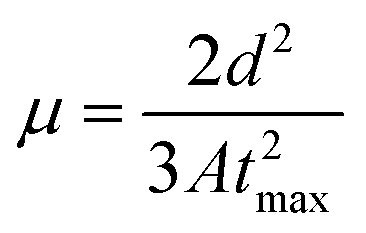New [1]benzothieno[3,2-b]benzothiophene-tetraoxide-based TADF emitters with a D–A–D structure for OLED applications†
Received
29th July 2024
, Accepted 16th October 2024
First published on 17th October 2024
Abstract
Luminescent organic molecules showing thermally activated delayed fluorescence (TADF) are appealing materials for high-efficiency OLEDs. Here, we report a new class of organic luminescent materials with TADF properties, and a D–A–D electronic structure based on [1]benzothieno[3,2-b]benzothiophene-tetraoxide BTBTOx4 as the acceptor unit A. Three donor units D were selected and coupled with BTBTOx4, using a straightforward synthetic protocol based on microwave-assisted Buchwald–Hartwig cross-coupling, to yield three organic luminescent molecules labelled PTz2-BTBTOx4, MPA2-BTBTOx4 and POCz2-BTBTOx4. Chemico-physical and structural properties were investigated by cyclic voltammetry, electrical measurements, crystallographic analysis, theoretical study and photophysical characterization. All three emitters showed high electrochemical stability with reversible oxidation waves. MPA2-BTBTOx4 was selected as the reference molecule for X-ray analysis, which revealed torsion angles of −59° and 86° between the donor (MPA) and acceptor (BTBTOx4) units supporting their appropriate structural configuration to have TADF properties. Photophysical studies highlighted a noteworthy increase in PL efficiency upon deoxygenation for all three compounds. The oxygen-induced quenching of delayed fluorescence and time-resolved photoluminescence studies supported the presence of the TADF properties, further corroborated for PTz2-BTBTOx4 and MPA2-BTBTOx4 by DFT studies. Preliminary steady-state photophysical studies were also carried out on neat films of all three emitters, revealing a pronounced self-quenching of photoluminescence for PTz2-BTBTOx4 and MPA2-BTBTOx4 and a minimal self-quenching for POCz2-BTBTOx4, which maintains a high ΦPL (22%) comparable to that in Zeonex and half of that in PMMA. As a proof of concept, the three emitting molecules were tested as neat films in simple-structure OLED devices. In accordance with the photoluminescence data, POCz2-BTBTOx4, thanks to its sterically bulky structure, retains a good emission capacity even in a neat film and was also selected as an active matrix to build OLED devices by using two different deposition techniques: inkjet-printing and spin coating.
1. Introduction
Organic light emitting diodes (OLEDs) represent a successful technology with applications in flat panel displays and solid-state lighting thanks to their excellent properties including facile fabrication, lightweight construction, low cost, facile colour tunability, low turn-on voltages and high flexibility.1–3 Commercial products such as smartphones and televisions based on this technology are currently present on the market. However, continuous progress is still necessary and existing open challenges include mainly enhancement of luminescence yields, saving operating energy, cheaper and easier preparation of organic emitting materials, lower costs and simplification of device architecture. First-generation OLEDs based on fluorescent emitters are limited to 25% of internal EL quantum efficiency (IQE), since they are able to harvest only singlet excitons. In phosphorescent materials,4–6 heavy metal atoms are required to ensure strong spin–orbit coupling (SOC), facilitating efficient intersystem crossing (ISC) between singlet (S1) and triplet (T1) electronic states, thereby allowing triplet excitons to become emissive. However, these systems typically rely on the use of precious metals such as Ir and Pt, and hence metal-free alternatives are highly desirable. Moreover, compared with fluorescent materials, the stability of phosphorescent materials7–9 is often unsatisfactory. On the other hand, TADF materials10–12 are purely organic compounds, characterized by the presence of efficient reverse intersystem crossing (RISC) between the nearly degenerate S1 and T1 excited electronic states. The singlet–triplet energy barrier (ΔEST) is comparable with the kBT at room temperature, allowing the RISC process to occur. A very small ΔEST in TADF materials is generally achieved using a twisted donor–acceptor (D–A) architecture, where D is an electron-donor unit and A is an electron-acceptor unit, characterized by the presence of a charge transfer (CT) state where the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) are separated on different moieties of the molecule. Furthermore, in TADF materials, the presence of 1CT states (vibronically coupled with 3LE states), or in some cases n–p* states, also allows increasing SOC, further facilitating the ISC/RISC process. The strategy to harvest emission from the triplet excitons appears to be the most promising, due to these materials being generally metal-free and usually easy to synthesize and process, as well as relatively low-cost. A common molecular design strategy to obtain TADF materials includes a D–π–A structural architecture, with π being a π-conjugated bridge.13–19 Other architectures include D–σ–A configurations,20–22 with σ being a non-conjugated linker or a fused polycyclic heteroaromatic structure.23–25 Since the pioneering work of Adachi et al.10 in exploiting the TADF properties in metal-free organic materials for OLEDs, a large number of TADF organic materials have been reported in the literature. These are most often based on D–A,26–30 D–A–D,31–34 D–A–D′,35–39 D–A–A40–42 and D–A–A′32 architectures. On the other hand, the electronic features affect the twisted D–A framework of TADF materials, hence the amplitude of ΔEST and therefore the final efficiency in devices. Chi et al.43 reported interesting results in the study of D–A structures based on dibenzo[a,c]phenazine as an electron acceptor and triphenylamine as an electron donor. To gain insights into the structure–property correlation, they synthesized three molecules with different frameworks of D–A, D–A–D and D–A–A and analysed the effects on photophysical and electroluminescence properties originated from the structural variations of the basic D–A framework. The results show that the introduction of an additional donor or acceptor unit reduces ΔEST, promotes RISC and enhances the photoluminescence quantum yield, providing useful insight into the rational molecular design of efficient TADF materials. From a synthetic standpoint TADF emitters with a D–A–D framework are usually easier to design and prepare and the challenge of creating new efficient TADF emitters based on simple molecular structures that are synthesizable by low cost enabling technologies is still open. Furthermore, the chemical structure of organic emitting materials plays a key role in the processing conditions and then in the complexity of the OLED structure. Organic materials processable from solution for neat-film-based OLEDs are highly desirable as they allow fabrication of simplified final devices based on a reduced number of components and simple processing conditions.44 Aryl-sulfone-derivatives have been widely investigated as suitable electron-acceptor units in TADF emitters.15,44–53 They are very attractive in the D–A–D framework and the corresponding TADF emitters also show very good performance in non-doped devices, highlighting the high potential of pristine films for OLED application. Aryl-sulfone planar units based on heteroaryl fused rings have also been investigated as a structural strategy to reduce exciton annihilation by increasing the rigidity of the electron-acceptor unit A.54–58 [1]Benzothieno[3,2-b]benzothiophene BTBT is a very intriguing polycyclic heteroaromatic planar framework, mainly investigated in field effect transistors58–61 for its excellent charge mobility and, to a lesser extent, as a π-bridge in DSSC solar cells.62 This moiety can be obtained in high yield with a simple synthetic protocol suitable for large-scale production. At the same time, the two thienyl rings can be easily oxidized to yield the S,S-tetraoxide derivative, converting the molecule into an electron-acceptor unit that is potentially useful in D–A–D TADF systems. Herein we report the results of our studies on three new TADF emitters with a D–A–D structure, based on [1]benzothieno[3,2-b]benzothiophene-tetraoxide BTBT-Ox4 as the electron-acceptor unit A and three different electron-donor units D. Phenothiazine, dimethoxyphenylamine and a carbazole derivative were selected as donor units D to prepare the final materials. New BTBT-Ox4-based small molecules were prepared following a straightforward synthetic protocol based on the microwave-assisted Buchwald–Hartwig reaction. They were then characterized by cyclic voltammetry, photo-CELIV measurements, crystallographic analysis, and theoretical and photophysical studies. Photophysical studies revealed the TADF properties in this new class of organic emitting materials and as a proof of concept they were tested as neat emissive layers in solution-processed OLED devices.
2. Results and discussion
2.1 Synthetic procedure
Scheme 1 shows the retrosynthetic procedure followed to prepare the final emitting materials (PTz2-BTBTOx4, MPA2-BTBTOx4, and POCz2-BTBTOx4). The synthesis was accomplished by the Hartwig–Buchwald amination of Br2-BTBTOx4 with two equivalents of the corresponding donor units, i.e. phenothiazine (PTz) for PTz2-BTBTOx4, 4,4′-dimethoxydiphenylamine (MPA) for MPA2-BTBTOx4 and a carbazole derivative (POCz) for POCz2-BTBTOx4. Pd2dba3·CHCl3 and P(t-Bu)3 were used as the catalytic system, sodium tert-butylate as the base and toluene as the solvent. The reactions were performed in a microwave reactor at 110 °C for 1 h. Compound Br2-BTBTOx4 was synthesized by oxidation of 2,7-dibromo-[1]benzothieno[3,2-b]benzothiophene Br2-BTBT63 with hydrogen peroxide in chloroform and trifluoroacetic acid.
 |
| | Scheme 1 Retrosynthetic pathway towards organic emitting materials POCz2-BTBTOx4, PTz2-BTBTOx4 and MPA2-BTBTOx4. | |
All details about the synthetic procedures and chemico-structural characterization are reported in the ESI.† The synthetic protocol is very straightforward and the final step to obtain the emitting materials can be performed in a short time and with high yield using the microwave-assisted Buchwald–Hartwig reaction. Electronic and chemico-structural characteristics determined the choice of donor units, starting from phenothiazine (PTz) to 4,4′-dimethoxydiphenylamine (MPA), to the carbazole derivative POCz. Phenothiazine is a heterocycle compound with a strong electron-donor behaviour and a non-planar butterfly shaped bent structure.64–66 4,4′-Dimethoxydiphenylamine is a strong donor unit widely used in D–π–D molecular semiconductors,67–72 characterized by a non-planar and less rigid structure than the phenothiazine. The carbazole derivative POCz, bis-3,6-(4-octyloxyphenyl)-carbazole, was designed as the donor unit with a more complex molecular structure, combining the carbazole with two octyloxyphenyl units able to enhance the electron donor power of carbazole and at the same time to modulate, by steric effects of aliphatic chains, the aggregation properties in the solid state of the final emitting material.
2.2 Cyclic voltammetry
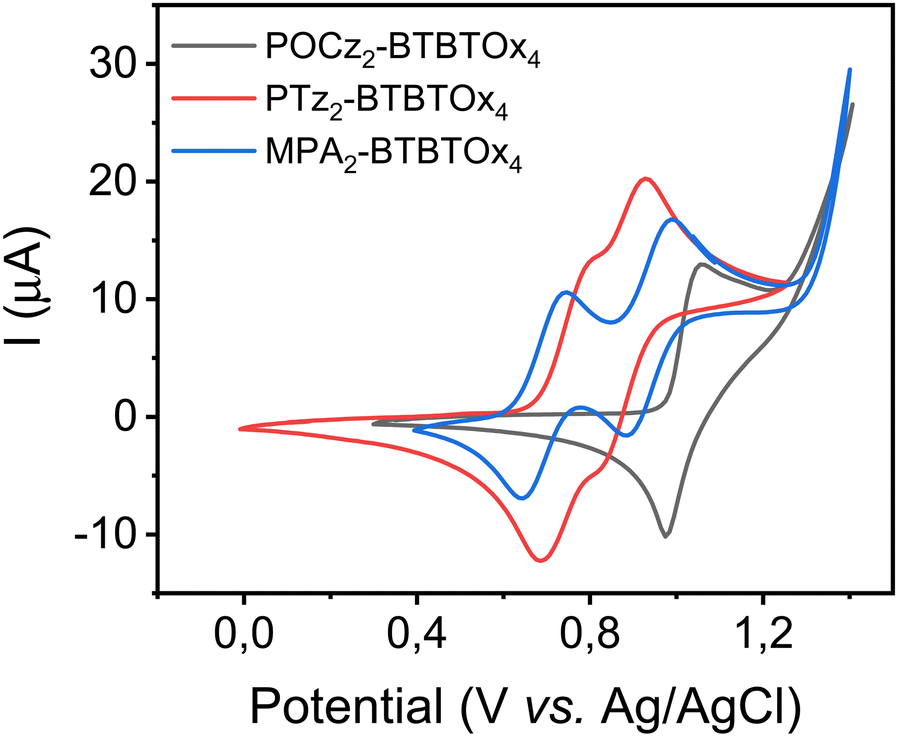
We investigated the electrochemical properties of the BTBTOx4-based emitters BTB by cyclic voltammetry (CV) measurements (Fig. 1).
 |
| | Fig. 1 CV curves of organic emitting materials POCz2-BTBTOx4, PTz2-BTBTOx4 and MPA2-BTBTOx4. | |
All three compounds show two redox oxidation processes, and the values were found to be 1.00 and 1.06 V for POCz2-BTBTOx4, 0.80 and 0.92 V for PTz2-BTBTOx4 and 0.73 and 0.97 V for MPA2-BTBTOx4vs. Ag/AgCl, respectively. The highest occupied molecular orbital (HOMO) energy levels were estimated to be −5.70, −5.42 and −5.35 eV for POCz2-BTBTOx4, PTz2-BTBTOx4 and MPA2-BTBTOx4, respectively. The lowest unoccupied molecular orbital (LUMO) energy levels were calculated from the HOMO levels and the optical energy gaps, and found to be −3.04, −2.70 and −2.97 eV for POCz2-BTBTOx4, PTz2-BTBTOx4 and MPA2-BTBTOx4, respectively. The data are reported in Table 1.
