
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Quantification of microplastic targets in environmental matrices using pyrolysis-gas chromatography-mass spectrometry†
Rebecca H.
Peel
 ab,
Charlotte E. M.
Lloyd
ac,
Stephen J.
Roberts
b,
B. D. A.
Naafs
a and
Ian D.
Bull
*a
ab,
Charlotte E. M.
Lloyd
ac,
Stephen J.
Roberts
b,
B. D. A.
Naafs
a and
Ian D.
Bull
*a
aOrganic Geochemistry Unit (OGU), School of Chemistry, University of Bristol, Cantock's Close, Bristol BS8 1TS, UK. E-mail: ian.d.bull@bristol.ac.uk
bBritish Antarctic Survey (BAS), High Cross, Madingly Road, Cambridge CB3 0ET, UK
cSchool of Geographical Sciences, University of Bristol, Bristol BS8 1SS, UK
First published on 8th November 2024
Abstract
Microplastic pollution is a growing environmental problem. Consequently, an emerging area of research is the analysis of these micro-particles, to identify the distribution and impacts of plastic in the environment. This paper details the development and application of a pyrolysis-gas chromatography-mass spectrometry (Py-GC-MS) method for the quantification of microplastic pollution in terrestrial samples. Initial analysis of plastic standards using Py-GC-MS revealed diagnostic pyrolytic products, which were utilised alongside internal standards and linear regression to create calibrations for each studied synthetic plastic. A microplastic extraction protocol for soils and sediments was developed, namely an overnight density separation with wet peroxide digestion, and its efficacy confirmed through spiking and recovery experiments. Matrix effects were observed for PE, PS and PVC, highlighting the need to use multiple diagnostic compounds per plastic, where possible. Overall, these findings demonstrate that Py-GC-MS can be successfully applied for the determination of microplastic concentrations in terrestrial samples, with a view to establishing effective mitigation strategies.
Environmental significanceThe majority of plastic pollution originates on land and therefore, with regard to mitigation, the terrestrial environment needs to be of primary focus. Particularly when considering microplastic, tackling plastic pollution at the source is crucial, before smaller and more damaging particles can develop. However, our knowledge and understanding of the spatial and temporal profile of terrestrial microplastic is still lacking and, without improvement, mitigation efforts cannot successfully target plastic pollution at the source. Furthermore, vast quantities of plastic also end up in terrestrial environments and the impact on these ecosystems is only just being realised. There is a real need, therefore, to quantify terrestrial polymer contamination and determine source-to-sink litter pathways around the world. |
1. Introduction
The impacts of microplastic on organisms and the environment is largely unknown.1–3 Microplastic is ubiquitous throughout environmental matrices, impacting organisms ranging from those that are unicellular to mammalian. In addition, they exist with a whole host of different chemical and physical properties.4 There are therefore thousands of different potential ecotoxicological studies to determine the impact of a given plastic on a given organism under specific conditions.5 Establishing the extent of microplastic pollution not only helps us ascertain how important determining its impact is, but it also enables us to focus our efforts on organisms that are likely to be the most affected, using environmentally-relevant polymers and concentrations.6 In addition, determining environmental microplastic concentrations both spatially and temporally facilitates the identification of sources of microplastic pollution to the environment. Thus, it is possible to establish where mitigation efforts are best placed.There are several approaches to identify and quantify microplastic in environmental samples.7 Firstly, the microplastic must be extracted from a complex natural matrix, but once this has been achieved, it can be analysed in several ways. Spectroscopic methods are the most common techniques,7,8 however, few studies using such approaches produce mass-related quantitative results. Whilst particle count is an important metric in the context of biological impacts, mass is the most relevant parameter when considering sources, pathways and flux. Due to the small size of the particles found, determining their mass gravimetrically on an analytical balance is challenging and imprecise. Pyrolysis is, potentially, a better option as it provides not only mass-based data, but also polymer-specific information. Pyrolysis also requires less sample pre-treatment than Fourier-transform infrared spectroscopy (FT-IR) and Raman analysis with protocols relying less heavily on the manual selection of particles, thereby reducing the potential for human error.
Historically it has been challenging to quantify using pyrolysis, and consequently it has predominantly been used for qualitative and semi-quantitative work.7,9 The process is inherently complex as a wide range of pyrolytic products are produced in almost all environmental samples.10 This results in matrix effects that can heavily impact the peak areas of the target compounds. For example, internal surfaces can become coated with heavier breakdown products that condense within the instrument, affecting the way that other pyrolytic products interact with these surfaces.10,11 Generated artefacts may also coelute or affect the volatility of other pyrolysis products.12 Another major factor affecting the reliable analysis of complex samples is variability in pyrolytic breakdown.13,14 Small changes in the analytical conditions can affect the composition of pyrolytic products, making quantification challenging and necessitating time-consuming optimisation of a given pyrolysis set-up, alongside external calibration.15
Recent advances, such as improvements in instrument design, have enhanced our ability to quantify using pyrolysis, with studies demonstrating low limits of detection (LOD < 1 μg).11,16 Nonetheless, very few papers have utilised Py-GC-MS for the analysis of microplastic in environmental samples, with only one study incorporating internal standards in their calibration.10 Since one of the challenges of pyrolysis is to overcome matrix effects from the wide range of generated species, the use of internal standards can help to account for them, enabling more accurate quantification.14,17
To analyse terrestrial microplastic, particles are usually extracted from their soil or sediment matrix.18,19 Depending on sample type, there are various methods utilised for microplastic extraction.20,21 For samples containing inorganic sediment, particle isolation is achieved via a density separation. Here the sample is suspended in a salt solution and left to settle out; the dense, inorganic material falls to the bottom, while the less dense microplastic floats. The choice of salt solution is somewhat contentious, however, as cost, efficacy and environmental factors are considered.22 As an inexpensive and environmentally-friendly option, sodium chloride (NaCl) is widely used, but it has attracted criticism for not providing a dense enough solution to extract all plastics.23,24 Denser salt solutions such as sodium iodide (NaI) or zinc chloride (ZnCl2) have been deployed as alternatives, however cost and toxicity concerns make them less desirable.22 As a compromise, many studies use a hybrid approach with an initial density separation using an inexpensive, low-density salt, followed by extraction with NaI or ZnCl2.25 More recently, potassium formate (CHKO2) has been suggested as an eco-friendly, medium-density option.26
Due to its low density, organic matter is not removed during density separation, thus necessitating further sample processing. Not only is this required for volume reduction, but organic matter is also amenable to pyrolysis, and therefore will interfere with pyrolytic characterisation of microplastic. Again, there are many methods for the removal of organic material.19,24 Typically, a peroxide digestion is performed,7,18 but this has a variety of limitations. It can degrade or completely digest, the microplastic particles as well as the targeted organics; it can bleach particles, masking their identity during visual inspections, and lastly its use is restricted in many labs. Alternatives such as acid and base digestions are also not recommended, as they have been shown to have similar disadvantages and are less effective at removing organic matter.27 Studies using very organic rich samples may benefit from the use of enzymes for their digestion, although the protocol for this is more involved, leading to a greater risk of contamination or sample loss.28,29 Restrictions around enzyme use also limit their application. The order in which these steps are undertaken, and the manner in which they are performed, is also undecided.23,25 Nuelle et al. determined that treatment with hydrogen peroxide was the most effective means of reducing the amount of organic matter with minimal impacts to the plastic particles.27 Therefore, this was investigated here, using various workflow designs.
Based on the literature, it may be hypothesised that Py-GC-MS can be successfully employed to qualify and quantify microplastic in complex environmental samples. The aim of this study is to develop a successful methodology using a pyrolysis-based analytical platform, to quantify microplastic in complex environmental matrices such as soil and sediment. Calibration, using internal standards, will be investigated for environmentally-relevant polymers to ascertain the potential for quantification, and to determine the most effective workflow for future applications. Several extraction protocols will also be explored to determine the most effective method to separate microplastic from beach sediment and soil, focussing on density separation and the various methods for the removal of organic material. The chosen protocol will be validated by spike and recovery experiments to determine efficacy and to verify that it is appropriate for the analysis of terrestrial samples.
