
From experimental studies to computational approaches: recent trends in designing novel therapeutics for amyloidogenesis
Pooja
Ghosh
 *a,
Agnibin
Kundu
b and
Debabani
Ganguly
*c
*a,
Agnibin
Kundu
b and
Debabani
Ganguly
*c
aCentre for Interdisciplinary Sciences, JIS Institute of Advanced Studies & Research (JISIASR) Kolkata, JIS University, GP Block, Sector-5, Salt Lake, Kolkata 700091, West Bengal, India. E-mail: poojaghosh@jisiasr.org
bDepartment of Medicine, District Hospital Howrah, 10, Biplabi Haren Ghosh Sarani Lane, Howrah 711101, West Bengal, India
cCentre for Health Science & Technology, JIS Institute of Advanced Studies & Research (JISIASR) Kolkata, JIS University, GP Block, Sector-5, Salt Lake, Kolkata 700091, West Bengal, India. E-mail: debabani@jisiasr.org
First published on 12th December 2024
Abstract
Amyloidosis is a condition marked by misfolded proteins that build up in tissues and eventually destroy organs. It has been connected to a number of fatal illnesses, including non-neuropathic and neurodegenerative conditions, which in turn have a significant influence on the worldwide health sector. The inability to identify the underlying etiology of amyloidosis has hampered efforts to find a treatment for the condition. Despite the identification of a multitude of putative pathogenic variables that may operate independently or in combination, the molecular mechanisms responsible for the development and progression of the disease remain unclear. A thorough investigation into protein aggregation and the impacts of toxic aggregated species will help to clarify the cytotoxicity of aggregation-mediated cellular apoptosis and lay the groundwork for future studies aimed at creating effective treatments and medications. This review article provides a thorough summary of the combination of various experimental and computational approaches to modulate amyloid aggregation. Further, an overview of the latest developments of novel therapeutic agents is given, along with a discussion of the possible obstacles and viewpoints on this developing field. We believe that the information provided by this review will help scientists create innovative treatment strategies that affect the way proteins aggregate.
1. Introduction
Currently, the biological phenomenon that is being explored the most is the aggregation of proteins and peptides into amyloid. The formation of aberrant protein aggregates in the brain caused by this misfolding may interfere with regular cellular processes and hasten the progression of various disorders. These pathologies are commonly known as protein misfolding disorders (PMDs) or amyloidosis which includes various types of localized and systemic diseases such as insulin-derived amyloidosis, frontotemporal dementia, diabetes type 2, Alzheimer's, Parkinson's, and Huntington's diseases, as well as spongiform encephalopathies, oncogenic disorders, etc. These disorders can have a variety of causes for protein misfolding, such as environmental stress, genetic mutations, or a mix of the two. Once misfolded, the proteins frequently set off a series of events that aggregate additional misfolded proteins, further impairing brain tissue and ultimately resulting in the symptoms associated with these illnesses.To date, various therapeutic approaches have been designed in order to impede the amyloid aggregation pathway.1,2 Finding substances that interfere with the protein aggregation pathway by interacting with the amyloidogenic protein or peptide, boosting protein stability, avoiding misfolding of proteins, blocking protein self-assembly, and preventing the disintegration of protein fibres is one of the significant methods for preventing amyloidosis. In this regard, numerous therapeutic substances, including vitamins,3 polyphenols,4 carbohydrates, antibodies,5 lipids,6 metal chelators7 and nanomaterials,8 have been found to be highly effective in inhibiting the aggregation of amyloidogenic proteins and peptides and the production of amyloid.9 Despite persistent efforts, the fundamental mechanism of amyloidogenesis still remains unclear.
A number of reviews, concentrating on small molecule inhibitors of protein aggregation, have been written previously on the topic of protein aggregation, its molecular aspects, and ways for inhibiting it.10,11 For instance, Xiang et al. reviewed the phenomena of protein aggregation, focusing on natural products that are employed to suppress this process.12 Similar to this, Liang and Li have reported a review article highlighting the role of peptide-based inhibitors13 and Dhouafli and coworkers showed the potential of phenolic compounds14 in suppressing the protein aggregation process. Recent research has shown that polymers are effective substitutes for small molecules in the suppression of protein aggregation.15 They can effectively prevent protein denaturation and promote refolding, which makes them a valuable tool in the fight against aggregation-related problems. Considering the importance of polymers, various review articles have been published focusing on their significant roles in suppressing amyloidogenesis.16,17 Nonetheless, the majority of review articles that have been published focus on either computational tools or experimental biophysical methodologies to explore the amyloid fibrillation process. However, there are several studies that have integrated both experimental and computational methods to design novel therapeutic approaches.18,19 In this regard, most recently, Chari et al. have investigated the impact of Hsc70 structural variants on preventing amylin aggregation using both experimental and computational tools.20 By combining experimental and theoretical results, this report establishes a mechanism via which the interaction of Hsc70 with human islet amyloid polypeptide (hIAPP) monomeric species breaks protein–protein bonds, mainly by protecting the β-sheet edges of the Hsc70-β-sandwich. Through blocking the exposed edges of the β-sandwich, especially at the β5–β8 area along the α-helix interface, the unique conformational dynamics of Hsc70's α-helices may improve hIAPP binding. As a result, fibril development was inhibited. Even though several research articles have been published, to the best of our knowledge, there is limited review in the literature21,22 that focuses in-depth on both the computational and experimental approaches for designing novel therapeutics for amyloidosis. Therefore, it is necessary to do a thorough assessment that covers all aspects of protein aggregation, including its causes, effects, and current inhibitors, with a particular emphasis on the combination of both computational and experimental approaches.
In the current review article, our main goal is to present the state of recent developments in therapeutic strategies for amyloidogenesis. Special emphasis is given on the broad applicability of experimental and computational approaches that are developed for designing novel anti-amyloidogenic drugs in regulating the process of protein fibrillation. In the initial part of the review article, we have provided a general overview of protein misfolding and the structural model of amyloid fibrils, followed by a discussion of various factors and driving forces that affect the protein aggregation pathway. Furthermore, we have summarized a list of computational and experimental biophysical techniques that are majorly employed for probing amyloid aggregation. We have also highlighted the application of computational approaches for studying protein aggregation in human disorders along with a detailed discussion on the structural effects of various types of anti-amyloidogenic drugs explored so far. Finally, a brief illustration of the difficulties and potential for this field's future has been provided at the end of the article.
2. A general overview of the amyloid protein aggregation process
2.1 Protein misfolding and structural models of amyloid fibrils
Protein folding is a spontaneous and robust process to ensure protein function within the cell. Multiple quality control pathways are present in the endoplasmic reticulum (ER) to guarantee the proper folding such as enzymes, posttranslational modification, molecular chaperones, formation of disulfide bonds, degradation of misfolded proteins, etc. In the cell, protein degradation and folding are also carefully balanced. However, in spite of all protections (quality control pathways), dysfunctional interactions often result in misfolding and aggregation and thus perturb the assigned cellular function followed by lethal amyloidosis diseases such as Alzheimer's, Parkinson's, Huntington's diseases, etc. At the initial stage, a few peptides assemble through the interaction between their exposed hydrophobic patches to form water-soluble dimers – which is known as nucleation – followed by the growth of larger and insoluble aggregates or oligomers. In the nucleation step, peptides are interconnected by the formation of intermolecular β-sheets with intermolecular main chain hydrogen bonding,23 and these are called cross β-sheets where β-sheets are placed perpendicular to the fibril axis. Several copies of oligomers stick together to form protofibrils followed by the formation of amyloid fibrils (Fig. 1) of 7–12 nm in diameter.24,25 These oligomers are more toxic than the fibrils. Also, the misfolded proteins can exist as amorphous aggregates, which are granular, less ordered and unstructured without having any ordered intermolecular interaction26 and preferably not greater than ∼1 μm in size.27 Both oligomers and fibrils are toxic in amorphous aggregates.28Fig. 2 represents the schematic overview of diverse structures formed by a polypeptide chain. Among the various aggregates, amyloid fibrils are the most usual type. These fibrils disrupt the natural function of the cell. The loss-of-function and/or gain-in-toxicity lead to neurodegenerative and systemic diseases. Not only the Aβ peptide, but some other proteins also form amyloid fibrils with similar morphology, cross-β structure, alternative polar and hydrophobic interaction along the fibril axis, rigid structure and also resistance to denaturation and degradation.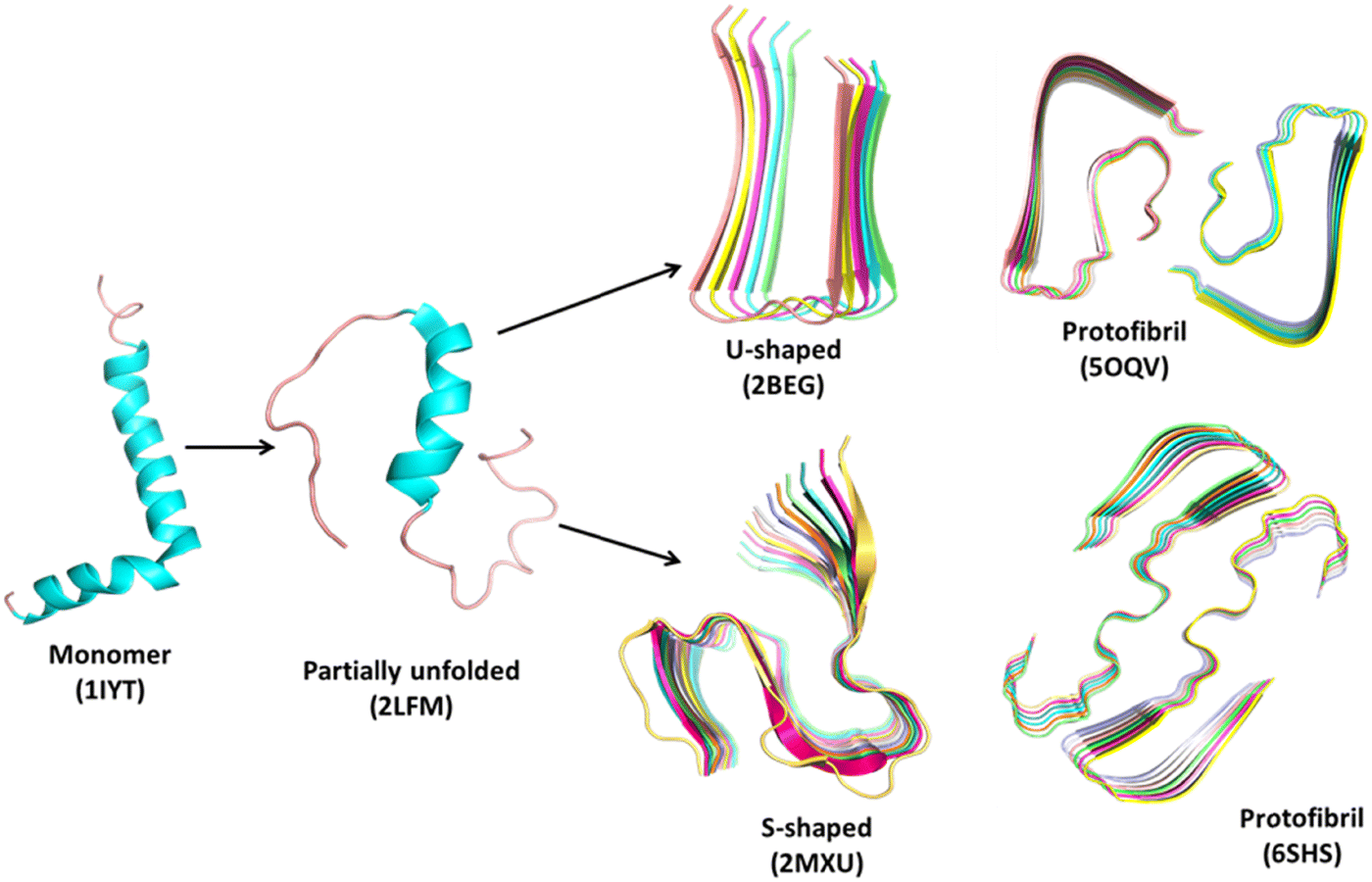 | ||
| Fig. 1 Representative structures of the amyloid aggregation pathway using Aβ-peptide as a model system. The structures are obtained from the Protein Data Bank (https://www.rcsb.org). The monomer (1IYT)29 and partially unfolded (2LFM)30 form, both U-shaped (2BEG)31 and S-shaped (2MXU)32 forms of oligomers and respective protofibrils (5OQV33 and 6SHS34) are shown in a cartoon representation. PDB codes of the downloaded structures are given in parenthesis. | ||
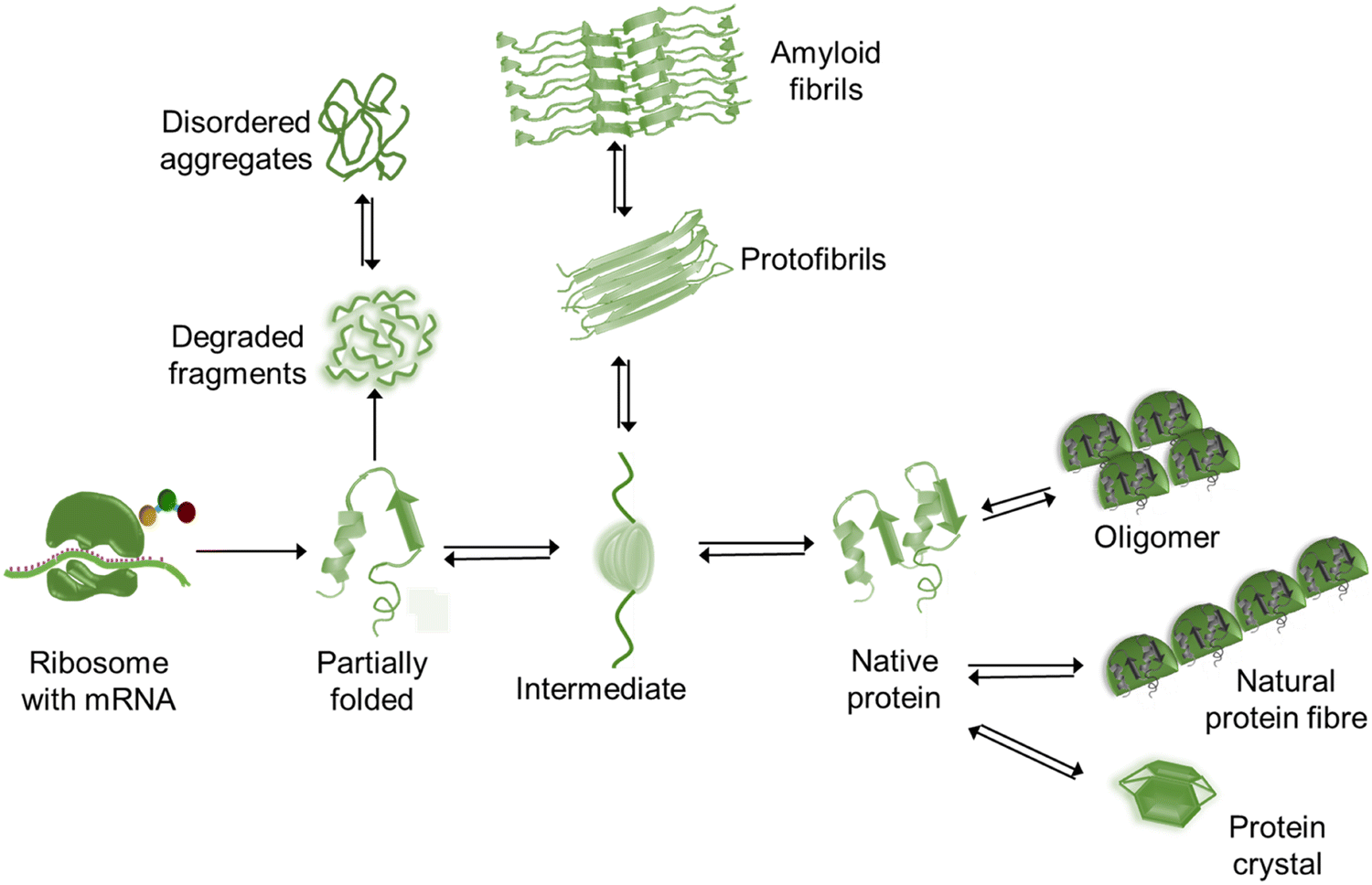 | ||
| Fig. 2 A schematic overview of diverse structures formed by a polypeptide chain. | ||
The aggregation events include primary nucleation, elongation, secondary nucleation and fragmentation (Fig. 3).35 Primary nucleation is a stage when monomers in solution interact to form soluble aggregates. In elongation the length of such aggregates increases due to their interaction with another monomer. Thus, elongation is the vital step of aggregation. When the surface of the soluble aggregates binds with other monomers the event is named the secondary nucleation. Molecular interactions such as salt bridges, trapped water molecules by van der Waals forces, and hydrophobic patches usually enhance the possibility of the secondary nucleation. Finally, in fragmentation the existing fibrils break down to multiple fibrils and as mentioned before, the soluble oligomers are more neurotoxic than the fibrils.36
 | ||
| Fig. 3 Schematic representation of the aggregation mechanism:37,38 (a) primary nucleation, (b) elongation, (c) secondary nucleation, and (d) fragmentation. Blue and red rings represent monomers and aggregates respectively. | ||
2.2 Different types of systemic and localized amyloidogenic diseases
Over the past few decades, amyloid protein aggregation has been linked to more than fifty human diseases, making it one of the most fascinating new frontiers in the pharmaceutical and biomedical field.39,40 These lethal human disorders are referred to as protein misfolding disorders (PMDs) or amyloidosis. Amyloidosis is mainly categorized as systemic and localized amyloidosis. The most common organs to be affected by systemic amyloidosis are the heart, kidneys, and nervous system. This can lead to peripheral and autonomic neuropathies, nephrotic syndrome, renal failure, and congestive heart failure. On the other hand, in the case of localized amyloidosis, as the amyloids are accumulated at the production site, only one organ is affected. Alzheimer's disease is considered as one of the most common and well understood forms of localized amyloidosis. The Aβ peptide, the primary constituent of amyloid plaques in Alzheimer's disease, causes synaptic damage, diffuse neuronal dysfunction, and neuronal cellular apoptosis. Immunoglobulin light chains secreted by plasma cells on mucosal surfaces misfold and deposit locally in localized AL amyloidosis, obstructing or creating a mass effect in the tracheobronchial tree, bladder, ureter, or breast. Amyloid deposition in the pancreas or at the site of insulin delivery can be caused by misfolding of the islet amyloid polypeptide or high-dose exogenous insulin. Table 1 represents the different types of localized and systemic amyloidosis and their respective precursor protein/peptide (Fig. 4).| Type of disease | Precursor protein/peptide |
|---|---|
| Localized amyloidosis | |
| Injection-localized amyloidosis | Insulin |
| Type II diabetes | Islet amyloid polypeptide (IAPP) |
| Cutaneous lichen amyloidosis | Keratins |
| Corneal amyloidosis associated with trichiasis | Lactoferrin |
| Pituitary prolactinoma | Prolactin |
| Cataract | γ-Crystallins |
| Medullary carcinoma of the thyroid | Calcitonin |
| Aortic medial amyloidosis | Medin |
| Alzheimer's disease | Amyloid β peptide |
| Parkinson's disease | α-Synuclein |
| Frontotemporal dementia | Tau |
| Huntington's disease | Huntington protein |
| Amyotrophic lateral sclerosis | Superoxide dismutase 1 |
| Spongiform encephalopathy (mad cow disease) | Prion protein |
| Systemic amyloidosis | |
| (AL) Light chain amyloidosis | Immunoglobulin light chains or fragments |
| (AH) Heavy chain amyloidosis | Immunoglobulin heavy chain |
| (ATRR) Familial amyloidotic polyneuropathy 1 | Mutant transthyretin |
| (ATRR) Senile systemic amyloidosis | Wild-type transthyretin |
| (Aβ2M) Haemodialysis related amyloidosis | β2-Microglobulin |
| Hereditary systemic amyloidosis/(ALys) lysozyme amyloidosis | Mutant lysozyme |
| Oncogenic | p53 amyloid aggregation |
| (AA) Secondary amyloidosis | Serum amyloid A protein |
| Finnish hereditary amyloidosis | Fragments of mutant gelsolin |
| (ACys) Cystatin amyloidosis | Cystatin C |
| (Afib) Fibrinogen amyloidosis | Variants of fibrinogen α-chain |
| (ABriPP) BriPP amyloidosis | BriPP |
| (ApoAI) ApoAI amyloidosis | Fragments of apolipoprotein AI |
| (ApoAII) ApoAII amyloidosis | Fragments of apolipoprotein AII |
| (ApoAIV) ApoAII amyloidosis | Fragments of apolipoprotein AIV |
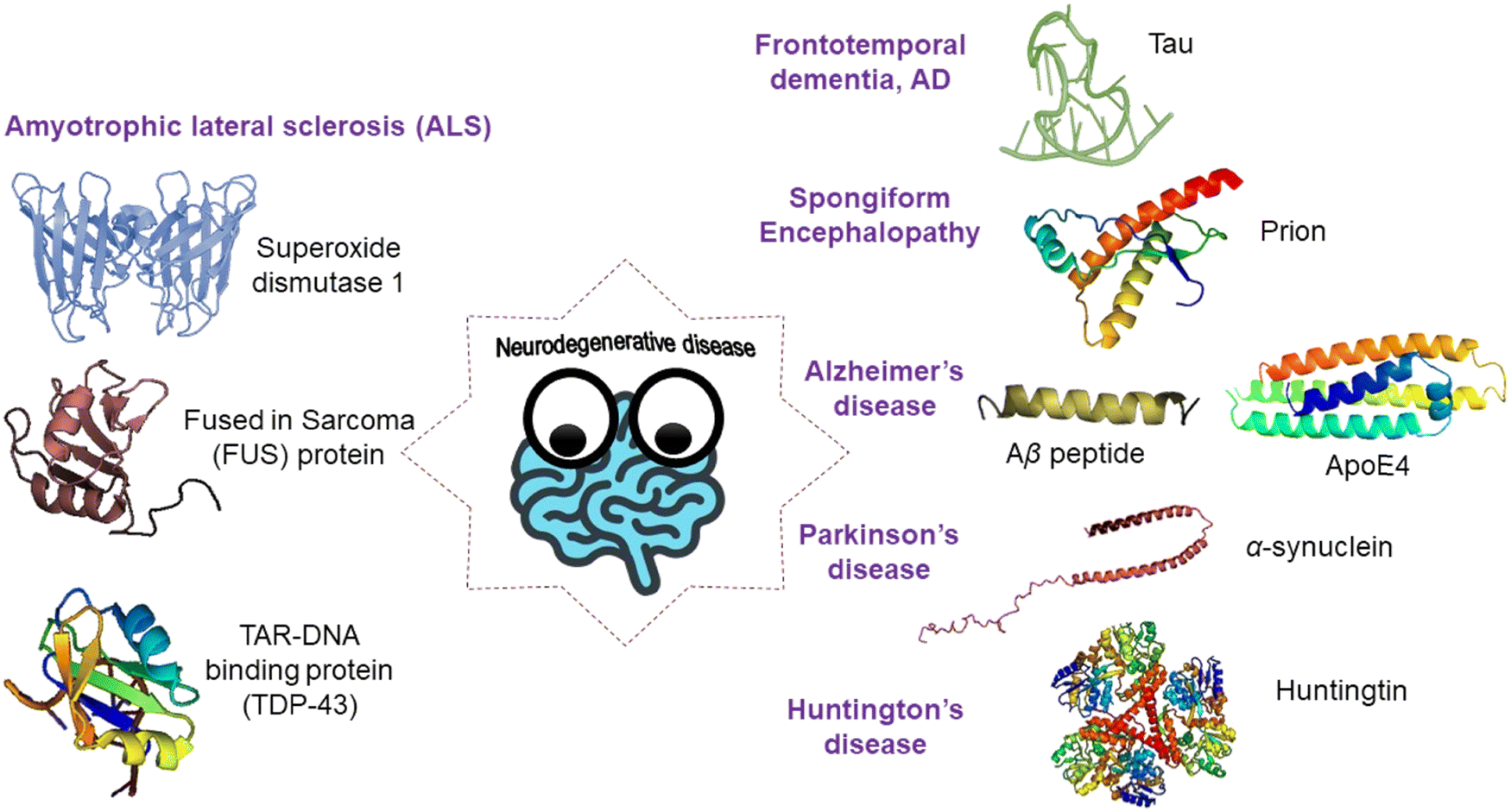 | ||
| Fig. 4 A schematic illustration of various types of amyloidogenic disorders and the proteins/peptides associated with them. | ||
3. Characterization techniques to probe amyloid aggregation
3.1 Computational techniques to probe amyloid aggregation
