
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhancing molecular aggregation and decreasing the optical gap by a dual-additive to reduce the energy loss of all-polymer organic solar cells†
Jiaqi
Pan
ab,
Qiang
Zhang
a,
Jian
Guan
ab,
Hanyue
Gao
ab,
Luzhuo
Li
ab,
Rui
Zhang
 c,
Hongxiang
Li
d,
Xinhong
Yu
a and
Yanchun
Han
*ab
c,
Hongxiang
Li
d,
Xinhong
Yu
a and
Yanchun
Han
*ab
aState Key Laboratory of Polymer Physics and Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun 130022, P. R. China. E-mail: ychan@ciac.ac.cn
bSchool of Applied Chemistry and Engineering, University of Science and Technology of China, Hefei 230026, P. R. China
cDepartment of Physics, Chemistry and Biology (IFM), Linköping University, Linköping, Sweden
dCollege of Polymer Science and Engineering State Key Laboratory of Polymer Materials Engineering, Sichuan University, Chengdu 610065, China
First published on 18th November 2024
Abstract
All-polymer solar cells (all-PSCs) have attracted significant attention due to their unique advantages, such as intrinsic stretchability and high thermal stability. However, in the PM6:PYIT system, controlling the phase-separated aggregation size of the acceptor PYIT is particularly crucial because of the difficulties in choosing suitable solvents. High-boiling-point solvents such as chlorobenzene may lead to excessive aggregation of the acceptor due to the slow solvent evaporation, and low-boiling-point solvents like chloroform (CF) may result in small aggregation sizes of the acceptor due to its fast evaporation. To optimize the aggregation size of the acceptor PYIT, we employ a strategy of using two high-boiling-point solvent additives (diphenyl ether: DPE and chloronaphthalene: CN) to prolong the aggregation and film formation time of the acceptor PYIT. This result shows that the aggregation state of PYIT is optimized effectively after the evaporation of CF, with the slow evaporation process of DPE and CN acting synergistically. Moreover, DPE with lower surface energy tends to make PM6 aggregate more in the bottom anode region, while CN with higher surface energy drives PYIT to aggregate upwards, forming a vertical phase separation distribution structure, which is conducive to efficient exciton dissociation and charge transport. Further analysis indicates that the introduction of the dual-additive leads to a corresponding increase in the external quantum efficiency (EQE) spectrum and an effective reduction in the absorption bandgap (Eg) of the solar cells. Consequently, the PCE of the optimized device increases notably from 14.58% to 16.67% and a reduction in Eloss from 0.500 eV to 0.476 eV by adding 0.6 vol% of 1-CN and 0.4 vol% of DPE simultaneously is also observed.
1. Introduction
All polymer solar cells (all-PSCs) have attracted significant interest due to their unique material composition and performance characteristics. The donor and acceptor molecules in all-PSCs are composed of polymers, which offers advantages such as light weight,1 semi-transparency2,3 and large-area fabrication.4 Furthermore, the energy levels of donors or acceptors can be altered by adjusting the main and side chain structures of polymer molecules.5–8 This leads to the achievement of a high open-circuit voltage (VOC).9 Additionally, polymer molecules exhibit strong absorbance in the visible and near-infrared regions, enhancing power conversion efficiency (PCE).10 In addition, the complexity of the polymer molecular structure results in the multiplicity of moving units, which enables the blended film to possess better morphological stability.11,12 This characteristic is crucial for ensuring the long-term stable operation of solar cells.13–16 Through precise control of the molecular structure and composition of the polymers, as well as optimization of the device fabrication process, the PCE of all-PSCs has been significantly improved to over 16%.17–20 However, in all-polymer blend systems, the complex interactions between the components make it difficult to precisely control the compatibility of the donor and acceptor after mixing, thereby making it challenging to form a suitable phase-separated size in the active layer.21–23 Furthermore, many polymer acceptors have lower crystallinity, which affects charge carrier mobility and creates a barrier to improving the electrical performance.19 Moreover, the thermodynamic differences between polymer donors and acceptors can also cause unfavorable changes in molecular orientation and order at the D/A interface in the active layer of all-PSCs, further restricting charge delocalization and increasing energy loss (Eloss) in the devices.18,24–26The morphology of the active layer has a crucial impact on the PCE of all-PSCs. The quality of the morphology directly affects key processes such as exciton generation, dissociation, charge transport, and collection.27,28 Therefore, optimizing the morphology of the active layer is one of the important ways to improve the performance of all-PSCs.29 Current literature research has shown that for all-PSCs, the desired active layer film morphology should have suitable phase-separated domains, an appropriate D/A interfacial area, and a favorable face-on molecular crystalline orientation.13,18,19,30–32 In optimizing the preparation process of all-PSC active layers, researchers have developed a series of post-processing strategies focused on regulating the dynamic behavior of thin film drying. The utilization of solvent additives emerges as a crucial processing strategy for optimizing the performance of all-PSCs. For instance, chloronaphthalene (CN) has demonstrated remarkable effectiveness in regulating the molecular aggregation behavior during solution evaporation.21,33 Li et al. demonstrated that the incorporation of CN into the PBDB-T:PYF-T system notably optimizes the π–π stacking structure of polymer molecules and promotes the formation of a vertical phase separation morphology, which is crucial for enhancing the fill factor (FF) of the device.34 Additionally, the introduction of CN has also been found to effectively reduce the energy disorder in the device, thereby significantly reducing non-radiative recombination energy loss and further enhancing the VOC. Furthermore, studies have shown that polymer donors and polymer acceptors possess the potential to serve as solid additives. Ma et al.35 introduced 0.7 vol% CN and 3 wt% N2200 (as solid polymer additives) to the PM6:PYIT system and discovered that this strategy can improve the PCE of all-PSCs from 14.93% to 16.03%.36 Experimental characterization of the active layer film morphology reveals that the introduction of additives successfully modifies the molecular aggregation in the active layer. The molecular aggregation changes from a “broad and short” shape to a “narrow and tall” shape. This transition in molecular packing reduces the charge recombination probability, enhances the charge carrier transport rate, and reduces the Eloss.
In the pursuit of high-efficiency all-PSCs, the regulation of the active layer morphology and film formation kinetics remains a key challenge that restricts further enhancement of PCE. Recently, numerous studies have been made to optimize the active layer morphology by altering processing conditions. However, most of these studies primarily focused on characterizing the morphological outcomes before and after changes in processing methods, without delving deeply into the specific mechanisms of morphology formation and the detailed kinetics of the film formation process. To gain a deeper understanding of the relationship between the active layer morphology and performance of all-polymer solar cells, this article focuses on the impact of a dual-additive on the film formation kinetics of all-polymer systems. In particular, we selected the PM6:PYIT system as our research model. Given the rapid evaporation characteristics of CF, it can lead to insufficient molecular aggregation, resulting in a smaller phase separation size in the final film, which poses obstacles to effective charge transport and extraction. To overcome this challenge, we have employed high-boiling-point solvent additives, diphenyl ether (DPE) and CN, which can continue to promote the crystallization growth of PYIT after the evaporation of CF, providing more time for the tight aggregation of polymer molecules and their migration towards their respective electrode regions. By introducing a dual-additive to influence the film-forming dynamics, we aim to precisely regulate the crystallization and phase separation behavior of donor and acceptor molecules. This approach aims to construct an interpenetrating network structure with high crystallinity and improved vertical phase distribution, ultimately reducing recombination rates, extending carrier lifetimes, and enhancing the overall performance of the device.