Table 1 Electrochemical properties and corresponding energy levelsa of POCz2-BTBTOx4, PTz2-BTBTOx4 and MPA2-BTBTOx4
| Compound |
E
1/2(1) (mV) |
E
1/2(2) (mV) |
E
HOMO
(eV) |
E
LUMO
(eV) |
E
optg![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) e (eV) e (eV) |
|
c = 1 × 10−3 in CH2Cl2/TBAPF6 (0.1 M), half-wave potentials calculated from the CVs of Fig. 1 and referenced to the Fc+/Fc couple (E1/2 = 434 mV) at 50 mV s−1.
The potentials of POCz2-BTBTOx4 were estimated by differential pulse voltammetry (DPV, see Fig. S4.4, ESI). DPV condition: modulation amplitude 5 mV; modulation time 0.05 s; interval time (Δt) 0.5 s; step potential (Estep) 0.005 V; scan rate ν = Estep/Δt 0.01 V s−1.
E
HOMO = −(Eonsetox1 + 516) (eV).
E
LUMO = EHOMO − Eoptg (eV).
E
optg was estimated by absorption onset.
|
|
POCz2-BTBTOx4
|
566 |
627b |
−5.70 |
−3.04 |
2.67 |
|
PTz2-BTBTOx4
|
367 |
487 |
−5.42 |
−2.70 |
2.72 |
|
MPA2-BTBTOx4
|
296 |
536 |
−5.35 |
−2.97 |
2.39 |
2.3 Charge carrier mobility
In the present work we used the photo-CELIV technique that has been developed and used for measuring the charge mobility of organic layers in the direction perpendicular to the film,73 which is the direction most relevant for photovoltaic applications or light emitting diodes.74,75 In Fig. 2 the photo-CELIV responses of an ITO/POCz2-BTBTOx4 or PTz2-BTBTOx4 or MPA2-BTBTOx4/Al device are shown. Transients were acquired by optically pumping the sample at a wavelength of 450 nm and detected as a voltage drop by using an external load. A wavelength of 450 nm was chosen to provide a bulk photo-excitation for an accurate measurement of the mobility.76 The time delay, maximum ramp voltage, pulse duration, and the external resistor were varied accordingly in order to optimize the signal to noise ratio.77 The photo-CELIV response is characterized by a sharp increase, rectangular current transient (J0), and a slowly growing current (ΔJ) whose maximum depends on the applied ramp voltage. By using peak time (tmax) values we were able to determine the carrier mobility according to the equation below, which is valid in the case of volume photogeneration and a drift current maximum (ΔJ) lower than the displacement current (J0):78
where d is the sample thickness and A is the ramp speed (voltage/ramp duration). In the present case, the PTz2-BTBTOx4 compound is characterized by the lower mobility value 5.2 × 10−8 cm2 V−1 s−1, meanwhile both MPA2-BTBTOx4 and POCz2-BTBTOx4 have similar mobility 1.1 × 10−6 cm2 V−1 s−1, at a speed of around 2 × 103 (V s−1). Regarding the latter case, we can clearly observe that the higher PLQY correlates with a good charge carrier mobility, making POCz2-BTBTOx4 a suitable candidate for OLED applications.
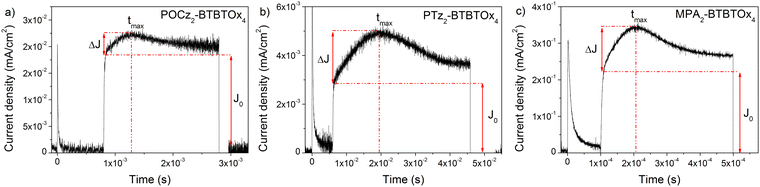 |
| | Fig. 2 Photo-CELIV transients on ITO/POCz2-BTBTOx4 or PTz2-BTBTOx4 or MPA2-BTBTOx4/Al devices. Measurements were performed in vacuum, at room temperature, at a ramp speed of around 2 × 103 (V s−1). | |
2.4 X-ray powder diffraction (XRPD) analysis
XRPD patterns were collected using a Rigaku RINT2500 rotating anode diffractometer (50 kV, 200 mA) equipped with a silicon strip Rigaku D/teX Ultra detector. An asymmetric Johansson Ge(111) crystal was used to select the monochromatic Cu Kα1 radiation (λ = 1.54056 Å). Measurements were executed in transmission mode by introducing the sample in a Lindemann glass capillary tube with a diameter of 0.5 mm. The XRPD patterns were recorded in the 2θ range of 4–60° for POCz2-BTBTOx4 and PTz2-BTBTOx4 and of 6–100° for MPA2-BTBTOx4, by step scanning, using 2θ increments of 0.02° and a fixed counting time of 2 s per step. The diffraction pattern of POCz2-BTBTOx4 revealed the amorphous nature of the material (see Fig. S5.1, ESI†), that, consequently, could not be characterized via an ab initio structure solution process. To check the novelty and phase purity of PTz2-BTBTOx4 and MPA2-BTBTOx4, a qualitative analysis using the powder diffraction data was carried out via the software QUALX2.0,79 aimed at identifying the presence of one (or more) known crystal phase stored in the POW_COD database.79 The qualitative analysis revealed that the two compounds were unpublished so far. For both, the structure solution process was attempted by EXPO;80 the full pathway (i.e., indexing, space group determination, extraction of the integrated intensities, structure solution and Rietveld refinement) was successful for MPA2-BTBTOx4 and did not succeed for PTz2-BTBTOx4 due to the lower quality and resolution of the diffraction data in this case. MPA2-BTBTOx4 crystallized in the centrosymmetric space group P21/c (Table S5.1, ESI†). The asymmetric unit, consisting of 27 non-H atoms (chemical formula C21H17NO4S), was characterized by a planar core (including the aromatic ring C13–C14–C16–C18–C17–C15 and three additional atoms, i.e., S1, N1 and C21, see Fig. S5.2, ESI†), and two out-of-plane methoxy-phenyl groups (torsion angles C1–C2–N1–C13 and C13–N1–C7–C8 equal to −59° and 86°, respectively, see Fig. S5.2 and Table S5.2, ESI†). A view of the refined crystal structure determined by EXPO is provided in Fig. 3, showing the asymmetric unit (i.e., half a molecule, colour setting by atomic species) and its symmetry equivalent unit (half a molecule, monochromatic blue colour) completing the butterfly-shape molecule. Details on the ab initio structure solution process are accompanied by two tables providing the main crystallographic data (Table S5.1, ESI†), the refined fractional atomic coordinates and isotropic displacement parameter, bond distances and angles and torsion angles (Table S5.2, ESI†), which are all supplied in the ESI.†
 |
| | Fig. 3
MPA2-BTBTOx4: a view of the asymmetric unit (half a molecule, drawn on the left hand side), showing the atom-labelling scheme and the colour setting by atomic species (i.e., yellow – S, red – O, light blue – N, and grey – C); the other half of the molecule is shown in blue and consists of symmetry equivalent atoms, whose labelling is identical to that of the corresponding symmetry independent atoms. H atoms are omitted for clarity. Mercury81 software was used to generate this graphic. | |
The crystal architecture was stabilized by a large number and variety of non-covalent intermolecular interactions acting cooperatively82–87 (see Fig. S5.3 and additional details described in the ESI†).
The weakness of the interactions between non-parallel rings is highlighted by the Hirshfeld surface generated by the software CrystalExporer,88 providing a useful tool for a three-dimensional representation of the main intermolecular interactions in the crystal (see Fig. S5.4, i.e., the Hirshfeld surface mapped over dnorm, ESI†).
2.5 DFT results
Density functional theory (DFT) and time-dependent DFT (TD-DFT) can provide useful insights for understanding of photophysical characteristics of luminescent molecules. In this work we study the ground (S0) as well as the singlet (S1) excited state geometries of POCz2-BTBTOx4, PTz2-BTBTOx4 and MPA2-BTBTOx4 to shed some light onto the delayed fluorescent properties of these luminophores. The geometry optimisations were performed using the PBE0/def2-svp89 level of theory, while single point energy calculations used CAM-B3LYP/ZORA-def2-TZVP89 following similar calculations recently performed for Ir(III) complexes.90 For this task we used the Orca 5.0.3 software package.91,92
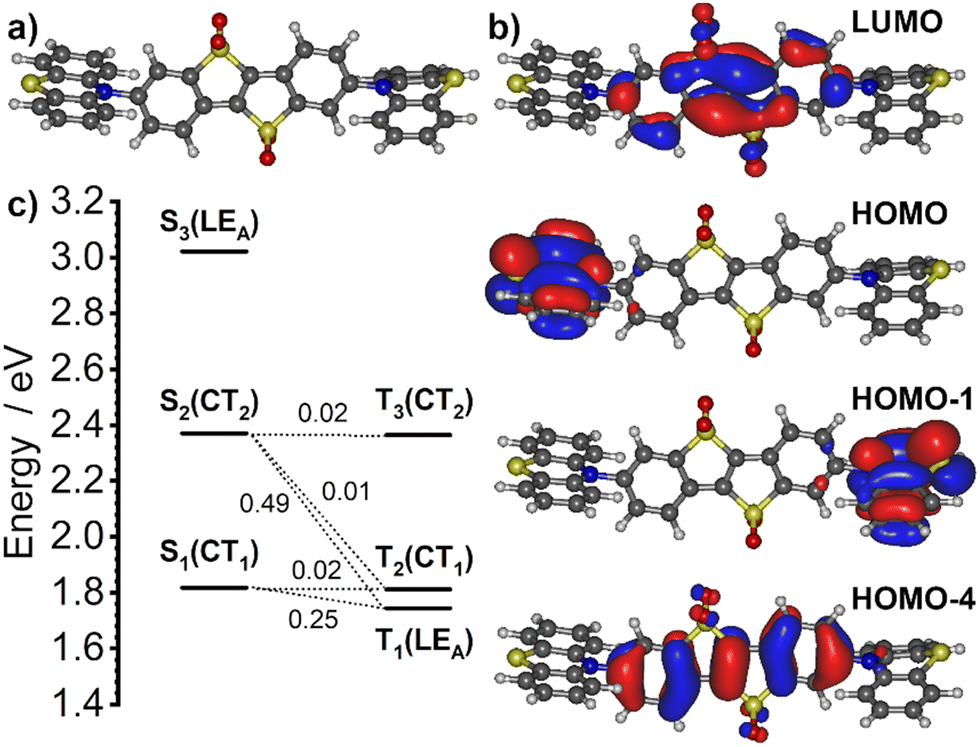
At first, we study the ground (S0) and excited state (S1) geometries of POCz2-BTBTOx4, PTz2-BTBTOx4 and MPA2-BTBTOx4 (Fig. 4). It is immediately apparent that the central acceptor core is flat, with the >SO2 oxygen atoms being the only moieties out of plane. There are no fundamental structural differences between the S0 and S1 geometries, except perhaps for the relative orientation of donors with respect to the acceptor. At S0 geometries both donors retain generally similar conformations against the acceptor core making them roughly equivalent. This equivalent function of donors is also reflected in the symmetric distribution of HOMOs in the ground state (Fig. 5). This equivalence or symmetry is broken in the S1 geometry, where the two donors show evidently different conformations from one to another: the first one retaining a relatively similar orientation to that in the ground state and the other being twisted around the C–N axis, or deformed. In POCz2-BTBTOx4 and MPA2-BTBTOx4 this is expressed by a larger D–A angle, leading to a more orthogonal configuration, and therefore a lower energy CT state (S1) energy associated with this donor. A CT state associated with the other donor is also present at a higher energy (i.e. S2). In the ground state geometry, the first singlet excitation (S0 → S1) is dominated by the HOMO → LUMO transition and can be assigned a CT (PTz2-BTBTOx4) or hybrid CT + LE (POCz2-BTBTOx4, MPA2-BTBTOx4) character. Interestingly, in PTz2-BTBTOx4 the configuration of both phenothiazines in the ground state is equatorial, while in the S1 state one of the donors rearranges into a planar configuration. The said planar configuration serves as a stronger donor and contributes to the formation of the S1 state, while the other phenothiazine unit in the equatorial configuration contributes to the S2. In both configurations however the D–A form a roughly orthogonal arrangement. Donor–acceptor systems featuring phenothiazine are notorious for displaying dual emissive behaviours.15,93,94 In the case of PTz2-BTBTOx4 two conformers of phenothiazine, equatorial and planar, coexist in one S1 geometry giving rise to distinctive singlet CT excited states associated with either one of the two donors, as mentioned above. We hypothesize that this evident potential to switch between equatorial and planar geometries in the excited state may give rise to dual emissive behaviour in PTz2-BTBTOx4. Although the phenothiazine donors in PTz2-BTBTOx4 are shown to both be in their quasi-equatorial configuration in the S0 this does not preclude higher energy conformers from being present, such as the quasi-axial forms.