The aggregation of amyloid proteins into fibrils is considered as the key factor of neurodegenerative diseases. Understanding the mechanism of formation of insoluble amyloid aggregates needs extensive research in the field. However, experiments on suitable protein models demand a significant amount of money, time and sample. The computational technique is considered as a crucial tool to not only understand the mechanism but also to design the therapeutic strategies to combat the lethal neurodegenerative diseases.41 Among the various approaches, molecular dynamics (MD) simulation, all-atom, coarse-grained and multi-scale, is an emerging tool to model and study various aggregations to find the related function.42 It spans a considerable amount of time to record substantial oligomer formation events in the brain. All-atom molecular dynamics simulations are computationally expensive, whereas low-resolution coarse-grained models are employed to explore the nanosecond to microsecond timescale, allowing for the observation of transient events.43 Importantly, such a low-resolution coarse-grained model fails to capture certain features including hydrogen bonding, electrostatic interactions, stacking interactions, the effect of side chains, etc. On the other hand, multi-scale MD simulation for protein aggregation effectively navigates multiple potential energy surfaces, transitioning from low-resolution model to more detailed descriptions,44 enabling fast and nearly accurate exploration of the system. The choice of the force field is extremely important to get the accurate MD simulation prediction. In addition to the MARTINI force field, Man et al.45 reported that other recently developed all atom force fields, such as AMBER99-ILDN, AMBER14SB, CHARMM22, CHARMM36, and CHARMM36m, are more appropriate to realistically model amyloid peptides. The nature of the monomer and the conformational ensemble of the aggregates for recognizing the events of the aggregation are also investigated from the MD simulation. The protein sequence has a vital impact on aggregation. The protein misfolding or unfolding is nothing but the loss of native intramolecular contacts and the formation of some non-native contacts. β-Aggregation enhances in the presence of β-branched hydrophobic residues – valine and isoleucine – and aromatic residues – tryptophan and phenylalanine, with a low net charge. Aggregation could be inhibited by replacement of nonpolar residues with polar residues. The aggregation propensity depends on the sequence of the peptide. Such specific composition of amino acids or the pattern of amino acids initiating the aggregation is called the aggregation prone region (APR). This region is identified based on the properties of the amino acids such as polarity, hydrophobicity, size, structure, aromaticity, gravity, beta-sheet propensity, etc. Mutations in the APR can often inhibit aggregation. A vast number of APR and aggregation propensity predictor tools, both sequence-based and structure-based, are available and well-reviewed earlier.41,46 The sequence-based predictors, e.g. AGGRESCAN, WALTZ, FoldAmyloid, TANGO, etc., are designed based on the physicochemical properties of the amino acids, the sequence pattern, secondary structure propensities, intramolecular and pair-wise contact, etc. The structure-based predictors are developed based on the solvent accessible surface area and the overall structural information of the protein obtained from the MD simulation.46 Other internal regulations, e.g. post-translational modification (PTM), can also control the protein conformation and thus the aggregation propensities.Protein-aggregation databases are also available which curate detailed information available from the experiments. Such databases are immensely useful to predict the chance and the propensity of aggregation of any protein. A few of such recent databases are ZipperDB,47 WALTZ-DB 2.0,48 CPAD 2.0,49 AL-Base,50 Aggrescan3D 2.0,51 A3D-MODB,52etc. The rate of aggregation under the experimental condition is an important feature for studying protein aggregation and therapeutics. The aggregation kinetics depends on the point mutation, polarity, hydrophobicity, secondary structural propensity of amino acids, experimental conditions (such as pH, temperature, ionic strength), etc. There are a few recent tools available to estimate the aggregation kinetics, namely, AggreRATE-Disc,53 AggreRATE-Pred,54 AbsoluRATE,55etc. AggreRATE-Disc is a sequence-based predictor, which uses machine learning to estimate the change in aggregation rate upon point mutation. AggreRATE-Pred is a structure-based predictor developed by the same group for estimating the rate of aggregation. AbsoluRATE is another tool for predicting absolute rates of protein aggregation under physiological conditions. All such techniques are extremely useful to tackle protein aggregation by explaining its mechanism and kinetics computationally in addition to the experimental research.
3.2 Experimental biophysical methods to study protein aggregation
Apart from the computational methods, a variety of biophysical techniques are typically used to characterize amyloid protein aggregates in order to identify aggregated proteins and look into the possible effects of various treatments on protein aggregation. Even though numerous studies have been conducted employing sophisticated and varied ways to identify protein fibrillation, it is imperative to condense and validate the methodologies for protein fibrillation detection using readily available laboratory resources, as opposed to employing intricate procedures. Here, we have selected a few popular biophysical methods that are frequently used to probe amyloid aggregation.Thioflavin T (ThT) is a commonly used indicator dye for in vitro detection of amyloid fibrils.56 After being excited at 450 nm, this dye binds to amyloid fibrils with structures rich in β-sheets and produces a fluorescence signal at about 482 nm. The rotational immobilisation of the core C–C bond between the benzothiazole and aniline rings has been identified as the mechanism responsible for the increase of fluorescence following binding to amyloid.57 Congo red (CR) is another amyloid specific marker dye that is widely used for sensing protein amyloid fibrils. At lower concentrations and neutral pH, its absorption spectra in aqueous solution have a peak absorption at 490 nm, resulting in a red hue solution. The conformational restriction of CR molecules occurs when they are coupled to β-sheet-rich fibrils, resulting in the adoption of a particular orientation where their long axis is parallel to the fibril axis. Furthermore, the hydrophobic properties of aggregated proteins and the nature of folding/unfolding intermediates can be distinguished using Nile red, 8-anilino-1-naphthalenesulfonic acid (ANS), and the dimeric form of ANS, namely, 4,4′-bis-1-anilinonaphthalene-8-sulfonate (bis-ANS). Additionally, structural transitions in proteins can be observed using UV-Visible spectroscopy.58 Tryptophan residues are typically employed to track structural alterations in proteins because of the absorption phenomenon that occurs naturally. The most significant chromophore found in a protein's UV region is tryptophan. However, tyrosine and phenylalanine have a smaller relative contribution to intrinsic fluorescence. Tryptophan's indole ring and tyrosine's phenol ring make these residues sensitive to solvent polarity, which can be utilised to monitor absorbance and identify protein aggregation and unfolding.
A promising biophysical tool for tracking secondary and tertiary structural changes in proteins/peptides is circular dichroism (CD) spectroscopy,59 which is the most widely used approach in protein chemistry and structural biology. Due to the presence of the amide chromophore in the peptide bonds, CD signals in the far UV range between 250 and 190 nm can provide secondary structural information about proteins. The CD signals in this wavelength range are caused by two different types of electron transitions: π → π* transitions at ∼208 and 190 nm, and a n → π* transition at ∼222 nm. In addition to identifying the secondary structure content of proteins, far UV-CD spectroscopy can track the folding and unfolding of proteins. Furthermore, protein aromatic residues’ environment-dependent CD spectra in the near UV region (250–300 nm) provide qualitative tertiary structural information. Synchrotron radiation CD (SRCD) spectroscopy is another novel method for characterizing the protein folding state. With the presence of absorbing materials (buffers, salts, etc.), SRCD expands the capabilities of traditional CD spectroscopy by speeding up data acquisition, boosting the signal-to-noise ratio, and extending the spectral range. Additionally, through the use of other spectroscopic techniques like Raman and Fourier-transform infrared (FTIR) spectroscopy, one can ascertain the relative predominance of the various secondary structures in a protein.
In addition to molecular spectroscopic characterization, the morphology and fibrillar growth can be understood through microscopic examination. In this regard, a variety of microscopic techniques, including atomic force microscopy (AFM), field emission scanning electron microscopy (FESEM), scanning electron microscopy (SEM), transmission electron microscopy (TEM), high-resolution transmission electron microscopy (HRTEM), fluorescence microscopy, and confocal microscopy, were used by researchers to examine the morphology and fibrillar growth. Owing to the quick and easy sample preparation, TEM imaging is a visually appealing and trustworthy technique for verifying the existence of protein aggregates. In most cases, heavy metal staining such as with uranyl acetate or lead citrate is necessary to improve biological sample contrast for TEM imaging. On the other hand, AFM imaging does not require any staining, and the imaging parameters are more closely aligned with the in vivo setting. Moreover, the most straightforward method of determining a protein's aggregation status is to measure its particle size or molecular weight. Protein aggregation-related changes in particle size have been widely studied using dynamic light scattering (DLS), which measures particle size distributions in the nanometre to micrometre range. Polyacrylamide gel electrophoresis (PAGE) is another useful method for assessing the aggregation of protein. This technique works effectively for detecting aggregates with disulfide connections in both reduced and nonreduced circumstances. More recent research has used cryo-electron microscopy, solid state NMR, and X-ray crystallography to provide extremely comprehensive structural data regarding the molecular architecture and composition of fibrils.
4. Application of computational approaches for studying protein aggregation in human disorders
4.1 Amyloid peptide and Alzheimer's disease
Alzheimer's disease (AD) is a slowly progressive neurodegenerative disease caused by the accumulation of extracellular amyloid-β (Aβ) peptide plaques in the brain and neurofibrillary tangles60,61 of tau protein. Both plaques and tangles are insoluble and highly ordered aggregates. In the AD brain, Aβ and tau help each other to convert from the normal to toxic state via a feedback loop.62The Aβ peptide is a 40 to 42 residue peptide (Aβ42 or Aβ40), cleaved from its transmembrane amyloid precursor protein (APP) by β-secretases and γ-secretases.63 Mutations nearby the cleavage region of the APP gene raise the ratio between Aβ42 and Aβ40, triggering the early development of AD.64 The Aβ42 variant30 is the dominant biological component of AD and a biomarker in serum and cerebrospinal fluid for AD detection.65 Aβ amyloid oligomers contain a very dynamic β-sheet structure with conformational plasticity66,67 and form a cross-β-sheet structure of two intermolecular β-sheets, β1 (residue range: 12–24 in Aβ40 and 18–26 in Aβ42) and β2 (residue range: 30–40 in Aβ40 and 31–42 in Aβ42).68 Strong intermolecular interactions among the above-mentioned aggregation prone domains are the key point of the self-assembly of peptides. The N- and C-terminus of Aβ42 adopt L and S like shape respectively, giving overall LS shaped fibrils.33 Aβ40 dimers are stabilized by intermolecular hydrophobic and/or π–π stacking interactions between the hydrophobic core and the N-terminal domain and Aβ42 dimers on the other hand are stabilized by interactions between the central hydrophobic core and the C-terminal domain.69 The C-terminal domain of the peptide promotes fibril formation due to its solvent accessibility.70,71
Numerous computational studies on AD hypothesis, structure, stability, and disruption of toxic aggregates and the potential treatment of the disease are published over the year.70,72–75 A number of extensive reviews76–78 have discussed the MD simulation approaches towards understanding the mechanism of Aβ aggregation. However, the computational cost related to the aggregation of Aβ42 peptides is high. It is worth using a coarse-grained low-resolution model to study the fibril formation of any small to medium length peptide. Very recently, Katila et al.79 explained that an oscillating oriented external electric field (OEEF) not only prevents the formation of Aβ plaques but also breaks the plaques noninvasively using 1–1.5 μs long molecular dynamics simulation. Also, two non-equilibrium MD simulations showed that ultrasonic waves and infrared laser irradiation can disrupt the amyloid fibrils80 Upon the application of ultrasonic waves the fibrils are broken down by cavitation, whereas under infrared laser irradiation the hydrogen bonds between carbonyl and amine groups forming the cross beta-sheets are broken and they can re-form again after the irradiation. Instead, if a water molecule forms the hydrogen bonds, then the chance of reconstituting the cross β-sheet by forming H-bonds does not exist. Ma et al. exploited coarse-grained molecular dynamics simulation81 to model the S-shaped polymorphic structure of the fibril and then to explore the free energy of binding of the monomer to the surface of the existing fibril, fibril stability, heterogeneity and helical twisting of protofilaments. Such monomer binding finally leads to secondary nucleation and elongation. The authors used GPU-enabled coarse-grained protein force field openAWSEM82 for long MD simulation of a relatively large system. Pasieka et al.83 used molecular docking to find how 1-benzylamino-2-hydroxyalkyl derivatives affect the aggregation kinetics of Aβ40.
During Aβ aggregation, β-hairpin formation initiates intermolecular β-sheet structures. An insightful review78 on MD simulations of Aβ40 peptides and Aβ(16–22) fragments in the air–water interface and on the GM1 clusters of the neuronal cell membrane explained the interplay of secondary structures in aggregate formation. As the peptide is formed by both hydrophilic and hydrophobic amino acids, the peptide forms more beta-hairpin structure at the interface accelerating aggregation of Aβ oligomers.84 Full length Aβ40 interacts with GM1-glycan clusters through its HHQ (residue range: 13–15) segment and also initiates the formation of an α-helix while interacting with GM1-glycan.85 A couple of recent MD simulations from various articles also reported polyphenols as potential inhibitors of Aβ (residue range: 16–22) aggregation.86–89 The salt bridges and hydrogen bonds within the region are disrupted as polyphenols interact by forming hydrogen bonds with both donors and acceptors, leading to the destabilization of β-sheet formation.