2. Results and discussions
2.1 Device performance
The chemical structures of polymer donor poly{[4,8-bis[5-(2-ethylhexyl)-4-fluoro-2-thienyl]benzo[1,2-b:4,5-b′]-dithiophene-2,6-diyl]-alt-[2,5-thiophenediyl[5,7-bis(2-ethylhexyl)-4,8-dioxo-4H,8H-benzo[1,2-c:4,5-c′]dithiophene-1,3-diyl]]} (PM6), polymer acceptor poly [2,2′-((2Z,2′Z)-((12,13-bis(2-octyldodecyl)-3,9-diundecyl-12,13-dihydro-[1,2,5]thiadiazolo[3,4-e]thieno[2′′,3′′:4′,5′]thieno[2′,3′:4,5]pyrrolo[3,2-g]thieno[2′,3′:4,5]thieno[3,2-b]indole-2,10-diyl)bis(methanylylidene))bis(5,6-difluoro-3-oxo-2,3-dihydro-1H-indene-2,1-diylidene))dimalononitrile-co-2,5-thiophene] (PYIT) and the solvent additives DPE and 1-CN used in this study are shown in Fig. 1(a). The energy level information of PM6 and PYIT is shown in Fig. 1(b). The highest occupied molecular orbital (EHOMO) levels of PM6 and PYIT are −5.30 and −5.56 eV, respectively. The lowest unoccupied molecular orbital (ELUMO) levels of PM6 and PYIT are −3.76 and −3.95 eV, respectively. There is a suitable energy level alignment between PM6 and PYIT, which is conducive to exciton dissociation and charge transfer. We also evaluated the optical properties of PM6 and PYIT; Fig. 1(c) shows the normalized UV-vis absorption spectra of neat PM6 and PYIT films. The donor PM6 exhibits a strong absorption peak near 600 nm, while the acceptor PYIT shows strong absorption near 810 nm. It can be observed that the absorption ranges of donor and acceptor molecules are complementary. | ||
| Fig. 1 (a) Chemical structures of donor PM6, acceptor PYIT, solvent additives DPE and 1-CN. (b) Energy level diagram of PM6 and PYIT. (c) Normalized UV-vis absorption spectra of pristine PM6 and PYIT films. (d) J–V curves and (e) EQE curves for the optimal binary OSCs. | ||
We fabricated bulk heterojunction polymer solar cells (BHJ-PSCs) with a standard structure of ITO/PEDOT:PSS/active layer/PDINN/Ag. To refine the morphology of the active layer, we employed high boiling point (BP) solvent additives, DPE (BP: 259 °C) and CN (BP: 263 °C). To assess the influence of these additives on the photovoltaic performance of the PSCs, we prepared devices with different treatments: no additive, single additive, and dual-additive.
The current density–voltage (J–V) curves were measured under standard illumination conditions using an AM 1.5G solar simulator (Newport) and are presented in Fig. 1(d). Table 1 provides a comprehensive summary of the key photovoltaic parameters for each device configuration. As shown in Table 1, the addition of CN can increase the FF of the PM6:PYIT binary devices, but it leads to a decrease in the VOC value. On the other hand, the addition of additive DPE can further boost the high VOC value of PM6:PYIT binary devices, but it results in a reduction of the FF. In order to find a more suitable amount of additives to be added, devices with different amounts of CN and DPE were manufactured. Table S1 (ESI†) summarizes the photovoltaic performance parameters of devices obtained by adding different additives in varying proportions and Fig. S1 (ESI†) shows the J–V curves of devices treated with different additives. After a series of comparative experiments, 0.6 vol% CN and 0.4 vol% DPE were ultimately chosen as the optimal additive combination. The optimal PCE of the binary PM6:PYIT device without additives is 14.58%, with a VOC of 0.943 V, a short-circuit current density (JSC) of 24.40 mA cm−2 and a FF of 63.30%. When 0.6 vol% CN and 0.4 vol% DPE were simultaneously added, the PCE of the binary device was remarkably increased to 16.67%, and both the VOC and FF were improved. The optimal binary device treated with a dual-additive achieved a high VOC of 0.949 V, FF of 70.26% and a JSC of 24.99 mA cm−2. The increase in PCE value of organic solar cells under the action of a dual-additive exceeds 10%. The results clearly indicate that the use of dual-additive treatment significantly enhances the storage stability of the organic solar cells (OSCs). As demonstrated in Fig. S2 (ESI†), the device without any additive treatment suffered a significant drop in performance, retaining only 80.6% of its initial efficiency after 192 hours of storage. In contrast, the device treated with a dual-additive maintained a remarkably higher efficiency of 88.6% under the same storage conditions. To further investigate the specific impact of a dual-additive on the photophysical processes of the device, conducting a series of optical tests is a reasonable and necessary step.
| Active layer | J SC (mA cm−2) | J calSC (mA cm−2) | V OC (V) | FF (%) | PCE (%) |
|---|---|---|---|---|---|
| (The data were obtained from 6 devices). | |||||
| PM6:PYIT | 24.40 (24.26 ± 0.23) | 23.40 | 0.943 (0.944 ± 0.001) | 63.30 (63.11 ± 0.17) | 14.58 (14.47 ± 0.13) |
| PM6:PYIT + 1.0 vol% CN | 21.71 (21.70 ± 0.05) | 20.83 | 0.934 (0.934 ± 0.005) | 71.12 (71.02 ± 0.08) | 14.42 (14.40 ± 0.01) |
| PM6:PYIT + 1.0 vol% DPE | 23.39 (23.23 ± 0.17) | 22.80 | 0.949 (0.948 ± 0.001) | 56.17 (55.94 ± 0.13) | 12.48 (12.34 ± 0.14) |
| PM6:PYIT + 0.6 vol% CN + 0.4 vol% DPE | 24.99 (24.82 ± 0.18) | 23.78 | 0.949 (0.946 ± 0.002) | 70.26 (70.27 ± 0.42) | 16.67 (16.50 ± 0.14) |
To understand the impact of different processing techniques on the charge collection efficiency in the OSCs, a functional plot was drawn between photocurrent density (Jph) and effective voltage (Veff) to investigate the extraction characteristics of the devices towards photogenerated charges.37,38 The Jph value is obtained from the formula Jph = JL − JD, where JL represents the current density under illuminated conditions, and JD represents the current density under dark conditions. The Veff is calculated using the formula Veff = V0 − Va, where V0 is the voltage value when Jph is equal to 0, and Va is the value of the applied voltage. Under the condition of device short-circuit, the charge collection efficiency (P) can be calculated by the formula P = Jph/Jsat. As shown in Fig. 2(a), the charge collection efficiency values of organic solar cell devices before and after dual-additive treatment are 95.3% and 96.6%, respectively. The OSC devices treated with the dual-additive have higher charge collection efficiency. The charge mobility of the device also has a significant impact on the energy conversion efficiency of OSCs. The electron mobility (μe) and hole mobility (μh) of devices were measured under dark conditions using the space-charge-limited current (SCLC) method,39 as shown in Fig. 3(b). For the PM6:PYIT system without additive treatment, the μe and μh of the device are 4.63 × 10−4 cm2 V−1 s−1 and 5.43 × 10−4 cm2 V−1 s−1, respectively. However, after dual-additive treatment, the μe and μh of the device can be enhanced to 4.97 × 10−4 cm2 V−1 s−1 and 5.66 × 10−4 cm2 V−1 s−1, respectively. The closer the ratio of μh/μe is to 1, the more balanced the transport of electrons and holes in the OSC, which is beneficial for improving the PCE. The calculation results show that for the PM6:PYIT system, the value decreased from 1.17 to 1.13 after the dual-additive treatment. The synergistic effect of the dual-additive indicates that the all-polymer system device has achieved balanced charge transport characteristics, which is beneficial for improving device performance.