 |
| | Fig. 4 Comparison between calculated ground (S0) and excited state (S1) geometries. | |
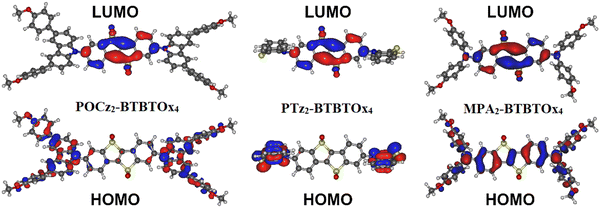 |
| | Fig. 5 HOMO and LUMO isosurfaces calculated at the S0 geometry of POCz2-BTBTOx4, PTz2-BTBTOx4 and MPA2-BTBTOx4. | |
TD-DFT singlet and triplet excitation energies and the respective orbital pairing calculated at the S1 geometry are shown in Table S6.1 (ESI†), in Fig. 6 for PTz2-BTBTOx4 and in Fig. S6.1 and S6.2 (ESI†). The general picture of the excited states is very similar in all three emitters with the T1 state being generally of 3LEA (excited state localised on the acceptor) nature, while the S1 state is associated with an intramolecular charge transfer (1CT) character. One may note S1 and S2 are associated with distinctive CT transitions involving different donor moieties. The ΔEST calculated directly from the S1 and T1 energies are: 0.53 eV in POCz2-BTBTOx4, 0.07 eV in PTz2-BTBTOx4, and 0.59 eV in MPA2-BTBTOx4. Given the small ΔEST in PTz2-BTBTOx4 one may reasonably expect this emitter to display TADF. At first the ΔEST for POCz2-BTBTOx4 and MPA2-BTBTOx4 appear too large to support potential TADF behaviour. However, a closer look at the reasons for the magnitude of ΔEST opens an alternative pathway to interpret these results. At the minimal S1 geometry of PTz2-BTBTOx4 the donor and acceptor are nearly completely decoupled, hence the T1 represents a ‘pure’ 3LEA state with an energy of 1.75 eV. The optimised S1 geometry of POCz2-BTBTOx4 and MPA2-BTBTOx4 however shows a partial conjugation between the donor and acceptor. In this case the T1 energy drops ever so slightly to 1.74 eV in POCz2-BTBTOx4 and more significantly to 1.51 eV in MPA2-BTBTOx4. We believe that, however, the influence of the partial donor–acceptor conjugation may be exaggerated in this case with respect to the real system. For example, the donors will be allowed to freely rotate around the C–N bonds due to the small activation energy for this motion. We believe that the contribution of configurations with more decoupled donor and acceptor pairs appearing in real systems may lead to the T1 energy being closer to that of a fully decoupled system than that determined in this calculation. By using the energy of a ‘pure’ 3LEA state with an energy of 1.75 eV from PTz2-BTBTOx4 we estimate ΔEST of 0.51 eV for POCz2-BTBTOx4 and 0.35 eV for MPA2-BTBTOx4, hence demonstrating that TADF may be possible at least in MPA2-BTBTOx4. Spin–orbit coupling matrix elements (SOCME) between the lowest singlet and triplet states are shown in Table S6.2 (ESI†). They clearly demonstrate potential pathways for ISC and RISC within these systems. For example, in PTz2-BTBTOx4 the 〈T1|ĤSO|S1〉 and 〈T1|ĤSO|S2〉 are ∼0.2–0.5 cm−1, one order of magnitude larger than 〈T2|ĤSO|S1〉 and 〈T2|ĤSO|S2〉. In this case the role of the T1 (3LEA) is necessary to promote RISC to the 1CT state. Somewhat similarly, the 〈T1|ĤSO|S1〉 and 〈T1|ĤSO|S2〉 in POCz2-BTBTOx4 and MPA2-BTBTOx4 are larger than other pairings due to the analogous orbital nature of these two states as in PTz2-BTBTOx4.
 |
| | Fig. 6 TD-DFT calculations for PTz2-BTBTOx4 at S1 geometry: (a) structure in the S1 state; (b) isosurfaces of the relevant molecular orbitals; (c) excited state energy diagram with the orbital character of each of the excited states; the values and dotted lines represent SOCME in cm−1 between the indicated states. | |
2.6 Photophysical properties
In this section, we explore the photophysical characteristics of PTz2-BTBTOx4, MPA2-BTBTOx4 and POCz2-BTBTOx4, aiming to understand how different donor units influence their optical behaviour and thus their potential as TADF materials in OLEDs. Specifically, the three donor units, phenothiazine (PTz), dimethoxyphenylamine (MPA) and carbazole (Cz) derivative (POCz) exhibit different electron-donating and steric behaviours. We conducted a comprehensive study of these compounds using UV-vis absorption, steady-state and time-resolved fluorescence spectroscopy. The initial investigation involved studying the photophysical properties in diluted solution (10−5 M) in three solvents with different polarity: cyclohexane (CH), toluene (Tol) and dichloromethane (DCM). Then, we moved to the solid-state (as isolated molecules) by means of two polymer matrices with different polarity, namely zeonex and poly(methyl methacrylate) (PMMA). Finally, the steady-state photophysical properties of the neat films (drop casting) were also explored. A summary of the photophysical results for all three compounds can be found in Tables 3 and 4.
2.6.1 Absorption spectra.
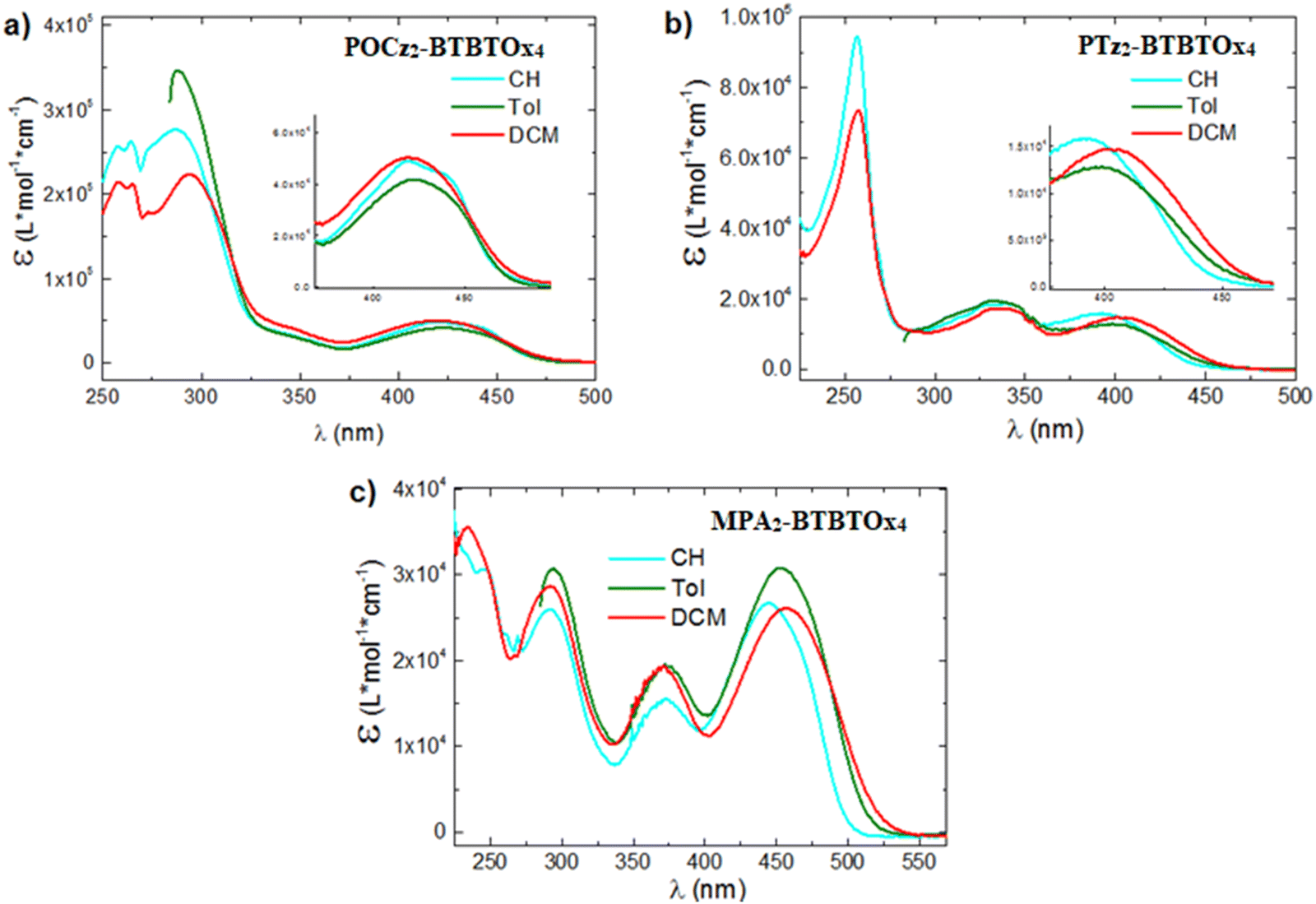
The UV-vis absorption spectra of the three compounds PTz2-BTBTOx4, MPA2-BTBTOx4 and POCz2-BTBTOx4 were recorded in diluted solutions (10−5 M) of three solvents with different polarity: cyclohexane (CH), toluene (Tol) and dichloromethane (DCM) (Fig. 7). In all systems, the lowest energy band is attributed to Intramolecular Charge Transfer (ICT) processes between the donor (D) and the acceptor (A) units. This finding is consistent with the TD-DFT calculations since it is identified as a CT or CT + LE HOMO–LUMO transition between the donor and acceptor units (Table S6.1, ESI†). Specifically, we observed that the CT band appears Gaussian in all cases, except for POCz2-BTBTOx4 in CH, where it is well-resolved indicating the presence of a hybrid localized charge transfer (HLCT or CT + LE) state, again in line with calculations. By increasing the solvent polarity, a red-shifted Gaussian emission is observed for all molecules, confirming the CT character of this transition. This shift is attributed to dipolar interactions with the solvent, favouring the stabilization of the CT state. Conversely, the peaks at higher energy remain unaffected by polarity and are associated with locally excited (LE) transitions on the donor or acceptor unit. Specifically, for POCz2-BTBTOx4, the absorption band at ∼290 nm is assigned to the π–π* localized absorption of Cz, as well as that at 330 nm.95 For PTz2-BTBTOx4, the absorption bands at λmax = 315 and 330 nm, along with the shoulder at 290 nm, are assigned to the localized absorption of phenothiazine.96 In the case of MPA2-BTBTOx4, at higher energy, two bands are observed at 280 and 375 nm, corresponding to LE transitions of dimethoxyphenylamine. Lastly, in the deep UV region, the transitions between 225 and 260 nm are associated with LE states of the acceptors for all the systems.
