’Amore and S. F. Badylak, J. Biomed. Mater. Res., Part A, 2016, 104, 957–965 CrossRef CAS.
- S. Biswas, A. Vasudevan, N. Yadav, S. Yadav, P. Rawal, I. Kaur, D. M. Tripathi, S. Kaur and V. S. Chauhan, ACS Appl. Bio Mater., 2022, 5, 4354–4365 CrossRef CAS.
- A. Vasudevan, N. Majumder, I. Sharma, I. Kaur, S. Sundarrajan, J. R. Venugopal, P. Vijayaraghavan, N. Singh, S. Ramakrishna, S. Ghosh, D. M. Tripathi and S. Kaur, ACS Biomater. Sci. Eng., 2023, 9, 6357–6368 CrossRef CAS PubMed.
- M. Krüger, R. A. Samsom, L. A. Oosterhoff, M. E. van Wolferen, H. S. Kooistra, N. Geijsen, L. C. Penning, L. M. Kock, P. Sainz-Arnal, P. M. Baptista and B. Spee, J. Cell. Mol. Med., 2022, 26, 4949 CrossRef.
- A. Subramanian, P. Tamayo, V. K. Mootha, S. Mukherjee, B. L. Ebert, M. A. Gillette, A. Paulovich, S. L. Pomeroy, T. R. Golub, E. S. Lander and J. P. Mesirov, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 15545–15550 CrossRef CAS.
- M. Kanehisa and S. Goto, Nucleic Acids Res., 2000, 28, 27 CrossRef CAS PubMed.
- A. Liberzon, C. Birger, H. Thorvaldsdóttir, M. Ghandi, J. P. Mesirov and P. Tamayo, Cell Syst., 2015, 1, 417–425 CrossRef CAS PubMed.
- S. Tyanova, T. Temu, P. Sinitcyn, A. Carlson, M. Y. Hein, T. Geiger, M. Mann and J. Cox, Nat. Methods, 2016, 13(9), 731–740 CrossRef CAS PubMed.
- L. T. Saldin, M. C. Cramer, S. S. Velankar, L. J. White and S. F. Badylak, Acta Biomater., 2017, 49, 1–15 CrossRef CAS.
- M. W. Tibbitt and K. S. Anseth, Biotechnol. Bioeng., 2009, 103, 655 CrossRef CAS PubMed.
- J. S. Lee, J. Shin, H. M. Park, Y. G. Kim, B. G. Kim, J. W. Oh and S. W. Cho, Biomacromolecules, 2014, 15, 206–218 CrossRef CAS PubMed.
- K. H. Hussein, K. M. Park, L. Yu, H. H. Kwak and H. M. Woo, Mater. Sci. Eng., C, 2020, 116, 111160 CrossRef CAS.
- M. Liu, X. Zeng, C. Ma, H. Yi, Z. Ali, X. Mou, S. Li, Y. Deng and N. He, Bone Res., 2017, 5(1), 1–20 CrossRef.
- T. Hoshiba, H. Lu, N. Kawazoe and G. Chen, Expert Opin. Biol. Ther., 2010, 10, 1717–1728 CrossRef CAS.
- X. Zhang and J. Dong, Biochem. Biophys. Res. Commun., 2015, 456, 938–944 CrossRef CAS PubMed.
- J. X. Jiang and N. J. Török, Curr. Pathobiol. Rep., 2013, 1, 215–223 CrossRef.
- M. E. Guicciardi and G. J. Gores, Semin. Liver Dis., 2010, 30, 402 CrossRef CAS PubMed.
- M. R. Elliott, F. B. Chekeni, P. C. Trampont, E. R. Lazarowski, A. Kadl, S. F. Walk, D. Park, R. I. Woodson, M. Ostankovich, P. Sharma, J. J. Lysiak, T. K. Harden, N. Leitinger and K. S. Ravichandran, Nature, 2009, 461, 282 CrossRef CAS PubMed.
- T. Tadokoro, S. Murata, M. Kato, Y. Ueno, T. Tsuchida, A. Okumura, Y. Kuse, T. Konno, Y. Uchida, Y. Yamakawa, M. Zushi, M. Yajima, T. Kobayashi, S. Hasegawa, Y. Kawakatsu-Hatada, Y. Hayashi, S. Osakabe, T. Maeda, K. Kimura, A. Mori, M. Tanaka, Y. Kamishibahara, M. Matsuo, Y. Z. Nie, S. Okamoto, T. Oba, N. Tanimizu and H. Taniguchi, Sci. Transl. Med., 2024, 16, eadg0338 CrossRef CAS PubMed.
- P. Liu, Y. Mao, Y. Xie, J. Wei and J. Yao, Stem Cell Res. Ther., 2022, 13(1), 1–20 CrossRef.