 | ||
| Fig. 2 (a) Jphversus Veff of the devices. (b) Hole mobility and electron mobility of the devices. (c) JSC and (d) VOC dependence on the light intensity of OSCs. Normalized (e) TPC and (f) TPV data for the devices. | ||
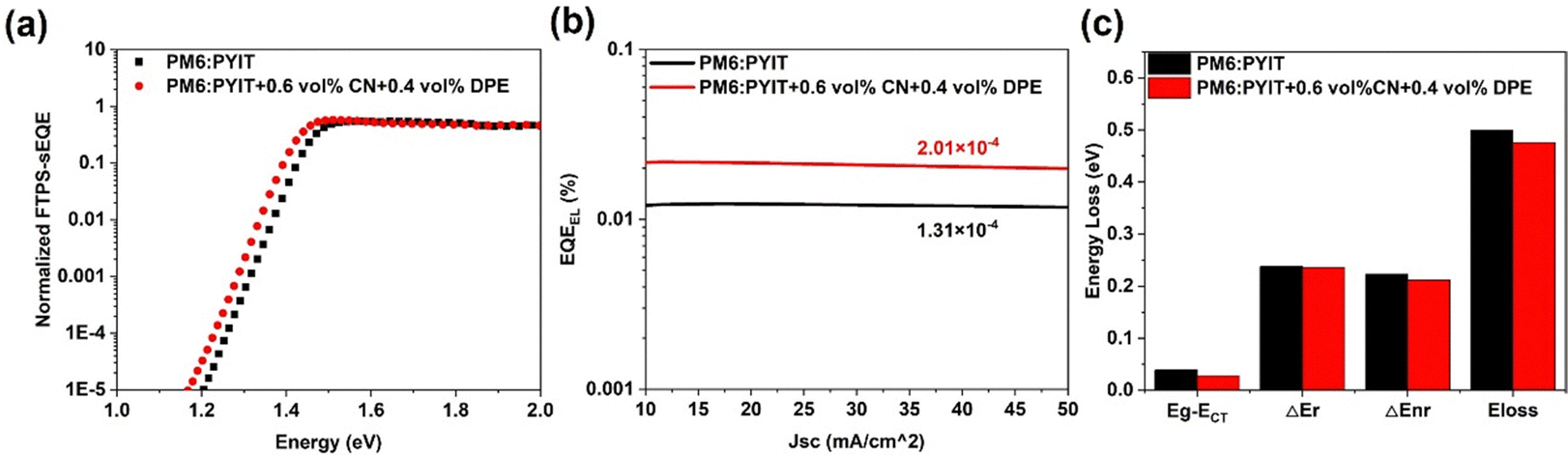 | ||
| Fig. 3 (a) FTPS-EQE spectra; (b) EQEEL spectra and (c) energy loss data of the PM6:PYIT devices before and after dual-additive treatment. | ||
The light intensity-dependent tests of the current and voltage are used to explore the influence of a dual-additive on the device's recombination process. Fig. 2(c) and (d) show the variation curves of JSC and VOC with Plight, respectively. The relationship between JSC and Plight can be represented by formula JSC ∝ Pαlight; the closer the α value is to 1, the lower the degree of bimolecular recombination in the active layer.40 The test results demonstrate that after the addition of dual-additive, the α value of the device increased from 0.975 in the untreated device to 0.987, suggesting that the addition of a dual-additive effectively suppresses the bimolecular recombination process in the device. In the relationship formula VOC ∝ (nkT/q)ln(Plight), when the value of n approaches 1, it indicates a weaker trap-assisted Shockley–Read–Hall (SRH) recombination in the devices.41 The initial device's n value is 1.391, and after the addition of 0.6 vol% CN and 0.4 vol% DPE, the n value of the device becomes 1.264. This indicates that the synergistic effect of CN and DPE can effectively reduce trap recombination in the PM6:PYIT all-polymer system.
After excitons dissociate into electrons and holes, there is always a recombination process for carriers before they are effectively extracted by the electrodes during the transport process. Next, the effects of the addition of dual-additive on the carrier recombination dynamics in the photoelectric conversion process will be tested through transient photocurrent (TPC) and transient photovoltage (TPV) measurements.29,42 Under short-circuit conditions, the charge extraction time of the PM6:PYIT device without any additives was fitted to be 0.35 μs. After adding 0.6 vol% CN and 0.4 vol% DPE simultaneously, the charge extraction time of the device decreased to 0.28 μs. The findings clearly demonstrate that the combined effect of the dual-additive significantly enhances the charge extraction process within the device, resulting in increased EQE response values and an improved FF. Under simulated sunlight irradiation, analysis of the TPV decay curve reveals that the carrier lifetime of the non-additive device stands at 3.24 μs. However, upon the incorporation of the dual-additive, this value rises to 4.72 μs. Consequently, the simultaneous introduction of CN and DPE into the PM6:PYIT system effectively mitigates recombination losses within the binary blend system, ultimately leading to an enhancement in both the device's FF and PCE.
2.2 Reduction of optical gap and energy loss
In order to clearly explain the reason why the dual-additive causes high VOC in OSC, the energy loss (Eloss) in the binary device was analyzed. The total Eloss is divided into three parts: ΔECT = Eg − ECT (charge separation loss), ΔEr (radiative recombination loss) and ΔEnr (non-radiative recombination loss).The changes in Eg after the addition of different additives are shown in Fig. S3 (ESI†). The results show that both single additive and dual-additive treatments can reduce the absorption bandgap of the binary PM6:PYIT device. Without additive treatment, the measured Eg for the PM6:PYIT system is 1.443 eV, corresponding to an Eloss of 0.500 eV. With the synergistic effect of the dual-additives, the PM6:PYIT binary system's Eg is reduced to 1.425 eV, and the VOC value also increases from 0.943 V to 0.949 V. As a result, the device's Eloss is reduced from 0.500 eV to 0.476 eV. When 1.0 vol% CN was added, although the device's Eg reduced from 1.443 eV to 1.436 eV, the VOC value also decreased to 0.934 V. This leads to an increase of the final Eloss from 0.500 eV to 0.502 eV, which may be due to the changes in the morphology of the active layer. On the other hand, when 1.0 vol% DPE was added, the device's Eg was reduced to 1.430 eV and the Eloss is reduced to 0.481 eV. Eg often represents the minimum energy required for the material to absorb photons and undergo electron transitions.43 The decrease in Eg indicates that the dual-additive can promote the utilization of photons in the OSC, thereby improving the PCE, even with the incident light remaining unchanged.