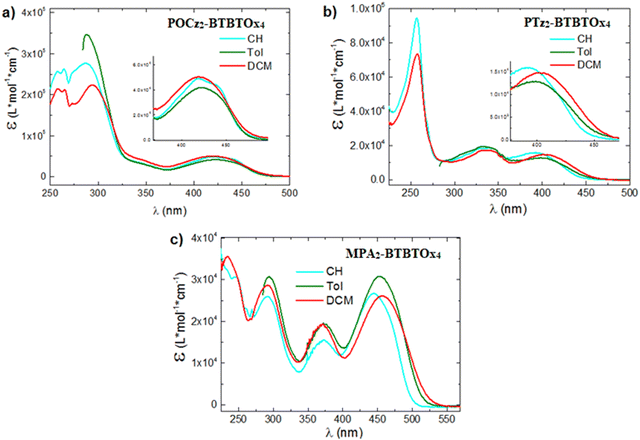 |
| | Fig. 7 UV-vis absorption spectra in CH (10−5 M, cyan), in Tol (10−5 M, green) and DCM (10−5 M, red) of (a) POCz2-BTBTOx4, (b) PTz2-BTBTOx4, and (c) MPA2-BTBTOx4. In toluene, the UV-vis absorption below 285 nm is not reported since it is obscured by the solvent UV-vis absorption cut-off wavelength. | |
2.6.2 Solution PL (steady-state and time-resolved).
The photoluminescence (PL) spectra in diluted solutions measured in three solvents with increasing polarity (CH, Tol and DCM), reveal distinct patterns among POCz2-BTBTOx4, PTz2-BTBTOx4 and MPA2-BTBTOx4 (Fig. 8). Notably, POCz2-BTBTOx4 and PTz2-BTBTOx4 exhibit two distinct bands (except in DCM where PTz2-BTBTOx4 displays only one band), while MPA2-BTBTOx4 shows a single band in all the three solvents. For all the samples, the low-lying band is the most intense and displays positive solvatochromic behaviour, typical for donor–acceptor (D–A) systems. In CH, its structured profile suggests a hybrid local and charge transfer (HLCT) character, while in toluene, the band becomes Gaussian and is red shifted due to the increased solvent polarity that stabilizes the CT state. Further increasing the polarity in DCM results in a red shift of the CT band for POCz2-BTBTOx4 and MPA2-BTBTOx4. For PTz2-BTBTOx4 a strong stabilization effect is responsible for the extinction of the CT emission. Concerning the higher-energy band, especially in POCz2-BTBTOx4 (the inset in Fig. 8a), it remains unperturbed by solvent polarity, indicating locally excited (LE) character.
 |
| | Fig. 8 Normalized PL spectra in CH (10−5 M, cyan), Tol (10−5 M, green) and DCM (10−5 M, red) of (a) POCz2-BTBTOx4, inset: normalized higher energy band, (b) PTz2-BTBTOx4, and (c) MPA2-BTBTOx4. | |
For deeper insights into the photophysical mechanism, decay profiles (Fig. S7.1, ESI†) were acquired, and lifetimes were determined (Table 2). We note that, in the cases of POCz2-BTBTOx4 in CH and DCM, and MPA2-BTBTOx4 in DCM, the decay collected for the lower energy band exhibits a bi-exponential decay, with the longer component assigned to the CT state, while the shorter one can be assigned to a HLCT state. In the other cases, i.e. for all systems in Tol, for PTz2-BTBTOx4 in CH and DCM and for MPA2-BTBTOx4 in CH, the decay collected for the lower energy band exhibited a mono-exponential course, originating from the CT state. For POCz2-BTBTOx4 and PTz2-BTBTOx4 we also collected the decay for the LE band (higher energy, Fig. S7.1, ESI†), nevertheless, since the excitation is at 405 nm we acquired the decay at 430–450 nm and thus the double lifetime observed in some depends on the tail of the CT band (Table 2). The photoluminescence quantum yields (ΦPL) were determined for all three compounds (Table 2). The values reduce upon increasing solvent polarity, which can be assigned to polarity-related stabilization of the excited state. The higher ΦPL in CH is attributed to the presence of a HLCT transition which, in comparison to the CT transition, displays a larger HOMO–LUMO overlap and hence higher oscillator strength f(S1 → S0). Notably, ΦPL exceeds 50% for POCz2-BTBTOx4 and MPA2-BTBTOx4, while for PTz2-BTBTOx4 it reaches only 3%, indicating a reduction in the donor–acceptor conjugation in the last example and thus of the transition oscillator strength. Furthermore, the effect of oxygen quenching on quantum yields and lifetimes was tested for all three compounds. However, after deoxygenation, none of the compounds showed intensity variation with long-lived emission, ruling out the possibility of TADF activity in solution.
Table 2 Photophysical data of POCz2-BTBTOx4, PTz2-BTBTOx4 and MPA2-BTBTOx4 in CH, Tol and DCM
|
|
CH |
Tol |
DCM |
|
Φ
PL (%) |
τ (ns) |
Φ
PL (%) |
τ (ns) |
Φ
PL (%) |
τ (ns) |
|
POCz2-BTBTOx4
|
68.8 |
λ
Em = 430 nm |
51.2 |
λ
Em = 430 nm |
3.8 |
λ
Em = 440 nm |
| 2.8(25%), 3.8(75%) |
1.0(7%), 5.4(93%) |
1.8(32%), 5.4(68%) |
|
λ
Em = 500 nm |
λ
Em = 510 nm |
λ
Em = 600 nm |
| 2.7(23%), 4.9(77%) |
5.5(100%) |
1.4(94%), 5.4(6%) |
|
|
|
PTz2-BTBTOx4
|
2.7 |
λ
Em = 450 nm |
1.2 |
λ
Em = 450 nm |
0.3 |
λ
Em = 450 nm |
| 3.2(100%) |
2.5(100%) |
1.3(60%), 4.2(40%) |
|
λ
Em = 570 nm |
λ
Em = 600 nm |
λ
Em = 530 nm |
| 5.1(100%) |
4.1(100%) |
4.0(100%) |
|
|
|
MPA2-BTBTOx4
|
59.6 |
λ
Em = 517 nm |
55.6 |
λ
Em = 550 nm |
11.2 |
λ
Em = 600 nm |
| 4.1(100%) |
4.5(100%) |
1.7(95%) 3.9(5%) |
2.6.3 Steady-state and time-resolved PL emission properties in solid polymer thin film matrices.
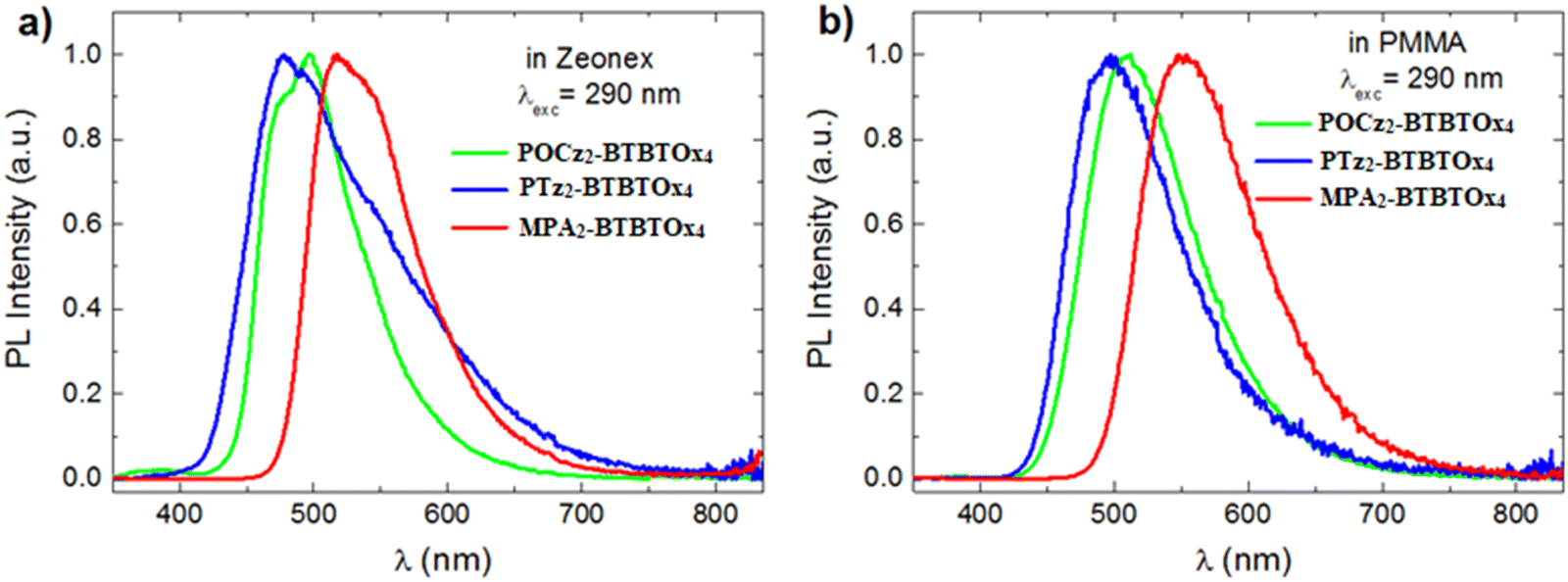
We also explored the photoluminescence features of POCz2-BTBTOx4, PTz2-BTBTOx4 and MPA2-BTBTOx4 in the solid state by dispersing the samples at a low concentration (0.1% by weight) in two different polarity polymer matrices, namely Zeonex and PMMA. The steady-state emission of all the three compounds reveals distinct behaviours (Fig. 9). In Zeonex, the profile appears resolved with HLCT character while, in PMMA, it becomes Gaussian due to its higher polarity stabilizing the CT state. A noteworthy observation is the bathochromic shift of the PL emission of MPA2-BTBTOx4 in both polymers compared to POCz2-BTBTOx4 and PTz2-BTBTOx4. We can also observe that PTz2-BTBTOx4 in Zeonex exhibits a broadened emission compared to PMMA, with the formation of a new shoulder at 550 nm, suggesting the presence of multiple emissive states.