4.2 α-Synuclein and Parkinson's disease
Parkinson's disease (PD) is another deadly neurodegenerative disease characterized by both physical and neuropsychiatric symptoms. In PD, neurons in the brain die slowly, causing the loss of dopaminergic neurons of the substantia nigra. A low level of dopamine causes irregular functioning of the brain which affects the nervous system and the parts of the body controlled by the nerves. The cause of PD is unknown and is a topic of current research. However, several changes have been identified in the PD brain. The presence of Lewy bodies, the abnormal clumps of proteins found in brain cells, is the hallmark of all synucleinopathies including PD. In the PD brain the major substances in the Lewy body are the aggregates of α-synuclein, a protein transcribed by the SNCA (Synuclein Alpha) gene. This is a small (14 kb) protein90 comprised of three different domains,91 namely, N-terminal amphipathic helix (residue range: 1–60), non-amyloid component (NAC) domain (residue range: 61–95), and disordered C-terminal acidic domain (residue range: 96–140). The N-terminal helix anchors α-synuclein to membranes, the central NAC determines the membrane binding affinity and the disordered C-terminal acidic domain is weakly associated with the membrane. α-Synuclein is a well-studied protein both experimentally and computationally. It is an intrinsically disordered protein, which adopts an ordered fibrillar structure upon aggregation. The pathway or the mechanism of α-synuclein from normal monomers to toxic aggregates is still unclear and constitutes a thrust area of active research. The intrinsically disordered nature and high conformational dynamics make it very challenging to track the actual mechanism of aggregation by both in vitro and in silico methods. The truncation of the C-terminal domain enhances the aggregation of α-synuclein.92,93 Balupuri et al.94 used all-atom molecular dynamics simulation and found the development of an α-strand intermediate in the NAC region followed by the formation of α-sheet which facilitates the early aggregation. Uversky et al.95 claimed experimentally the presence of a partially folded intermediate in α-synuclein aggregation by either a decrease in pH or an increase in temperature. Yu et al.96 confirmed the formation of β-hairpin in the region of amino acids ranging between 38 and 53 in the N-terminus using coarse-grained molecular dynamics simulation. An important finding from the same group was two mutations Ala30Pro and Ala53Thr that enhance the formation of β-hairpin. Yu et al.96 also found that Gly47Val blocks β-hairpin formation, retarding further aggregation. Zhang et al.97 recently used an atomistic discrete MD simulation of both alpha-synuclein monomers and dimers to report the creation of a helical structure around 8–32 residues in the N-terminal area with β-sheets in residues 35–56 of the N-terminal domain and residues 61–95 situated in the NAC. These two beta-sheet regions play a pivotal role in amyloid-like aggregation.98 The unstructured C-terminal domain wrapped the aggregated parts formed by the other two domains. Dimerization however enhances the aggregation propensity while decreasing the intra-peptide interactions. Another study recently revealed that the prevention of α-synuclein fibril formation could be possible by applying an external electric field using molecular dynamics simulations.99 A report revealed the effect of a crowded cellular environment during the self-assembly of α-synuclein using coarse-grained MD simulation with the MARTINI3 force field.100 The study claimed that both crowed and saline environment facilitated the liquid–liquid phase separation of α-synuclein. The peptide starts to aggregate at 500 μM which is the reported critical concentration for LLPS of α-synuclein. The key finding of this study is the possibility of adopting perpendicular orientation of the disordered peptide chains to minimize C-terminal inter-chain electrostatic repulsion. Being intrinsically disordered is indeed a challenge to underpin the mechanism of α-synuclein self-assembly. For MD simulation, the main concern is to use and develop force fields that critically capture the transient intermediates of fibril formation. A recent MD simulation of the NAC domain of α-synuclein used the ff14SB AMBER force field and its ff14IDPSFF variation with the grid-based energy correction map (CMAP) to capture both α-helix and β-sheet intermediates of the fibrillation process.101 A combined multiscale simulation and experimental study102 of a membrane–α-synuclein model explored the loss of the helical structure (residues 65–70) near the NAC region and gradual formation of β-sheets. Zhao et al.103 explored the misfolding mechanism of α-synuclein using three different models of fibril structures available in the Protein Data Bank (https://www.rcsb.org) by conventional and steered MD simulation and observed different contribution of van der Waals and electrostatic interactions in each system. A recent combined spectroscopy and molecular dynamics simulation confirmed the presence of the subpopulation of stable local structures of the α-synuclein monomer which was hypothesized to form oligomers and fibrils.1044.3 Huntingtin and Huntington's disease
In Huntington's disease (HD) the nerve cells in the brain gradually become dysfunctional and die with time and the patient faces an uncontrolled body movement. It is a genetically inherited neurodegenerative disease, originating from the abnormal expansion of the cytosine–adenine–guanine (CAG) tri-nucleotide repeat within exon-1 of the HTT gene, also known as the IT15 (interesting transcript 15) gene, encoding the huntingtin protein (348 kDa),105 expressed in all mammalian cells. In nerve cells in the brain, it is expressed in higher concentration. CAG-expansion mutations in the HTT gene result in a polyglutamine (polyQ) tract at the N-terminus of the huntingtin protein that possesses above a critical threshold of ∼35 glutamine residues.106,107 The protein has three domains, namely, N17 (first 17 N-terminal residues), polyQ tract, and two proline rich domains (PRD). Together these domains form toxic fibrils where dynamic proline-rich domains form a bristle-like structure accessing the surface, and the polyQ and N17 regions form the core of the bottlebrush.108 The misfolding of the mutant HTT protein leads to accumulation of amyloid aggregates in the brain. The polyglutamine tract at the N-terminal forms highly ordered insoluble amyloid fibrils with high β-sheet content in the cerebral cortex and in the striatum.109 Higher the length of the polyQ tract, higher the β-sheet content in the structure and hence the aggregation propensity.110,111 In addition, the N17 domain and the polyP rich domain are also essential for the aggregation.112 The N17 domain is intrinsically disordered and adopts a wide variety of conformational states, but initiates and accelerates the aggregation by forming amphipathic alpha helix rich oligomers.113 The proline-rich domain on the other hand is known to decrease the aggregation rate without altering the basic mechanism. MD simulation with electron paramagnetic resonance and solid-state NMR described that both proline-rich domains fold into a polyproline (polyP) II helical conformation.114 It has been studied well that the difference in the toxicity of HTT aggregates grossly depends on the different structure and dynamics of proline-rich domains.115However, the molecular and cellular mechanisms of the occurrence of disease are still unclear. Miller et al.116 suggested the accumulation of insoluble aggregates in the brain. Other groups suggested that the interactions of monomers or the oligomers of HTT with other proteins by polyQ tracts eventually lead to loss of neurons.117 With the help of both experiments and MD simulation, another group118 found that the wild type Siah1-interacting protein lowers the protein levels encoded by exon 1 wild type and mutated HTT, which greatly regulates the aggregation.
4.4 p53 amyloid aggregation in cancer
p53 acts as the main coordinator of cellular stress control. All p53 isoforms bind to DNA and act as a signalling hub by involving in three main functions namely apoptosis, growth arrest and DNA damage repair thereby controlling the cell cycle growth.119–122 Native p53 remains in a dormant state in normal cells by continuous degradation mediated by MDM2 binding.123 p53 often undergoes various post-translational modifications and mutations. Cellular stress, e.g., DNA damage, induces phosphorylation at numerous sites in p53, which weakens MDM2 binding and elevates the p53 level in the cell. Such changes may introduce conformational modulations, which in turn not only affect the interactions between the p53 domain and its binding partner(s), but also control the inter-domain interactions.p53 has a long inseparable relationship with maturation and advancement of cancer. Mutations in p53 domains occur in more than 50% of cancer. According to the IARC TP53 mutation database124 ∼90% of such mutations are detected in the core domain (DNA binding domain) of p53.125 Hence p53 is one of the most important proteins126 in cancer research. There are six cancer related hotspot mutations, namely, R175H, G245S, R248Q, R248W, R273H, and R282W, reported in the DNA binding domain along with other lethal mutations. These mutations may cause structural modification or conformational fluctuations, which in turn cause loss of function of p53 as a tumor suppressor. Among the cancer mutations, a few are responsible for p53 aggregation in the cell. It has been well established to date that p53 aggregates in vitro127,128 causing loss of function as a “cellular gatekeeper” and gain of tumor oncogenic function. Among the various aggregates, prion-like amyloid fibrils are the most common type. These fibrils accumulate in the cytoplasm and nucleus and disrupt the normal cellular function. Among many, one of the causes of p53 inactivation is mutation-driven aggregation. The mutations R110L, R110P, R175H, R248Q, R249S, P250L, E258V, R282W and many more destabilize the DNA binding domain as reported previously129 and initiate aggregation.130 The conformational changes due to mutation may expose the deep routed hydrophobic beta strands among which the 250–257 region, PILTIITL, acts as a nucleating agent. Experimental and computational study suggested that the aggregation in the core domain is amyloidogenic type, rich in β-sheet like conformers. Saha et al.131 recently detected a novel hotspot A138V in pancreatic ductal adenocarcinoma. Singh et al.132 have also reported P152L cancer mutations that up-regulate certain cellular pathways related to tumorigenesis and metastasis. The two new mutations reported by both groups may lead to destabilization of the DNA binding domain, potentially through aggregation or other mechanisms that require further investigation, both experimentally and computationally. R175H promotes pancreatic cancer133 and also initiates aggregation.134 R248Q aggregates at pH 7.2 in vitro.135 Levy et al.136 found strong correlation between R248Q mutation and amyloid-like aggregation in breast cancer cells. Other two mutations R282W and E258V form larger aggregates in vitro.130,137 The authors also suggested that this oncogenic gain of function destabilizes the DBD structure by increasing the accumulation of p53 in cells. This accumulation is probably achieved by the exposure of the deep routed hydrophobic beta strand [S9, residue range: 250–257, PILTIITL] of DBD, which acts as a nucleating agent. Recent computer simulation of the DBD of p53 revealed a region spanning the S6–S7 turn, which is modulated in the presence of the above-mentioned mutations and may cause destabilization of the DBD.138 This information from Pradhan et al.138 is very useful for the advance therapeutic research. The C-terminal tetramerization domain of p53 possesses the aggregation nucleating region (residues 327–332). There are a few mutations, namely, G334V, R337H, R337C, L344P, L330H, R337L, R342P, E349D, etc., reported so far that cause tumorigenesis.139 These mutations destabilize tetramerization assembly and then disrupt the DNA binding ability and transactivation activity. Two mutations outside this range, G334V and R337H, are reported to initiate the aggregation in vitro. The aggregation in the C-terminal tetramerization domain is reported as less amyloidogenic.140 There is an urgent need for research on p53 for a detailed understanding of the mechanism or pathway of p53 inactivation due to mutations. However, mutation is not solely responsible for loss of p53 function, but mutation-driven aggregation also claims substantial contribution towards p53 inactivation. Such aggregations are oncogenic and hence lethal. Thus, investigation of the cause of such aggregations upon specific mutations is crucial. Despite lots of study, both computational and experimental, the mechanism of such aggregation is still unclear and a subject of active research.
5. Various types of therapeutic agents as amyloidogenic inhibitors
5.1 Impact of small molecules
Over the past few years, numerous small molecules have been screened to explore their efficacy in controlling the amyloid fibrillation process and improve the therapeutic strategies of amyloidogenic disorders. The majority of naturally occurring substances that are tested are polyphenols, and the most significant polyphenols include curcumin,141 epigallocatechin gallate (EGCG),142 myricetin,143 quercetin,144 baicalein,145 resveratrol,146 gallic acid,147 ferulic acid,148 caffeic acid,149etc. Curcumin is one of the most explored polyphenols in the biomedical field due to its unique molecular structure. Mounting evidence indicates that curcumin may inhibit the accumulation of proteins linked to a number of neurological disorders, including Alzheimer's, Parkinson's and Huntington's disease.150 It can bind to protein aggregation inclusions and traverse the blood–brain barrier (BBB), making it a promising treatment candidate for neurodegenerative disorders. According to molecular dynamics simulation study, curcumin has the potential to impact the elongation as well as the primary nucleation stage of amyloid fibril formation.151,152 Furthermore, curcumin can be functionalized with other nanomaterials to fabricate new, multifunctional therapeutic agents that successfully restore memory deficits in mice, lower the oxidative stress damage caused by amyloid deposition, and dramatically decrease the burden of amyloid plaques in APP/PS1 transgenic mice (Fig. 5).153 Another natural substance that has drawn special interest for targeting amyloid fibrillization is EGCG, which is readily available and has a minimal toxicity.154 Li et al. used the replica exchange molecular dynamics simulation (REMD) technique to explore the impact of EGCG on the amyloid-beta and human islet amyloid polypeptide (hIAPP) aggregation process.155 The findings revealed that EGCG efficiently reduces intra- and inter-chain contacts in the co-aggregates by focusing on the polar and aromatic residues of Aβ and hIAPP through hydrogen bonding, π–π stacking, and cationic–π interactions. Three primary strategies are majorly used by EGCG to suppress amyloid aggregation. The first involves directly binding to oligomeric species to destroy their structure. Restructuring oligomers and altering their structure is the second strategy and the third strategy involves chelating metal ions to reduce their toxicity.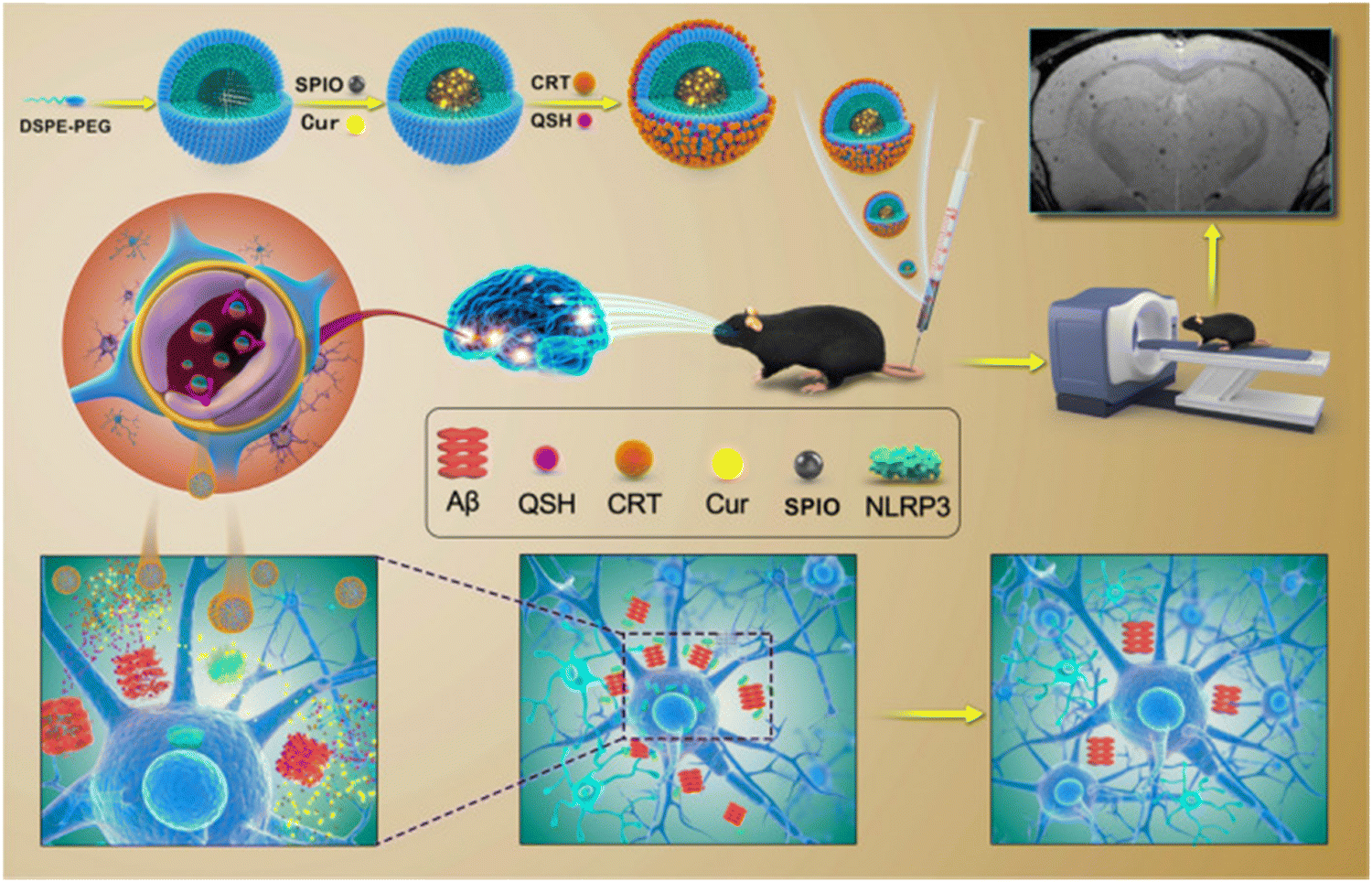 | ||
| Fig. 5 Schematic representation of the extremely sensitive curcumin-conjugated nanotheranostic platform for the detection of amyloid-beta plaques by magnetic resonance imaging and reversing cognitive deficits of AD through NLRP3-inhibition. Adapted from ref. 153. Copyright (2022) BMC, Part of Springer Nature. | ||
Apart from these, surfactants are highly valuable in controlling the process of protein fibrillation because their micelles can resemble the membrane environments found in biological systems. Hydrophobic and electrostatic interactions are typically involved in the production of amyloid fibrils generated by surfactants. In this regard, Siposova and coworkers have investigated the role of non-ionic detergent dodecyl maltoside (DDM), two phospholipids, namely, 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) and 1,2-dihexanoyl-sn-glycero-3-phosphocholine (DHPC), and the detergent–phospholipid mixtures on the insulin aggregation process using both experimental and computational tools.156 According to the molecular modelling, the phospholipids and DDM occupy the same binding sites. DDM participates more in hydrogen bonding than DHPC or DMPC because it contains maltose with multiple oxygen atoms (hydroxylic, glycosidic, and ring). Hydrophobic force of interactions plays a crucial role in keeping phospholipids and DDM stable in their binding locations. Additionally, low molecular weight substances known as “osmolytes,” which include amino acids, sugars and polyhydric alcohols, are created by the cell in times of stress and have been demonstrated to prevent numerous protein aggregation processes.157 The hydration mechanism along with the solvophobic effect plays a crucial role in stabilizing the protein structure and prevention of aggregation. Venkatraman and coworkers investigated the inhibitory as well as amyloid dissociation properties of four osmolytes such as betaine, sarcosine, raffinose, and taurine in an in vitro transforming growth factor-beta induced (TGFBI) peptide aggregation model.158 Interestingly, the osmolytes showed significant efficacy in suppressing and disintegrating amyloid fibrils originating from TGFBIp. Additionally, the osmolytes showed no harmful effects on human corneal fibroblast cells in culture, suggesting that they might be a helpful treatment option for people with TGFBIp corneal dystrophies.
Furthermore, anti-Aβ monoclonal antibodies have also attracted a lot of attention due to their capacity to both break pre-existing aggregates and suppress amyloid-beta fibrillogenesis.159,160 Depending on the precise target, monoclonal antibodies (mAbs) may be utilized to target amyloid fibrils, plasma cell clones, or misfolded amyloidogenic precursors in cardiac amyloidosis (CA) to reduce amyloid elimination through a variety of methods.161,162 Further research indicates that mAbs against cardiac amyloid primarily trigger an immunological response, which is thereafter cleared by phagocytic cells.163 In treating AL amyloidosis, daratumumab has demonstrated outstanding efficacy when used as a combination therapy as well as a monotherapy. Later, in 2021, the Food and Drug Administration (FDA) expedited approval of daratumumab in conjunction with CyBorD for the treatment of AL amyloidosis. According to Sevigny and coworkers, aducanumab, a human monoclonal antibody, can specifically target the accumulated peptide and reduce the amyloid-beta plaques in AD.164 Later, El-Agnaf et al. designed and fabricated antibodies that detected different oligomers (syn-O1, -O2, and -O4) and fibrillar (syn-F1 and -F2) forms of α-syn in a differentiated manner, and interestingly the antibodies syn-O1, -O4, and -F1 were shown to be the most efficient in avoiding neurodegeneration and lowering the buildup of α-syn oligomers in various brain areas.165 In a recent article, Gupta and coworkers have developed single-chain variable fragments (scFvs) in order to combat fibrillar α-syn, a hypothesized disease-relevant variant of α-syn.166 Interestingly, scFvs have been shown in vitro to diminish the generation of insoluble α-syn phosphorylated at Ser-129 (pS129-α-syn), reduce α-syn seed-induced cytotoxicity in a cell model of Parkinson's disease, and prevent the aggregation seeding in a time-dependent way. Various anti-Aβ drugs such as acenumab, lecanemab, gantenerumab, donanemab, β-site Aβ precursor protein cleaving enzyme-1 (BACE1), and BACE2 are presently undergoing clinical studies.167 However, following years of unsuccessful clinical studies, two medications for moderate cognitive impairment (MCI) to early, mild stages of AD have been approved by the FDA. These are lecanemab (Leqembi®) and aducanemab (Aduhelm®), with aducanemab obtaining expedited approval.168,169 Conversely, some mAbs evaluated in Phase III studies, like solanezumab, especially target amyloid-beta monomers, while others, like bapineuzumab and crenezumab, do not distinguish between various amyloid-beta types.170,171
Besides, in vitro and in vivo studies have demonstrated the effective inhibition of the amyloid fibrillation process by a range of quinones, including mitoquinone, embelin, geldanamycin, naphthoquinones, phenanthraquinones, benzoquinones, anthraquinones, coenzyme Q10, and their derivative equivalents.172,173 The asymmetric dipole that exists in the quinone molecule is the primary component that causes quinone to interact with amyloidogenic peptides and proteins. Furthermore, metal ions and metal chelators play a critical role in modulating the amyloid aggregation process.174 Transition metal ions such as Cu and Zn have been found in numerous instances to have significant effects on both the stabilization of soluble hazardous peptide aggregates and the amyloid fibrillation process.175 By creating an aggregation-inert complex, metal ion coordination can also postpone the amyloid beta peptide's self-assembly. In a recent article, Iscen and coworkers have designed and fabricated a cobalt(III) Schiff base complex and further explored its efficacy in preventing the amyloid fibrillation process using both experimental and computational approaches.176 Molecular dynamics simulations of monomeric and pentameric amyloid beta indicate the reduced β-sheet structure development, destabilization of preexisting β-sheets, and aggregation suppression. Overall, this investigation shows the beneficial pharmacological action of the cobalt complex, and these results are in line with the experimental results. With all this evidence, it can be concluded that small molecules possess the capacity to function as effective inhibitors of protein misfolding and aggregation.