2. Methods
2.1. Analysis of polymer standards
Standards of eight common plastics were purchased from Goodfellow Ltd [polyamide 6 (PA6), polycarbonate (PC), polyethylene terephthalate (PET), polypropylene (PP) and polyvinyl chloride (PVC)] and Sigma Aldrich [polyethylene (PE), polymethyl methacrylate (PMMA) and polystyrene (PS)] to determine specific diagnostic moieties for each polymer. These are required to identify the specific plastics present in complex environmental samples.11 PET, PMMA, PS and PVC were purchased as powders, whilst PA6, PC, PE and PP were bought as granules. The latter four standards were cut using a scalpel to obtain particles of the required mass (see calibration details, below), before adding them to quartz pyrolysis tubes. Powders were added directly using a small spatula or syringe needle. Each standard was pyrolysed with and without the addition of tetramethylammonium hydroxide (TMAH) and the subsequent pyrograms compared with an in-house library and those from the literature.10,11,23,30,31 The National Institute of Standards and Technology (NIST08) database was also used for compound identification. Diagnostic compounds were selected for each polymer standard based on their specificity and abundance, as described by Fischer and Scholz-Böttcher.11Calibration was performed by weighing between 3 and 107 μg of polymer standard directly into quartz pyrolysis tubes using a Mettler Toledo XP6 microbalance. This range is restricted at the upper end by the limits of the pyrolyser and at the lower end by the sensitivity of the microbalance. Eight different internal standards were tested for their use in aiding quantification: 5α-androstane, bisphenol A-d10, 5β-cholanic acid, 9-fluorenone and pyrene-d10, each were introduced as 20 μL at 0.1 μg μL−1 in methanol, whilst 20 μL of anthracene-d10, 1,2-dibromobenzene and naphthalene-d8 were added at 0.1 μg μL−1 in n-hexane. The internal standard solutions were added directly to the quartz pyrolysis tubes up to 1 hour before pyrolysis and left for solvent to evaporate at room temperature. 9-Fluorenone, anthracene-d10, 5α-androstane and pyrene-d10 were selected as the internal standards for subsequent use and were added to each calibration sample as described above. Calibration models were created for each plastic standard, using the mass-filtered peak areas of selected diagnostic compounds and linear regression. Various combinations of diagnostic compound, diagnostic ion and internal standard were investigated to determine the most appropriate calibration method for each plastic. The coefficient of determination (R2) and the process standard deviation (sx0) were calculated in each case to enable comparison.11 See Tables 1 and 2 for the diagnostic compounds selected for each polymer. For the selected models, limit of detection (LOD) and limit of quantification (LOQ) were calculated by extrapolating the signal-to-noise (S![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :N) ratio from the lowest three calibration points across all calibrations for each polymer, using a S:N ratio of 3 for LOD and 10 for LOQ.32 95% quantification and prediction bands were determined and plotted for the selected calibration method for each plastic.11
:N) ratio from the lowest three calibration points across all calibrations for each polymer, using a S:N ratio of 3 for LOD and 10 for LOQ.32 95% quantification and prediction bands were determined and plotted for the selected calibration method for each plastic.11
| Polymer | Pyrolytic product | No internal standard | 9-Fluoren-one | Anthracene-d10 | 5α-Andros-tane | Pyrene-d10 |
|---|---|---|---|---|---|---|
| PA6 | ε-Caprolactam | 0.93 | 0.83 | 0.94 | 0.82 | 0.94 |
| PC | p-Tertbutylphenol | 0.95 | 0.57 | 0.95 | 0.60 | 0.96 |
| p-Isopropenylphenol | 0.46 | 0.84 | 0.44 | 0.81 | 0.45 | |
| Bisphenol A | 0.52 | 0.46 | 0.59 | 0.13 | 0.46 | |
| PE | C22–C24 terminal dienes | 0.98 | 0.74 | 0.94 | 0.56 | 0.91 |
| C22–C24 terminal alkenes | 0.96 | 0.74 | 0.92 | 0.53 | 0.89 | |
| C22–C24 alkanes | 0.94 | 0.83 | 0.95 | 0.72 | 0.95 | |
| PET | Divinyl terephthalate | 0.83 | 0.76 | 0.93 | 0.44 | 0.92 |
| 2-(Benzoyloxy)ethyl vinyl terephthalate | 0.92 | 0.71 | 0.97 | 0.38 | 0.95 | |
| PMMA | Methyl methacrylate | 0.87 | 0.25 | 0.91 | 0.03 | 0.89 |
| PP | 2,4-Dimethylhept-1-ene | 0.80 | 0.50 | 0.60 | 0.44 | 0.80 |
| 2,4,6,8-Tetramethylundec-1-enes | 0.96 | 0.92 | 0.91 | 0.93 | 0.97 | |
| PS | But-3-ene-1,3-diyldibenzene | 0.85 | 0.82 | 0.92 | 0.79 | 0.84 |
| Hex-5-ene-1,3,5-triyltribenzene | 0.95 | 0.92 | 0.93 | 0.92 | 0.94 | |
| PVC | Methylnaphthalenes | 0.89 | 0.46 | 0.90 | 0.47 | 0.93 |
| Fluorene | 0.89 | 0.47 | 0.93 | 0.49 | 0.95 | |
| Anthracene | 0.92 | 0.46 | 0.90 | 0.46 | 0.90 |
| Polymer | Pyrolytic product | Molecular ion/m/z | Diagnostic ion/m/z | Retention time/min | LOD/μg | LOQ/μg |
|---|---|---|---|---|---|---|
| PA6 | ε-Caprolactam | 113 | 113, 85 | 25.73 | 0.041 | 0.137 |
| PC | p-Tertbutylphenol | 150 | 135, 107 | 26.84 | 0.014 | 0.048 |
| p-Isopropenylphenol | 134 | 134, 119, 91 | 27.08 | 0.045 | 0.150 | |
| Bisphenol A | 228 | 213, 119 | 49.88 | 0.017 | 0.056 | |
| PE | C22–C24 terminal dienes | 306, 320, 334 | 55, 82, 96, 69 | 51.31, 53.41, 55.44 | 0.682, 0.515, 0.458 | 2.273, 1.717, 1.527 |
| C22–C24 terminal alkenes | 308, 322, 336 | 97, 83, 57, 69 | 51.50, 53.59, 55.60 | 0.174, 0.169, 0.121 | 0.579, 0.564, 0.403 | |
| C22–C24 alkanes | 310, 324, 338 | 57, 71, 85 | 51.68, 53.76, 55.76 | 0.212, 0.238, 0.187 | 0.707, 0.795, 0.624 | |
| PET | Divinyl terephthalate | 218 | 175, 104, 76 | 35.28 | 0.080 | 0.266 |
| 2-(Benzoyloxy)ethyl vinyl terephthalate | 340 | 297, 149, 105 | 58.71 | 0.085 | 0.282 | |
| PMMA | Methyl methacrylate | 100 | 100, 99 | 7.30 | 0.004 | 0.013 |
| PP | 2,4-Dimethylhept-1-ene | 126 | 70, 55, 57 | 10.47 | 0.023 | 0.078 |
| 2,4,6,8-Tetramethylundec-1-ene (isotactic, heterotactic and syndiotactic) | 210 | 69, 57, 55 | 28.09, 28.37, 28.66 | 0.090, 0.636, 0.159 | 0.302, 2.120, 0.531 | |
| PS | But-3-ene-1,3-diyldibenzene (styrene dimer) | 208 | 91, 130 | 39.53 | 0.008 | 0.027 |
| Hex-5-ene-1,3,5-triyltribenzene (styrene trimer) | 312 | 91, 117 | 55.86 | 0.004 | 0.012 | |
| PVC | Chlorobenzene | 112 | 112, 77 | 10.72 | 2.967 | 9.889 |
| Methylnaphthalenes | 142 | 142, 141, 115 | 26.89, 27.41 | 0.564, 0.567 | 1.881, 1.890 | |
| Fluorene | 166 | 165, 166 | 36.12 | 0.401 | 1.336 | |
| Anthracene | 178 | 178, 176 | 41.38 | 0.193 | 0.642 |
2.2. Spiked sample matrix
Several protocols for the extraction of microplastics from sediments and soils were developed. To test the extraction efficiency of each one, they were conducted on a spiked inorganic sediment matrix (sand) and a more organic-rich soil matrix. Firstly, the sand was furnaced at 450 °C for 4 hours, to remove any potential plastic contamination. However, this could not be replicated for the soil matrix due to its organic content. Instead, the soil was density separated with water to remove any light microplastic, and mixed with 50% furnaced sand to create an organic-rich matrix. Then, approximately 50 g of sand or soil matrix was weighed into a furnaced glass sample vial or jar and between 5 and 100 μg of up to three different plastic standards added. An additional PET standard (granule, Goodfellow Ltd) was also procured and used in the spike and recovery experiment detailed in Section 2.2.2.3. Blanks of furnaced sand were extracted alongside the spiked samples in each case to establish whether any contamination was introduced during sample preparation. Each sample was then thoroughly mixed and freeze dried ready for subsequent analysis.Each spiked sand or soil sample (approximately 50 g) was transferred from its sample jar to a furnaced 250 mL glass beaker. Approximately 100 mL of 1.6 g mL−1 CHKO2 (aq) was added to the beaker and the sample thoroughly mixed with a solvent-cleaned metal spatula. The supernatant was then transferred via pouring to a funnel equipped with latex tubing and a clamp and any immediately settled material was drained back into the beaker. A further aliquot of CHKO2 (aq) was added and the separation repeated. The beaker was thoroughly rinsed with CHKO2 (aq) to ensure any microplastic stuck to the sides was washed into the funnel. The density separation was left to settle overnight. All settled material was drained and the remaining solution filtered through 13 mm GF/A Whatman filters. Multiple filters were used to prevent overloading. The funnel, filters and surrounding glassware were thoroughly rinsed with double-distilled water to ensure all microplastic particles were transferred to the filter paper. All filters were dried at 50 °C overnight before being stored in covered, furnaced, glass Petri dishes.