 |
| | Fig. 9 Normalized PL spectra of POCz2-BTBTOx4, PTz2-BTBTOx4 and MPA2-BTBTOx4 in: (a) Zeonex (0.1 wt%) and (b) PMMA (0.1 wt%). | |
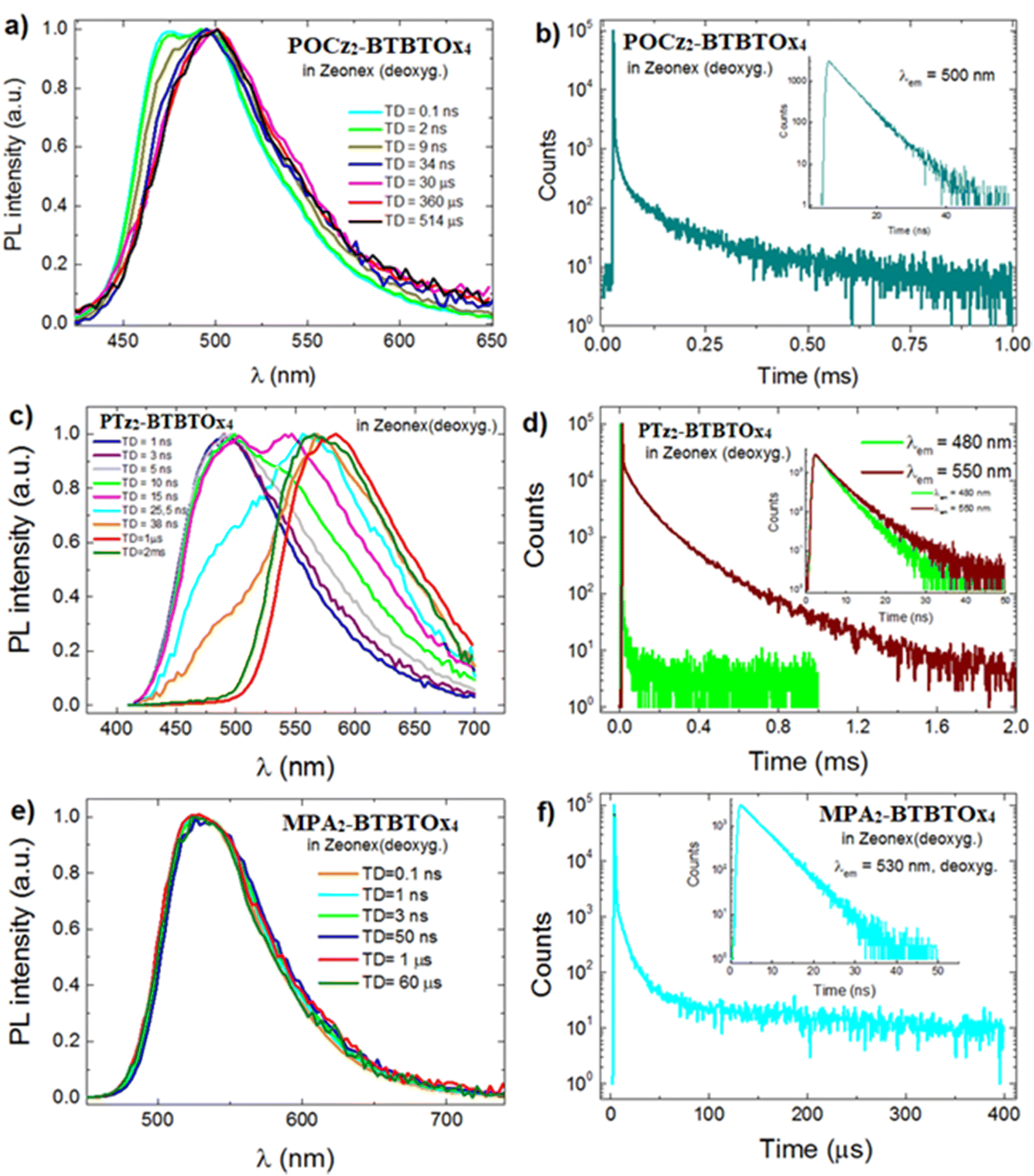
The photophysical properties of all the three compounds in Zeonex were also investigated under deoxygenated conditions. In Fig. S7.2 (ESI†) we can note that while the emission spectrum of POCz2-BTBTOx4 and MPA2-BTBTOx4 remains almost unchanged, PTz2-BTBTOx4 in Zeonex reveals an additional intense band at lower energy. Further experiments, using various excitation wavelengths and doping concentrations (0.1, 1 and 5 wt%) (Fig. S7.3, ESI†), demonstrate that the lower energy PL band in PTz2-BTBTOx4 is affected by these parameters. Notably, the lower energy PL band gradually diminishes as the excitation shifts to longer wavelengths (λex in Fig. S7.3, ESI†), resulting in fully Gaussian emission at λex = 400 nm. This behaviour can be observed at all doping concentrations, with the lower energy band being markedly more pronounced at the highest load. Based on these findings, we can identify two distinct emissive species. Specifically, it exhibits emission varying with excitation wavelength: one PL band evident for λexc (below 400 nm) and another for λexc = 300–350 nm. According to the DFT calculations, these two species can be potentially attributed to the two different phenothiazine conformers on the same molecule (planar and equatorial), aligning with previous reports on phenothiazine-substituted compounds.15,93,94 The increased presence of conformers at higher doping concentrations indicates that intermolecular interactions occurring in the condensed phase are crucial for the formation of lower energy emissive conformers. As shown in Table 3, POCz2-BTBTOx4 and PTz2-BTBTOx4 in PMMA display double the PLQY values in air compared to the same conditions in Zeonex, while that for MPA2-BTBTOx4 in air is very similar in both polymers. Furthermore, when comparing the ΦPL of POCz2-BTBTOx4 in polymers with that in dilute solutions, it is evident that the ΦPL is higher in CH and Tol than in both polymers, particularly in Zeonex. This potentially suggests an aggregation-caused quenching (ACQ) effect, which is more pronounced in Zeonex, probably due to the lower compatibility of this polymer with POCz2-BTBTOx4. On the other hand, PTz2-BTBTOx4 displays a larger ΦPL in both polymer matrices, especially in PMMA, than in solution, indicating that rigido-chromic effects dominate. Finally, for MPA2-BTBTOx4ΦPL remains almost unchanged from solution to both polymer matrices, and this effect may be attributed to the presence of bulky dimethoxyphenylamine donors, which likely prevent both π–π stacking and other non-radiative deactivation processes. We also studied photoluminescence quenching in both polymer matrices for all three emitters. Interestingly, after deoxygenation, the PL intensity increases for all three compounds in both polymer matrices (Table 3), thus suggesting that all three materials may present TADF properties. In particular, for PTz2-BTBTOx4, the ΦPL in Zeonex films gradually decreases with increasing doping concentration. Still, the ratio ΦairPL/ΦdeoxPL (Table S7.2, ESI†) is higher for 0.1 wt% film upon deoxygenation, suggesting that around a concentration of 0.1 wt% in Zeonex, the formation of species with delayed fluorescence (DF) properties is favoured. We conducted time-resolved PL studies of all three compounds in deoxygenated thin film in Zeonex, where the ΦPL in deoxygenated conditions is higher, and the resulting decay lifetimes are shown in Table 3. Additionally, we performed time-resolved emission spectroscopy (TRES) mode measurements, allowing the reconstruction of individual spectra at specific time delays (TD) (see the photophysical data in the ESI,† for details), for all three compounds in Zeonex (0.1 wt%). The time-resolved spectra at rt for all three compounds depicted in Fig. 10, display distinct behaviours among the three systems.
Table 3 Photophysical results for POCz2-BTBTOx4, PTz2-BTBTOx4 and MPA2-BTBTOx4 in PMMA and Zeonex
|
|
PMMA |
Zeonex |
| %ΦPL air/deox. |
τ
1/τ2 (ns) deox. |
τ
1/τ2 (ms) deox. |
Φ
PL air/deox. |
τ
1/τ2 (ns) deox. |
τ
1/τ2 (μs) air/deox. |
| air = in aerated conditions; deox = deoxygenated conditions; τav = average lifetime. |
|
POCz2-BTBTOx4
|
44/59 |
λ
em = 500 nm |
λ
em = 500 nm |
25/76 |
λ
em = 500 nm |
λ
em = 500 nm |
| 8.3(100%) |
0.3(18%), 1.7(82%) |
4.1(59%), 6.0(41%) |
36.4(36%) |
|
|
τ
av = 1.7 |
τ
av = 5.4 |
194.1(64%) |
|
|
|
|
τ
av = 179 |
|
|
|
PTz2-BTBTOx4
|
18/30 |
λ
em = 490 nm |
λ
em = 490 nm |
10/30 |
λ
em = 480 nm |
λ
em = 480 nm |
| 8.6(100%) |
0.5(38%), 3.1(61%) |
1.9(20%), 4.2(80%) |
NO DF |
|
|
τ
av = 2.9 |
τ
av = 4.0 |
λ
em = 550 nm |
|
|
|
λ
em = 550 nm |
87.4(61%), 253.3(39%) |
|
|
|
2.9(44%), 5.8(56%) |
τ
av = 195 |
|
|
|
τ
av = 5.0 |
|
|
|
|
MPA2-BTBTOx4
|
58/85 |
λ
em = 560 nm |
λ
em = 560 nm |
60/93 |
λ
em = 530 nm |
λ
em = 530 nm |
| 6.2(100%) |
0.3(25%), 1.7(75%) |
4.9(100%)/5.0(100%) |
7.4(71%), 88.5(29%) |
|
|
τ
av = 1.6 |
τ
av = 5.0 |
τ
av = 74.7 |
Table 4 Photophysical data for POCz2-BTBTOx4, PTz2-BTBTOx4 and MPA2-BTBTOx4 in neat film
| Compound |
Absorption (λmax, nm) |
Emission (λmax, nm) |
% ΦPL |
|
POCz2-BTBTOx4
|
267, 296, 347(s), 439 |
560 |
22 |
|
PTz2-BTBTOx4
|
262, 341, 416, 600 |
516, 630 |
1.2 |
|
MPA2-BTBTOx4
|
293, 372, 467, 632 |
593 |
1.3 |
 |
| | Fig. 10 (a) Time-resolved emission spectra in Zeonex (0.1 wt%) of (a) POCz2-BTBTOx4, (c) PTz2-BTBTOx4, and (e) MPA2-BTBTOx4. PL decay in Zeonex (0.1 wt%) in the microsecond scale (insets: decay in the nanosecond scale) of (b) POCz2-BTBTOx4, (d) PTz2-BTBTOx4, and (f) MPA2-BTBTOx4. | |
Specifically, for POCz2-BTBTOx4 (Fig. 10a), the high-energy emission peak is visible only in the early times, attributed to a locally excited state (1LE) from direct excitation and emission from the D or A moiety. At longer time delays (TD = 30 ns), the initial 1LE emission disappears, and only the lower energy band attributed to CT emission remains invariant throughout the decay. This indicates luminescence from the singlet state for both prompt and delayed components, suggesting TADF origin of this emission.97,98 For MPA2-BTBTOx4 (Fig. 10e), the emission spectra appear more Gaussian-shaped, remain invariant throughout the decay, suggesting TADF emission for this molecule in Zeonex at room temperature. For both POCz2-BTBTOx4 and MPA2-BTBTOx4, PL decays were collected, and lifetimes were measured (Table 3), showing two characteristic average lifetimes (τav): one in the nano-second regime attributed to the prompt fluorescence (PF) and the other in the micro-/milli-second one due to TADF. However, as observed in the decay curves (Fig. 10b and f), it is important to note that for both POCz2-BTBTOx4 and MPA2-BTBTOx4, the contribution of the TADF appears rather insignificant. This is clearly visible through the relative counts of the longer component in the decay, below 103, more than two orders of magnitude lower than the PF component. Additionally, a higher laser power is required to observe the DF component, especially in POCz2-BTBTOx4. These observations are consistent with the singlet–triplet energy gap obtained from the DFT calculations, which is particularly high for POCz2-BTBTOx4. Conversely, the analysis of spectra acquired for PTz2-BTBTOx4 (Fig. 10c) reveals a different behaviour for different time delays. At early times (0–2 ns), the emission arises entirely from the higher energy emissive state (λmax = 480 nm), while the emission associated with the low-energy emissive state (570 nm) appears after 5 ns, becoming dominant at TD = 25 ns. Upon increasing delay time (after 1 μs), only the state centred at 570 nm remains invariant, with the spectral shape almost invariant, even with an increased delay time up to a few milliseconds. This behaviour is attributed to the presence of TADF, observed in the low-energy emissive state only,97,98 which is attributed, according to DFT calculations, to the conformer with phenothiazine in the planar configuration. Additional confirmations come from the decay profiles (Fig. 10d) collected at both emission peaks (λ = 480 nm and 570 nm, respectively), where a strong long-lived DF component (i.e. significantly more intense than in the case of the other two emitters) is present only for collection at 570 nm, while for collection at 480 nm, only a short decay typical of prompt fluorescence is detected. This pattern implies that at 0.1 wt% concentration, the higher energy emissive state (with phenothiazine equatorial) is more abundant, overshadowing the prompt fluorescence of the low-energy emissive state. Nonetheless, with longer TD, the low-energy emissive associated to the planar phenothiazine conformer, becomes discernible since it is the sole emitting state exhibiting delayed fluorescence. We have also carried out time-resolved experiments at 77 K (Fig. S7.5 and S7.6, ESI†), which allow us to gain more insight into the photophysics of these materials. However, we were unable to obtain reliable results for MPA2-BTBTOx4 and POCz2-BTBTOx4. This is certainly related to the very weak delayed fluorescence in the two molecules and could be due either to an inefficient ISC or to the presence of non-radiative deactivation channels that are also efficient at 77 K. On the other hand, we were able to observe phosphorescence for PTz2-BTBTOx4 allowing us to estimate a ΔEST of 0.1 eV (Fig. S7.5, ESI†) between the triplet and the singlet resulting from the energetically lower photoluminescence component exhibiting TADF. We have also determined the KRISC to be on the order of 104 s−1. For the method previously used by some of the current authors99KRISC = 0.6 × 104 s−1, while for a more simplified approach,100KRISC = 0.6 × 104 s−1. Due to the overall extremely low TADF intensity in the other two emitters we were unable to determine the KRISC values. In conclusion, the photophysical study is in agreement with TD-DFT calculations. For example, the best compound in terms of TADF properties is PTz2-BTBTOx4, while MPA2-BTBTOx4 presents less impressive TADF behaviour. At the same time, POCz2-BTBTOx4 shows the worst TADF properties. This ordering agrees with the ΔEST values predicted by TD-DFT.
2.6.4 Steady-state photophysical properties in neat films.
In this section we present a short study of the photophysical properties of the three emitting materials in neat films to assess their potential as self-hosted emitters in OLED devices. First, their UV-Vis properties were investigated, as shown in Fig. S7.4 (ESI†), in comparison to the isolated molecules in CH solution. While the higher energy transitions associated with the LE states retain their location found in solvents, all pure films show a clear bathochromic shift in the lower energy band associated with the CT transition. This red shift is consistent with the stabilizing effect induced by the dipoles of neighbouring molecules in the neat film. Notably, an additional weak broad band at lower energy is observed, in both POCz2-BTBTOx4 and PTz2-BTBTOx4 neat films, possibly indicating the formation of new aggregate-induced electronic states.
The PL spectra of the three emitting materials in neat films (Fig. 11) were compared with those of isolated molecules dispersed in polymer matrices (Zeonex and PMMA, Fig. 9). POCz2-BTBTOx4 and MPA2-BTBTOx4 neat films show Gaussian shaped emissions with a bathochromic shift of about 50 nm compared to PMMA. In addition, POCz2-BTBTOx4 shows a tail in the red region of the PL (700–800 nm), possibly associated with emission from aggregates. Conversely, the PTz2-BTBTOx4 neat film shows two distinct bands, one less intense at higher energy (525 nm) and another much more intense at 625 nm, probably due to different molecular conformers of the phenothiazine unit. The PL of the lower energy conformer is more intense in the neat film due to favoured Förster energy transfer between neighbouring molecules. We also determined the ΦPL of compounds in neat films which is summarized in Table 5. Only POCz2-BTBTOx4 maintains a good ΦPL in neat film (22%), compared to that in Zeonex, and half of that in PMMA, indicating minimal self-quenching. On the other hand, self-quenching is pronounced in PTz2-BTBTOx4 and MPA2-BTBTOx4, with ΦPL dropping to around 1% in neat film. The bulky POCz moieties linked to the BTBTOx4 in POCz2-BTBTOx4 likely prevent self-quenching in the solid state by hindering close packing of molecules, thereby reducing non-radiative pathways via strong π–π stacking interactions. Based on these observations, POCz2-BTBTOx4 is the only emitting molecule suitable for use in neat film form for self-hosted OLED devices. Furthermore, the neat phase can be used to achieve more red-shifted electroluminescence.