5.2 Effect of nanomaterials
As nanotechnology has advanced over the past few decades, a wide range of nanomaterials have been developed, fabricated, and used in various sectors including physics, environmental research, pharmaceutical field and biomedical field.177,178 The outstanding biocompatibility, consistent physicochemical characteristics, and adaptability in synthesis and modification of nanomaterials make them highly promising for addressing the difficulties encountered in the existing applications of therapeutic and diagnostic approaches. In this milieu, considerable effort has been made in this direction to consider ways to improve the effectiveness of amyloidosis treatment from a nanomaterial's viewpoint. Nanomaterials are highly advantageous in targeting amyloid plaques as they can easily pass the blood–brain barrier (BBB), affect amyloid fibril nucleation and cause disintegration of mature fibrils.2,179 Furthermore, due to the fascinating characteristics of certain nanoparticles, they are highly responsive to temperature, light, ultrasound, electricity, and magnetism. These capabilities have led to the gradual development and application of nanomaterials in the study of neurodegenerative illnesses.180Different types of nanomaterials have been fabricated so far to modulate the aggregation of amyloid peptides based on their main composition and dimensions. These include zero-dimensional (0D) nanomaterials, one-dimensional (1D) nanomaterials, two-dimensional (2D) nanomaterials, metal–organic frameworks (MOFs), and self-assembled nanomaterials. In the realm of protein aggregation, zero-dimensional (0D) nanomaterials such as carbon-based nanomaterials,181 inorganic quantum dots,182 organic quantum dots,183 gold nanoclusters,184 gold nanoparticles (GNPs), and metal oxide nanoparticles185 have garnered a lot of attention recently. Numerous researchers have demonstrated that gold nanoparticles (AuNPs) can penetrate the blood–brain barrier, prevent the amyloid beta peptide from aggregating, and degrade Aβ aggregates according to their size, shape, surface charge, functionality, and concentration.186 In a recent article, Yang et al. designed a hybrid nanomaterial, AuNPs@PEG@MIL-101, which showed intriguing properties for possible therapeutic uses in AD through modifying and targeting Aβ aggregation.187 The findings revealed that the nanomaterial may lessen the amount of Aβ40 immobilised on the cell membrane and lower intracellular Aβ40 aggregation. AuNPs@PEG@MIL-101 also showed a protective effect against Aβ40-induced microtubular defects and membrane disruption. The possibility of treating AD using a novel tetrahydroacridine derivative (CHDA) conjugated with AuNPs is investigated in the work by Mojzych et al.188 through acetylcholinesterase inhibition, CHDA's adsorption onto gold surfaces and coupling with AuNPs which in turn enhance its therapeutic potential for the treatment of AD. Furthermore, in another recent article, Mukherjee and Sarkar reported a facile one-pot microwave-assisted synthesis of highly water-soluble carbon quantum dots (CQDs) and investigated their anti-amyloidogenic properties using hen egg-white lysozyme (HEWL) as a model protein, as well as their possible clinical application against protein-linked neurological disorders.189 Apart from these, metal oxide nanoparticles such as ferric oxide nanoparticles (Fe3O4 NPs),190 zinc oxide nanoparticles (ZnO NPs),191 and cerium oxide nanoparticles (CeO2 NPs)192 showed significant potential in controlling the amyloid aggregation process. The exponential rise in research in this area suggests that nanomaterials for controlling amyloid aggregation associated with Alzheimer's disease are not only a hot issue for future study but also have a wide range of possible applications.
Not only zero-dimensional (0D) nanomaterials, but also one-dimensional (1D) nanomaterials such as carbon nanotubes and gold nanorods exhibit remarkable efficacy in modulating the amyloid fibrillation process.193,194 In this regard, Mo et al. have examined the role of hydroxylated single-walled carbon nanotubes (SWCNT-OHs) in human islet amyloid polypeptide (hIAPP) aggregation using a combination of computational and experimental tools.195 In-depth examinations of the interactions between hIAPP and SWCNT-OH show that the inter- and intrapeptide interactions that are essential for the production of β-sheets are considerably weakened by van der Waals, hydrogen bonding, and π-stacking interactions between hIAPP and SWCNT-OH. Overall, the combined experimental and computational findings demonstrate SWCNT-OH's inhibitory action and mechanism against hIAPP aggregation, offering fresh insights for the creation of potential future medications to treat type 2 diabetes. Later, Liu and coworkers have explored the effect of SWCNT-OH on the cytotoxicity and fibrillogenesis of amyloid beta 1–42.196 The results revealed that SWCNT-OH dose-dependently reduces the Aβ42 fibrillation process and disintegrates produced amyloid fibrils (Fig. 6). Additionally, the proportion of hydroxyl groups in SWCNT-OH plays a critical role in its ability to inhibit the Aβ42 fibrillation process. Recently, in another work, a neurotheranostic nanosystem was developed using gold nanorods (GNRs) that functions as a delivery system for therapeutic peptides and as an anti-amyloid agent for AD and can be identified in vivo for the diagnostic purpose of microcomputed tomography (micro-CT).197 Additionally, multiwalled carbon nanotubes (MWCNTs) have numerous benefits over single-walled carbon nanotubes (SWCNTs), including reduced product costs, superior chemical stability, and the capacity to adsorb drugs.198 Considering the advantages of MWCNTs, a system of phospholipid and polysorbate-coated MWCNTs loaded with berberine (BRB) was developed by Lohan et al.199 which enhanced the targeting and imaging capabilities of CNTs as well as demonstrated a notable improvement in memory function. Moreover, metal–organic frameworks (MOFs) have garnered a lot of interest in the biomedical field because of their porous architecture, large surface area, tuneable size, high biocompatibility, and high loading ability. Due to these fascinating physiognomies, materials based on MOFs have been shown to have potential use in the diagnosis and treatment of a number of brain diseases and disorders.200 In this regard, Wang et al. designed and fabricated a porphyrinic MOF-based nanoprobe which in the presence of near infrared light (NIR) prevents amyloid beta aggregation and reduces the amyloid-induced toxicity.201 In another recent article, Kowalczyk and coworkers investigated the dual role of Prussian blue (PB), a subclass of the MOF family, in preventing amyloid beta (1–40) fibrillation and chelating Cu2+.202 Interestingly, the findings revealed that PB nanoparticles (PBNPs) have the ability to decrease the production of typical amyloid-β fibres, which can be identified by ThT fluorescence, and to restore the typical amyloid fibrillation pathway by sequestering or chelating copper, which is present in high concentrations in senile plaques. Most recently, Andrikopoulos and coworkers have developed a biocompatible metal–phenolic network (MPN) using physiological zinc(II) to coordinate a polyphenol EGCG scaffold.203 Interestingly, after adhering to Au NPs, the MPN@AuNP nanostructure demonstrated exceptional strength against amyloid toxicity and Aβ aggregation in vitro. When compared to EGCG alone, MPN@AuNP performed better than EGCG@AuNP because of its porosity and consequently higher surface area. Discrete molecular dynamics simulations were used to further investigate the structure and dynamics of amyloid beta aggregation controlled by the MPN, while density functional theory calculations were used to decipher the atomic detail of Zn(II)–EGCG coordination. While the discovery of nanoparticles has opened a new avenue for the treatment of amyloidogenic disorders, more work is still urgently required, and other innovative nanomaterials should be examined.
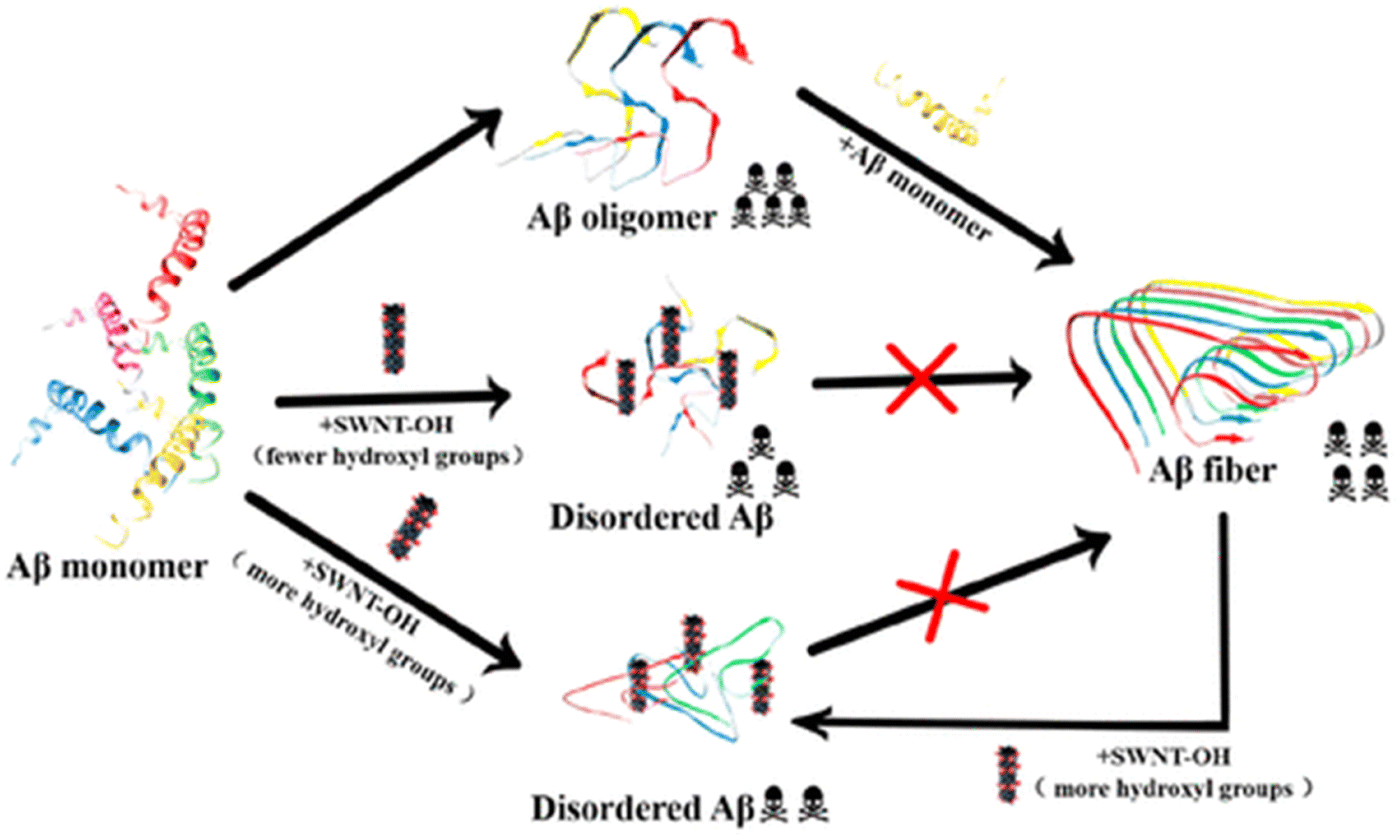 | ||
| Fig. 6 Schematic diagram of inhibition of Aβ42 fibrillogenesis by hydroxylated single-walled carbon nanotubes. Adapted from ref. 196. Copyright (2019) American Chemical Society. | ||
5.3 Synthetic supramolecular approach
The synthetic supramolecular approach has also become a potentially useful tool for altering the protein aggregation process. The formation of novel supramolecular structures involves the integration of two or more chemical entities through host–guest complexation. Over the past few decades, a number of macrocycles and their derivatives have been produced, including cyclodextrins (CDs), calixarenes (CAs), and cucurbiturils (CBs). Under aqueous conditions, these macrocyclic compounds typically have distinctive hydrophobic cavities that serve as hosts for encasing and binding various hydrophobic guest molecules. Their inclusion also depends critically on other forces including hydrogen bonding, electrostatic contact, and molecule size or shape matching. There are various benefits of these macrocyclic molecules: they are (a) highly stable because their skeletons are stiffer than those of flexible peptides; (b) substantially more specific because of greater binding surfaces than those of tiny molecules; (c) primarily aqueous-soluble and biocompatible; and (d) easily functionalized to boost specificity and affinity for the guest. Considering the fascinating physiognomies of macrocycles, numerous macrocycles have been widely fabricated to control the amyloid fibrillation process. Among different macrocycles, phenol containing “chalice-like” macrocycles, calix[n]arenes (n = 4, 5, 6, 8), are one of the most explored supramolecular scaffolds. In this regard, Shinde et al. reported a facile supramolecular approach for impeding the insulin fibrillation process.204 The results revealed that water soluble p-sulfonatocalix[4/6]arene macrocyclic hosts (SCX4/6) may efficiently suppress insulin fibril formation and have the ability to destroy matured amyloid fibrils (Fig. 7).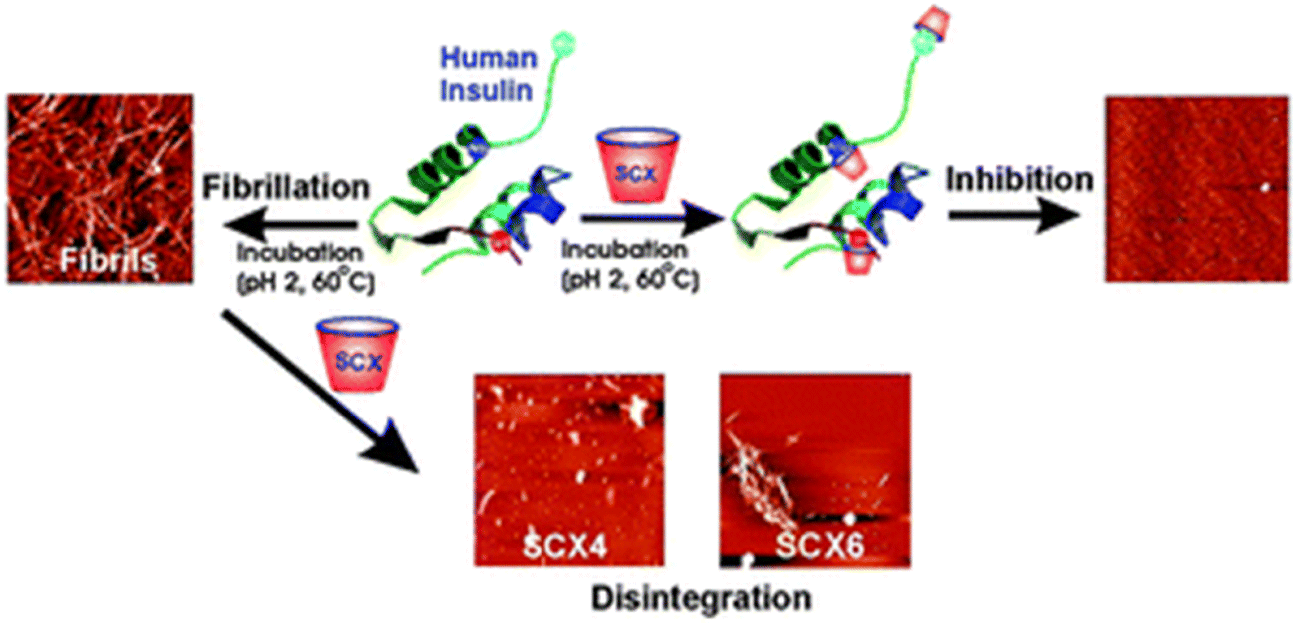 | ||
| Fig. 7 A graphical illustration of prevention of the insulin fibrillation process by p-sulfonatocalix[4/6]arene macrocyclic hosts. Adapted from ref. 204. Copyright (2016) Royal Society of Chemistry. | ||
Furthermore, a combination of computational and experimental techniques was employed to investigate the effect of resorcinarene in inhibiting amyloid beta fibrillation in vitro.205 In another work, Geng et al. fabricated NIR-responsive nanoparticles (NPs) comprised of 4-(dodecyloxy) benzamide-terminated methoxy poly(ethylene glycol), poly-5,5′-(2,5-bis(π 2-octyldodecyl)) 3,6-di(thiophen-2-yl)-2,5-dihydropyrrolo[3,4-c]pyrrole-1,4-dione and amphiphilic guanidinocalix[5]arene (GC5A).2 In the hippocampal regions of AD mice, NPs efficiently crossed the BBB after being exposed to NIR light. This prevented amyloid beta 1–42 from aggregating and caused mature fibrils to disintegrate. Cyclodextrins (CD) are another well-explored supramolecular scaffold that are widely employed for regulating the amyloid fibrillation process. In this regard, Oliveri and coworkers examined the inhibitory effects of functionalizing CD with various moieties on amyloid beta (1–42) aggregation.206 Interestingly, β-CD functionalized with 5-carboxy-8-hydroxyquinoline inhibited the amyloid aggregation process dose-dependently. β-CD functionalized with 5-carboxy-8-hydroxyquinoline at the 6-position exhibited more inhibition than the corresponding 3-functionalized isomer. Later, the same group fabricated a β-CD functionalized porphyrin moiety (CDTHPP) and evaluated its potency in suppressing Aβ42 aggregation.207 The results revealed that CDTHPP binds with the aggregation-prone region (LVFF) of Aβ42 through host–guest interactions and π–π stacking interaction. Furthermore, Xu et al. developed a supramolecular strategy utilizing multivalency to suppress amyloid fibrillation (Fig. 8).208 A vesicular assembly with two different types of host–guest recognition sites on the surface for heteromultivalent binding of various amino acid residues of peptides and proteins was formed by a mixture of macrocyclic cyclodextrin (CD) and calixarene (CA) amphiphiles. The co-assembly's ability to break down matured amyloid fibrils into smaller, non-toxic forms is crucial from a therapeutic perspective. For neuronal cell lines, the CD–CA co-assembly is less harmful and shields cells from the cytotoxicity caused by amyloid aggregation. Besides, there have also been reports of crown ethers modulating the aggregation of amyloid proteins.209 Yokoyama and Mizuguchi have shown the therapeutic efficacy of crown ethers as inhibitors of transthyretin (TTR) amyloidogenesis, based on X-ray crystallographic study, chemical cross-linking assay, and competitive binding experiment with fluorescent probes.210 Overall, the host–guest method has a tremendous deal of potential for therapeutic applications because of its ability to detect proteins and reduce amyloid toxicity. We think these methods will help us find potential anti-amyloid medications to treat amyloidogenic disorders in the near future.