The soils had more floating matter than the pure sand samples, which could not necessarily be contained on a filter paper. In these cases, the floating matter was also transferred to the glass Petri dish and dried overnight, ready for subsequent organic matter digestion.
2.2.2.1. Organic matter digestion in a crucible. Following the density separation, the spiked soils were subjected to a wet peroxide digestion. Here, the dried filter papers and floating matter were transferred to a sintered glass crucible (porosity grade 5, 1.0–1.6 μm pore size) and Fenton's reagent added. Fenton's reagent comprised equal parts hydrogen peroxide (H2O2, 30% in water) and iron(II) sulfate solution [Fe(II)SO4 (aq), 50 mM] and was added in 5 mL aliquots until all organic matter had been digested (as indicated by a lack of effervescence upon addition). The solution was then removed using a vacuum and the crucible and its contents thoroughly rinsed with double-distilled water, before being transferred to a covered, furnaced, glass Petri dish and dried overnight at 50 °C.
2.2.2.2. Organic matter digestion in a filter apparatus. In this method the dried filters and floating matter from the spiked soils were transferred to a furnaced glass filter holder, equipped with a pre-combusted 25 mm GF/C Whatman filter. Fenton's reagent was added in 5 mL aliquots, as above, until there was no further effervescence upon addition. The solution was then vacuum filtered and the filter paper thoroughly rinsed with double-distilled water. Subsequently, the filter holder was thoroughly rinsed onto a clean filter and all filters and floating matter were transferred to a covered, furnaced, glass Petri dish and dried overnight at 50 °C.
2.2.2.3. Organic matter digestion in a beaker. After density separation, the dried filters and floating matter from the spiked soils were transferred to furnaced 100 mL beakers. Here, Fenton's reagent was added in 5 mL aliquots until there was no further effervescence upon addition. Between additions the beakers were mixed on a shaker table to ensure all matter stayed in contact with the solution. The contents of each beaker were then filtered through pre-combusted GF/C Whatman filters and thoroughly rinsed with double-distilled water. As above, the filter holder was additionally rinsed onto a clean filter paper and everything transferred to a covered, furnaced, glass Petri dish for drying at 50 °C overnight.
Py-GC-MS analyses were performed using a Chemical Data Systems (CDS) 6200 pyroprobe pyrolyser equipped with a DISC module and autosampler and a Tenax-TA trap, attached to a ThermoScientific Trace 1310 gas chromatograph (GC, ThermoScientific, Hemel Hempstead, UK) with an Agilent HP-1 fused capillary column. The column comprised 100% dimethylpolysiloxane, with a 60 m × 0.32 mm i.d. and a 0.25 μm film thickness. The GC was coupled, via a heated transfer line (310 °C), to a ThermoScientific single quadrupole mass spectrometer (MS). Pyrolysis was performed in a quartz tube, positioned inside a platinum filament coil, at 610 °C for 20 s before being flushed by a helium carrier gas (30 mL min−1) to a Tenax-TA trap held at 50 °C. After pyrolysis, the trap was heated to 300 °C, and this temperature was maintained for 4 min. The trap contents were flushed through a 310 °C transfer line to the GC. Injection into the GC occurred using a split–splitless injector with split ratio of 20:1, maintained at a temperature of 310 °C with a 2 mL min−1 helium carrier gas flow. The ramped temperature cycle of the GC oven began at 40 °C for 4 min, before linearly increasing to 310 °C at 4 °C min−1, where the temperature was maintained for 15 min. The MS operated in electron ionisation (EI) mode with an electron energy of 70 eV, and scanning at 5 scan per s from m/z 50–650, with a 2–7 min filament delay. The EI source was held at 300 °C. A summary of the pyrolytic conditions used can be found in Table S1 (ESI†). Thermally assisted hydrolysis and methylation (THM) was performed by adding 10 μL of TMAH (25% in water) directly into the quartz pyrolysis tube and allowing it to evaporate at room temperature for an hour before pyrolysis.
3. Results and discussion
3.1. Analysis of polymer standards by Py-GC-MS
The primary thermal degradation pathways for polymeric compounds are: chain scission, unzipping and side-group scission.34 PP and PE are examples of polymers that primarily undergo chain scission where the main chain-bonds are broken to create terminal radicals. Stabilisation via hydrogen abstraction results in the formation of a terminal alkene and an alkane.35 Subsequent chain scission at the saturated part of the chain produces either two terminal alkenes or an alkane and a terminal alkadiene. In the case of PP this is a series of methyl branched terminal alkenes and dienes, whilst PE pyrolyses to give a characteristic series of terminal dienes, terminal alkenes and alkanes. The addition of TMAH did not affect the pyrolytic behaviour of PP or PE. As PP's most abundant pyrolytic products, 2,4-dimethylhept-1-ene and the three 2,4,6,8-tetramethylundec-1-enes were selected as its diagnostic moieties. The choice for PE is slightly more difficult, as dienes, alkenes and alkanes are also generated on pyrolysis of many natural materials, such as fats and waxes.11,36 In general, these interferences decrease with increasing chain length, as the natural distributions differ to those of PE. Thus, the C22, C23 and C24 terminal dienes, alkenes and alkanes have been selected as diagnostic compounds for PE.11
PMMA, PS and PA6 largely pyrolyse into their monomer units (methyl methacrylate, styrene and ε-caprolactam, respectively), in a process known as unzipping.34,35 No change in the thermal decomposition of PMMA or PS was observed after THM, and therefore the abundant monomer, methyl methacrylate, was chosen as the diagnostic species for PMMA. Styrene, however, is also a pyrolytic product of PVC and PET and some natural products, including chitin.37 Hence, it is not suitable as a diagnostic compound. The other major pyrolytic products, formed via chain scission, are the styrene dimer and trimer. Whilst these are less abundant, neither have been observed in pyrograms of biopolymers and thus are suitable diagnostic moieties.11 In the case of PA6, the monomer unit becomes methylated after the addition of TMAH, producing N-methyl caprolactam. In the literature N-methyl caprolactam has been chosen as the diagnostic species for PA6,11 however it has low abundance here, due to the incomplete methylation of ε-caprolactam. As a result, ε-caprolactam was chosen as the diagnostic compound. This highlights one of the potential problems of using derivatising agents, as varying degrees of derivatisation can alter the peak areas of selected pyrolytic products, and thus impact quantification.