As shown in Fig. S4 (ESI†), by simultaneously fitting the sEQE (spectral external quantum efficiency) and EL (electroluminescence) curves of the device, we can obtain the ECT value. The ECT values of devices without and with dual-additive are calculated to be 1.404 eV and 1.397 eV, respectively. ΔEnr is linearly related to the natural logarithm of EQEEL, and can be obtained from the formula ΔEnr = −kT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln(EQEEL).44,45 As shown in Fig. 3(b), the EQEEL of the PM6:PYIT device without any additive is 1.31 × 10−4. When 0.6 vol% CN and 0.4 vol% DPE were added, the EQEEL of the device increases to 2.01 × 10−4. The detailed values of Eloss for different parts are provided in Table 2.
ln(EQEEL).44,45 As shown in Fig. 3(b), the EQEEL of the PM6:PYIT device without any additive is 1.31 × 10−4. When 0.6 vol% CN and 0.4 vol% DPE were added, the EQEEL of the device increases to 2.01 × 10−4. The detailed values of Eloss for different parts are provided in Table 2.
| Device | E loss (ev) | E g (ev) | E CT (eV) | ΔECT (ev) | ΔEr (eV) | ΔEnr (eV) | EQEEL |
|---|---|---|---|---|---|---|---|
| Control | 0.500 | 1.443 | 1.404 | 0.039 | 0.238 | 0.223 | 1.31 × 10−4 |
| Dual-additive | 0.476 | 1.425 | 1.397 | 0.028 | 0.236 | 0.212 | 2.01 × 10−4 |
In summary, the introduction of CN and DPE can improve the exciton dissociation efficiency, carrier lifetime and charge mobility. This leads to a more balanced charge transfer in the OSC. Additionally, the dual-additive treatment helps in reducing the Eg, which contributes to lower Eloss. Since the important optoelectronic conversion processes occur in the active layer of the OSC, various morphological characterizations will be used below to explain the fundamental reasons behind the improved performance of the OSC.
2.3 Morphology and film-forming dynamics
To investigate the impact of the introduction of the dual-additive on the morphology and molecular aggregation state of the active layer, atomic force microscopy (AFM), transmission electron microscopy (TEM) and UV-vis absorption were employed. Grazing-incidence wide-angle X-ray scattering (GIWAXS) was used to observe changes in the crystalline structure of active layer. Additionally, in situ UV-vis absorption spectroscopy was used to provide the film-forming dynamics changes of active layer after the addition of the dual-additive.As shown in Fig. 4(a) and (b), it is clear that the root-mean-square roughness (RMS) value of PM6:PYIT neat film can be increased after the introduction of the dual-additive. In the original PM6:PYIT thin film morphology, the active layer was observed to be very uniform, without the presence of large aggregates. The RMS value was measured to be 1.09 nm. After the addition of the 1.0 vol% CN additive, the RMS value of the thin film decreased to 0.96 nm, as shown in Fig. S5(a) (ESI†). The film treated with DPE exhibits a larger RMS value (1.17 nm) compared to the original film, indicating a slightly increased phase separation size. However, this treatment also disrupts the original fibrous structure, which leads to a decrease in the JSC and FF.
 | ||
| Fig. 4 The AFM images of the PM6:PYIT blend film (a) without additives and (b) with a dual-additive; the TEM images of the PM6:PYIT blend film (c) without additives and (d) with a dual-additive; (e) the change in the intensity ratio of the absorption peaks between acceptor and donor molecules with etching depth; and (f) mechanism diagram of the active layer phase separation morphology. | ||
When delving into the reasons behind our choice of the dual-additive combination of DPE and CN, we need to consider multiple dimensions. Firstly, through detailed performance analysis, we observed that the introduction of the CN additive significantly enhanced the FF of the device. However, CN also caused a slight decrease in the VOC. Furthermore, morphological characterization techniques revealed the positive impact of the CN additive on the miscibility between materials. In particular, the addition of CN effectively increased the compatibility between PM6 and PYIT, which, although leading to a red shift in their UV-vis absorption spectra, indicating an increase in aggregation, also resulted in a slight reduction in the phase domain size of the film at the microscopic level, accompanied by a decrease in the RMS value. This improvement in microstructure promoted the formation of a more uniform donor–acceptor interpenetrating network, thereby enhancing the FF of the device.
However, when we introduced the DPE additive into the film, the situation changed. Although the DPE-treated film exhibited a greater increase in phase domain size, this increase exceeded the ideal range, leading to the destruction of the original fiber-like network structure. This structural change directly affected the performance of the device, manifesting as a decrease in the JSC and FF values. This indicates that while DPE can maintain a sufficient donor–acceptor interface to promote exciton dissociation, the morphology of the film it forms is not conducive to effective charge transport and collection, thus only bringing about a slight increase in VOC. Based on the above analysis, we proposed a dual-additive strategy. This strategy aims to achieve comprehensive enhancement in three key photoelectric parameters of the device through the synergistic effect of CN and DPE. The introduction of CN promotes the formation of a more uniform conductive interpenetrating network, which is beneficial for the enhancement of FF. Meanwhile, DPE ensures a sufficient donor–acceptor interface, providing conditions for exciton dissociation. Through the combined use of these two additives, we expect to further enhance JSC and FF without sacrificing VOC, thus optimizing the overall performance of the device.
After the addition of the dual-additive, the RMS value of the PM6:PYIT film increased to 1.14 nm. Furthermore, from the images, it can be observed that the fibrous cluster structure of the film is well maintained. The increase in RMS value also indicates an enhancement in the aggregation of donor and acceptor. The original phase separation size was too small, resulting in an increased probability of charge recombination. However, after the CN+DPE treatment, the active layer achieved an appropriate phase separation size, leading to an optimal interface area. This is beneficial for exciton dissociation, as well as the transport and extraction of charges.
The morphology changes of the PM6 and PYIT neat films under the introduction of a single additive are shown in Fig. S6(a) and (b) (ESI†). It can be observed that CN has a stronger impact on PYIT, while DPE has a stronger impact on PM6. From Fig. 4(c) and (d) TEM images, we can clearly observe the significant changes in the PM6:PYIT thin film before and after the introduction of the dual-additive. In the original binary film, the donor and acceptor materials are mixed relatively uniformly, with almost no obvious block-like aggregations, indicating good compatibility between the components within the film without additives. However, after the addition of dual-additive, the film morphology undergoes a significant change, with larger areas of black aggregations appearing, reflecting the effective aggregation of the acceptor PYIT after additive treatment.