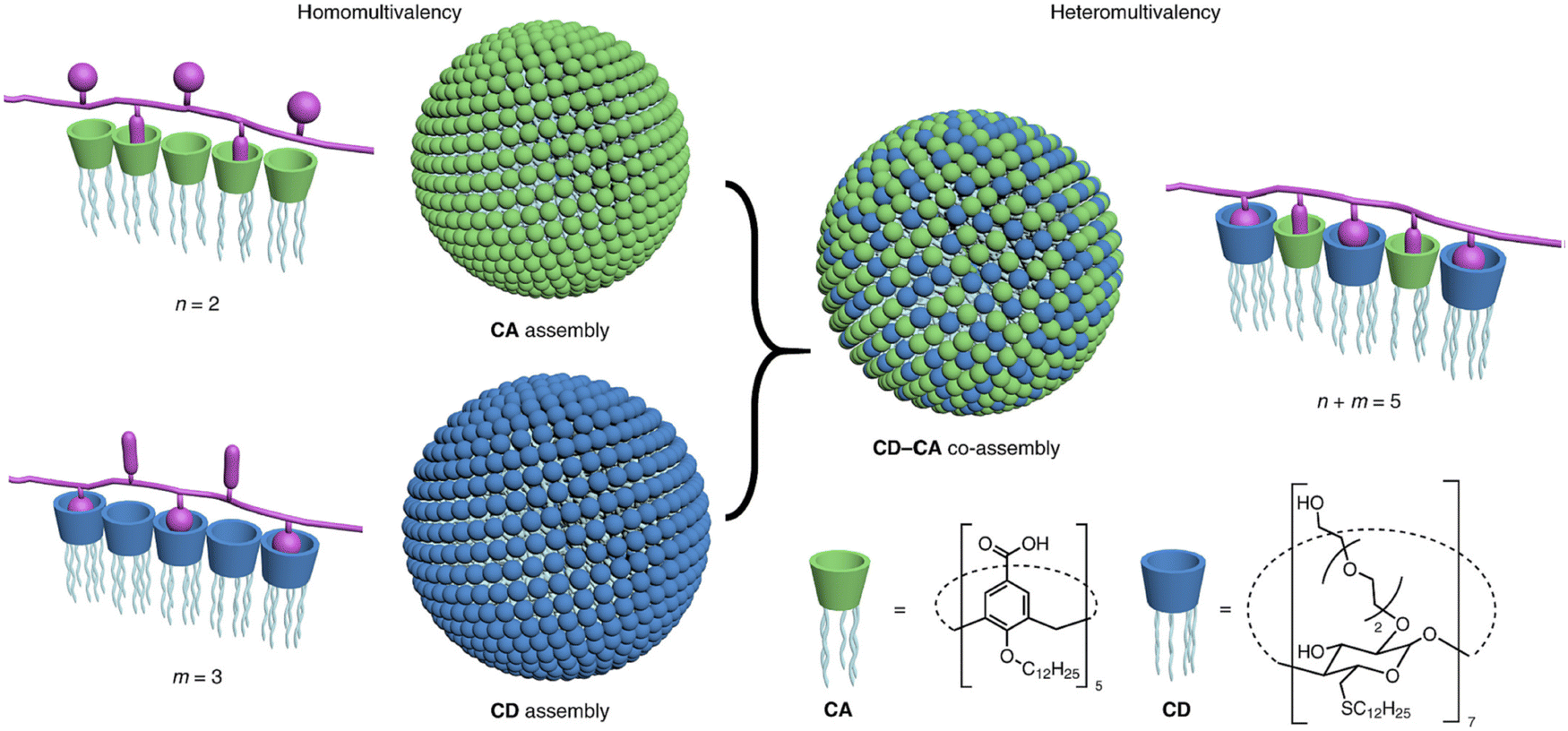 | ||
| Fig. 8 Schematic representation of amphiphilic cyclodextrin (CD) and calixarene (CA) based co-assembly mediated heteromultivalent peptide recognition. Adapted from ref. 208. Copyright (2019) Springer Nature Limited. | ||
5.4 Peptide/peptidomimetic inhibitors
Peptide-based inhibitors have been increasingly explored as potential therapeutics for protein misfolding disorders because of their good biocompatibility, specificity and minimal toxicity.211 Several studies in the literature have documented the anti-amyloidogenic potential of rationally designed short peptides.212,213 Compared to small molecules, the use of short peptide-based inhibitors as anti-amyloidogenic agents is highly preferable due to their multifarious fascinating properties such as the following: (i) peptides possess a high degree of chemical and biological diversity; (ii) peptides can be rationally fabricated by using the necessary knowledge about the target molecule's sequence; and (iii) peptides can be modified to increase their affinity, specificity, and proteolytic resistance. There have been several documented methods for rationalising short peptides, such as altering side groups or peptide termini, or selecting based on the protein/peptide's amyloidogenic sequence. One of the first peptide leads for anti-amyloid medications was the pentapeptide LPFFD, which strongly inhibited amyloid beta fibrillogenesis in both in vitro and in vivo rat models.214 Later, Lin and coworkers fabricated aggregation core-based peptide HKQLPFFEED based on the structure and hydrophobic property of the amyloid beta fragment, which has been shown to prevent amyloid aggregation.215 The low bioavailability and protease stability of short peptides, however, limit their potential therapeutic uses. Furthermore, these limitations have been improved by a number of modification techniques, including the addition of peptidomimetics and N- or C-terminal functionalization.174 In a recent study, it has been demonstrated that α-syn aggregation can be inhibited by the short peptide SuMO1 (15–55), which is generated from ubiquitin-related modifier 1 (SuMO1) and targets two SuMO-interacting motifs inside the N-term area bordering the NAC.216 The diphenylalanine (FF) motif, which is widely recognised for its origin from the amyloid beta peptide linked to Alzheimer's disease, was first discovered in 2003 as the peptide's minimalistic sequence with a significant tendency towards self-association.217 Considering the importance of the FF motif, Yao and coworkers have conjugated FF with ferrocene and explored their role in inhibiting the insulin fibrillation process.218 Moreover, Zhang et al. designed and fabricated a ferrocene modified tripeptide and studied its kinetics as well as mechanistic pathway of amending the insulin fibrillation process.219 Additionally, cyclic peptides (CPs) and their derivatives also act as a novel class of potent inhibitors that reduce aggregation cytotoxicity and stop amyloid protein aggregation.220,221Considering the importance of peptide-based inhibitors in the protein aggregation field, most recently our group proposed a novel method for creating and synthesizing amyloid interaction surfaces employing segments [VF(Aβ(18–19))/FF(Aβ(19–20))/LVF(Aβ(17–19))/LVFF(Aβ(17–20))] generated from the amyloid-promoting sequence of amyloid β-peptide.222 These segments are coupled with side-chain proline containing methacrylate polymers, which show decreased cytotoxicity of amyloid aggregations and act as strong lysozyme amyloidosis inhibitors (Fig. 9). A thorough spectroscopic, microscopic, and computational analysis revealed that the LVFF-conjugated polymer was one of the most effective inhibitors, considerably suppressing lysozyme amyloidosis. These findings present new opportunities for the treatment of lysozyme amyloidosis by revealing the synergistic interaction of side-chain proline-based polymers and the amyloid β-peptide produced from the “segment.” Therefore, all the above findings conclude that peptide-based inhibitors show a lot of promise and might be a wise option for modulating the treatment of amyloidogenic diseases.
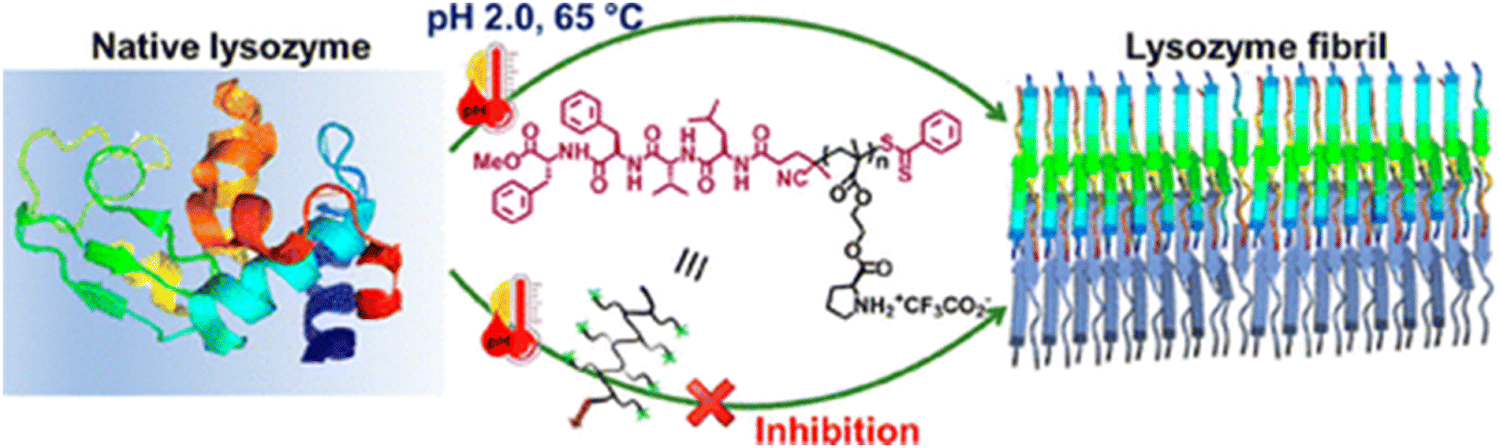 | ||
| Fig. 9 A schematic representation of lysozyme fibril inhibition by peptide-based inhibitors. Adapted from ref. 222. Copyright (2024) American Chemical Society. | ||
5.5 Polymeric materials
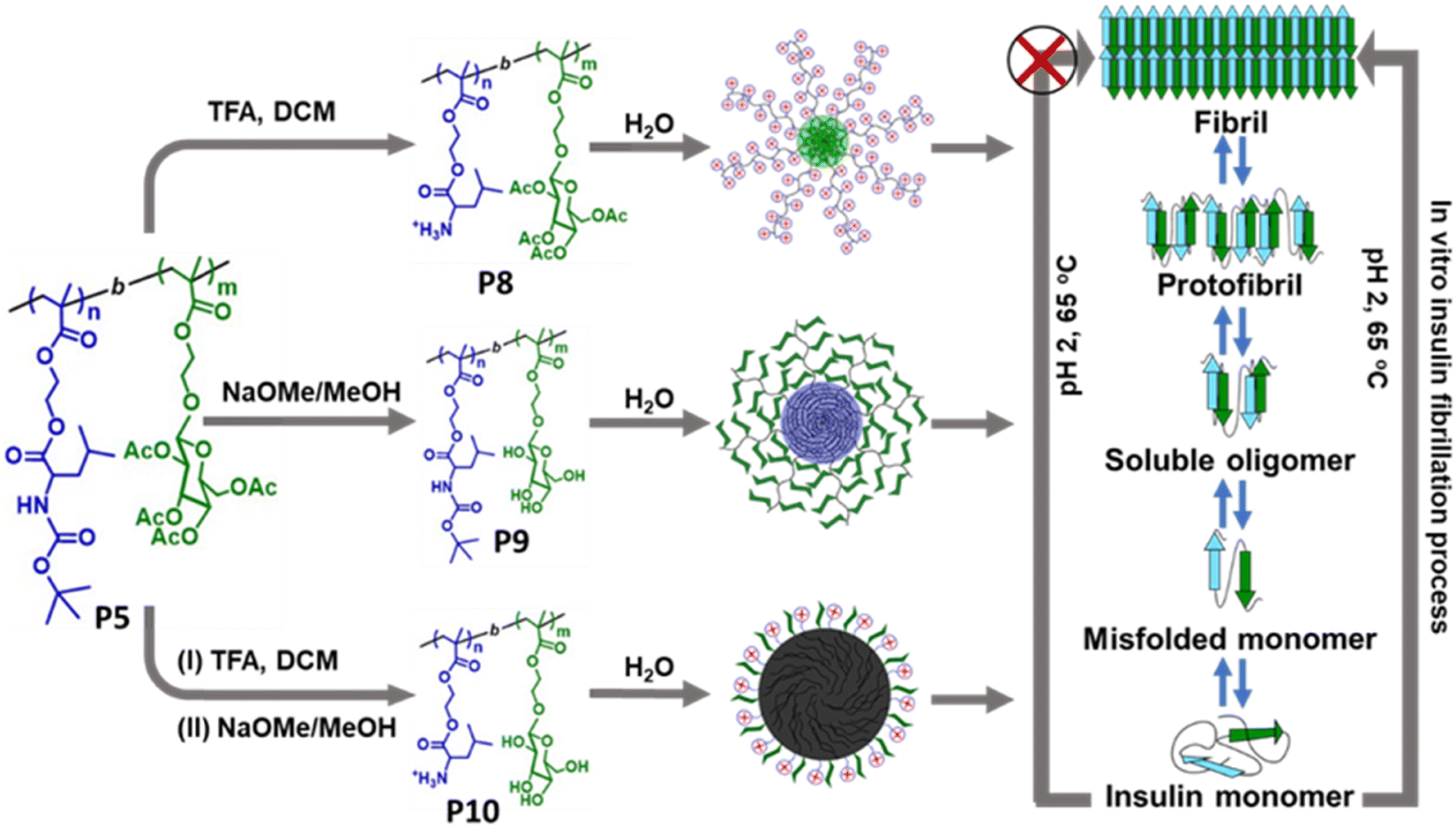
Researchers are becoming interested in polymeric materials due of their notable characteristics, which include high porosity, mechanical qualities, increased surface to volume ratio, controlled degradation rate, biodegradability, and biocompatibility. In recent years, a number of research teams have concentrated on designing innovative polymeric materials with the ability to both regulate and inhibit the development of amyloid fibrils and amyloidogenic diseases. It is important to consider the general structural characteristics required to prevent the production of amyloid fibrils while developing polymeric amyloidogenic inhibitors. The polymers had a range of impacts on the amyloid aggregation process, contingent on the sorts of functional groups they included, including hydrophobic functional groups, hydrophilic moieties, charged groups, degree of polymerization, etc.223,224 In this regard, numerous polymers, including side-chain amino acid containing polymers, amphiphilic polymers, zwitterionic polymers, antioxidant polymers, glycopolymers, hyperbranched/star polymers and polymeric nanomaterials, showed varying effects in regulating the process of amyloid fibrillation.Because of their many intriguing advantages, polymers containing amino acids have shown great utility in the medical field, including the potential for straightforward chemical modifications, enhanced biological activity, and the ability to interact with genes and proteins. Given the significance of polymers containing side-chain amino acids, various research groups have begun to look into how these polymers can help cure amyloidogenic illnesses. In this regard, Palmal et al. fabricated side-chain histidine containing polymeric nanoparticles, which were able to completely halt the formation of amyloid fibrils.223 They also discovered that hydrophobic moieties and surfaces with both positive and negative charges are essential for totally delaying the aggregation process. Moreover, in order to develop efficient treatments that can manage the amyloidogenesis process and prevent the formation of fibrils, our group synthesized a side-chain proline (Pro) containing homopolymer and a block copolymer using the reversible addition–fragmentation chain transfer (RAFT) polymerization technique and further investigated their essential function in the in vitro insulin fibrillation process.225 Using numerous biophysical tools, we revealed that Pro-based polymers can significantly inhibit the insulin fibrillation process. Despite not being able to postpone the lag phase time, the polymers are quite effective at reducing the degree of fibrillation, according to ThT fluorescence kinetic data. The lag phase of the fibrillation pathway cannot be delayed by Pro-based polymers because of the electrostatic repulsion between the comparable charges, which prevents the polymers from forming strong interactions with insulin during the lag phase. On the other hand, it is anticipated that during the growth phase, the polymers are adhered to the fibrillary ends and stop the fibrils from expanding further. The primary driving force underlying the inhibitory process is the hydrophobic contact, as confirmed by studies of Nile red fluorescence and Tyr fluorescence assay. Furthermore, ITC investigations suggest that polar contacts, in addition to hydrophobic interactions, may potentially be accountable for the inhibitory process. Later, we fabricated side-chain cholic acid containing cationic, anionic, and neutral polymers to explore the impact of cholic acid-based charge variable polymers in modulating the insulin fibrillation process.226
The field of biological applications has recently shown a great deal of interest in glycopolymers.227 In this regard, Das et al. have fabricated glycopolymers with varying molecular weights using the aqueous reversible addition–fragmentation chain transfer (aRAFT) polymerization process.228 Using polyacrylamide gel electrophoresis (PAGE) and thioflavin-T fluorescence as models, the polymers were utilized to examine how saccharide unit type and molecular weight affected amyloid-beta aggregation. Interestingly, the findings revealed that the propagation process of amyloid formation was not significantly affected by the other glycopolymers or the negative control, but it was by the high molecular weight (∼350 DP) glucose-containing glycopolymers, which encouraged the development of soluble oligomers of Aβ and limited the production of fibrils. Hitherto, researchers have examined the effect of various sugars and sugar mixes in preventing the development of α-lactalbumin (α-LA) fibrils.229 The suppression of α-LA aggregation was caused by hydrogen bonding between these sugar osmolytes. However, numerous observations revealed that the interactive nature between sugar and proteins is very weak; nevertheless, this interaction can be strengthened by a multivalent effect. As a result, several polymeric substances, including glycopolymers, glycopeptides, and glycodendrimers, have been identified.230,231 Numerous interactions, including hydrogen bonding, hydrophobic and electrostatic interactions, and others, were crucial in inhibiting the process of insulin amyloid formation. In order to integrate all of these interactions into a single system and benefit from the “glycocluster effect” caused by sugar moieties, Bera et al. created block copolymers in this instance that are made up of pendant glucose moieties and amino acid leucine (Fig. 10).232 Interestingly, the results revealed that upon binding to preformed oligomeric species and active nuclei via interactions such as hydrogen bonding–hydrophobic, electrostatic–hydrophobic–hydrogen bonding, and hydrogen bonding–hydrophobic–electrostatic, the polymeric aggregates successfully prevent the formation of elongated fibrils. In another article, Dey and coworkers have used a combination of RAFT polymerization and living cationic polymerization methods to synthesize a class of amphiphilic diblock copolymers with a hydrophilic block with sugar pendants and a hydrophobic polyisobutylene segment to evaluate their possible role in the suppression of insulin fibrillation.233 The glucose moiety and the hydrophobic region both significantly contribute to the insulin aggregation process, as the ThT kinetic assay amply illustrated. Furthermore, the CD study made it clear that while the polymers can slowdown the fibrillation process, they are unable to convert pre-formed fibrils into native form of insulin.
 | ||
| Fig. 10 Schematic representation of the glycopolymeric nanoaggregates for preventing the insulin fibrillation process. Adapted from ref. 232. Copyright 2023 Royal Society of Chemistry. | ||
Besides amino acid-based polymers and glycopolymers, grafting various antioxidant chemicals onto polymers has garnered significant attention from researchers in recent years for potential medical applications due to their distinct biological properties and chemical tunability. Numerous substances with antioxidant properties have already been shown to reduce the oxidative stress caused by amyloid aggregates.234 Nonetheless, their effectiveness is sometimes questioned due to their high volatility, low bioavailability, and quick rate of body clearance, which actually have a therapeutic effect. In this regard, antioxidant polymers are of utmost interest since they can be easily tailored to improve their therapeutic efficiency in a number of ways, including functional group modification, optimization of molecular weight, increasing aqueous solubility, control of the architecture, etc. Based on the fascinating physiognomies and advantages of antioxidant polymers, in our recent article, we have fabricated side-chain lipoic acid (LA) and dihydrolipoic acid (DHLA) containing antioxidant polymers and further evaluated their efficacy in controlling the insulin fibrillation process.235 Interestingly, the synthesized polymers prevent the fibrillation process by prolonging the lag period, and Tyr/NR fluorescence assay results showed a decrease in fibrillation propensity because of the polymers’ favourable hydrophobic contact with the insulin fibrils. Additionally, the polymers exhibit no toxicity, have the capacity to consume reactive oxygen species (ROS), and are very successful in shielding cells from the harm that insulin fibrillar aggregates can produce.
Apart from these polymeric materials, polymeric nanomaterials also showed great promise in terms of their capacity to modify surfaces, their numerous functional group variations, biocompatible nature, and structural flexibility. In this milieu, Meesaragandla and coworkers have fabricated biopolymer functionalized gold nanoparticles (Au NPs) and further evaluated their efficiency in modulating the insulin aggregation process.236 Most recently, Bera and coworkers have synthesized polymer-coated silver nanoparticles containing alanine, leucine and phenylalanine in the side-chain to examine their potency in regulating the formation of insulin fibrils.237 It is interesting to note that amino acid-based polymer-coated silver nanoparticles can stop insulin fibrillation by prolonging the lag phase period, according to ThT kinetic data. Native insulin is adsorbed on the surface of NPs during the nucleation phase, which prevents insulin monomers from approaching, inhibiting the formation of nuclei and subsequently impeding the production of protofibrils as well as matured amyloid fibrils.
Furthermore, polymeric micelles have been used in numerous bio-applications recently because of their good biocompatibility, superior pharmacokinetics, adherence to biosurfaces, targetability, and endurance. Considering the importance of polymeric micelles, Huang et al. have successfully designed and fabricated mixed-shell polymeric micelles (MSPMs), a new artificial molecular chaperone that may effectively prevent AD.238 By utilising their biocompatibility, selectivity for aberrant proteins, and extended blood circulation, the MSPMs can preserve amyloid-beta homeostasis through a dual mechanism that involves preventing amyloid fibrillation and promoting amyloid aggregate clearance, and concurrently decreasing amyloid-mediated neuro-cytotoxicity. The optimal therapeutic impact of MSPMs depends on the hydrophilic/hydrophobic functional groups that are balanced on their surface. Later, Zhang and coworkers created a brand-new photodynamic micelle that inhibits and photodegrades amyloid aggregation.239 The micelles produce ROS in response to irradiation of a 655 nm laser, which breaks down the hazardous oligomeric species and protofibrils. Additionally, the chlorin e6 (Ce6) micelles loaded with Tanshinone I (TAS) effectively photodegrade protofibrils, prevent amyloid fibrillation and offer an optional insight for AD therapy. In another article, reactive oxygen species (ROS)-generating cores and effective thermoresponsive surfaces were combined to create conjugated polymer-based thermoresponsive micelles (CPMs).240 At physiological temperature, the CPMs showed a strong ability to trap the hazardous amyloid-beta aggregates. On the other hand, in the presence of white-light irradiation, ROS was produced in the CPMs and the poisonous amyloid aggregates were effectively disassembled by the ROS oxidation, which resulted in the proper balance of amyloid-beta between aggregation and disaggregation and decreased the cytotoxicity caused by amyloid aggregates. Most recently, Bera and coworkers have looked into the possible function of several fatty acid (FA)-based polymeric micelles in preventing insulin fibril formation (Fig. 11).241 The findings demonstrate that the presence of FA-based polymeric micelles significantly reduces the amount of fibrillation and delays the fibrillation kinetics. Besides, as demonstrated by the Tyr fluorescence assay and NR fluorescence measurements, the hydrophobic interaction between the exposed hydrophobic amino acid residues of insulin and the hydrophobic FA chains present in the micellar core is thought to be one of the main causes of the micelle's higher inhibitory rate. It is noteworthy that the synthesized polymeric micelles have the ability to break down the mature insulin fibrils and so reduce the cytotoxicity caused by fibrils. Hence, by utilizing all the aforementioned properties, it is possible to design novel polymeric scaffolds that will be useful in the future for controlling amyloidogenic disorders.