Side-group scission occurs when the polymer has easily cleavable bonds to the side groups, such as in PVC.34 HCl is easily lost to give almost exclusively aromatic pyrolytic products, with the most prominent peaks corresponding to toluene and naphthalene.38 These species are not impacted by the addition of TMAH, although its presence is responsible for the elution of trimethylamine at 16–30 min. The choice of diagnostic moiety for PVC is difficult, as its pyrolysis products are either not specific or not abundant. The two methylnaphthalene isomers have been selected as a compromise, as they are thought to be specific to PVC and are relatively abundant when the signal from both species is combined.
During pyrolysis, PET and PC mainly cleave along their backbones, forming a variety of aromatic carbonyls and alcohols, respectively. After THM, additional pyrolytic products were also observed, namely dimethyl terephthalate for PET and p-methoxy-tert-butylbenzene and 2,2-bis(4′-methoxyphenyl)propane (dimethyl bisphenol A) in the pyrogram of PC. If TMAH is to be used, these two species will be used as diagnostic compounds. If not, divinyl terephthalate and 2-(benzoyloxy)ethyl vinyl terephthalate have been selected as the diagnostic compounds for PET and p-tertbutylphenol, p-isopropenylphenol and bisphenol A selected for PC.
After THM, additional pyrolytic products were observed in the pyrograms of PA6, PC and PET, whilst the other polymers remain unaffected. Whilst the THM pyrogram of PET contains the most abundant product, this is not the case for PC. The peak corresponding to bisphenol A was greatly reduced by the addition of TMAH, due to the production of 2,2-bis(4′-methoxyphenyl)propane. Since there is also a low abundance of the styrene trimer after addition of TMAH to PS, it can be concluded that THM is likely not a beneficial approach for future analyses.
The other potential interference comes from co-polymeric materials. They are very difficult to identify, and their pyrolytic products may contribute to the signal of one of the diagnostic compounds identified above. Thus, it cannot be determined, for example, whether the styrene oligomers detected are from styrene acrylonitrile (SAN), acrylonitrile butadiene styrene (ABS) or from PS.11 Related polymers, such as alkylated PMMA, will also amplify the diagnostic ion peak for PMMA and thus the quantity of polymer measured. The same applies for the inclusion of polybutylene terephthalate (PBT) in the qualification/quantification of PET.
Various internal standards were investigated to determine their suitability for microplastic quantification. As a variety of different plastics and diagnostic moieties were investigated, a mixture of structurally dissimilar internal standards is desirable, to replicate the behaviour of different pyrolytic products. Initially bisphenol A-d10, 5β-cholanic acid, 1,2-dibromobenzene and naphthalene-d8 were investigated but proved unsuccessful. Therefore, a mixture of 5α-androstane, anthracene-d10, 9-fluorenone and pyrene-d10 were chosen and added to each sample and calibration run. In some cases, this improved the linearity of the calibration, whilst for others it was notably worse (Table 1). The most likely explanation for the poor correlation when using 9-fluorenone or 5α-androstane as the internal standard is lack of compound stability. Since the pyrolysis tubes are left uncovered on the autosampler after internal standard addition, this suggests that these standards were evaporating or degrading after addition and are therefore not suitable for quantification.
Therefore, the proposed ions and diagnostic compounds for the calibration of PA6, PC, PE, PET, PMMA, PP, PS and PVC are listed in Table 2. See Fig. 1 for the selected calibration model for each plastic.
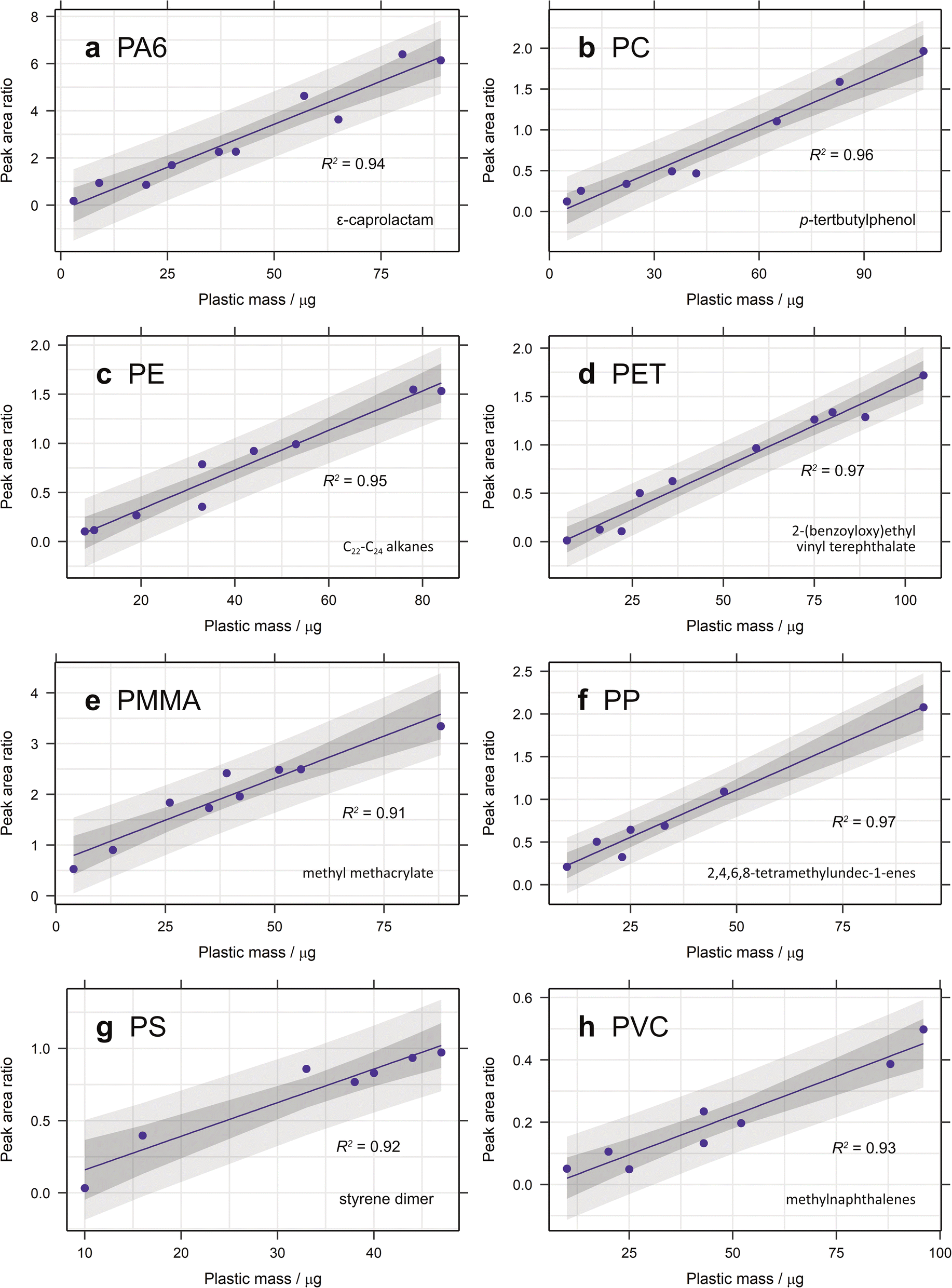 | ||
| Fig. 1 Calibration curves for (a) PA6, (b) PC, (c) PE, (d) PET, (e) PMMA, (f) PP, (g) PS and (h) PVC created from the ion chromatogram peak area ratio of each selected diagnostic compound with anthracene-d10 or pyrene-d10 as the internal standard and 95% confidence and prediction bands plotted. | ||
3.1.2.1. Calibration range. The calibration range used here is restricted by the limits of the pyrolyser and the sensitivity of the microbalance used for polymer addition. The largest amount of organic material that can be pyrolysed is approximately 100 μg, to prevent overloading the system. Some samples of larger polymer masses were analysed and their peak areas added to the calibration curves (Fig. 2). In general, these did not fit with the linear regression established, demonstrating the lack of linearity at higher masses. Carry over between samples is also a problem when analysing samples containing more pyrolysable material, resulting in more blanks between sample runs and increasing analysis time. The lowest point in each calibration is mostly limited by the sensitivity of the microbalance (1 μg) and the user's practical ability to accurately weigh out very small amounts of standard. Realistically, this is about 2–3 μg (depending on the standard) and this forms the lower limit of the calibration range. In future studies, homogenously spiking diatomaceous earth with ground plastic standard could be useful means of extending this calibration range. The extrapolated limit of detection (LOD) and limit of quantification (LOQ) are very low for the selected diagnostic compounds for most polymers, with values below 1 μg in almost all cases (Table 2). For PMMA the LOD is as low as 0.004 μg, demonstrating the sensitivity of this technique. For PP and PVC, however, the LOQ is 2.12 and 1.89 μg, respectively. Nevertheless, given that the lowest point in the calibration curve is unlikely to be this low, these limits should not pose a problem.