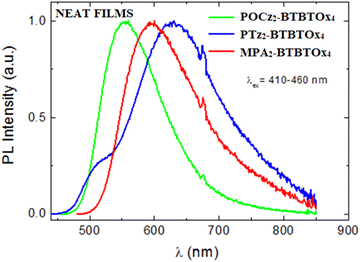 |
| | Fig. 11 Normalized PL spectra of POCz2-BTBTOx4, PTz2-BTBTOx4 and MPA2-BTBTOx4 in neat films. | |
2.7 OLED devices
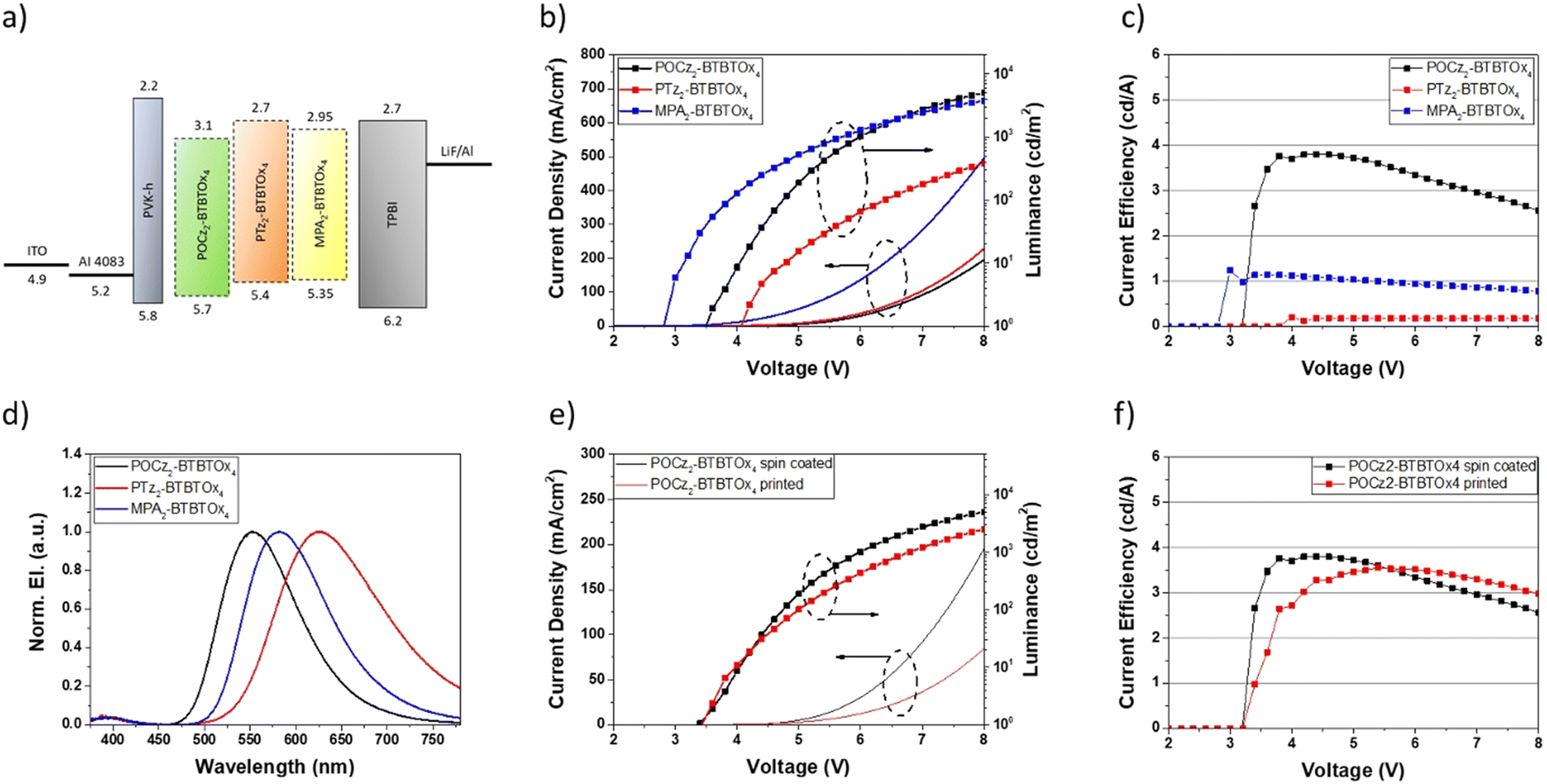
Proof of concept OLED devices with a simple structure were fabricated using PTz2-BTBTOx4, MPA2-BTBTOx4 and POCz2-BTBTOx4 in the emitting layer. In particular, these materials have been tested as neat emitting layers, avoiding the use of a host material and thus simplifying the device architecture. The OLEDs consist of indium-tin-oxide (ITO) as the anode, poly(3,4-ethylenedioxythiophene) doped with poly(styrene sulfonate) (PEDOT:PSS, 35 nm) as the hole transport layer (HTL), poly(9-vinylcarbazole) (PVK) as the electron blocking layer (EBL, 15 nm), an emitting layer (EML, 35 nm – PTz2-BTBTOx4 or MPA2-BTBTOx4 or POCz2-BTBTOx4), 1,3,5-tris(1-phenyl-1H-benzimidazol-2-yl)benzene (TPBi) as the electron transport layer (ETL, 50 nm), and LiF (1 nm) and Al (100 nm) as the cathode. The device architecture is shown in Fig. 12a. A summary of all OLED characteristics is reported in Table 5.
 |
| | Fig. 12 Device architecture and characterization: (a) energy level diagram for the fabricated OLEDs using POCz2-BTBTOx4 or PTz2-BTBTOx4 or MPA2-BTBTOx4 as the EML; (b) device current density and luminance for the three different emitting layers; (c) the current efficiency of OLEDs; (d) electroluminescence spectra; (e) current density and luminance comparison between the OLEDs with spin coated and printed POCz2-BTBTOx4; (f) comparison of current efficiency for spin coated and printed POCz2-BTBTOx4-based OLEDs. | |
Table 5 Summary of the main OLED characteristics, including external quantum efficiency (EQE) and exciton utilization efficiency (EUE)
| Emitting layer |
Current density (mA cm−2) at max efficiency |
Max luminance (cd m−2) |
Current efficiency (cd A−1) @ 100 cd m−2 |
Current efficiency (cd A−1) @ 1000 cd m−2 |
Turn on voltage (@ 10 cd m−2) |
Peak wavelength (nm) |
EML ΦPL (%) |
EQE (%) |
EUE (%) |
|
POCz2-BTBTOx4 spin coated |
0.48@4.2 V |
5016@8 V |
3.8 |
3.35 |
4 V |
553 |
28.9 |
1.19 |
20.6 |
|
POCz2-BTBTOx4 printed |
5.5@5.5 V |
2524@10 V |
3.46 |
3.35 |
4 V |
553 |
25.8 |
1.01 |
19.6 |
|
PTz2-BTBTOx4 spin coated |
1.23@4 V |
381@10 V |
0.17 |
n.a. |
4.8 V |
624 |
2.1 |
0.14 |
33.3 |
|
MPA2-BTBTOx4 spin coated |
0.47@3 V |
3733@7 V |
1.13 |
0.97 |
3.1 V |
583 |
8.52 |
0.45 |
26.7 |
A comparison of the performance of the three OLEDs shows that the MPA2-BTBTOx4-based device has a higher current density then that using PTz2-BTBTOx4 and POCz2-BTBTOx4 in the emitting layer (Fig. 12b). The maximum luminance values are very similar for the devices based on MPA2-BTBTOx4 and POCz2-BTBTOx4, while they are considerably lower for the device based on PTz2-BTBTOx4. This results in higher efficiency for the POCz2-BTBTOx4-based device, with a maximum current efficiency of about 4 cd A−1 compared to significantly lower values for PTz2-BTBTOx4 and MPA2-BTBTOx4, of 0.17 and 1.1 cd A−1, respectively (Fig. 12c). Our results are in line with the photophysical study of PTz2-BTBTOx4, MPA2-BTBTOx4 and POCz2-BTBTOx4 in neat film (Fig. 12 and Table 5). POCz2-BTBTOx4 exhibits the highest photoluminescence quantum yield in neat film (around 20%), while PTz2-BTBTOx4 and MPA2-BTBTOx4 undergo significant self-quenching phenomena, with quantum yields in the order of a few percent. The difference in the turn on voltage values of these devices is probably related to a different charge balance, due to the combined effect of the energy levels and the peculiar charge carrier mobilities of the active materials. These results agree with the charge carrier mobility of our emitters, which is higher for MPA2-BTBTOx4 and POCz2-BTBTOx4 (1.1 × 10−6 cm2 V−1 s−1) and about two orders of magnitude lower for PTz2-BTBTOx4 (5.2 × 10−8 cm2 V−1 s−1). The electroluminescence spectra are also shown in Fig. 12d and the peak emission wavelengths confirm the trend observed in the photoluminescence spectra of the neat film (Fig. 11). Taking into account our results, the best performing material POCz2-BTBTOx4 was also tested with an inkjet-printed emissive layer. The ink formulation was selected according to our recent work.101 A comparison between the devices obtained by spin coating and inkjet printing the active layer is shown in Fig. 12e and f. As reported in our previous work, solution processing of EML (spin coating or printing) can cause a slight redissolution of the underlying PVK-based EBL layer, resulting in a blurred/mixed interface between the EBL and the EML. This, therefore, affects the electrical characteristics of the printed device, introducing slight differences between OLEDs with printed and spin coated EML. Indeed, we note that the current density is lower for the printed EML (Fig. 12e), and similarly, the luminance is also lower than that with the spin coated EML. This is probably due to a slight difference in the total thickness of the PVK-EML bilayer, as a consequence of the different deposition technique. As a result, the efficiency of the device with the spin coated active layer is slightly higher than one with the printed film, and the maximum efficiency of the latter is shifted to higher voltages. In any case, the performances of the two devices are comparable, demonstrating the quality of the printing process for the deposition of active layers in non-doped OLED devices. Table 5 also reports the external quantum efficiency (EQE) and exciton utilization efficiency (EUE, ηr) values for each device (calculated as described in the ESI,† Section 8, OLED devices). EQE and EUE values are in line with the photophysical study of PTz2-BTBTOx4, MPA2-BTBTOx4 and POCz2-BTBTOx4. As expected, the photoluminescence quantum yields of EMLs are higher than those measured for neat films, with a particularly strong self-quenching in the case of PTz2-BTBTOx4. Although from the EQE point of view PTz2-BTBTOx4 and MPA2-BTBTOx4 show the lowest performances (0.14% and 0.45%, respectively) compared to POCz2-BTBTOx4 (1.19%), the related EUE values exceed the spin-statistic limitation of 25% for fluorescent materials. This suggests the presence of a triplet-harvesting mechanism in line with TADF behavior in these two materials, in agreement with our photophysical study. The electroluminescence spectra are shown in Fig. 12d, and the peak emission wavelengths are in line with the photoluminescence spectra of the neat film (Fig. 11). As already highlighted, the wet deposition process of the EML causes a partial redissolution of the PVK. This is confirmed by the blue shift of the EL peaks with respect to the PL peaks of the neat films, for all materials, due to the presence of PVK, which dilutes the active materials inside the EMLs. All the EL spectra show a weak band at around 400 nm, attributed to PVK or TPBi emission. In our opinion, considering that EMLs are virtually non-doped the presence of such a peak, highlights a slight imbalance in the charge injection, which causes excitonic recombination near one of the interfaces between the EML and the HTL (or ETL).