Subsequently, the depth-dependent absorption spectroscopy technique was used to investigate the mechanism of the dual-additive in the vertical phase separation of the active layer. Fig. S7 (ESI†) shows the film-depth-dependent-light absorption spectroscopy of no additive and dual-additive-treated PM6:PYIT blend films. As shown in Fig. 4(e), we got the distribution of the acceptor and donor molecules in the vertical direction by comparing the ratios of their absorption peak intensities at different depths in the film. The results indicate that the ratio of the absorption peak intensities of the acceptor and donor at the bottom of the film, after treatment with the dual-additive, is 0.66, which is lower than the 0.77 of the film without additive treatment. These data suggest that the introduction of the dual-additive promotes the aggregation of donor molecules in the bottom anode region. Furthermore, we observed that donor molecules tend to aggregate in the anode region, while acceptor molecules tend to be distributed in the cathode region (like the morphology shown in Fig. 4(f)), further demonstrating the effectiveness of the dual-additive in optimizing the vertical phase distribution, thereby promoting efficient charge transport and extraction in the device.46,47
Regarding the reason why the addition of the dual-additive improves the vertical phase separation morphology of the active layer, we propose the following hypothesis. During the solution film formation process, the difference in surface energy has a significant impact on the molecular movement and the ability to wet the substrate. We measured the surface energies of PM6 and PYIT, which were 32.7 mN m−1 and 40.2 mN m−1, respectively (as shown in Fig. S8, ESI†). Meanwhile, the surface energies of the liquid additives DPE and CN are 36.4 mN m−1 and 42.9 mN m−1, respectively. Based on the principle of compatibility in blend systems, the interaction between DPE and PM6 is stronger, while the interaction between CN and PYIT is stronger. Under the influence of surface energy, liquids with lower surface energy (or solids with higher surface energy) are more conducive to spreading, while the opposite is unfavorable. Therefore, liquids with lower surface energy (DPE) are more likely to wet the substrate and spread, while liquids with higher surface energy (CN) are less likely to wet the substrate and tend to move upward. Consequently, under the dual-additive effect, PM6 and PYIT are more concentrated in the bottom and top regions of the active layer, resulting in an optimized vertical phase separation morphology.
In order to further verify the accuracy of the influence of additives on the observed morphological changes, we conducted UV-vis absorption spectroscopy analysis. First, we examined the spectral changes in neat PM6 films after treatment with different additives. As shown in Fig. S9(a) (ESI†), for the PM6 film without additive treatment, the A0–0 peak and A0–1 peak were located at 610 nm and 578 nm, respectively, and the intensities of these two peaks were almost equal. After the introduction of CN, no significant changes were observed in the shape or absorption intensity, with only a slight red shift in peak position, indicating that CN had a minor impact on the aggregation state of PM6 molecules. However, after the introduction of DPE, both the A0–0 and A0–1 peaks of PM6 were noticeably enhanced, especially with a larger change in the A0–0 peak. By calculating the peak intensity ratio of A0–0/A0–1, we found that this ratio increased significantly, directly reflecting a tighter aggregation of PM6 molecules under the influence of the DPE additive.
Fig. S9(b) (ESI†) provides a detailed display of the UV-vis absorption spectra of the pure acceptor PYIT film under different additive treatment conditions. In the case of no additive treatment, the absorption peak of PYIT is located at 802 nm. Furthermore, when treated with DPE, only a slight red shift in the absorption peak of PYIT was observed. However, when the 1-CN additive was introduced, a significant red shift in the absorption peak of PYIT was observed, with the peak shifting from the original 802 nm to 821 nm. This significant change indicates that compared to DPE, 1-CN has a more significant impact on the acceptor PYIT, promoting a tighter aggregation of PYIT molecules. Additionally, we also noticed that under 1-CN treatment, the absorption tail curve of PYIT showed a steeper trend, further implying that 1-CN helps to orderly arrange the PYIT molecules.
Fig. S10 (ESI†) shows the UV-vis spectra of the PM6:PYIT blended film under different additive treatment conditions. From the absorption spectrum of the original PM6:PYIT blended film, it can be found that PM6 and PYIT can form good light absorption complementarity. The absorption peak of PM6 is located near 618 nm, and the original absorption peak of PYIT is located at 801 nm. After adding 1.0 vol% solvent additive CN, the donor and acceptor peaks were shifted to 630 nm and 817 nm, respectively, indicating that CN increased the degree of aggregation of the donor and acceptor. After adding 1.0 vol% solvent additive DPE, the peak positions of the donor and acceptor were 634 nm and 813 nm, respectively. After the dual-additive treatment, the absorption peaks of the donor and acceptor in the active layer film both showed a larger red shift compared to the single additive treatment, indicating that the dual-additive treatment increased the degree of aggregation of the donor and acceptor. The detailed information of the absorption peak changes is summarized in Table S2 (ESI†).
These results provide strong spectroscopic evidence for understanding the impact of additives on the morphology and performance of the material. However, the surface morphology of the film cannot represent the internal aggregation characteristics. Next, we use grazing incidence wide-angle X-ray scattering (GIWAXS) to study the effect of additives on the crystallization state of the film. As shown in Fig. S11(a) (ESI†), the pristine PM6 film exhibited a distinct (100) diffraction peak in the out-of-plane direction, and the position of the diffraction peak is at 0.32 Å−1. After adding 1.0 vol% DPE, the (100) diffraction peak position of PM6 increased to 0.33 Å−1, and the CCL value increased from 58.83 Å to 59.77 Å. For the acceptor condition, as shown in Fig. S10(b) (ESI†), the acceptor PYIT has a relatively distinct (010) diffraction peak in the out-of-plane direction, and the position of the π–π stacking diffraction peak is at 1.59 Å−1. After adding 1.0 vol% CN, the π–π stacking diffraction peak shifts to 1.60 Å−1, and the CCL value increases from 11.11 Å to 11.47 Å. The larger diffraction peak position and CCL values indicate that the stacking of PM6 and PYIT becomes more compact after the addition of DPE and CN, and the crystal size increases, which is consistent with the conclusion of the previous UV-vis absorption spectrum that the additive enhances molecular aggregation. Fig. 5(a) shows the two-dimensional images of PM6:PYIT before and after single additive and dual-additive treatments, the crystal coherence length (CCL) values of the acceptor (010) diffraction peak in the out-of-plane direction are summarized in Table S3 (ESI†). When no additives were added, the (010) peak position corresponding to the acceptor molecule in PM6:PYIT was at 1.58 Å−1, and the CCL value was 14.36 Å. After adding 1.0 vol% CN, the (010) diffraction peak changed to 1.60 Å−1, and the CCL value increased to 15.56 Å. After adding 1.0 vol% DPE, the (010) diffraction peak of the film was at 1.60 Å−1, and the calculated CCL value of the diffraction peak was 15.43 Å. When 0.6 vol% CN and 0.4 vol% DPE were added simultaneously, the (010) peak appeared at 1.64 Å−1, and the CCL value increased to 21.66 Å. This indicates that the synergistic effect of a dual-additive effectively enhances the aggregation of acceptor molecules, increases the size of phase separation, and obtains a more suitable phase separation morphology. In addition, we also tested the changes in donor crystallinity with the addition of different additives, and the data are summarized in Table S4 (ESI†). We found that the addition of a dual-additive also contributed to the improvement of donor crystallinity.
 | ||
| Fig. 5 (a) GIWAXS patterns of the PM6:PYIT blend films processed with different additives. (b) One dimensional GIWAXS line-cut (out-of-plane) profiles of blend films. | ||
Through the above characterization results and descriptions, the addition of a dual-additive has successfully increased the aggregation of both donor and acceptor, and enhanced their crystalline size, ultimately leading to an increase in the aggregation size of the PM6:PYIT active layer film from its initially undersized state to an appropriate size. In order to further investigate the regulatory mechanism of dual-additive on the morphology and crystallization of the active layer, the in situ UV-vis absorption spectroscopy will be employed to explore the film formation kinetics of the active layer before and after dual-additive treatment. The corresponding film formation kinetics processes before and after dual-additive treatment are illustrated in Fig. 6.
 | ||
| Fig. 6 (a) The absorption peak position of PYIT as a function of time. In situ absorption spectra of binary films (b) without additives and (c) with dual-additive during the film-formation process. | ||
From the graph, it can be observed that in the film formation process of the PM6:PYIT system without any additives, there is not a significant change in the absorption peak of donor PM6 over time, with only a slight increase in absorption intensity. However, the absorption peak of PYIT gradually undergoes a redshift with the increasing film formation. In the case of the blend system treated with a dual-additive, similar to the untreated system, the changes in the absorption peak of the donor PM6 are less pronounced compared to those of the acceptor PYIT during the film formation process.