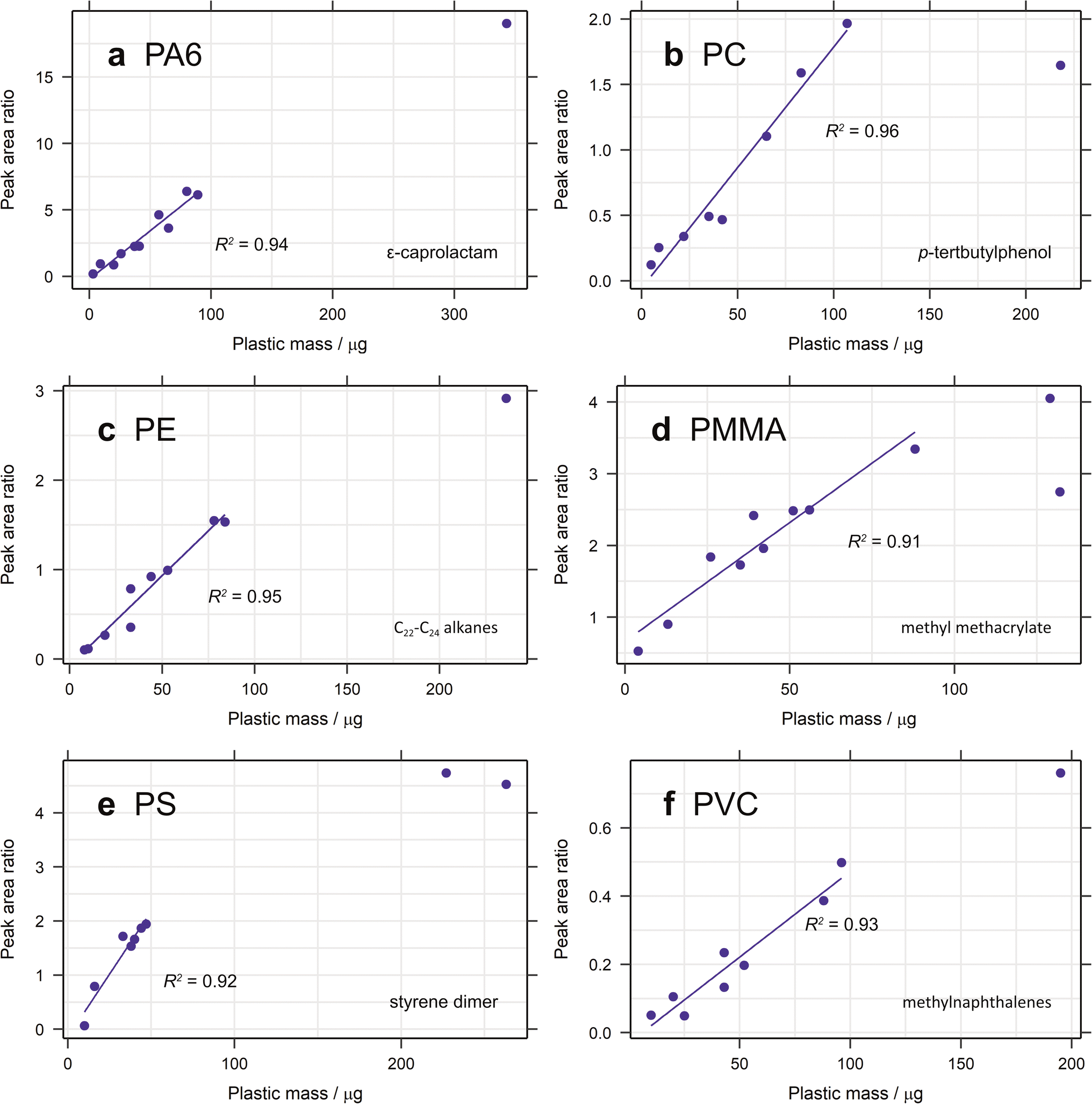 | ||
| Fig. 2 Calibration curves for (a) PA6, (b) PC, (c) PE, (d) PMMA, (e) PS and (f) PVC created from the ion chromatogram peak area ratio of each selected diagnostic compound with anthracene-d10 or pyrene-d10 as the internal standard, with additional standards outside the calibration range added. | ||
3.1.2.2. Calibration stability. Validation standards were run after the initial calibration to test the stability of the calibration over time. Whilst some validations fitted within the prediction bands, others fell outside of the acceptable range, necessitating new calibrations for subsequent samples. For several plastics these differed substantially to the original calibrations. One reason for this could be changes to the internal surfaces of the pyrolyser and GC over time.10,39 The peak areas observed for a given pyrolytic product are affected by its interactions with these surfaces, which in turn are impacted by the contents of samples previously analysed by the instrument. Therefore, the type of samples run between calibrations could influence their values. The resultant instrument maintenance will also have an effect. Differing compounds will be affected differently by this, which could explain why some calibrations are more dissimilar than others. All of this highlights the importance of validation standards and regular calibration. It is therefore recommended that a new calibration be run before each batch of samples.
3.2. Developing a method for the extraction of microplastic from soil and sediment
Most of the pyrolytic breakdown products of PVC are commonly found in the pyrolysis of natural materials.11,40 This makes it challenging to select specific diagnostic compounds for PVC identification and quantification in environmental samples. Benzene40,41 and methylnaphthalenes10,11 have been used for quantification in the literature, but neither are very specific. While chlorobenzene is specific to PVC, its abundance is too low to make it suitable for quantification. Nevertheless, it can still be used to confirm the presence of PVC, as has been done in the literature.40,41 Other abundant peaks in the pyrogram of PVC include biphenyl, fluorene and anthracene. Unfortunately, biphenyl is also a pyrolytic product of PET and PS, and while fluorene and anthracene both calibrate well (Table 1), they were still observed in blanks with an organic-rich matrix. The choice is therefore somewhat of a compromise, and the most appropriate diagnostic compound to use will be sample dependent. Alongside chlorobenzene, all three diagnostic species should still be used to confirm the presence of PVC.
3.2.2.1. Density separation. Initially, the density separation method was tested on spiked, furnaced sand, where it proved to be effective at extracting most plastics (Fig. 3a). All values fell within the prediction bands for each calibration, with the exception of one data point for PE and all those for PET (Fig. S1, ESI†). One explanation for PET not having an acceptable recovery could be electrostatic effects. Since the PET standard used here is a fine powder, the high surface area of the particles makes them susceptible to such effects. The other impact of the small particle size is that they are more difficult to retain when filtering. The pore size of GF/A and GF/C Whatman filters is 1.6 μm and 1.2 μm, respectively, giving a limit to how small a particle can be preserved. The other plastic standards that are powders have higher particle sizes and hence are not affected. A different PET standard is therefore required to test this and future extraction methods.