- A. Ghavamzadeh, M. Sotoudeh, A. P. Hashemi Taheri, K. Alimoghaddam, H. Pashaiefar, M. Jalili, F. Shahi, M. Jahani and M. Yaghmaie, Ann. Hematol., 2018, 97, 327–334 CrossRef CAS PubMed.
- D. van der Helm, M. C. Barnhoorn, E. S. M. de Jonge-Muller, I. Molendijk, L. J. A. C. Hawinkels, M. J. Coenraad, B. van Hoek and H. W. Verspaget, J. Cell. Mol. Med., 2019, 23, 6238–6250 CrossRef CAS.
- M. Huch, C. Dorrell, S. F. Boj, J. H. Van Es, V. S. W. Li, M. Van De Wetering, T. Sato, K. Hamer, N. Sasaki, M. J. Finegold, A. Haft, R. G. Vries, M. Grompe and H. Clevers, Nature, 2013, 494(7436), 247–250 CrossRef CAS.
- F. Sampaziotis, D. Muraro, O. C. Tysoe, S. Sawiak, T. E. Beach, E. M. Godfrey, S. S. Upponi, T. Brevini, B. T. Wesley, J. Garcia-Bernardo, K. Mahbubani, G. Canu, R. Gieseck, N. L. Berntsen, V. L. Mulcahy, K. Crick, C. Fear, S. Robinson, L. Swift, L. Gambardella, J. Bargehr, D. Ortmann, S. E. Brown, A. Osnato, M. P. Murphy, G. Corbett, W. T. H. Gelson, G. F. Mells, P. Humphreys, S. E. Davies, I. Amin, P. Gibbs, S. Sinha, S. A. Teichmann, A. J. Butler, T. C. See, E. Melum, C. J. E. Watson, K. Saeb-Parsy and L. Vallier, Science, 2021, 371, 839–846 CrossRef CAS PubMed.
- S. Wang, X. Wang, Z. Tan, Y. Su, J. Liu, M. Chang, F. Yan, J. Chen, T. Chen, C. Li, J. Hu and Y. Wang, Cell Res., 2019, 29(12), 1009–1026 CrossRef CAS PubMed.
- H. Yang, L. Sun, Y. Pang, D. Hu, H. Xu, S. Mao, W. Peng, Y. Wang, Y. Xu, Y. C. Zheng, S. Du, H. Zhao, T. Chi, X. Lu, X. Sang, S. Zhong, X. Wang, H. Zhang, P. Huang, W. Sun and Y. Mao, Gut, 2021, 70, 567–574 CrossRef CAS.
- H. Hu, H. Gehart, B. Artegiani, C. LÖpez-Iglesias, F. Dekkers, O. Basak, J. van Es, S. M. Chuva de Sousa Lopes, H. Begthel, J. Korving, M. van den Born, C. Zou, C. Quirk, L. Chiriboga, C. M. Rice, S. Ma, A. Rios, P. J. Peters, Y. P. de Jong and H. Clevers, Cell, 2018, 175, 1591–1606.e19 CrossRef CAS.
- G. Xie, Y. Song, N. Li, Z. Zhang, X. Wang, Y. Liu, S. Jiao, M. Wei, B. Yu, Y. Wang, H. Wang and A. Qu, Hepatobiliary Surg. Nutr., 2022, 11, 199–211 CrossRef PubMed.
- K. Yanger, Y. Zong, L. R. Maggs, S. N. Shapira, R. Maddipati, N. M. Aiello, S. N. Thung, R. G. Wells, L. E. Greenbaum and B. Z. Stanger, Genes Dev., 2013, 27, 719–724 CrossRef CAS PubMed.
- S. Sekiya and A. Suzuki, Am. J. Pathol., 2014, 184, 1468–1478 CrossRef CAS.
- T. Katsuda, M. Kawamata, K. Hagiwara, R. U. Takahashi, Y. Yamamoto, F. D. Camargo and T. Ochiya, Cell Stem Cell, 2017, 20, 41–55 CrossRef CAS PubMed.
- S. Park, S. In Hwang, J. Kim, S. Hwang, S. Kang, S. Yang, J. Kim, W. Kang, K. H. Kim, D. W. Han and Y. H. Paik, Stem Cell Res. Ther., 2019, 10, 1–11 CrossRef.
- B. D. Tarlow, C. Pelz, W. E. Naugler, L. Wakefield, E. M. Wilson, M. J. Finegold and M. Grompe, Cell Stem Cell, 2014, 15, 605–618 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: All supporting data and detailed methods. Fig. S1–S6 and Table S1. See DOI: https://doi.org/10.1039/d4tb01503g |
| ‡ Equal author contribution. |
| This journal is © The Royal Society of Chemistry 2025 |