3. Conclusion
In summary, we designed and synthesized three new organic luminescent molecules with a D–A–D electronic structure and based on the [1]benzothieno[3,2-b]benzothiophene-tetraoxide BTBTOx4 as the acceptor unit A. Three donor units D with different electron-donating capabilities and structural complexity were selected to be coupled with the acceptor A by the microwave-assisted Buchwald–Hartwig reaction, yielding PTz2-BTBTOx4, MPA2-BTBTOx4 and POCz2-BTBTOx4. Cyclic voltammetry and charge carrier mobility measurements were carried out to investigate the electrical properties of compounds, X-ray analysis was performed for determining the crystal structure. UV-vis absorption, steady-state fluorescence and time resolved fluorescence spectroscopy were used to investigate photophysical properties of the three organic emitters studied in this work. Theoretical studies were carried out to aid the photophysical characterization. OLED devices were fabricated to analyse the luminescence properties of the new organic emitters as self-hosted active matrices. All three organic emitters show high electrochemical stability with reversible oxidation waves. MPA2-BTBTOx4, which can be obtained with a suitable degree of crystallinity, was selected as a reference molecule for X-ray analysis. Crystallographic data reveal a butterfly-shape molecule with a planar core (BTBTOx4) and out-of-plane p-methoxyphenyl groups of the donor unit (DMeOPA), with torsion angles (C1–C2–N1–C13 and C13–N1–C7–C8) of −59° and 86°, respectively. Steady-state and time-resolved photoluminescence studies in polymer matrices (Zeonex and PMMA) revealed a noteworthy increase in PL efficiency upon deoxygenation for all three organic emitters, especially in PMMA. We demonstrate the pronounced TADF properties of the three studied luminescent molecules representing a new class of organic emitting materials. We carried out preliminary studies of photophysical properties in neat films, with the aim of assessing the potential of this class of compounds as emitters in solution-processed self-hosted OLEDs. In PTz2-BTBTOx4 and MPA2-BTBTOx4 a pronounced self-quenching can be observed with a ΦPL as low as 1%. In contrast, POCz2-BTBTOx4 reveals a minimal self-quenching with a capability to maintain a good ΦPL (22%), comparable to that in Zeonex and half of that in PMMA. As a proof of concept, we tested the new organic compounds in neat films serving as an active matrix in simple self-hosted OLED devices, evaluating their optoelectronic characteristics and exciton utilization efficiencies. Although the performances of OLEDs are not in line with the state of the art, the purpose of our work was to demonstrate the possibility of employing this new acceptor as a building block in new emitters used in printed devices. Starting from this study, we plan to develop more efficient printable systems, with higher photoluminescence quantum yields and lower self-quenching, to be used as a neat film EML, thereby simplifying the device structure and minimizing the production costs. Indeed, these are key factors in developing fabrication processes towards solution-based techniques.
Author contributions
M. Montrone: synthesis and characterization of organic emitters. A. Maggiore: design of photophysical experiments, supervision of the photophysical study and data analysis, writing the manuscript. A. Moliterni: X-ray data analysis, writing the manuscript. P. Pander: computational analysis, supplementary photophysical studies, writing the manuscript. M. Pugliese: OLED device fabrication and characterization, writing the manuscript. A. L. Capodilupo: CV analysis and chemico-structural characterization. S. Gambino: charge carrier mobility measurements, writing the manuscript. C. T. Prontera: OLED device fabrication. V. Valenzano: photophysical experiments. F. Mariano: OLED device fabrication. G. Accorsi: supervision of the photophysical data. T. Sibillano: X-ray data collection, writing of the manuscript. C. Giannini: supervision of X-ray data analysis and writing of the manuscript. G. Gigli: found acquisition, conceptualization and supervision of physical data. A. Cardone: design of organic emitters, original draft, writing review & editing. V. Maiorano: supervision, review and funding acquisition. All authors participated in the general discussion.
Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data of BDMeOPA-BTBTOx4 are deposited at the Cambridge Crystallographic Data Centre, under deposition number CCDC 2362085 and can be obtained free of charge viahttps://www.ccdc.cam.ac.uk/structures/.†
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by: (a) MUR, project PON ARS01_00951_V0584, D.D.n. 2051 del 02/08/2018, titled: “ECOTEC – Smart and eco-sustainable fibres and tissues for technical clothing and high fashion”; (b) MUR, under the complementary actions to the NRRP (PNC0000007) “Fit4MedRob-Fit for Medical Robotics” Grant (contract number CUP B53C22006960001); European Union-NextGenerationEU (National Sustainable Mobility Center CN00000023, Italian Ministry of University and Research Decree no. 1033 – 17/06/2022, Spoke 11 – Innovative Materials & Lightweighting); (c) Project “Nano Foundries and Fine Analysis – Digital Infrastructure (NFFA-DI)” (CUPB53C22004310006), supported by the Italian Ministry of Research (MUR) in the framework of the National Recovery and Resilience Plan (NRRP), funded by the European Union – NextGenerationEU; (d) PNRR MUR Project (Integrated Infrastructure Initiative in Photonic and Quantum Sciences)” (I-PHOQS, N. IR0000016). This work made use of the Hamilton High Performance Computing Service of Durham University. A. Moliterni, T. Sibillano and C. Giannini gratefully thank F. Baldassarre for his technical assistance.
References
- C. W. Tang and S. A. VanSlyke, Appl. Phys. Lett., 1987, 51, 913–915 CrossRef CAS.
- J. H. Burroughes, D. D. Bradley, A. R. Brown, R. N. Marks, K. Mackay, R. H. Friend, P. L. Burns and A. B. Holmes, Nature, 1990, 347, 539–541 CrossRef CAS.
- S. Kim, H.-J. Kwon, S. Lee, H. Shim, Y. Chun, W. Choi, J. Kwack, D. Han, M. Song, S. Kim, S. Mohammadi, I. Kee and S. Y. Lee, Adv. Mater., 2011, 23, 3511–3516 CrossRef CAS PubMed.
- M. A. Baldo, D. F. O’Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Nature, 1998, 395, 151–154 CrossRef CAS.
- C. Adachi, M. A. Baldo, S. R. Forrest and M. E. Thompson, Appl. Phys. Lett., 2000, 77, 904–906 CrossRef CAS.
- C. Adachi, M. A. Baldo, M. E. Thompson and S. R. Forrest, J. Appl. Phys., 2001, 90, 5048–5051 CrossRef CAS.
- M. V. Werrett, G. S. Huff, S. Muzzioli, V. Fiorini, S. Zacchini, B. W. Shelton, A. Maggiore, J. M. Malicka, M. Cocchi, K. C. Gordon, S. Stagni and M. Massi, Dalton Trans., 2014, 44, 8379–8393 RSC.
- A. Maggiore, M. Puglie, F. Di Maria, G. Accorsi, M. Gazzano, E. Fabiano, V. Tasco, M. Esposito, M. Cuscunà, L. Blasi, A. Capodilupo, G. Ciccarella, G. Gigli and V. Maiorano, Inorg. Chem., 2016, 55, 6532–6538 CrossRef CAS.
- R. Ragni, V. Maiorano, M. Pugliese, A. Maggiore, E. Orselli, F. Babudri, G. Gigli, L. De Cola and G. M. Farinola, Synth. Met., 2017, 227, 148–155 CrossRef.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS.
- Q. Zhang, B. Li, S. Huang, H. Nomura, H. Tanaka and C. Adachi, Nat. Photonics, 2014, 8, 326–332 CrossRef CAS.
- S. Hirata, Y. Sakai, K. Masui, H. Tanaka, S. Y. Lee, H. Nomura, N. Nakamura, M. Yasumatsu, H. Nakanotani, Q. Zhang, K. Shizu, H. Miyazaki and C. Adachi, Nat. Mater., 2015, 14, 330–336 CrossRef CAS PubMed.
- T. A. Lin, T. Chatterjee, W. L. Tsai, W. K. Lee, M. J. Wu, M. Jiao, K. C. Pan, C. L. Yi, C. L. Chung, K. T. Wong and C. C. Wu, Adv. Mater., 2016, 28, 6976–6983 CrossRef CAS.
- W. Zeng, H. Y. Lai, W. K. Lee, M. Jiao, Y. J. Shiu, C. Zhong, S. Gong, T. Zhou, G. Xie, M. Sarma, K. T. Wong, C. C. Wu and C. Yang, Adv. Mater., 2018, 30, 1704961 CrossRef PubMed.
- H. Tanaka, K. Shizu, H. Nakanotani and C. Adachi, J. Phys. Chem. C, 2014, 118, 15985–15994 CrossRef CAS.
- H. Kaji, H. Suzuki, T. Fukushima, K. Shizu, K. Suzuki, S. Kubo, T. Komino, H. Oiwa, F. Suzuki, A. Wakamiya, Y. Murata and C. Adachi, Nat. Commun., 2015, 6, 8476–8484 CrossRef CAS PubMed.
- L. Gan, Z. Xu, Z. Wang, B. Li, W. Li, X. Cai, K. Liu, Q. Liang and S. J. Su, Adv. Funct. Mater., 2019, 29, 1808088 CrossRef.
- Y.-J. Yu, F.-M. Liu, X.-Y. Meng, L.-Y. Ding, L.-S. Liao and Z.-Q. Jiang, Chem. – Eur. J., 2023, 29, e202202628 CrossRef CAS.
- D. Zhong, Y. Yu, L. Yue, X. Yang, L. Ma, G. Zhou and Z. Wu, Chem. Eng. J., 2021, 413, 127445 CrossRef CAS.
- Y. Li, J.-J. Liang, H.-C. Li, L.-S. Cui, M.-K. Fung, S. Barlow, S. R. Marder, C. Adachi, Z.-Q. Jiang and L.-S. Liao, J. Mater. Chem. C, 2018, 6, 5536–5541 RSC.
- S. J. Woo, Y. Kim, S. K. Kwon, Y. H. Kim and J. J. Kim, ACS Appl. Mater. Interfaces, 2019, 11, 7199–7207 CrossRef CAS.
- L. S. Cui, H. Nomura, Y. Geng, J. U. Kim, H. Nakanotani and C. Adachi, Angew. Chem., Int. Ed., 2017, 56, 1571–1575 CrossRef CAS PubMed.
- K. L. Woon, C. L. Yi, K. C. Pan, M. K. Etherington, C. C. Wu, K. T. Wong and A. P. Monkman, J. Phys. Chem. C, 2019, 123, 12400–12410 CrossRef CAS.
- X. Wang, J. Hu, J. Lv, Q. Yang, H. Tian, S. Shao, L. Wang, X. Jing and F. Wang, Angew. Chem., Int. Ed., 2021, 60, 16585–16593 CrossRef CAS.
- M. Auffray, U. Balijapalli, J.-C. Ribierre, Y. Tsuchiya and C. Adachi, Chem. Lett., 2020, 49, 932–935 CrossRef CAS.
- G. Yang, Y. Ran, Y. Wu, M. Chen, Z. Bin and J. You, Aggregate, 2021, 3, e127 CrossRef.
- F. M. Xie, H. Z. Li, G. L. Dai, Y. Q. Li, T. Cheng, M. Xie, J. X. Tang and X. Zhao, ACS Appl. Mater. Interfaces, 2019, 11, 26144–26151 CrossRef CAS.
- W. Li, B. Li, X. Cai, L. Gan, Z. Xu, W. Li, K. Liu, D. Chen and S. J. Su, Angew. Chem., Int. Ed., 2019, 58, 11301–11305 CrossRef CAS.
- A. Maggiore, X. Tan, A. Brosseau, A. Danos, F. Miomandre, A. P. Monkman, P. Audebert and G. Clavier, Phys. Chem. Chem. Phys., 2022, 24, 17770–17781 RSC.
- X. Chen, S. Liu, Y. Sun, D. Zhong, Z. Feng, X. Yang, B. Su, Y. Sun, G. Zhou, B. Jiao and Z. Wu, Mater.
Chem. Front., 2023, 7, 1841–1854 RSC.
- Y. Chen, D. Zhang, Y. Zhang, X. Zeng, T. Huang, Z. Liu, G. Li and L. Duan, Adv. Mater., 2021, 33, 2103293 CrossRef CAS.
- H. Zhang, G. Li, X. Guo, K. Zhang, B. Zhang, X. Guo, Y. Li, J. Fan, Z. Wang, D. Ma and B. Z. Tang, Angew. Chem., Int. Ed., 2021, 60, 23635–23640 CrossRef.
- D. Ma, T. Liu, Z. Chen, Z. Yang, J. Zhao, Z. Yang, Y. Zhang and Z. Chi, CCS Chem., 2022, 4, 1284–1294 CrossRef.
- D. Zhong, Y. Yu, D. Song, X. Yang, Y. Zhang, X. Chen, G. Zhou and Z. Wu, ACS Appl. Mater. Interfaces, 2019, 11, 27112–27124 CrossRef CAS PubMed.
- W. Wei, Z. Yang, X. Chen, T. Liu, Z. Mao, J. Zhao and Z. Chi, J. Mater. Chem. C, 2020, 8, 3663–3668 RSC.
- Z. Yang, Z. Mao, C. Xu, X. Chen, J. Zhao, Z. Yang, Y. Zhang, W. Wu, S. Jiao, Y. Liu, M. P. Aldred and Z. Chi, Chem. Sci., 2019, 10, 8129–8134 RSC.