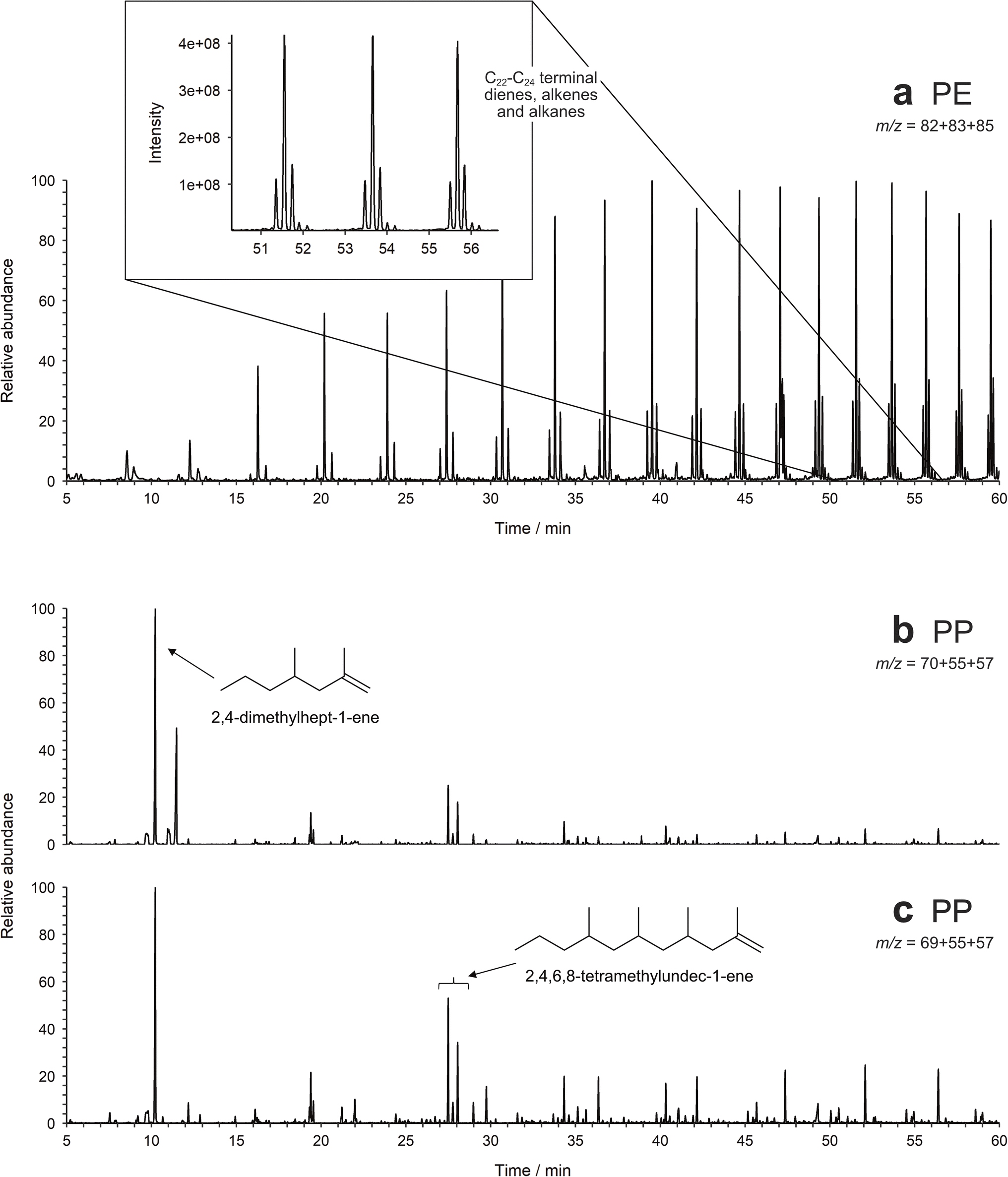 | ||
| Fig. 3 Partial Py-GC-MS chromatograms of (a) sand spiked with PE, extracted using density separation via overnight settling, filtered by m/z 82 + 83 + 85, and (b) and (c) soil spiked with PP, extracted using density separation via overnight settling and hydrogen peroxide digestion in a beaker, filtered by (b) m/z 70 + 55 + 57 and (c) m/z 69 + 55 + 57. | ||
3.2.2.2. Organic matter digestion. The density separation in Section 3.2.2.1 was initially tested in isolation on spiked samples with an inorganic matrix, to determine the efficacy of this part of the method. Now that a successful density separation protocol has been established, the method was applied to spiked samples with a higher organic matter content. For these samples an organic matter digestion was also required, in our case utilising Fenton's reagent.
To limit the amount of glassware used, thereby minimising sample losses and contamination, the organic matter digestion was first performed in a crucible, as utilised by Fischer and Scholz-Böttcher.10 Overall, this method did not appear to be very effective. Samples still contained large quantities of organic material (determined via visual inspection), causing them to be split between more pyrolysis runs, increasing run time, error and the LOD. The main reason for the incomplete digestion was difficulty in ensuring that the organic matter stayed in contact with the peroxide solution, since it drained through the crucible. It therefore needed to be replenished more, making the method wasteful, time-consuming and ineffective. The second challenge with this method was transferring the matter quantitatively from the crucible into a Petri dish. It was challenging to ensure than the smallest particles were removed, without damaging the sintered glass. It was therefore not surprising that the recovery of plastic from this method was poor, with only 60% of the spiked soils containing the plastic of interest.
To reduce the impact of matrix effects, a more effective method for removing organic matter is required. Since one problem with the crucible method is the loss of microplastic during transfer from the crucible, the digestion was instead repeated in a filter funnel. Here, everything could be washed directly onto the filter paper, facilitating easy transfer to a Petri dish. In addition, more Fenton's reagent was retained as there was a smaller surface area to drain through. There was a concern that Fenton's reagent might affect the filter paper's structural integrity, causing the loss of microplastic particles. Nevertheless, the method proved to be effective at recovering the microplastic added to the soil samples (Fig. S2, ESI†). The spiked plastic was observed within the prediction bands in all but two samples, with only PE and PS each missing from one sample. There were some problems with Fenton's reagent leaking out of the side of the filter holder after being left to digest overnight. Additionally, as for the crucible method above, repeated applications of Fenton's reagent were required to maintain the digestion conditions, as it constantly drained through the filter. Therefore, this method was deemed to be quick and effective, as it reduces transfer between glassware and the potential for contamination and particle losses, but only for samples with low amounts of organic matter.
For more organic-rich samples, a new approach was required and the more traditional method of performing the digestion in a beaker was used. Whilst it was more difficult to rinse the beaker to ensure all matter was transferred to the filter apparatus, there was no danger of particle losses through the filter during digestion and the sample remained in contact with the Fenton's reagent for the entire digestion period. The method is also effective (Fig. 3b and 4), popular in literature and easy to perform in bulk.7,42 The recovery of most plastics is slightly lower than when no organic matter digestion is used, however, therefore it is recommended that organic matter digestion should only be applied when necessary. A new PET standard, purchased as a granule, was also utilised in this spike and recovery experiment. As can be seen in Fig. 4, the recovered quantity of PET falls within the prediction bands, demonstrating that it likely is the small particle size of the previous standard that prevented its recovery. Indeed, this illustrates that PET can be extracted and quantified using this method, should the particle size be above the pore size of the filter paper.