Therefore, the changes in the peak position of the acceptor were extracted under both conditions to characterize the crystallization kinetics changes during the film formation process of PYIT. By tracking the evolution of the absorption peak position of PYIT with film formation time, the film formation kinetics process can be roughly divided into three stages: (1) the initial solvent evaporation stage, where the peak position of the PYIT molecule shows little variation (blue region); (2) as the solvent further evaporates to reach the point of saturation solubility, the PYIT absorption peak begins to redshift, indicating the aggregation and crystallization film formation stage of the PYIT molecules (pink region); and (3) subsequently, as the solvent completely evaporates, the PYIT absorption peak position stabilizes, representing the thin film formation stage (yellow region). In the PM6:PYIT system without additives, the absorption peak position of PYIT gradually increases from 0.5 s and then stabilizes after 2.8 s, suggesting an aggregation film deposition time of 2.3 s for PYIT. After adding 0.6 vol% CN and 0.4 vol% DPE, the absorption peak of PYIT starts to change from 0.3 s and stabilizes after 3.8 s. Therefore, the addition of a dual-additive increases the overall aggregation and crystallization film formation time for the acceptor PYIT, allowing the PYIT molecules more time to form a better-ordered structure induced by the additives, improving the morphology of the active layer phase separation and enhancing the efficiency of charge transport.
2.4 The relationship between morphological changes and performance improvement
Based on the comprehensive characterization results, we speculate that the dual high-boiling-point additives can continuously interact with donor and acceptor molecules after the evaporation of the main solvent CF, producing the following effects: Firstly, they prolong the aggregation and film-forming time of PYIT molecules, contributing to the formation of a more stable molecular aggregation structure. Secondly, these additives simultaneously induce a redshift in the absorption spectra of both the donor and acceptor, enhancing their aggregation and increasing the phase domain size of the donor and acceptor, thereby reducing the probability of charge recombination. During the film-forming process, the action of surface energy prompts the directed movement of donor and acceptor molecules. In particular, DPE prompts the donor molecules to move towards the bottom anode, while CN prompts PYIT to move towards the top cathode. This ordered molecular arrangement facilitates charge transport and extraction. In the original PM6:PYIT system, the molecular arrangement is disordered, and the aggregation size is small, which is unfavorable for charge transport and extraction. However, under the effect of the dual-additive, molecular aggregation is significantly enhanced, the phase domain size is increased, and the face-on orientation is strengthened. Furthermore, a vertical phase separation structure that is more conducive to charge extraction and transport, as well as a better interpenetrating network structure between donor and acceptor is formed. While the increased molecular aggregation promoted the enlargement of phase separation size, the reduced charge extraction time obtained from TPC tests indicates that the addition of a dual-additive facilitated the faster dissociation of excitons. Additionally, due to the increased crystallinity and improved vertical phase separation structure, the carrier lifetime increased from 3.24 μs to 4.72 μs in TPV tests. This enhancement in carrier lifetime is primarily attributed to the improved morphology of the active layer, which reduces the probability of charge recombination (as shown in Fig. 7). Consequently, the formation of better vertical phase separation facilitates more efficient charge transport, leading to an increase in carrier lifetime. These changes collectively enhance the performance of the device.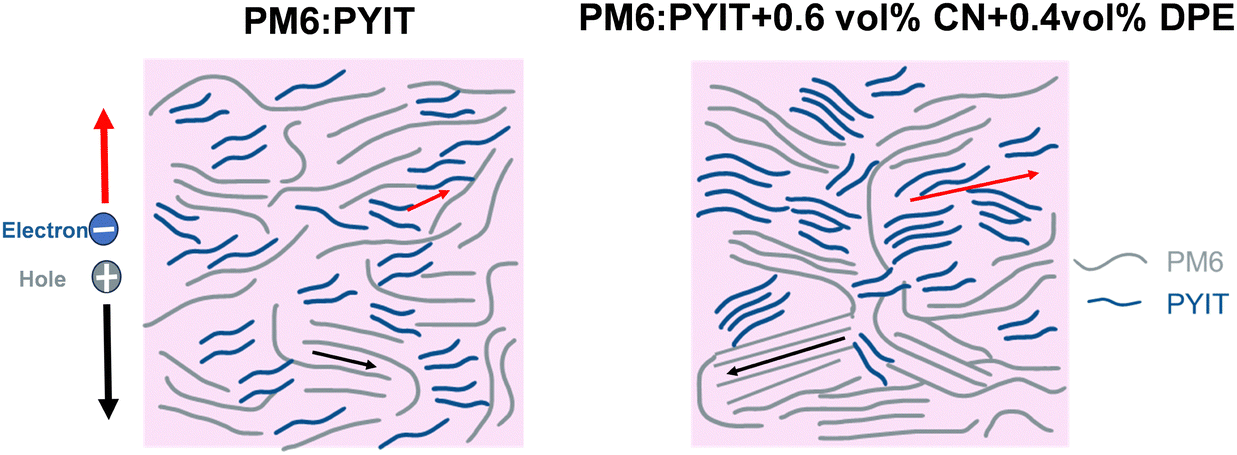 | ||
| Fig. 7 Hypothetical diagram of changes in morphology and molecular aggregation state of the active layer. | ||
3. Conclusions
In summary, we have proposed a dual-additive strategy to enhance the molecular aggregation and decrease the optical gap to reduce the energy loss of all-polymer organic solar cells. The high boiling point solvent additives DPE and CN effectively address the issue of the excessively short aggregation time caused by the rapid evaporation of low-boiling-point CF. After the evaporation of the primary solvent, the additives continue to act on the polymer molecules, allowing PYIT to achieve a longer aggregation and film formation time as well as an extended period of migration and aggregation towards the top. Furthermore, the dual-additive system successfully enhances the EQE and UV-vis absorption spectrum, resulting in improved photon utilization efficiency of the device and a reduction in the absorption bandgap (Eg). The optimized morphology of the active layer also effectively suppresses the recombination process, increasing the carrier lifetime from 3.24 μs to 4.72 μs. After the synergistic action of adding 0.6 vol% CN and 0.4 vol% DPE dual-liquid-additive, the device's bandgap decreases from 1.443 eV to 1.425 eV. The Eloss decreases from 0.500 eV to 0.476 eV, non-radiative energy recombination decreases from 0.223 eV to 0.212 eV, and the optimal PCE increases from 14.58% to 16.67%. The FF also shows significant improvement, increasing from 63.30% to 70.26%. Our work proposes a new dual-additive combination that can improve the morphology of the all-polymer system active layer and enhance the PCE of all-PSCs.4. Experimental section
4.1. Materials
PM6 and PYIT were all purchased from Derthon Optoelectronics Materials Science Technology Co Ltd. The Mn of PM6 is 31.6 kDa, the Mw is 61.4 kDa, and the polydispersity index (PDI) is 1.93. The Mn of polymer acceptor PYIT is 5.4 kDa, the Mw is 10.2 kDa, and the PDI is 1.86. 1-Chloronaphthalene (CN) was purchased from Tokyo Chemical Industry and diphenyl ether (DPE) was purchased from Sigma-Aldrich. All materials are directly used without purification.4.2. Device fabrication
The binary solar cells were fabricated using a conventional structure of ITO/PEDOT:PSS/active layer/PDINN/Ag. The patterned ITO glass substrates (15 Ω) were cleaned in an ultrasonic bath using a detergent, deionized water, acetone and isopropyl alcohol for 20 min each, and then blow-dried by high-purity nitrogen. All the pre-cleaned substrates were treated in an ultraviolet ozone generator for 25 min, then a ∼20 nm thick PEDOT:PSS layer was deposited by spin-coating under 5000 rpm for 30 s and annealed at 140 °C for 20 min in the atmosphere. Then ITO substrates with PEDOT:PSS films were transferred into a N2-filled glovebox. The PM6:PYIT blend with a 1:1.2 weight ratio was dissolved in chloroform at a donor polymer weight concentration of 7 mg mL−1. The solution was stirred overnight in the glove box at 50 °C. Before spin-coating to form a film, the solution was divided into several bottles of same volume. 1.0 vol% CN, 1.0 vol% DPE, and 0.6 vol% CN + 0.4 vol% DPE (the ratio of dual additives used in devices with optimized best performance) were added to the PM6:PYIT solutions, respectively. After adding the additives, the mixed solution was stirred for half an hour under the conditions of room temperature and 200 rpm. Before spin-coating to form a film, the stirring was turned off and let to stand for 20 minutes before using. The blend solution was spin-coated on PEDOT:PSS at 2500 rpm for 40 s to obtain a film of about 100 nm. It was then thermally annealed at 100 °C for 2 min. Subsequently, the electron transport layer of PDINN was deposited by spin-coating the solution in methanol (1 mg mL−1) at 3000 rpm for 30 s. Finally, a 150 nm Ag layer was deposited by thermal evaporation through a shadow mask under a vacuum of 1 × 10−4 Pa.