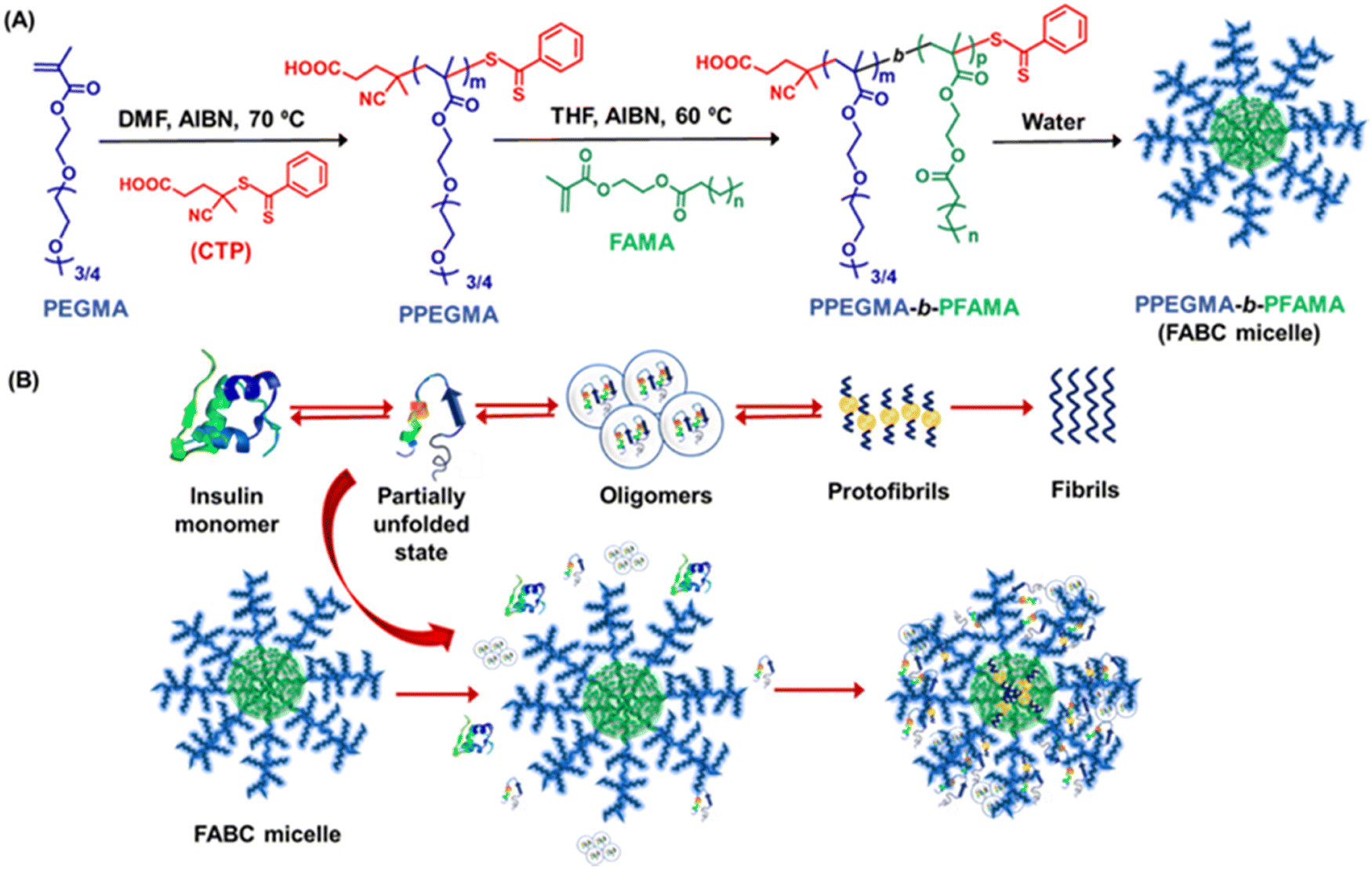 | ||
| Fig. 11 (A) Synthetic scheme of side-chain FA-based polymeric micelles and (B) plausible mechanistic pathway of suppression of the insulin fibrillation process by FABC micelles. Adapted from ref. 241. Copyright 2022 Royal Society of Chemistry. | ||
6. Clinical trials, challenges and potential difficulties
Globally, amyloidogenic disorders are a significant medical, societal, and financial burden. As per the 2022 World Alzheimer Report, over 55 million individuals globally suffer from AD or associated disorders; this figure is expected to rise to 82 million by 2030 and 138 million by 2050. Additionally, the prevalence of diabetes mellitus is predicted to rise to 643 million people (11.3%) by 2030 and 783 million people (12.2%) by 2045, which is of concern. In this regard, numerous strategies have been tried or are being studied to target amyloid-beta production, clearance, or aggregation.242,243 The majority of medications studied in clinical trials have been monoclonal antibodies (mAb) based on immunotherapy that target a single protein, mainly amyloid, with a failure rate of over 100%. Most clinical studies focus on early detection of AD and intervention with AD therapeutic medicines at early/mild to moderate phases of the disorder.244 There are 211 interventional studies filed under AD that are eligible out of the 3388 AD clinical trials. Over the past few decades, pharmacological trials targeting amyloid-beta have failed, because of either side effects or ineffectiveness.245 In order to decrease the amyloid-beta peptide in the brain and stop the development of AD, anti-Aβ medications such as solanezumab, bapineuzumab, lecanemab, aducanumab, gantenerumab, crenezumab, donanemab, etc have been developed and studied. In 2021, the Food and Drug Administration (FDA) controversially gave aducanumab its first expedited approval. Due to low uptake, aducanumab has not been used much in clinical settings, and one of the two-phase IV trials was discontinued.246,247 Biogen, a pharmaceutical company, declared in January 2024 that it was cancelling its sales license for aducanumab in the US.248 Another drug that has received traditional approval from the FDA is lecanemab.249 However, the European Medicines Agency (EMA) suggested in late July 2024 that lecanemab's marketing authorization be denied due to an unfavourable clinical risk.250 Both solanezumab and bapineuzumab's phase III trials were terminated because they failed to satisfy their primary goals.251 The lack of an efficient delivery method has been attributed to previous anti-amyloid treatments’ poor BBB penetration. Crenezumab, gantenerumab, and donanemab, however, are still being studied.252 Additional instances of amyloid beta peptide-targeting medications that were unsuccessful because of their ineffectiveness include the BACE inhibitors verubecestat,253 lanabecestat254 and atabecestat.255 Despite demonstrating a mechanism of action by reducing the plasma and candidate cerebrospinal fluid (CSF) biomarkers Aβ1–40 and Aβ1–42, these medications that target BACE-1 failed to show a therapeutic effect.The majority of Aβ-targeting pharmacological studies have failed because of the loss of Aβ physiological homeostasis, inadequate specificity and translational models, and inability to deliver within the optimal therapeutic window.256 Looking ahead, we believe that the following factors can be taken into account for the continued development of novel anti-amyloid inhibitors. First, the mechanism of the amyloid aggregation process is still complicated, and thus represents one of the major challenges to create preventative and therapeutic approaches. Furthermore, in animal tests, some potential medications displayed notable activity, but in clinical trials, they did not meet expectations. This is because of the genetic background difference between animal models and clinical trials. Hence, it is vital to elucidate the pathophysiology of AD in order to create more accurate models based on the similarities between human and animals and decrease the gap between fundamental research and clinical practice.257 In order to halt the progression of AD, identifying patients who have preclinical AD is essential which can be based on biomarkers but the design of novel biomarkers is considered as one of the major challenges. Targeting many pathways and diseases, combination therapy would be tailored to each patient and guided by biomarkers. Despite the fact that a number of vaccines, small compounds, and nutritional supplements are currently undergoing clinical trials, more research is required to understand the molecular underpinnings behind the intricate pathogenic process of amyloidogenic disorders.
7. Concluding remarks and future outlook
This review article provided a thorough summary of the most current studies using different therapeutic agents to modify amyloid aggregation linked to amyloidogenic disorders, such as AD, PD and HD. The primary causes of protein fibrillation include a variety of interaction mechanisms, including hydrophobic interaction, electrostatic interaction, hydrogen bonding, metal ion chelation and π–π stacking interaction. By utilizing these interactions during the fabrication and application phases, anti-amyloid therapeutic agents can prevent amyloid aggregation. Previous literature findings have shown that, depending on their chemical physiologies and mechanistic pathways, several therapeutic agents either accelerate or inhibit the fibrillation process. Mounting evidence indicates that among the various small molecules, polyphenols have drawn special interest for targeting the amyloid fibrillation process and their anti-amyloid behaviour varies as different polyphenols have varied numbers of phenolic OH groups and variable structure–activity correlations. In addition to small molecules, the researchers have examined the effectiveness of various polymeric materials in modifying the aggregation process. Based on the different architectures, degrees of polymerization, surface charges, functional groups and chemical properties of polymers, they exhibited miscellaneous effects on the amyloid fibrillation pathway. The results of numerous literature publications suggest that there is much space for the development of next-generation therapeutic drugs, which could significantly improve the way amyloidogenic disorders are treated. Thus, the synthesis of diverse therapeutic drugs and the investigation of their critical uses in the management of amyloidogenic disorders present exceptional prospects along with new problems.Despite the fact that several anti-amyloid agents have been fabricated, there is still much space for the development of novel therapeutic drugs. First, although many inhibitors have been developed, it is still unknown exactly which molecular mechanism these anti-amyloid agents use to slow down the aggregation process. Therefore, resolving this issue is essential for enabling the development of novel inhibitors for enhanced therapeutic uses. Besides, there are very few approved and commercially available medications for the treatment of amyloidogenic disorders because there aren’t enough in vivo studies. Therefore, more in vivo studies, including long-term toxicity evaluations utilizing animal models, are required to obtain a thorough understanding of the mechanism of action of the anti-amyloidogenic inhibitors and to expedite the rate of their clinical approval. In the future, we believe that the above aspects should be taken into account for the development of more efficacious small molecule-based and polymer-based anti-amyloid inhibitors.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
The authors declare that they have no known competing financial interests that could have appeared to influence the work reported in this paper.Acknowledgements
PG acknowledges the Department of Science & Technology and Biotechnology, Government of West Bengal (DSTBT, GoWB, Project ID: 1368/RND/CS23/Nil/Nov-2023/1/1), for financial support.References
- H. Jung, Y. J. Chung, R. Wilton, C. H. Lee, B. I. Lee, J. Lim, H. Lee, J.-H. Choi, H. Kang, B. Lee, E. A. Rozhkova, C. B. Park and J. Lee, Adv. Funct. Mater., 2020, 30, 1910475 CrossRef CAS.
- H. Geng, Y.-C. Pan, R. Zhang, D. Gao, Z. Wang, B. Li, N. Li, D.-S. Guo and C. Xing, Adv. Funct. Mater., 2021, 31, 2102953 CrossRef CAS.
- S. Andrade, J. A. Loureiro and M. C. Pereira, ACS Chem. Neurosci., 2021, 12, 2491–2502 CrossRef CAS.
- B. Mirzaei-Behbahani, A. A. Meratan, B. Moosakhani, M. Mohammad-Zaheri, Z. Mousavi-Jarrahi, N. Nikfarjam, M. B. Shahsavani and A. A. Saboury, Sci. Rep., 2024, 14, 3907 CrossRef CAS.
- A. G. Kreutzer, C. M. T. Parrocha, S. Haerianardakani, G. Guaglianone, J. T. Nguyen, M. N. Diab, W. Yong, M. Perez-Rosendahl, E. Head and J. S. Nowick, ACS Cent. Sci., 2024, 10, 104–121 CrossRef CAS PubMed.
- A. F. Chaparro Sosa, S. M. de Oliveira da Silva, G. P. Morgan, D. K. Schwartz and J. L. Kaar, J. Phys. Chem. Lett., 2020, 11, 7417–7422 CrossRef CAS PubMed.
- H. Yasir Khan, A. Ahmad, M. Nadir Hassan, Y. Hasan Khan, F. Arjmand and R. Hasan Khan, Coord. Chem. Rev., 2024, 501, 215580 CrossRef CAS.
- X. Shao, C. Yan, C. Wang, C. Wang, Y. Cao, Y. Zhou, P. Guan, X. Hu, W. Zhu and S. Ding, Nanoscale Adv., 2023, 5, 46–80 RSC.
- M. P. T. Prabhu and N. Sarkar, Biophys. Chem., 2022, 280, 106714 CrossRef CAS PubMed.
- G. F. Martins and N. Galamba, Crit. Rev. Biochem. Mol. Biol., 2023, 58, 50–80 CrossRef CAS PubMed.
- R. Rajan, S. Ahmed, N. Sharma, N. Kumar, A. Debas and K. Matsumura, Mater. Adv., 2021, 2, 1139–1176 RSC.
- L. Xiang, Y. Wang, S. Liu, B. Liu, X. Jin and X. Cao, Int. J. Mol. Sci., 2023, 24, 11275 CrossRef CAS PubMed.
- C.-Q. Liang and Y.-M. Li, Curr. Opin. Chem. Biol., 2021, 64, 124–130 CrossRef CAS PubMed.
- Z. Dhouafli, K. Cuanalo-Contreras, E. A. Hayouni, C. E. Mays, C. Soto and I. Moreno-Gonzalez, Cell. Mol. Life Sci., 2018, 75, 3521–3538 CrossRef CAS PubMed.
- P. Ghosh and P. De, ACS Appl. Bio Mater., 2020, 3, 6598–6625 CrossRef CAS PubMed.
- P. Ghosh, A. Bera, P. Bhadury and P. De, ACS Chem. Neurosci., 2021, 12, 1737–1748 CrossRef CAS.
- P. Ghosh, A. Mondal and P. De, Current Indian Sci., 2024, 2, 1–15 Search PubMed.
- B. Ren, R. Hu, M. Zhang, Y. Liu, L. Xu, B. Jiang, J. Ma, B. Ma, R. Nussinov and J. Zheng, Methods Mol. Biol., 2018, 1777, 429–447 CrossRef CAS PubMed.
- X. Xiao, A. S. Robang, S. Sarma, J. V. Le, M. E. Helmicki, M. J. Lambert, R. Guerrero-Ferreira, J. Arboleda-Echavarria, A. K. Paravastu and C. K. Hall, PNAS Nexus, 2022, 1, pgac263 CrossRef.
- A. Chaari, N. Saikia, P. Paul, M. Yousef, F. Ding and M. Ladjimi, Biophys. Chem., 2024, 309, 107235 CrossRef CAS.
- A. P. Kumar, S. Lee and S. Lukman, Curr. Drug Targets, 2019, 20, 1680–1694 CrossRef CAS.
- P. H. Nguyen, A. Ramamoorthy, B. R. Sahoo, J. Zheng, P. Faller, J. E. Straub, L. Dominguez, J. E. Shea, N. V. Dokholyan, A. De Simone, B. Ma, R. Nussinov, S. Najafi, S. T. Ngo, A. Loquet, M. Chiricotto, P. Ganguly, J. McCarty, M. S. Li, C. Hall, Y. Wang, Y. Miller, S. Melchionna, B. Habenstein, S. Timr, J. Chen, B. Hnath, B. Strodel, R. Kayed, S. Lesné, G. Wei, F. Sterpone, A. J. Doig and P. Derreumaux, Chem. Rev., 2021, 121, 2545–2647 CrossRef CAS.
- F. Chiti and C. M. Dobson, Annu. Rev. Biochem., 2006, 75, 333–366 CrossRef CAS.
- A. Mitraki and J. King, Bio/Technology, 1989, 7, 690–697 CAS.
- P. Salahuddin, R. H. Khan, M. Furkan, V. N. Uversky, Z. Islam and M. T. Fatima, Int. J. Biol. Macromol., 2021, 186, 580–590 CrossRef CAS PubMed.
- K. L. Zapadka, F. J. Becher, A. L. Gomes Dos Santos and S. E. Jackson, Interface Focus, 2017, 7, 20170030 CrossRef.
- Y. Kuroda, Biophys. Rev., 2022, 14, 1495–1501 CrossRef CAS PubMed.
- V. Bellotti and F. Chiti, Curr. Opin. Struct. Biol., 2008, 18, 771–779 CrossRef CAS PubMed.
- O. Crescenzi, S. Tomaselli, R. Guerrini, S. Salvadori, A. M. D’Ursi, P. A. Temussi and D. Picone, Eur. J. Biochem., 2002, 269, 5642–5648 CrossRef CAS.
- S. Vivekanandan, J. R. Brender, S. Y. Lee and A. Ramamoorthy, Biochem. Biophys. Res. Commun., 2011, 411, 312–316 CrossRef CAS PubMed.
- T. Lührs, C. Ritter, M. Adrian, D. Riek-Loher, B. Bohrmann, H. Döbeli, D. Schubert and R. Riek, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 17342–17347 CrossRef.
- Y. Xiao, B. Ma, D. McElheny, S. Parthasarathy, F. Long, M. Hoshi, R. Nussinov and Y. Ishii, Nat. Struct. Mol. Biol., 2015, 22, 499–505 CrossRef CAS.
- L. Gremer, D. Schölzel, C. Schenk, E. Reinartz, J. Labahn, R. B. G. Ravelli, M. Tusche, C. Lopez-Iglesias, W. Hoyer, H. Heise, D. Willbold and G. F. Schröder, Science, 2017, 358, 116–119 CrossRef CAS PubMed.
- M. Kollmer, W. Close, L. Funk, J. Rasmussen, A. Bsoul, A. Schierhorn, M. Schmidt, C. J. Sigurdson, M. Jucker and M. Fändrich, Nat. Commun., 2019, 10, 4760 CrossRef PubMed.
- S. Giorgetti, C. Greco, P. Tortora and F. A. Aprile, Int. J. Mol. Sci., 2018, 19, 2677 CrossRef.
- R. Cascella, A. Bigi, N. Cremades and C. Cecchi, Cell. Mol. Life Sci., 2022, 79, 174 CrossRef CAS PubMed.
- R. Zeineddine and J. J. Yerbury, Front. Physiol., 2015, 6, 277 Search PubMed.
- K. Pagano, S. Tomaselli, H. Molinari and L. Ragona, Front. Neurosci., 2020, 14, 619667 CrossRef PubMed.
- M. Fändrich, Cell. Mol. Life Sci., 2007, 64, 2066–2078 CrossRef PubMed.
- P. Ghosh, A. Bera and P. De, J. Indian Chem. Soc., 2021, 98, 100011 CrossRef CAS.
- S. Navarro and S. Ventura, Curr. Opin. Struct. Biol., 2022, 73, 102343 CrossRef CAS PubMed.
- R. L. Redler, D. Shirvanyants, O. Dagliyan, F. Ding, D. N. Kim, P. Kota, E. A. Proctor, S. Ramachandran, A. Tandon and N. V. Dokholyan, J. Mol. Cell Biol., 2014, 6, 104–115 CrossRef CAS PubMed.
- L. Borges-Araújo, G. P. Pereira, M. Valério and P. C. T. Souza, Biochim. Biophys. Acta, Proteins Proteomics, 2024, 1872, 141014 CrossRef.
- C. L. Avila, N. J. Drechsel, R. Alcántara and J. Villà-Freixa, Curr. Protein Pept. Sci., 2011, 12, 221–234 CrossRef CAS PubMed.
- V. H. Man, X. He, P. Derreumaux, B. Ji, X.-Q. Xie, P. H. Nguyen and J. Wang, J. Chem. Theory Comput., 2019, 15, 1440–1452 CrossRef CAS.
- R. Prabakaran, P. Rawat, A. M. Thangakani, S. Kumar and M. M. Gromiha, Biophys. Rev., 2021, 13, 71–89 CrossRef CAS.
- L. Goldschmidt, P. K. Teng, R. Riek and D. Eisenberg, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 3487–3492 CrossRef CAS.
- N. Louros, K. Konstantoulea, M. De Vleeschouwer, M. Ramakers, J. Schymkowitz and F. Rousseau, Nucleic Acids Res., 2020, 48, D389–D393 CrossRef CAS PubMed.
- P. Rawat, R. Prabakaran, R. Sakthivel, A. Mary Thangakani, S. Kumar and M. M. Gromiha, Amyloid, 2020, 27, 128–133 CrossRef CAS.
- K. Bodi, T. Prokaeva, B. Spencer, M. Eberhard, L. H. Connors and D. C. Seldin, Amyloid, 2009, 16, 1–8 CrossRef CAS PubMed.
- R. Zambrano, M. Jamroz, A. Szczasiuk, J. Pujols, S. Kmiecik and S. Ventura, Nucleic Acids Res., 2015, 43, W306–W313 CrossRef CAS.