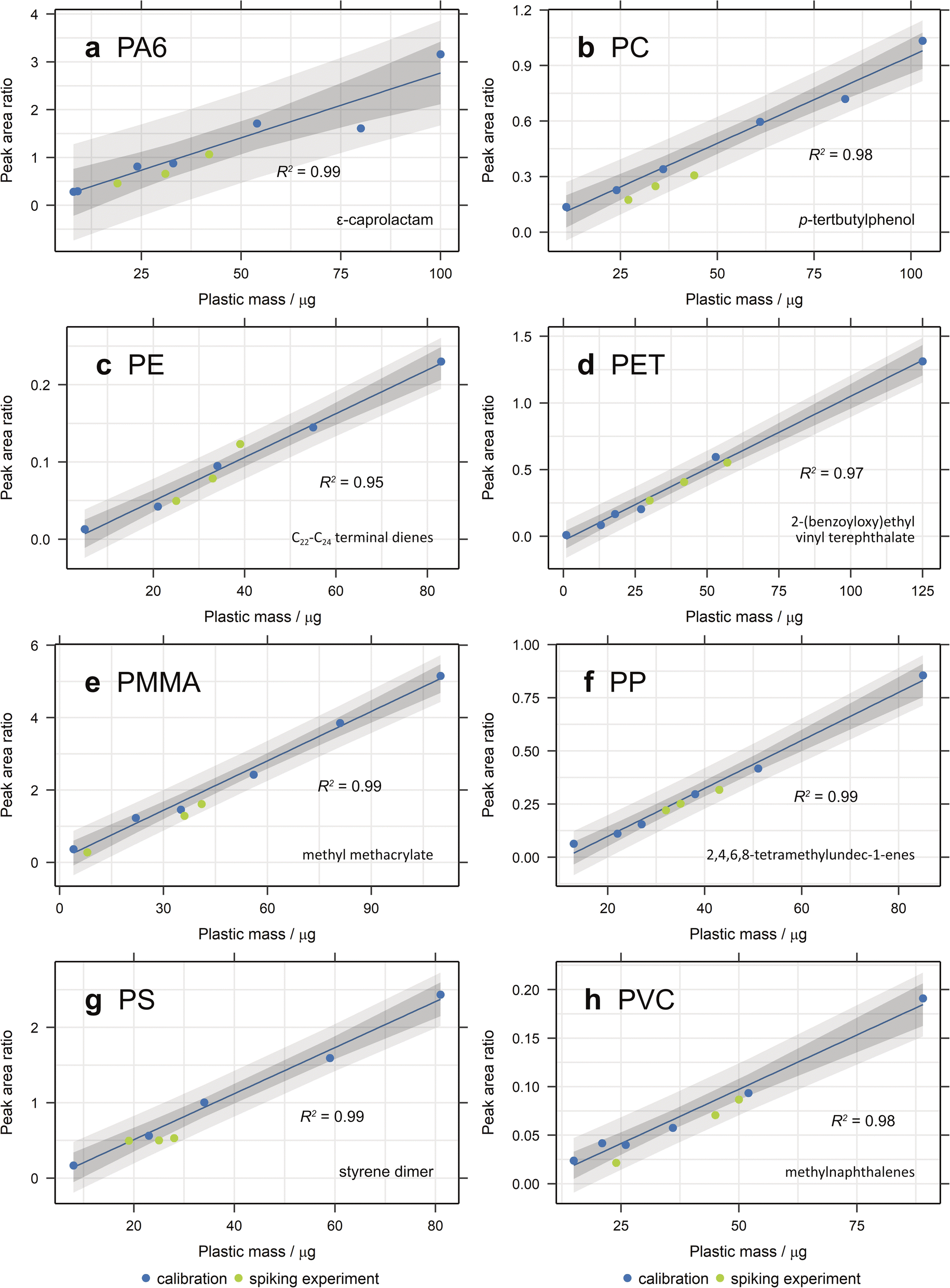 | ||
| Fig. 4 Recovery of (a) PA6, (b) PC, (c) PE, (d) PET, (e) PMMA, (f) PP, (g) PS and (h) PVC from spiked samples after density separation via overnight settling and hydrogen peroxide digestion in a beaker. Calibration curves were created from the ion chromatogram peak area ratio of each selected diagnostic compound with anthracene-d10 or pyrene-d10 as the internal standard and 95% confidence and prediction bands plotted. | ||
4. Conclusion
There are numerous techniques applied to the analysis of microplastic, each with its challenges and disadvantages, with accurate quantification being of particular difficulty. Py-GC-MS is increasingly being utilised for microplastic identification in complex environmental samples, and its true potential is only just being realised. Specific and abundant diagnostic moieties have been identified for the eight polymer types analysed, suggesting that they will be identifiable in more complex media. Since the addition of TMAH appears to decrease the detection sensitivity of PC and PS, THM should not be used. There is also the potential for incomplete methylation with TMAH, which can impact the accuracy of quantification. Of the different types of plastics investigated, all have at least one diagnostic compound that demonstrated linearity with the mass of polymer, suggesting that quantitative analysis is possible. Internal standardisation also improved on these calibrations.A variety of microplastic extraction methods were tested here, with a view to determining an effective extraction protocol for microplastic from terrestrial samples. An appropriate method for inorganic and organic matter removal was determined, specifically density separation via overnight settling, followed by hydrogen peroxide digestion in a beaker. Organic matter digestion in the filter apparatus also proved effective when only a small quantity of organic matter needs digesting. These methods were verified through the use of spiked sand and soils. Limitations in the selection of appropriate diagnostic compounds were identified for some plastics, with matrix interferences affecting PE, PS and PVC. New diagnostic compounds were chosen for both PE and PVC, although the choice for PVC is sample dependent. The presence of some plastic diagnostic compounds as natural pyrolysis products emphasises the need to use all identified diagnostic compounds of a given plastic to confirm its identity in a sample pyrogram. The application of these pyrolytic and extraction protocols to spiked samples demonstrates the applicability of the developed method to detect the presence of microplastics in environmental samples.
Data availability
The data supporting this article are either part of the main article or have been included as part of the ESI.† Raw data files used to produce the results are freely available on request and will be retained by the University of Bristol for 10+ years in line with UKRI guidelines.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was possible due to funding awarded to R. H. P. from the Natural Environment Research Council (NERC) GW4+ Doctoral Training Partnership (NE/S007504/1). NERC are also thanked for partial funding of the National Environment Isotope Facility (NEIF; NE/V003917/1). We are grateful to A. Kuhl and H. Whelton (University of Bristol, UK) for their maintenance of instruments within NEIF.References
- M. C. Rillig, R. Ingraffia and A. A. D. Machado, Microplastic Incorporation into Soil in Agroecosystems, Front. Plant Sci., 2017, 8, 1805 CrossRef PubMed.
- F. Corradini, P. Meza, R. Eguiluz, F. Casado, E. Huerta-Lwanga and V. Geissen, Evidence of microplastic accumulation in agricultural soils from sewage sludge disposal, Sci. Total Environ., 2019, 671, 411–420 CrossRef CAS PubMed.
- X. Mao, Y. Xu, Z. Cheng, Y. Yang, Z. Guan, L. Jiang and K. Tang, The impact of microplastic pollution on ecological environment: a review, FBL, 2022, 27, 46 CAS.
- S. Lambert, C. Scherer and M. Wagner, Ecotoxicity testing of microplastics: considering the heterogeneity of physicochemical properties, Integrated Environ. Assess. Manag., 2017, 13, 470–475 CrossRef CAS.
- L. Van Cauwenberghe, L. Devriese, F. Galgani, J. Robbens and C. R. Janssen, Microplastics in sediments: a review of techniques, occurrence and effects, Mar. Environ. Res., 2015, 111, 5–17 CrossRef CAS.
- A. A. Koelmans, P. E. Redondo-Hasselerharm, N. H. M. Nor, V. N. de Ruijter, S. M. Mintenig and M. Kooi, Risk assessment of microplastic particles, Nat. Rev. Mater., 2022, 7, 138–152 CrossRef.
- A. B. Silva, A. S. Bastos, C. I. L. Justino, J. P. da Costa, A. C. Duarte and T. A. P. Rocha-Santos, Microplastics in the environment: challenges in analytical chemistry - a review, Anal. Chim. Acta, 2018, 1017, 1–19 CrossRef CAS PubMed.
- G. Renner, T. C. Schmidt and J. Schram, Analytical methodologies for monitoring micro(nano)plastics: Which are fit for purpose?, Curr. Opin. Environ. Sci. Health, 2018, 1, 55–61 CrossRef.
- F. Akoueson, C. Chbib, S. Monchy, I. Paul-Pont, P. Doyen, A. Dehaut and G. Duflos, Identification and quantification of plastic additives using pyrolysis-GC/MS: a review, Sci. Total Environ., 2021, 773, 145073 CrossRef CAS.
- M. Fischer and B. M. Scholz-Böttcher, Microplastics analysis in environmental samples-recent pyrolysis-gas chromatography-mass spectrometry method improvements to increase the reliability of mass-related data, Anal. Methods, 2019, 11, 2489–2497 RSC.
- M. Fischer and B. M. Scholz-Böttcher, Simultaneous Trace Identification and Quantification of Common Types of Microplastics in Environmental Samples by Pyrolysis-Gas Chromatography-Mass Spectrometry, Environ. Sci. Technol., 2017, 51, 5052–5060 CrossRef CAS PubMed.