- X. Zeng, Y.-H. Huang, S. Gong, P. Li, W.-K. Lee, X. Xiao, Y. Zhang, C. Zhong, C.-C. Wu and C. Yang, Mater. Horiz., 2021, 8, 2286–2292 RSC.
- Y. Liu, J. Yang, Z. Mao, X. Chen, Z. Yang, X. Ge, X. Peng, J. Zhao, S. J. Su and Z. Chi, ACS Appl. Mater. Interfaces, 2022, 14, 33606–33613 CrossRef CAS PubMed.
- A. Maggiore, Y. Qu, R. Guillot, P. Pander, M. Vasylieva, P. Data, F. B. Dias, P. Audebert, G. Clavier and F. Miomandre, J. Phys. Chem. B, 2022, 126, 2740–2753 CrossRef CAS PubMed.
- W. Ma, Z. Bin, G. Yang, J. Liu and J. You, Angew. Chem., Int. Ed., 2022, 61, e202116681 CrossRef CAS PubMed.
- L. Yao, S. Zhang, R. Wang, W. Li, F. Shen, B. Yang and Y. Ma, Angew. Chem., Int. Ed., 2014, 53, 2119–2123 CrossRef CAS.
- K. H. Kim, C. K. Moon, J. H. Lee, S. Y. Kim and J. J. Kim, Adv. Mater., 2014, 26, 3844–3847 CrossRef CAS PubMed.
- Y. Liu, J. Yang, Z. Mao, D. Ma, Y. Wang, J. Zhao, S.-J. Su and Z. Chi, Adv. Opt. Mater., 2023, 11, 2201695 CrossRef CAS.
- M. Cinquino, M. Pugliese, C. T. Prontera, F. Mariano, A. Zizzari, G. Maiorano, A. Maggiore, R. Manfredi, C. Mello, I. Tarantini, G. Gigli and V. Maiorano, J. Sci.: Adv. Mater. Devices, 2024, 9, 100707 CAS.
- F. B. Dias, J. Santos, D. R. Graves, P. Data, R. S. Nobuyasu, M. A. Fox, A. S. Batsanov, T. Palmeira, M. N. Berberan-Santos, M. R. Bryce and A. P. Monkaman, Adv. Sci., 2016, 3, 1600080 CrossRef PubMed.
- S. Wu, M. Aonuma, Q. Zhang, S. Huang, T. Nakagawa, K. Kuwabara and C. Adachi, J. Mater. Chem. C, 2014, 2, 421–424 RSC.
- Q. Zhang, J. Li, K. Shizu, S. Huang, S. Hirata, H. Miyazaki and C. Adachi, J. Am. Chem. Soc., 2012, 134, 14706–14709 CrossRef CAS.
- Q. Zhang, D. Tsang, H. Kuwabara, Y. Hatae, B. Li, T. Takahashi, S. Y. Lee, T. Yasuda and C. Adachi, Adv. Mater., 2015, 27, 2096–2100 CrossRef CAS PubMed.
- J. Li, R. Zhang, Z. Wang, B. Zhao, J. Xie, F. Zhang, H. Wang and K. Guo, Adv. Opt. Mater., 2018, 6, 1701256 CrossRef.
- X. Ban, B. Lin, W. Jiang and Y. Sun, Chem. – Asian J., 2017, 12, 216–223 CrossRef CAS PubMed.
- H. Lee, W. Song and J. Y. Lee, Org. Electron., 2016, 29, 22–26 CrossRef.
- J. Luo, S. Gong, Y. Gu, T. Chen, Y. Li, C. Zhong, G. Xie and C. Yang, J. Mater. Chem. C, 2016, 4, 2442–2446 RSC.
- X. Ban, W. Jiang, K. Sun, B. Lin and Y. Sun, ACS Appl. Mater. Interfaces, 2017, 9, 7339–7346 CrossRef CAS.
- K. Sun, W. Jiang, X. Ban, B. Huang, Z. Zhang, M. Ye and Y. Sun, RSC Adv., 2016, 6, 22137–22143 RSC.
- S. Xiang, Z. Huang, S. Sun, X. Lv, L. Fan, S. Ye, H. Chen, R. Guo and L. Wang, J. Mater. Chem. C, 2018, 6, 11436–11443 RSC.
- R. S. Nobuyasu, Z. Ren, G. C. Griffiths, A. S. Batsanov, P. Data, S. Yan, A. P. Monkman, M. R. Bryce and F. B. Dias, Adv. Opt. Mater., 2016, 4, 597–607 CrossRef CAS.
- S. Xiang, R. Guo, Z. Huang, X. Lv, S. Sun, H. Chen, Q. Zhang and L. Wang, Dyes Pigm., 2019, 170, 107636 CrossRef CAS.
- Y. Li, T. H. Chen, M. L. Huang, Y. Gu, S. L. Gong, G. H. Xie and C. L. Yang, J. Mater. Chem. C, 2017, 5, 3480–3487 RSC.
- H. Ebata, T. Izawa, E. Miyazaki, K. Takimiya, M. Ikeda, H. Kuwabara and T. Yui, J. Am. Chem. Soc., 2007, 129, 15732–15733 CrossRef CAS.
- K. Takimiya, H. Ebata, K. Sakamoto, T. Izawa, T. Otsubo and Y. Kunugi, J. Am. Chem. Soc., 2006, 128, 12604–12605 CrossRef CAS PubMed.
- T. Izawa, E. Miyazaki and K. Takimiya, Adv. Mater., 2008, 20, 3388–3392 CrossRef CAS.
- A. L. Capodilupo, E. Fabiano, L. De Marco, G. Ciccarella, G. Gigli, C. Martinelli and A. Cardone, J. Org. Chem., 2016, 81, 3235–3245 CrossRef CAS.
- S. H. Zherdeva, A. Barudi, Y. A. Zheltov and B. I. Stepanov, Zh. Org. Khim., 1980, 16, 383–390 Search PubMed.
- D.-G. Chen, Y. Chen, C.-H. Wu, Y.-A. Chen, M.-C. Chen, J.-A. Lin, C.-Y. Huang, J. Su, H. Tian and P.-T. Chou, Angew. Chem., Int. Ed., 2019, 8, 13297–13301 CrossRef.
- F. Khan and R. Misra, J. Mater. Chem. C, 2023, 11, 2786–2825 RSC.
- P. S. Gangdhar, G. Reddy, S. Prasanthjumar and L. Giribabu, Phys. Chem. Chem. Phys., 2021, 23, 14969–14996 RSC.
- X. Liu, C. Huang and M. Li, Dyes Pigm., 2017, 139, 283–291 CrossRef CAS.
- N. Xu, A. Zheng, Y. Wei, Y. Yuan, J. Zhang, M. Lei and P. Wang, Chem. Sci., 2020, 11, 3418–3426 RSC.
- K. Rakstys, S. Paek, G. Grancini, P. Gao, V. Jankauskas, A. M. Asiri and M. K. Nazeeruddin, ChemSusChem, 2017, 10, 3825–3832 CrossRef CAS PubMed.
- A. Cardone and A. L. Capodilupo, Materials, 2022, 15, 6333 CrossRef CAS.
- A. L. Capodilupo, F. Manni, G. A. Corrente, G. Accorsi, E. Fabiano, A. Cardone, R. Giannuzzi, A. Beneduci and G. Gigli, Dyes Pigm., 2020, 177, 108325 CrossRef CAS.
- A. L. Capodilupo, L. De Marco, G. A. Corrente, R. Giannuzzi, E. Fabiano, A. Cardone, G. Gigli and G. Ciccarella, Dyes Pigm., 2016, 130, 79–89 CrossRef CAS.
- G. Juška, K. Arlauskas, N. Nekrašas, J. Stuchlik, X. Niquille and N. Wyrsch, J. Non-Cryst. Solids, 2002, 299–302, 375–379 CrossRef.
- I. A. Wright, P. J. Skabara, J. C. Forgie, A. L. Kanibolotsky, B. González, S. J. Coles, S. Gambino and I. D. W. Samuel, J. Mater. Chem., 2011, 21, 1462–1469 RSC.
- M. Leoncini, A. L. Capodilupo, D. Altamura, C. Giannini, G. Accorsi, E. Fabiano, A. Rizzo, G. Gigli and S. Gambino, J. Mater. Chem. C, 2022, 10, 5981–5993 RSC.
- O. Semeniuk, G. Juška, J. O. Oelerich, M. Wiemer, S. D. Baranovskii and A. Reznik, Sci. Rep., 2016, 6, 33359 CrossRef CAS PubMed.
- M. T. Neukom, N. A. Reinke and B. Ruhstaller, Sol. Energy, 2011, 85, 1250–1256 CrossRef CAS.
-
O. Grynko, G. Juška and A. Reznik, in Photoconductivity and Photoconductive Materials, ed. S. O. Kasap, Wiley, 2022, pp. 339–368 Search PubMed.
- A. Altomare, N. Corriero, C. Cuocci, A. Falcicchio, A. Moliterni and R. Rizzi, J. Appl. Crystallogr., 2015, 48, 598–603 CrossRef CAS.
- A. Altomare, C. Cuocci, C. Giacovazzo, A. Moliterni, R. Rizzi, N. Corriero and A. Falcicchio, J. Appl. Crystallogr., 2013, 46, 1231–1235 CrossRef CAS.
- C. F. Macrae, I. Sovago, S. J. Cottrell, P. T. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens, M. Towler and P. A. Wood, J. Appl. Crystallogr., 2020, 53, 226–235 CrossRef CAS PubMed.
- G. R. Desiraju, Acc. Chem. Res., 1991, 24, 290–296 CrossRef CAS.
- C. Janiak, J. Chem. Soc., Dalton Trans., 2000, 3885–3896 RSC.
- E. A. Meyer, R. K. Castellano and F. Diederich, Angew. Chem., Int. Ed., 2003, 42, 1210–1250 CrossRef CAS PubMed.
- W. B. Motherwell, R. B. Moreno, I. Pavlakos, J. R. T. Arendorf, T. Arif, G. J. Tizzard, S. J. Coles and A. E. Aliev, Angew. Chem., Int. Ed., 2018, 57, 1193–1198 CrossRef CAS.
- A. Moliterni, D. Altamura, R. Lassandro, V. Olieric, G. Ferri, F. Cardarelli, A. Camposeo, D. Pisignano, J. E. Anthony and C. Giannini, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2020, 76, 427–435 CrossRef CAS PubMed.
- J.-L. Wang, J.-S. Xu, D.-Y. Wang, H. Wang, Z.-T. Li and D.-W. Zhang, CrystEngComm, 2014, 16, 2078–2084 RSC.
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, J. Appl. Crystallogr., 2012, 54, 1006–1011 CrossRef.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- P. Pander, A. V. Zaytsev, A. Sil, G. V. Baryshnikov, F. Siddique, J. A. Gareth Williams, F. B. Dias and V. N. Kozhevnikov, Chem. Sci., 2023, 14, 13934–13943 RSC.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1606 Search PubMed.
- M. Okazaki, Y. Takeda, P. Data, P. Pander, H. Higginbotham, A. P. Monkman and S. Minakata, Chem. Sci., 2017, 8, 2677–2686 RSC.
- J. Zhang, M. Zhu, Dr. Y. Lu, X. Zhang, S. Xiaom, H. Lan and T. Yi, Chem. – Eur. J., 2022, 28, e202200458 CrossRef CAS PubMed.
- S. M. Bonesi and R. Erra-Balsells, J. Lumin., 2001, 93, 51–74 CrossRef CAS.
- J. A. VanAllan, G. A. Reinolds and R. E. Adel, J. Org. Chem., 1962, 27, 1659–1664 CrossRef CAS.
- F. B. Dias, T. J. Penfold and A. P. Monkman, Methods Appl. Fluoresc., 2017, 5, 012001 CrossRef PubMed.
- A. Maggiore, Y. Qu, P. Pander, F. B. Dias, G. Claiver, R. Guillot, D. Altamura, C. Giannini, V. Maiorano, P. Audebert and F. Miomandre, J. Mater. Chem. C, 2024, 12, 7943–7955 RSC.
- S. Montanaro, P. Pander, J.-R. Mistry, M. R. J. Elsegood, S. J. Teat, A. D. Bond, I. A. Wright, D. G. Congrave and M. K. Etherington, J. Mater. Chem. C, 2022, 10, 6306–6313 RSC.
- F. B. Dias, Philos. Trans. R. Soc., A, 2015, 373, 20140447 CrossRef.
- M. Cinquino, C. C. T. Prontera, A. Maggiore, A. Zizzari, M. Pugliese, F. Mariano, V. Valenzano, I. E. Palamà, R. Manfredi, G. Gigli and V. Maiorano, Adv. Electron. Mater., 2024, 10, 2300358 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b,
Anna
Moliterni
*b,
Anna
Moliterni