- A. E. Badaczewska-Dawid, A. Kuriata, C. Pintado-Grima, J. Garcia-Pardo, M. Burdukiewicz, V. Iglesias, S. Kmiecik and S. Ventura, Nucleic Acids Res., 2024, 52, D360–D367 CrossRef PubMed.
- P. Rawat, S. Kumar and M. Michael Gromiha, Int. J. Biol. Macromol., 2018, 118, 1157–1167 CrossRef CAS PubMed.
- P. Rawat, R. Prabakaran, S. Kumar and M. M. Gromiha, Bioinformatics, 2019, 36, 1439–1444 CrossRef.
- P. Rawat, R. Prabakaran, S. Kumar and M. M. Gromiha, BBiochim. Biophys. Acta, Proteins Proteomics, 2021, 1869, 140682 CrossRef CAS PubMed.
- M. Biancalana, K. Makabe, A. Koide and S. Koide, J. Mol. Biol., 2009, 385, 1052–1063 CrossRef CAS PubMed.
- A. Srivastava, P. K. Singh, M. Kumbhakar, T. Mukherjee, S. Chattopadyay, H. Pal and S. Nath, Chem. – Eur. J., 2010, 16, 9257–9263 CrossRef CAS PubMed.
- P. Sindrewicz, X. Li, E. A. Yates, J. E. Turnbull, L. Y. Lian and L.-G. Yu, Sci. Rep., 2019, 9, 11851 CrossRef PubMed.
- C. R. Cantor and P. R. Schimmel, Biophysical Chemistry, W. H. Freeman and Company, New York, 11th edn, 2004 Search PubMed.
- A. T. Petkova, Y. Ishii, J. J. Balbach, O. N. Antzutkin, R. D. Leapman, F. Delaglio and R. Tycko, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 16742–16747 CrossRef CAS PubMed.
- V. J. De-Paula, M. Radanovic, B. S. Diniz and O. V. Forlenza, Subcell. Biochem., 2012, 65, 329–352 CAS.
- G. S. Bloom, JAMA Neurol., 2014, 71, 505–508 CrossRef PubMed.
- J. Nunan and D. H. Small, FEBS Lett., 2000, 483, 6–10 CrossRef CAS PubMed.
- G.-f Chen, T.-h Xu, Y. Yan, Y.-r Zhou, Y. Jiang, K. Melcher and H. E. Xu, Acta Pharmacol. Sin., 2017, 38, 1205–1235 CrossRef CAS PubMed.
- S. A. Funke, E. Birkmann and D. Willbold, Curr. Alzheimer Res., 2009, 6, 285–289 CrossRef CAS PubMed.
- Y. Zhang, M. Hashemi, Z. Lv and Y. L. Lyubchenko, Nanoscale, 2016, 8, 18928–18937 RSC.
- W. Zheng, M. Y. Tsai, M. Chen and P. G. Wolynes, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 11835–11840 CrossRef CAS.
- H. Okumura and S. G. Itoh, Molecules, 2022, 27, 2483 CrossRef CAS PubMed.
- M. Hashemi, Y. Zhang, Z. Lv and Y. L. Lyubchenko, Nanoscale Adv., 2019, 1, 3892–3899 RSC.
- B. Barz, Q. Liao and B. Strodel, J. Am. Chem. Soc., 2018, 140, 319–327 CrossRef CAS PubMed.
- B. Mehrazma and A. Rauk, J. Phys. Chem. A, 2019, 123, 4658–4670 CrossRef CAS PubMed.
- S. Sasmal, N. Schwierz and T. Head-Gordon, J. Phys. Chem. B, 2016, 120, 12088–12097 CrossRef CAS.
- N. Schwierz, C. V. Frost, P. L. Geissler and M. Zacharias, J. Am. Chem. Soc., 2016, 138, 527–539 CrossRef CAS PubMed.
- C. R. Watts, A. J. Gregory, C. P. Frisbie and S. Lovas, Proteins, 2017, 85, 1024–1045 CrossRef CAS.
- H. Okumura and S. G. Itoh, J. Chem. Phys., 2020, 152, 095101 CrossRef CAS PubMed.
- G. Grasso and A. Danani, Adv. Phys.: X, 2020, 5, 1770627 CAS.
- S. G. Itoh and H. Okumura, Int. J. Mol. Sci., 2021, 22, 1859 CrossRef CAS.
- Y. Tachi, S. G. Itoh and H. Okumura, Biophys. Physicobiol., 2022, 19, 1–18 Search PubMed.
- S. Kalita, H. Bergman, K. D. Dubey and S. Shaik, J. Am. Chem. Soc., 2023, 145, 3543–3553 CrossRef CAS PubMed.
- H. Okumura and S. G. Itoh, J. Am. Chem. Soc., 2014, 136, 10549–10552 CrossRef CAS PubMed.
- Y. W. Ma, T. Y. Lin and M. Y. Tsai, Front. Mol. Biosci., 2021, 8, 719320 CrossRef CAS.
- W. Lu, C. Bueno, N. P. Schafer, J. Moller, S. Jin, X. Chen, M. Chen, X. Gu, A. Davtyan, J. J. de Pablo and P. G. Wolynes, PLoS Comput. Biol., 2021, 17, e1008308 CrossRef CAS.
- A. Pasieka, D. Panek, N. Szałaj, A. Espargaró, A. Więckowska, B. Malawska, R. Sabaté and M. Bajda, ACS Chem. Neurosci., 2021, 12, 2057–2068 CrossRef CAS PubMed.
- S. G. Itoh, M. Yagi-Utsumi, K. Kato and H. Okumura, J. Phys. Chem. B, 2019, 123, 160–169 CrossRef CAS.
- Y. Tachi, Y. Okamoto and H. Okumura, Sci. Rep., 2019, 9, 6853 CrossRef PubMed.
- D. Fukuhara, S. G. Itoh and H. Okumura, Biophys. Physicobiol., 2023, 20, e200045 CAS.
- H. Okumura, J. Phys. Chem. B, 2023, 127, 10931–10940 CrossRef CAS.
- Y.-L. Han, H.-H. Yin, C. Xiao, M. T. Bernards, Y. He and Y.-X. Guan, ACS Chem. Neurosci., 2023, 14, 4051–4061 CrossRef CAS.
- L. L. N. Ngoc, S. G. Itoh, P. Sompornpisut and H. Okumura, Chem. Phys. Lett., 2020, 758, 137913 CrossRef CAS.
- T. S. Ulmer, A. Bax, N. B. Cole and R. L. Nussbaum, J. Biol. Chem., 2005, 280, 9595–9603 CrossRef CAS.
- G. Fusco, A. De Simone, T. Gopinath, V. Vostrikov, M. Vendruscolo, C. M. Dobson and G. Veglia, Nat. Commun., 2014, 5, 3827 CrossRef CAS.
- I. M. van der Wateren, T. P. J. Knowles, A. K. Buell, C. M. Dobson and C. Galvagnion, Chem. Sci., 2018, 9, 5506–5516 RSC.
- X. Ni, R. P. McGlinchey, J. Jiang and J. C. Lee, J. Mol. Biol., 2019, 431, 3913–3919 CrossRef CAS PubMed.
- A. Balupuri, K.-E. Choi and N. S. Kang, Sci. Rep., 2019, 9, 59 CrossRef.
- V. N. Uversky, J. Li and A. L. Fink, J. Biol. Chem., 2001, 276, 10737–10744 CrossRef CAS PubMed.
- H. Yu, W. Han, W. Ma and K. Schulten, J. Chem. Phys., 2015, 143, 243142 CrossRef PubMed.
- Y. Zhang, Y. Wang, Y. Liu, G. Wei, F. Ding and Y. Sun, ACS Chem. Neurosci., 2022, 13, 3126–3137 CrossRef CAS PubMed.
- S. D. Khare, P. Chinchilla and J. Baum, Curr. Opin. Struct. Biol., 2023, 80, 102579 CrossRef CAS.
- J. Razzokov, S. Fazliev, M. Makhkamov, P. Marimuthu, A. Baev and E. Kurganov, Int. J. Mol. Sci., 2023, 24, 6312 CrossRef CAS PubMed.
- P. C. T. Souza, R. Alessandri, J. Barnoud, S. Thallmair, I. Faustino, F. Grünewald, I. Patmanidis, H. Abdizadeh, B. M. H. Bruininks, T. A. Wassenaar, P. C. Kroon, J. Melcr, V. Nieto, V. Corradi, H. M. Khan, J. Domański, M. Javanainen, H. Martinez-Seara, N. Reuter, R. B. Best, I. Vattulainen, L. Monticelli, X. Periole, D. P. Tieleman, A. H. de Vries and S. J. Marrink, Nat. Methods, 2021, 18, 382–388 CrossRef CAS.
- C. Privat, S. Madurga, F. Mas and J. Rubio-Martinez, Phys. Chem. Chem. Phys., 2022, 24, 18841–18853 RSC.
- S. T. A. Amos, T. C. Schwarz, J. Shi, B. P. Cossins, T. S. Baker, R. J. Taylor, R. Konrat and M. S. P. Sansom, J. Phys. Chem. B, 2021, 125, 2929–2941 CrossRef CAS.
- N. Zhao, Q. Zhang, F. Yu, X. Yao and H. Liu, Biomolecules, 2023, 13, 682 CrossRef CAS PubMed.
- J. Chen, S. Zaer, P. Drori, J. Zamel, K. Joron, N. Kalisman, E. Lerner and N. V. Dokholyan, Structure, 2021, 29, 1048–1064 CrossRef CAS PubMed.
- R. J. Harding, J. C. Deme, J. F. Hevler, S. Tamara, A. Lemak, J. P. Cantle, M. M. Szewczyk, N. Begeja, S. Goss, X. Zuo, P. Loppnau, A. Seitova, A. Hutchinson, L. Fan, R. Truant, M. Schapira, J. B. Carroll, A. J. R. Heck, S. M. Lea and C. H. Arrowsmith, Commun. Biol., 2021, 4, 1374 CrossRef CAS.
- F. Saudou and S. Humbert, Neuron, 2016, 89, 910–926 CrossRef CAS.
- R. J. Harding, P. Loppnau, S. Ackloo, A. Lemak, A. Hutchinson, B. Hunt, A. S. Holehouse, J. C. Ho, L. Fan, L. Toledo-Sherman, A. Seitova and C. H. Arrowsmith, J. Biol. Chem., 2019, 294, 6986–7001 CrossRef CAS.
- J. M. Isas, R. Langen and A. B. Siemer, Biochemistry, 2015, 54, 3942–3949 CrossRef CAS.
- M. E. MacDonald, S. Gines, J. F. Gusella and V. C. Wheeler, NeuroMol. Med., 2003, 4, 7–20 CrossRef CAS.
- A. K. Thakur, M. Jayaraman, R. Mishra, M. Thakur, V. M. Chellgren, I.-J. L. Byeon, D. H. Anjum, R. Kodali, T. P. Creamer, J. F. Conway, A. M. Gronenborn and R. Wetzel, Nat. Struct. Mol. Biol., 2009, 16, 380–389 CrossRef CAS.
- M. Khaled, B. Strodel and A. Sayyed-Ahmad, Front. Mol. Biosci., 2023, 10, 1143353 CrossRef CAS PubMed.
- S. N. Moldovean and V. Chiş, ACS Chem. Neurosci., 2020, 11, 105–120 CrossRef CAS.
- S. Vieweg, A.-L. Mahul-Mellier, F. S. Ruggeri, N. Riguet, S. M. DeGuire, A. Chiki, U. Cendrowska, G. Dietler and H. A. Lashuel, J. Mol. Biol., 2021, 433, 167222 CrossRef CAS.
- A. S. Falk, J. M. Bravo-Arredondo, J. Varkey, S. Pacheco, R. Langen and A. B. Siemer, Biophys. J., 2020, 119, 2019–2028 CrossRef CAS PubMed.
- J. Mario Isas, N. K. Pandey, H. Xu, K. Teranishi, A. K. Okada, E. K. Fultz, A. Rawat, A. Applebaum, F. Meier, J. Chen, R. Langen and A. B. Siemer, Nat. Commun., 2021, 12, 4272 CrossRef CAS.
- J. Miller, M. Arrasate, E. Brooks, C. P. Libeu, J. Legleiter, D. Hatters, J. Curtis, K. Cheung, P. Krishnan, S. Mitra, K. Widjaja, B. A. Shaby, G. P. Lotz, Y. Newhouse, E. J. Mitchell, A. Osmand, M. Gray, V. Thulasiramin, F. Saudou, M. Segal, X. W. Yang, E. Masliah, L. M. Thompson, P. J. Muchowski, K. H. Weisgraber and S. Finkbeiner, Nat. Chem. Biol., 2011, 7, 925–934 CrossRef CAS.
- I. Gkekas, A. Gioran, M. K. Boziki, N. Grigoriadis, N. Chondrogianni and S. Petrakis, Antioxidants, 2021, 10, 1450 CrossRef CAS PubMed.
- E. Latoszek, M. Wiweger, J. Ludwiczak, S. Dunin-Horkawicz, J. Kuznicki and M. Czeredys, Cell Biosci., 2022, 12, 34 CrossRef CAS PubMed.
- K. S. Yee and K. H. Vousden, Carcinogenesis, 2005, 26, 1317–1322 CrossRef PubMed.
- T. Riley, E. Sontag, P. Chen and A. Levine, Nat. Rev. Mol. Cell Biol., 2008, 9, 402–412 CrossRef CAS PubMed.
- D. R. Green and G. Kroemer, Nature, 2009, 458, 1127–1130 CrossRef CAS PubMed.
- J. T. Zilfou and S. W. Lowe, Cold Spring Harbor Perspect. Biol., 2009, 1, a001883 Search PubMed.
- T. Ozaki and A. Nakagawara, Cancers, 2011, 3, 994–1013 CrossRef CAS.
- M. Olivier, R. Eeles, M. Hollstein, M. A. Khan, C. C. Harris and P. Hainaut, Hum. Mutat., 2002, 19, 607–614 CrossRef CAS PubMed.
- M. Olivier, M. Hollstein and P. Hainaut, Cold Spring Harbor Perspect. Biol., 2010, 2, a001008 Search PubMed.
- Y. Chen, X. Zhang, A. C. Dantas Machado, Y. Ding, Z. Chen, P. Z. Qin, R. Rohs and L. Chen, Nucleic Acids Res., 2013, 41, 8368–8376 CrossRef CAS.
- D. Ishimaru, L. R. Andrade, L. S. Teixeira, P. A. Quesado, L. M. Maiolino, P. M. Lopez, Y. Cordeiro, L. T. Costa, W. M. Heckl, G. Weissmüller, D. Foguel and J. L. Silva, Biochemistry, 2003, 42, 9022–9027 CrossRef CAS PubMed.
- Y. Higashimoto, Y. Asanomi, S. Takakusagi, M. S. Lewis, K. Uosaki, S. R. Durell, C. W. Anderson, E. Appella and K. Sakaguchi, Biochemistry, 2006, 45, 1608–1619 CrossRef CAS PubMed.
- H. C. Ang, A. C. Joerger, S. Mayer and A. R. Fersht, J. Biol. Chem., 2006, 281, 21934–21941 CrossRef CAS PubMed.
- J. Xu, J. Reumers, J. R. Couceiro, F. De Smet, R. Gallardo, S. Rudyak, A. Cornelis, J. Rozenski, A. Zwolinska, J.-C. Marine, D. Lambrechts, Y.-A. Suh, F. Rousseau and J. Schymkowitz, Nat. Chem. Biol., 2011, 7, 285–295 CrossRef CAS.
- G. Saha, R. Singh, A. Mandal, S. Das, E. Chattopadhyay, P. Panja, P. Roy, N. DeSarkar, S. Gulati, S. Ghatak, S. Ghosh, S. Banerjee, B. Roy, S. Ghosh, D. Chaudhuri, N. Arora, N. K. Biswas and N. Sikdar, Mol. Med., 2020, 26, 59 CAS.
- S. Singh, M. Kumar, S. Kumar, S. Sen, P. Upadhyay, S. Bhattacharjee, N. M, V. S. Tomar, S. Roy, A. Dutt and T. K. Kundu, J. Biol. Chem., 2019, 294, 14081–14095 CrossRef CAS PubMed.
- K. Polireddy, K. Singh, M. Pruski, N. C. Jones, N. V. Manisundaram, P. Ponnela, M. Ouellette, G. Van Buren, M. Younes, J. S. Bynon, W. A. Dar and J. M. Bailey, Cancer Lett., 2019, 453, 122–130 CrossRef CAS.
- J. S. Butler and S. N. Loh, Biochemistry, 2003, 42, 2396–2403 CrossRef CAS PubMed.
- A. P. Ano Bom, L. P. Rangel, D. C. Costa, G. A. de Oliveira, D. Sanches, C. A. Braga, L. M. Gava, C. H. Ramos, A. O. Cepeda, A. C. Stumbo, C. V. De Moura Gallo, Y. Cordeiro and J. L. Silva, J. Biol. Chem., 2012, 287, 28152–28162 CrossRef CAS PubMed.
- C. B. Levy, A. C. Stumbo, A. P. Ano Bom, E. A. Portari, Y. Cordeiro, J. L. Silva and C. V. De Moura-Gallo, Int. J. Biochem. Cell Biol., 2011, 43, 60–64 CrossRef CAS PubMed.
- Y. Zhang, S. V. Coillie, J. Y. Fang and J. Xu, Oncogenesis, 2016, 5, e196 CrossRef CAS PubMed.
- M. R. Pradhan, J. W. Siau, S. Kannan, M. N. Nguyen, Z. Ouaray, C. K. Kwoh, D. P. Lane, F. Ghadessy and C. S. Verma, Nucleic Acids Res., 2019, 47, 1637–1652 CrossRef CAS PubMed.
- R. Kamada, T. Nomura, C. W. Anderson and K. Sakaguchi, J. Biol. Chem., 2011, 286, 252–258 CrossRef CAS PubMed.
- S. Ghosh, D. Ghosh, S. Ranganathan, A. Anoop, P. SK, N. N. Jha, R. Padinhateeri and S. K. Maji, Biochemistry, 2014, 53, 5995–6010 CrossRef CAS PubMed.
- Z. Chen, D. Xia, X. Liang, Q. Liu, J. Li, Q. Li and M. Dong, Mater. Today Phys., 2024, 44, 101436 CrossRef CAS.
- Z. Yang, Y. Yao, Y. Zhou, X. Li, Y. Tang and G. Wei, Int. J. Biol. Macromol., 2023, 230, 123194 CrossRef CAS.
- K. P. Prajapati, A. P. Singh, K. Dubey, M. Ansari, M. Temgire, B. G. Anand and K. Kar, Colloids Surf., B, 2020, 186, 110640 CrossRef CAS.
- A. Alghamdi, D. J. S. Birch, V. Vyshemirsky and O. J. Rolinski, J. Phys. Chem. B, 2022, 126, 7229–7237 CrossRef CAS PubMed.
- S. K. Sonawane, V. N. Uversky and S. Chinnathambi, Cell Commun. Signaling, 2021, 19, 16 CrossRef CAS PubMed.
- K. Cieślik-Boczula and P. Trombik, Int. J. Biol. Macromol., 2019, 125, 630–641 CrossRef.
- A. Sakalauskas, M. Ziaunys and V. Smirnovas, Sci. Rep., 2020, 10, 14466 CrossRef CAS.
- S. Malik, F. Nabi, M. K. Siddiqi, A. Masroor, M. Hisamuddin, T. I. Chandel, N. Majid and R. H. Khan, J. Mol. Liq., 2023, 389, 122944 CrossRef CAS.
- S. Andrade, J. A. Loureiro and M. C. Pereira, Int. J. Biol. Macromol., 2021, 190, 853–861 CrossRef CAS PubMed.
- P. H. Reddy, M. Manczak, X. Yin, M. C. Grady, A. Mitchell, R. Kandimalla and C. S. Kuruva, J. Invest. Med., 2016, 64, 1220–1234 CrossRef PubMed.
- J. M. Jakubowski, A. A. Orr, D. A. Le and P. Tamamis, J. Chem. Inf. Model., 2020, 60, 289–305 CrossRef CAS PubMed.
- I. Doytchinova, M. Atanasova, E. Salamanova, S. Ivanov and I. Dimitrov, Biomolecules, 2020, 10, 1323 CrossRef CAS PubMed.