- G. S. Groenewold, K. M. Johnson, S. C. Fox, C. Rae, C. A. Zarzana, B. R. Kersten, S. M. Rowe, T. L. Westover, G. L. Gresham, R. M. Emerson and A. N. Hoover, Pyrolysis Two-Dimensional GC-MS of Miscanthus Biomass: Quantitative Measurement Using an Internal Standard Method, Energy Fuel, 2017, 31, 1620–1630 CrossRef CAS.
- G. S. Groenewold, B. Hodges, A. N. Hoover, C. L. Li, C. A. Zarzana, K. Rigg and A. E. Ray, Signatures of Biologically Driven Hemicellulose Modification Quantified by Analytical Pyrolysis Coupled with Multidimensional Gas Chromatography Mass Spectrometry, ACS Sustain. Chem. Eng., 2020, 8, 1989–1997 CrossRef CAS.
- S. C. Moldoveanu, Pyrolysis GC/MS, present and future - (recent past and present needs), J. Microcolumn Sep., 2001, 13, 102–125 CrossRef CAS.
- J. C. J. Bart, Polymer/additive analysis by flash pyrolysis techniques, J. Anal. Appl. Pyrol., 2001, 58, 3–28 CrossRef.
- A. Gomiero, K. B. Øysæd, T. Agustsson, N. van Hoytema, T. van Thiel and F. Grati, First record of characterization, concentration and distribution of microplastics in coastal sediments of an urban fjord in south west Norway using a thermal degradation method, Chemosphere, 2019, 227, 705–714 CrossRef CAS PubMed.
- M. F. Silva, M. T. Domenech-Carbo, L. Fuster-Lopez, S. Martin-Rey and M. F. Mecklenburg, Determination of the plasticizer content in poly(vinyl acetate) paint medium by pyrolysis-silylation-gas chromatography-mass spectrometry, J. Anal. Appl. Pyrol., 2009, 85, 487–491 CrossRef CAS.
- D. He, Y. Luo, S. Lu, M. Liu, Y. Song and L. Lei, Microplastics in soils: analytical methods, pollution characteristics and ecological risks, Trac. Trends Anal. Chem., 2018, 109, 163–172 CrossRef CAS.
- F. Corradini, H. Bartholomeus, E. H. Lwanga, H. Gertsen and V. Geissen, Predicting soil microplastic concentration using vis-NIR spectroscopy, Sci. Total Environ., 2019, 650, 922–932 CrossRef CAS PubMed.
- J. S. Hanvey, P. J. Lewis, J. L. Lavers, N. D. Crosbie, K. Pozo and B. O. Clarke, A review of analytical techniques for quantifying microplastics in sediments, Anal. Methods, 2017, 9, 1369–1383 RSC.
- M. Blasing and W. Amelung, Plastics in soil: analytical methods and possible sources, Sci. Total Environ., 2018, 612, 422–435 CrossRef PubMed.
- J. Pinto da Costa, V. Reis, A. Paço, M. Costa, A. C. Duarte and T. Rocha-Santos, Micro(nano)plastics – analytical challenges towards risk evaluation, Trac. Trends Anal. Chem., 2019, 111, 173–184 CrossRef CAS.
- E. Hendrickson, E. C. Minor and K. Schreiner, Microplastic Abundance and Composition in Western Lake Superior As Determined via Microscopy, Pyr-GC/MS, and FTIR, Environ. Sci. Technol., 2018, 52, 1787–1796 CrossRef CAS.
- R. Hurley, J. Woodward and J. J. Rothwell, Microplastic contamination of river beds significantly reduced by catchment-wide flooding, Nat. Geosci., 2018, 11, 251–257 CrossRef CAS.
- J. Masura, J. Baker, G. Foster and C. Arthur, Laboratory Methods for the Analysis of Microplastics in the Marine Environment, NOAA Marine Debris Program National, 2015, 1–39 Search PubMed.
- Y. Fan, K. Zheng, Z. Zhu, G. Chen and X. Peng, Distribution, sedimentary record, and persistence of microplastics in the Pearl River catchment, China, Environ. Pollut., 2019, 251, 862–870 CrossRef CAS PubMed.
- M. T. Nuelle, J. H. Dekiff, D. Remy and E. Fries, A new analytical approach for monitoring microplastics in marine sediments, Environ. Pollut., 2014, 184, 161–169 CrossRef CAS PubMed.
- M. Cole, H. Webb, P. K. Lindeque, E. S. Fileman, C. Halsband and T. S. Galloway, Isolation of microplastics in biota-rich seawater samples and marine organisms, Sci. Rep., 2014, 4, 4528 CrossRef PubMed.
- W. Courtene-Jones, B. Quinn, F. Murphy, S. F. Gary and B. E. Narayanaswamy, Optimisation of enzymatic digestion and validation of specimen preservation methods for the analysis of ingested microplastics, Anal. Methods, 2017, 9, 1437–1445 RSC.
- E. Dümichen, P. Eisentraut, C. G. Bannick, A. K. Barthel, R. Senz and U. Braun, Fast identification of microplastics in complex environmental samples by a thermal degradation method, Chemosphere, 2017, 174, 572–584 CrossRef.
- L. Hermabessiere, C. Himber, B. Boricaud, M. Kazour, R. Amara, A. L. Cassone, M. Laurentie, I. Paul-Pont, P. Soudant, A. Dehaut and G. Duflos, Optimization, performance, and application of a pyrolysis-GC/MS method for the identification of microplastics, Anal. Bioanal. Chem., 2018, 410, 6663–6676 CrossRef CAS.
- A. Gomiero, K. B. Øysæd, L. Palmas and G. Skogerbø, Application of GCMS-pyrolysis to estimate the levels of microplastics in a drinking water supply system, J. Hazard. Mater., 2021, 416, 125708 CrossRef CAS.
- F. Stock, C. Kochleus, B. Bänsch-Baltruschat, N. Brennholt and G. Reifferscheid, Sampling techniques and preparation methods for microplastic analyses in the aquatic environment – a review, Trac. Trends Anal. Chem., 2019, 113, 84–92 CrossRef CAS.
- T. P. Wampler, Introduction to pyrolysis-capillary gas chromatography, J. Chromatogr., A, 1999, 842, 207–220 CrossRef CAS PubMed.
- T. P. Wampler, Applied Pyrolysis Handbook, 2nd edn, 2007 Search PubMed.
- J. David, Z. Steinmetz, J. Kučerík and G. E. Schaumann, Quantitative Analysis of Poly(ethylene terephthalate) Microplastics in Soil via Thermogravimetry-Mass Spectrometry, Anal. Chem., 2018, 90, 8793–8799 CrossRef CAS PubMed.
- D. Fabbri, C. Trombini and I. Vassura, Analysis of Polystyrene in Polluted Sediments by Pyrolysis-Gas Chromatography-Mass Spectrometry, J. Chromatogr. Sci., 1998, 36, 600–604 CAS.
- E. P. Chang and R. Salovey, Pyrolysis of Polyvinyl-Chloride, J. Polym. Sci. Pol. Chem., 1974, 12, 2927–2941 CrossRef CAS.
- M. E. Seeley and J. M. Lynch, Previous successes and untapped potential of pyrolysis-GC/MS for the analysis of plastic pollution, Anal. Bioanal. Chem., 2023, 415, 2873–2890 CrossRef CAS PubMed.
- D. Fabbri, D. Tartari and C. Trombini, Analysis of poly(vinyl chloride) and other polymers in sediments and suspended matter of a coastal lagoon by pyrolysis-gas chromatography-mass spectrometry, Anal. Chim. Acta, 2000, 413, 3–11 CrossRef CAS.
- D. Fabbri, Use of pyrolysis-gas chromatography/mass spectrometry to study environmental pollution caused by synthetic polymers: a case study: the Ravenna Lagoon, J. Anal. Appl. Pyrol., 2001, 58–59, 361–370 CrossRef CAS.
- A. A. Horton, A. Walton, D. J. Spurgeon, E. Lahive and C. Svendsen, Microplastics in freshwater and terrestrial environments: evaluating the current understanding to identify the knowledge gaps and future research priorities, Sci. Total Environ., 2017, 586, 127–141 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4va00269e |
| This journal is © The Royal Society of Chemistry 2025 |