To precisely determine the optimal proportion of additives, we have carefully planned and implemented a series of detailed experiments. In these experiments, we keep the total volume fraction of additives constant at 1.0 vol% to ensure the consistency and comparability of experimental conditions. In the experiments of dual-additive combinations, we determined the impact of three combinations of CN addition on device performance, where the CN concentration was 0.3 vol%, 0.6 vol%, and 0.9 vol% respectively, thus obtaining the optimal combination of additive dosage.
4.3. Characterization
Data availability
The authors confirm that the data supporting the findings of this study are available within the article and ESI.†Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was partly funded by the Ministry of Science and Technology (No. 2022YFB4200400) and the National Natural Science Foundation of China (52394274). The authors gratefully acknowledge the cooperation of the beamline scientists at the BSRF-1W1A beamline.References
- X. Zheng, L. Zuo, K. Yan, S. Shan, T. Chen, G. Ding, B. Xu, X. Yang, J. Hou, M. Shi and H. Chen, Energy Environ. Sci., 2023, 16, 2284–2294 RSC.
- W. Liu, S. Sun, L. Zhou, Y. Cui, W. Zhang, J. Hou, F. Liu, S. Xu and X. Zhu, Angew. Chem., Int. Ed., 2022, 61, e202116111 CrossRef CAS.
- S.-Y. Chang, P. Cheng, G. Li and Y. Yang, Joule, 2018, 2, 1039–1054 CrossRef CAS.
- J. Liu, J. Deng, Y. Zhu, X. Geng, L. Zhang, S. Y. Jeong, D. Zhou, H. Y. Woo, D. Chen, F. Wu and L. Chen, Adv. Mater., 2023, 35, 2208008 CrossRef CAS.
- B. Walker, J. H. Liu, C. Kim, G. C. Welch, J. K. Park, J. Lin, P. Zalar, C. M. Proctor, J. H. Seo, G. C. Bazan and T. Q. Nguyen, Energy Environ. Sci., 2013, 6, 952–962 RSC.
- K. D. Deshmukh, T. Qin, J. K. Gallaher, A. C. Y. Liu, E. Gann, K. O’Donnell, L. Thomsen, J. M. Hodgkiss, S. E. Watkins and C. R. McNeill, Energy Environ. Sci., 2015, 8, 332–342 RSC.
- S. Xiao, Q. Zhang and W. You, Adv. Mater., 2017, 29, 1601391 CrossRef.
- M. Zhang, X. Guo, W. Ma, H. Ade and J. Hou, Adv. Mater., 2015, 27, 4655–4660 CrossRef CAS.
- X. Long, Z. Ding, C. Dou, J. Zhang, J. Liu and L. Wang, Adv. Mater., 2016, 28, 6504–6508 CrossRef CAS PubMed.
- L.-Y. Xu, W. Wang, X. Yang, S. Wang, Y. Shao, M. Chen, R. Sun and J. Min, Nat. Commun., 2024, 15, 1248 CrossRef CAS PubMed.
- Y. Xu, J. Yuan, S. Liang, J.-D. Chen, Y. Xia, B. W. Larson, Y. Wang, G. M. Su, Y. Zhang, C. Cui, M. Wang, H. Zhao and W. Ma, ACS Energy Lett., 2019, 4, 2277–2286 CrossRef CAS.
- R. Sun, T. Wang, Q. Fan, M. Wu, X. Yang, X. Wu, Y. Yu, X. Xia, F. Cui, J. Wan, X. Lu, X. Hao, A. K. Y. Jen, E. Spiecker and J. Min, Joule, 2023, 7, 221–237 CrossRef CAS.
- W. Zhang, C. Sun, I. Angunawela, L. Meng, S. Qin, L. Zhou, S. Li, H. Zhuo, G. Yang, Z.-G. Zhang, H. Ade and Y. Li, Adv. Mater., 2022, 34, 2108749 CrossRef CAS PubMed.
- X. Li, X. Duan, Z. Liang, L. Yan, Y. Yang, J. Qiao, X. Hao, C. Zhang, J. Zhang, Y. Li, F. Huang and Y. Sun, Adv. Energy Mater., 2022, 12, 2103684 CrossRef CAS.
- B. Sharma, Y. Sarothia, R. Singh, Z. Kan, P. E. Keivanidis and J. Jacob, Polym. Int., 2016, 65, 57–65 CrossRef CAS.
- Z. Luo, T. Liu, R. Ma, Y. Xiao, L. Zhan, G. Zhang, H. Sun, F. Ni, G. Chai, J. Wang, C. Zhong, Y. Zou, X. Guo, X. Lu, H. Chen, H. Yan and C. Yang, Adv. Mater., 2020, 32, 2005942 CrossRef CAS PubMed.
- W. Xu, M. Zhang, X. Ma, X. Zhu, S. Y. Jeong, H. Y. Woo, J. Zhang, W. Du, J. Wang, X. Liu and F. Zhang, Adv. Funct. Mater., 2023, 33, 2215204 CrossRef CAS.
- B. Li, X. Zhang, Z. Wu, J. Yang, B. Liu, Q. Liao, J. Wang, K. Feng, R. Chen, H. Y. Woo, F. Ye, L. Niu, X. Guo and H. Sun, Sci. China: Chem., 2022, 65, 1157–1163 CrossRef CAS.