- Y. Ruan, Y. Xiong, W. Fang, Q. Yu, Y. Mai, Z. Cao, K. Wang, M. Lei, J. Xu, Y. Liu, X. Zhang, W. Liao and J. Liu, J. Nanobiotechnol., 2022, 20, 322 CrossRef CAS.
- B. N. Singh, S. Shankar and R. K. Srivastava, Biochem. Pharmacol., 2011, 82, 1807–1821 CrossRef CAS PubMed.
- X. Li, Y. Zhang, Z. Yang, S. Zhang and L. Zhang, Int. J. Mol. Sci., 2024, 25, 1636 CrossRef CAS PubMed.
- K. Siposova, T. Kozar, M. Stupakova and A. Musatov, Colloids Surf., B, 2021, 197, 111428 CrossRef CAS.
- F. Macchi, M. Eisenkolb, H. Kiefer and D. E. Otzen, Int. J. Mol. Sci., 2012, 13, 3801–3819 CrossRef CAS.
- A. Venkatraman, E. Murugan, S. J. Lin, G. S. L. Peh, L. Rajamani and J. S. Mehta, Sci. Rep., 2020, 10, 4011 CrossRef CAS PubMed.
- B. Solomon, R. Koppel, E. Hanan and T. Katzav, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 452–455 CrossRef CAS.
- D. Frenkel, O. Katz and B. Solomon, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 11455–11459 CrossRef CAS.
- R. Perneczky, G. Dom, A. Chan, P. Falkai and C. Bassetti, Eur. J. Neurol., 2024, 31, e16049 CrossRef.
- J. Zhou, Y. Li, J. Geng, H. Zhou, L. Liu and X. Peng, J. Cardiovasc. Pharmacol., 2023, 82, 427–437 CrossRef CAS PubMed.
- M. Nuvolone, A. Nevone and G. Merlini, BioDrugs, 2022, 36, 591–608 CrossRef CAS.
- J. Sevigny, P. Chiao, T. Bussière, P. H. Weinreb, L. Williams, M. Maier, R. Dunstan, S. Salloway, T. Chen, Y. Ling, J. O’Gorman, F. Qian, M. Arastu, M. Li, S. Chollate, M. S. Brennan, O. Quintero-Monzon, R. H. Scannevin, H. M. Arnold, T. Engber, K. Rhodes, J. Ferrero, Y. Hang, A. Mikulskis, J. Grimm, C. Hock, R. M. Nitsch and A. Sandrock, Nature, 2016, 537, 50–56 CrossRef CAS PubMed.
- O. El-Agnaf, C. Overk, E. Rockenstein, M. Mante, J. Florio, A. Adame, N. Vaikath, N. Majbour, S.-J. Lee, C. Kim, E. Masliah and R. A. Rissman, Neurobiol. Dis., 2017, 104, 85–96 CrossRef CAS PubMed.
- V. Gupta, S. Salim, I. Hmila, N. N. Vaikath, I. P. Sudhakaran, S. S. Ghanem, N. K. Majbour, S. A. Abdulla, M. M. Emara, H. B. Abdesselem, T. Lukacsovich, D. Erskine and O. M. A. El-Agnaf, Sci. Rep., 2020, 10, 8137 CrossRef CAS PubMed.
- W. Wu, Y. Ji, Z. Wang, X. Wu, J. Li, F. Gu, Z. Chen and Z. Wang, Eur. J. Med. Res., 2023, 28, 544 CrossRef CAS.
- C. H. v Dyck, C. J. Swanson, P. Aisen, R. J. Bateman, C. Chen, M. Gee, M. Kanekiyo, D. Li, L. Reyderman, S. Cohen, L. Froelich, S. Katayama, M. Sabbagh, B. Vellas, D. Watson, S. Dhadda, M. Irizarry, L. D. Kramer and T. Iwatsubo, N. Engl. J. Med., 2023, 388, 9–21 CrossRef.
- B. Dunn, P. Stein and P. Cavazzoni, JAMA Intern. Med., 2021, 181, 1276–1278 CrossRef.
- S. S. Plotkin and N. R. Cashman, Neurobiol. Dis., 2020, 144, 105010 CrossRef CAS.
- L. Söderberg, M. Johannesson, P. Nygren, H. Laudon, F. Eriksson, G. Osswald, C. Möller and L. Lannfelt, Neurotherapeutics, 2023, 20, 195–206 CrossRef.
- Á. Cores, N. Carmona-Zafra, J. Clerigué, M. Villacampa and J. C. Menéndez, Antioxidants, 2023, 12, 1464 CrossRef.
- M. Campora, V. Francesconi, S. Schenone, B. Tasso and M. Tonelli, Pharmaceuticals, 2021, 14, 33 CrossRef CAS.
- K. Rajasekhar, C. Madhu and T. Govindaraju, ACS Chem. Neurosci., 2016, 7, 1300–1310 CrossRef CAS PubMed.
- M. Rana and A. K. Sharma, Metallomics, 2019, 11, 64–84 CrossRef CAS PubMed.
- A. Iscen, C. R. Brue, K. F. Roberts, J. Kim, G. C. Schatz and T. J. Meade, J. Am. Chem. Soc., 2019, 141, 16685–16695 CrossRef CAS PubMed.
- P. Ghosh, S. Bag, S. Parveen, E. Subramani, K. Chaudhury and S. Dasgupta, ACS Omega, 2022, 7, 7931–7944 CrossRef CAS.
- P. Ghosh, S. Chaudhury, S. Parveen and S. Dasgupta, AsiaChem Magazine, 2023, 3, 66–77 Search PubMed.
- A. Li, J. Tyson, S. Patel, M. Patel, S. Katakam, X. Mao and W. He, Front. Bioeng. Biotechnol., 2021, 9, 672594 CrossRef PubMed.
- P. C. Ke, E. H. Pilkington, Y. Sun, I. Javed, A. Kakinen, G. Peng, F. Ding and T. P. Davis, Adv. Mater., 2020, 32, e1901690 CrossRef PubMed.
- K. Tak, R. Sharma, V. Dave, S. Jain and S. Sharma, ACS Chem. Neurosci., 2020, 11, 3741–3748 CrossRef CAS.
- S. Wang, C. Li, Y. Xia, S. Chen, J. Robert, X. Banquy, R. Huang, W. Qi, Z. He and R. Su, iScience, 2020, 23, 101044 CrossRef CAS.
- E. Damian Guerrero, A. M. Lopez-Velazquez, J. Ahlawat and M. Narayan, ACS Appl. Nano Mater., 2021, 4, 2423–2433 CrossRef PubMed.
- G. Gao, M. Zhang, D. Gong, R. Chen, X. Hu and T. Sun, Nanoscale, 2017, 9, 4107–4113 RSC.
- M. Li, P. Shi, C. Xu, J. Ren and X. Qu, Chem. Sci., 2013, 4, 2536–2542 RSC.
- Y. Kim, J.-H. Park, H. Lee and J.-M. Nam, Sci. Rep., 2016, 6, 19548 CrossRef CAS PubMed.
- L. Yang, Y. Chen, Z. Jia, X. Yuan and J. Liu, J. Mater. Chem. B, 2023, 11, 4453–4463 RSC.
- I. Mojzych, A. Zawadzka, K. Kaczyńska, P. Wojciechowski, D. Zając, M. Chotkowski, K. Wiktorska, J. K. Maurin and M. Mazur, Phys. Chem. Chem. Phys., 2023, 25, 16796–16806 RSC.
- A. Mukherjee and N. Sarkar, Mater. Adv., 2023, 4, 2106–2118 RSC.
- M. Mahmoudi, F. Quinlan-Pluck, M. P. Monopoli, S. Sheibani, H. Vali, K. A. Dawson and I. Lynch, ACS Chem. Neurosci., 2013, 4, 475–485 CrossRef CAS.
- A. Sharma and K. S. Ghosh, Bionanoscience, 2023, 13, 1243–1249 CrossRef.
- X. Yao, Y. Guan, J. Wang and D. Wang, Heliyon, 2024, 10, e21789 CrossRef CAS.
- J. Luo, S. K. T. S. Wärmländer, C.-H. Yu, K. Muhammad, A. Gräslund and J. Pieter Abrahams, Nanoscale, 2014, 6, 6720–6726 RSC.
- D. Lin, R. He, S. Li, Y. Xu, J. Wang, G. Wei, M. Ji and X. Yang, ACS Chem. Neurosci., 2016, 7, 1728–1736 CrossRef CAS PubMed.
- Y. Mo, S. Brahmachari, J. Lei, S. Gilead, Y. Tang, E. Gazit and G. Wei, ACS Chem. Neurosci., 2018, 9, 2741–2752 CrossRef CAS PubMed.
- F. Liu, W. Wang, J. Sang, L. Jia and F. Lu, ACS Chem. Neurosci., 2019, 10, 588–598 CrossRef CAS.
- F. Morales-Zavala, P. Jara-Guajardo, D. Chamorro, A. L. Riveros, A. Chandia-Cristi, N. Salgado, P. Pismante, E. Giralt, M. Sánchez-Navarro, E. Araya, R. Vasquez, G. Acosta, F. Albericio, A. Alvarez R and M. J. Kogan, Biomater. Sci., 2021, 9, 4178–4190 RSC.
- J. H. Lucas, Q. Wang, T. Muthumalage and I. Rahman, Toxics, 2021, 9, 144 CrossRef CAS.
- S. Lohan, K. Raza, S. K. Mehta, G. K. Bhatti, S. Saini and B. Singh, Int. J. Pharm., 2017, 530, 263–278 CrossRef CAS.
- M. Bazi Alahri, R. Arshadizadeh, M. Raeisi, M. Khatami, M. Sadat Sajadi, W. Kamal Abdelbasset, R. Akhmadeev and S. Iravani, Inorg. Chem. Commun., 2021, 134, 108997 CrossRef CAS.
- J. Wang, Y. Fan, Y. Tan, X. Zhao, Y. Zhang, C. Cheng and M. Yang, ACS Appl. Mater. Interfaces, 2018, 10, 36615–36621 CrossRef CAS.
- J. Kowalczyk, E. Grapsi, A. Espargaró, A. B. Caballero, J. Juárez-Jiménez, M. A. Busquets, P. Gamez, R. Sabate and J. Estelrich, Biomacromolecules, 2021, 22, 430–440 CrossRef CAS.
- N. Andrikopoulos, Y. Li, A. Nandakumar, J. F. Quinn, T. P. Davis, F. Ding, N. Saikia and P. C. Ke, ACS Appl. Mater. Interfaces, 2023, 15, 7777–7792 CrossRef CAS.
- M. N. Shinde, N. Barooah, A. C. Bhasikuttan and J. Mohanty, Chem. Commun., 2016, 52, 2992–2995 RSC.
- X. Han, J. Park, W. Wu, A. Malagon, L. Wang, E. Vargas, A. Wikramanayake, K. N. Houk and R. M. Leblanc, Chem. Sci., 2017, 8, 2003–2009 RSC.
- V. Oliveri, F. Bellia and G. Vecchio, ChemistrySelect, 2017, 2, 655–659 CrossRef CAS.
- V. Oliveri, S. Zimbone, M. L. Giuffrida, F. Bellia, M. F. Tomasello and G. Vecchio, Chemistry, 2018, 24, 6349–6353 CrossRef CAS PubMed.
- Z. Xu, S. Jia, W. Wang, Z. Yuan, B. Jan Ravoo and D.-S. Guo, Nat. Chem., 2019, 11, 86–93 CrossRef CAS.
- Y. Tian, X. Zhang, Y. Li, T. M. Shoup, X. Teng, D. R. Elmaleh, A. Moore and C. Ran, Chem. Commun., 2014, 50, 15792–15795 RSC.
- T. Yokoyama and M. Mizuguchi, J. Med. Chem., 2019, 62, 2076–2082 CrossRef CAS.
- S. Brahmachari, A. Paul, D. Segal and E. Gazit, Future Med. Chem., 2017, 9, 797–810 CrossRef CAS.
- Y. Wu, S. Guo, K. Wang and J. Kang, Front. Aging Neurosci., 2023, 15, 1139418 CrossRef CAS PubMed.
- B. Rosetti and S. Marchesan, Int. J. Mol. Sci., 2023, 24, 1306 CrossRef CAS PubMed.
- C. Soto, M. S. Kindy, M. Baumann and B. Frangione, Biochem. Biophys. Res. Commun., 1996, 226, 672–680 CrossRef CAS.
- L. X. Lin, X. Y. Bo, Y. Z. Tan, F. X. Sun, M. Song, J. Zhao, Z. H. Ma, M. Li, K. J. Zheng and S. M. Xu, PLoS One, 2014, 9, e112052 CrossRef.
- Z. Liang, H. Y. E. Chan, M. M. Lee and M. K. Chan, Cell Chem. Biol., 2021, 28, 180–190 CrossRef CAS PubMed.
- M. Reches and E. Gazit, Science, 2003, 300, 625–627 CrossRef CAS PubMed.
- P. Yao, J. Zhang, S. You, W. Qi, R. Su and Z. He, J. Mater. Chem. B, 2020, 8, 3076–3086 RSC.
- J. Zhang, P. Yao, S. You, W. Qi, R. Su and Z. He, J. Mater. Chem. B, 2022, 10, 7780–7788 RSC.
- J. Luo and J. P. Abrahams, Chem. – Eur. J., 2014, 20, 2410–2419 CrossRef CAS.
- C. B. Est, P. Mangrolia and R. M. Murphy, Protein Eng., Des. Sel., 2019, 32, 47–57 CrossRef.
- K. Nayak, P. Ghosh, S. Barman, B. Sudhamalla, P. Theato and P. De, Bioconjugate Chem., 2024, 35, 312–323 CrossRef CAS PubMed.
- S. Palmal, N. R. Jana and N. R. Jana, J. Phys. Chem. C, 2014, 118, 21630–21638 CrossRef CAS.
- Z. Evgrafova, B. Voigt, A. H. Roos, G. Hause, D. Hinderberger, J. Balbach and W. H. Binder, Phys. Chem. Chem. Phys., 2019, 21, 20999–21006 RSC.
- P. Ghosh, A. Bera, A. Ghosh, P. Bhadury and P. De, ACS Appl. Bio Mater., 2020, 3, 5407–5419 CrossRef CAS PubMed.
- A. Bera, S. Sahoo, K. Goswami, S. K. Das, P. Ghosh and P. De, Biomacromolecules, 2021, 22, 4833–4845 CrossRef CAS.
- I. Pramudya and H. Chung, Biomater. Sci., 2019, 7, 4848–4872 RSC.
- P. K. Das, D. N. Dean, A. L. Fogel, F. Liu, B. A. Abel, C. L. McCormick, E. Kharlampieva, V. Rangachari and S. E. Morgan, Biomacromolecules, 2017, 18, 3359–3366 CrossRef CAS PubMed.
- S. Bashir, A. Shamsi, F. Ahmad, M. I. Hassan, M. A. Kamal and A. Islam, ACS Omega, 2020, 5, 26871–26882 CrossRef CAS.
- R. Roy, K. Pradhan, J. Khan, G. Das, N. Mukherjee, D. Das and S. Ghosh, ACS Omega, 2020, 5, 18628–18641 CrossRef CAS.
- O. Klementieva, E. Aso, D. Filippini, N. Benseny-Cases, M. Carmona, S. Juvés, D. Appelhans, J. Cladera and I. Ferrer, Biomacromolecules, 2013, 14, 3570–3580 CrossRef CAS.
- A. Bera, P. Ghosh, S. Barman, S. Bhattacharya, B. Sudhamalla, K. Goswami and P. De, Biomater. Sci., 2023, 11, 3574–3588 RSC.
- A. Dey, U. Haldar, T. Rajasekhar, P. Ghosh, R. Faust and P. De, J. Mater. Chem. B, 2022, 10, 9446–9456 RSC.
- A. Mondal, M. E. H. Khan, P. Ghosh and P. De, J. Mol. Eng. Mater., 2021, 09, 2140001 CrossRef CAS.
- A. Bera, P. Ghosh, S. Ghosh, A. Mukherjee and P. De, Macromol. Biosci., 2023, 23, e2300100 CrossRef.
- B. Meesaragandla, S. Karanth, U. Janke and M. Delcea, Sci. Rep., 2020, 10, 7862 CrossRef CAS.
- A. Bera, P. Ghosh, K. Goswami and P. De, ACS Appl. Nano Mater., 2023, 6, 8705–8716 CrossRef CAS.
- F. Huang, J. Wang, A. Qu, L. Shen, J. Liu, J. Liu, Z. Zhang, Y. An and L. Shi, Angew. Chem., Int. Ed., 2014, 53, 8985–8990 CrossRef CAS PubMed.
- J. Zhang, J. Liu, Y. Zhu, Z. Xu, J. Xu, T. Wang, H. Yu and W. Zhang, Chem. Commun., 2016, 52, 12044–12047 RSC.
- H. Geng, H. Yuan, L. Qiu, D. Gao, Y. Cheng and C. Xing, J. Mater. Chem. B, 2020, 8, 10126–10135 RSC.
- A. Bera, D. Mukhopadhyay, K. Goswami, P. Ghosh, R. De and P. De, Biomater. Sci., 2022, 10, 3466–3479 RSC.
- M. Ankarcrona, B. Winblad, C. Monteiro, C. Fearns, E. T. Powers, J. Johansson, G. T. Westermark, J. Presto, B. G. Ericzon and J. W. Kelly, J. Intern. Med., 2016, 280, 177–202 CrossRef CAS.
- J. Hardy and C. Mummery, Brain, 2023, 146, 1240–1242 CrossRef.
- O. Langford, R. Raman, R. A. Sperling, J. Cummings, C. K. Sun, G. Jimenez-Maggiora, P. S. Aisen and M. C. Donohue, J. Prev. Alzheimers Dis., 2020, 7, 213–218 CAS.
- J. Tcw, L. Qian, N. H. Pipalia, M. J. Chao, S. A. Liang, Y. Shi, B. R. Jain, S. E. Bertelsen, M. Kapoor, E. Marcora, E. Sikora, E. J. Andrews, A. C. Martini, C. M. Karch, E. Head, D. M. Holtzman, B. Zhang, M. Wang, F. R. Maxfield, W. W. Poon and A. M. Goate, Cell, 2022, 185, 2213–2233 CrossRef CAS.
- ClinicalTrials.gov. An observational study of aducanumab-avwa in participants with Alzheimer's disease in the US., https://clinicaltrials.gov/study/NCT05097131, (accessed September 5, 2023, 2023).
- ClinicalTrials.gov. A study to verify the clinical benefit of aducanumab in participants with early Alzheimer's disease (ENVISION). https://clinicaltrials.gov/study/NCT05310071, (accessed September 5, 2023, 2023).
- Biogen. Biogen to realign resources for Alzheimer's disease franchise. https://investors.biogen.com/news-releases/news-release-details/biogen-realign-resources-alzheimers-diseasefranchise#:~:text=Biogen%20has%20recorded%20, (accessed March 8, 2024, 2024).
- FDA, Leqembi prescribing information, https://www.accessdata.fda.gov/drugsatfda_d, (accessed September 5, 2023, 2023).
- European Medicines Agency. Leqembi (Lecanemab), https://www.ema.europa.eu/en/medicines/human, (accessed July 30, 2024, 2024).
- Y. Li, S. M. Laws, L. A. Miles, J. S. Wiley, X. Huang, C. L. Masters and B. J. Gu, Cell. Mol. Life Sci., 2021, 78, 7397–7426 CrossRef CAS.
- K. W. Park, Nucl. Med. Mol. Imaging, 2024, 58, 227–236 CrossRef CAS PubMed.
- M. F. Egan, J. Kost, T. Voss, Y. Mukai, P. S. Aisen, J. L. Cummings, P. N. Tariot, B. Vellas, C. H. V. Dyck, M. Boada, Y. Zhang, W. Li, C. Furtek, E. Mahoney, L. H. Mozley, Y. Mo, C. Sur and D. Michelson, N. Engl. J. Med., 2019, 380, 1408–1420 CrossRef CAS PubMed.
- T. Burki, Lancet, 2018, 391, 2486 CrossRef.
- D. Henley, N. Raghavan, R. Sperling, P. Aisen, R. Raman and G. Romano, N. Engl. J. Med., 2019, 380, 1483–1485 CrossRef CAS.
- Y. Zhang, H. Chen, R. Li, K. Sterling and W. Song, Signal Transduction Targeted Ther., 2023, 8, 248 CrossRef CAS.
- T. Wu, D. Lin, Y. Cheng, S. Jiang, M. W. Riaz, N. Fu, C. Mou, M. Ye and Y. Zheng, Aging Dis., 2022, 13, 1745–1758 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2025 |