- M. An, Q. Bai, S. Y. Jeong, J. Ding, C. Zhao, B. Liu, Q. Liang, Y. Wang, G. Zhang, H. Y. Woo, X. Qiu, L. Niu, X. Guo and H. Sun, Adv. Energy Mater., 2023, 13, 2301110 CrossRef CAS.
- X. Yang, R. Sun, Y. Wang, M. Chen, X. Xia, X. Lu, G. Lu and J. Min, Adv. Mater., 2023, 35, 2209350 CrossRef CAS.
- G. Li, R. Zhu and Y. Yang, Nat. Photonics, 2012, 6, 153–161 CrossRef CAS.
- G. Wang, F. S. Melkonyan, A. Facchetti and T. J. Marks, Angew. Chem., Int. Ed., 2019, 58, 4129–4142 CrossRef CAS.
- Y. Yan, Y. Liu, J. Zhang, Q. Zhang and Y. Han, J. Mater. Chem. C, 2021, 9, 3835–3845 RSC.
- K. Zhou, Y. Wu, Y. Liu, X. Zhou, L. Zhang and W. Ma, ACS Energy Lett., 2019, 4, 1057–1064 CrossRef CAS.
- R. Ma, K. Zhou, Y. Sun, T. Liu, Y. Kan, Y. Xiao, T. A. Dela Peña, Y. Li, X. Zou, Z. Xing, Z. Luo, K. S. Wong, X. Lu, L. Ye, H. Yan and K. Gao, Matter, 2022, 5, 725–734 CrossRef CAS.
- J. Pan, J. Guan, Z. Wang, R. Zhang, Y. Fu, X. Yu, Q. Zhang and Y. Han, J. Mater. Chem. C, 2024, 12, 4142–4156 RSC.
- Y. Liu, Q. Zhang, J. Guan, J. Xue, X. Yu, F. Wu, W. Ma and Y. Han, ACS Appl. Mater. Interfaces, 2022, 14, 44685–44696 CrossRef CAS.
- Y. Liu, T. Zhang, R. Zhang, J. Guan, J. Pan, X. Yu, Q. Zhang and Y. Han, Macromol. Rapid Commun., 2023, 44, 2300338 CrossRef CAS.
- Z. Li, F. Gao, N. C. Greenham and C. R. McNeill, Adv. Funct. Mater., 2011, 21, 1419–1431 CrossRef CAS.
- J. Yang, B. Liu, J.-W. Lee, Y. Wang, H. Sun, Z. Chen, Q. Bai, B. J. Kim, Y. Jiang, L. Niu and X. Guo, Chin. J. Chem., 2022, 40, 2900–2908 CrossRef CAS.
- B. Liu, H. Sun, J.-W. Lee, Z. Jiang, J. Qiao, J. Wang, J. Yang, K. Feng, Q. Liao, M. An, B. Li, D. Han, B. Xu, H. Lian, L. Niu, B. J. Kim and X. Guo, Nat. Commun., 2023, 14, 967 CrossRef CAS PubMed.
- L. Jin, R. Ma, H. Liu, W. Xu, Z. Luo, T. Liu, W. Su, Y. Li, R. Lu, X. Lu, H. Yan, B. Z. Tang and T. Yang, ACS Appl. Mater. Interfaces, 2021, 13, 34301–34307 CrossRef CAS.
- Q. Bai, Q. Liang, Q. Liu, B. Liu, X. Guo, L. Niu and H. Sun, Chin. J. Chem., 2023, 41, 3714–3728 CrossRef CAS.
- Z. Li, Y. Liang, X. Qian, L. Ying and Y. Cao, Chem. Eng. J., 2022, 446, 136877 CrossRef CAS.
- R. Ma, J. Yu, T. Liu, G. Zhang, Y. Xiao, Z. Luo, G. Chai, Y. Chen, Q. Fan, W. Su, G. Li, E. Wang, X. Lu, F. Gao, B. Tang and H. Yan, Aggregate, 2022, 3, e58 CrossRef CAS.
- P. Q. Bi, S. Q. Zhang, T. Xiao, M. H. Cui, Z. H. Chen, J. Z. Ren, C. C. Qin, G. H. Lu, X. T. Hao and J. H. Hou, Sci. China: Chem., 2021, 64, 599–607 CrossRef CAS.
- D. Bartesaghi, I. d C. Pérez, J. Kniepert, S. Roland, M. Turbiez, D. Neher and L. J. A. Koster, Nat. Commun., 2015, 6, 7083 CrossRef CAS.
- X. Zhang, N. Yao, R. Wang, Y. Li, D. Zhang, G. Wu, J. Zhou, X. Li, H. Zhang, J. Zhang, Z. Wei, C. Zhang, H. Zhou, F. Zhang and Y. Zhang, Nano Energy, 2020, 75, 105032 CrossRef CAS.
- A. Carbone, B. K. Kotowska and D. Kotowski, Phys. Rev. Lett., 2005, 95, 236601 CrossRef CAS PubMed.
- X. Ma, Q. An, O. A. Ibraikulov, P. Lévêque, T. Heiser, N. Leclerc, X. Zhang and F. Zhang, J. Mater. Chem. A, 2020, 8, 1265–1272 RSC.
- L. Zhan, S. Li, T.-K. Lau, Y. Cui, X. Lu, M. Shi, C.-Z. Li, H. Li, J. Hou and H. Chen, Energy Environ. Sci., 2020, 13, 635–645 RSC.
- X. Song, K. Zhang, R. Guo, K. Sun, Z. Zhou, S. Huang, L. Huber, M. Reus, J. Zhou, M. Schwartzkopf, S. V. Roth, W. Liu, Y. Liu, W. Zhu and P. Müller-Buschbaum, Adv. Mater., 2022, 34, 2200907 CrossRef CAS.
- Y. Wang, D. Qian, Y. Cui, H. Zhang, J. Hou, K. Vandewal, T. Kirchartz and F. Gao, Adv. Energy Mater., 2018, 8, 1801352 CrossRef.
- T. Kirchartz, A. Helbig, W. Reetz, M. Reuter, J. H. Werner and U. Rau, Prog. Photovoltaics, 2009, 17, 394–402 CAS.
- B. Geffroy, P. le Roy and C. Prat, Polym. Int., 2006, 55, 572–582 CrossRef CAS.
- R. Sun, J. Guo, Q. Wu, Z. Zhang, W. Yang, J. Guo, M. Shi, Y. Zhang, S. Kahmann, L. Ye, X. Jiao, M. A. Loi, Q. Shen, H. Ade, W. Tang, C. J. Brabec and J. Min, Energy Environ. Sci., 2019, 12, 3118–3132 RSC.
- K. Jiang, J. Zhang, Z. Peng, F. Lin, S. Wu, Z. Li, Y. Chen, H. Yan, H. Ade, Z. Zhu and A. K. Y. Jen, Nat. Commun., 2021, 12, 468 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details about the fabrication of OSCs, characterization methods, water contact angle data, J–V curves for OSCs with different additives, stability curves of binary devices, normalized absorption spectra, AFM images, and GIWAXS patterns of neat and blend films. See DOI: https://doi.org/10.1039/d4tc03309d |
| This journal is © The Royal Society of Chemistry 2025 |